Chapter 156 Primary Vitreoretinal Lymphoma
Introduction
PCNSL is a variant of extranodal non-Hodgkin lymphoma (NHL) that is predominantly a high-grade B-cell malignancy associated with a median survival ranging from 1–8 years depending on factors such as age and Karnofsky performance status.1 It originates in the brain parenchyma, spinal cord, leptomeninges, and eyes.2–4 Formerly used descriptors such as “reticulum cell sarcoma” and “microgliomatosis” are no longer preferred as both misleadingly imply that the lymphoma arises from transformed reticulum or microglial cells.5,6
Epidemiology
PCNSL accounts for 1–2% of all cases of lymphoma and approximately 3–5% of primary tumors of the central nervous system.7–9 From 1973–1997, the incidence of PCNSL increased threefold, partially owing to the corresponding rise in individuals with human immunodeficiency virus (HIV) infection and possibly due to improved diagnosis and/or detection leading to more cases being ascertained.7 Rates have since stabilized with an age-adjusted incidence of PCNSL in the USA of 4.8 per million population reported in 2002.7 PCNSL is therefore a rare disease affecting fewer than 2000 individuals annually in the USA.4
While PVRL is frequently seen in the setting of PCNSL, the exact incidence is unknown. Between 1999 and 2002, approximately 100 new cases of PVRL were reported in the USA.10 The association between PVRL and PCNSL is variable, with CNS disease manifesting prior to, following, or occurring simultaneously with ocular presentation. Approximately 25% of patients with PCNSL will have concomitant PVRL.3 In contrast, 56–85% of individuals with PVRL will ultimately develop central nervous system involvement.11–14 Among immunocompetent individuals, the peak incidence of PVRL occurs between ages 50 and 70 years. In the immunocompromised population, PVRL occurs in younger individuals.15–17 There are no known racial or ethnic associations. In a large, multicenter retrospective review of 221 patients with PCNSL with intraocular involvement, a slight female predominance (57%) was observed.18
Etiology and pathogenesis
PCNSL is believed to originate from late-germinal center or post-germinal center lymphoid cells, however the neurotropic mechanism by which these cells localize to the CNS remains uncertain.19 It has been hypothesized that the trafficking of lymphoma cells from the brain to the eye and vice versa involves either invasion of the optic nerve, seeding through shared venous drainage of the brain and eye, or common integrin expression of both organs.4 While there are no known risk factors for the development of PCNSL in immunocompetent individuals, immunosuppression secondary to HIV, congenital immunodeficiency, and iatrogenic immunosuppression are risk factors.20 PCNSL develops in as many as 6% of patients with acquired immune deficiency syndrome (AIDS).21,22 Epstein–Barr virus infection of B-lymphocytes in the absence of T-suppressor lymphocytes results in uncontrolled lymphocytic proliferation. Rare cases of PVRL may be secondary to human T-cell lymphotropic virus type 1 (HTLV-1) infection.23
Clinical findings
Ophthalmic findings
While individuals may be asymptomatic, more than half present with painless, decreased visual acuity or floaters.13,24,25 Of those who are asymptomatic, many are diagnosed when ophthalmic screening examination is performed for individuals with known PCNSL. The clinical findings are bilateral in 80% of cases, but are frequently asymmetric.11
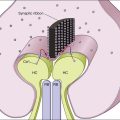
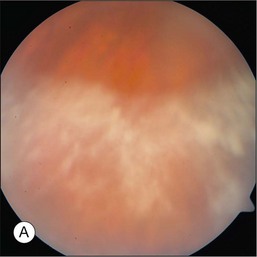
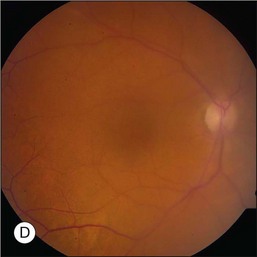
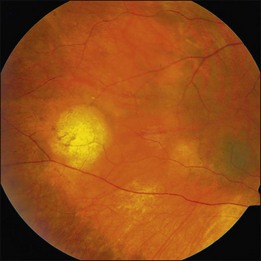
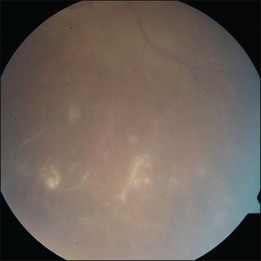
The hallmark of PVRL is the presence of fine vitreous cells or clumps of cells and sub-retinal pigment epithelium (RPE) deposits comprised of aggregated lymphoma cells (Fig. 156.1).26–28 When present, these lesions are considered pathognomonic.28 Anterior segment findings including keratic precipitates, iris nodules, aqueous cells, and flare are frequently encountered but are nonspecific.17,25 On fundus examination, focal, multifocal, or diffuse choroidal, retinal, or chorioretinal infiltrates can be seen in the presence or absence of vitreous cells (Fig. 156.2).26,29 Other less commonly reported findings include: perivasculitis (Fig. 156.3),13 retinal artery occlusion,30 exudative retinal detachment,31 multifocal “punched-out” lesions at the level of the RPE,32 and optic atrophy.33
Central nervous system findings
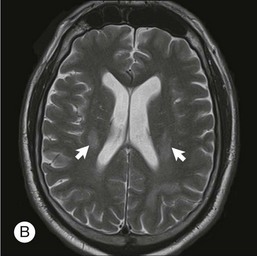
PCNSL is an aggressive malignancy and the diagnosis is generally established within several months of the onset of symptoms. This is in sharp contrast to PVRL where the diagnosis is often delayed, by as long as 2 years according to one tertiary care center, secondary to being misdiagnosed as posterior uveitis or other masquerading diseases.25 In PCNSL, personality changes are a common presenting symptom as the frontal lobe is the most frequent region of brain parenchymal involvement. Seizures are a rare feature of this disease.
The lesions in PCNSL tend to be periventricular in location thus allowing access to the cerebrospinal fluid (CSF) and leptomeninges. Leptomeningeal involvement occurs in approximately 40% of cases.34 Rarely, PCNSL limited to the spinal cord is observed.35 The lesions can be multifocal, particularly in immunocompromised individuals.
Diagnosis
In the absence of prior CNS disease, the diagnosis of PVRL is based upon histopathological and cytological features. Several techniques exist for biopsy to establish the diagnosis including vitreous biopsy, retinal biopsy, and subretinal biopsy. Most commonly, diagnostic 23-gauge pars plana vitrectomy is performed. Proper surgical techniques and handling are critical as aspirates are generally of low cellularity and fragile lymphoma cells are prone to lysis during sample collection. Techniques vary by center and expertise; however, it is generally recommended that an undiluted vitreous sample of approximately 1–2 mL be collected prior to the start of the saline infusion during vitrectomy.20,36 Following collection of the first sample, the infusion fluid is started, and a second diluted vitreous specimen using gentle vitreous cutting can be collected in a separate syringe.37 Some centers also submit the vitreous cassette as a third sample.38 It is recommended that samples be delivered to the laboratory, without fixative, within one hour of surgery.36,39 Repeated vitreous biopsies are commonly performed in order to establish the diagnosis. In a study of 20 patients with PVRL, three eyes required more than one vitreous biopsy before the diagnosis was confirmed.40 More recently, there has been an interest in using 25-gauge sutureless vitrectomy for diagnostic purposes and for improved patient comfort and decreased operative times. This technique has been used with success in some centers.38
When chorioretinal or subretinal lesions are present, a retinal or subretinal biopsy may be performed. A subretinal biopsy technique using a standard three-port pars plana vitrectomy approach has been described.41 An initial core vitrectomy is performed allowing access to the subretinal infiltrate. Vitreous separation is then induced and thorough vitrectomy is performed over the biopsy site. An incision in the overlying retina just large enough to allow entrance of the vitreous cutter is then made. Suction tubing is then inserted through the retinectomy incision and with gentle cutting action, several samples are obtained. Subretinal aspirates should be placed in a mild cytofixative, such as herpes-glutamic acid buffer mediated organic solvent protection effect (HOPE) fixation or CytoLyt® (Cytyc Corporation).36 In a series of 84 patients who underwent pars plana vitrectomy, additional chorioretinal biopsy with extended immunohistochemistry and polymerase chain reaction (PCR) gene rearrangement studies were performed in three patients after vitrectomy was nondiagnostic. This technique yielded a definitive diagnosis of PVRL in each case.42
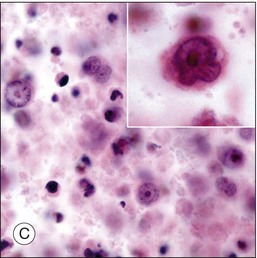
Although diagnostic techniques vary depending upon local expertise, cytology remains the first-line for diagnosis. Access to a pathologist or cytopathologist with experience in ophthalmic diagnosis is essential. The majority of cases of PVRL are diffuse large B-cell lymphomas. In a multicenter retrospective review of 221 individuals with histologically diagnosed PCNSL with ocular involvement, the subtype was large B-cell in 73%, T-cell in 2%, and not specified in 25%.18 The cytologic features are characteristic. PVRL cells are typically 2–4 times larger than normal lymphocytes, are pleomorphic, and have scant cytoplasm.43 The nuclei may be round, oval, or indented, with conspicuous nuclear membranes, occasional fingerlike protrusions, and multiple, prominent, eccentrically located nucleoli. Mitoses are frequently observed.24 With the use of electron microscopy, intranuclear inclusions, cytoplasmic crystalloids, pseudopodal extensions of the cytoplasm, cytosomes, and autophagic vacuoles can be identified.44
As vitreous biopsy is not diagnostic in all cases, supplemental techniques can be helpful in confirming the diagnosis of PVRL. Immunohistochemistry is useful for identifying markers for leukocytes (CD45), B-cells (CD20, CD79a, PAX-5), T-cells (CD45RO), and macrophages (CD68).36 Additionally, clonality can be established with the use of antibodies directed against κ and λ light chains.14,43,45 Flow cytometry provides a means of quantitatively assessing the proportion of cells in a given sample that demonstrate these immunohistochemical markers. PCR gene rearrangement studies can detect monoclonality of the heavy chain variable (V), diversity (D), and joining (J) immunoglobulin gene segments and are therefore helpful in establishing the diagnosis, however most vitreous biopsy samples are inadequate for PCR.46,47 Evaluation for gene rearrangement by PCR is most successful in tissue biopsy specimens in which DNA has been isolated by laser capture microdissection.46,48 Measurement of IL-6 and IL-10 in either aqueous or vitreous fluid can also facilitate diagnosis, although an elevated IL-10/IL-6 ratio is not specific for PVRL.49 Measurement of these cytokines has no role in the routine evaluation of suspected PVRL.
Central nervous system involvement
Due to the high correlation between PVRL and PCNSL, all patients diagnosed with ophthalmic disease should be evaluated by an experienced neuro-oncologist. This evaluation should include neuroimaging and CSF studies. Gadolinium-enhanced MRI of the brain is the imaging modality of choice in evaluating individuals with suspected PCNSL. High-volume (>10 cc) lumbar puncture for protein, glucose, cytology, and flow cytometry is particularly important as leptomeningeal involvement is present in up to 40% of individuals with PCNSL.34 CSF can demonstrate lymphocytic pleocytosis, elevated protein concentration, and low or normal glucose levels. Malignant cells in the CSF are diagnostic for PCNSL. Flow cytometry, however, is the most sensitive and specific marker of CNS lymphoma.50 Additional diagnostic procedures should include CT scans of the chest, abdomen, and pelvis; testicular ultrasound in elderly men; and HIV testing.
Differential diagnosis
Delay in diagnosis of PVRL is common due to the nonspecific ophthalmic manifestations that can masquerade as inflammatory, other neoplastic, and infectious conditions. In one series of 32 patients with histologically confirmed PVRL, the average interval between the onset of ocular symptoms and diagnosis was 21 months.14 The differential diagnosis includes diseases characterized by chronic anterior and posterior uveitis including sarcoidosis, syphilis, and tuberculosis, birdshot retinochoroidopathy, multifocal chorioretinitis, acute posterior multifocal placoid pigmentary epitheliopathy, serpiginous choroiditis, and punctate inner choroidopathy.51
Treatment
Ophthalmic treatment
Consensus guidelines for treatment selection for PVRL have not been firmly established and therefore depend upon patient factors as well as local expertise. When disease is unilateral and limited to the eye, local delivery of intravitreal chemotherapy with either methotrexate or rituximab has been shown to be effective and well tolerated.52–55 Methotrexate as single agent intravitreal chemotherapy for PVRL was first reported in seven eyes of four patients in 1997. All patients achieved complete remission without serious ocular side-effects.52 In a larger series of 26 eyes in 16 patients with PVRL at two centers, methotrexate (400 µg/0.1 mL saline) was injected intravitreally twice weekly as an induction phase, then weekly for 1 month in one center and weekly for 2 months in the other center as consolidation therapy, followed by once monthly for 1 year as maintenance therapy. All eyes achieved remission following a maximum of 12 injections. During a median follow-up of 18.5 months, a total of three patients from the first center developed relapsed PVRL and were treated again following the same protocol with complete remission.55 The largest series reported to date included 44 eyes in 26 patients treated with the same induction–consolidation–maintenance regimen. Clinical remission was achieved following an average of 6.4 injections and 95% of eyes required 13 injections or less to be free of clinically detectable disease.56 None of the patients in this series developed relapse after a follow-up period ranging from 41 to 107 months.56 The most commonly reported ocular side effects associated with intravitreal injection of methotrexate include cataract, conjunctival hyperemia, and transient keratopathy ranging from mild punctuate epithelial erosions to severe epitheliopathy.55,56 Symptoms typically resolved when intravitreal injections were no more than once monthly.55,56 Early evidence suggests that rituximab may have fewer side-effects and require fewer injections to achieve remission, however further investigation is required.53,57 Larger, collaborative studies are required to firmly establish guidelines regarding dose, frequency of administration, and duration of treatment.
Prior to acceptance of intravitreal chemotherapy for PVRL, bilateral external beam radiotherapy (EBRT) was widely employed. EBRT remains an important therapy, particularly in patients with bilateral involvement, for those who may not tolerate intravitreal chemotherapy, and in individuals who find it difficult to return for multiple injections (Fig. 156.4). EBRT to both eyes is more commonly performed due to the high incidence of bilateral involvement, however in patients with unilateral disease, EBRT limited to the affected eye is preferred.
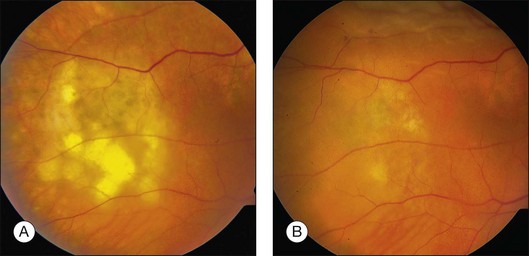
Radiation doses have varied between centers and have ranged from 30 Gy to 50 Gy with an average dose of approximately 40 Gy given in fractions of 1.5–2.0 Gy.13,58–63 Response to radiation and relapse rates are difficult to assess due to differences in standard practices and because patients are frequently treated with combinations of therapy. In one series of 19 eyes treated by radiation, nine achieved clinical remission although relapse eventually occurred in two eyes.58 In a second series of 13 patients treated by radiotherapy with or without chemotherapy, three had only a partial response and two developed recurrent disease.61 In another series of 21 eyes in 12 patients, initial treatment included EBRT and systemic chemotherapy (six patients), systemic chemotherapy alone (four patients), EBRT alone (one patient), and no treatment (one patient). No ocular recurrence was observed in those receiving EBRT. Two patients who did not receive EBRT suffered ocular relapse.63 Radiation-related side-effects include optic neuropathy, retinopathy, conjunctivitis, dry eyes, cataracts, and glaucoma.64 In most cases, EBRT is reserved for individuals under age 65 years due to the high incidence of neurotoxicity.
Central nervous system treatment
Historically, WBRT was treatment of choice for patients with PCNSL. While WBRT prolonged median survival from 4 months in untreated patients to approximately 12–18 months65, it was also associated with significant neurotoxicity in older individuals.66 Currently, most individuals with PCNSL are treated with systemic chemotherapy. High-dose methotrexate (MTX) is most commonly used, either as a single agent or as part of a combination regimen. High doses (3–8 g/m2) are required to reach cytotoxic concentrations in the CSF and to treat occult leptomeningeal disease.9 Other regimens include high-dose cytarabine (HDAC) and ifosfamide.18 Alternative methods of chemotherapy delivery include high-dose chemotherapy with autologous stem cell rescue and intra-arterial chemotherapy with blood–brain barrier disruption (BBBD) using mannitol infusion.65 BBBD is associated with a high rate of maculopathy.9 In selected patients with positive CSF cytology, intrathecal (IT) methotrexate should be included in the chemotherapy regimen. IT delivery of methotrexate is also used in some individuals who are receiving lower systemic doses of methotrexate over longer periods of time.9 When both ophthalmic and CNS disease are present, a combined therapeutic approach must be taken. Collaboration between ophthalmologists and neuro-oncologists is essential.
Prognosis
Unfortunately, the majority of patients (56–85%) with PVRL subsequently develop CNS disease.11,12,14,67 In a large, multicenter, retrospective study of 221 immunocompetent patients with CNS lymphoma with ocular involvement, the median progression-free survival and overall survival in patients was reported to be 18 and 31 months, respectively.18 Age less than 60 years at the time of diagnosis and high initial performance status are recognized as favorable prognostic factors in PCNSL.68,69 Conversely, involvement of the brainstem and leptomeninges portends a poor prognosis.69 The presence or absence of retinal involvement in the setting of existing CNS disease does not appear to be an influential factor.69 While large retrospective studies have shown that ocular treatment appears to improve disease control, no overall survival benefit of ocular therapy has been proven.18
1 Abrey LE, Ben-Porat L, Panageas KS, et al. Primary central nervous system lymphoma: the Memorial Sloan-Kettering Cancer Center prognostic model. J Clin Oncol. 2006;24:5711–5715.
2 Henry JM, Heffner RR, Jr., Dillard SH, et al. Primary malignant lymphomas of the central nervous system. Cancer. 1974;34:1293–1302.
3 Hochberg FH, Miller DC. Primary central nervous system lymphoma. J Neurosurg. 1988;68:835–853.
4 Pe’er J, Hochberg FH, Foster CS. Clinical review: treatment of vitreoretinal lymphoma. Ocul Immunol Inflamm. 2009;17:299–306.
5 Kinney TD, Adam RD. Reticulum cell sarcoma of the brain. Arch Neurol Psychiatr. 1943;50:552–564.
6 Russell DS, Marshall AHE, Smith FB. Microgliomatosis. Brain. 1948;71:1–15.
7 Olson JE, Janney CA, Rao RD, et al. The continuing increase in the incidence of primary central nervous system non-Hodgkin lymphoma: a surveillance, epidemiology, and end results analysis. Cancer. 2002;95:1504–1510.
8 Gerstner ER, Batchelor TT. Primary central nervous system lymphoma. Arch Neurol. 2010;67:291–297.
9 Ahluwalia MS, Peereboom DM. Primary central nervous system lymphoma. Curr Treat Options Neurol. 2010;12:347–359.
10 Chan CC, Buggage RR, Nussenblatt RB. Intraocular lymphoma. Curr Opin Ophthalmol. 2002;13:411–418.
11 Peterson K, Gordon KB, Heinemann MH, et al. The clinical spectrum of ocular lymphoma. Cancer. 1993;72:843–849.
12 Cassoux N, Merle-Beral H, Leblond V, et al. Ocular and central nervous system lymphoma: clinical features and diagnosis. Ocul Immunol Inflamm. 2000;8:243–250.
13 Char DH, Ljung BM, Miller T, et al. Primary intraocular lymphoma (ocular reticulum cell sarcoma) diagnosis and management. Ophthalmology. 1988;95:625–630.
14 Freeman LN, Schachat AP, Knox DL, et al. Clinical features, laboratory investigations, and survival in ocular reticulum cell sarcoma. Ophthalmology. 1987;94:1631–1639.
15 Babu K, Murthy KR, Krishnakumar S. Two successive ocular malignancies in the same eye of a HIV-positive patient: a case report. Ocul Immunol Inflamm. 2010;18:101–103.
16 Mathai A, Lall A, Jain R, et al. Systemic non-Hodgkin’s lymphoma masquerading as Vogt-Koyanagi-Harada disease in an HIV-positive patient. Clin Experiment Ophthalmol. 2006;34:280–282.
17 Rajagopal R, Harbour JW. Diagnostic testing and treatment choices in primary vitreoretinal lymphoma. Retina. 2011;31:435–440.
18 Grimm SA, McCannel CA, Omuro AM, et al. Primary CNS lymphoma with intraocular involvement: International PCNSL Collaborative Group Report. Neurology. 2008;71:1355–1360.
19 DeAngelis LM. Primary central nervous system lymphoma. Curr opinion in neurology. 1999;12:687–691.
20 Singh AD, Lewis H, Schachat AP. Primary lymphoma of the central nervous system. Ophthalmol Clin North Am. 2005;18:199–207.
21 Stanton CA, Sloan B, 3rd., Slusher MM, et al. Acquired immunodeficiency syndrome-related primary intraocular lymphoma. Arch Ophthalmol. 1992;110:1614–1617.
22 Mittra RA, Pulido JS, Hanson GA, et al. Primary ocular Epstein-Barr virus-associated non-Hodgkin’s lymphoma in a patient with AIDS: a clinicopathologic report. Retina. 1999;19:45–50.
23 Marshall AG, Pawson R, Thom M, et al. HTLV-I associated primary CNS T-cell lymphoma. J Neurol Sci. 1998;158:226–231.
24 Whitcup SM, de Smet MD, Rubin BI, et al. Intraocular lymphoma. Clinical and histopathologic diagnosis. Ophthalmology. 1993;100:1399–1406.
25 Akpek EK, Ahmed I, Hochberg FH, et al. Intraocular-central nervous system lymphoma: clinical features, diagnosis, and outcomes. Ophthalmology. 1999;106:1805–1810.
26 Ridley ME, McDonald HR, Sternberg P, et al. Retinal manifestations of ocular lymphoma (reticulum cell sarcoma). Ophthalmology. 1992;99:1153–1161.
27 Dean JM, Novak MA, Chan CC, et al. Tumor detachments of the retinal pigment epithelium in ocular/ central nervous system lymphoma. Retina. 1996;16:47–56.
28 Gass JD, Sever RJ, Grizzard WS, et al. Multifocal pigment epithelial detachments by reticulum cell sarcoma. A characteristic funduscopic picture. Retina. 1984;4:135–143.
29 Corriveau C, Easterbrook M, Payne D. Lymphoma simulating uveitis (masquerade syndrome). Can J Ophthalmol. 1986;21:144–149.
30 Gass JD, Trattler HL. Retinal artery obstruction and atheromas associated with non-Hodgkin’s large cell lymphoma (reticulum cell sarcoma). Arch Ophthalmol. 1991;109:1134–1139.
31 Michelson JB, Michelson PE, Bordin GM, et al. Ocular reticulum cell sarcoma. Presentation as retinal detachment with demonstration of monoclonal immunoglobulin light chains on the vitreous cells. Arch Ophthalmol. 1981;99:1409–1411.
32 Lang GK, Surer JL, Green WR, et al. Ocular reticulum cell sarcoma. Clinicopathologic correlation of a case with multifocal lesions. Retina. 1985;5:79–86.
33 Purvin V, Van Dyk HJ. Primary reticulum cell sarcoma of the brain presenting as steroid-responsive optic neuropathy. J Clin Neuroophthalmol. 1984;4:15–23.
34 Balmaceda C, Gaynor JJ, Sun M, et al. Leptomeningeal tumor in primary central nervous system lymphoma: recognition, significance, and implications. Ann Neurol. 1995;38:202–209.
35 Hautzer NW, Aiyesimoju A, Robitaille Y. “Primary” spinal intramedullary lymphomas: a review. Ann Neurol. 1983;14:62–66.
36 Coupland SE. Vitreous biopsy: specimen preparation and interpretation. Acta Cytol. 2012;21:61–71.
37 Margolis R, Brasil OF, Lowder CY, et al. Vitrectomy for the diagnosis and management of uveitis of unknown cause. Ophthalmology. 2007;114:1893–1897.
38 Yeh S, Weichel ED, Faia LJ, et al. 25-Gauge transconjunctival sutureless vitrectomy for the diagnosis of intraocular lymphoma. Br J Ophthalmol. 2010;94:633–638.
39 Rishi K, Font RL, Chevez-Barrios P. Diagnostic yield of liquid-based cytology, immunophenotyping and molecular techniques in lymphomas and other entities in vitrectomy specimens. Invest Ophthalmol Vis Sci. 2004;45:1072.
40 Katai N, Kuroiwa S, Fujimori K, et al. Diagnosis of intraocular lymphoma by polymerase chain reaction. Graefes Arch Clin Exp Ophthalmol. 1997;235:431–436.
41 Bechrakis NE, Foerster MH, Bornfeld N. Biopsy in indeterminate intraocular tumors. Ophthalmology. 2002;109:235–242.
42 Coupland SE, Bechrakis NE, Anastassiou G, et al. Evaluation of vitrectomy specimens and chorioretinal biopsies in the diagnosis of primary intraocular lymphoma in patients with Masquerade syndrome. Graefes Arch Clin Exp Ophthalmol. 2003;241:860–870.
43 Farkas T, Harbour JW, Davila RM. Cytologic diagnosis of intraocular lymphoma in vitreous aspirates. Acta Cytol. 2004;48:487–491.
44 Kim EW, Zakov ZN, Albert DM, et al. Intraocular reticulum cell sarcoma: a case report and literature review. Graefes Arch Clin Exp Ophthalmol. 1979;209:167–178.
45 Zaldivar RA, Martin DF, Holden JT, et al. Primary intraocular lymphoma: clinical, cytologic, and flow cytometric analysis. Ophthalmology. 2004;111:1762–1767.
46 Chan CC, Shen D, Nussenblatt RB, et al. Detection of molecular changes in primary intraocular lymphoma by microdissection and polymerase chain reaction. Diagnostic molecular pathology. Am J Surg Pathol, part B. 1998;7:63–64.
47 Chan CC, Gonzales JA. Classification of lymphomas. Primary intraocular lymphoma, 1st ed. Hackensack, NJ: World Scientific; 2007.
48 Shen DF, Zhuang Z, LeHoang P, et al. Utility of microdissection and polymerase chain reaction for the detection of immunoglobulin gene rearrangement and translocation in primary intraocular lymphoma. Ophthalmology. 1998;105:1664–1669.
49 Akpek EK, Maca SM, Christen WG, et al. Elevated vitreous interleukin-10 level is not diagnostic of intraocular-central nervous system lymphoma. Ophthalmology. 1999;106:2291–2295.
50 Ahluwalia MS, Wallace PK, Peereboom DM. Flow cytometry as a diagnostic tool in lymphomatous or leukemic meningitis: ready for prime time? Cancer. 2011. Oct 24: 22025088 [Epub ahead of print]
51 Singh AD, Lewis H, et al. Lymphoma of the retina and CNS. In: Singh A, Damato BE, Pe’er J, et al. Clinical ophthalmic oncology. Philadelphia: Elsevier; 2007:372–377.
52 Fishburne BC, Wilson DJ, Rosenbaum JT, et al. Intravitreal methotrexate as an adjunctive treatment of intraocular lymphoma. Arch Ophthalmol. 1997;115:1152–1156.
53 Kitzmann AS, Pulido JS, Mohney BG, et al. Intraocular use of rituximab. Eye (Lond). 2007;21:1524–1527.
54 Itty S, Pulido JS. Rituximab for intraocular lymphoma. Retina. 2009;29:129–132.
55 Smith JR, Rosenbaum JT, Wilson DJ, et al. Role of intravitreal methotrexate in the management of primary central nervous system lymphoma with ocular involvement. Ophthalmology. 2002;109:1709–1716.
56 Frenkel S, Hendler K, Siegal T, et al. Intravitreal methotrexate for treating vitreoretinal lymphoma: 10 years of experience. Br J Ophthalmol. 2008;92:383–388.
57 Ohguro N, Hashida N, Tano Y. Effect of intravitreous rituximab injections in patients with recurrent ocular lesions associated with central nervous system lymphoma. Arch Ophthalmol. 2008;126:1002–1003.
58 Margolis L, Fraser R, Lichter A, et al. The role of radiation therapy in the management of ocular reticulum cell sarcoma. Cancer. 1980;45:688–692.
59 Siegel MJ, Dalton J, Friedman AH, et al. Ten-year experience with primary ocular ‘reticulum cell sarcoma’ (large cell non-Hodgkin’s lymphoma). Br J Ophthalmol. 1989;73:342–346.
60 Hoffman PM, McKelvie P, Hall AJ, et al. Intraocular lymphoma: a series of 14 patients with clinicopathological features and treatment outcomes. Eye (Lond). 2003;17:513–521.
61 Hormigo A, Abrey L, Heinemann MH, et al. Ocular presentation of primary central nervous system lymphoma: diagnosis and treatment. Br J Haematol. 2004;126:202–208.
62 Isobe K, Ejima Y, Tokumaru S, et al. Treatment of primary intraocular lymphoma with radiation therapy: a multi-institutional survey in Japan. Leuk Lymphoma. 2006;47:1800–1805.
63 Berenbom A, Davila RM, Lin HS, et al. Treatment outcomes for primary intraocular lymphoma: implications for external beam radiotherapy. Eye (Lond). 2007;21:1198–1201.
64 Jahnke K, Thiel E, Abrey LE, et al. Diagnosis and management of primary intraocular lymphoma: an update. Clin Ophthalmol. 2007;1:247–258.
65 Deangelis LM, Hormigo A. Treatment of primary central nervous system lymphoma. Semin Oncol. 2004;31:684–692.
66 Correa DD, DeAngelis LM, Shi W, et al. Cognitive functions in survivors of primary central nervous system lymphoma. Neurology. 2004;62:548–555.
67 Char DH, Ljung BM, Deschenes J, et al. Intraocular lymphoma: immunological and cytological analysis. Br J Ophthalmol. 1988;72:905–911.
68 Nasir S, DeAngelis LM. Update on the management of primary CNS lymphoma. Oncology (Williston Park). 2000;14:228–244.
69 Blay JY, Conroy T, Chevreau C, et al. High-dose methotrexate for the treatment of primary cerebral lymphomas: analysis of survival and late neurologic toxicity in a retrospective series. J Clin Oncol. 1998;16:864–871.