49
Primary Immunodeficiencies
• The genetic basis has been determined for >150 primary immunodeficiencies.
• Many immunodeficiency syndromes present with dermatologic findings that can facilitate early diagnosis (Table 49.1; Figs. 49.1–49.8); these features may be divided into three categories:
Table 49.1
Cutaneous findings in primary immunodeficiency (ID) disorders.
Infectious conditions are in darker blue shading and non-infectious conditions in lighter blue shading.
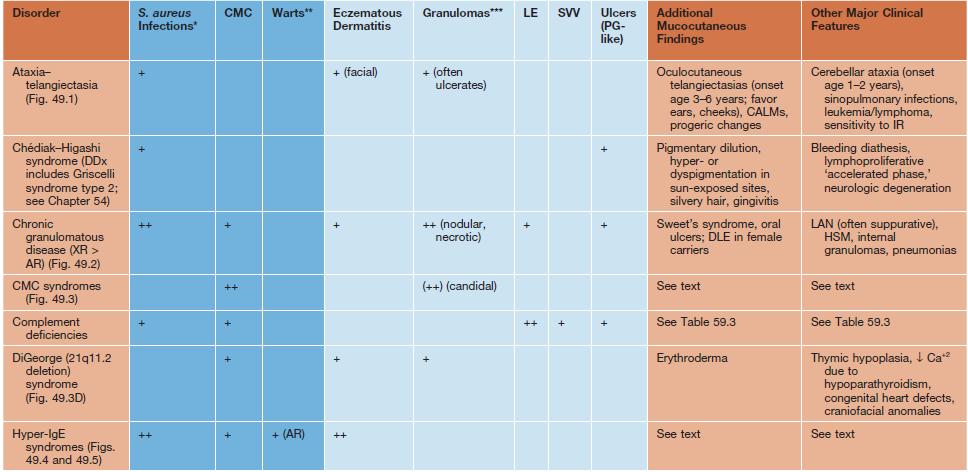
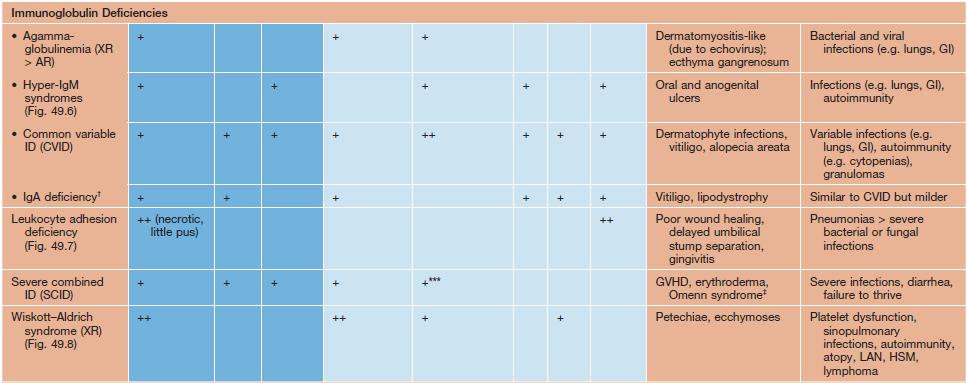

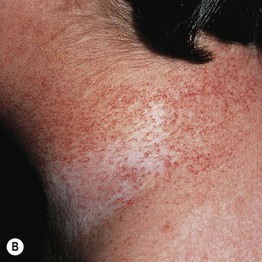
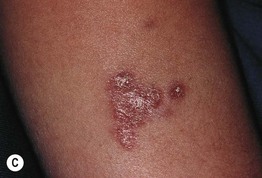
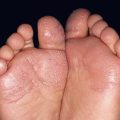
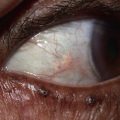
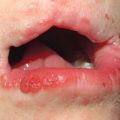
Fig. 49.1 Ataxia–telangiectasia. A Characteristic linear telangiectasias on the bulbar conjunctivae. B Extensive telangiectasias on the neck of a young woman. C Persistent granulomatous plaques on the leg of a child. These lesions often ulcerate and are difficult to manage. A, Courtesy, Jean L. Bolognia, MD; B, C, Courtesy, Amy Paller, MD.
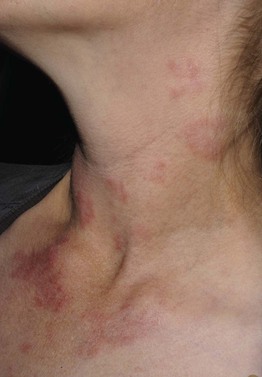
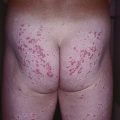
Fig. 49.2 Chronic granulomatous disease. Lupus erythematosus-like annular plaques on the neck. Courtesy, Edward Cowen, MD.
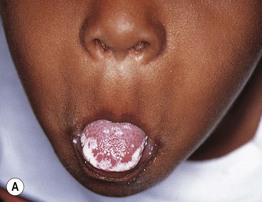
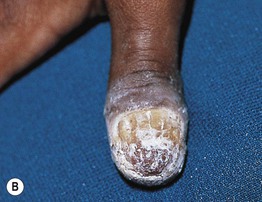
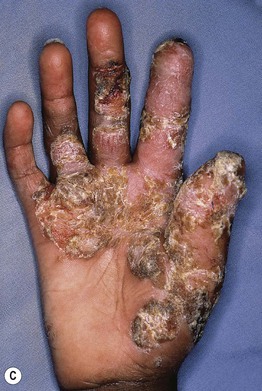
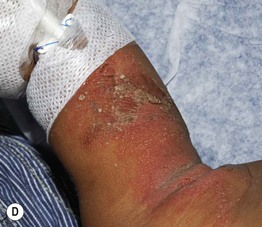
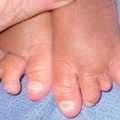
Fig. 49.3 Chronic mucocutaneous candidiasis (CMC). Non-syndromic CMC presenting as (A) extensive, recalcitrant thrush on the tongue of a 5-year-old child; (B) marked onychodystrophy with significant paronychial swelling and erythema. C Non-syndromic CMC presenting as crusted granulomatous plaques on the palm. D Recurrent cutaneous candidiasis in an infant with DiGeorge syndrome. Note the erythema, pustules, and scale-crust near the site of an intravenous line on the arm. A, B, Courtesy, Amy Paller, MD. D, Courtesy, Julie V. Schaffer, MD.
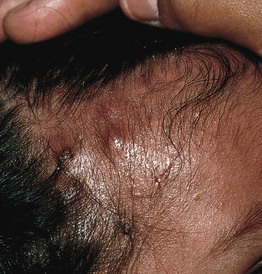
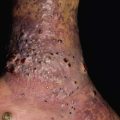
Fig. 49.4 Classic hyperimmunoglobulin E syndrome. Several mildly erythematous, slightly purulent ‘cold’ abscesses on the forehead and scalp of an infant. Courtesy, Amy Paller, MD.
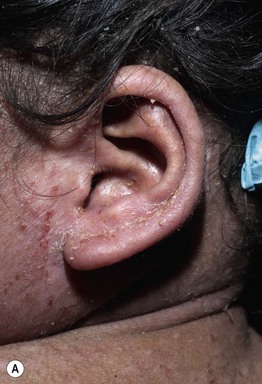
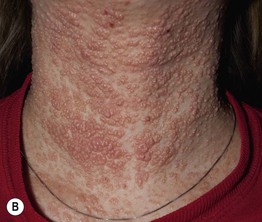
Fig. 49.5 Autosomal recessive hyperimmunoglobulin E syndrome due to DOCK8 deficiency. A Widespread eczematous dermatitis. B Extensive molluscum contagiosum. Courtesy, Edward Cowen, MD.
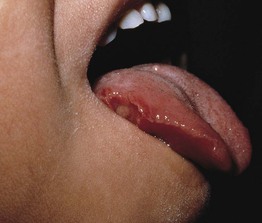
Fig. 49.6 Hyperimmunoglobulin M syndrome. Painful oral ulceration. Reprinted with permission from Schachner L, Hansen R (Eds.). Pediatric Dermatology, 4th edn. London: Mosby, 2011.
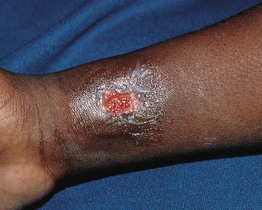
Fig. 49.7 Leukocyte adhesion deficiency type I. This 7-year-old boy was scratched by his sister, resulting in a large gaping wound that healed poorly. Reprinted with permission from Schachner L, Hansen R (Eds.). Pediatric Dermatology, 4th edn. London: Mosby, 2011.
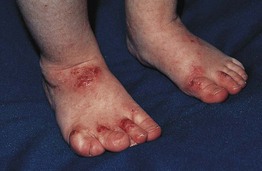
Fig. 49.8 Wiskott–Aldrich syndrome. This young boy presented with widespread, severe atopic dermatitis. Following successful treatment with hematopoietic stem cell transplantation, his dermatitis virtually cleared. Reprinted with permission from Schachner L, Hansen R (Eds.). Pediatric Dermatology, 4th edn. London: Mosby, 2011.
• In addition to extracutaneous infections with unusual organisms and increased frequency (e.g. pneumonia ≥2 times or otitis media ≥4 times yearly) or severity, signs of immunodeficiency in children may include failure to thrive, chronic diarrhea, lymphadenopathy (or lack of expected lymph nodes), and hepatosplenomegaly.
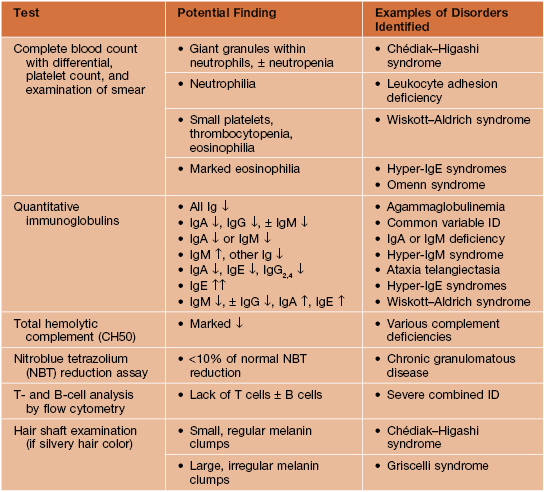
• Initial laboratory evaluation for patients suspected to have an immunodeficiency is outlined in Table 49.2.
Table 49.2
Initial laboratory evaluation for a patient suspected to have a primary immunodeficiency (ID).
Chronic Mucocutaneous Candidiasis (CMC)
• Group of immunodeficiencies characterized by recurrent and severe infections of the skin, nails, and mucous membranes with Candida albicans, together with variable autoimmunity and susceptibility to other infections (see Table 49.1).
• Clinical manifestations include recalcitrant oral thrush, dystrophic nails, and granulomatous plaques with scale-crust favoring the scalp, face, and skin folds (see Fig. 49.3).
• The autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy syndrome (APECED) is caused by mutations (usually with autosomal recessive inheritance) in the autoimmune regulator gene (AIRE), which result in failure to delete autoreactive T cells in the thymus; patients present with a variety of autoimmune disorders (e.g. hypoparathyroidism, hypoadrenocorticism, alopecia areata) in addition to CMC.
Complement Disorders
• Clinical manifestations of various complement deficiencies are presented in Table 49.3.
Table 49.3
Complement disorders.
Unless otherwise specified, refers to homozygous/biallelic deficiencies.
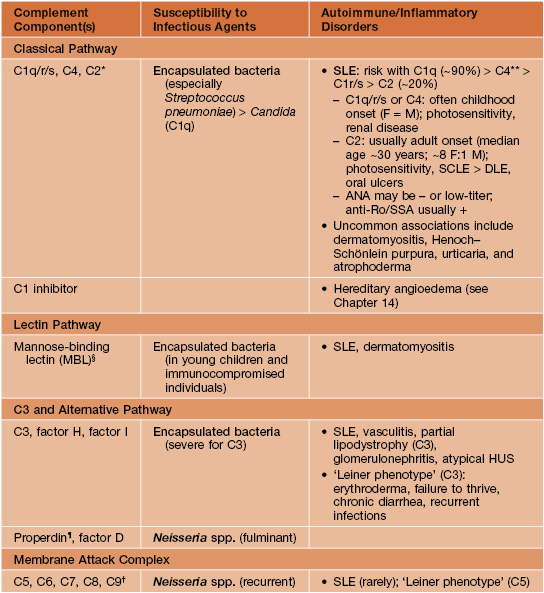
* C2 deficiency is the most common homozygous complement disorder.
** For complete deficiency of C4A and C4B; homozygous C4A deficiency is associated with an increased (but lower) risk of SLE.
§ Very low penetrance.
¶ X-linked recessive, so occurs primarily in male patients.
† Prevalence of ~1:1000 in Japan; milder than other etiologies.
DLE, discoid lupus erythematosus; F, female; HUS, hemolytic–uremic syndrome; M, male; SCLE, subacute cutaneous lupus erythematosus.
Hyperimmunoglobulin E Syndromes (HIESs)
• HIESs feature eczematous dermatitis, recurrent staphylococcal skin (including ‘cold’ abscesses and impetigo) and respiratory tract infections, and markedly elevated serum IgE levels (usually > 2000 IU/ml) (see Figs. 49.4 and 49.5A).
• Classic HIES has autosomal dominant inheritance and is caused by mutations in the STAT3 gene, which encodes a signaling protein that promotes production of cytokines (e.g. IL-6, IL-10, IL-17, IL-22) important to fighting infections at epithelial surfaces and controlling inflammation.
• An autosomal recessive form of HIES (AR-HIES) can be caused by mutations in the dedicator of cytokinesis 8 (DOCK8) gene.
– Characterized by severe viral (e.g. warts, molluscum contagiosum, HSV, VZV; see Fig. 49.5B) and opportunistic infections, other atopic manifestations, CNS vasculitis, and increased risk of mucocutaneous SCC as well as lymphoma.
For further information see Ch. 60. From Dermatology, Third Edition.