PART 15: Immune-Mediated, Inflammatory, and Rheumatologic Disorders
SECTION 1 |
THE IMMUNE SYSTEM IN HEALTH AND DISEASE |
372e |
Introduction to the Immune System |
DEFINITIONS
• Adaptive immune system—recently evolved system of immune responses mediated by T and B lymphocytes. Immune responses by these cells are based on specific antigen recognition by clonotypic receptors that are products of genes that rearrange during development and throughout the life of the organism. Additional cells of the adaptive immune system include various types of antigen-presenting cells.
• Antibody—B cell–produced molecules encoded by genes that re-arrange during B cell development consisting of immunoglobulin heavy and light chains that together form the central component of the B cell receptor for antigen. Antibody can exist as B cell–surface antigen-recognition molecules or as secreted molecules in plasma and other body fluids.
• Antigens—foreign or self-molecules that are recognized by the adaptive and innate immune systems resulting in immune cell triggering, T cell activation, and/or B cell antibody production.
• Antimicrobial peptides—small peptides <100 amino acids in length that are produced by cells of the innate immune system and have anti-infectious agent activity.
• Apoptosis—the process of programmed cell death whereby signaling through various “death receptors” on the surface of cells (e.g., tumor necrosis factor [TNF] receptors, CD95) leads to a signaling cascade that involves activation of the caspase family of molecules and leads to DNA cleavage and cell death. Apoptosis, which does not lead to induction of inordinate inflammation, is to be contrasted with cell necrosis, which does lead to induction of inflammatory responses.
• Autoimmune diseases—diseases such as systemic lupus erythematosus and rheumatoid arthritis in which cells of the adaptive immune system such as autoreactive T and B cells become overreactive and produce self-reactive T cell and antibody responses.
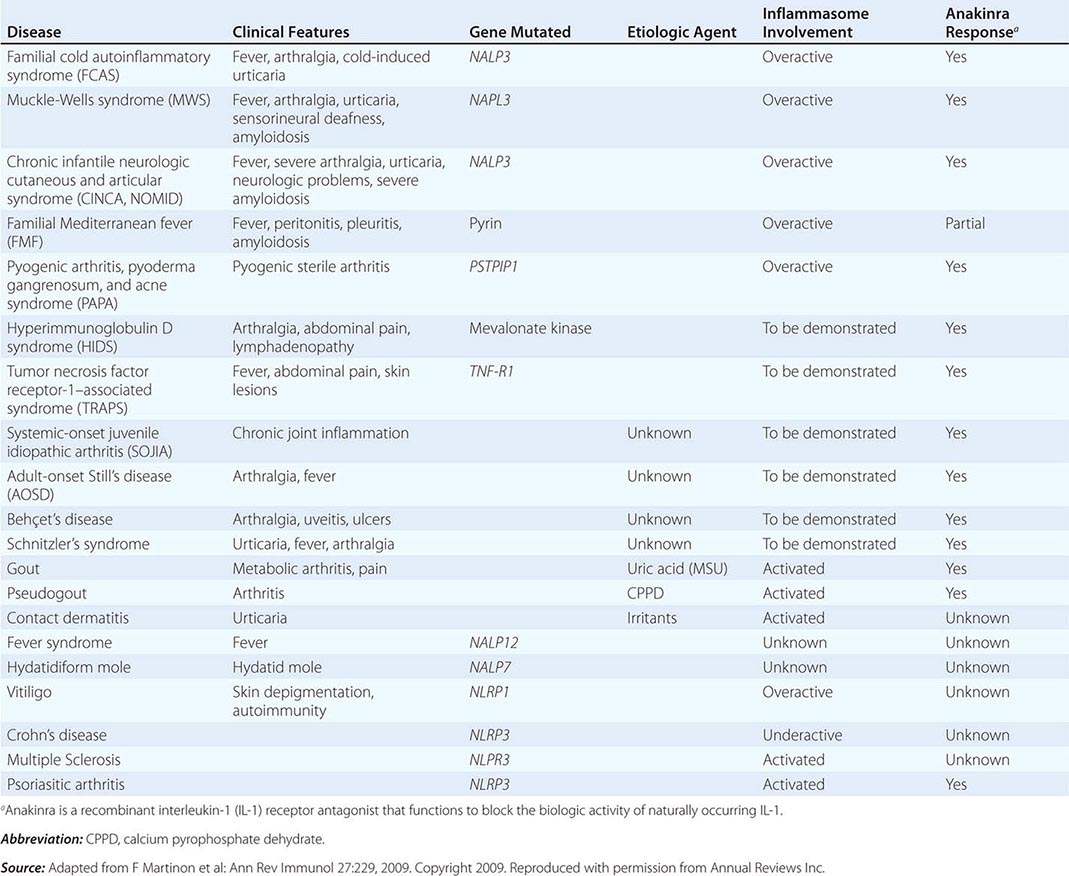
• Autoinflammatory diseases—hereditary disorders such as hereditary periodic fevers (HPFs) characterized by recurrent episodes of severe inflammation and fever due to mutations in controls of the innate inflammatory response, i.e., the inflammasome (see below and Table 372e-6). Patients with HPFs also have rashes and serosal and joint inflammation, and some can have neurologic symptoms. Autoinflammatory diseases are different from autoimmune diseases in that evidence for activation of adaptive immune cells such as autoreactive B cells is not present.
• B cell receptor for antigen—complex of surface molecules that rearrange during postnatal B cell development, made up of surface immunoglobulin (Ig) and associated Ig αβ chain molecules that recognize nominal antigen via Ig heavy- and light-chain variable regions, and signal the B cell to terminally differentiate to make antigen-specific antibody.
• B lymphocytes—bone marrow-derived or bursal-equivalent lymphocytes that express surface immunoglobulin (the B cell receptor for antigen) and secrete specific antibody after interaction with antigen.
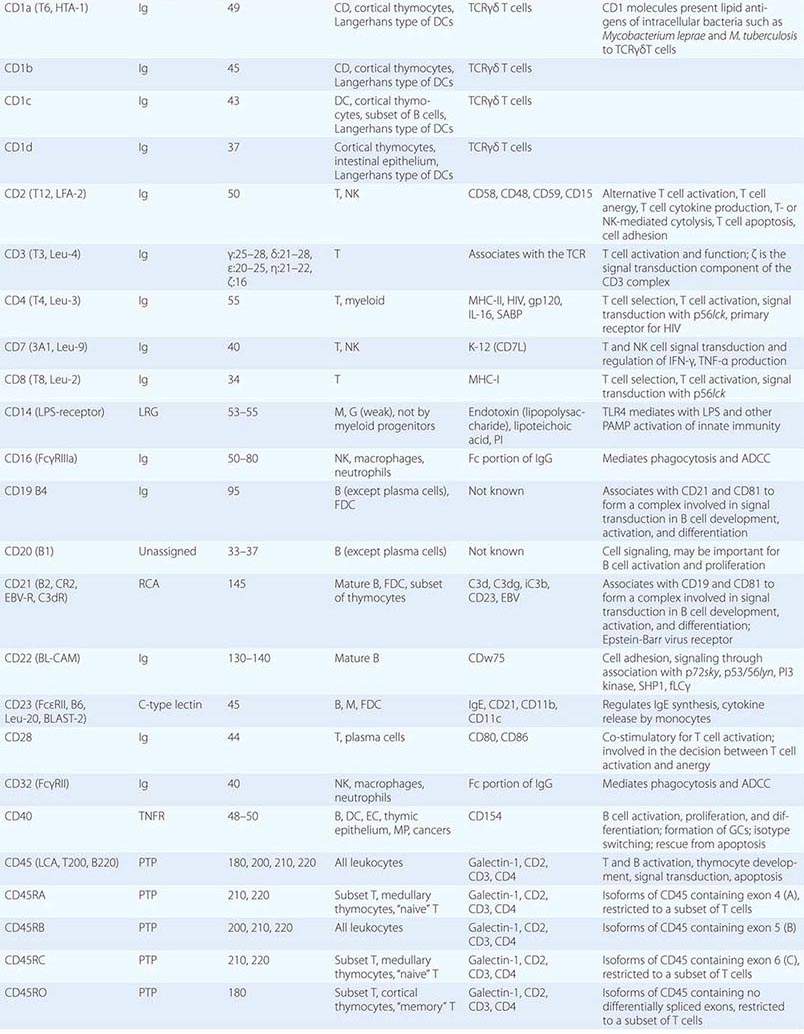
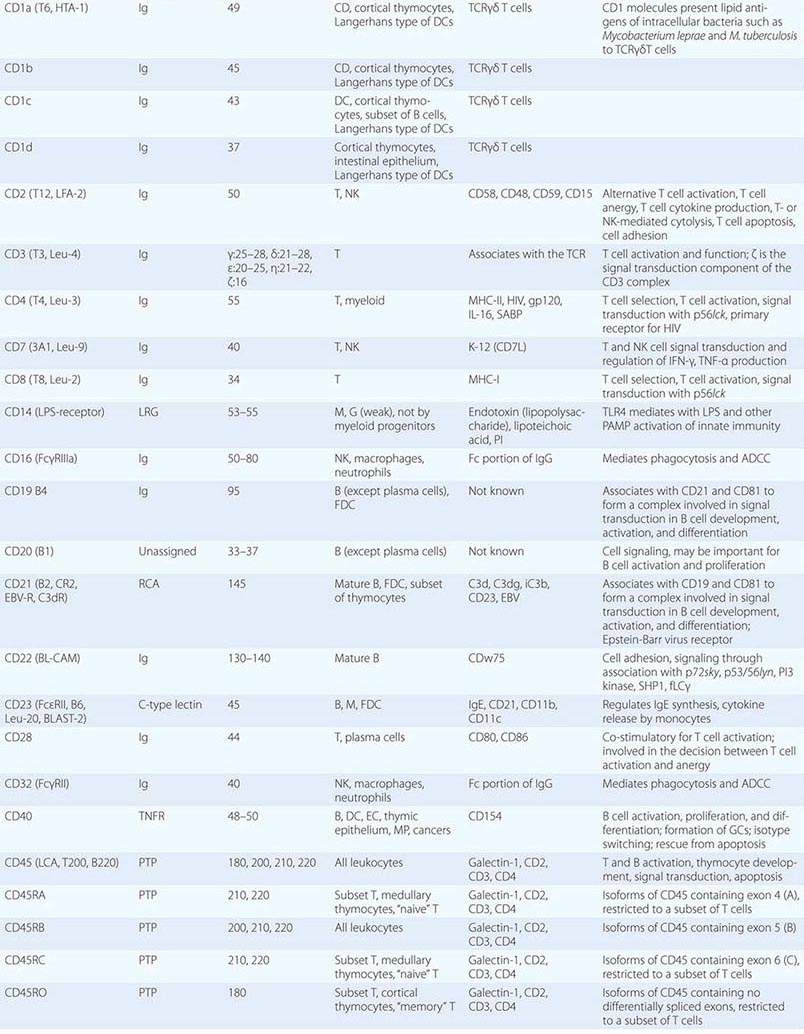
• CD classification of human lymphocyte differentiation antigens—the development of monoclonal antibody technology led to the discovery of a large number of new leukocyte surface molecules. In 1982, the First International Workshop on Leukocyte Differentiation Antigens was held to establish a nomenclature for cell-surface molecules of human leukocytes. From this and subsequent leukocyte differentiation workshops has come the cluster of differentiation (CD) classification of leukocyte antigens.
• Chemokines—soluble molecules that direct and determine immune cell movement and circulation pathways.
• Complement—cascading series of plasma enzymes and effector proteins whose function is to lyse pathogens and/or target them to be phagocytized by neutrophils and monocyte/macrophage lineage cells of the reticuloendothelial system.
• Co-stimulatory molecules—molecules of antigen-presenting cells (such as B7-1 and B7-2 or CD40) that lead to T cell activation when bound by ligands on activated T cells (such as CD28 or CD40 ligand).
• Cytokines—soluble proteins that interact with specific cellular receptors that are involved in the regulation of the growth and activation of immune cells and mediate normal and pathologic inflammatory and immune responses.
• Dendritic cells—myeloid and/or lymphoid lineage antigen-presenting cells of the adaptive immune system. Immature dendritic cells, or dendritic cell precursors, are key components of the innate immune system by responding to infections with production of high levels of cytokines. Dendritic cells are key initiators both of innate immune responses via cytokine production and of adaptive immune responses via presentation of antigen to T lymphocytes.
• Ig Fc receptors—receptors found on the surface of certain cells including B cells, natural killer cells, macrophages, neutrophils, and mast cells. Fc receptors bind to antibodies that have attached to invading pathogen-infected cells. They stimulate cytotoxic cells to destroy microbe-infected cells through a mechanism known as antibody-dependent cell-mediated cytotoxicity (ADCC). Examples of important Fc receptors include CD16 (FcγRIIIa), CD23 (FcεR), CD32 (FcγRII), CD64 (FcγRI), and CD89 (FcαR).
• Inflammasome—large cytoplasmic complexes of intracellular proteins that link the sensing of microbial products and cellular stress to the proteolytic activation of interleukin (IL)-1β and IL-18 inflammatory cytokines. Activation of molecules in the inflammasome is a key step in the response of the innate immune system for intracellular recognition of microbial and other danger signals in both health and pathologic states.
• Innate immune system—ancient immune recognition system of host cells bearing germline-encoded pattern recognition receptors that recognize pathogens and trigger a variety of mechanisms of pathogen elimination. Cells of the innate immune system include natural killer cell lymphocytes, monocytes/macrophages, dendritic cells, neutrophils, basophils, eosinophils, tissue mast cells, and epithelial cells.
• Large granular lymphocytes—lymphocytes of the innate immune system with azurophilic cytotoxic granules that have natural killer cell activity capable of killing foreign and host cells with few or no self-major histocompatibility complex (MHC) class I molecules.
• Natural killer (NK) cells—large granular lymphocytes that kill target cells expressing few or no human leukocyte antigen (HLA) class I molecules, such as malignantly transformed cells and virally infected cells. NK cells express receptors that inhibit killer cell function when self-MHC class I is present.
• NK T cells—innate-like lymphocytes that use an invariant T cell receptor (TCR)-α chain combined with a limited set of TCR-β chains and coexpress receptors commonly found on NK cells. NK T cells recognize lipid antigens of bacterial, viral, fungal, and protozoal infectious agents.
• Pathogen-associated molecular patterns (PAMPs)—Invariant molecular structures expressed by large groups of microorganisms that are recognized by host cellular pattern recognition receptors in the mediation of innate immunity.
• Pattern recognition receptors (PRRs)—germline-encoded receptors expressed by cells of the innate immune system that recognize PAMPs.
• Polyreactive natural antibodies—preexisting low-affinity antibodies produced by B cells that cross-react with multiple antigens and are available at the time of infection to bind to and coat the invading pathogen and harness innate responses to slow the infection until an adaptive high-affinity protective antibody response can be made.
• T cell exhaustion—state of T cells when the persistence of antigen disrupts memory T cell function, resulting in defects in memory T cell responses. Most frequently occurs in malignancies and in chronic viral infections such as HIV-1 and hepatitis C.
• T cell receptor (TCR) for antigen—complex of surface molecules that rearrange during postnatal T cell development made up of clonotypic TCR-α and -β chains that are associated with the CD3 complex composed of invariant γ, δ, ε, ζ, and η chains. TCR-α and -β chains recognize peptide fragments of protein antigen physically bound in antigen-presenting cell MHC class I or II molecules, leading to signaling via the CD3 complex to mediate effector functions.
• T follicular helper T cells (Tfh)—CD4 T cells in B cell follicle germinal centers that produce IL-4 and IL-21 and drive B cell development and affinity maturation in peripheral lymphoid tissues such as lymph node and spleen.
• TH17 T cells—CD4 T cells that secrete IL-17, IL-22, and IL-26 and play roles in autoimmune inflammatory disorders as well as defend against bacterial and fungal pathogens.
• T lymphocytes—thymus-derived lymphocytes that mediate adaptive cellular immune responses including T helper, T regulatory, and cytotoxic T lymphocyte effector cell functions.
• Tolerance—B and T cell nonresponsiveness to antigens that results from encounter with foreign or self-antigens by B and T lymphocytes in the absence of expression of antigen-presenting cell co-stimulatory molecules. Tolerance to antigens may be induced and maintained by multiple mechanisms either centrally (in the thymus for T cells or bone marrow for B cells) or peripherally at sites throughout the peripheral immune system.
INTRODUCTION
The human immune system has evolved over millions of years from both invertebrate and vertebrate organisms to develop sophisticated defense mechanisms to protect the host from microbes and their virulence factors. The normal immune system has three key properties: a highly diverse repertoire of antigen receptors that enables recognition of a nearly infinite range of pathogens; immune memory, to mount rapid recall immune responses; and immunologic tolerance, to avoid immune damage to normal self-tissues. From invertebrates, humans have inherited the innate immune system, an ancient defense system that uses germline-encoded proteins to recognize pathogens. Cells of the innate immune system, such as macrophages, dendritic cells, and NK lymphocytes, recognize PAMPs that are highly conserved among many microbes and use a diverse set of PRR molecules. Important components of the recognition of microbes by the innate immune system include recognition by germline-encoded host molecules, recognition of key microbe virulence factors but not recognition of self-molecules, and nonrecognition of benign foreign molecules or microbes. Upon contact with pathogens, macrophages and NK cells may kill pathogens directly or, in concert with dendritic cells, may activate a series of events that both slow the infection and recruit the more recently evolved arm of the human immune system, the adaptive immune system.
Adaptive immunity is found only in vertebrates and is based on the generation of antigen receptors on T and B lymphocytes by gene rearrangements, such that individual T or B cells express unique antigen receptors on their surface capable of specifically recognizing diverse antigens of the myriad infectious agents in the environment. Coupled with finely tuned specific recognition mechanisms that maintain tolerance (nonreactivity) to self-antigens, T and B lymphocytes bring both specificity and immune memory to vertebrate host defenses.
This chapter describes the cellular components, key molecules (Table 372e-1), and mechanisms that make up the innate and adaptive immune systems and describes how adaptive immunity is recruited to the defense of the host by innate immune responses. An appreciation of the cellular and molecular bases of innate and adaptive immune responses is critical to understanding the pathogenesis of inflammatory, autoimmune, infectious, and immunodeficiency diseases.
|
HUMAN LEUKOCYTE SURFACE ANTIGENS—THE CD CLASSIFICATION OF LEUKOCYTE DIFFERENTIATION ANTIGENS |
THE INNATE IMMUNE SYSTEM
All multicellular organisms, including humans, have developed the use of a limited number of surface and intracellular germline-encoded molecules that recognize large groups of pathogens. Because of the myriad human pathogens, host molecules of the human innate immune system sense “danger signals” and either recognize PAMPs, the common molecular structures shared by many pathogens, or recognize host cell molecules produced in response to infection such as heat shock proteins and fragments of the extracellular matrix. PAMPs must be conserved structures vital to pathogen virulence and survival, such as bacterial endotoxin, so that pathogens cannot mutate molecules of PAMPs to evade human innate immune responses. PRRs are host proteins of the innate immune system that recognize PAMPs as host danger signal molecules (Tables 372e-2 and 372e-3). Thus, recognition of pathogen molecules by hematopoietic and nonhematopoietic cell types leads to activation/production of the complement cascade, cytokines, and antimicrobial peptides as effector molecules. In addition, pathogen PAMPs as host danger signal molecules activate dendritic cells to mature and to express molecules on the dendritic cell surface that optimize antigen presentation to respond to foreign antigens.
|
MAJOR COMPONENTS OF THE INNATE IMMUNE SYSTEM |
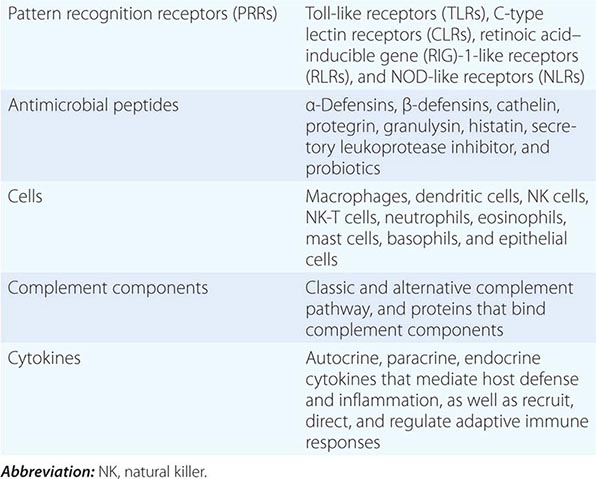
PATTERN RECOGNITION
Major PRR families of proteins include transmembrane proteins, such as the Toll-like receptors (TLRs) and C-type lectin receptors (CLRs), and cytoplasmic proteins, such as the retinoic acid–inducible gene (RIG)-1-like receptors (RLRs) and NOD-like receptors (NLRs) (Table 372e-3). A major group of PRR collagenous glycoproteins with C-type lectin domains are termed collectins and include the serum protein mannose-binding lectin (MBL). MBL and other collectins, as well as two other protein families—the pentraxins (such as C-reactive protein and serum amyloid P) and macrophage scavenger receptors—all have the property of opsonizing (coating) bacteria for phagocytosis by macrophages and can also activate the complement cascade to lyse bacteria. Integrins are cell-surface adhesion molecules that affect attachment between cells and the extracellular matrix and mediate signal transduction that reflects the chemical composition of the cell environment. For example, integrins signal after cells bind bacterial lipopolysaccharide (LPS) and activate phagocytic cells to ingest pathogens.
|
PATTERN RECOGNITION RECEPTORS (PRRs) AND THEIR LIGANDS |
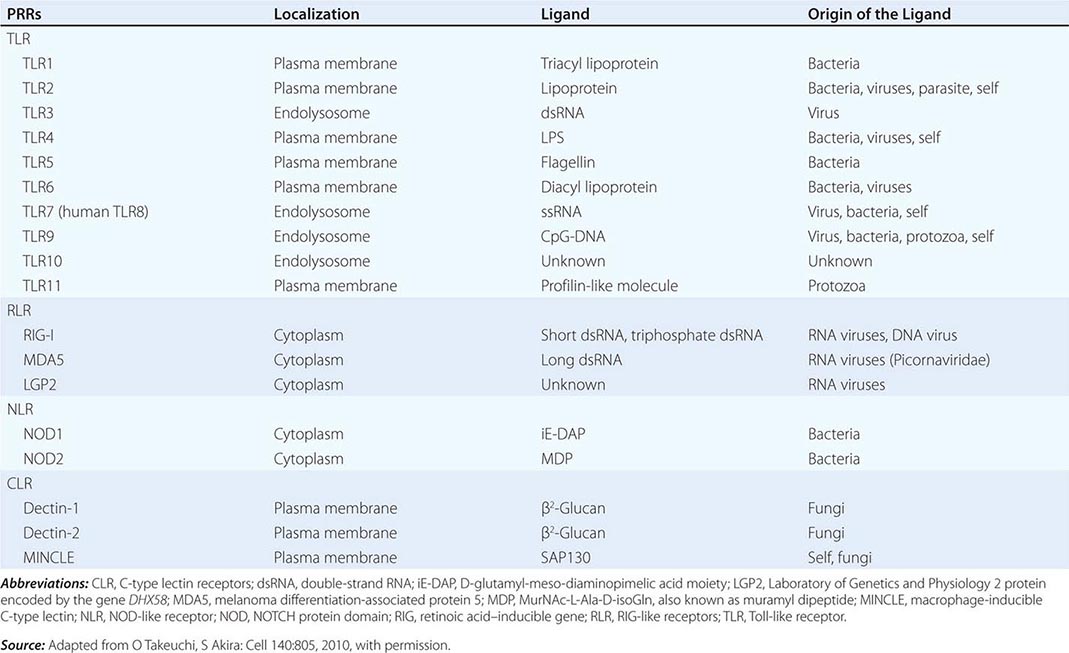
There are multiple connections between the innate and adaptive immune systems; these include (1) a plasma protein, LPS-binding protein, that binds and transfers LPS to the macrophage LPS receptor, CD14; (2) a human family of proteins called Toll-like receptor proteins (TLRs), some of which are associated with CD14, bind LPS, and signal epithelial cells, dendritic cells, and macrophages to produce cytokines and upregulate cell-surface molecules that signal the initiation of adaptive immune responses (Fig. 372e-1, Tables 372e-3 and 372e-4), and (3) families of intracellular microbial sensors called NLRs and RLRs. Proteins in the Toll family can be expressed on macrophages, dendritic cells, and B cells as well as on a variety of nonhematopoietic cell types, including respiratory epithelial cells. Eleven TLRs have been identified in humans, and 13 TLRs have been identified in mice (Tables 372e-4 and 372e-5). Upon ligation, TLRs activate a series of intracellular events that lead to the killing of bacteria- and viral-infected cells as well as to the recruitment and ultimate activation of antigen-specific T and B lymphocytes (Fig. 372e-1). Importantly, signaling by massive amounts of LPS through TLR4 leads to the release of large amounts of cytokines that mediate LPS-induced shock. Mutations in TLR4 proteins in mice protect from LPS shock, and TLR mutations in humans protect from LPS-induced inflammatory diseases such as LPS-induced asthma (Fig. 372e-1).
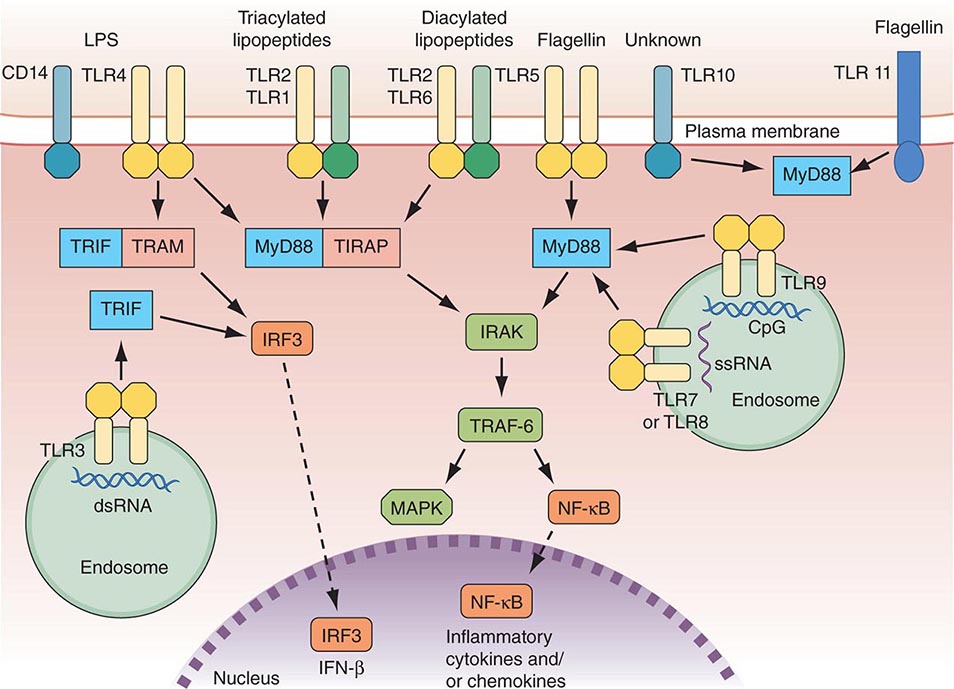
FIGURE 372e-1 Overview of major TLR signaling pathways. All TLRs signal through MyD88, with the exception of TLR3. TLR4 and the TLR2 subfamily (TLR1, TLR2, TLR6) also engage TIRAP. TLR3 signals through TRIF. TRIF is also used in conjunction with TRAM in the TLR4-MyD88-independent pathway. Dashed arrows indicate translocation into the nucleus. dsRNA, double-strand RNA; IFN, interferon; IRF3, interferon regulatory factor 3; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinases; NF-κB, nuclear factor-κB; ssRNA, single-strand RNA; TLR, Toll-like receptor. (Adapted from D van Duin et al: Trends Immunol 27:49, 2006, with permission.)
|
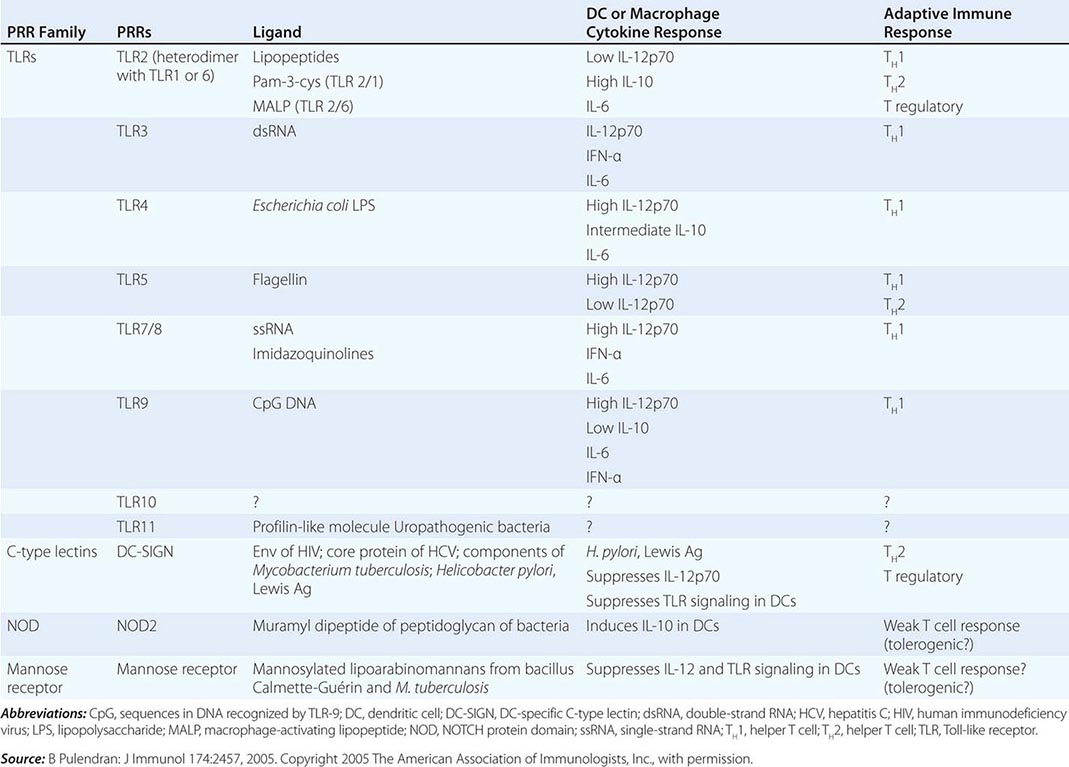
THE ROLE OF PATTERN RECOGNITION RECEPTORS (PRRs) IN MODULATION OF ADAPTIVE IMMUNE RESPONSES |
|
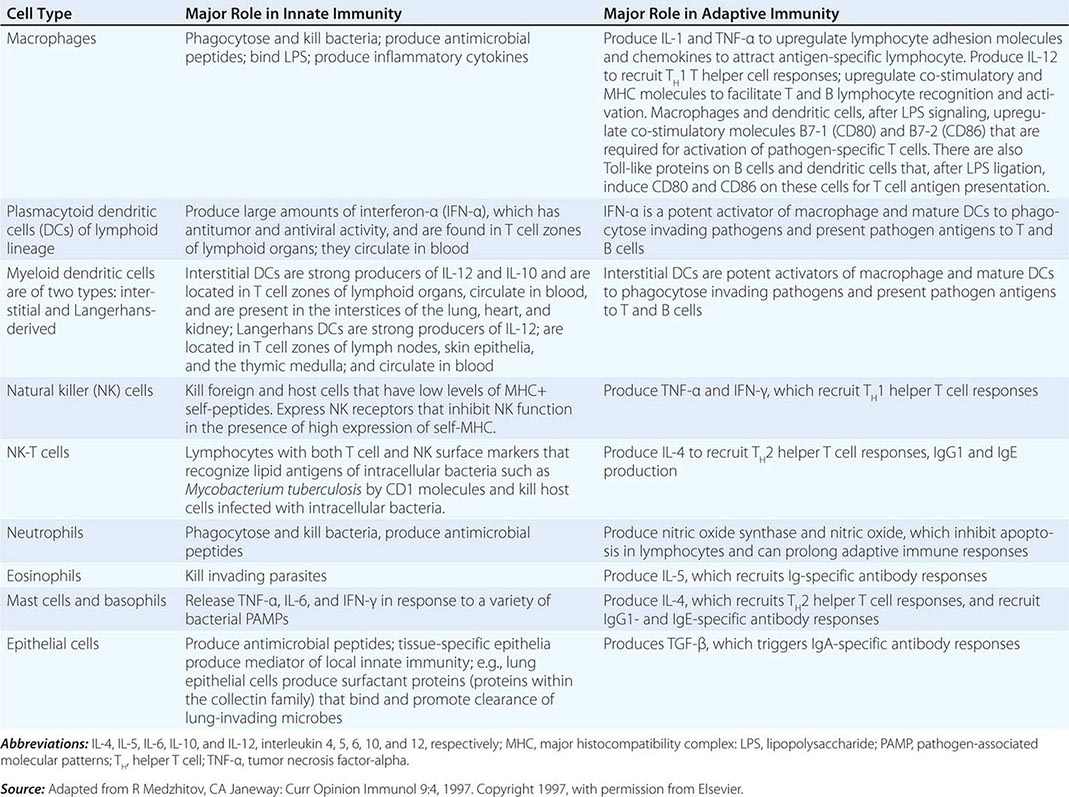
CELLS OF THE INNATE IMMUNE SYSTEM AND THEIR MAJOR ROLES IN TRIGGERING ADAPTIVE IMMUNITY |
Two other families of cytoplasmic PRRs are the NLRs and the RLRs. These families, unlike the TLRs, are composed primarily of soluble intracellular proteins that scan host cell cytoplasm for intracellular pathogens (Tables 372e-2 and 372e-3).
The intracellular microbial sensors, NLRs, after triggering, form large cytoplasmic complexes termed inflammasomes, which are aggregates of molecules including NOD-like receptor pyrin (NLRP) proteins that are members of the NLR family (Table 372e-3). Inflammasomes activate inflammatory caspases and IL-1β in the presence of nonbacterial danger signals (cell stress) and bacterial PAMPs. Mutations in inflammasome proteins can lead to chronic inflammation in a group of periodic febrile diseases called autoinflammatory syndromes (Table 372e-6).
|
DISEASES ASSOCIATED WITH INFLAMMASOME ACTIVITY |
EFFECTOR CELLS OF INNATE IMMUNITY
Cells of the innate immune system and their roles in the first line of host defense are listed in Table 372e-5. Equally important as their roles in the mediation of innate immune responses are the roles that each cell type plays in recruiting T and B lymphocytes of the adaptive immune system to engage in specific pathogen responses.
Monocytes-Macrophages Monocytes arise from precursor cells within bone marrow (Fig. 372e-2) and circulate with a half-life ranging from 1 to 3 days. Monocytes leave the peripheral circulation via capillaries and migration into a vast extravascular cellular pool. Tissue macrophages arise from monocytes that have migrated out of the circulation and by in situ proliferation of macrophage precursors in tissue. Common locations where tissue macrophages (and certain of their specialized forms) are found are lymph node, spleen, bone marrow, perivascular connective tissue, serous cavities such as the peritoneum, pleura, skin connective tissue, lung (alveolar macrophages), liver (Kupffer cells), bone (osteoclasts), central nervous system (microglia cells), and synovium (type A lining cells).
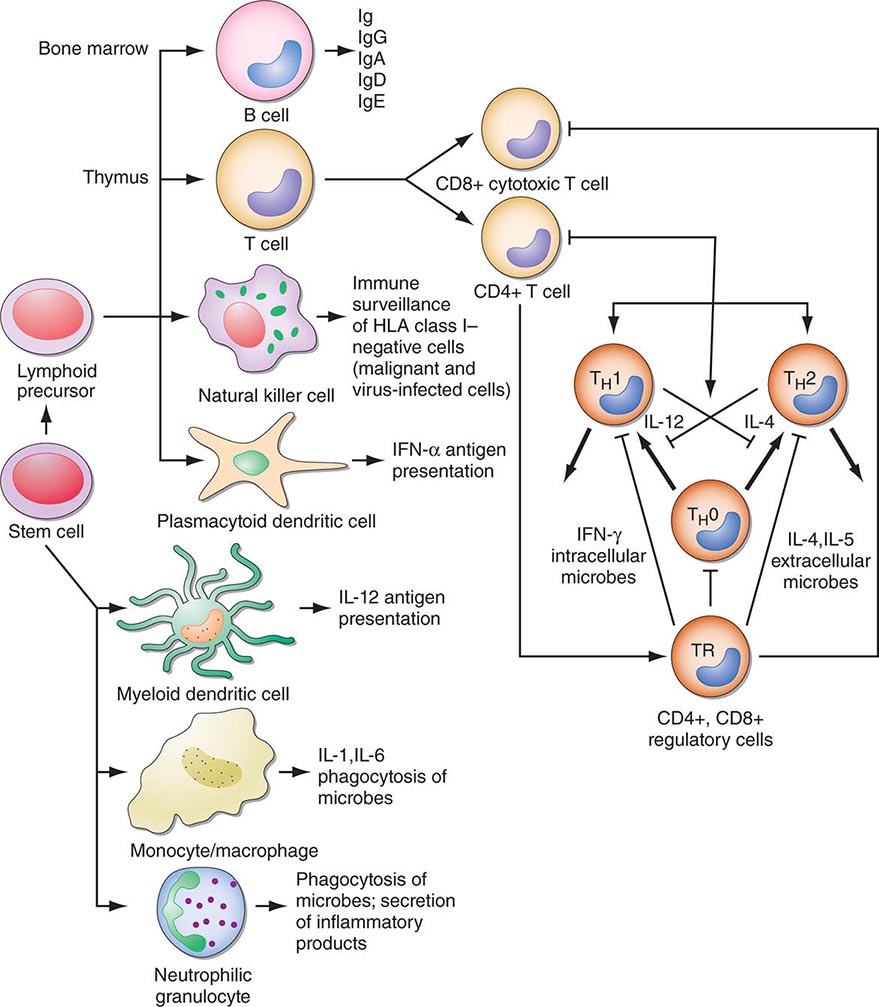
FIGURE 372e-2 Schematic model of intercellular interactions of adaptive immune system cells. In this figure, the arrows denote that cells develop from precursor cells or produce cytokines or antibodies; lines ending with bars indicate suppressive intercellular interactions. Stem cells differentiate into either T cells, antigen-presenting dendritic cells, natural killer cells, macrophages, granulocytes, or B cells. Foreign antigen is processed by dendritic cells, and peptide fragments of foreign antigen are presented to CD4+ and/or CD8+ T cells. CD8+ T cell activation leads to induction of cytotoxic T lymphocyte (CTL) or killer T cell generation, as well as induction of cytokine-producing CD8+ cytotoxic T cells. For antibody production against the same antigen, active antigen is bound to sIg within the B cell receptor complex and drives B cell maturation into plasma cells that secrete Ig. TH1 or TH2 CD4+ T cells producing interleukin (IL) 4, IL-5, or interferon (IFN) γ regulate the Ig class switching and determine the type of antibody produced. TH17 cells secrete IL-17, IL-22, IL-26, which contribute to host defense against extracellular bacteria and fungi, particularly at mucosal surfaces. CD4+, CD25+ T regulatory cells produce IL-10 and downregulate T and B cell responses once the microbe has been eliminated. GM-CSF, granulocyte-macrophage colony-stimulating factor; TNF, tumor necrosis factor.
In general, monocytes-macrophages are on the first line of defense associated with innate immunity and ingest and destroy microorganisms through the release of toxic products such as hydrogen peroxide (H2O2) and nitric oxide (NO). Inflammatory mediators produced by macrophages attract additional effector cells such as neutrophils to the site of infection. Macrophage mediators include prostaglandins; leukotrienes; platelet activating factor; cytokines such as IL-1, TNF-α, IL-6, and IL-12; and chemokines (Tables 372e-7 to 372e-9).
|
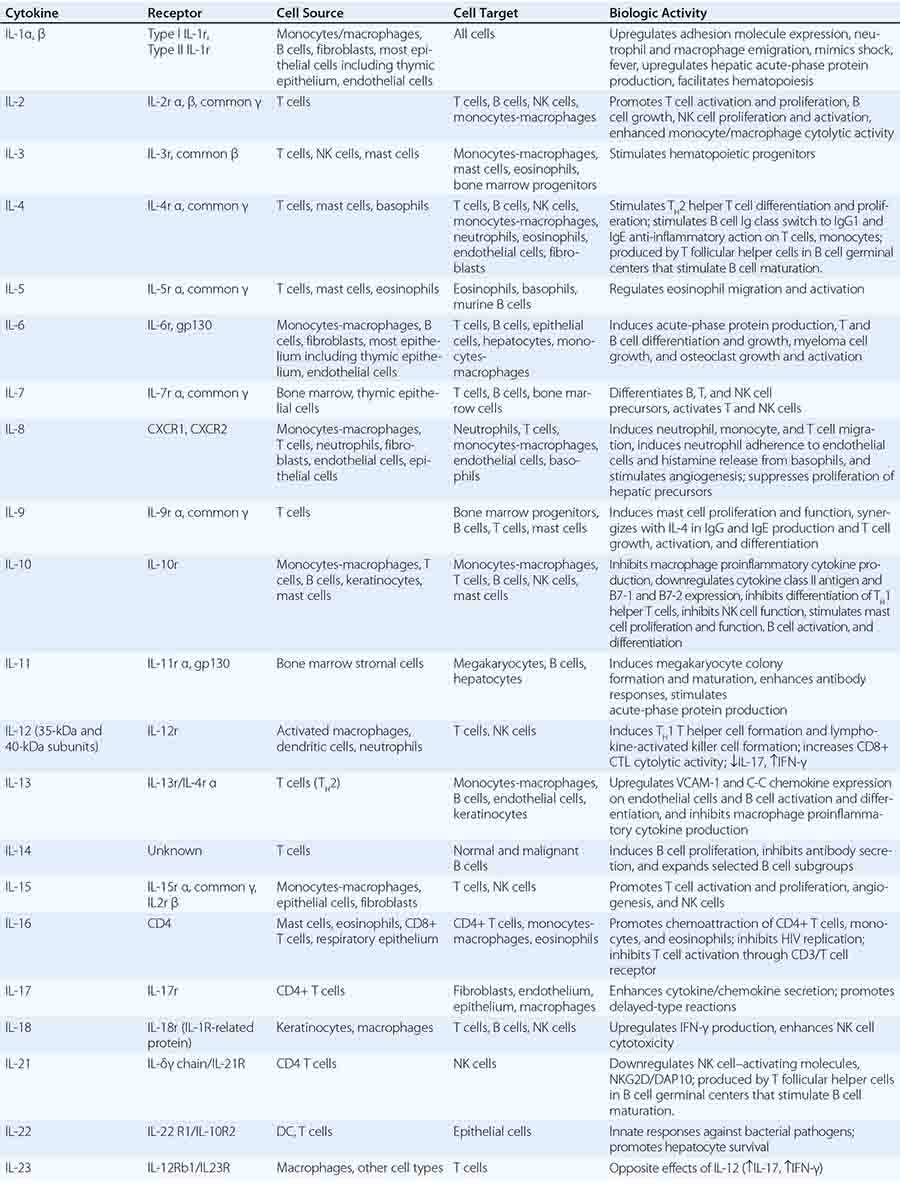
CYTOKINES AND CYTOKINE RECEPTORS |
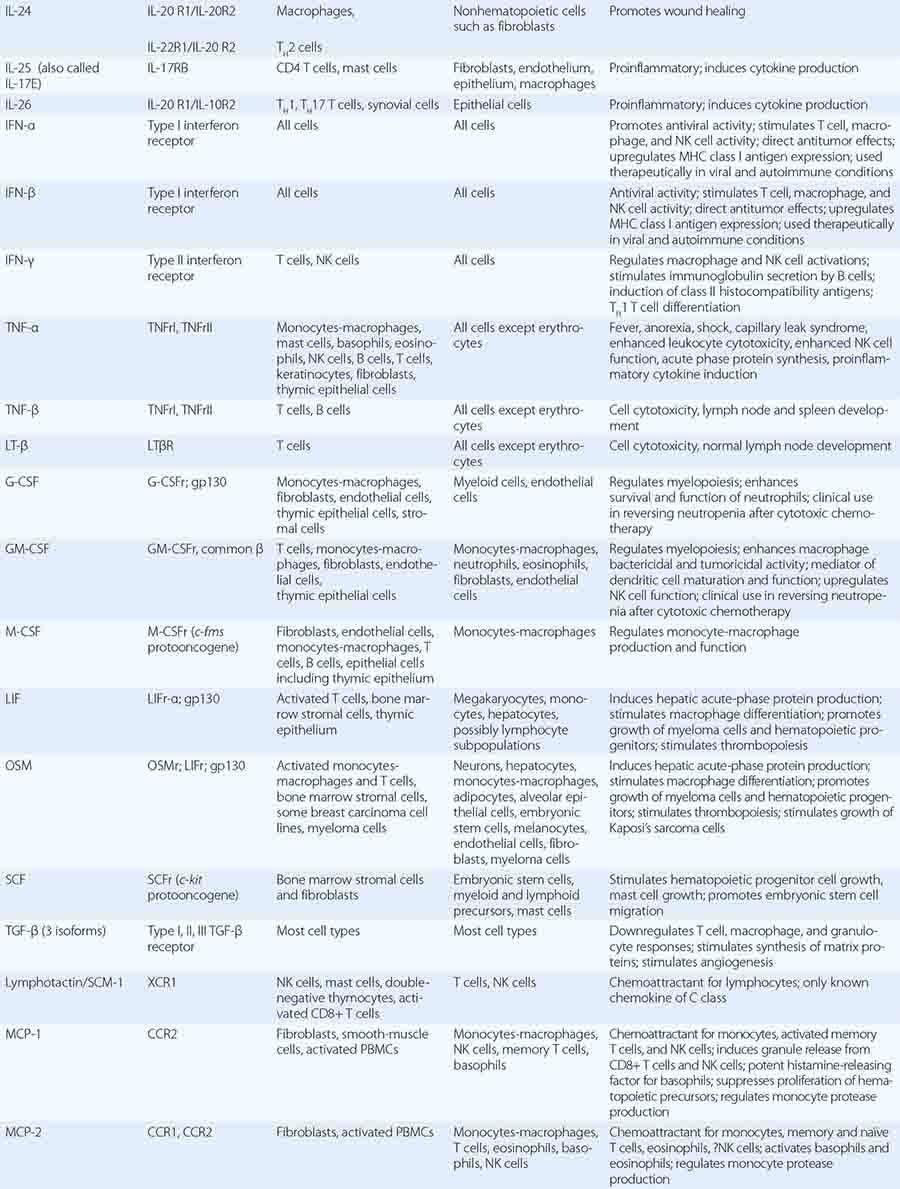
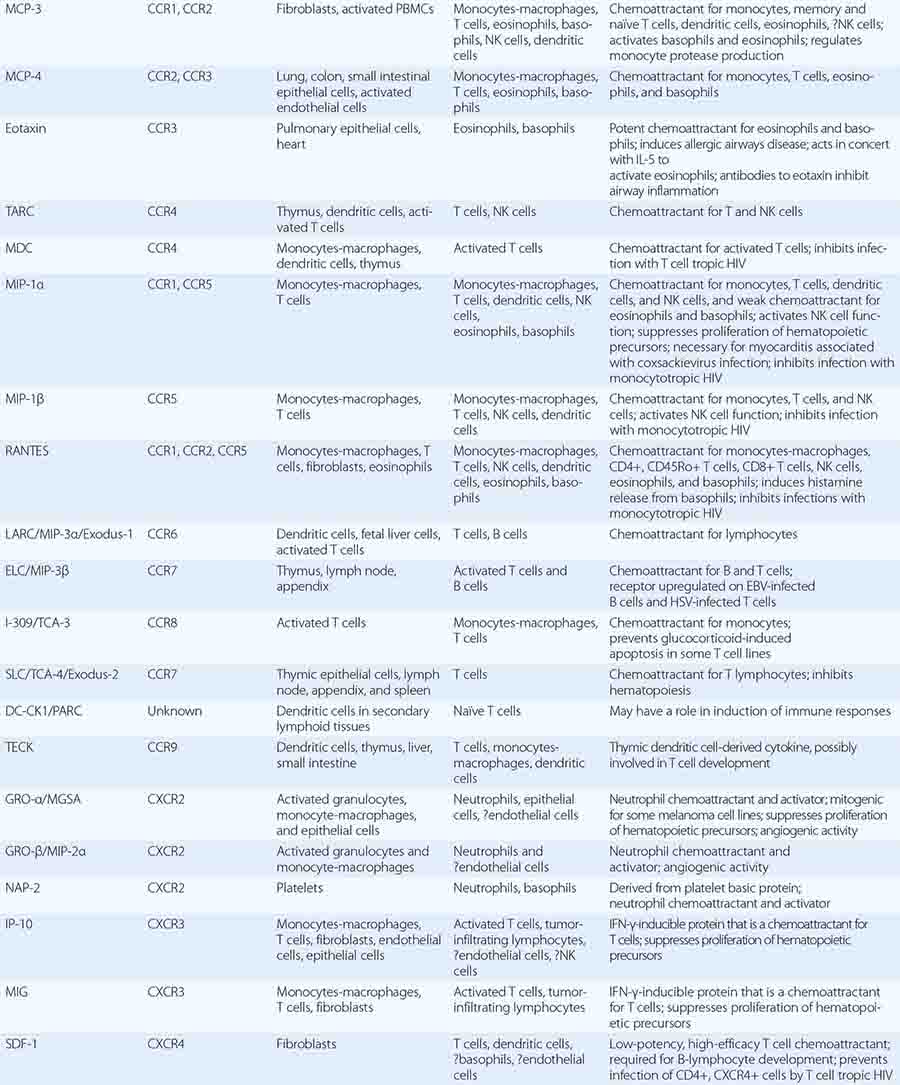

|
CC, CXC1, CX3, C1, AND XC FAMILIES OF CHEMOKINES AND CHEMOKINE RECEPTORS |
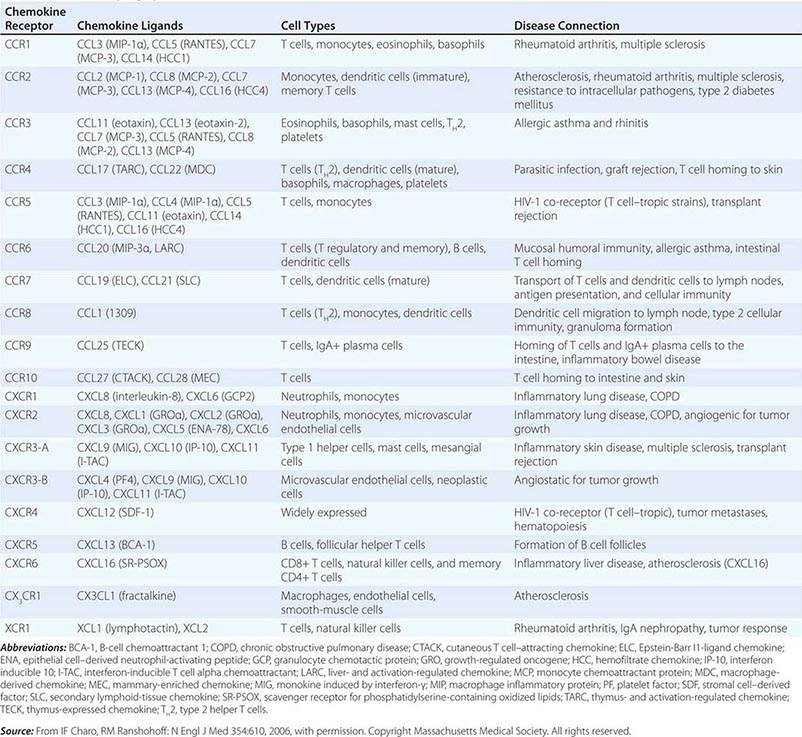
|
CYTOKINE FAMILIES GROUPED BY STRUCTURAL SIMILARITY |
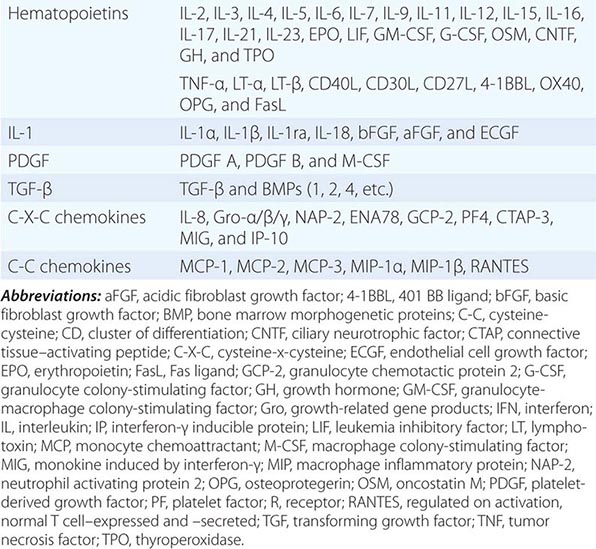
Although monocytes-macrophages were originally thought to be the major antigen-presenting cells (APCs) of the immune system, it is now clear that cell types called dendritic cells are the most potent and effective APCs in the body (see below). Monocytes-macrophages mediate innate immune effector functions such as destruction of antibody-coated bacteria, tumor cells, or even normal hematopoietic cells in certain types of autoimmune cytopenias. Monocytes-macrophages ingest bacteria or are infected by viruses, and in doing so, they frequently undergo programmed cell death or apoptosis. Macrophages that are infected by intracellular infectious agents are recognized by dendritic cells as infected and apoptotic cells and are phagocytosed by dendritic cells. In this manner, dendritic cells “cross-present” infectious agent antigens of macrophages to T cells. Activated macrophages can also mediate antigen-nonspecific lytic activity and eliminate cell types such as tumor cells in the absence of antibody. This activity is largely mediated by cytokines (i.e., TNF-α and IL-1). Monocytes-macrophages express lineage-specific molecules (e.g., the cell-surface LPS receptor, CD14) as well as surface receptors for a number of molecules, including the Fc region of IgG, activated complement components, and various cytokines (Table 372e-7).
Dendritic Cells Human dendritic cells (DCs) contain several subsets, including myeloid DCs and plasmacytoid DCs. Myeloid DCs can differentiate into either macrophages-monocytes or tissue-specific DCs. In contrast to myeloid DCs, plasmacytoid DCs are inefficient APCs but are potent producers of type I interferon (IFN) (e.g., IFN-α) in response to viral infections. The maturation of DCs is regulated through cell-to-cell contact and soluble factors, and DCs attract immune effectors through secretion of chemokines. When DCs come in contact with bacterial products, viral proteins, or host proteins released as danger signals from distressed host cells (Figs. 372e-2 and 372e-3), infectious agent molecules bind to various TLRs and activate DCs to release cytokines and chemokines that drive cells of the innate immune system to become activated to respond to the invading organism, and recruit T and B cells of the adaptive immune system to respond. Plasmacytoid DCs produce antiviral IFN-α that activates NK cell killing of pathogen-infected cells; IFN-α also activates T cells to mature into antipathogen cytotoxic (killer) T cells. Following contact with pathogens, both plasmacytoid and myeloid DCs produce chemokines that attract helper and cytotoxic T cells, B cells, polymorphonuclear cells, and naïve and memory T cells as well as regulatory T cells to ultimately dampen the immune response once the pathogen is controlled. TLR engagement on DCs upregulates MHC class II, B7-1 (CD80), and B7-2 (CD86), which enhance DC-specific antigen presentation and induce cytokine production (Table 372e-7). Thus, DCs are important bridges between early (innate) and later (adaptive) immunity. DCs also modulate and determine the types of immune responses induced by pathogens via the TLRs expressed on DCs (TLR7–9 on plasmacytoid DCs, TLR4 on monocytoid DCs) and via the TLR adapter proteins that are induced to associate with TLRs (Fig. 372e-1, Table 372e-4). In addition, other PRRs, such as C-type lectins, NLRs, and mannose receptors, upon ligation by pathogen products, activate cells of the adaptive immune system and, like TLR stimulation, by a variety of factors, determine the type and quality of the adaptive immune response that is triggered (Table 372e-4).
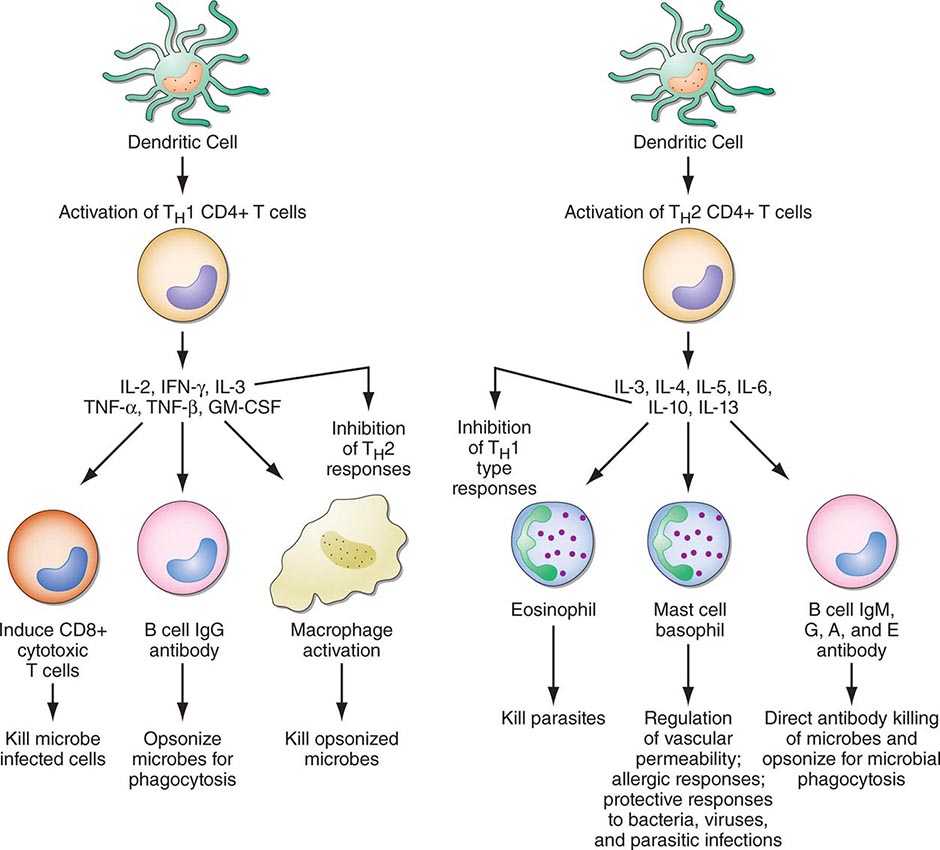
FIGURE 372e-3 CD4+ helper T1 (TH1) cells and TH2 T cells secrete distinct but overlapping sets of cytokines. TH1 CD4+ cells are frequently activated in immune and inflammatory reactions against intracellular bacteria or viruses, whereas TH2 CD4+ cells are frequently activated for certain types of antibody production against parasites and extracellular encapsulated bacteria; they are also activated in allergic diseases. GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; TNF, tumor necrosis factor. (Adapted from S Romagnani: CD4 effector cells, in Inflammation: Basic Principles and Clinical Correlates, 3rd ed, J Gallin, R Snyderman [eds]. Philadelphia, Lippincott Williams & Wilkins, 1999, p 177; with permission.)
Large Granular Lymphocytes/Natural Killer Cells Large granular lymphocytes (LGLs) or NK cells account for ~5–15% of peripheral blood lymphocytes. NK cells are nonadherent, nonphagocytic cells with large azurophilic cytoplasmic granules. NK cells express surface receptors for the Fc portion of IgG (FcR) (CD16) and for NCAM-I (CD56), and many NK cells express T lineage markers, particularly CD8, and proliferate in response to IL-2. NK cells arise in both bone marrow and thymic microenvironments.
Functionally, NK cells share features with both monocytes-macrophages and neutrophils in that they mediate both ADCC and NK cell activity. ADCC is the binding of an opsonized (antibody-coated) target cell to an Fc receptor-bearing effector cell via the Fc region of antibody, resulting in lysis of the target by the effector cell. NK cell cytotoxicity is the nonimmune (i.e., effector cell never having had previous contact with the target), MHC-unrestricted, non-antibody-mediated killing of target cells, which are usually malignant cell types, transplanted foreign cells, or virus-infected cells. Thus, NK cell cytotoxicity may play an important role in immune surveillance and destruction of malignant and virus-infected host cells. NK cell hyporesponsiveness is also observed in patients with Chédiak-Higashi syndrome, an autosomal recessive disease associated with fusion of cytoplasmic granules and defective degranulation of neutrophil lysosomes.
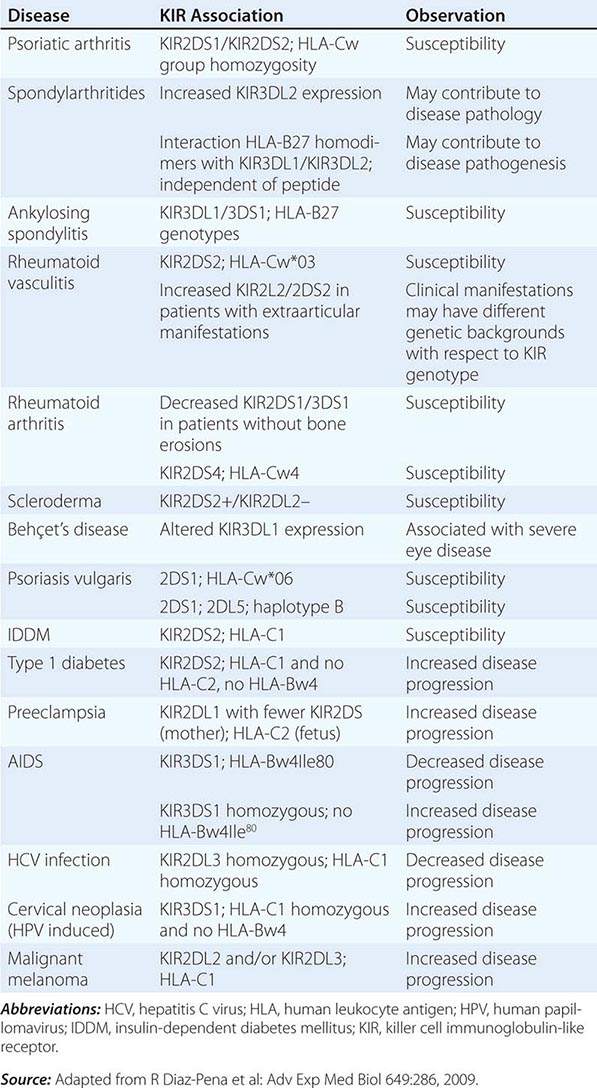
NK cells have a variety of surface receptors that have inhibitory or activating functions and belong to two structural families. These families include the immunoglobulin superfamily and the lectin-like type II transmembrane proteins. NK immunoglobulin superfamily receptors include the killer cell immunoglobulin-like activating or inhibitory receptors (KIRs), many of which have been shown to have HLA class I ligands. The KIRs are made up proteins with either two (KIR2D) or three (KIR3D) extracellular immunoglobulin domains (D). Moreover, their nomenclature designates their function as either inhibitory KIRs with a long (L) cytoplasmic tail and immunoreceptor tyrosine-based inhibitory motif (ITIM) (KIRDL) or activating KIRs with a short (S) cytoplasmic tail (KIRDS). NK cell inactivation by KIRs is a central mechanism to prevent damage to normal host cells. Genetic studies have demonstrated the association of KIRs with viral infection outcome and autoimmune disease (Table 372e-10).
|
ASSOCIATION OF KIRs WITH DISEASE |
In addition to the KIRs, a second set of immunoglobulin superfamily receptors includes the natural cytotoxicity receptors (NCRs), which include NKp46, NKp30, and NKp44. These receptors help to mediate NK cell activation against target cells. The ligands to which NCRs bind on target cells have been recently recognized to be comprised of molecules of pathogens such as influenza, vaccinia, and malaria as well as host molecules expressed on tumor cells.
NK cell signaling is, therefore, a highly coordinated series of inhibiting and activating signals that prevent NK cells from responding to uninfected, nonmalignant self-cells; however, they are activated to attack malignant and virally infected cells (Fig. 372e-4). Recent evidence suggests that NK cells, although not possessing rearranging immune recognition genes, may be able to mediate recall for NK cell responses to viruses and for immune responses such as contact hypersensitivity.
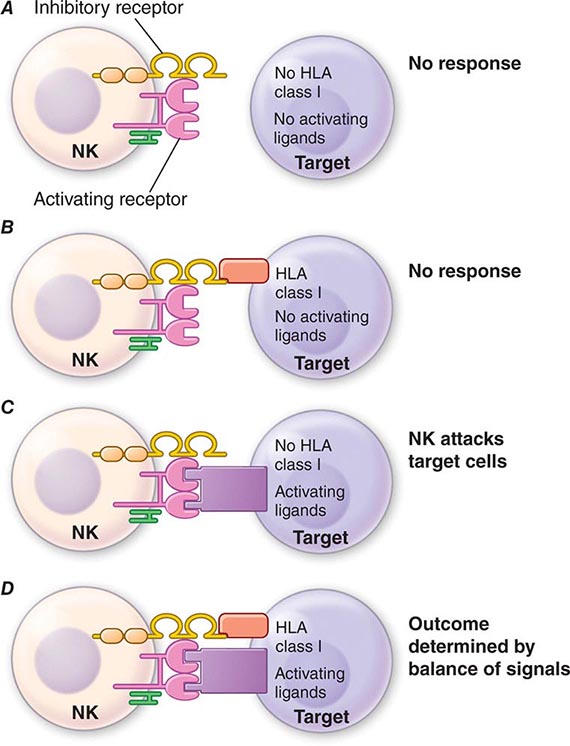
FIGURE 372e-4 Encounters between NK cells: potential targets and possible outcomes. The amount of activating and inhibitory receptors on the NK cells and the amount of ligands on the target cell, as well as the qualitative differences in the signals transduced, determine the extent of the NK response. A. When target cells have no HLA class I or activating ligands, NK cells cannot kill target cells. B. When target cells bear self-HLA, NK cells cannot kill targets. C. When target cells are pathogen infected and have downregulated HLA and express activating ligands, NK cells kill target cells. D. When NK cells encounter targets with both self-HLA and activating receptors, then the level of target killing is determined by the balance of inhibitory and activating signals to the NK cell. HLA, human leukocyte antigen; NK, natural killer. (Adapted from L Lanier: Annu Rev Immunol 23:225, 2005; reproduced with permission from Annual Reviews Inc. Copyright 2011 by Annual Reviews Inc.)
Some NK cells express CD3 and invariant TCR-α chains and are termed NK T cells. TCRs of NK T cells recognize lipid molecules of intracellular bacteria when presented in the context of CD1d molecules on APCs. Upon activation, NK T cells secrete effector cytokines such as IL-4 and IFN-γ. This mode of recognition of intracellular bacteria such as Listeria monocytogenes and Mycobacterium tuberculosis by NK T cells leads to induction of activation of DCs and is thought to be an important innate defense mechanism against these organisms.
The receptors for the Fc portion of IgG (FcγRs) are present on NK cells, B cells, macrophages, neutrophils, and mast cells and mediate interactions of IgG with antibody-coated target cells, such as virally infected cells. Antibody-NK interaction via antibody Fc and NK cell FcR links the adaptive and innate immune systems and regulates the mediation of IgG antibody effector functions such as ADCC. There are both activation and inhibitory FcγRs. Activation FcRs, such as FcγRI (CD64), FcγRII (CD32), and FcγRII (CD64), are characterized by the presence of an immunoreceptor tyrosine-based activating motif (ITAM) sequence, whereas inhibitory FcRs, such as FcγRIIb, contain an immunoreceptor tyrosine-based inhibitory motif (ITIM) sequence. There is evidence that dysregulation in IgG-FcγR interactions plays a role in arthritis, multiple sclerosis, and systemic lupus erythematosus.
Neutrophils, Eosinophils, and Basophils Granulocytes are present in nearly all forms of inflammation and are amplifiers and effectors of innate immune responses (Figs. 372e-2 and 372e-3). Unchecked accumulation and activation of granulocytes can lead to host tissue damage, as seen in neutrophil- and eosinophil-mediated systemic necrotizing vasculitis. Granulocytes are derived from stem cells in bone marrow. Each type of granulocyte (neutrophil, eosinophil, or basophil) is derived from a different subclass of progenitor cell that is stimulated to proliferate by colony-stimulating factors (Table 372e-7). During terminal maturation of granulocytes, class-specific nuclear morphology and cytoplasmic granules appear that allow for histologic identification of granulocyte type.
Neutrophils express Fc receptor IIIa for IgG (CD16) as well as receptors for activated complement components (C3b or CD35). Upon interaction of neutrophils with antibody-coated (opsonized) bacteria or immune complexes, azurophilic granules (containing myeloperoxidase, lysozyme, elastase, and other enzymes) and specific granules (containing lactoferrin, lysozyme, collagenase, and other enzymes) are released, and microbicidal superoxide radicals (O2–) are generated at the neutrophil surface. The generation of superoxide leads to inflammation by direct injury to tissue and by alteration of macromolecules such as collagen and DNA.
Eosinophils express Fc receptor II for IgG (CD32) and are potent cytotoxic effector cells for various parasitic organisms. In Nippostrongylus brasiliensis helminth infection, eosinophils are important cytotoxic effector cells for removal of these parasites. Key to regulation of eosinophil cytotoxicity to N. brasiliensis worms are antigen-specific T helper cells that produce IL-4, thus providing an example of regulation of innate immune responses by adaptive immunity antigen-specific T cells. Intracytoplasmic contents of eosinophils, such as major basic protein, eosinophil cationic protein, and eosinophil-derived neurotoxin, are capable of directly damaging tissues and may be responsible in part for the organ system dysfunction in the hypereosinophilic syndromes (Chap. 80). Because the eosinophil granule contains anti-inflammatory types of enzymes (histaminase, arylsulfatase, phospholipase D), eosinophils may homeostatically downregulate or terminate ongoing inflammatory responses.
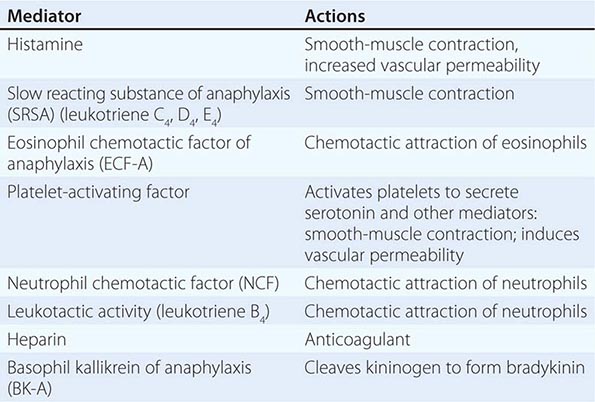
Basophils and tissue mast cells are potent reservoirs of cytokines such as IL-4 and can respond to bacteria and viruses with antipathogen cytokine production through multiple TLRs expressed on their surface. Mast cells and basophils can also mediate immunity through the binding of antipathogen antibodies. This is a particularly important host defense mechanism against parasitic diseases. Basophils express high-affinity surface receptors for IgE (FcεRII) (CD23) and, upon cross-linking of basophil-bound IgE by antigen, can release histamine, eosinophil chemotactic factor of anaphylaxis, and neutral protease—all mediators of allergic immediate (anaphylaxis) hypersensitivity responses (Table 372e-11). In addition, basophils express surface receptors for activated complement components (C3a, C5a), through which mediator release can be directly affected. Thus, basophils, like most cells of the immune system, can be activated in the service of host defense against pathogens, or they can be activated for mediation release and cause pathogenic responses in allergic and inflammatory diseases. For further discussion of tissue mast cells, see Chap. 376.
|
EXAMPLES OF MEDIATORS RELEASED FROM IMMUNE CELLS AND BASOPHILS |
The Complement System The complement system, an important soluble component of the innate immune system, is a series of plasma enzymes, regulatory proteins, and proteins that are activated in a cascading fashion, resulting in cell lysis. There are four pathways of the complement system: the classic activation pathway activated by antigen/antibody immune complexes, the mannose-binding lectin (MBL) (a serum collectin; Table 372e-3) activation pathway activated by microbes with terminal mannose groups, the alternative activation pathway activated by microbes or tumor cells, and the terminal pathway that is common to the first three pathways and leads to the membrane attack complex that lyses cells (Fig. 372e-5). The series of enzymes of the complement system are serine proteases.
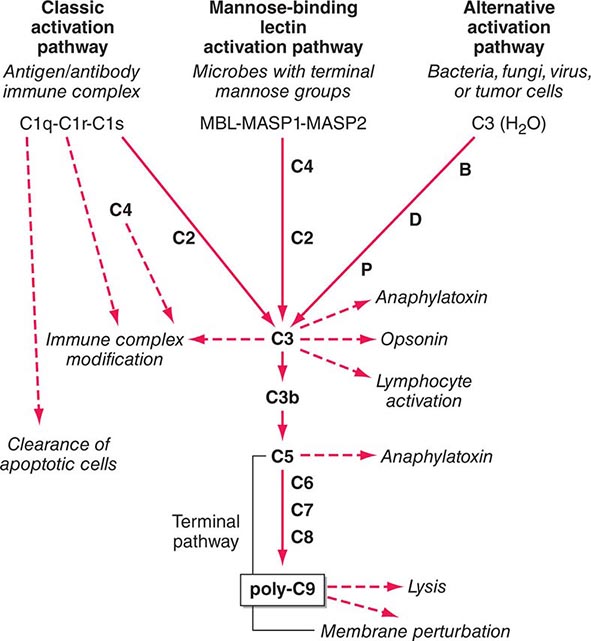
FIGURE 372e-5 The four pathways and the effector mechanisms of the complement system. Dashed arrows indicate the functions of pathway components. (After BJ Morley, MJ Walport: The Complement Facts Books. London, Academic Press, 2000, Chap. 2; with permission. Copyright Academic Press, London, 2000.)
Activation of the classic complement pathway via immune complex binding to C1q links the innate and adaptive immune systems via specific antibody in the immune complex. The alternative complement activation pathway is antibody-independent and is activated by binding of C3 directly to pathogens and “altered self” such as tumor cells. In the renal glomerular inflammatory disease IgA nephropathy, IgA activates the alternative complement pathway and causes glomerular damage and decreased renal function. Activation of the classic complement pathway via C1, C4, and C2 and activation of the alternative pathway via factor D, C3, and factor B both lead to cleavage and activation of C3. C3 activation fragments, when bound to target surfaces such as bacteria and other foreign antigens, are critical for opsonization (coating by antibody and complement) in preparation for phagocytosis. The MBL pathway substitutes MBL-associated serine proteases (MASPs) 1 and 2 for C1q, C1r, and C1s to activate C4. The MBL activation pathway is activated by mannose on the surface of bacteria and viruses.
The three pathways of complement activation all converge on the final common terminal pathway. C3 cleavage by each pathway results in activation of C5, C6, C7, C8, and C9, resulting in the membrane attack complex that physically inserts into the membranes of target cells or bacteria and lyses them.
Thus, complement activation is a critical component of innate immunity for responding to microbial infection. The functional consequences of complement activation by the three initiating pathways and the terminal pathway are shown in Fig. 372e-5. In general the cleavage products of complement components facilitate microbe or damaged cell clearance (C1q, C4, C3), promote activation and enhancement of inflammation (anaphylatoxins, C3a, C5a), and promote microbe or opsonized cell lysis (membrane attack complex).
CYTOKINES
Cytokines are soluble proteins produced by a wide variety of cell types (Tables 372e-7 to 372e-9). They are critical for both normal innate and adaptive immune responses, and their expression may be perturbed in most immune, inflammatory, and infectious disease states.
Cytokines are involved in the regulation of the growth, development, and activation of immune system cells and in the mediation of the inflammatory response. In general, cytokines are characterized by considerable redundancy; different cytokines have similar functions. In addition, many cytokines are pleiotropic in that they are capable of acting on many different cell types. This pleiotropism results from the expression on multiple cell types of receptors for the same cytokine (see below), leading to the formation of “cytokine networks.” The action of cytokines may be (1) autocrine when the target cell is the same cell that secretes the cytokine, (2) paracrine when the target cell is nearby, and (3) endocrine when the cytokine is secreted into the circulation and acts distal to the source.
Cytokines have been named based on presumed targets or based on presumed functions. Those cytokines that are thought to primarily target leukocytes have been named interleukins (IL-1, -2, -3, etc.). Many cytokines that were originally described as having a certain function have retained those names (e.g., granulocyte colony-stimulating factor [G-CSF]). Cytokines belong in general to three major structural families: the hematopoietin family; the TNF, IL-1, platelet-derived growth factor (PDGF), and transforming growth factor (TGF) β families; and the CXC and C-C chemokine families (Table 372e-9). Chemokines are cytokines that regulate cell movement and trafficking; they act through G protein-coupled receptors and have a distinctive three-dimensional structure. IL-8 is the only chemokine that early on was named an IL (Table 372e-7).
In general, cytokines exert their effects by influencing gene activation that results in cellular activation, growth, differentiation, functional cell-surface molecule expression, and cellular effector function. In this regard, cytokines can have dramatic effects on the regulation of immune responses and the pathogenesis of a variety of diseases. Indeed, T cells have been categorized on the basis of the pattern of cytokines that they secrete, which results in either humoral immune response (TH2) or cell-mediated immune response (TH1). A third type of T helper cell is the TH17 cell that contributes to host defense against extracellular bacteria and fungi, particularly at mucosal sites (Fig. 372e-2).
Cytokine receptors can be grouped into five general families based on similarities in their extracellular amino acid sequences and conserved structural domains. The immunoglobulin (Ig) superfamily represents a large number of cell-surface and secreted proteins. The IL-1 receptors (type 1, type 2) are examples of cytokine receptors with extracellular Ig domains.
The hallmark of the hematopoietic growth factor (type 1) receptor family is that the extracellular regions of each receptor contain two conserved motifs. One motif, located at the N terminus, is rich in cysteine residues. The other motif is located at the C terminus proximal to the transmembrane region and comprises five amino acid residues, tryptophan-serine-X-tryptophan-serine (WSXWS). This family can be grouped on the basis of the number of receptor subunits they have and on the utilization of shared subunits. A number of cytokine receptors, i.e., IL-6, IL-11, IL-12, and leukemia inhibitory factor, are paired with gp130. There is also a common 150-kDa subunit shared by IL-3, IL-5, and granulocyte-macrophage colony-stimulating factor (GM-CSF) receptors. The gamma chain (γc) of the IL-2 receptor is common to the IL-2, IL-4, IL-7, IL-9, and IL-15 receptors. Thus, the specific cytokine receptor is responsible for ligand-specific binding, whereas the subunits such as gp130, the 150-kDa subunit, and γc are important in signal transduction. The γc gene is on the × chromosome, and mutations in the γc protein result in the X-linked form of severe combined immune deficiency syndrome (X-SCID) (Chap. 374).
The members of the interferon (type II) receptor family include the receptors for IFN-γ and -β, which share a similar 210-amino-acid binding domain with conserved cysteine pairs at both the amino and carboxy termini. The members of the TNF (type III) receptor family share a common binding domain composed of repeated cysteine-rich regions. Members of this family include the p55 and p75 receptors for TNF (TNF-R1 and TNF-R2, respectively); CD40 antigen, which is an important B cell-surface marker involved in immunoglobulin isotype switching; fas/Apo-1, whose triggering induces apoptosis; CD27 and CD30, which are found on activated T cells and B cells; and nerve growth factor receptor.
The common motif for the seven transmembrane helix family was originally found in receptors linked to GTP-binding proteins. This family includes receptors for chemokines (Table 372e-8), β-adrenergic receptors, and retinal rhodopsin. It is important to note that two members of the chemokine receptor family, CXC chemokine receptor type 4 (CXCR4) and β chemokine receptor type 5 (CCR5), have been found to serve as the two major co-receptors for binding and entry of HIV into CD4-expressing host cells (Chap. 226).
Significant advances have been made in defining the signaling pathways through which cytokines exert their intracellular effects. The Janus family of protein tyrosine kinases (JAK) is a critical element involved in signaling via the hematopoietin receptors. Four JAK kinases, JAK1, JAK2, JAK3, and Tyk2, preferentially bind different cytokine receptor subunits. Cytokine binding to its receptor brings the cytokine receptor subunits into apposition and allows a pair of JAKs to transphosphorylate and activate one another. The JAKs then phosphorylate the receptor on the tyrosine residues and allow signaling molecules to bind to the receptor, whereby the signaling molecules become phosphorylated. Signaling molecules bind the receptor because they have domains (SH2, or src homology 2 domains) that can bind phosphorylated tyrosine residues. There are a number of these important signaling molecules that bind the receptor, such as the adapter molecule SHC, which can couple the receptor to the activation of the mitogen-activated protein kinase pathway. In addition, an important class of substrate of the JAKs is the signal transducers and activators of transcription (STAT) family of transcription factors. STATs have SH2 domains that enable them to bind to phosphorylated receptors, where they are then phosphorylated by the JAKs. It appears that different STATs have specificity for different receptor subunits. The STATs then dissociate from the receptor and translocate to the nucleus, bind to DNA motifs that they recognize, and regulate gene expression. The STATs preferentially bind DNA motifs that are slightly different from one another and thereby control transcription of specific genes. The importance of this pathway is particularly relevant to lymphoid development. Mutations of JAK3 itself also result in a disorder identical to X-SCID; however, because JAK3 is found on chromosome 19 and not on the × chromosome, JAK3 deficiency occurs in boys and girls (Chap. 374).
THE ADAPTIVE IMMUNE SYSTEM
Adaptive immunity is characterized by antigen-specific responses to a foreign antigen or pathogen. A key feature of adaptive immunity is that following the initial contact with antigen (immunologic priming), subsequent antigen exposure leads to more rapid and vigorous immune responses (immunologic memory). The adaptive immune system consists of dual limbs of cellular and humoral immunity. The principal effectors of cellular immunity are T lymphocytes, whereas the principal effectors of humoral immunity are B lymphocytes. Both B and T lymphocytes derive from a common stem cell (Fig. 372e-6).
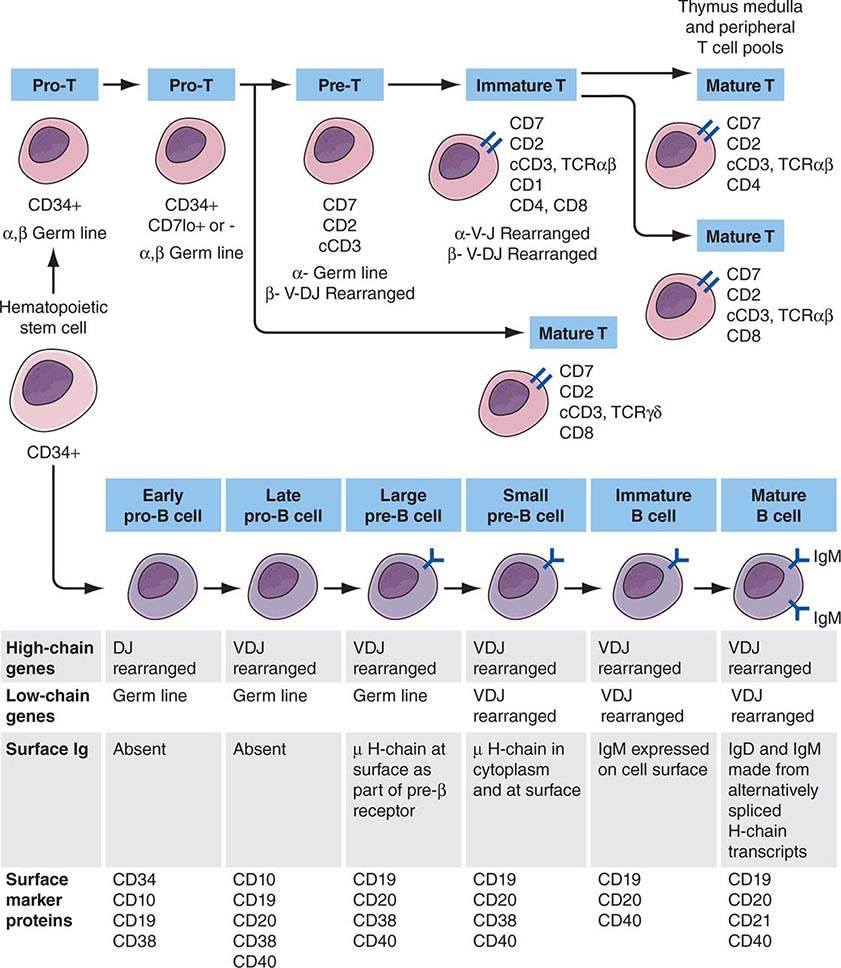
FIGURE 372e-6 Development stages of T and B cells. Elements of the developing T and B cell receptor for antigen are shown schematically. The classification into the various stages of B cell development is primarily defined by rearrangement of the immunoglobulin (Ig) heavy (H) and light (L) chain genes and by the absence or presence of specific surface markers. The classification of stages of T cell development is primarily defined by cell-surface marker protein expression (sCD3, surface CD3 expression; cCD3, cytoplasmic CD3 expression; TCR, T cell receptor). (Adapted from CA Janeway et al [eds]: Immunobiology. The Immune Systemic Health and Disease, 4th ed. New York, Garland, 1999; with permission.)
The proportion and distribution of immunocompetent cells in various tissues reflect cell traffic, homing patterns, and functional capabilities. Bone marrow is the major site of maturation of B cells, monocytes-macrophages, DCs, and granulocytes and contains pluripotent stem cells that, under the influence of various colony-stimulating factors, are capable of giving rise to all hematopoietic cell types. T cell precursors also arise from hematopoietic stem cells and home to the thymus for maturation. Mature T lymphocytes, B lymphocytes, monocytes, and DCs enter the circulation and home to peripheral lymphoid organs (lymph nodes, spleen) and mucosal surface-associated lymphoid tissue (gut, genitourinary, and respiratory tracts) as well as the skin and mucous membranes and await activation by foreign antigen.
T Cells The pool of effector T cells is established in the thymus early in life and is maintained throughout life both by new T cell production in the thymus and by antigen-driven expansion of virgin peripheral T cells into “memory” T cells that reside in peripheral lymphoid organs. The thymus exports ~2% of the total number of thymocytes per day throughout life, with the total number of daily thymic emigrants decreasing by ~3% per year during the first four decades of life.
Mature T lymphocytes constitute 70–80% of normal peripheral blood lymphocytes (only 2% of the total-body lymphocytes are contained in peripheral blood), 90% of thoracic duct lymphocytes, 30–40% of lymph node cells, and 20–30% of spleen lymphoid cells. In lymph nodes, T cells occupy deep paracortical areas around B cell germinal centers, and in the spleen, they are located in periarteriolar areas of white pulp (Chap. 79). T cells are the primary effectors of cell-mediated immunity, with subsets of T cells maturing into CD8+ cytotoxic T cells capable of lysis of virus-infected or foreign cells (short-lived effector T cells) and CD4+ T cells capable of T cell help for CD8+ T cell and B cell development. Two populations of long-lived memory T cells are triggered by infections: effector memory and central memory T cells. Effector memory T cells reside in nonlymphoid organs and respond rapidly to repeated pathogenic infections with cytokine production and cytotoxic functions to kill virus-infected cells. Central memory T cells home to lymphoid organs where they replenish long- and short-lived and effector memory T cells as needed.
In general, CD4+ T cells are the primary regulatory cells of T and B lymphocyte and monocyte function by the production of cytokines and by direct cell contact (Fig. 372e-2). In addition, T cells regulate erythroid cell maturation in bone marrow and, through cell contact (CD40 ligand), have an important role in activation of B cells and induction of Ig isotype switching. Considerable evidence now exists that colonization of the gut by commensal bacteria (the gut microbiome) is responsible for expansion of the peripheral CD4+ T cell compartment in normal children and adults.
Human T cells express cell-surface proteins that mark stages of intrathymic T cell maturation or identify specific functional subpopulations of mature T cells. Many of these molecules mediate or participate in important T cell functions (Table 372e-1, Fig. 372e-6).
The earliest identifiable T cell precursors in bone marrow are CD34+ pro-T cells (i.e., cells in which TCR genes are neither rearranged nor expressed). In the thymus, CD34+ T cell precursors begin cytoplasmic (c) synthesis of components of the CD3 complex of TCR-associated molecules (Fig. 372e-6). Within T cell precursors, TCR for antigen gene rearrangement yields two T cell lineages, expressing either TCR-αβ chains or TCR-γδ chains. T cells expressing the TCR-αβ chains constitute the majority of peripheral T cells in blood, lymph node, and spleen and terminally differentiate into either CD4+ or CD8+ cells. Cells expressing TCR-γδ chains circulate as a minor population in blood; their functions, although not fully understood, have been postulated to be those of immune surveillance at epithelial surfaces and cellular defenses against mycobacterial organisms and other intracellular bacteria through recognition of bacterial lipids.
In the thymus, the recognition of self-peptides on thymic epithelial cells, thymic macrophages, and DCs plays an important role in shaping the T cell repertoire to recognize foreign antigen (positive selection) and in eliminating highly autoreactive T cells (negative selection). As immature cortical thymocytes begin to express surface TCR for antigen, autoreactive thymocytes are destroyed (negative selection), thymocytes with TCRs capable of interacting with foreign antigen peptides in the context of self-MHC antigens are activated and develop to maturity (positive selection), and thymocytes with TCRs that are incapable of binding to self-MHC antigens die of attrition (no selection). Mature thymocytes that are positively selected are either CD4+ helper T cells or MHC class II–restricted cytotoxic (killer) T cells, or they are CD8+ T cells destined to become MHC class I–restricted cytotoxic T cells. MHC class I– or class II–restricted means that T cells recognize antigen peptide fragments only when they are presented in the antigen-recognition site of a class I or class II MHC molecule, respectively (Chap. 373e).
After thymocyte maturation and selection, CD4 and CD8 thymocytes leave the thymus and migrate to the peripheral immune system. The thymus continues to be a contributor to the peripheral immune system well into adult life, both normally and when the peripheral T cell pool is damaged, such as occurs in AIDS and cancer chemotherapy.
MOLECULAR BASIS OF T CELL RECOGNITION OF ANTIGEN The TCR for antigen is a complex of molecules consisting of an antigen-binding heterodimer of either αβ or γδ chains noncovalently linked with five CD3 subunits (γ, δ, ε, ζ, and η) (Fig. 372e-7). The CD3 ζ chains are either disulfide-linked homodimers (CD3-ζ2) or disulfide-linked heterodimers composed of one ζ chain and one η chain. TCR-αβ or TCR-γδ molecules must be associated with CD3 molecules to be inserted into the T cell-surface membrane, TCRα being paired with TCR-β and TCR-γ being paired with TCR-δ. Molecules of the CD3 complex mediate transduction of T cell activation signals via TCRs, whereas TCR-α and -β or -γ and -δ molecules combine to form the TCR antigen-binding site.
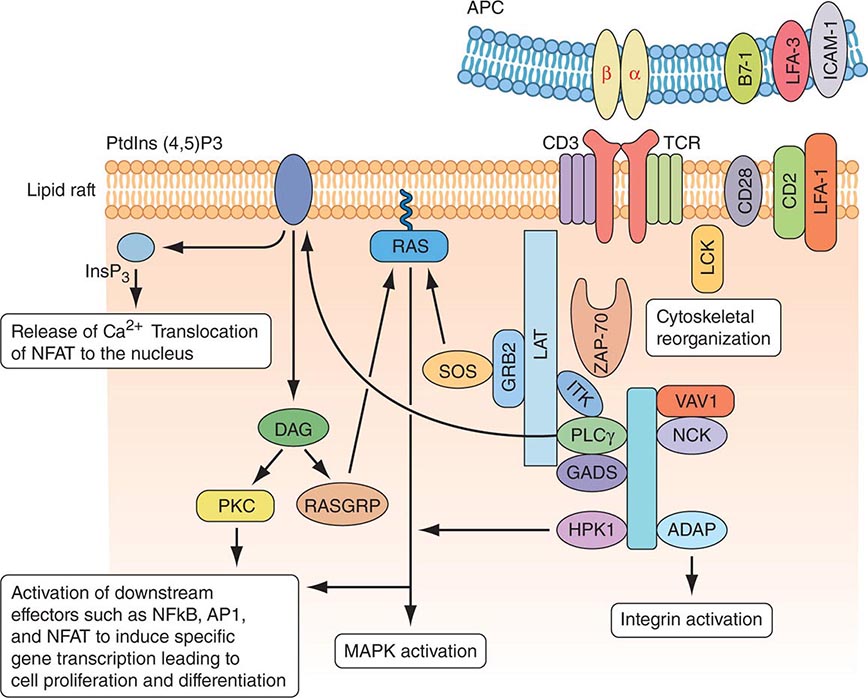
FIGURE 372e-7 Signaling through the T cell receptor. Activation signals are mediated via immunoreceptor tyrosine-based activation (ITAM) sequences in LAT and CD3 chains (blue bars) that bind to enzymes and transduce activation signals to the nucleus via the indicated intracellular activation pathways. Ligation of the T cell receptor (TCR) by MHC complexed with antigen results in sequential activation of LCK and γ-chain-associated protein kinase of 70 kDa (ZAP-70). ZAP-70 phosphorylates several downstream targets, including LAT (linker for activation of T cells) and SLP76 (SCR homology 2 [SH2] domain-containing leukocyte protein of 76 kDa). SLP76 is recruited to membrane-bound LAT through its constitutive interaction with GADS (GRB2-related adaptor protein). Together, SLP76 and LAT nucleate a multimolecular signaling complex, which induces a host of downstream responses, including calcium flux, mitogen-activated protein kinase (MAPK) activation, integrin activation, and cytoskeletal reorganization. APC, antigen-presenting cell. (Adapted from GA Koretzky et al: Nat Rev Immunol 6:67, 2006; with permission from Macmillan Publishers Ltd. Copyright 2006.)
The α, β, γ, and δ TCR for antigen molecules have amino acid sequence homology and structural similarities to immunoglobulin heavy and light chains and are members of the immunoglobulin gene superfamily of molecules. The genes encoding TCR molecules are encoded as clusters of gene segments that rearrange during the course of T cell maturation. This creates an efficient and compact mechanism for housing the diversity requirements of antigen receptor molecules. The TCR-α chain is on chromosome 14 and consists of a series of V (variable), J (joining), and C (constant) regions. The TCR-β chain is on chromosome 7 and consists of multiple V, D (diversity), J, and C TCR-β loci. The TCR-γ chain is on chromosome 7, and the TCR-δ chain is in the middle of the TCR-α locus on chromosome 14. Thus, molecules of the TCR for antigen have constant (framework) and variable regions, and the gene segments encoding the α, β, γ, and δ chains of these molecules are recombined and selected in the thymus, culminating in synthesis of the completed molecule. In both T and B cell precursors (see below), DNA rearrangements of antigen receptor genes involve the same enzymes, recombinase activating gene (RAG) 1 and RAG2, both DNA-dependent protein kinases.
TCR diversity is created by the different V, D, and J segments that are possible for each receptor chain by the many permutations of V, D, and J segment combinations, by “N-region diversification” due to the addition of nucleotides at the junction of rearranged gene segments, and by the pairing of individual chains to form a TCR dimer. As T cells mature in the thymus, the repertoire of antigen-reactive T cells is modified by selection processes that eliminate many autoreactive T cells, enhance the proliferation of cells that function appropriately with self-MHC molecules and antigen, and allow T cells with nonproductive TCR rearrangements to die.
TCR-αβ cells do not recognize native protein or carbohydrate antigens. Instead, T cells recognize only short (~9–13 amino acids) peptide fragments derived from protein antigens taken up or produced in APCs. Foreign antigens may be taken up by endocytosis into acidified intracellular vesicles or by phagocytosis and degraded into small peptides that associate with MHC class II molecules (exogenous antigen-presentation pathway). Other foreign antigens arise endogenously in the cytosol (such as from replicating viruses) and are broken down into small peptides that associate with MHC class I molecules (endogenous antigen-presenting pathway). Thus, APCs proteolytically degrade foreign proteins and display peptide fragments embedded in the MHC class I or II antigen-recognition site on the MHC molecule surface, where foreign peptide fragments are available to bind to TCR-αβ or TCR-γδ chains of reactive T cells. CD4 molecules act as adhesives and, by direct binding to MHC class II (DR, DQ, or DP) molecules, stabilize the interaction of TCR with peptide antigen (Fig. 372e-7). Similarly, CD8 molecules also act as adhesives to stabilize the TCR-antigen interaction by direct CD8 molecule binding to MHC class I (A, B, or C) molecules.
Antigens that arise in the cytosol and are processed via the endogenous antigen-presentation pathway are cleaved into small peptides by a complex of proteases called the proteasome. From the proteasome, antigen peptide fragments are transported from the cytosol into the lumen of the endoplasmic reticulum by a heterodimeric complex termed transporters associated with antigen processing, or TAP proteins. There, MHC class I molecules in the endoplasmic reticulum membrane physically associate with processed cytosolic peptides. Following peptide association with class I molecules, peptide-class I complexes are exported to the Golgi apparatus, and then to the cell surface, for recognition by CD8+ T cells.
Antigens taken up from the extracellular space via endocytosis into intracellular acidified vesicles are degraded by vesicle proteases into peptide fragments. Intracellular vesicles containing MHC class II molecules fuse with peptide-containing vesicles, thus allowing peptide fragments to physically bind to MHC class II molecules. Peptide-MHC class II complexes are then transported to the cell surface for recognition by CD4+ T cells (Chap. 373e).
Whereas it is generally agreed that the TCR-αβ receptor recognizes peptide antigens in the context of MHC class I or class II molecules, lipids in the cell wall of intracellular bacteria such as M. tuberculosis can also be presented to a wide variety of T cells, including subsets of TCR-γδ T cells, and a subset of CD8+ TCR-αβ T cells. Importantly, bacterial lipid antigens are not presented in the context of MHC class I or II molecules, but rather are presented in the context of MHC-related CD1 molecules. Some γδ T cells that recognize lipid antigens via CD1 molecules have very restricted TCR usage, do not need antigen priming to respond to bacterial lipids, and may actually be a form of innate rather than acquired immunity to intracellular bacteria.
Just as foreign antigens are degraded and their peptide fragments presented in the context of MHC class I or class II molecules on APCs, endogenous self-proteins also are degraded, and self-peptide fragments are presented to T cells in the context of MHC class I or class II molecules on APCs. In peripheral lymphoid organs, there are T cells that are capable of recognizing self-protein fragments but normally are anergic or tolerant, i.e., nonresponsive to self-antigenic stimulation, due to lack of self-antigen upregulating APC co-stimulatory molecules such as B7-1 (CD80) and B7-2 (CD86) (see below).
Once engagement of mature T cell TCR by foreign peptide occurs in the context of self-MHC class I or class II molecules, binding of non-antigen-specific adhesion ligand pairs such as CD54-CD11/CD18 and CD58-CD2 stabilizes MHC peptide-TCR binding, and the expression of these adhesion molecules is upregulated (Fig. 372e-7). Once antigen ligation of the TCR occurs, the T cell membrane is partitioned into lipid membrane microdomains, or lipid rafts, that coalesce the key signaling molecules TCR/CD3 complex, CD28, CD2, LAT (linker for activation of T cells), intracellular activated (dephosphorylated) src family protein tyrosine kinases (PTKs), and the key CD3ζ-associated protein-70 (ZAP-70) PTK (Fig. 372e-7). Importantly, during T cell activation, the CD45 molecule, with protein tyrosine phosphatase activity, is partitioned away from the TCR complex to allow activating phosphorylation events to occur. The coalescence of signaling molecules of activated T lymphocytes in microdomains has suggested that T cell-APC interactions can be considered immunologic synapses, analogous in function to neuronal synapses.
After TCR-MHC binding is stabilized, activation signals are transmitted through the cell to the nucleus and lead to the expression of gene products important in mediating the wide diversity of T cell functions such as the secretion of IL-2. The TCR does not have intrinsic signaling activity but is linked to a variety of signaling pathways via immunoreceptor tyrosine-based activation motifs (ITAMs) expressed on the various CD3 chains that bind to proteins that mediate signal transduction. Each of the pathways results in the activation of particular transcription factors that control the expression of cytokine and cytokine receptor genes. Thus, antigen-MHC binding to the TCR induces the activation of the src family of PTKs, fyn and lck (lck is associated with CD4 or CD8 co-stimulatory molecules); phosphorylation of CD3ζ chain; activation of the related tyrosine kinases ZAP-70 and syk; and downstream activation of the calcium-dependent calcineurin pathway, the ras pathway, and the protein kinase C pathway. Each of these pathways leads to activation of specific families of transcription factors (including NF-AT, fos and jun, and rel/NF-κB) that form heteromultimers capable of inducing expression of IL-2, IL-2 receptor, IL-4, TNF-α, and other T cell mediators.
In addition to the signals delivered to the T cell from the TCR complex and CD4 and CD8, molecules on the T cell, such as CD28 and inducible co-stimulator (ICOS), and molecules on DCs, such as B7-1 (CD80) and B7-2 (CD86), also deliver important co-stimulatory signals that upregulate T cell cytokine production and are essential for T cell activation. If signaling through CD28 or ICOS does not occur, or if CD28 is blocked, the T cell becomes anergic rather than activated (see “Immune Tolerance and Autoimmunity” below). CTLA-4 (CD152) is similar to CD28 in its ability to bind CD80 and CD86. Unlike CD28, CTLA-4 transmits an inhibitory signal to T cells, acting as an off switch.
T CELL EXHAUSTION IN VIRAL INFECTIONS AND CANCER In chronic viral infections such as HIV-1, hepatitis C virus, and hepatitis B virus and in chronic malignancies, the persistence of antigen disrupts memory T cell function, resulting in defects in memory T cell responses. This has been defined as T cell exhaustion and is associated with T cell programmed cell death protein 1 (PD-1) (CD279) expression. Exhausted T cells have compromised proliferation and lose the ability to produce effector molecules, like IL-2, TNF-α, and IFN-γ. PD-1 downregulates T cell responses and is associated with T cell exhaustion and disease progression. For this reason, inhibition of T cell PD-1 activity to enhance effector T cell function is being explored as a target for immunotherapy in both viral infections and certain malignancies.
T CELL SUPERANTIGENS Conventional antigens bind to MHC class I or II molecules in the groove of the αβ heterodimer and bind to T cells via the V regions of the TCR-α and -β chains. In contrast, superantigens bind directly to the lateral portion of the TCR-β chain and MHC class II β chain and stimulate T cells based solely on the Vβ gene segment used independent of the D, J, and Vα sequences present. Superantigens are protein molecules capable of activating up to 20% of the peripheral T cell pool, whereas conventional antigens activate <1 in 10,000 T cells. T cell superantigens include staphylococcal enterotoxins and other bacterial products. Superantigen stimulation of human peripheral T cells occurs in the clinical setting of staphylococcal toxic shock syndrome, leading to massive overproduction of T cell cytokines that leads to hypotension and shock (Chap. 172).
B CELLS Mature B cells constitute 10–15% of human peripheral blood lymphocytes, 20–30% of lymph node cells, 50% of splenic lymphocytes, and ~10% of bone marrow lymphocytes. B cells express on their surface intramembrane immunoglobulin (Ig) molecules that function as B cell receptors (BCRs) for antigen in a complex of Ig-associated α and β signaling molecules with properties similar to those described in T cells (Fig. 372e-8). Unlike T cells, which recognize only processed peptide fragments of conventional antigens embedded in the notches of MHC class I and class II antigens of APCs, B cells are capable of recognizing and proliferating to whole unprocessed native antigens via antigen binding to B cell–surface Ig (sIg) receptors. B cells also express surface receptors for the Fc region of IgG molecules (CD32) as well as receptors for activated complement components (C3d or CD21, C3b or CD35). The primary function of B cells is to produce antibodies. B cells also serve as APCs and are highly efficient at antigen processing. Their antigen-presenting function is enhanced by a variety of cytokines. Mature B cells are derived from bone marrow precursor cells that arise continuously throughout life (Fig. 372e-6).
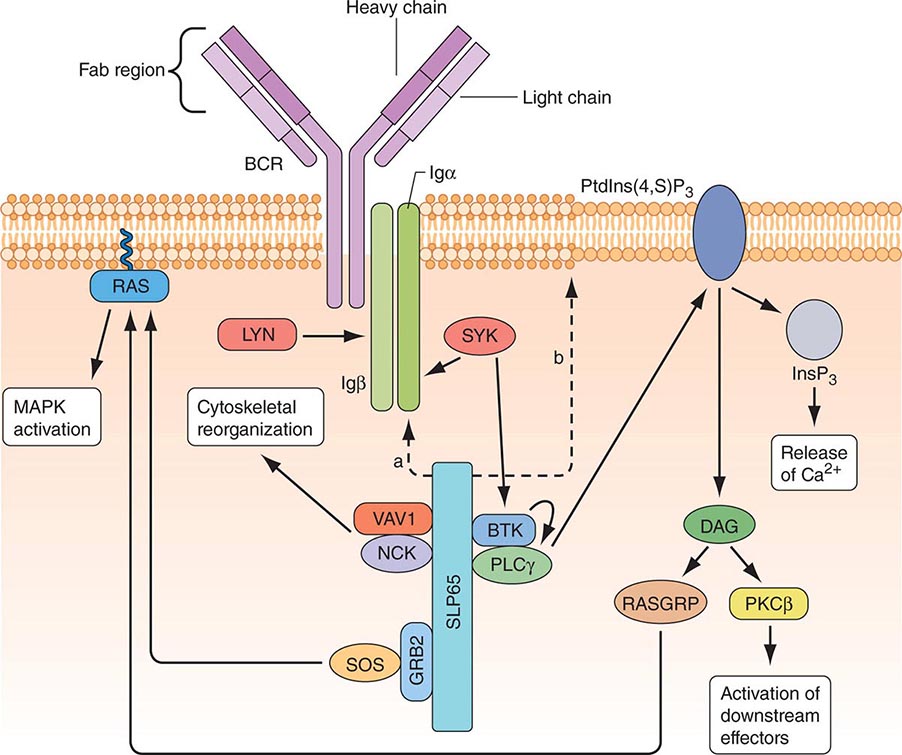
FIGURE 372e-8 B cell receptor (BCR) activation results in the sequential activation of protein tyrosine kinases, which results in the formation of a signaling complex and activation of downstream pathways as shown. Whereas SLP76 is recruited to the membrane through GADS and LAT, the mechanism of SLP65 recruitment is unclear. Studies have indicated two mechanisms: (a) direct binding by the SH2 domain of SLP65 to immunoglobulin (Ig) of the BCR complex or (b) membrane recruitment through a leucine zipper in the amino terminus of SLP65 and an unknown binding partner. ADAP, adhesion- and degranulation-promoting adaptor protein; AP1, activator protein 1; BTK, Bruton’s tyrosine kinase; DAG, diacylglycerol; GRB2, growth factor receptor-bound protein 2; HPK1, hematopoietic progenitor kinase 1; InsP3, inositol-1,4,5-trisphosphate; ITK, interleukin-2-inducible T cell kinase; NCK, noncatalytic region of tyrosine kinase; NF-B, nuclear factor B; PKC, protein kinase C; PLC, phospholipase C; PtdIns(4,5)P2, phosphatidylinositol-4,5-bisphosphate; RASGRP, RAS guanyl-releasing protein; SOS, son of sevenless homologue; SYK, spleen tyrosine kinase. (Adapted from GA Koretzky et al: Nat Rev Immunol 6:67, 2006; with permission from Macmillan Publishers Ltd. Copyright 2006.)
B lymphocyte development can be separated into antigen-independent and antigen-dependent phases. Antigen-independent B cell development occurs in primary lymphoid organs and includes all stages of B cell maturation up to the sIg+ mature B cell. Antigen-dependent B cell maturation is driven by the interaction of antigen with the mature B cell sIg, leading to memory B cell induction, Ig class switching, and plasma cell formation. Antigen-dependent stages of B cell maturation occur in secondary lymphoid organs, including lymph node, spleen, and gut Peyer’s patches. In contrast to the T cell repertoire that is generated intrathymically before contact with foreign antigen, the repertoire of B cells expressing diverse antigen-reactive sites is modified by further alteration of Ig genes after stimulation by antigen—a process called somatic hypermutation—that occurs in lymph node germinal centers.
During B cell development, diversity of the antigen-binding variable region of Ig is generated by an ordered set of Ig gene rearrangements that are similar to the rearrangements undergone by TCR α, β, γ, and δ genes. For the heavy chain, there is first a rearrangement of D segments to J segments, followed by a second rearrangement between a V gene segment and the newly formed D-J sequence; the C segment is aligned to the V-D-J complex to yield a functional Ig heavy chain gene (V-D-J-C). During later stages, a functional κ or γ light chain gene is generated by rearrangement of a V segment to a J segment, ultimately yielding an intact Ig molecule composed of heavy and light chains.
The process of Ig gene rearrangement is regulated and results in a single antibody specificity produced by each B cell, with each Ig molecule comprising one type of heavy chain and one type of light chain. Although each B cell contains two copies of Ig light and heavy chain genes, only one gene of each type is productively rearranged and expressed in each B cell, a process termed allelic exclusion.
There are ~300 Vκ genes and 5 Jκ genes, resulting in the pairing of Vκ and Jκ genes to create >1500 different kappa light chain combinations. There are ~70 Vλ genes and 4Jλ genes for >280 different lambda light chain combinations. The number of distinct light chains that can be generated is increased by somatic mutations within the V and J genes, thus creating large numbers of possible specificities from a limited amount of germline genetic information. As noted above, in heavy chain Ig gene rearrangement, the VH domain is created by the joining of three types of germline genes called VH, DH, and JH, thus allowing for even greater diversity in the variable region of heavy chains than of light chains.
The most immature B cell precursors (early pro-B cells) lack cytoplasmic Ig (cIg) and sIg (Fig. 372e-6). The large pre-B cell is marked by the acquisition of the surface pre-BCR composed of μ heavy (H) chains and a pre-B light chain, termed ψLC. ψLC is a surrogate light chain receptor encoded by the nonrearranged V pre-B and the γ5 light chain locus (the pre-BCR). Pro- and pre-B cells are driven to proliferate and mature by signals from bone marrow stroma—in particular, IL-7. Light chain rearrangement occurs in the small pre-B cell stage such that the full BCR is expressed at the immature B cell stage. Immature B cells have rearranged Ig light chain genes and express sIgM. As immature B cells develop into mature B cells, sIgD is expressed as well as sIgM. At this point, B lineage development in bone marrow is complete, and B cells exit into the peripheral circulation and migrate to secondary lymphoid organs to encounter specific antigens.
Random rearrangements of Ig genes occasionally generate self-reactive antibodies, and mechanisms must be in place to correct these mistakes. One such mechanism is BCR editing, whereby autoreactive BCRs are mutated to not react with self-antigens. If receptor editing is unsuccessful in eliminating autoreactive B cells, then autoreactive B cells undergo negative selection in the bone marrow through induction of apoptosis after BCR engagement of self-antigen.
After leaving the bone marrow, B cells populate peripheral B cell sites, such as lymph node and spleen, and await contact with foreign antigens that react with each B cell’s clonotypic receptor. Antigen-driven B cell activation occurs through the BCR, and a process known as somatic hypermutation takes place whereby point mutations in rearranged H- and L-genes give rise to mutant sIg molecules, some of which bind antigen better than the original sIg molecules. Somatic hypermutation, therefore, is a process whereby memory B cells in peripheral lymph organs have the best binding, or the highest-affinity antibodies. This overall process of generating the best antibodies is called affinity maturation of antibody.
Lymphocytes that synthesize IgG, IgA, and IgE are derived from sIgM+, sIgD+ mature B cells. Ig class switching occurs in lymph node and other peripheral lymphoid tissue germinal centers. CD40 on B cells and CD40 ligand on T cells constitute a critical co-stimulatory receptor-ligand pair of immune-stimulatory molecules. Pairs of CD40+ B cells and CD40 ligand+ T cells bind and drive B cell Ig class switching via T cell-produced cytokines such as IL-4 and TGF-β. IL-1, -2, -4, -5, and -6 synergize to drive mature B cells to proliferate and differentiate into Ig-secreting cells.
Humoral Mediators of Adaptive Immunity: Immunoglobulins Immunoglobulins are the products of differentiated B cells and mediate the humoral arm of the immune response. The primary functions of antibodies are to bind specifically to antigen and bring about the inactivation or removal of the offending toxin, microbe, parasite, or other foreign substance from the body. The structural basis of Ig molecule function and Ig gene organization has provided insight into the role of antibodies in normal protective immunity, pathologic immune-mediated damage by immune complexes, and autoantibody formation against host determinants.
All immunoglobulins have the basic structure of two heavy and two light chains (Fig. 372e-8). Immunoglobulin isotype (i.e., G, M, A, D, E) is determined by the type of Ig heavy chain present. IgG and IgA isotypes can be divided further into subclasses (G1, G2, G3, G4, and A1, A2) based on specific antigenic determinants on Ig heavy chains. The characteristics of human immunoglobulins are outlined in Table 372e-12. The four chains are covalently linked by disulfide bonds. Each chain is made up of a V region and C regions (also called domains), themselves made up of units of ~110 amino acids. Light chains have one variable (VL) and one constant (CL) unit; heavy chains have one variable unit (VH) and three or four constant (CH) units, depending on isotype. As the name suggests, the constant, or C, regions of Ig molecules are made up of homologous sequences and share the same primary structure as all other Ig chains of the same isotype and subclass. Constant regions are involved in biologic functions of Ig molecules. The CH2 domain of IgG and the CH4 units of IgM are involved with the binding of the C1q portion of C1 during complement activation. The CH region at the carboxy-terminal end of the IgG molecule, the Fc region, binds to surface Fc receptors (CD16, CD32, CD64) of macrophages, DCs, NK cells, B cells, neutrophils, and eosinophils. The Fc of IgA binds to FcαR (CD89), and the Fc of IgE binds to FcεR (CD23).
|
PHYSICAL, CHEMICAL, AND BIOLOGIC PROPERTIES OF HUMAN IMMUNOGLOBULINS |
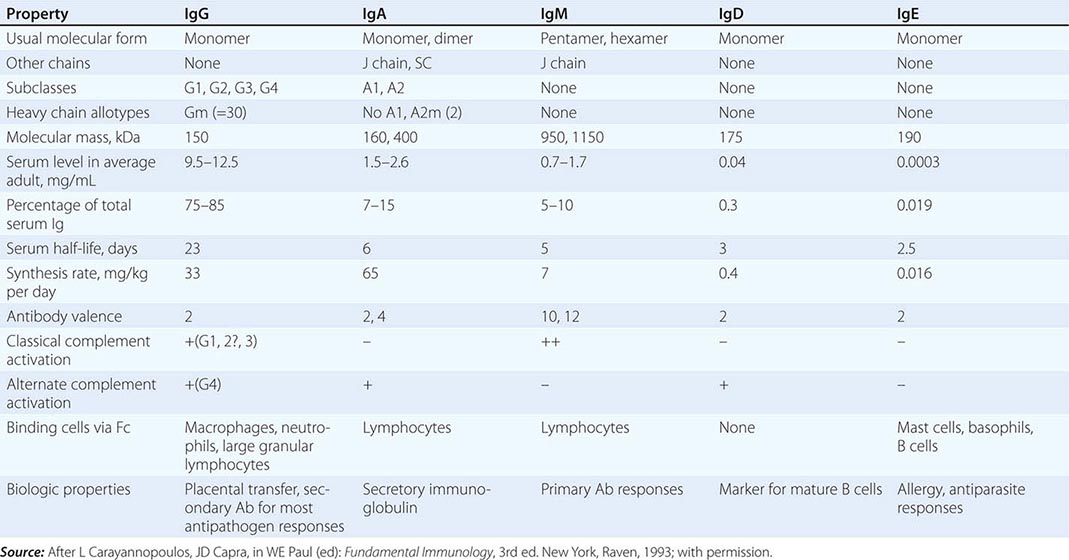
Variable regions (VL and VH) constitute the antibody-binding (Fab) region of the molecule. Within the VL and VH regions are hypervariable regions (extreme sequence variability) that constitute the antigen-binding site unique to each Ig molecule. The idiotype is defined as the specific region of the Fab portion of the Ig molecule to which antigen binds. Antibodies against the idiotype portion of an antibody molecule are called anti-idiotype antibodies. The formation of such antibodies in vivo during a normal B cell antibody response may generate a negative (or “off”) signal to B cells to terminate antibody production.
IgG constitutes ~75–85% of total serum immunoglobulin. The four IgG subclasses are numbered in order of their level in serum, IgG1 being found in greatest amounts and IgG4 the least. IgG subclasses have clinical relevance in their varying ability to bind macrophage and neutrophil Fc receptors and to activate complement (Table 372e-12). Moreover, selective deficiencies of certain IgG subclasses give rise to clinical syndromes in which the patient is inordinately susceptible to bacterial infections. IgG antibodies are frequently the predominant antibody made after rechallenge of the host with antigen (secondary antibody response).
IgM antibodies normally circulate as a 950-kDa pentamer with 160-kDa bivalent monomers joined by a molecule called the J chain, a 15-kDa nonimmunoglobulin molecule that also effects polymerization of IgA molecules. IgM is the first immunoglobulin to appear in the immune response (primary antibody response) and is the initial type of antibody made by neonates. Membrane IgM in the monomeric form also functions as a major antigen receptor on the surface of mature B cells (Table 372e-12). IgM is an important component of immune complexes in autoimmune diseases. For example, IgM antibodies against IgG molecules (rheumatoid factors) are present in high titers in rheumatoid arthritis, other collagen diseases, and some infectious diseases (subacute bacterial endocarditis).
IgA constitutes only 7–15% of total serum immunoglobulin but is the predominant class of immunoglobulin in secretions. IgA in secretions (tears, saliva, nasal secretions, gastrointestinal tract fluid, and human milk) is in the form of secretory IgA (sIgA), a polymer consisting of two IgA monomers, a joining molecule, again called the J chain, and a glycoprotein called the secretory protein. Of the two IgA subclasses, IgA1 is primarily found in serum, whereas IgA2 is more prevalent in secretions. IgA fixes complement via the alternative complement pathway and has potent antiviral activity in humans by prevention of virus binding to respiratory and gastrointestinal epithelial cells.
IgD is found in minute quantities in serum and, together with IgM, is a major receptor for antigen on the naïve B cell surface. IgE, which is present in serum in very low concentrations, is the major class of immunoglobulin involved in arming mast cells and basophils by binding to these cells via the Fc region. Antigen cross-linking of IgE molecules on basophil and mast cell surfaces results in release of mediators of the immediate hypersensitivity (allergic) response (Table 372e-12).
CELLULAR INTERACTIONS IN REGULATION OF NORMAL IMMUNE RESPONSES
The net result of activation of the humoral (B cell) and cellular (T cell) arms of the adaptive immune system by foreign antigen is the elimination of antigen directly by specific effector T cells or in concert with specific antibody. Figure 372e-2 is a simplified schematic diagram of the T and B cell responses indicating some of these cellular interactions.
The expression of adaptive immune cell function is the result of a complex series of immunoregulatory events that occur in phases. Both T and B lymphocytes mediate immune functions, and each of these cell types, when given appropriate signals, passes through stages, from activation and induction through proliferation, differentiation, and ultimately effector functions. The effector function expressed may be at the end point of a response, such as secretion of antibody by a differentiated plasma cell, or it might serve a regulatory function that modulates other functions, such as is seen with CD4+ and CD8+ T lymphocytes that modulate both differentiation of B cells and activation of CD8+ cytotoxic T cells.
CD4 helper T cells can be subdivided on the basis of cytokines produced (Fig. 372e-2). Activated TH1-type helper T cells secrete IL-2, IFN-γ, IL-3, TNF-α, GM-CSF, and TNF-β, whereas activated TH2-type helper T cells secrete IL-3, -4, -5, -6, -10, and -13. TH1 CD4+ T cells, through elaboration of IFN-γ, have a central role in mediating intracellular killing by a variety of pathogens. TH1 CD4+ T cells also provide T cell help for generation of cytotoxic T cells and some types of opsonizing antibody, and they generally respond to antigens that lead to delayed hypersensitivity types of immune responses for many intracellular viruses and bacteria (such as HIV or M. tuberculosis). In contrast, TH2 cells have a primary role in regulatory humoral immunity and isotype switching. TH2 cells, through production of IL-4 and IL-10, have a regulatory role in limiting proinflammatory responses mediated by TH1 cells (Fig. 372e-2). In addition, TH2 CD4+ T cells provide help to B cells for specific Ig production and respond to antigens that require high antibody levels for foreign antigen elimination (extracellular encapsulated bacteria such as Streptococcus pneumoniae and certain parasite infections). A new subset of the TH family has been described, termed TH17, characterized as cells that secrete cytokines such as IL-17, -22, and -26. TH17 cells have been shown to play a role in autoimmune inflammatory disorders in addition to defense against extracellular bacteria and fungi, particularly at mucosal surfaces. In summary, the type of T cell response generated in an immune response is determined by the microbe PAMPs presented to the DCs, the TLRs on the DCs that become activated, the types of DCs that are activated, and the cytokines that are produced (Table 372e-4). Commonly, myeloid DCs produce IL-12 and activate TH1 T cell responses that result in IFN-γ and cytotoxic T cell induction, and plasmacytoid DCs produce IFN-α and lead to TH2 responses that result in IL-4 production and enhanced antibody responses.
As shown in Figs. 372e-2 and 372e-3, upon activation by DCs, T cell subsets that produce IL-2, IL-3, IFN-γ, and/or IL-4, -5, -6, -10, and -13 are generated and exert positive and negative influences on effector T and B cells. For B cells, trophic effects are mediated by a variety of cytokines, particularly T cell–derived IL-3, -4, -5, and -6, that act at sequential stages of B cell maturation, resulting in B cell proliferation, differentiation, and ultimately antibody secretion. For cytotoxic T cells, trophic factors include inducer T cell secretion of IL-2, IFN-γ, and IL-12.
An important type of immunomodulatory T cell that controls immune responses is CD4+ and CD8+ T regulatory cells. These cells constitutively express the α chain of the IL-2 receptor (CD25), produce large amounts of IL-10, and can suppress both T and B cell responses. T regulatory cells are induced by immature DCs and play key roles in maintaining tolerance to self-antigens in the periphery. Loss of T regulatory cells is the cause of organ-specific autoimmune disease in mice such as autoimmune thyroiditis, adrenalitis, and oophoritis (see “Immune Tolerance and Autoimmunity” below). T regulatory cells also play key roles in controlling the magnitude and duration of immune responses to microbes. Normally, after the initial immune response to a microbe has eliminated the invader, T regulatory cells are activated to suppress the antimicrobe response and prevent host injury. Some microbes have adapted to induce T regulatory cell activation at the site of infection to promote parasite infection and survival. In Leishmania infection, the parasite locally induces T regulatory cell accumulation at skin infection sites that dampens anti-Leishmania T cell responses and prevents elimination of the parasite. It is thought that many chronic infections such as by M. tuberculosis are associated with abnormal T regulatory cell activation that prevents elimination of the microbe.
Although B cells recognize native antigen via B cell–surface Ig receptors, B cells require T cell help to produce high-affinity antibody of multiple isotypes that are the most effective in eliminating foreign antigen. This T cell dependence likely functions in the regulation of B cell responses and in protection against excessive autoantibody production. T cell–B cell interactions that lead to high-affinity antibody production require (1) processing of native antigen by B cells and expression of peptide fragments on the B cell surface for presentation to TH cells, (2) the ligation of B cells by both the TCR complex and the CD40 ligand, (3) induction of the process termed antibody isotype switching in antigen-specific B cell clones, and (4) induction of the process of affinity maturation of antibody in the germinal centers of B cell follicles of lymph node and spleen.
Naïve B cells express cell-surface IgD and IgM, and initial contact of naïve B cells with antigen is via binding of native antigen to B cell–surface IgM. T cell cytokines, released following TH2 cell contact with B cells or by a “bystander” effect, induce changes in Ig gene conformation that promote recombination of Ig genes. These events then result in the “switching” of expression of heavy chain exons in a triggered B cell, leading to the secretion of IgG, IgA, or, in some cases, IgE antibody with the same V region antigen specificity as the original IgM antibody, for response to a wide variety of extracellular bacteria, protozoa, and helminths. CD40 ligand expression by activated T cells is critical for induction of B cell antibody isotype switching and for B cell responsiveness to cytokines. Patients with mutations in T cell CD40 ligand have B cells that are unable to undergo isotype switching, resulting in lack of memory B cell generation and the immunodeficiency syndrome of X-linked hyper-IgM syndrome (Chap. 374).
IMMUNE TOLERANCE AND AUTOIMMUNITY
Immune tolerance is defined as the absence of activation of pathogenic autoreactivity. Autoimmune diseases are syndromes caused by the activation of T or B cells or both, with no evidence of other causes such as infections or malignancies (Chap. 377e). Once thought to be mutually exclusive, immune tolerance and autoimmunity are now both recognized to be present normally in health; when abnormal, they represent extremes from the normal state. For example, it is now known that low levels of autoreactivity of T and B cells with self-antigens in the periphery are critical to their survival. Similarly, low levels of autoreactivity and thymocyte recognition of self-antigens in the thymus are the mechanisms whereby (1) normal T cells are positively selected to survive and leave the thymus to respond to foreign microbes in the periphery and (2) T cells highly reactive to self-antigens are negatively selected and die to prevent overly self-reactive T cells from getting into the periphery (central tolerance). However, not all self-antigens are expressed in the thymus to delete highly self-reactive T cells, and there are mechanisms for peripheral tolerance induction of T cells as well. Unlike the presentation of microbial antigens by mature DCs, the presentation of self-antigens by immature DCs neither activates nor matures the DCs to express high levels of co-stimulatory molecules such as B7-1 (CD80) or B7-2 (CD86). When peripheral T cells are stimulated by DCs expressing self-antigens in the context of HLA molecules, sufficient stimulation of T cells occurs to keep them alive, but otherwise they remain anergic, or nonresponsive, until they contact a DC with high levels of co-stimulatory molecules expressing microbial antigens. In the latter setting, normal T cells then become activated to respond to the microbe. If B cells have high-self-reactivity BCRs, they normally undergo either deletion in the bone marrow or receptor editing to express a less autoreactive receptor. Although many autoimmune diseases are characterized by abnormal or pathogenic autoantibody production (Table 372e-13), most autoimmune diseases are caused by a combination of excess T and B cell reactivity.
|
RECOMBINANT OR PURIFIED AUTOANTIGENS RECOGNIZED BY AUTOANTIBODIES ASSOCIATED WITH HUMAN AUTOIMMUNE DISORDERS |
Multiple factors contribute to the genesis of clinical autoimmune disease syndromes, including genetic susceptibility (Table 372e-13), environmental immune stimulants such as drugs (e.g., procainamide and phenytoin [Dilantin] with drug-induced systemic lupus erythematosus), infectious agent triggers (such as Epstein-Barr virus and autoantibody production against red blood cells and platelets), and loss of T regulatory cells (leading to thyroiditis, adrenalitis, and oophoritis).
Immunity at Mucosal Surfaces Mucosa covering the respiratory, digestive, and urogenital tracts; the eye conjunctiva; the inner ear; and the ducts of all exocrine glands contain cells of the innate and adaptive mucosal immune system that protect these surfaces against pathogens. In the healthy adult, mucosa-associated lymphoid tissue (MALT) contains 80% of all immune cells within the body and constitutes the largest mammalian lymphoid organ system.
MALT has three main functions: (1) to protect the mucous membranes from invasive pathogens; (2) to prevent uptake of foreign antigens from food, commensal organisms, and airborne pathogens and particulate matter; and (3) to prevent pathologic immune responses from foreign antigens if they do cross the mucosal barriers of the body (Fig. 372e-9).
FIGURE 372e-9 Increased epithelial permeability may be important in the development of chronic gut T cell–mediated inflammation. CD4 T cells activated by gut antigens in Peyer’s patches migrate to the lamina propria (LP). In healthy individuals, these cells die by apoptosis. Increased epithelial permeability may allow sufficient antigen to enter the LP to trigger T cell activation, breaking tolerance mediated by immunosuppressive cytokines and perhaps T regulatory cells. Proinflammatory cytokines then further increase epithelial permeability, setting up a vicious cycle of chronic inflammation. (From TT MacDonald et al: Science 307:1920, 2005; with permission.)
MALT is a compartmentalized system of immune cells that functions independently from systemic immune organs. Whereas the systemic immune organs are essentially sterile under normal conditions and respond vigorously to pathogens, MALT immune cells are continuously bathed in foreign proteins and commensal bacteria, and they must select those pathogenic antigens that must be eliminated. MALT contains anatomically defined foci of immune cells in the intestine, tonsil, appendix, and peribronchial areas that are inductive sites for mucosal immune responses. From these sites, immune T and B cells migrate to effector sites in mucosal parenchyma and exocrine glands where mucosal immune cells eliminate pathogen-infected cells. In addition to mucosal immune responses, all mucosal sites have strong mechanical and chemical barriers and cleansing functions to repel pathogens.
Key components of MALT include specialized epithelial cells called “membrane” or “M” cells that take up antigens and deliver them to DCs or other APCs. Effector cells in MALT include B cells producing antipathogen neutralizing antibodies of secretory IgA as well as IgG isotype, T cells producing similar cytokines as in systemic immune system response, and T helper and cytotoxic T cells that respond to pathogen-infected cells.
Secretory IgA is produced in amounts of >50 mg/kg of body weight per 24 h and functions to inhibit bacterial adhesion, inhibit macromolecule absorption in the gut, neutralize viruses, and enhance antigen elimination in tissue through binding to IgA and receptor-mediated transport of immune complexes through epithelial cells.
Recent studies have demonstrated the importance of commensal gut and other mucosal bacteria to the health of the human immune system. Normal commensal flora induces anti-inflammatory events in the gut and protects epithelial cells from pathogens through TLRs and other PRR signaling. When the gut is depleted of normal commensal flora, the immune system becomes abnormal, with loss of TH1 T cell function. Restoration of the normal gut flora can reestablish the balance in T helper cell ratios characteristic of the normal immune system. When the gut barrier is intact, either antigens do not transverse the gut epithelium or, when pathogens are present, a self-limited, protective MALT immune response eliminates the pathogen (Fig. 372e-9). However, when the gut barrier breaks down, immune responses to commensal flora antigens can cause inflammatory bowel diseases such as Crohn’s disease and, perhaps, ulcerative colitis (Fig. 372e-9) (Chap. 351). Uncontrolled MALT immune responses to food antigens, such as gluten, can cause celiac disease (Chap. 351).
THE CELLULAR AND MOLECULAR CONTROL OF PROGRAMMED CELL DEATH
The process of apoptosis (programmed cell death) plays a crucial role in regulating normal immune responses to antigen. In general, a wide variety of stimuli trigger one of several apoptotic pathways to eliminate microbe-infected cells, eliminate cells with damaged DNA, or eliminate activated immune cells that are no longer needed (Fig. 372e-10). The largest known family of “death receptors” is the TNF receptor (TNF-R) family (TNF-R1, TNF-R2, Fas [CD95], death receptor 3 [DR3], death receptor 4 [DR4; TNF-related apoptosis-including ligand receptor 1, or TRAIL-R1], and death receptor 5 [DR5, TRAIL-R2]); their ligands are all in the TNF-α family. Binding of ligands to these death receptors leads to a signaling cascade that involves activation of the caspase family of molecules that leads to DNA cleavage and cell death. Two other pathways of programmed cell death involve nuclear p53 in the elimination of cells with abnormal DNA and mitochondrial cytochrome c to induce cell death in damaged cells (Fig. 372e-10). A number of human diseases have now been described that result from, or are associated with, mutated apoptosis genes (Table 372e-14). These include mutations in the Fas and Fas ligand genes in autoimmune and lymphoproliferation syndromes, and multiple associations of mutations in genes in the apoptotic pathway with malignant syndromes.
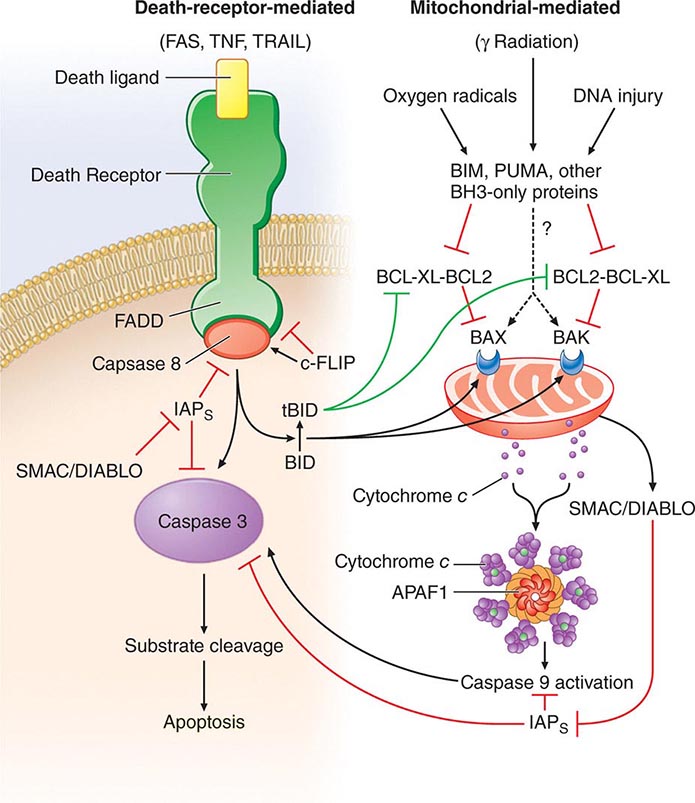
FIGURE 372e-10 Pathways of cellular apoptosis. There are two major pathways of apoptosis: the death-receptor pathway, which is mediated by activation of death receptors, and the BCL2-regulated mitochondrial pathway, which is mediated by noxious stimuli that ultimately lead to mitochondrial injury. Ligation of death receptors recruits the adaptor protein FAS-associated death domain (FADD). FADD in turn recruits caspase 8, which ultimately activates caspase 3, the key “executioner” caspase. Cellular FLICE-inhibitory protein (c-FLIP) can either inhibit or potentiate binding of FADD and caspase 8, depending on its concentration. In the intrinsic pathway, proapoptotic BH3 proteins are activated by noxious stimuli, which interact with and inhibit antiapoptotic BCL2 or BCL-XL. Thus, BAX and BAK are free to induce mitochondrial permeabilization with release of cytochrome c, which ultimately results in the activation of caspase 9 through the apoptosome. Caspase 9 then activates caspase 3. SMAC/DIABLO is also released after mitochondrial permeabilization and acts to block the action of inhibitors of apoptosis protein (IAPs), which inhibit caspase activation. There is potential cross-talk between the two pathways, which is mediated by the truncated form of BID (tBID) that is produced by caspase 8–mediated BID cleavage; tBID acts to inhibit the BCL2-BCL-XL pathway and to activate BAX and BAK. There is debate (indicated by the question mark) as to whether proapoptotic BH3 molecules (e.g., BIM and PUMA) act directly on BAX and BAK to induce mitochondrial permeability or whether they act only on BCL2-BCL-XL. APAF1, apoptotic protease-activating factor 1; BH3, BCL homologue; TNF, tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand. (From RS Hotchkiss et al: N Engl J Med 361:1570, 2009; with permission.)
|
IMMUNE SYSTEM MOLECULE DEFECTS IN ANIMALS OR HUMANS THAT CAUSE AUTOIMMUNE OR MALIGNANT SYNDROMES |
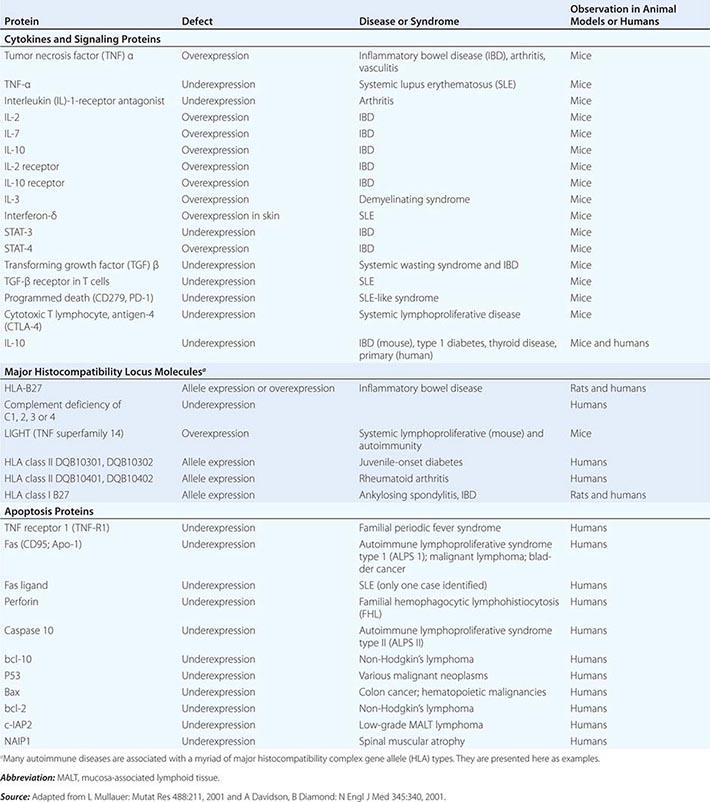
MECHANISMS OF IMMUNE-MEDIATED DAMAGE TO MICROBES OR HOST TISSUES
Several responses by the host innate and adaptive immune systems to foreign microbes culminate in rapid and efficient elimination of microbes. In these scenarios, the classic weapons of the adaptive immune system (T cells, B cells) interface with cells (macrophages, DCs, NK cells, neutrophils, eosinophils, basophils) and soluble products (microbial peptides, pentraxins, complement and coagulation systems) of the innate immune system (Chaps. 80 and 376).
There are five general phases of host defenses: (1) migration of leukocytes to sites of antigen localization; (2) antigen-nonspecific recognition of pathogens by macrophages and other cells and systems of the innate immune system; (3) specific recognition of foreign antigens mediated by T and B lymphocytes; (4) amplification of the inflammatory response with recruitment of specific and nonspecific effector cells by complement components, cytokines, kinins, arachidonic acid metabolites, and mast cell–basophil products; and (5) macrophage, neutrophil, and lymphocyte participation in destruction of antigen with ultimate removal of antigen particles by phagocytosis (by macrophages or neutrophils) or by direct cytotoxic mechanisms (involving macrophages, neutrophils, DCs, and lymphocytes). Under normal circumstances, orderly progression of host defenses through these phases results in a well-controlled immune and inflammatory response that protects the host from the offending antigen. However, dysfunction of any of the host defense systems can damage host tissue and produce clinical disease. Furthermore, for certain pathogens or antigens, the normal immune response itself might contribute substantially to the tissue damage. For example, the immune and inflammatory response in the brain to certain pathogens such as M. tuberculosis may be responsible for much of the morbidity rate of this disease in that organ system (Chap. 202). In addition, the morbidity rate associated with certain pneumonias such as that caused by Pneumocystis jiroveci may be associated more with inflammatory infiltrates than with the tissue-destructive effects of the microorganism itself (Chap. 244).
Molecular Basis of Lymphocyte–Endothelial Cell Interactions The control of lymphocyte circulatory patterns between the bloodstream and peripheral lymphoid organs operates at the level of lymphocyte–endothelial cell interactions to control the specificity of lymphocyte subset entry into organs. Similarly, lymphocyte–endothelial cell interactions regulate the entry of lymphocytes into inflamed tissue. Adhesion molecule expression on lymphocytes and endothelial cells regulates the retention and subsequent egress of lymphocytes within tissue sites of antigenic stimulation, delaying cell exit from tissue and preventing reentry into the circulating lymphocyte pool (Fig. 372e-11). All types of lymphocyte migration begin with lymphocyte attachment to specialized regions of vessels, termed high endothelial venules (HEVs). An important concept is that adhesion molecules do not generally bind their ligand until a conformational change (ligand activation) occurs in the adhesion molecule that allows ligand binding. Induction of a conformation-dependent determinant on an adhesion molecule can be accomplished by cytokines or via ligation of other adhesion molecules on the cell.
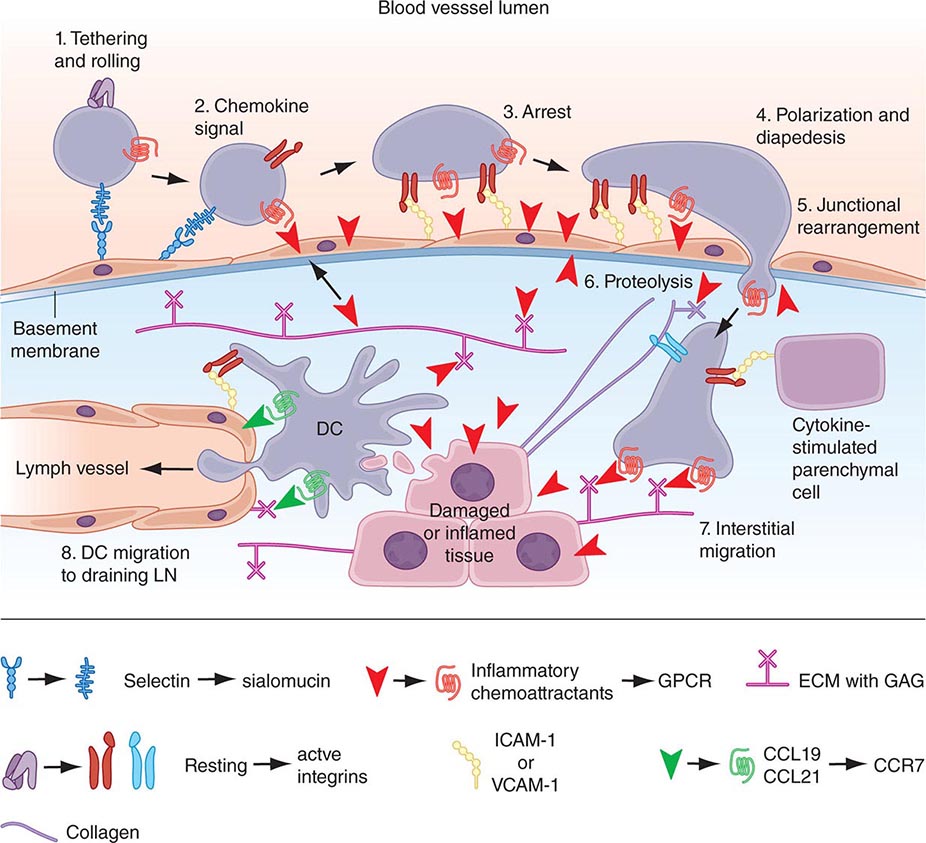
FIGURE 372e-11 Key migration steps of immune cells at sites of inflammation. Inflammation due to tissue damage or infection induces the release of cytokines (not shown) and inflammatory chemoattractants (red arrowheads) from distressed stromal cells and “professional” sentinels, such as mast cells and macrophages (not shown). The inflammatory signals induce upregulation of endothelial selectins and immunoglobulin “superfamily” members, particularly ICAM-1 and/or VCAM-1. Chemoattractants, particularly chemokines, are produced by or translocated across venular endothelial cells (red arrow) and are displayed in the lumen to rolling leukocytes. Those leukocytes that express the appropriate set of trafficking molecules undergo a multistep adhesion cascade (steps 1–3) and then polarize and move by diapedesis across the venular wall (steps 4 and 5). Diapedesis involves transient disassembly of endothelial junctions and penetration through the underlying basement membrane (step 6). Once in the extravascular (interstitial) space, the migrating cell uses different integrins to gain “footholds” on collagen fibers and other ECM molecules, such as laminin and fibronectin, and on inflammation-induced ICAM-1 on the surface of parenchymal cells (step 7). The migrating cell receives guidance cues from distinct sets of chemoattractants, particularly chemokines, which may be immobilized on glycosaminoglycans (GAG) that “decorate” many ECM molecules and stromal cells. Inflammatory signals also induce tissue dendritic cells (DCs) to undergo maturation. Once DCs process material from damaged tissues and invading pathogens, they upregulate CCR7, which allows them to enter draining lymph vessels that express the CCR7 ligand CCL21 (and CCL19). In lymph nodes (LNs), these antigen-loaded mature DCs activate naïve T cells and expand pools of effector lymphocytes, which enter the blood and migrate back to the site of inflammation. T cells in tissue also use this CCR7-dependent route to migrate from peripheral sites to draining lymph nodes through afferent lymphatics. (Adapted from AD Luster et al: Nat Immunol 6:1182, 2005; with permission from Macmillan Publishers Ltd. Copyright 2005.)
The first stage of lymphocyte–endothelial cell interactions, attachment and rolling, occurs when lymphocytes leave the stream of flowing blood cells in a postcapillary venule and roll along venule endothelial cells (Fig. 372e-11). Lymphocyte rolling is mediated by the L-selectin molecule (LECAM-1, LAM-1, CD62L) and slows cell transit time through venules, allowing time for activation of adherent cells.
The second stage of lymphocyte–endothelial cell interactions, firm adhesion with activation-dependent stable arrest, requires stimulation of lymphocytes by chemoattractants or by endothelial cell–derived cytokines. Cytokines thought to participate in adherent cell activation include members of the IL-8 family, platelet-activation factor, leukotriene B4, and C5a. In addition, HEVs express chemokines, SLC (CCL21) and ELC (CCL19), which participate in this process. Following activation by chemoattractants, lymphocytes shed L-selectin from the cell surface and upregulate cell CD11b/18 (MAC-1) or CD11a/18 (LFA-1) molecules, resulting in firm attachment of lymphocytes to HEVs.
Lymphocyte homing to peripheral lymph nodes involves adhesion of L-selectin to glycoprotein HEV ligands collectively referred to as peripheral node addressin (PNAd), whereas homing of lymphocytes to intestine Peyer’s patches primarily involves adhesion of the α4β7 integrin to mucosal addressin cell adhesion molecule-1 (MAdCAM-1) on the Peyer’s patch HEVs. However, for migration to mucosal Peyer’s patch lymphoid aggregates, naïve lymphocytes primarily use L-selectin, whereas memory lymphocytes use α4β7 integrin. α4β1 integrin (CD49d/CD29, VLA-4)–VCAM-1 interactions are important in the initial interaction of memory lymphocytes with HEVs of multiple organs in sites of inflammation (Table 372e-15).
|
TRAFFICKING MOLECULES INVOLVED IN INFLAMMATORY DISEASE PROCESSES |
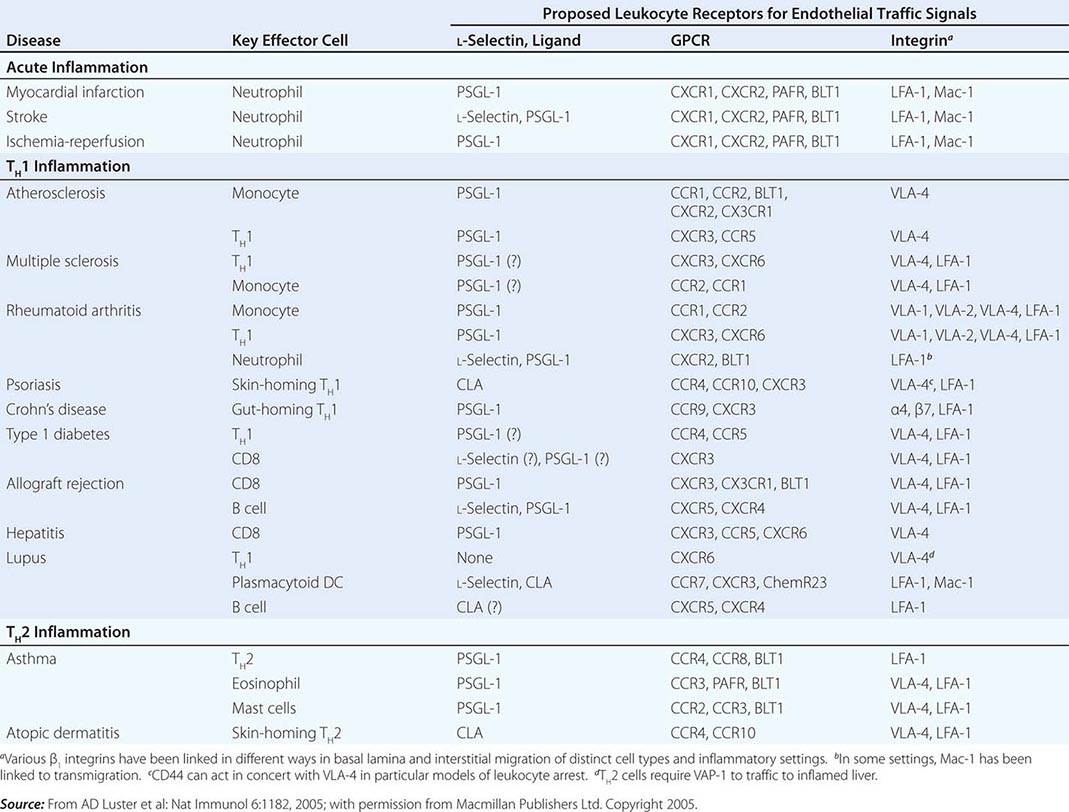
The third stage of leukocyte emigration in HEVs is sticking and arrest. Sticking of the lymphocyte to endothelial cells and arrest at the site of sticking are mediated predominantly by ligation of α1β2 integrin LFA-1 to the integrin ligand ICAM-1 on HEVs. Whereas the first three stages of lymphocyte attachment to HEVs take only a few seconds, the fourth stage of lymphocyte emigration, transendothelial migration, takes ~10 min. Although the molecular mechanisms that control lymphocyte transendothelial migration are not fully characterized, the HEV CD44 molecule and molecules of the HEV glycocalyx (extracellular matrix) are thought to play important regulatory roles in this process (Fig. 372e-11). Finally, expression of matrix metalloproteases capable of digesting the subendothelial basement membrane, rich in nonfibrillar collagen, appears to be required for the penetration of lymphoid cells into the extravascular sites.
Abnormal induction of HEV formation and use of the molecules discussed above have been implicated in the induction and maintenance of inflammation in a number of chronic inflammatory diseases. In animal models of type 1 diabetes mellitus, MAdCAM-1 and GlyCAM-1 have been shown to be highly expressed on HEVs in inflamed pancreatic islets, and treatment of these animals with inhibitors of L-selectin and α4 integrin function blocked the development of type 1 diabetes mellitus (Chap. 417). A similar role for abnormal induction of the adhesion molecules of lymphocyte emigration has been suggested in rheumatoid arthritis (Chap. 380), Hashimoto’s thyroiditis (Chap. 405), Graves’ disease (Chap. 405), multiple sclerosis (Chap. 458), Crohn’s disease (Chap. 351), and ulcerative colitis (Chap. 351).
Immune-Complex Formation Clearance of antigen by immune-complex formation between antigen, complement, and antibody is a highly effective mechanism of host defense. However, depending on the level of immune complexes formed and their physicochemical properties, immune complexes may or may not result in host and foreign cell damage. After antigen exposure, certain types of soluble antigen-antibody complexes freely circulate and, if not cleared by the reticuloendothelial system, can be deposited in blood vessel walls and in other tissues such as renal glomeruli and cause vasculitis or glomerulonephritis syndromes (Chaps. 338 and 385). Deficiencies of early complement components are associated with inefficient clearance of immune complexes and immune complex mediated tissue damage in autoimmune syndromes, whereas deficiencies of the later complement components are associated with susceptibility to recurrent Neisseria infections (Table 372e-16).
|
COMPLEMENT DEFICIENCIES AND ASSOCIATED DISEASES |
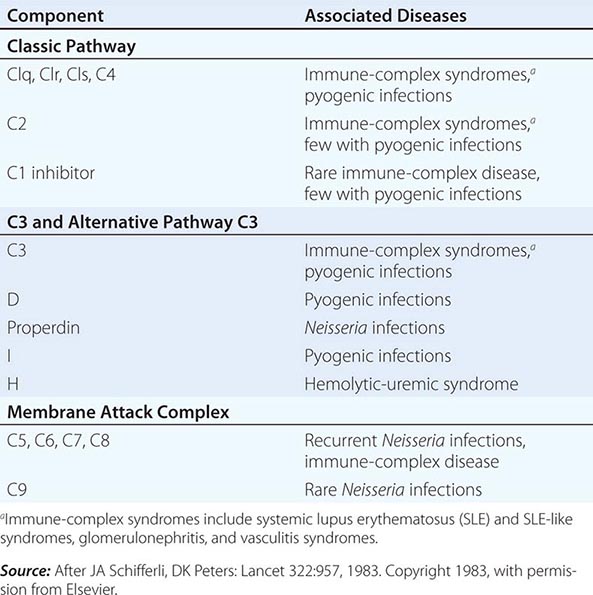
Immediate-Type Hypersensitivity Helper T cells that drive antiallergen IgE responses are usually TH2-type inducer T cells that secrete IL-4, IL-5, IL-6, and IL-10. Mast cells and basophils have high-affinity receptors for the Fc portion of IgE (FcRI), and cell-bound antiallergen IgE effectively “arms” basophils and mast cells. Mediator release is triggered by antigen (allergen) interaction with Fc receptor-bound IgE, and the mediators released are responsible for the pathophysiologic changes of allergic diseases (Table 372e-11). Mediators released from mast cells and basophils can be divided into three broad functional types: (1) those that increase vascular permeability and contract smooth muscle (histamine, platelet-activating factor, SRS-A, BK-A), (2) those that are chemotactic for or activate other inflammatory cells (ECF-A, NCF, leukotriene B4), and (3) those that modulate the release of other mediators (BK-A, platelet-activating factor) (Chap. 376).
Cytotoxic Reactions of Antibody In this type of immunologic injury, complement-fixing (C1-binding) antibodies against normal or foreign cells or tissues (IgM, IgG1, IgG2, IgG3) bind complement via the classic pathway and initiate a sequence of events similar to that initiated by immune-complex deposition, resulting in cell lysis or tissue injury. Examples of antibody-mediated cytotoxic reactions include red cell lysis in transfusion reactions, Goodpasture’s syndrome with anti–glomerular basement membrane antibody formation, and pemphigus vulgaris with antiepidermal antibodies inducing blistering skin disease.
Classic Delayed-Type Hypersensitivity Reactions Inflammatory reactions initiated by mononuclear leukocytes and not by antibody alone have been termed delayed-type hypersensitivity reactions. The term delayed has been used to contrast a secondary cellular response that appears 48–72 h after antigen exposure with an immediate hypersensitivity response generally seen within 12 h of antigen challenge and initiated by basophil mediator release or preformed antibody. For example, in an individual previously infected with M. tuberculosis organisms, intradermal placement of tuberculin purified protein derivative as a skin test challenge results in an indurated area of skin at 48–72 h, indicating previous exposure to tuberculosis.
The cellular events that result in classic delayed-type hypersensitivity responses are centered on T cells (predominantly, although not exclusively, IFN-γ, IL-2, and TNF-α-secreting TH1-type helper T cells) and macrophages. Recently NK cells have been suggested to play a major role in the form of delayed hypersensitivity that occurs following skin contact with immunogens. First, local immune and inflammatory responses at the site of foreign antigen upregulate endothelial cell adhesion molecule expression, promoting the accumulation of lymphocytes at the tissue site. In the general schemes outlined in Figs. 372e-2 and 372e-3, antigen is processed by DCs and presented to small numbers of CD4+ T cells expressing a TCR specific for the antigen. IL-12 produced by APCs induces T cells to produce IFN-γ (TH1 response). Macrophages frequently undergo epithelioid cell transformation and fuse to form multinucleated giant cells in response to IFN-γ. This type of mononuclear cell infiltrate is termed granulomatous inflammation. Examples of diseases in which delayed-type hypersensitivity plays a major role are fungal infections (histoplasmosis; Chap. 236), mycobacterial infections (tuberculosis, leprosy; Chaps. 202 and 203), chlamydial infections (lymphogranuloma venereum; Chap. 213), helminth infections (schistosomiasis; Chap. 259), reactions to toxins (berylliosis; Chap. 311), and hypersensitivity reactions to organic dusts (hypersensitivity pneumonitis; Chap. 310). In addition, delayed-type hypersensitivity responses play important roles in tissue damage in autoimmune diseases such as rheumatoid arthritis, temporal arteritis, and granulomatosis with polyangiitis (Wegener’s) (Chaps. 380 and 385).
CLINICAL EVALUATION OF IMMUNE FUNCTION
Clinical assessment of immunity requires investigation of the four major components of the immune system that participate in host defense and in the pathogenesis of autoimmune diseases: (1) humoral immunity (B cells); (2) cell-mediated immunity (T cells, monocytes); (3) phagocytic cells of the reticuloendothelial system (macrophages), as well as polymorphonuclear leukocytes; and (4) complement. Clinical problems that require an evaluation of immunity include chronic infections, recurrent infections, unusual infecting agents, and certain autoimmune syndromes. The type of clinical syndrome under evaluation can provide information regarding possible immune defects (Chap. 374). Defects in cellular immunity generally result in viral, mycobacterial, and fungal infections. An extreme example of deficiency in cellular immunity is AIDS (Chap. 226). Antibody deficiencies result in recurrent bacterial infections, frequently with organisms such as S. pneumoniae and Haemophilus influenzae (Chap. 374). Disorders of phagocyte function are frequently manifested by recurrent skin infections, often due to Staphylococcus aureus (Chap. 80). Finally, deficiencies of early and late complement components are associated with autoimmune phenomena and recurrent Neisseria infections (Table 372e-16). For further discussion of useful initial screening tests of immune function, see Chap. 374.
IMMUNOTHERAPY
Many therapies for autoimmune and inflammatory diseases involve the use of nonspecific immune-modulating or immunosuppressive agents such as glucocorticoids or cytotoxic drugs. The goal of development of new treatments for immune-mediated diseases is to design ways to specifically interrupt pathologic immune responses, leaving nonpathologic immune responses intact. Novel ways to interrupt pathologic immune responses that are under investigation include the use of anti-inflammatory cytokines or specific cytokine inhibitors as anti-inflammatory agents, the use of monoclonal antibodies against T or B lymphocytes as therapeutic agents, the use of intravenous Ig for certain infections and immune complex–mediated diseases, the use of specific cytokines to reconstitute components of the immune system, and bone marrow transplantation to replace the pathogenic immune system with a more normal immune system (Chaps. 80, 374, and 226). In particular, the use of a monoclonal antibody to B cells (rituximab, anti-CD20 MAb) is approved in the United States for the treatment of non-Hodgkin’s lymphoma (Chap. 134) and, in combination with methotrexate, for treatment of adult patients with severe rheumatoid arthritis resistant to TNF-α inhibitors (Chap. 380). The U.S. Food and Drug Administration (FDA) approved the use of CTLA-4 antibodies in 2010 to block T cell anergy for use in cancer immunotherapy, and it was the first agent to demonstrate survival benefit in patients with advanced melanoma. Early-stage clinical trials have now shown that PD-1 blockade to reverse T cell exhaustion can induce tumor regression.
Cell-based therapies have been studied for many years, including ex vivo activation of NK cells for reinfusion into patients with malignancies and DC therapy of ex vivo priming of DCs for enhanced presentation of cancer antigens, with reinfusion of primed DCs into the patient. One such strategy for DC therapy has been approved by the FDA for treatment of advanced prostate cancer.
Cytokines and Cytokine Inhibitors Several TNF inhibitors are used as biological therapies in the treatment of rheumatoid arthritis; these include monoclonal antibodies, TNF-R Fc fusion proteins, and Fab fragments. Use of anti-TNF-α antibody therapies such as adalimumab, infliximab, and golimumab has resulted in clinical improvement in patients with these diseases and has opened the way for targeting TNF-α to treat other severe forms of autoimmune and/or inflammatory disease. Blockage of TNF-α has been effective in rheumatoid arthritis, psoriasis, Crohn’s disease, and ankylosing spondylitis. Other cytokine inhibitors are recombinant soluble TNF-α receptor (R) fused to human Ig and anakinra (soluble IL-1 receptor antagonist, or IL-1ra). The treatment of autoinflammatory syndromes (Table 372e-6) with recombinant IL-1 receptor antagonist can prevent symptoms in these syndromes, because the overproduction of IL-1β is a hallmark of these diseases.
TNF-αR-Fc fusion protein (etanercept) and IL-1ra act to inhibit the activity of pathogenic cytokines in rheumatoid arthritis, i.e., TNF-α and IL-1, respectively. Similarly, anti-IL-6, IFN-β, and IL-11 act to inhibit pathogenic proinflammatory cytokines. Anti-IL-6 (tocilizumab) inhibits IL-6 activity, whereas IFN-β and IL-11 decrease IL-1 and TNF-α production.
Of particular note has been the successful use of IFN-γ in the treatment of the phagocytic cell defect in chronic granulomatous disease (Chap. 80).
Monoclonal Antibodies to T and B Cells The OKT3 MAb against human T cells has been used for several years as a T cell-specific immunosuppressive agent that can substitute for horse antithymocyte globulin (ATG) in the treatment of solid organ transplant rejection. OKT3 produces fewer allergic reactions than ATG but does induce human anti-mouse Ig antibody—thus limiting its use. Anti-CD4 MAb therapy has been used in trials to treat patients with rheumatoid arthritis. While inducing profound immunosuppression, anti-CD4 MAb treatment also induces susceptibility to severe infections. Treatment of patients with a MAb against the T cell molecule CD40 ligand (CD154) is under investigation to induce tolerance to organ transplants, with promising results reported in animal studies. Monoclonal antibodies to the CD25 (IL-2a) receptor (basiliximab) are being used for treatment of graft-versus-host disease in bone marrow transplantation, and anti-CD20 MAb (rituximab) is used to treat hematologic neoplasms, autoimmune diseases, kidney transplant rejection, and rheumatoid arthritis. The anti-IgE monoclonal antibody (omalizumab) is used for blocking antigen-specific IgE that causes hay fever and allergic rhinitis (Chap. 376); however, side effects of anti-IgE include increased risk of anaphylaxis. Studies have shown that TH17 cells, in addition to TH1, are mediators of inflammation in Crohn’s disease, and anti–IL-12/IL-23p40 antibody therapy has been studied as a treatment.
It is important to realize the potential risks of these immunosuppressive monoclonal antibodies. Natalizumab is a humanized IgG antibody against an α4 integrin that inhibits leukocyte migration into tissues and has been approved for treatment of multiple sclerosis in the United States. Both it and anti-CD20 (rituximab) have been associated with the onset of progressive multifocal leukoencephalopathy (PML)—a serious and usually fatal CNS infection caused by JC polyomavirus. Efalizumab, a humanized IgG monoclonal antibody previously approved for treatment of plaque psoriasis, has now been taken off the market due to reactivation of JC virus leading to fatal PML. Thus, use of any currently approved immunosuppressant immunotherapies should be undertaken with caution and with careful monitoring of patients according to FDA guidelines.
Tolerance Induction Specific immunotherapy has moved into a new era with the introduction of soluble CTLA-4 protein into clinical trials. Use of this molecule to block T cell activation via TCR/CD28 ligation during organ or bone marrow transplantation has showed promising results in animals and in early human clinical trials. Specifically, treatment of bone marrow with CTLA-4 protein reduces rejection of the graft in HLA-mismatched bone marrow transplantation. In addition, promising results with soluble CTLA-4 have been reported in the downmodulation of autoimmune T cell responses in the treatment of psoriasis; and it is being studied for treatment of systemic lupus erythematosus (Chap. 378).
Intravenous Immunoglobulin (IVIg) IVIg has been used successfully to block reticuloendothelial cell function and immune complex clearance in various immune cytopenias such as immune thrombocytopenia (Chap. 140). In addition, IVIg is useful for prevention of tissue damage in certain inflammatory syndromes such as Kawasaki disease (Chap. 385) and as Ig replacement therapy for certain types of immunoglobulin deficiencies (Chap. 374). In addition, controlled clinical trials support the use of IVIg in selected patients with graft-versus-host disease, multiple sclerosis, myasthenia gravis, Guillain-Barré syndrome, and chronic demyelinating polyneuropathy.
Stem Cell Transplantation Hematopoietic stem cell transplantation (SCT) is now being comprehensively studied to treat several autoimmune diseases, including systemic lupus erythematosus, multiple sclerosis, and scleroderma. The goal of immune reconstitution in autoimmune disease syndromes is to replace a dysfunctional immune system with a normally reactive immune cell repertoire. Preliminary results in patients with scleroderma and lupus have showed encouraging results. Controlled clinical trials in these three diseases are now being launched in the United States and Europe to compare the toxicity and efficacy of conventional immunosuppression therapy with that of myeloablative autologous SCT. Recently, SCT was used in the setting of HIV-1 infection. HIV-1 infection of CD4+ T cells requires the presence of surface CD4 receptor and the chemokine receptor 5 (CCR5) co-receptor. Studies have demonstrated that patients who are homozygous for a 32-bp deletion in the CCR5 allele do not express CD4+ T cell CCR5 and thus are resistant to HIV-1 infection with HIV-1 strains that use this co-receptor. Stem cells from a homozygous CCR5 delta32 donor were transplanted to an HIV-infected patient following standard conditioning for such transplants, and the patient has maintained long-term control of the virus without antiretrovirals. Thus, a number of recent insights into immune system function have spawned a new field of interventional immunotherapy and have enhanced the prospect for development of more specific and nontoxic therapies for immune and inflammatory diseases.
373e |
The Major Histocompatibility Complex |
THE HLA COMPLEX AND ITS PRODUCTS
The human major histocompatibility complex (MHC), commonly called the human leukocyte antigen (HLA) complex, is a 4-megabase (Mb) region on chromosome 6 (6p21.3) that is densely packed with expressed genes. The best known of these genes are the HLA class I and class II genes, whose products are critical for immunologic specificity and transplantation histocompatibility, and they play a major role in susceptibility to a number of autoimmune diseases. Many other genes in the HLA region are also essential to the innate and antigen-specific functioning of the immune system. The HLA region shows extensive conservation with the MHC of other mammals in terms of genomic organization, gene sequence, and protein structure and function.
The HLA class I genes are located in a 2-Mb stretch of DNA at the telomeric end of the HLA region (Fig. 373e-1). The classic (MHC class Ia) HLA-A, -B, and -C loci, the products of which are integral participants in the immune response to intracellular infections, tumors, and allografts, are expressed in all nucleated cells and are highly polymorphic in the population. Polymorphism refers to a high degree of allelic variation within a genetic locus that leads to extensive variation between different individuals expressing different alleles. More than 2000 alleles at HLA-A, nearly 3000 alleles at HLA-B, and more than 1700 at HLA-C have been identified in different human populations, making this the most highly polymorphic segment known within the human genome. Each of the alleles at these loci encodes a heavy chain (also called an α chain) that associates noncovalently with the nonpolymorphic light chain β2-microglobulin, encoded on chromosome 15.
FIGURE 373e-1 Physical map of the HLA region, showing the class I and class II loci, other immunologically important loci, and a sampling of other genes mapped to this region. Gene orientation is indicated by arrowheads. Scale is in kilobase (kb). The approximate genetic distance from DP to A is 3.2 cM. This includes 0.8 cM between A and B (including 0.2 cM between C and B), 0.4–0.8 cM between B and DR-DQ, and 1.6–2.0 cM between DR-DQ and DP.
The nomenclature of HLA genes and their products is based on a revised World Health Organization (WHO) nomenclature, in which alleles are given a single designation that indicates locus, allotype, and sequence-based subtype. For example, HLA-A*02:01 indicates subtype 1 of a group of alleles that encode HLA-A2 molecules. Subtypes that differ from each other at the nucleotide but not the amino acid sequence level are designated by an extra numeral (e.g., HLA-B*07:02:01 and HLA-B*07:02:02 are two variants of HLA-B*07:02, both encoding the same HLA-B7 molecule). The nomenclature of class II genes, discussed below, is made more complicated by the fact that both chains of a class II molecule are encoded by closely linked HLA-encoded loci, each of which may be polymorphic, and by the presence of differing numbers of isotypic DRB loci in different individuals. It has become clear that accurate HLA genotyping requires DNA sequence analysis, and the identification of alleles at the DNA sequence level has contributed greatly to the understanding of the role of HLA molecules as peptide-binding ligands, to the analysis of associations of HLA alleles with certain diseases, to the study of the population genetics of HLA, and to a clearer understanding of the contribution of HLA differences to allograft rejection and graft-versus-host disease. Current databases of HLA class I and class II sequences can be accessed by the Internet (e.g., from the IMGT/HLA Database, http://www.ebi.ac.uk/imgt/hla), and frequent updates of HLA gene lists are published in several journals.
The biologic significance of this MHC genetic diversity, resulting in extreme variation in the human population, is evident from the perspective of the structure of MHC molecules. As shown in Fig. 373e-2, the MHC class I and class II genes encode MHC molecules that bind small peptides, and together this complex (pMHC; peptide-MHC) forms the ligand for recognition by T lymphocytes, through the antigen-specific T cell receptor (TCR). There is a direct link between the genetic variation and this structural interaction: The allelic changes in genetic sequence result in diversification of the peptide-binding capabilities of each MHC molecule and in differences for specific TCR binding. Thus, different pMHC complexes bind different antigens and are targets for recognition by different T cells.
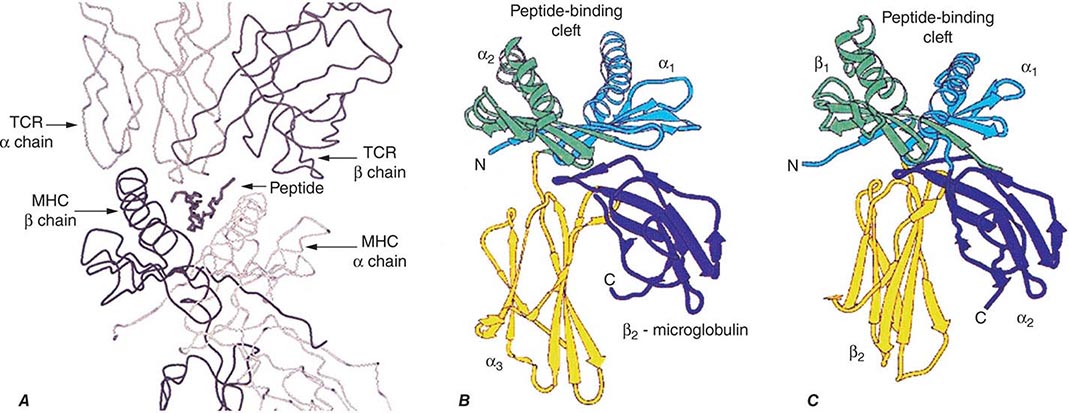
FIGURE 373e-2 A. The trimolecular complex of TCR (top), MHC molecule (bottom), and a bound peptide form the structural determinants of specific antigen recognition. Other panels (B and C) show the domain structure of MHC class I (B) and class II (C) molecules. The α1 and α2 domains of class I and the α1 and β1 domains of class II form a β-sheet platform that forms the floor of the peptide-binding groove, and α helices that form the sides of the groove. The α3 (B) and β2 domains (C) project from the cell surface and form the contact sites for CD8 and CD4, respectively. (Adapted from EL Reinherz et al: Science 286:1913, 1999; and C Janeway et al: Immunobiology Bookshelf, 2nd ed. Garland Publishing, New York, 1997; with permission.)
The class I MHC and class II MHC structures, shown in Fig. 373e-2B, C, are structurally closely related; however, there are a few key differences. While both bind peptides and present them to T cells, the binding pockets have different shapes, which influence the types of immune responses that result (discussed below). In addition, there are structural contact sites for T cell molecules known as CD8 and CD4, expressed on the class I or class II membrane-proximal domains, respectively. This ensures that when peptide antigens are presented by class I molecules, the responding T cells are predominantly of the CD8 class, and similarly, that T cells responding to class II pMHC complexes are predominantly CD4.
The nonclassic, or class Ib, MHC molecules, HLA-E, -F, and -G, are much less polymorphic than MHC Ia and appear to have distinct functions. The HLA-E molecule has a peptide repertoire displaying signal peptides cleaved from classic MHC class I molecules and is the major self-recognition target for the natural killer (NK) cell–inhibitory receptors NKG2A or NKG2C paired with CD94 (see below and Chap. 372e). This appears to be a function of immune surveillance, because loss of MHC class I signal peptides serves as a surrogate marker for injured or infected cells, leading to release of the inhibitory signal and subsequent activation of NK cells. HLA-E can also bind and present peptides to CD8 T cells, albeit with a limited scope, as only three HLA-E alleles are known. HLA-G is expressed mainly in stem cells and in extravillous trophoblasts, the fetal cell population directly in contact with maternal tissues. It binds a wide array of peptides, is expressed in six different alternatively spliced forms, and provides inhibitory signals to both NK cells and T cells, presumably in the service of maintaining maternofetal tolerance. Pathologic expression in cancer and infections may also deliver a similar inhibitory immunologic function; 16 HLA-G alleles have been identified. The protein product of HLA-F is found mainly intracellularly, and the function of this locus, which encodes four alleles but has multiple transcriptional variations, remains largely unknown.
Additional class I–like genes have been identified, some HLA-linked and some encoded on other chromosomes, that show only distant homology to the class Ia and Ib molecules but share the three-dimensional class I structure. Those on chromosome 6p21 include MIC-A and MIC-B, which are encoded centromeric to HLA-B, and HLA-HFE, located 3 to 4 cM (centi-Morgan) telomeric of HLA-F. MIC-A and MIC-B do not bind peptide but are expressed on gut and other epithelium in a stress-inducible manner and serve as activation signals for certain γδ T cells, NK cells, CD8 T cells, and activated macrophages, acting through the activating NKG2D receptors. Ninety-one MIC-A and 40 MIC-B alleles are known, and additional diversification comes from variable alanine repeat sequences in the transmembrane domain. Due to this structural diversity, MIC-A can be recognized as a foreign tissue target during organ transplantation, contributing to graft failure. HLA-HFE encodes the gene defective in hereditary hemochromatosis (Chap. 428). Among the non-HLA, class I–like genes, CD1 refers to a family of molecules that present glycolipids or other nonpeptide ligands to certain T cells, including T cells with NK activity; FcRn binds IgG within lysosomes and protects it from catabolism (Chap. 372e); and Zn-α2-glycoprotein 1 binds a nonpeptide ligand and promotes catabolism of triglycerides in adipose tissue. Like the HLA-A, -B, -C, -E, -F, and -G heavy chains, each of which forms a heterodimer with β2-microglobulin (Fig. 373e-2), the class I–like molecules, HLA-HFE, FcRn, and CD1 also bind to β2-microglobulin, but MIC-A, MIC-B, and Zn-α2-glycoprotein 1 do not.
The HLA class II region is also illustrated in Fig. 373e-1. Multiple class II genes are arrayed within the centromeric 1 Mb of the HLA region, forming distinct haplotypes. A haplotype refers to an array of alleles at polymorphic loci along a chromosomal segment. Multiple class II genes are present on a single haplotype, clustered into three major subregions: HLA-DR, -DQ, and -DP. Each of these subregions contains at least one functional alpha (A) locus and one functional beta (B) locus. Together these encode proteins that form the α and β polypeptide chains of a mature class II HLA molecule. Thus, the DRA and DRB genes encode an HLA-DR molecule; DQA and DQB genes encode HLA-DQ molecules; and DPA and DPB genes encode HLA-DP molecules. There are several DRB genes (DRB1, DRB2, DRB3, etc.), so that two expressed DR molecules are encoded on most haplotypes by combining the α-chain product of the DRA gene with separate β chains. More than 1000 alleles have been identified at the HLA-DRB1 locus, with most of the variation occurring within limited segments encoding residues that interact with antigens. Detailed analysis of sequences and population distribution of these alleles strongly suggest that this diversity is actively selected by environmental pressures associated with pathogen diversity. In the DQ region, both DQA1 and DQB1 are polymorphic, with 50 DQA1 alleles and over 300 DQB1 alleles. The current nomenclature is largely analogous to that discussed above for class I, using the convention “locus * allele.”
In addition to allelic polymorphism, products of different DQA alleles can, with some limitations, pair with products of different DQB alleles through both cis and trans pairing to create combinatorial complexity and expand the number of expressed class II molecules. Because of the enormous allelic diversity in the general population, most individuals are heterozygous at all of the class I and class II loci. Thus, most individuals express six classic class I molecules (two each of HLA-A, -B, and -C) and many class II molecules—two DP, two to four DR, and multiple DQ (both cis and trans dimers).
OTHER GENES IN THE MHC
In addition to the class I and class II genes themselves, there are numerous genes interspersed among the HLA loci that have interesting and important immunologic functions. Our current concept of the function of MHC genes now encompasses many of these additional genes, some of which are also highly polymorphic. Indeed, direct comparison of the complete DNA sequences for eight of the entire 4-Mb MHC regions from different haplotypes show >44,000 nucleotide variations, encoding an extremely high potential for biologic diversity, and at least 97 genes located in this region are known to have coding region sequence variation. Specific examples include the TAP and LMP genes, as discussed in more detail below, which encode molecules that participate in intermediate steps in the HLA class I biosynthetic pathway. Another set of HLA genes, DMA and DMB, perform an analogous function for the class II pathway. These genes encode an intracellular molecule that facilitates the proper complexing of HLA class II molecules with antigen (see below). The HLA class III region is a name given to a cluster of genes between the class I and class II complexes, which includes genes for the two closely related cytokines tumor necrosis factor (TNF)-α and lymphotoxin (TNF-β); the complement components C2, C4, and Bf; heat shock protein (HSP) 70; and the enzyme 21-hydroxylase.
The class I genes HLA-A, -B, and -C are expressed in all nucleated cells, although generally to a higher degree on leukocytes than on nonleukocytes. In contrast, the class II genes show a more restricted distribution: HLA-DR and HLA-DP genes are constitutively expressed on most cells of the myeloid cell lineage, whereas all three class II gene families (HLA-DR, -DQ, and -DP) are inducible by certain stimuli provided by inflammatory cytokines such as interferon γ. Within the lymphoid lineage, expression of these class II genes is constitutive on B cells and inducible on human T cells. Most endothelial and epithelial cells in the body, including the vascular endothelium and the intestinal epithelium, are also inducible for class II gene expression, and some cells show specialized expression, such as HLA-DQA2 and HLA-DQB2 on Langerhans cells. While somatic tissues normally express only class I and not class II genes, during times of local inflammation, they are recruited by cytokine stimuli to express class II genes as well, thereby becoming active participants in ongoing immune responses. Class II expression is controlled largely at the transcriptional level through a conserved set of promoter elements that interact with a protein known as CIITA. Cytokine-mediated induction of CIITA is a principal method by which tissue-specific expression of HLA gene expression is controlled. Other HLA genes involved in the immune response, such as TAP and LMP, are also susceptible to upregulation by signals such as interferon γ.
LINKAGE DISEQUILIBRIUM
In addition to extensive polymorphism at the class I and class II loci, another characteristic feature of the HLA complex is linkage disequilibrium. This is formally defined as a deviation from Hardy-Weinberg equilibrium for alleles at linked loci. This is reflected in the very low recombination rates between certain loci within the HLA complex. For example, recombination between DR and DQ loci is almost never observed in family studies, and characteristic haplotypes with particular arrays of DR and DQ alleles are found in every population. Similarly, the complement components C2, C4, and Bf are almost invariably inherited together, and the alleles at these loci are found in characteristic haplotypes. In contrast, there is a recombinational hotspot between DQ and DP, which are separated by 1–2 cM of genetic distance, despite their close physical proximity. Certain extended haplotypes encompassing the interval from DQ into the class I region are commonly found, the most notable being the haplotype DR3-B8-A1, which is found, in whole or in part, in 10–30% of northern European whites. It has been hypothesized that selective pressures may maintain linkage disequilibrium in HLA, but this remains to be determined. As discussed below under HLA and immunologic disease, one consequence of the phenomenon of linkage disequilibrium has been the resulting difficulty in assigning HLA-disease associations to a single allele at a single locus.
MHC STRUCTURE AND FUNCTION
Class I and class II molecules display a distinctive structural architecture, which contains specialized functional domains responsible for the unique genetic and immunologic properties of the HLA complex. The principal known function of both class I and class II HLA molecules is to bind antigenic peptides in order to present antigen to an appropriate T cell. The ability of a particular peptide to satisfactorily bind to an individual HLA molecule is a direct function of the molecular fit between the amino acid residues on the peptide with respect to the amino acid residues of the HLA molecule. The bound peptide forms a tertiary structure called the MHC-peptide complex, which communicates with T lymphocytes through binding to the TCR molecule. The first site of TCR-MHC-peptide interaction in the life of a T cell occurs in the thymus, where self-peptides are presented to developing thymocytes by MHC molecules expressed on thymic epithelium and hematopoietically derived antigen-presenting cells, which are primarily responsible for positive and negative selection, respectively (Chap. 372e). Thus, the population of MHC–T cell complexes expressed in the thymus shapes the TCR repertoire. Mature T cells encounter MHC molecules in the periphery both in the maintenance of tolerance (Chap. 377e) and in the initiation of immune responses. The MHC-peptide-TCR interaction is the central event in the initiation of most antigen-specific immune responses, since it is the structural determinant of the specificity. For potentially immunogenetic peptides, the ability of a given peptide to be generated and bound by an HLA molecule is a primary feature of whether or not an immune response to that peptide can be generated, and the repertoire of peptides that a particular individual’s HLA molecules can bind exerts a major influence over the specificity of that individual’s immune response.
When a TCR molecule binds to an HLA-peptide complex, it forms intermolecular contacts with both the antigenic peptide and with the HLA molecule itself. The outcome of this recognition event depends on the density and duration of the binding interaction, accounting for a dual specificity requirement for activation of the T cell. That is, the TCR must be specific both for the antigenic peptide and for the HLA molecule. The polymorphic nature of the presenting molecules, and the influence that this exerts on the peptide repertoire of each molecule, results in the phenomenon of MHC restriction of the T cell specificity for a given peptide. The binding of CD8 or CD4 molecules to the class I or class II molecule, respectively, also contributes to the interaction between T cell and the HLA-peptide complex, by providing for the selective activation of the appropriate T cell.
CLASS I STRUCTURE
(Fig. 373e-2B) As noted above, MHC class I molecules provide a cell-surface display of peptides derived from intracellular proteins, and they also provide the signal for self-recognition by NK cells. Surface-expressed class I molecules consist of an MHC-encoded 44-kD glycoprotein heavy chain, a non-MHC-encoded 12-kD light chain β2-microglobulin, and an antigenic peptide, typically 8–11 amino acids in length and derived from intracellularly produced protein. The heavy chain displays a prominent peptide-binding groove. In HLA-A and -B molecules, the groove is ∼3 nm in length by 1.2 nm in maximum width (30 Å × 12 Å), whereas it is apparently somewhat wider in HLA-C. Antigenic peptides are noncovalently bound in an extended conformation within the peptide-binding groove, with both N- and C-terminal ends anchored in pockets within the groove (A and F pockets, respectively) and, in many cases, with a prominent kink, or arch, approximately one-third of the way from the N-terminus that elevates the peptide main chain off the floor of the groove.
A remarkable property of peptide binding by MHC molecules is the ability to form highly stable complexes with a wide array of peptide sequences. This is accomplished by a combination of peptide sequence–independent and peptide sequence–dependent bonding. The former consists of hydrogen bond and van der Waals interactions between conserved residues in the peptide-binding groove and charged or polar atoms along the peptide backbone. The latter is dependent upon the six side pockets that are formed by the irregular surface produced by protrusion of amino acid side chains from within the binding groove. The side chains lining the pockets interact with some of the peptide side chains. The sequence polymorphism among different class I alleles and isotypes predominantly affects the residues that line these pockets, and the interactions of these residues with peptide residues constitute the sequence-dependent bonding that confers a particular sequence “motif” on the range of peptides that can bind any given MHC molecule.
CLASS I BIOSYNTHESIS
(Fig. 373e-3A) The biosynthesis of the classic MHC class I molecules reflects their role in presenting endogenous peptides. The heavy chain is cotranslationally inserted into the membrane of the endoplasmic reticulum (ER), where it becomes glycosylated and associates sequentially with the chaperone proteins calnexin and ERp57. It then forms a complex with β2-microglobulin, and this complex associates with the chaperone calreticulin and the MHC-encoded molecule tapasin, which physically links the class I complex to TAP, the MHC-encoded transporter associated with antigen processing. Meanwhile, peptides generated within the cytosol from intracellular proteins by the multisubunit, multicatalytic proteasome complex are actively transported into the ER by TAP, where they are trimmed by enzymes known as ER aminopeptidases. At this point, peptides with appropriate sequence complementarity bind specific class I molecules to form complete, folded heavy chain–β2-microglobulin–peptide trimer complexes. These are transported rapidly from the ER, through the cis– and trans-Golgi where the N-linked oligosaccharide is further processed, and thence to the cell surface.
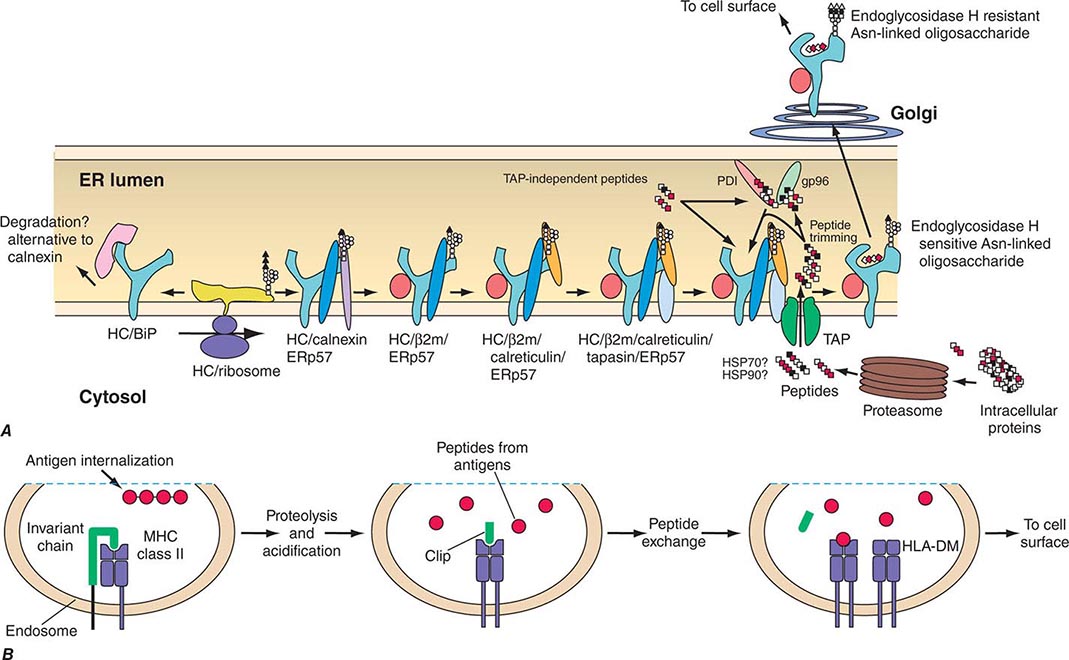
FIGURE 373e-3 Biosynthesis of class I (A) and class II (B) molecules. A. Nascent heavy chain (HC) becomes associated with β2-microglobulin (β2m) and peptide through interactions with a series of chaperones. Peptides generated by the proteasome are transported into the endoplasmic reticulum (ER) by TAP. Peptides undergo N-terminal trimming in the ER and become associated with chaperones, including gp96 and PDI. Once peptide binds to HC-β2m, the HC-β2m-peptide trimeric complex exits the ER and is transported by the secretory pathway to the cell surface. In the Golgi, the N-linked oligosaccharide undergoes maturation, with addition of sialic acid residues. Molecules are not necessarily drawn to scale. B. Pathway of HLA class II molecule assembly and antigen processing. After transport through the Golgi and post-Golgi compartment, the class II–invariant chain complex moves to an acidic endosome, where the invariant chain is proteolytically cleaved into fragments and displaced by antigenic peptides, facilitated by interactions with the DMA-DMB chaperone protein. This class II molecule–peptide complex is then transported to the cell surface.
Most of the peptides transported by TAP are produced in the cytosol by proteolytic cleavage of intracellular proteins by the multisubunit, multicatalytic proteasome, and inhibitors of the proteasome dramatically reduce expression of class I–presented antigenic peptides. A thiol-dependent oxidoreductase ERp57, which mediates disulfide bond rearrangements, also appears to play an important role in folding the class I–peptide complex into a stable multicomponent molecule. The MHC-encoded proteasome subunits LMP2 and LMP7 may influence the spectrum of peptides produced but are not essential for proteasome function.
CLASS I FUNCTION
Peptide Antigen Presentation On any given cell, a class I molecule occurs in 100,000–200,000 copies and binds several hundred to several thousand distinct peptide species. The vast majority of these peptides are self-peptides to which the host immune system is tolerant by one or more of the mechanisms that maintain tolerance (e.g., clonal deletion in the thymus or clonal anergy or clonal ignorance in the periphery [Chaps. 372e and 377e]). However, class I molecules bearing foreign peptides expressed in a permissive immunologic context activate CD8 T cells, which, if naïve, will then differentiate into cytolytic T lymphocytes (CTLs). These T cells and their progeny, through their αβ TCRs, are then capable of Fas/CD95- and/or perforin-mediated cytotoxicity and/or cytokine secretion (Chap. 372e) upon further encounter with the class I–peptide combination that originally activated it, or other structurally related class I–peptide complexes. As alluded to above, this phenomenon by which T cells recognize foreign antigens in the context of specific MHC alleles is termed MHC restriction, and the specific MHC molecule is termed the restriction element. The most common source of foreign peptides presented by class I molecules is viral infection, in the course of which peptides from viral proteins enter the class I pathway. The generation of a strong CTL response that destroys virally infected cells represents an important antigen-specific defense against many viral infections (Chap. 372e). In the case of some viral infections—hepatitis B, for example—CTL-induced target cell apoptosis is thought to be a more important mechanism of tissue damage than any direct cytopathic effect of the virus itself. The importance of the class I pathway in the defense against viral infection is underscored by the identification of a number of viral products that interfere with the normal class I biosynthetic pathway and thus block the immunogenetic expression of viral antigens.
Other examples of intracellularly generated peptides that can be presented by class I molecules in an immunogenic manner include peptides derived from nonviral intracellular infectious agents (e.g., Listeria, Plasmodium), tumor antigens, minor histocompatibility antigens, and certain autoantigens. There are also situations in which cell surface–expressed class I molecules are thought to acquire and present exogenously derived peptides.
HLA Class I Receptors and NK Cell Recognition (Chap. 372e) NK cells, which play an important role in innate immune responses, are activated to cytotoxicity and cytokine secretion by contact with cells that lack MHC class I expression, and NK cell activation is inhibited by cells that express MHC class I. In humans, the recognition of class I molecules by NK cells is carried out by three classes of receptor families, the killer cell–inhibitory cell receptor (KIR) family, the leukocyte Ig-like receptor (LIR) family, and the CD94/NKG2 family. The KIR family, also called CD158, is encoded on chromosome 19q13.4. KIR gene nomenclature is based on the number of domains (2D or 3D) and the presence of long (L) or short (S) cytoplasmic domains. The KIR2DL1 and S1 molecules primarily recognize alleles of HLA-C, which possess a lysine at position 80 (HLA-Cw2, -4, -5, and -6), whereas the KIR2DL2/S2 and KIR2DL3/S3 families primarily recognize alleles of HLA-C with asparagine at this position (HLA-Cw1, -3, -7, and -8). The KIR3DL1 and S1 molecules predominantly recognize HLA-B alleles that fall into the HLA-Bw4 class determined by residues 77–83 in the α1 domain of the heavy chain, whereas the KIR3DL2 molecule is an inhibitory receptor for HLA-A*03. One of the KIR products, KIR2DL4, is known to be an activating receptor for HLA-G. The most common KIR haplotype in whites contains one activating KIR and six inhibitory KIR genes, although there is a great deal of diversity in the population, with >100 different combinations. It appears that most individuals have at least one inhibitory KIR for a self-HLA class I molecule, providing a structural basis for NK cell target specificity, which helps prevent NK cells from attacking normal cells. The importance of KIR-HLA interactions to many immune responses is illustrated by studies associating KIR3DL1 or S1 with multiple sclerosis (Chap. 458), an autoimmune disease, but also with partial protection against HIV (Chap. 226), in both cases consistent with a role for HLA-KIR mediated NK activation. Studies also show an association of KIR2DS1 with protection from relapse following allogeneic bone marrow transplantation in acute myeloid leukemia when these inhibitory receptors in the donors do not recognize the recipient HLA-C.
The LIR gene family (CD85, also called ILT) is encoded centromeric of the KIR locus on 19q13.4, and it encodes a variety of inhibitory immunoglobulin-like receptors expressed on many lymphocyte and other hematopoietic lineages. Interaction of LIR-1 (ILT2) with NK or T cells inhibits activation and cytotoxicity, mediated by many different HLA class I molecules, including HLA-G. HLA-F also appears to interact with LIR molecules, although the functional context for this is not understood.
The third family of NK receptors for HLA is encoded in the NK complex on chromosome 12p12.3-13.1 and consists of CD94 and five NKG2 genes, A/B, C, E/H, D, and F. These molecules are C-type (calcium-binding) lectins, and most function as disulfide-bonded heterodimers between CD94 and one of the NKG2 glycoproteins. The principal ligand of CD94/NKG2A receptors is the HLA-E molecule, complexed to a peptide derived from the signal sequence of classic HLA class I molecules and HLA-G. Thus, analogous to the way in which KIR receptors recognize HLA-C, the NKG2 receptor monitors self–class I expression, albeit indirectly through peptide recognition in the context of HLA-E. NKG2C, -E, and -H appear to have similar specificities but act as activating receptors. NKG2D is expressed as a homodimer and functions as an activating receptor expressed on NK cells, γδ TCR T cells, and activated CD8 T cells. When complexed with an adaptor called DAP10, NKG2D recognizes MIC-A and MIC-B molecules and activates the cytolytic response. NKG2D also binds a class of molecules known as ULBP, structurally related to class I molecules but not encoded in the MHC. The function of NK cells in immune responses is discussed in Chap. 372e.
CLASS II STRUCTURE
(Fig. 373e-2C) A specialized functional architecture similar to that of the class I molecules can be seen in the example of a class II molecule depicted in Fig. 373e-2C, with an antigen-binding cleft arrayed above a supporting scaffold that extends the cleft toward the external cellular environment. However, in contrast to the HLA class I molecular structure, β2-microglobulin is not associated with class II molecules. Rather, the class II molecule is a heterodimer, composed of a 29-kD α chain and a 34-kD β chain. The amino-terminal domains of each chain form the antigen-binding elements that, like the class I molecule, cradle a bound peptide in a groove bounded by extended α-helical loops, one encoded by the A (α chain) gene and one by the B (β chain) gene. Like the class I groove, the class II antigen-binding groove is punctuated by pockets that contact the side chains of amino acid residues of the bound peptide, but unlike the class I groove, it is open at both ends. Therefore, peptides bound by class II molecules vary greatly in length, since both the N- and C-terminal ends of the peptides can extend through the open ends of this groove. Approximately 11 amino acids within the bound peptide form intimate contacts with the class II molecule itself, with backbone hydrogen bonds and specific side chain interactions combining to provide, respectively, stability and specificity to the binding (Fig. 373e-4).
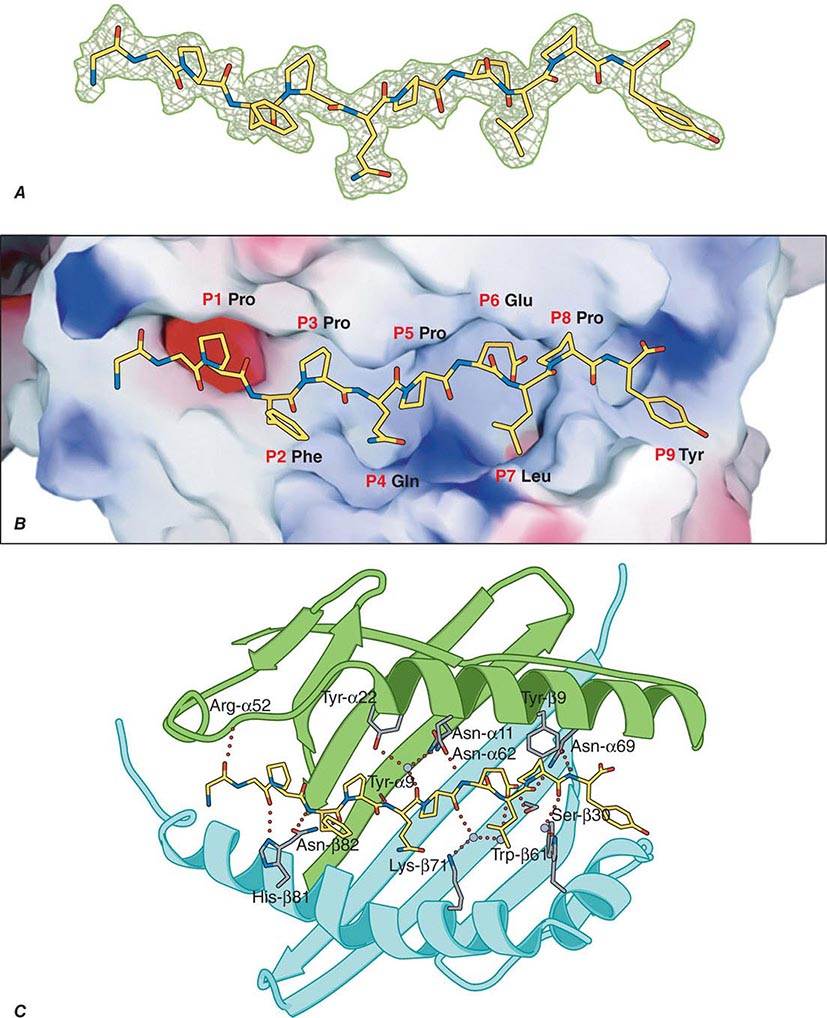
FIGURE 373e-4 Specific intermolecular interactions determine peptide binding to MHC class II molecules. A short peptide sequence derived from alpha-gliadin (A) is accommodated within the MHC class II binding groove by specific interactions between peptide side chains (the P1–P9 residues illustrated in B) and corresponding pockets in the MHC class II structure. The latter are determined by the genetic polymorphisms of the MHC gene, in this case encoding an HLA-DQ2 molecule (C). This shows the extensive hydrogen bond and salt bridge network, which tightly constrains the pMHC complex and presents the complex of antigen and restriction element for CD4 T cell recognition. (From C Kim et al: Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci USA 101:4175, 2004.)
The genetic polymorphisms that distinguish different class II genes correspond to changes in the amino acid composition of the class II molecule, and these variable sites are clustered predominantly around the pocket structures within the antigen-binding groove. As with class I, this is a critically important feature of the class II molecule, which explains how genetically different individuals have functionally different HLA molecules.
BIOSYNTHESIS AND FUNCTION OF CLASS II MOLECULES
(Fig. 373e-3B) The intracellular assembly of class II molecules occurs within a specialized compartmentalized pathway that differs dramatically from the class I pathway described above. As illustrated in Fig. 373e-3B, the class II molecule assembles in the ER in association with a chaperone molecule, known as the invariant chain. The invariant chain performs at least two roles. First, it binds to the class II molecule and blocks the peptide-binding groove, thus preventing antigenic peptides from binding. This role of the invariant chain appears to account for one of the important differences between class I and class II MHC pathways, since it can explain why class I molecules present endogenous peptides from proteins newly synthesized in the ER but class II molecules generally do not. Second, the invariant chain contains molecular localization signals that direct the class II molecule to traffic into post-Golgi compartments known as endosomes, which develop into specialized acidic compartments where proteases cleave the invariant chain, and antigenic peptides can now occupy the class II groove. The specificity and tissue distribution of these proteases appear to be an important way in which the immune system regulates access to the peptide-binding groove and T cells become exposed to specific self-antigens. Differences in protease expression in the thymus and in the periphery may in part determine which specific peptide sequences comprise the peripheral repertoire for T cell recognition. It is at this stage in the intracellular pathway, after cleavage of the invariant chain, that the MHC-encoded DM molecule catalytically facilitates the exchange of peptides within the class II groove to help optimize the specificity and stability of the MHC-peptide complex.
Once this MHC-peptide complex is deposited in the outer cell membrane, it becomes the target for T cell recognition via a specific TCR expressed on lymphocytes. Because the endosome environment contains internalized proteins retrieved from the extracellular environment, the class II–peptide complex often contains bound antigens that were originally derived from extracellular proteins. In this way, the class II peptide–loading pathway provides a mechanism for immune surveillance of the extracellular space. This appears to be an important feature that permits the class II molecule to bind foreign peptides, distinct from the endogenous pathway of class I–mediated presentation.
ROLE OF HLA IN TRANSPLANTATION
The development of modern clinical transplantation in the decades since the 1950s provided a major impetus for elucidation of the HLA system, as allograft survival is highest when donor and recipient are HLA-identical. Although many molecular events participate in transplantation rejection, allogeneic differences at class I and class II loci play a major role. Class I molecules can promote T cell responses in several different ways. In the cases of allografts in which the host and donor are mismatched at one or more class I loci, host T cells can be activated by classic direct alloreactivity, in which the antigen receptors on the host T cells react with the foreign class I molecule expressed on the allograft. In this situation, the response of any given TCR may be dominated by the allogeneic MHC molecule, the peptide bound to it, or some combination of the two. Another type of host anti-graft T cell response involves the uptake and processing of donor MHC antigens by host antigen-presenting cells and the subsequent presentation of the resulting peptides by host MHC molecules. This mechanism is termed indirect alloreactivity.
In the case of class I molecules on allografts that are shared by the host and the donor, a host T cell response may still be triggered because of peptides that are presented by the class I molecules of the graft but not of the host. The most common basis for the existence of these endogenous antigen peptides, called minor histocompatibility antigens, is a genetic difference between donor and host at a non-MHC locus encoding the structural gene for the protein from which the peptide is derived. These loci are termed minor histocompatibility loci, and nonidentical individuals typically differ at many such loci. CD4 T cells react to analogous class II variation, both direct and indirect, and class II differences alone are sufficient to drive allograft rejection.
ASSOCIATION OF HLA ALLELES WITH SUSCEPTIBILITY TO DISEASE
It has long been postulated that infectious agents provide the driving force for the allelic diversification seen in the HLA system. An important corollary of this hypothesis is that resistance to specific pathogens may differ between individuals, based on HLA genotype. Observations of specific HLA genes associated with resistance to malaria or dengue fever, persistence of hepatitis B, and to disease progression in HIV infection are consistent with this model. For example, failure to clear persistent hepatitis B or C viral infection may reflect the inability of particular HLA molecules to present viral antigens effectively to T cells. Similarly, both protective and susceptible HLA allelic associations have been described for human papilloma virus–associated cervical neoplasia, implicating the MHC as an influence in mediating viral clearance in this form of cancer.
Pathogen diversity is probably also the major selective pressure favoring HLA heterozygosity. The extraordinary scope of HLA allelic diversity increases the likelihood that most new pathogens will be recognized by some HLA molecules, helping to ensure immune fitness to the host. However, another consequence of diversification is that some alleles may become capable of recognition of “innocent bystander” molecules, including drugs, environmental molecules, and tissue-derived self-antigens. In a few instances, single HLA alleles display a strong selectivity for binding of a particular agent that accounts for a genetically determined response: Hypersensitivity to abacavir, an antiretroviral therapeutic, is directly linked to binding of abacavir in the antigen-binding pockets of HLA-B*57:01, where it is buried underneath antigenic peptides and distorts the landscape, changing T cell recognition specificity; an adverse drug reaction to abacavir is more than 500 times more likely to occur in persons with HLA-B*57:01 than in individuals without this HLA allele. Another example is chronic beryllium toxicity, which is linked to binding of beryllium by HLA-DP molecules with a specific glutamic acid polymorphic residue on the class II beta chain. Even in the case of more complex diseases, particular HLA alleles are strongly associated with certain inappropriate immune-mediated disease states, particularly for some common autoimmune disorders (Chap. 377e). By comparing allele frequencies in patients with any particular disease and in control populations, >100 such associations have been identified, some of which are listed in Table 373e-1. The strength of genetic association is reflected in the term relative risk, which is a statistical odds ratio representing the risk of disease for an individual carrying a particular genetic marker compared with the risk for individuals in that population without that marker. The nomenclature shown in Table 373e-1 reflects both the HLA serotype (e.g., DR3, DR4) and the HLA genotype (e.g., DRB1*03:01, DRB1*04:01). It very likely the class I and class II alleles themselves are the true susceptibility alleles for most of these associations. However, because of the extremely strong linkage disequilibrium between the DR and DQ loci, in some cases it has been difficult to determine the specific locus or combination of class II loci involved. In some cases, the susceptibility gene may be one of the HLA-linked genes located near the class I or class II region, but not the HLA gene itself, and in other cases, the susceptibility gene may be a non-HLA gene such as TNF-α, which is nearby. Indeed, since linkage disequilibrium of some haplotypes extends across large segments of the MHC region, it is quite possible that combinations of genes may account for the particular associations of HLA haplotypes with disease. For example, on some haplotypes associated with rheumatoid arthritis, both HLA-DRB1 alleles and a particular polymorphism associated with the TNF locus may be contributory to disease risk. Other candidates for similar epistatic effects include the IKBL gene and the MICA locus, potentially in combination with classic HLA class II risk alleles.
|
SIGNIFICANT HLA CLASS I AND CLASS II ASSOCIATIONS WITH DISEASE |
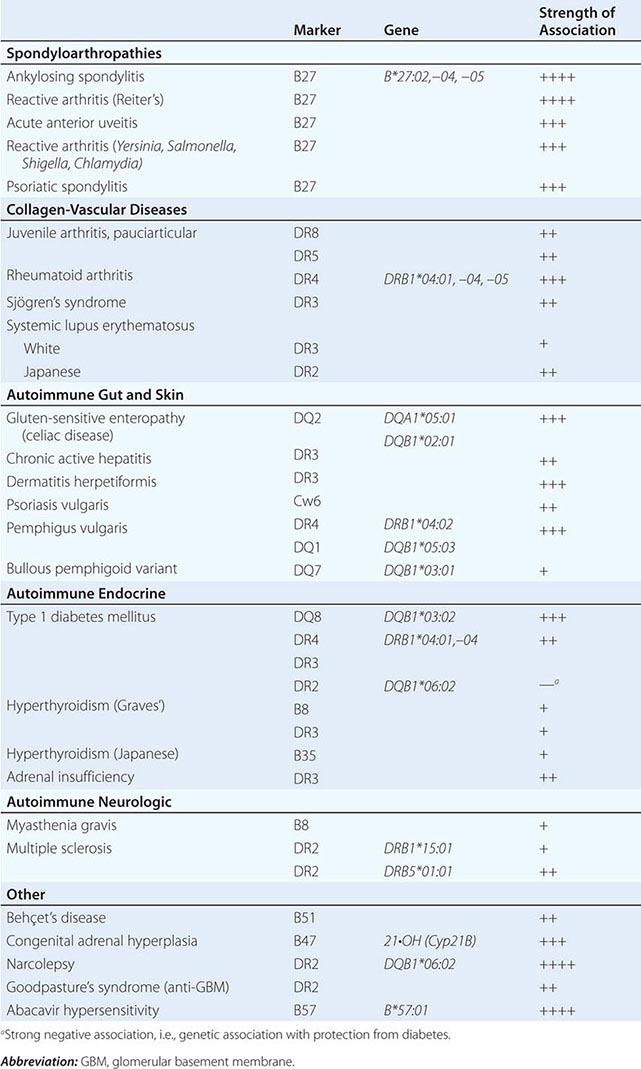
As might be predicted from the known function of the class I and class II gene products, almost all of the diseases associated with specific HLA alleles have an immunologic component to their pathogenesis. The recent development of soluble HLA-peptide recombinant molecules as biological probes of T cell function, often in multivalent complexes referred to as “MHC tetramers,” represents an opportunity to use HLA genetic associations to develop biomarkers for detection of early disease progression. However, it should be stressed that even the strong HLA associations with disease (those associations with relative risk of ≥10) implicate normal, rather than defective, alleles. Most individuals who carry these susceptibility genes do not express the associated disease; in this way, the particular HLA gene is permissive for disease but requires other environmental (e.g., the presence of specific antigens) or genetic factors for full penetrance. In each case studied, even in diseases with very strong HLA associations, the concordance of disease in monozygotic twins is higher than in HLA-identical dizygotic twins or other sibling pairs, indicating that non-HLA genes contribute to susceptibility and can significantly modify the risk attributable to HLA.
Another group of diseases is genetically linked to HLA, not because of the immunologic function of HLA alleles but rather because they are caused by autosomal dominant or recessive abnormal alleles at loci that happen to reside in or near the HLA region. Examples of these are 21-hydroxylase deficiency (Chap. 406), hemochromatosis (Chap. 428), and spinocerebellar ataxia (Chap. 452).
CLASS I ASSOCIATIONS WITH DISEASE
Although the associations of human disease with particular HLA alleles or haplotypes predominantly involve the class II region, there are also several prominent disease associations with class I alleles. These include the association of Behçet’s disease (Chap. 387) with HLA-B51, psoriasis vulgaris (Chap. 71) with HLA-Cw6, and, most notably, the spondyloarthritides (Chap. 384) with HLA-B27. Twenty-five HLA-B locus alleles, designated HLA-B*27:01–B*27:25, encode the family of B27 class I molecules. All of the subtypes share a common B pocket in the peptide-binding groove—a deep, negatively charged pocket that shows a strong preference for binding the arginine side chain. In addition, B27 is among the most negatively charged of HLA class I heavy chains, and the overall preference is for positively charged peptides. HLA-B*27:05 is the predominant subtype in whites and most other non-Asian populations, and this subtype is very highly associated with ankylosing spondylitis (AS) (Chap. 384), both in its idiopathic form and in association with chronic inflammatory bowel disease or psoriasis vulgaris. It is also associated with reactive arthritis (ReA) (Chap. 384), with other idiopathic forms of peripheral arthritis (undifferentiated spondyloarthropathy), and with recurrent acute anterior uveitis. B27 is found in 50–90% of individuals with these conditions, compared with a prevalence of ∼7% in North American whites.
It can be concluded that the B27 molecule itself is involved in disease pathogenesis, based on strong evidence from clinical epidemiology and on the occurrence of a spondyloarthropathy-like disease in HLA-B27 transgenic rats. The association of B27 with these diseases may derive from the specificity of a particular peptide or family of peptides bound to B27 or through another mechanism that is independent of the peptide specificity of B27. In particular, HLA-B27 has been shown to form heavy chain homodimers, utilizing the cysteine residue at position 67 of the B57 α chain, in the absence of β2-microglobulin. These homodimers are expressed on the surface of lymphocytes and monocytes from patients with AS, and receptors including KIR3DL1, KIR3DL2, and ILT4 (LILRB2) are capable of binding to them, promoting the activation and survival of cells expressing these receptors. Alternatively, this dimerization “misfolding” of B27 may initiate an intracellular stress signaling response, called the unfolded protein response (UPR), capable of modulating immune cell function, possibly in enthesial-resident T cells that act as sensors of damage and environmental stress.
CLASS II DISEASE ASSOCIATIONS
As can be seen in Table 373e-1, the majority of associations of HLA and disease are with class II alleles. Several diseases have complex HLA genetic associations.
Celiac Disease In the case of celiac disease (Chap. 349), it is probable that the HLA-DQ genes are the primary basis for the disease association. HLA-DQ genes present on both the celiac-associated DR3 and DR7 haplotypes include the DQB1*02:01 gene, and further detailed studies have documented a specific class II αβ dimer encoded by the DQA1*05:01 and DQB1*02:01 genes, which appears to account for most of the HLA genetic contribution to celiac disease susceptibility. This specific HLA association with celiac disease may have a straightforward explanation: Peptides derived from the wheat gluten component gliaden are bound to the molecule encoded by DQA1*05:01 and DQB1*02:01 and presented to T cells. Gliaden-derived peptides that are implicated in this immune activation bind the DQ class II dimer best when the peptide contains a glutamine to glutamic acid substitution. It has been proposed that tissue transglutaminase, an enzyme present at increased levels in the intestinal cells of celiac patients, converts glutamine to glutamic acid in gliadin, creating peptides that are capable of being bound by the DQ2 molecule and presented to T cells.
Pemphigus Vulgaris In the case of pemphigus vulgaris (Chap. 73), there are two HLA genes associated with disease, DRB1*04:02 and DQB1*05:03. Peptides derived from desmoglein-3, an epidermal autoantigen, bind to the DRB1*04:02– and DQB1*05:03-encoded HLA molecules, and this combination of specific peptide binding and disease-associated class II molecule is sufficient to stimulate desmoglein-specific T cells. A bullous pemphigoid clinical variant, not involving desmoglein recognition, has been found to be associated with HLA-DQB1*03:01.
Juvenile Arthritis Pauciarticular juvenile arthritis (Chap. 380) is an autoimmune disease associated with genes at the DRB1 locus and also with genes at the DPB1 locus. Patients with both DPB1*02:01 and a DRB1 susceptibility allele (usually DRB1*08 or –*05) have a higher relative risk than expected from the additive effect of those genes alone. In juvenile patients with rheumatoid factor–positive polyarticular disease, heterozygotes carrying both DRB1*04:01 and –*04:04 have a relative risk >100, reflecting an apparent synergy in individuals inheriting both of these susceptibility genes.
Type 1 Diabetes Mellitus Type 1 (autoimmune) diabetes mellitus (Chap. 417) is associated with MHC genes on more than one haplotype. The presence of both the DR3 and DR4 haplotypes in one individual confers a twentyfold increased risk for type 1 diabetes; the strongest single association is with DQB1*03:02, and all haplotypes that carry a DQB1*03:02 gene are associated with type 1 diabetes, whereas related haplotypes that carry a different DQB1 gene are not. However, the relative risk associated with inheritance of this gene can be modified, depending on other HLA genes present either on the same or a second haplotype. For example, the presence of a DR2-positive haplotype containing a DQB1*06:02 gene is associated with decreased risk. This gene, DQB1*06:02, is considered “protective” for type 1 diabetes. Even some DRB1 genes that can occur on the same haplotype as DQB1*03:02 may modulate risk, so that individuals with the DR4 haplotype that contains DRB1*04:03 are less susceptible to type 1 diabetes than individuals with other DR4-DQB1*03:02 haplotypes. There are some characteristic structural features of the diabetes-associated DQ molecule encoded by DQB1*03:02, particularly the capability for binding peptides that have negatively charged amino acids near their C-termini. This may indicate a role for specific antigenic peptides or T cell interactions in the immune response to islet-associated proteins.
Although the presence of a DR3 haplotype in combination with the DR4-DQB1*0302 haplotype is a very high-risk combination for diabetes susceptibility, the specific gene on the DR3 haplotype that is responsible for this synergy is not yet identified.
Rheumatoid Arthritis The HLA genes associated with rheumatoid arthritis (RA) (Chap. 380) encode a distinctive sequence of amino acids from codons 67–74 of the DRβ molecule: RA-associated class II molecules carry the sequence LeuLeuGluGlnArgArgAlaAla or LeuLeuGluGlnLysArgAlaAla in this region, whereas non-RA-associated genes carry one or more differences in this region. These residues form a portion of the molecule that lies in the middle of the α-helical portion of the DRB1-encoded class II molecule, termed the shared epitope.
The highest risk for susceptibility to RA comes in individuals who carry both a DRB1*04:01 and DRB1*04:04 gene. These DR4-positive RA-associated alleles with the shared epitope are most frequent among patients with more severe, erosive disease. Several mechanisms have been proposed that link the shared epitope to immune reactivity in RA. This portion of the class II molecule may allow preferential binding of an arthritogenic peptide, it may favor the expansion of a type of self-reactive T lymphocyte, or it may itself form part of the pMHC ligand recognized by TCR that initiates synovial tissue recognition.
MOLECULAR MECHANISMS FOR HLA-DISEASE ASSOCIATIONS
As noted above, HLA molecules play a key role in the selection and establishment of the antigen-specific T cell repertoire and a major role in the subsequent activation of those T cells during the initiation of an immune response. Precise genetic polymorphisms characteristic of individual alleles dictate the specificity of these interactions and thereby instruct and guide antigen-specific immune events. These same genetically determined pathways are therefore implicated in disease pathogenesis when specific HLA genes are responsible for autoimmune disease susceptibility.
The fate of developing T cells within the thymus is determined by the affinity of interaction between TCR and HLA molecules bearing self-peptides, and thus the particular HLA types of each individual control the precise specificity of the T cell repertoire (Chap. 372e). The primary basis for HLA-associated disease susceptibility may well lie within this thymic maturation pathway. The positive selection of potentially autoreactive T cells, based on the presence of specific HLA susceptibility genes, may establish the threshold for disease risk in a particular individual.
At the time of onset of a subsequent immune response, the primary role of the HLA molecule is to bind peptide and present it to antigen-specific T cells. The HLA complex can therefore be viewed as encoding genetic determinants of precise immunologic activation events. Antigenic peptides that bind particular HLA molecules are capable of stimulating T cell immune responses; peptides that do not bind are not presented to T cells and are not immunogenic. This genetic control of the immune response is mediated by the polymorphic sites within the HLA antigen–binding groove that interact with the bound peptides. In autoimmune and immune-mediated diseases, it is likely that specific tissue antigens that are targets for pathogenic lymphocytes are complexed with the HLA molecules encoded by specific susceptibility alleles. In autoimmune diseases with an infectious etiology, it is likely that immune responses to peptides derived from the initiating pathogen are bound and presented by particular HLA molecules to activate T lymphocytes that play a triggering or contributory role in disease pathogenesis. The concept that early events in disease initiation are triggered by specific HLA-peptide complexes offers some prospects for therapeutic intervention, since it may be possible to design compounds that interfere with the formation or function of specific HLA-peptide–TCR interactions.
When considering mechanisms of HLA associations with immune response and disease, it is well to remember that just as HLA genetics are complex, so are the mechanisms likely to be heterogeneous. Immune-mediated disease is a multistep process in which one of the HLA-associated functions is to establish a repertoire of potentially reactive T cells, whereas another HLA-associated function is to provide the essential peptide-binding specificity for T cell recognition. For diseases with multiple HLA genetic associations, it is possible that both of these interactions occur and synergize to advance an accelerated pathway of disease.
374 |
Primary Immune Deficiency Diseases |
Immunity is intrinsic to life and an important tool in the fight for survival against pathogenic microorganisms. The human immune system can be divided into two major components: the innate immune system and the adaptive immune system (Chap. 372e). The innate immune system provides the rapid triggering of inflammatory responses based on the recognition (at the cell surface or within cells) of either molecules expressed by microorganisms or molecules that serve as “danger signals” released by cells under attack. These receptor/ligand interactions trigger signaling events that ultimately lead to inflammation. Virtually all cell lineages (not just immune cells) are involved in innate immune responses; however, myeloid cells (i.e., neutrophils and macrophages) play a major role because of their phagocytic capacity. The adaptive immune system operates by clonal recognition of antigens followed by a dramatic expansion of antigen-reactive cells and execution of an immune effector program. Most of the effector cells die off rapidly, whereas memory cells persist. Although both T and B lymphocytes recognize distinct chemical moieties and execute distinct adaptive immune responses, the latter is largely dependent on the former in generating long-lived humoral immunity. Adaptive responses utilize components of the innate immune system; for example, the antigen-presentation capabilities of dendritic cells help to determine the type of effector response. Not surprisingly, immune responses are controlled by a series of regulatory mechanisms.
Hundreds of gene products have been characterized as effectors or mediators of the immune system (Chap. 372e). Whenever the expression or function of one of these products is genetically impaired (provided the function is nonredundant), a primary immunodeficiency (PID) occurs.
PIDs are genetic diseases with primarily Mendelian inheritance. More than 250 conditions have now been described, and deleterious mutations in approximately 210 genes have been identified. The overall prevalence of PIDs has been estimated in various countries at 5 per 100,000 individuals; however, given the difficulty in diagnosing these rare and complex diseases, this figure is probably an underestimate. PIDs can involve all possible aspects of immune responses, from innate through adaptive, cell differentiation, and effector function and regulation. For the sake of clarity, PIDs should be classified according to (1) the arm of the immune system that is defective and (2) the mechanism of the defect (when known). Table 374-1 classifies the most prevalent PIDs according to this manner of classification; however, one should bear in mind that the classification of PIDs sometimes involves arbitrary decisions because of overlap and, in some cases, lack of data.
|
CLASSIFICATION OF PRIMARY IMMUNE DEFICIENCY DISEASES |
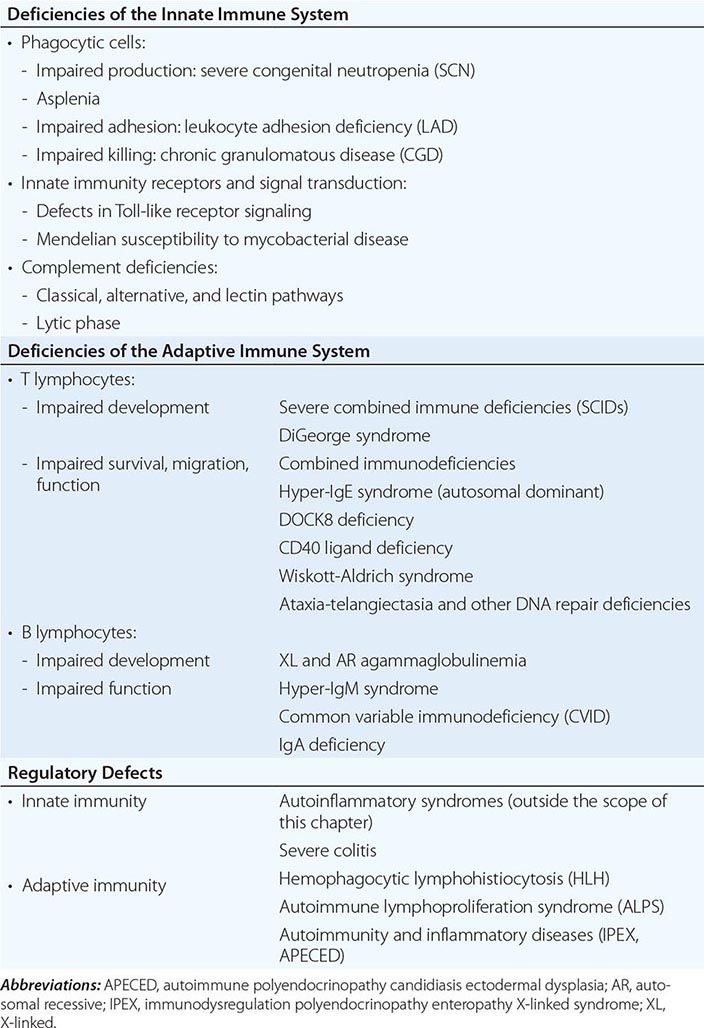
The consequences of PIDs vary widely as a function of the molecules that are defective. This concept translates into multiple levels of vulnerability to infection by pathogenic and opportunistic microorganisms, ranging from extremely broad (as in severe combined immunodeficiency [SCID]) to narrowly restricted to a single microorganism (as in Mendelian susceptibility to mycobacterial disease [MSMD]). The locations of the sites of infection and the causal microorganisms involved will thus help physicians arrive at proper diagnoses. PIDs can also lead to immunopathologic responses such as allergy (as in Wiskott-Aldrich syndrome), lymphoproliferation, and autoimmunity. A combination of recurrent infections, inflammation, and autoimmunity can be observed in a number of PIDs, thus creating obvious therapeutic challenges. Finally, some PIDs increase the risk of cancer, notably but not exclusively lymphocytic cancers, e.g., lymphoma.
DIAGNOSIS OF PRIMARY IMMUNODEFICIENCIES
The most frequent symptom prompting the diagnosis of a PID is the presence of recurrent or unusually severe infections. As mentioned above, recurrent allergic or autoimmune manifestations may also alert the physician to a possible diagnosis of PID. In such cases, a detailed account of the subject’s personal and family medical history should be obtained. It is of the utmost importance to gather as much medical information as possible on relatives and up to several generations of ancestors. In addition to the obvious focus on primary symptoms, the clinical examination should evaluate the size of lymphoid organs and, when appropriate, look for the characteristic signs of a number of complex syndromes that may be associated with a PID.
The performance of laboratory tests should be guided to some extent by the clinical findings. Infections of the respiratory tract (bronchi, sinuses) mostly suggest a defective antibody response. In general, invasive bacterial infections can result from complement deficiencies, signaling defects of innate immune responses, asplenia, or defective antibody responses. Viral infections, recurrent Candida infections, and opportunistic infections are generally suggestive of impaired T cell immunity. Skin infections and deep-seated abscesses primarily reflect innate immune defects (such as chronic granulomatous disease); however, they may also appear in the autosomal dominant hyper-IgE syndrome. Table 374-2 summarizes the laboratory tests that are most frequently used to diagnose a PID. More specific tests (notably genetic tests) are then used to make a definitive diagnosis.
|
TESTS MOST FREQUENTLY USED TO DIAGNOSE A PRIMARY IMMUNE DEFICIENCY (PID) |
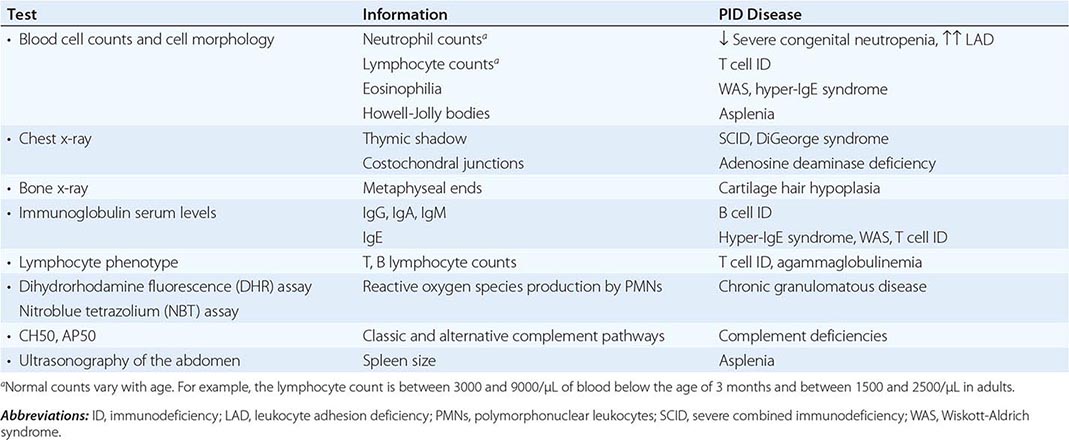
The PIDs discussed below have been grouped together according to the affected cells and the mechanisms involved (Table 374-1, Fig. 374-1).
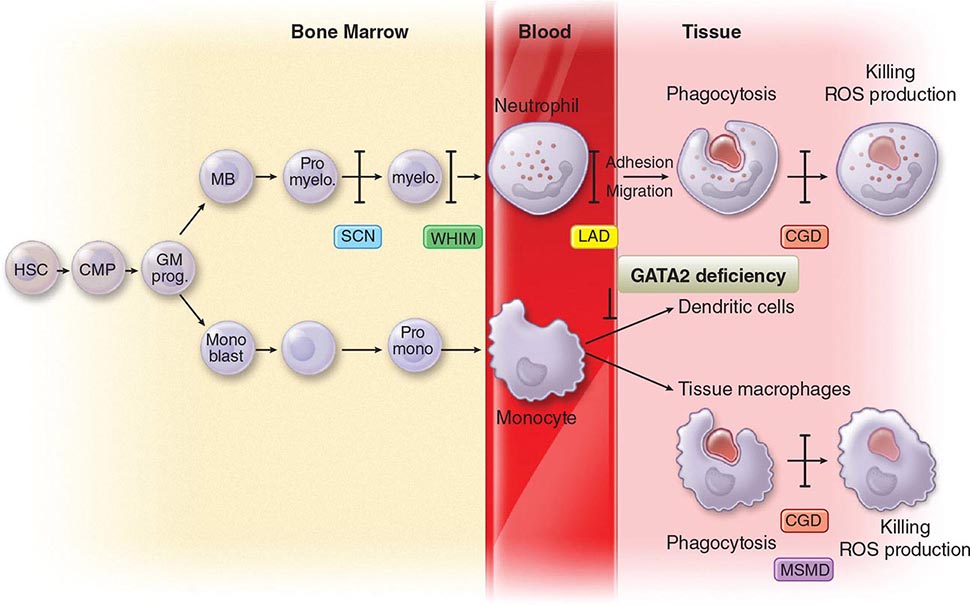
FIGURE 374-1 Differentiation of phagocytic cells and related primary immunodeficiencies (PIDs). Hematopoietic stem cells (HSCs) differentiate into common myeloid progenitors (CMPs) and then granulocyte-monocyte progenitors (GM-prog.), which, in turn, differentiate into neutrophils (MB: myeloblasts; Promyelo: promyelocytes; myelo: myelocytes) or monocytes (monoblasts and promonocytes). Upon activation, neutrophils adhere to the vascular endothelium, transmigrate, and phagocytose the targets. Reactive oxygen species (ROS) are delivered to the microorganism-containing phagosomes. Macrophages in tissues kill using the same mechanism. Following activation by interferon γ (not shown here), macrophages can be armed to kill intracellular pathogens such as mycobacteria. For sake of simplicity, not all cell differentiation stages are shown. The abbreviations for PIDs are contained in boxes placed at corresponding stages of the pathway. CGD, chronic granulomatous diseases; GATA2, zinc finger transcription factor; LAD, leukocyte adhesion deficiencies; MSMD, Mendelian susceptibility to mycobacterial disease; SCN, severe congenital neutropenia; WHIM, warts, hypogammaglobulinemia, infections, and myelokathexis.
PRIMARY IMMUNODEFICIENCIES OF THE INNATE IMMUNE SYSTEM
PIDs of the innate immune system are relatively rare and account for approximately 10% of all PIDs.
SEVERE CONGENITAL NEUTROPENIA
Severe congenital neutropenia (SCN) consists of a group of inherited diseases that are characterized by severely impaired neutrophil counts (<500 polymorphonuclear leukocytes [PMN]/μL of blood). The condition is usually manifested from birth. SCN may also be cyclic (with a 3-week periodicity), and other neutropenia syndromes can also be intermittent. Although the most frequent inheritance pattern for SCN is autosomal dominant, autosomal recessive and X-linked recessive conditions also exist. Bacterial infections at the interface between the body and the external milieu (e.g., the orifices, wounds, and the respiratory tract) are common manifestations. Bacterial infections can rapidly progress through soft tissue and are followed by dissemination in the bloodstream. Severe visceral fungal infections can also ensue. The absence of pus is a hallmark of this condition.
Diagnosis of SCN requires examination of the bone marrow. Most SCNs are associated with a block in granulopoiesis at the promyelocytic stage (Fig. 374-1). SCN has multiple etiologies, and to date, mutations in 11 different genes have been identified. Most of these mutations result in isolated SCN, whereas others are syndromic (Chap. 80). The most frequent forms of SCN are caused by the premature cell death of granulocyte precursors, as observed in deficiencies of GFI1, HAX1, and elastase 2 (ELANE), with the latter accounting for 50% of SCN sufferers. Certain ELANE mutations cause cyclic neutropenia syndrome. A gain-of-function mutation in the WASP gene (see the section on “Wiskott-Aldrich syndrome” below) causes X-linked SCN, which is also associated with monocytopenia.
As mentioned above, SCN exposes the patient to life-threatening, disseminated bacterial and fungal infections. Treatment requires careful hygiene measures, notably in infants. Later in life, special oral and dental care is essential, along with the prevention of bacterial infection by prophylactic administration of trimethoprim/sulfamethoxazole. Subcutaneous injection of the cytokine granulocyte colony-stimulating factor (G-CSF) usually improves neutrophil development and thus prevents infection in most SCN diseases. However, there are two caveats: (1) a few cases of SCN with ELANE mutation are refractory to G-CSF and may require curative treatment via allogeneic hematopoietic stem cell transplantation (HSCT); and (2) a subset of G-CSF-treated patients carrying ELANE mutations are at a greater risk of developing acute myelogenous leukemia associated (in most cases) with somatic gain-of-function mutations of the G-CSF receptor gene.
ASPLENIA
Primary failure of the development of a spleen is an extremely rare disease that can be either syndromic (in Ivemark syndrome) or isolated with an autosomal dominant expression; in the latter case, mutations in the ribosomal protein SA gene were recently found. Due to the absence of natural filtration of microbes in the blood, asplenia predisposes affected individuals to fulminant infections by encapsulated bacteria. Although most infections occur in the first years of life, cases may also arise in adulthood. The diagnosis is confirmed by abdominal ultrasonography and the detection of Howell-Jolly bodies in red blood cells. Effective prophylactic measures (twice-daily oral penicillin and appropriate vaccination programs) usually prevent fatal outcomes.
GATA2 DEFICIENCY
Recently an immunodeficiency combining monocytopenia and dendritic and lymphoid (B and natural killer [NK]) cell deficiency (DCML), also called monocytopenia with nontuberculous mycobacterial infections (mono-MAC), has been described as a consequence of a dominant mutation in the gene GATA2, a transcription factor involved in hematopoiesis. This condition also predisposes to lymphedema, myelodysplasia, and acute myeloid leukemia. Infections (bacterial and viral) are life-threatening, thus indicating, together with the malignant risk, HSCT.
LEUKOCYTE ADHESION DEFICIENCY (LAD)
Leukocyte adhesion deficiency (LAD) consists of three autosomal recessive conditions (LAD I, II, and III) (Chap. 80). The most frequent condition (LAD I) is caused by mutations in the β2 integrin gene; following leukocyte activation, β2 integrins mediate adhesion to inflamed endothelium expressing cognate ligands. LAD III results from a defect in a regulatory protein (kindlin, also known as Fermt 3) involved in activating the ligand affinity of β2 integrins. The extremely rare LAD II condition is the end result of a defect in selectin-mediated leukocyte rolling that occurs prior to β2 integrin binding. There is a primary defect in fucose transporter such that oligosaccharide selectin ligands are missing in this syndromic condition.
Given that neutrophils are not able to reach infected tissues, LAD renders the individual susceptible to bacterial and fungal infections in a way that is similar to that of patients with SCN. LAD also causes impaired wound healing and delayed loss of the umbilical cord. A diagnosis can be suspected in cases of pus-free skin/tissue infections and massive hyperleukocytosis (>30,000/μL) in the blood (mostly granulocytes). Patients with LAD III also develop bleeding because the β2 integrin in platelets is not functional. Use of immunofluorescence and functional assays to detect β2 integrin can help form a diagnosis. Severe forms of LAD may require HSCT, although gene therapy is also now being considered. Neutrophil-specific granule deficiency (a very rare condition caused by a mutation in the gene for transcription factor C/EBPα) results in a condition that is clinically similar to LAD.
CHRONIC GRANULOMATOUS DISEASES
Chronic granulomatous diseases (CGDs) are characterized by impaired phagocytic killing of microorganisms by neutrophils and macrophages (Chap. 80). The incidence is approximately 1 per 200,000 live births. About 70% of cases are associated with X-linked recessive inheritance versus autosomal inheritance in the remaining 30%. CGD causes deep-tissue bacterial and fungal abscesses in macrophage-rich organs such as the lymph nodes, liver, and lungs. Recurrent skin infections (such as folliculitis) are common and can prompt an early diagnosis of CGD. The infectious agents are typically catalase-positive bacteria (such as Staphylococcus aureus and Serratia marcescens) but also include Burkholderia cepacia, pathogenic mycobacteria (in certain regions of the world), and fungi (mainly filamentous molds, such as Aspergillus).
CGD is caused by defective production of reactive oxygen species (ROS) in the phagolysosome membrane following phagocytosis of microorganisms. It results from the lack of a component of NADPH oxidase (gp91phox or p22phox) or of the associated adapter/activating proteins (p47phox, p67phox, or p40phox) that mediate the transport of electrons into the phagolysosome for creating ROS by interaction with O2. Under normal circumstances, these ROS either directly kill engulfed microorganisms or enable the rise in pH needed to activate the phagosomal proteases that contribute to microbial killing. Diagnosis of CGD is based on assays of ROS production in neutrophils and monocytes (Table 374-2). As its name suggests, CGD is also a granulomatous disease. Macrophage-rich granulomas can often arise in the liver, spleen, and other organs. These are sterile granulomas that cause disease by obstruction (bladder, pylorus, etc.) or inflammation (colitis, restrictive lung disease).
The management of infections in patients with CGD can be a complex process. The treatment of bacterial infections is generally based on combination therapy with antibiotics that are able to penetrate into cells. The treatment of fungal infections requires aggressive, long-term use of antifungals. Inflammatory/granulomatous lesions are usually steroid-sensitive; however, glucocorticoids often contribute to the spread of infections. Hence, there is strong need for new therapeutic options in what is still a poorly understood disease.
The treatment of CGD mostly relies on preventing infections. It has been unambiguously demonstrated that prophylactic usage of trimethoprim/sulfamethoxazole is both well tolerated and highly effective in reducing the risk of bacterial infection. Daily administration of azole derivatives (notably itraconazole) also reduces the frequency of fungal complications. It has long been suggested that interferon γ administration is helpful, although medical experts continue to disagree over this controversial issue. Patients may do reasonably well with prophylaxis and careful management. However, other patients develop severe and persistent fungal infections and/or chronic inflammatory complications that ultimately require HSCT. The latter is an established curative approach for CGD; however, the risk-versus-benefit ratio must be carefully assessed on a case-by-case basis. Gene therapy approaches are also being evaluated.
MENDELIAN SUSCEPTIBILITY TO MYCOBACTERIAL DISEASE (MSMD)
This group of diseases is characterized by a defect in the interleukin-12 (IL-12)–interferon (IFN) γ axis (including IL-12p40, IL-12 receptor [R] β1, IFN-γ R1 and R2, STAT1, IRF8 and ISG515 deficiencies), which ultimately leads to impaired IFN-γ-dependent macrophage activation. Both recessive and dominant inheritance modes have been observed. The hallmark of this PID is a specific and narrow vulnerability to tuberculous and nontuberculous mycobacteria. The most severe phenotype (as observed in complete IFN-γ receptor deficiency) is characterized by disseminated infection that can be fatal even when aggressive and appropriate antimycobacterial therapy is applied. In addition to mycobacterial infections, MSMD patients (and particularly those with an IL-12/IL-12 R deficiency) are prone to developing Salmonella infections. Although MSMDs are very rare, they should be considered in any patient with persistent mycobacterial infection. Treatment with IFN-γ may efficiently bypass an IL-12/IL-12R deficiency.
TOLL-LIKE RECEPTOR (TLR) PATHWAY DEFICIENCIES
In a certain group of patients with early-onset, invasive Streptococcus pneumoniae infections or (less frequently) Staphylococcus aureus or other pyogenic infections, conventional screening for PIDs does not identify the cause of the defect in host defense. It has been established that these patients carry recessive mutations in genes that encode essential adaptor molecules (IRAK4 and MYD88) involved in the signaling pathways of the majority of known Toll-like receptors (TLRs) (Chap. 372e). Remarkably, susceptibility to infection appears to decrease after the first few years of life—perhaps an indication that adaptive immunity (once triggered by an initial microbial challenge) is then able to prevent recurrent infections.
Certain TLRs (TLR-3, -7, -8, and -9) are involved in the recognition of RNA and DNA and usually become engaged during viral infections. Very specific susceptibility to herpes simplex encephalitis has been described in patients with a deficiency in Unc93b (a molecule associated with TLR-3, -7, -8, and -9 required for correct subcellular localization), TLR-3, or associated signaling molecules TRIF, TBK1, and TRAF3, resulting in defective type I IFN production. The fact that no other TLR deficiencies have been found—despite extensive screening of patients with unexplained, recurrent infections—strongly suggests that these receptors are functionally redundant. Hypomorphic mutations in NEMO/IKK-γ (a member of the NF-κB complex, which is activated downstream of TLR receptors) lead to a complex, variable immunodeficiency and a number of associated features. Susceptibility to both invasive, pyogenic infections and mycobacteria may be observed in this particular setting.
COMPLEMENT DEFICIENCY
The complement system is composed of a complex cascade of plasma proteins (Chap. 372e) that leads to the deposition of C3b fragments on the surface of particles and the formation of immune complexes that can culminate in the activation of a lytic complex at the bacterial surface. C3 cleavage can be mediated via three pathways: the classic, alternate, and lectin pathways. C3b coats particles as part of the opsonization process that facilitates phagocytosis following binding to cognate receptors. A deficiency in any component of the classic pathway (C1q, C1r, C1s, C4, and C2) can predispose an individual to bacterial infections that are tissue-invasive or that occur in the respiratory tract. Likewise, a C3 deficiency or a deficiency in factor I (a protein that regulates C3 consumption, thus leading to a C3 deficiency due to its absence) also results in the same type of vulnerability to infection. It has recently been reported that a very rare deficiency in ficolin-3 predisposes affected individuals to bacterial infections. Deficiencies in the alternative pathway (factors D and properdin) are associated with the occurrence of invasive Neisseria infections.
Lastly, deficiencies of any complement component involved in the lytic phase (C5, C6, C7, C8, and, to a lesser extent, C9) predispose affected individuals to systemic infection by Neisseria. This is explained by the critical role of complement in the lysis of the thick cell wall possessed by this class of bacteria.
Diagnosis of a complement deficiency relies primarily on testing the status of the classic and alternate pathway via functional assays, i.e., the CH50 and AP50 tests, respectively. When either pathway is profoundly impaired, determination of the status of the relevant components in that pathway enables a precise diagnosis. Appropriate vaccinations and daily administration of oral penicillin are efficient means of preventing recurrent infections. It is noteworthy that several complement deficiencies (in the classic pathway and the lytic phase) may also predispose affected individuals to autoimmune diseases (notably systemic lupus erythematosus; Chap. 378).
PRIMARY IMMUNODEFICIENCIES OF THE ADAPTIVE IMMUNE SYSTEM
T LYMPHOCYTE DEFICIENCIES (TABLE 374-1, FIGS. 374-2 AND 374-3)
Given the central role of T lymphocytes in adaptive immune responses (Chap. 372e), PIDs involving T cells generally have severe pathologic consequences; this explains the poor overall prognosis and the need for early diagnosis and the early intervention with appropriate therapy. Several differentiation pathways of T cell effectors have been described, one or all of which may be affected by a given PID (Fig. 374-2). Follicular helper CD4+ T cells in germinal centers are required for T-dependent antibody production, including the generation of Ig class-switched, high-affinity antibodies. CD4+ TH1 cells provide cytokine-dependent (mostly IFN-γ-dependent) help to macrophages for intracellular killing of various microorganisms, including mycobacteria and Salmonella. CD4+ TH2 cells produce IL-4, IL-5, and IL-13 and thus recruit and activate eosinophils and other cells required to fight helminth infections. CD4+ TH17 cells produce IL-17 and IL-22 cytokines that recruit neutrophils to the skin and lungs to fight bacterial and fungal infections. Cytotoxic CD8+ T cells can kill infected cells, notably in the context of viral infections. In addition, certain T cell deficiencies predispose affected individuals to Pneumocystis jiroveci lung infections early in life and to chronic gut/biliary duct/liver infections by Cryptosporidium and related genera later on in life. Lastly, naturally occurring or induced regulatory T cells are essential for controlling inflammation (notably reactivity to commensal bacteria in the gut) and autoimmunity. The role of other T cell subsets with limited T cell receptor (TCR) diversity (such as γδTCR T cells or natural killer T [NKT] cells) in PIDs is less well known; however, these subsets can be defective in certain PIDs, and this finding can sometimes contribute to the diagnosis (e.g., NKT cell deficiency in X-linked proliferative syndrome). T cell deficiencies account for approximately 20% of all cases of PID.
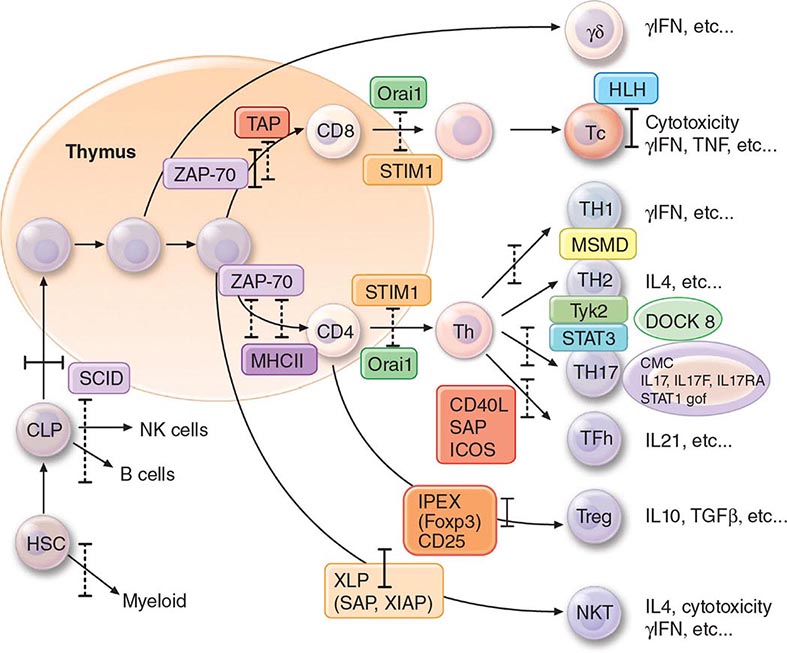
FIGURE 374-2 T cell differentiation, effector pathways, and related primary immunodeficiencies (PIDs). Hematopoietic stem cells (HSCs) differentiate into common lymphoid progenitors (CLPs), which, in turn, give rise to the T cell precursors that migrate to the thymus. The development of CD4+ and CD8+ T cells is shown. Known T cell effector pathways are indicated, i.e., γδ cells, cytotoxic T cells (Tc), TH1, TH2, TH17, TFh (follicular helper) CD4 effector T cells, regulatory T cells (Treg), and natural killer T cells (NKTs); abbreviations for PIDs are contained in boxes. Vertical bars indicate a complete deficiency; broken bars a partial deficiency. SCID, severe combined immunodeficiency; ZAP-70, zeta-associated protein deficiency; MHCII, major histocompatibility complex class II deficiency; TAP, TAP1 and TAP2 deficiencies; Orai1, STIM1 deficiencies; HLH, hematopoietic lymphohistiocytosis; MSMD, Mendelian susceptibility to mycobacterial disease; Tyk2, DOCK8, autosomal recessive form of hyper-IgE syndrome; STAT3, autosomal dominant form of hyper-IgE syndrome; IL17F, IL17RA, STAT1 (gof: gain of function), CMC (chronic mucocutaneous candidiasis), CD40L, ICOS, SAP deficiencies; IPEX, immunodysregulation polyendocrinopathy enteropathy X-linked syndrome; XLP, X-linked proliferative syndromes.
Severe Combined Immunodeficiencies Severe combined immunodeficiencies (SCIDs) constitute a group of rare PIDs characterized by a profound block in T cell development and thus the complete absence of these cells. The developmental block is always the consequence of an intrinsic deficiency. The incidence of SCID is estimated to be 1 in 50,000 live births. Given the severity of the T cell deficiency, clinical consequences occur early in life (usually within 3 to 6 months of birth). The most frequent clinical manifestations are recurrent oral candidiasis, failure to thrive, and protracted diarrhea and/or acute interstitial pneumonitis caused by Pneumocystis jiroveci (although the latter can also be observed in the first year of life in children with B cell deficiencies). Severe viral infections or invasive bacterial infections can also occur. Patients may also experience complications related to infections caused by live vaccines (notably bacille Calmette-Guérin [BCG]) that may lead not only to local and regional infection but also to disseminated infection manifested by fever, splenomegaly, and skin and lytic bone lesions. A scaly skin eruption can be observed in a context of maternal T cell engraftment (see below). A diagnosis of SCID can be suspected based on the patient’s clinical history and, possibly, a family history of deaths in very young children (suggestive of either X-linked or recessive inheritance). Lymphocytopenia is strongly suggestive of SCID in more than 90% of cases (Table 316-2). The absence of a thymic shadow on a chest x-ray can also be suggestive of SCID. An accurate diagnosis relies on precise determination of the number of circulating T, B, and NK lymphocytes and their subsets. T cell lymphopenia may be masked in some patients by the presence of maternal T cells (derived from maternal-fetal blood transfers) that cannot be eliminated. Although counts are usually low (<500/μL of blood), higher maternal T cell counts may, under some circumstances, initially mask the presence of SCID. Thus, screening for maternal cells by using adequate genetic markers should be performed whenever necessary. Inheritance pattern analysis and lymphocyte phenotyping can discriminate between various forms of SCID and provide guidance in the choice of accurate molecular diagnostic tests (see below). To date, five distinct causative mechanisms for SCID (Fig. 374-3) have been identified:
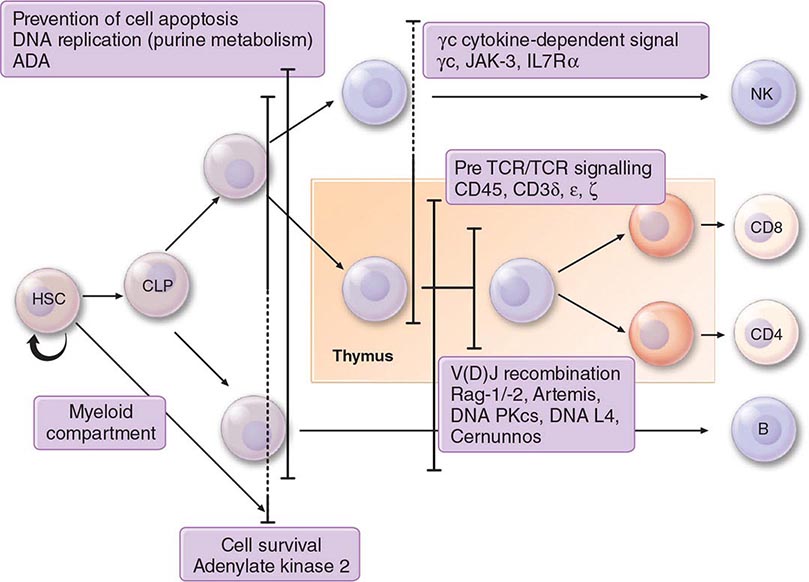
FIGURE 374-3 T cell differentiation and severe combined immunodeficiencies (SCIDs). The vertical bars indicate the five mechanisms currently known to lead to SCID. The names of deficient proteins are indicated in the boxes adjacent to the vertical bars. A broken line means that deficiency is partial or involves only some of the indicated immunodeficiencies. ADA, adenosine deaminase deficiency; CLPs, common lymphoid progenitors; DNAL4, DNA ligase 4; HSCs, hematopoietic stem cells; NKs, natural killer cells; TCR, T cell receptor.
SEVERE COMBINED IMMUNODEFICIENCY CAUSED BY A CYTOKINE-SIGNALING DEFICIENCY The most frequent SCID phenotype (accounting for 40–50% of all cases) is the absence of both T and NK cells. This outcome results from a deficiency in either the common γ chain (γc) receptor that is shared by several cytokine receptors (the IL-2, -4, -7, -9, -15, and -21 receptors) or Jak-associated kinase (JAK) 3 that binds to the cytoplasmic portion of the γc chain receptor and induces signal transduction following cytokine binding. The former form of SCID (γc deficiency) has an X-linked inheritance mode, while the second is autosomal recessive. A lack of the IL-7Rα chain (which, together with γc, forms the IL-7 receptor) induces a selective T cell deficiency.
PURINE METABOLISM DEFICIENCY Ten to 20% of SCID patients exhibit a deficiency in adenosine deaminase (ADA), an enzyme of purine metabolism that deaminates adenosine (ado) and deoxyadenosine (dAdo). An ADA deficiency results in the accumulation of ado and dAdo metabolites that induce premature cell death of lymphocyte progenitors. The condition results in the absence of B and NK lymphocytes as well as T cells. The clinical expression of complete ADA deficiency typically occurs very early in life. Since ADA is a ubiquitous enzyme, its deficiency can also cause bone dysplasia with abnormal costochondral junctions and metaphyses (found in 50% of cases) and neurologic defects. The very rare purine nucleoside phosphorylase (PNP) deficiency causes a profound although incomplete T cell deficiency that is often associated with severe neurologic impairments.
DEFECTIVE REARRANGEMENTS OF T AND B CELL RECEPTORS A series of SCID conditions are characterized by a selective deficiency in T and B lymphocytes with autosomal recessive inheritance. These conditions account for 20–30% of SCID cases and result from mutations in genes encoding proteins that mediate the recombination of V(D)J gene elements in T and B cell antigen receptor genes (required for the generation of diversity in antigen recognition). The main deficiencies involve RAG-1, RAG-2, DNA-dependent protein kinase, and Artemis. A less severe (albeit variable) immunologic phenotype can result from other deficiencies in the same pathway, i.e., DNA ligase 4 and Cernunnos deficiencies. Given that these latter factors are involved in DNA repair, these deficiencies also cause developmental defects.
DEFECTIVE (PRE-)T CELL RECEPTOR SIGNALING IN THE THYMUS A selective T cell defect can be caused by a series of rare deficiencies in molecules involved in signaling via the pre-TCR or the TCR. These include deficiencies in CD3 subunits associated with the (pre-)TCR (i.e., CD3δ, ε, and ζ) and CD45.
RETICULAR DYSGENESIS Reticular dysgenesis is an extremely rare form of SCID that causes T and NK deficiencies with severe neutropenia and sensorineural deafness. It results from an adenylate kinase 2 deficiency.
Patients with SCID require appropriate care with aggressive anti-infective therapies, immunoglobulin replacement, and (when necessary) parenteral nutrition support. In most cases, curative treatment relies on HSCT. Today, HSCT provides a very high curative potential for SCID patients who are otherwise in reasonably good condition. In this regard, neonatal screening, based on quantification of T cell receptor excision circles (TRECs) on a Guthrie card sample, is being developed. Gene therapy has been found to be successful for cases of X-linked SCID (γc deficiency) and SCID caused by an ADA deficiency, although toxicity has become an issue in the treatment of the former disease that may now be overcome by use of newly generated vectors. Lastly, a third option for the treatment of ADA deficiency consists of enzyme substitution with a pegylated enzyme.
Thymic Defects A profound T cell defect can also result from faulty development of the thymus, as is most often observed in rare cases of DiGeorge syndrome—a relatively common condition leading to a constellation of developmental defects. In approximately 1% of such cases, the thymus is completely absent, leading to virtually no mature T cells. However, expansion of oligoclonal T cells can occur and is associated with skin lesions. Diagnosis (using immunofluorescence in situ hybridization) is based on the identification of a hemizygous deletion in the long arm of chromosome 22. To recover the capability for T cell differentiation, these cases require a thymic graft. CHARGE (coloboma of the eye, heart anomaly, choanal atresia, retardation, genital and ear anomalies) syndrome (CHD7 deficiency) is a less frequent cause of impaired thymus development. Lastly, the very rare “nude” defect is characterized by the absence of both hair and the thymus.
Omenn Syndrome Omenn syndrome consists of a subset of T cell deficiencies that present with a unique phenotype, including early-onset erythrodermia, alopecia, hepatosplenomegaly, and failure to thrive. These patients usually display T cell lymphocytosis, eosinophilia, and low B cell counts. It has been found that the T cells of these patients exhibit a low TCR heterogeneity. This peculiar syndrome is the consequence of hypomorphic mutations in genes usually associated with SCID, i.e., RAG-1, RAG-2, or (less frequently) Artemis or IL-7Rα. The impaired homeostasis of differentiating T cells thus causes this immune system–associated disease. These patients are very fragile, requiring simultaneous anti-infective therapy, nutritional support, and immunosuppression. HSCT provides a curative approach.
Functional T Cell Defects (Fig. 374-2) A subset of T cell PIDs with autosomal inheritance is characterized by partially preserved T cell differentiation but defective activation resulting in abnormal effector function. There are many causes of these defects, but all lead to susceptibility to viral and opportunistic infections, chronic diarrhea, and failure to thrive, with onset during childhood. Careful phenotyping and in vitro functional assays are required to identify these diseases, the best characterized of which are the following.
ZETA-ASSOCIATED PROTEIN 70 (ZAP70) DEFICIENCY Zeta-associated protein 70 (ZAP70) is recruited to the TCR following antigen recognition. A ZAP70 deficiency leads typically to an almost complete absence of CD8+ T cells; CD4+ T cells are present but cannot be activated in vitro by TCR stimulation.
CALCIUM SIGNALING DEFECTS A small number of patients have been reported who exhibit a profound defect in in vitro T and B cell activation as a result of defective antigen receptor-mediated Ca2+ influx. This defect is caused by a mutation in the calcium channel gene (ORAI-) or its activator (STIM-1). It is noteworthy that these patients are also prone to autoimmune manifestations (blood cytopenias) and exhibit a nonprogressive muscle disease.
HUMAN LEUKOCYTE ANTIGEN (HLA) CLASS II DEFICIENCY Defective expression of HLA class II molecules is the hallmark of a group of four recessive genetic defects all of which affect molecules (RFX5, RFXAP, RFXANK, and CIITA) involved in the transactivation of the genes coding for HLA class II. As a result, low but variable CD4+ T cell counts are observed in addition to defective antigen-specific T and B cell responses. These patients are particularly susceptible to herpesvirus, adenovirus, and enterovirus infections and chronic gut/liver Cryptosporidium infections.
HLA CLASS I DEFICIENCY Defective expression of molecules involved in antigen presentation by HLA class I molecules (i.e., TAP-1, TAP-2, and Tapasin) leads to reduced CD8+ T cell counts, loss of HLA class I antigen expression, and a particular phenotype consisting of chronic obstructive pulmonary disease and severe vasculitis.
OTHER DEFECTS A variety of other T cell PIDs have been described, some of which are associated with a precise molecular defect (e.g., IL-2-inducible T cell kinase [ITK] deficiency, IL-21 receptor deficiency, CARD11 deficiency). These conditions are also characterized by profound vulnerability to infections, such as severe Epstein-Barr virus (EBV)–induced B cell proliferation and autoimmune disorders in ITK deficiency. Milder phenotypes are associated with CD8 and CD3γ deficiencies.
HSCT is indicated for most of these diseases, although the prognosis is worse than in SCID because many patients are chronically infected at the time of diagnosis. Fairly aggressive immunosuppression and myeloablation may be necessary to achieve engraftment of allogeneic stem cells.
T Cell Primary Immunodeficiencies with DNA Repair Defects This is a group of PIDs characterized by a combination of T and B cell defects of variable intensity, together with a number of nonimmunologic features resulting from DNA fragility. The autosomal recessive disorder ataxia-telangiectasia (AT) is the most frequently encountered condition in this group. It has an incidence of 1:40,000 live births and causes B cell defects (low IgA, IgG2 deficiency, and low antibody production), which often require immunoglobulin replacement. AT is associated with a progressive T cell immunodeficiency. As the name suggests, the hallmark features of AT are telangiectasia and cerebellar ataxia. The latter manifestations may not be detectable before the age of 3–4 years, so that AT should be considered in young children with IgA deficiency and recurrent and problematic infections. Diagnosis is based on a cytogenetic analysis showing excessive chromosomal rearrangements (mostly affecting chromosomes 7 and 14) in lymphocytes. AT is caused by a mutation in the gene encoding the ATM protein—a kinase that plays an important role in the detection and repair of DNA lesions (or cell death if the lesions are too numerous) by triggering several different pathways. Overall, AT is a progressive disease that carries a very high risk of lymphoma, leukemia, and (during adulthood) carcinomas. A variant of AT (“AT-like disease”) is caused by mutation in the MRE11 gene.
Nijmegen breakage syndrome (NBS) is a less common condition that also results from chromosome instability (with the same cytogenetic abnormalities as in AT). NBS is characterized by a severe T and B cell combined immune deficiency with autosomal recessive inheritance. Individuals with NBS exhibit microcephaly and a bird-like face, but have neither ataxia nor telangiectasia. The risk of malignancies is very high. NBS results from a deficiency in nibrin (NBSI, a protein associated with MRE11 and Rad50 that is involved in checking DNA lesions) caused by hypomorphic mutations.
Severe forms of dyskeratosis congenita (also known as Hoyeraal-Hreidarsson syndrome) combine a progressive immunodeficiency that can also include an absence of B and NK lymphocytes, progressive bone marrow failure, microcephaly, in utero growth retardation, and gastrointestinal disease. The disease can be X-linked or, more rarely, autosomal recessive. It is caused by the mutation of genes encoding telomere maintenance proteins, including dyskerin (DKC1).
Finally, immunodeficiency with centromeric and facial anomalies (ICF) is a complex syndrome of autosomal recessive inheritance that variably combines a mild T cell immune deficiency with a more severe B cell immune deficiency, coarse face, digestive disease, and mild mental retardation. A diagnostic feature is the detection by cytogenetic analysis of multiradial aspects in multiple chromosomes (most frequently 1, 9, and 16) corresponding to an abnormal DNA structure secondary to defective DNA methylation. It is the consequence of a deficiency in the DNA methyltransferase DNMT3B, or ZBTB24.
T Cell Primary Immunodeficiencies with Hyper-IgE Several T cell PIDs are associated with elevated serum IgE levels (as in Omenn syndrome). A condition sometimes referred to as autosomal recessive hyper-IgE syndrome is notably characterized by recurrent bacterial infections in the skin and respiratory tract and severe skin and mucosal infections by pox viruses and human papillomaviruses, together with severe allergic manifestations. T and B lymphocyte counts are low. Mutations in the DOCK8 gene have been found in most of these patients. This condition is an indication for HSCT.
A very rare, related condition with autosomal recessive inheritance that causes a similar susceptibility to infection with various microbes (see above), including mycobacteria, reportedly results from a deficiency in Tyk-2, a JAK family kinase involved in the signaling of many different cytokine receptors.
Autosomal Dominant Hyper-IgE Syndrome This unique condition, the autosomal dominant hyper-IgE syndrome, is usually diagnosed by the combination of recurrent skin and lung infections that can be complicated by pneumatoceles. Infections are caused by pyogenic bacteria and fungi. Several other manifestations characterize hyper-IgE syndrome, including facial dysmorphy, defective loss of primary teeth, hyperextensibility, scoliosis, and osteoporosis. Elevated serum IgE levels are typical of this syndrome. Defective TH17 effector responses have been shown to account at least in part for the specific patterns of susceptibility to particular microbes. This condition is caused by a heterozygous (dominant) mutation in the gene encoding the transcription factor STAT3 that is required in a number of signaling pathways following binding of cytokine to cytokine receptors (such as that of IL-6 and the IL-6 receptor). It also results in partially defective antibody production because of defective IL-21R signaling. Hence, immunoglobulin substitution can be considered as prophylaxis of bacterial infections.
Cartilage Hair Hypoplasia The autosomal recessive cartilage hair hypoplasia (CHH) disease is characterized by short-limb dwarfism, metaphyseal dysostosis, and sparse hair, together with a combined T and B cell PID of extremely variable intensity (ranging from quasi-SCID to no clinically significant immune defects). The condition can predispose to erythroblastopenia, autoimmunity, and tumors. It is caused by mutations in the RMRP gene for a noncoding ribosome-associated RNA.
CD40 Ligand and CD40 Deficiencies Hyper-IgM syndrome (HIGM) is a well-known PID that is usually classified as a B cell immune deficiency (see Fig. 374-4 and below). It results from defective immunoglobulin class switch recombination (CSR) in germinal centers and leads to profound deficiency in production of IgG, IgA, and IgE (although IgM production is maintained). Approximately half of HIGM sufferers are also prone to opportunistic infections, e.g., interstitial pneumonitis caused by Pneumocystis jiroveci (in young children), protracted diarrhea and cholangitis caused by Cryptosporidium, and infection of the brain with Toxoplasma gondii.
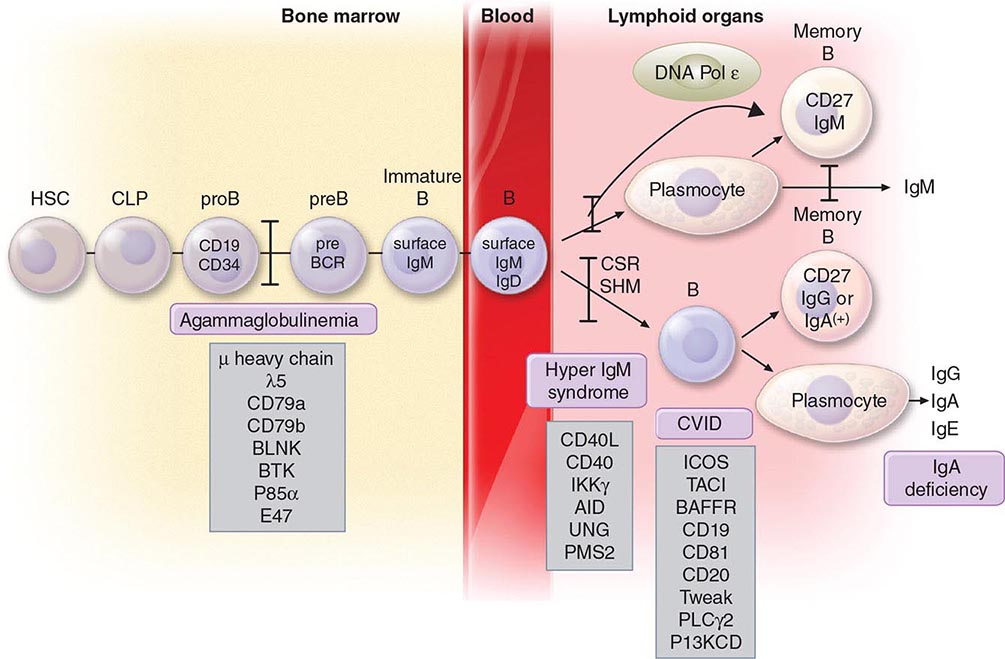
FIGURE 374-4 B cell differentiation and related primary immunodeficiencies (PIDs). Hematopoietic stem cells (HSCs) differentiate into common lymphoid progenitors (CLPs), which give rise to pre-B cells. The B cell differentiation pathway goes through the pre–B cell stage (expression of the μ heavy chain and surrogate light chain), the immature B cell stage (expression of surface IgM), and the mature B cell stage (expression of surface IgM and IgD). The main phenotypic characteristics of these cells are indicated. In lymphoid organs, B cells can differentiate into plasma cells and produce IgM or undergo (in germinal centers) Ig class switch recombination (CSR) and somatic mutation of the variable region of V genes (SHM) that enable selection of high-affinity antibodies. These B cells produce antibodies of various isotypes and generate memory B cells. PIDs are indicated in the purple boxes. CVID, common variable immunodeficiency.
In the majority of cases, this condition has an X-linked inheritance and is caused by a deficiency in CD40 ligand (L). CD40L induces signaling events in B cells that are necessary for both CSR and adequate activation of other CD40-expressing cells that are involved in innate immune responses against the above-mentioned microorganisms. More rarely, the condition is caused by a deficiency in CD40 itself. The poorer prognosis of CD40L and CD40 deficiencies (relative to most other HIGM conditions) implies that (1) thorough investigations have to be performed in all cases of HIGM and (2) potentially curative HSCT should be discussed on a case-by-case basis for this group of patients.
Wiskott-Aldrich Syndrome Wiskott-Aldrich syndrome (WAS) is a complex, recessive, X-linked disease with an incidence of approximately 1 in 200,000 live births. It is caused by mutations in the WASP gene that affect not only T lymphocytes but also the other lymphocyte subsets, dendritic cells, and platelets. WAS is typically characterized by the following clinical manifestations: recurrent bacterial infections, eczema, and bleeding caused by thrombocytopenia. However, these manifestations are highly variable—mostly as a consequence of the many different WASP mutations that have been observed. Null mutations predispose affected individuals to invasive and bronchopulmonary infections, viral infections, severe eczema, and autoimmune manifestations. The latter include autoantibody-mediated blood cytopenia, glomerulonephritis, skin and visceral vasculitis (including brain vasculitis), erythema nodosum, and arthritis. Another possible consequence of WAS is lymphoma, which may be virally induced (e.g., by EBV or Kaposi’s sarcoma–associated herpesvirus). Thrombocytopenia can be severe and compounded by the peripheral destruction of platelets associated with autoimmune disorders. Hypomorphic mutations usually lead to milder outcomes that are generally limited to thrombocytopenia. It is noteworthy that even patients with “isolated” X-linked thrombocytopenia can develop severe autoimmune disease or lymphoma later in life. The immunologic workup is not very informative; there can be a relative CD8+ T cell deficiency, frequently accompanied by low serum IgM levels and decreased antigen-specific antibody responses. A typical feature is reduced-sized platelets on a blood smear. Diagnosis is based on intracellular immunofluorescence analysis of WAS protein (WASp) expression in blood cells. WASp regulates the actin cytoskeleton and thus plays an important role in many lymphocyte functions, including cell adhesion and migration and the formation of synapses between antigen-presenting and target cells. Predisposition to autoimmune disorders is in part related to defective regulatory T cells. The treatment of WAS should match the severity of disease expression. Prophylactic antibiotics, immunoglobulin G (IgG) supplementation, and careful topical treatment of eczema are indicated. Although splenectomy improves platelet count in a majority of cases, this intervention is associated with a significant risk of infection (both before and after HSCT). Allogeneic HSCT is curative, with fairly good results overall. Gene therapy trials are also under way. A similar condition has been reported in a girl with a deficiency in the Wiskott-Aldrich interacting protein (WIP).
A few other complex PIDs are worth mentioning. Sp110 deficiency causes a T cell PID with liver venoocclusive disease and hypogammaglobulinemia. Chronic mucocutaneous candidiasis (CMC) is a heterogeneous disease, considering the different inheritance patterns that have been observed. In some cases, chronic candidiasis is associated with late-onset bronchopulmonary infections, bronchiectasis, and brain aneurysms. Moderate forms of CMC are related to autoimmunity and AIRE deficiency (see below). In this setting, predisposition to Candida infection is associated with the detection of autoantibodies to TH17 cytokines. Recently, deficiencies in IL-17F and IL-17 receptor A and, above all, gain-of-function mutations in STAT1 have been found to be associated with CMC. In all cases, CMC is related to defective TH17 function. Innate immunodeficiency in CARD9 also predisposes to chronic invasive fungal infection.
B LYMPHOCYTE DEFICIENCIES (TABLE 374-1, FIG. 374-4)
Deficiencies that predominantly affect B lymphocytes are the most frequent PIDs and account for 60–70% of all cases. B lymphocytes make antibodies. Pentameric IgMs are found in the vascular compartment and are also secreted at mucosal surfaces. IgG antibodies diffuse freely into extravascular spaces, whereas IgA antibodies are produced and secreted predominantly from mucosa-associated lymphoid tissues. Although Ig isotypes have distinct effector functions, including Fc receptor–mediated and (indirectly) C3 receptor–dependent phagocytosis of microorganisms, they share the ability to recognize and neutralize a given pathogen. Defective antibody production therefore allows the establishment of invasive, pyogenic bacterial infections as well as recurrent sinus and pulmonary infections (mostly caused by Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis, and, less frequently, gram-negative bacteria). If left untreated, recurrent bronchial infections lead to bronchiectasis and, ultimately, cor pulmonale and death. Parasitic infections such as caused by Giardia lamblia and bacterial infections caused by Helicobacter and Campylobacter of the gut are also observed. A complete lack of antibody production (namely agammaglobulinemia) can also predispose affected individuals to severe, chronic, disseminated enteroviral infections causing meningoencephalitis, hepatitis, and a dermatomyositis-like disease.
Even with the most profound of B cell deficiencies, infections rarely occur before the age of 6 months; this is because of transient protection provided by the transplacental passage of immunoglobulins during the last trimester of pregnancy. Conversely, a genetically nonimmunodeficient child born to a mother with hypogammaglobulinemia is, in the absence of maternal Ig substitution, usually prone to severe bacterial infections in utero and for several months after birth.
Diagnosis of B cell PIDs relies on the determination of serum Ig levels (Table 316-2). Determination of antibody production following immunization with tetanus toxoid vaccine or nonconjugated pneumococcal polysaccharide antigens can also help diagnose more subtle deficiencies. Another useful test is B cell phenotype determination in switched μ–δ– CD27+ and nonswitched memory B cells (μ+δ+ CD27+). In agammaglobulinemic patients, examination of bone marrow B cell precursors (Fig. 374-4) can help obtain a precise diagnosis and guide the choice of genetic tests.
Agammaglobulinemia Agammaglobulinemia is characterized by a profound defect in B cell development (<1% of the normal B cell blood count). In most patients, very low residual Ig isotypes can be detected in the serum. In 85% of cases, agammaglobulinemia is caused by a mutation in the BTK gene that is located on the × chromosome. The BTK gene product is a kinase that participates in (pre) B cell receptor signaling. When the kinase is defective, there is a block (albeit a leaky one) at the pre-B to B cell stage (Fig. 374-4). Detection of BTK by intracellular immunofluorescence of monocytes, and lack thereof in patients with X-linked agammaglobulinemia, is a useful diagnostic test. Not all of the mutations in BTK result in agammaglobulinemia, since some patients have a milder form of hypogammaglobulinemia and low but detectable B cell counts. These cases should not be confused with common variable immunodeficiency (CVID, see below). About 10% of agammaglobulinemia cases are caused by alterations in genes encoding elements of the pre-B cell receptor, i.e., the μ heavy chain, the λ5 surrogate light chain, Igα or Igβ, the scaffold protein BLNK, and the p85 α subunit of phosphatidylinositol 3 phosphate kinase (P13K). In 5% of cases, the defect is unknown. It is noteworthy that agammaglobulinemia can be observed in patients with ICF syndrome, despite the presence of normal peripheral B cell counts. Lastly, agammaglobulinemia can be a manifestation of a myelodysplastic syndrome (associated or not with neutropenia). Treatment of agammaglobulinemic patients is based on immunoglobulin replacement (see below). Profound hypogammaglobulinemia is also observed in adults, in association with thymoma.
Hyper-IgM (HIGM) Syndromes HIGM is a rare B cell PID characterized by defective Ig CSR. It results in very low serum levels of IgG and IgA and elevated or normal serum IgM levels. The clinical severity is similar to that seen in agammaglobulinemia, although chronic lung disease and sinusitis are less frequent and enteroviral infections are uncommon. As discussed above, a diagnosis of HIGM involves screening for an X-linked CD40L deficiency and an autosomal recessive CD40 deficiency, which affect both B and T cells. In 50% of cases affecting only B cells, these isolated HIGM syndromes result from mutations in the gene encoding activation-induced deaminase, the protein that induces CSR in B cell germinal centers. These patients usually have enlarged lymphoid organs. In the other 50% of cases, the etiology is unknown (except for rare UNG and PMS2 deficiencies). Furthermore, IgM-mediated autoimmunity and lymphomas can occur in HIGM syndrome. It is noteworthy that HIGM can result from fetal rubella syndrome or can be a predominant immunologic feature of other PIDs, such as the immunodeficiency associated with ectodermic anhydrotic hypoplasia X-linked NEMO deficiency and the combined T and B cell PIDs caused by DNA repair defects such as AT and Cernunnos deficiency.
Common Variable Immunodeficiency (CVID) CVID is an ill-defined condition characterized by low serum levels of one or more Ig isotypes. Its prevalence is estimated to be 1 in 20,000. The condition is recognized predominantly in adults, although clinical manifestations can occur earlier in life. Hypogammaglobulinemia is associated with at least partially defective antibody production in response to vaccine antigens. B lymphocyte counts are often normal but can be low. Besides infections, CVID patients may develop lymphoproliferation (splenomegaly), granulomatous lesions, colitis, antibody-mediated autoimmune disease, and lymphomas. A family history is found in 10% of cases. A clear-cut dominant inheritance pattern is found in some families, whereas recessive inheritance is observed more rarely. In most cases, no molecular cause can be identified. A small number of patients in Germany were found to carry mutations in the ICOS gene encoding a T cell-membrane protein that contributes to B cell activation and survival. In 10% of patients with CVID, monoallelic or biallelic mutations of the gene encoding TACI (a member of the tumor necrosis factor [TNF] receptor family that is expressed on B cells) have been found. In fact, heterozygous TACI mutations correspond to a genetic susceptibility factor, since similar heterozygous mutations are found in 1% of controls. The BAFF receptor was found to be defective in a kindred with CVID, although not all individuals carrying the mutation have CVID.
Recently a group of patients with hypogammaglobulinemia and lymphoproliferation were shown to exhibit dominant gain of function mutations in the PIK3CD gene encoding the p110δ form of P13 kinase. A diagnosis of CVID should be made after excluding the presence of hypomorphic mutations associated with agammaglobulinemia or more subtle T cell defects; this is particularly the case in children. It is possible that many cases of CVID result from a constellation of factors, rather than a single genetic defect. Recently, rare cases of hypogammaglobulinemia were found to be associated with CD19 and CD81 deficiencies. These patients have B cells that can be identified by typing for other B cell markers. Hypogammaglobulinemia can be associated with neutropenia and lymphopenia in the warts, hypogammaglobulinemia, infections, and myelokathexis syndrome (WHIM) caused by dominant gain-of-function mutation of CXCR4, resulting in cell retention in the bone marrow.
Selective Ig Isotype Deficiencies IgA deficiency and CVID represent polar ends of a clinical spectrum due to the same underlying gene defect(s) in a large subset of these patients. IgA deficiency is the most common PID; it can be found in 1 in every 600 individuals. It is asymptomatic in most cases; however, individuals may present with increased numbers of acute and chronic respiratory infections that may lead to bronchiectasis. In addition, over their lifetime, these patients experience an increased susceptibility to drug allergies, atopic disorders, and autoimmune diseases. Symptomatic IgA deficiency is probably related to CVID, since it can be found in relatives of patients with CVID. Furthermore, IgA deficiency may progress to CVID. It is thus important to assess serum Ig levels in IgA-deficient patients (especially when infections occur frequently) in order to detect changes that should prompt the initiation of immunoglobulin replacement. Selective IgG2 (+G4) deficiency (which in some cases may be associated with IgA deficiency) can also result in recurrent sinopulmonary infections and should thus be specifically sought in this clinical setting. These conditions are ill-defined and often transient during childhood. A pathophysiologic explanation has not been found.
Selective Antibody Deficiency to Polysaccharide Antigens Some patients with normal serum Ig levels are prone to S. pneumoniae and H. influenzae infections of the respiratory tract. Defective production of antibodies against polysaccharide antigens (such as those in the S. pneumoniae cell wall) can be observed and is probably causative. This condition may correspond to a defect in marginal zone B cells, a B cell subpopulation involved in T-independent antibody responses.
Immunoglobulin Replacement IgG antibodies have a half-life of 21–28 days. Thus, injection of plasma-derived polyclonal IgG containing a myriad of high-affinity antibodies can provide protection against disease-causing microorganisms in patients with defective IgG antibody production. This form of therapy should not be based on laboratory data alone (i.e., IgG and/or antibody deficiency) but should be guided by the occurrence or not of infections; otherwise, patients might be subjected to unjustified IgG infusions. Immunoglobulin replacement can be performed by IV or subcutaneous routes. In the former case, injections have to be repeated every 3–4 weeks, with a residual target level of 800 mg/mL in patients who had very low IgG levels prior to therapy. Subcutaneous injections are typically performed once a week, although the frequency can be adjusted on a case-by-case basis. A trough level of 800 mg/mL is desirable. Whatever the mode of administration, the main goal is to reduce the frequency of the respiratory tract infections and prevent chronic lung and sinus disease. The two routes appear to be equally safe and efficacious, and so the choice should be left to the preference of the patient.
In patients with chronic lung disease, chest physical therapy with good pulmonary toilet and the cyclic use of antibiotics are also needed. Immunoglobulin replacement is well tolerated by most patients, although the selection of the best-tolerated Ig preparation may be necessary in certain cases. Since IgG preparations contain a small proportion of IgAs, caution should be taken in patients with residual antibody production capacity and a complete IgA deficiency, as these subjects may develop anti-IgA antibodies that can trigger anaphylactic shock. These patients should be treated with IgA-free IgG preparations. Immunoglobulin replacement is a lifelong therapy; its rationale and procedures have to be fully understood and mastered by the patient and his or her family in order to guarantee the strict observance required for efficacy.
PRIMARY IMMUNODEFICIENCIES AFFECTING REGULATORY PATHWAYS (TABLE 374-1)
An increasing number of PIDs have been found to cause homeostatic dysregulation of the immune system, either alone or in association with increased vulnerability to infections. Defects of this type affecting the innate immune system and autoinflammatory syndromes will not be covered in this chapter. However, three specific entities (hemophagocytic lymphohistiocytosis, lymphoproliferation, and autoimmunity) will be described below.
HEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS
Hemophagocytic lymphohistiocytosis (HLH) is characterized by an unremitting activation of CD8+ T lymphocytes and macrophages that leads to organ damage (notably in the liver, bone marrow, and central nervous system). This syndrome results from a broad set of inherited diseases, all of which impair T and NK lymphocyte cytotoxicity. The manifestations of HLH are often induced by a viral infection. EBV is the most frequent trigger. In severe forms of HLH, disease onset may start during the first year of life or even (in rare cases) at birth.
Diagnosis relies on the identification of the characteristic symptoms of HLH (fever, hepatosplenomegaly, edema, neurologic diseases, blood cytopenia, increased liver enzymes, hypofibrinogenemia, high triglyceride levels, elevated markers of T cell activation, and hemophagocytic features in the bone marrow or cerebrospinal fluid). Functional assays of postactivation cytotoxic granule exocytosis (CD107 fluorescence at the cell membrane) can suggest genetically determined HLH. The conditions can be classified into three subsets:
1. Familial HLH with autosomal recessive inheritance, including perforin deficiency (30% of cases) that can be recognized by assessing intracellular perforin expression; Munc13-4 deficiency (30% of cases); syntaxin 11 deficiency (10% of cases); Munc18-2 deficiency (20% of cases); and a few residual cases that lack a known molecular defect.
2. HLH with partial albinism. Three conditions combine HLH and abnormal pigmentation, where hair examination can help in the diagnosis: Chédiak-Higashi syndrome, Griscelli syndrome, and Hermansky Pudlak syndrome type II. Chédiak-Higashi syndrome is also characterized by the presence of giant lysosomes within leukocytes (Chap. 80), in addition to a primary neurologic disorder with slow progression of symptoms over time.
3. X-linked proliferative syndrome (XLP) is characterized in most patients by the induction of HLH following EBV infection, while other patients develop progressive hypogammaglobulinemia similar to what is observed in CVID and/or certain lymphomas. XLP is caused by a mutation in the SH2DIA gene that encodes the adaptor protein SAP (associated with a SLAM family receptor). Several immunologic abnormalities have been described, including low 2B4-mediated NK cell cytotoxicity, impaired differentiation of NKT cells, defective antigen-induced T cell death, and defective T cell helper activity for B cells. A related disorder (XLP2) has recently been described. It is also X-linked and induces HLH (frequently after EBV infection), although the clinical manifestation may be less pronounced. The condition is associated with a deficiency of the antiapoptotic molecule XIAP. The pathophysiology of XLP2 and its relationship to XLP1 remain unclear.
HLH is a life-threatening complication. The treatment of this condition requires aggressive immunosuppression with either the cytotoxic agent etoposide or anti–T cell antibodies. Once remission has been achieved, HSCT should be performed, since it provides the only curative form of therapy.
AUTOIMMUNE LYMPHOPROLIFERATIVE SYNDROME
Autoimmune lymphoproliferative syndrome (ALPS) is characterized by nonmalignant T and B lymphoproliferation causing splenomegaly and enlarged lymph nodes; 70% of patients also display autoimmune manifestations such as autoimmune cytopenias, Guillain-Barré syndrome, uveitis, and hepatitis (Chaps. 79 and 372e). A hallmark of ALPS is the presence of CD4–CD8–TCRαβ+ T cells (2–50%) in the blood of affected individuals. Hypergammaglobulinemia involving IgG and IgA is also frequently observed. The syndrome is caused by a defect in Fas-mediated apoptosis of lymphocytes, which can thus accumulate and mediate autoimmunity. Furthermore, ALPS can lead to malignancies.
Most patients carry a heterozygous mutation in the gene encoding Fas that is characterized by dominant inheritance and variable penetrance, depending on the nature of the mutation. A rare and severe form of the disease with early onset can be observed in patients carrying a biallelic mutation of Fas, which profoundly impairs the protein’s expression and/or function. Fas-ligand, caspase 10, caspase 8, and neuroblastoma RAS viral oncogene homologue (NRAS) mutations have also been reported in a few cases of ALPS. Many cases of ALPS have not been precisely delineated at the molecular level. A B cell–predominant ALPS has recently been found associated with a protein kinase Cδ gene mutation. Treatment of ALPS is essentially based on the use of proapoptotic drugs, which need to be carefully administered in order to avoid toxicity.
COLITIS, AUTOIMMUNITY, AND PRIMARY IMMUNODEFICIENCIES
Several PIDs (most of which are T cell–related) can cause severe gut inflammation. The prototypic example is immunodysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX), characterized by a widespread inflammatory enteropathy, food intolerance, skin rashes, autoimmune cytopenias, and diabetes. The syndrome is caused by loss-of-function mutations in the gene encoding the transcription factor FOXP3, which is required for the acquisition of effector function by regulatory T cells. In most cases of IPEX, CD4+CD25+ regulatory T cells are absent from the blood. This condition has a poor prognosis and requires aggressive immunosuppression. The only possible curative approach is allogeneic HSCT. IPEX-like syndromes that lack a FOXP3 mutation have also been described. In some cases, a CD25 deficiency has been found. Defective CD25 expression also impairs regulatory cell expansion/function. This functional T cell deficiency means that CD25-deficient patients are also at increased risk of opportunistic infections. It is noteworthy that abnormalities in regulatory T cells have been described in other PID settings, such as in Omenn syndrome, STAT5b deficiency, STIM1 (Ca flux) deficiency, and WAS; these abnormalities may account (at least in part) for the occurrence of inflammation and autoimmunity. The autoimmune features observed in a small fraction of patients with DiGeorge syndrome may have the same cause. Recently, severe inflammatory gut disease has been described in patients with a deficiency in the IL-10 receptor or IL-10.
A distinct autoimmune entity is observed in autoimmune polyendocrinopathy candidiasis ectodermal dysplasia (APECED) syndrome, which is characterized by autosomal recessive inheritance. It consists of multiple autoimmune manifestations that can affect solid organs in general and endocrine glands in particular. Mild, chronic Candida infection is often associated with this syndrome. The condition is due to mutations in the autoimmune regulator (AIRE) gene and results in impaired thymic expression of self-antigens by medullary epithelial cells and impaired negative selection of self-reactive T cells that leads to autoimmune manifestations.
A combination of hypogammaglobulinemia, autoantibody production, cold-induced urticaria or skin granulomas, or autoinflammation has been reported, and has been termed the PLCγ2-associated antibody deficiency and immune dysregulation (PLAID or APLAID).
CONCLUSION
The variety and complexity of the clinical manifestations of the many different PIDs strongly indicate that it is important to raise awareness of these diseases. Indeed, early diagnosis is essential for establishing an appropriate therapeutic regimen. Hence, patients with suspected PIDs must always be referred to experienced clinical centers that are able to perform appropriate molecular and genetic tests. A precise molecular diagnosis is not only necessary for initiating the most suitable treatment, but is also important for genetic counseling and prenatal diagnosis.
One pitfall that may hamper diagnosis is the high variability that is associated with many PIDs. Variable disease expression can result from the differing consequences of various mutations associated with a given condition, as exemplified by WAS and, to a lesser extent, X-linked agammaglobulinemia (XLA). There can also be effects of modifier genes (as also suspected in XLA) and environmental factors such as EBV infection that can be the main trigger of disease in XLP conditions. Furthermore, it has recently been established that somatic mutations in an affected gene can attenuate the phenotype of a number of T cell PIDs. This has been described for ADA deficiency, X-linked SCID, RAG deficiencies, NF-κB essential modulator (NEMO) deficiency, and, most frequently, WAS. In contrast, somatic mutations can create disease states analogous to PID, as reported for ALPS. Lastly, cytokine-neutralizing autoantibodies can mimic a PID, as shown for IFN-γ.
Many aspects of the pathophysiology of PIDs are still unknown, and the disease-causing gene mutations have not been identified in all cases (as illustrated by CVID and IgA deficiency). However, our medical understanding of PIDs has now reached the stage where scientifically based approaches to the diagnosis and treatment of these diseases can be implemented.