[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 7
Primary Hyperparathyroidism
Natural History Without Surgery
Randomized Studies of the Natural History of Asymptomatic Primary Hyperparathyroidism
Primary hyperparathyroidism (PHPT) is characterized by hypercalcemia and elevated parathyroid hormone (PTH) levels. The disease today bears little resemblance to the severe disorder of “stones, bones, and groans” described by Fuller Albright and others in the 1930s.1–6 Osteitis fibrosa cystica was the hallmark of classic PHPT. Radiography of the skeleton showed brown tumors of the long bones, subperiosteal bone resorption, distal tapering of the clavicles and phalanges, and “salt-and-pepper”–appearing erosions of the skull on radiograph (Fig. 7-1).7 Nephrocalcinosis was present in 80% of patients, and neuromuscular dysfunction with muscle weakness was also common. With the advent of the automated serum chemistry autoanalyzer in the 1970s, the diagnosis of PHPT became much more common, with a four- to fivefold increase in incidence.8–10 Classic symptomatology, concomitantly, became much less common. In the United States and elsewhere in the developed world, symptomatic PHPT is now the exception rather than the rule, with more than three-fourths of patients having no symptoms attributable to their disease, making PHPT a disease which has “evolved” from its classic presentation (Table 7-1).11–14 Nephrolithiasis is still seen, although much less frequently than in the past. Now, radiologically evident bone disease is rare, but bone involvement is readily detected by bone mass measurement. This chapter describes the clinical picture of PHPT as it presents today, how it can be differentiated from other causes of hypercalcemia, and its clinical course. Issues in management, many of which are still unresolved, are also addressed. A detailed discussion of our current understanding of the etiology of this disease can be found in Chapter 8.
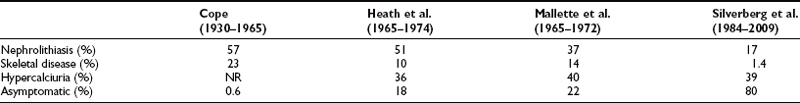
FIGURE 7-1 Radiologic representation of osteitis fibrosa cystica in classic primary hyperparathyroidism. A, Salt-and-pepper skull. B, Cystic bone disease of the clavicle. C, Subperiosteal bone resorption of the digits. D, Cortical erosions.
Pathology
By far the most common lesion found in patients with PHPT is the solitary parathyroid adenoma, occurring in 80% of patients.7,14 Several risk factors have been identified in the development of PHPT. These include a history of neck irradiation16 and prolonged use of lithium therapy for affective disorders.17–18 While in most cases a single adenoma is found, multiple parathyroid adenomas have been reported in 2% to 4% of cases.19 These may be familial or sporadic. Parathyroid adenomas can be discovered in many unexpected anatomic locations (see Chapter 5). Embryonal migration patterns of parathyroid tissue account for a plethora of possible sites of ectopic parathyroid adenomas. The most common sites are within the thyroid gland, the superior mediastinum, and within the thymus.20 Occasionally, the adenoma may ultimately be identified in the retroesophageal space, the pharynx, the lateral neck, and even the alimentary submucosa of the esophagus.21–23 On histologic examination, most parathyroid adenomas are encapsulated and composed of parathyroid chief cells. Adenomas containing mainly oxyphilic or oncocytic cells are rare but can give rise to clinical PHPT.
Clinical Presentation
The incidence of PHPT has changed dramatically.8,9,24,25 Prior to the advent of the multichannel autoanalyzer in the early 1970s, Heath et al.8 reported an incidence of 7.8 cases per 100,000 persons in Rochester, Minnesota. With the introduction of routine calcium measurements in the mid-1970s, this rate rose precipitously to 51.1 cases per 100,000 in the same community. Once prevalent cases were diagnosed (the “sweeping” effect), the incidence declined to approximately 27 per 100,000 persons per year. A report from Rochester, Minnesota, suggested that newly diagnosed cases of PHPT have been declining continuously since the mid-1970s.24 The decline in incidence is not explained by a change in the use of multichannel chemical screening because, in the United States, it is only in the very late 1990s that use of this technique became limited. Moreover, such declines in incidence are not apparent at other medical centers. It is possible that the special demographics of Rochester, Minnesota, combined with the rather complete discovery of PHPT in a population that receives virtually all of its care in one system (ideal epidemiologic surveillance) would naturally be associated with declining numbers for years thereafter. The analogy here might be to overfishing a small pond. Unrelated to the Rochester, Minnesota, experience, the United States may soon experience a decline in new cases of PHPT because multichannel screening is now more limited. On the other hand, greater appreciation of the potential of parathyroid hormone to be a catabolic force in postmenopausal women with osteoporosis has led to measurement of PTH even in subjects who do not have hypercalcemia. This trend has led to the emergence of a new entity, normocalcemic PHPT. A number of factors and forces are thus likely to influence the incidence of PHPT in the future (see later discussion).
Clinical Features
PHPT occurs predominantly in individuals in their middle years, with a peak incidence between ages 50 and 60 years. However, the disease is seen at all ages. Women are affected more frequently than men, in a ratio of approximately 3 : 1. At the time of diagnosis, most patients with PHPT do not have classic symptoms or signs associated with disease. Kidney stones are uncommon, and fractures are rare.14 Diseases associated epidemiologically with PHPT have included hypertension,26–28 peptic ulcer disease, gout, or pseudogout.29,30 Some concomitant disorders such as hypertension are commonly seen, but it is not established that any of these associated disorders are etiologically linked to the disease. The only exception is the MEN syndromes, in which MEN1 is often seen with peptic ulcer disease, and MEN2 may be associated with a pheochromocytoma. Constitutional complaints such as weakness, easy fatigability, depression, and intellectual weariness are seen with some regularity (see later discussion).31
Differential Diagnosis
The diagnosis of PHPT is made when hypercalcemia and elevated PTH levels are present. The other major cause of hypercalcemia, malignancy, is readily distinguished from PHPT. Patients with hypercalcemia of malignancy typically have advanced disease that has already been diagnosed. An exception is multiple myeloma, in which hypercalcemia can be the initial manifestation. These diseases can be easily distinguished; PTH levels are suppressed in multiple myeloma. The differential diagnosis of hypercalcemia, however, includes a number of other etiologies, including rare ones.32
Improved testing for PTH, especially immunoradiometric (IRMA) and immunochemiluminometric (ICMA) assay, has facilitated the distinction between PHPT and hypercalcemia of malignancy. In recent years, it has become clear that the “intact” immunoradiometric assay for PTH (“intact” IRMA) may significantly overestimate the concentration of biologically active parathyroid hormone. In 1998, Lepage et al.33 demonstrated a large non-(1–84) PTH fragment that comigrated with a large amino-terminally truncated fragment (PTH[7–84]) and had substantial cross-reactivity in commercially available IRMAs. This large, inactive moiety constituted as much as 50% (20% to 90%) of immunoreactivity by IRMA for PTH in individuals with chronic renal failure.34 Recognition of this molecule led to the development of a new IRMA utilizing affinity purified polyclonal antibodies to PTH(39–84) and to the extreme N-terminal amino acid regions, PTH(1–4).35,36 This assay detects only the full-length PTH molecule, PTH(1–84). It does not detect the large inactive fragment the normally circulates This assay has clear utility in uremic patients, in whom the “intact” IRMA has been shown to considerably overestimate elevations in biologically active hormone concentration.33,37,38 In PHPT, it is less clear that this assay will aid in the diagnostic evaluation.
A small percentage of patients with PHPT have PTH levels that are within the normal reference range as measured either by the classic IRMA or the newer PTH(1–84) assay. In these patients, levels tend to be in the upper range of normal. In PHPT, such values, although within the normal range, are clearly abnormal in a hypercalcemic setting. This is even more evident in patients younger than 45 years of age. Because PTH levels normally rise with age, the broad normal range for the older IRMA (10 to 65 pg/mL) reflects values for the entire population. In the younger-than-45 individual, one expects a narrower, lower normal range (10 to 45 pg/mL), so hypercalcemia and a PTH level of 50 pg/mL is distinctly abnormal. Occasionally, in either a younger or older patient, the PTH level as measured by the established IRMA will not even be in the upper end of the normal range but as low as 30 pg/mL. Such unusual examples generally require a more careful consideration of other causes of hypercalcemia. But in the end, these individuals are likely to have PHPT also because non–PTH dependent hypercalcemia should suppress the PTH concentration to levels that are either undetectable or at the lower limits of the reference range. Souberbielle et al.39 have illustrated that the normal range is very much a function of whether or not the reference population is or is not vitamin D deficient. When vitamin D deficient individuals were excluded, the upper limit of the PTH reference interval decreased from 65 to 46 pg/mL. When vitamin D–deficient individuals were excluded from the subjects used to establish a reference interval for “whole PTH,” the upper limit decreased from 44 to 34 ng/L.
Ninety percent of patients with hypercalcemia will be shown either to have PHPT or malignancy. Although there are many other causes of hypercalcemia (Table 7-2), they constitute only approximately 10% of the hypercalcemic population. Here also, the PTH assay is useful. With the exception of lithium and thiazide use and familial hypocalciuric hypercalcemia (FHH), virtually all other causes of hypercalcemia are associated with suppressed levels of PTH. If the patient can be safely withdrawn from lithium or thiazide, this should be attempted. Serum calcium and PTH levels are then reassessed 3 months later. If the serum calcium and PTH levels continue to be elevated, the diagnosis of PHPT is made. While patients can generally be readily withdrawn from a thiazide diuretic, patient safety must be the first consideration in any decision to withdraw lithium therapy. The recent emergence of a number of alternative therapeutic approaches may make this option realistic. FHH is differentiated from PHPT by (1) family history, (2) markedly lowered urinary calcium excretion, and (3) the specific gene abnormality (see Chapter 8).
Table 7-2
Rarely, a patient with malignancy will be shown to have elevated PTH levels due to ectopic secretion of native PTH from the tumor itself.32 Much more commonly, the malignancy is associated with the secretion of parathyroid hormone–related protein (PTHrP), a molecule that does not cross-react in the IRMA and ICMA assay. Finally, it is possible that a malignancy is present in association with PHPT. When the PTH level is elevated in someone with a malignancy, this is more likely to be the case than a true ectopic PTH syndrome.
Using the third-generation assay for PTH(1–84), a second molecular form of PTH(1–84) that is immunologically intact at both extremes has been identified. This molecule reacts only poorly in second-generation PTH assays. This molecular species represents less than 10% of the immunoreactivity in normal individuals and up to 15% in renal failure patients. It has been shown to be overexpressed, however, in a limited number of patients with a severe form of PHPT or with parathyroid cancer.41
With these points in mind, patients with elevated PTH levels and normal calcium levels have been reported. These subjects do not have serum calcium levels that extend occasionally into the abnormal range nor do they have any of the known secondary causes for an elevated PTH level. An explanation for why this entity is being seen today may reside in the fact that endocrinologists and other osteoporosis specialists currently evaluate the skeletal status of women at risk for osteoporosis not only with determination of bone density but also with calciotropic hormone measurements. These patients may represent the earliest manifestations of PHPT, a “forme fruste” of the disease. That these patients should exist is not surprising, insofar as clinical manifestations of PHPT are already present when the disorder is commonly diagnosed with hypercalcemia.42,43 One would expect the earliest phase of this disease to be characterized by elevated PTH levels in the absence of hypercalcemia. During this clinically silent period, the patient would not come to medical attention because the serum calcium is normal. However, if these patients were to have PTH levels measured, one might expect to discover them. Several reports describing these individuals have recently been reported, with several patients progressing to overt hypercalcemia while under observation.44–46 Frankly low or low-normal serum calcium concentrations suggest an adaptive response to hypocalcemia with high PTH levels. Secondary hyperparathyroidism can be seen in patients with renal insufficiency, malabsorption, or any of the other vitamin D–deficiency states. Rarely, patients with PHPT and coexisting vitamin D deficiency will present with low calcium concentration. PTH levels are high. In such patients, correction of the vitamin D deficiency is associated with a rise in serum calcium concentration into the hypercalcemic range.47
Other Biochemical Features
In PHPT, serum phosphorus tends to be in the lower range of normal, but frank hypophosphatemia is present in less than a fourth of patients. The hypophosphatemia, when present, represents the phosphaturic actions of PTH. Average total urinary calcium excretion is at the upper end of the normal range, with about 40% of all patients having hypercalciuria. Serum 25(OH)D levels tend to be in the lower end of the normal range. While mean values of 1,25(OH)2D3 are in the high-normal range, approximately a third of patients have frankly elevated levels of 1,25(OH)2D3.48 This pattern is due to the actions of PTH to facilitate the conversion of 25(OH)D to 1,25(OH)2D. A mild hyperchloremia is seen occasionally, due to the effect of PTH on renal acid-base balance. A typical biochemical profile is shown in Table 7-3.
Table 7-3
Biochemical Profile in Primary Hyperparathyroidism (n = 137)
| Patients (Mean ± SEM) | Normal Range | |
| Serum calcium | 10.7 ± 0.1 mg/dL | 8.2–10.2 mg/dL |
| Serum phosphorus | 2.8 ± 0.1 mg/dL | 2.5–4.5 mg/dL |
| Total alkaline phosphatase | 114 ± 5 IU/L | <100 IU/L |
| Serum magnesium | 2.0 ± 0.1 mg/dL | 1.8–2.4 mg/dL |
| PTH (IRMA) | 119 ± 7 pg/mL | 10–65 pg/mL |
| 25(OH)D | 19 ± 1 ng/mL | 30–100 ng/mL |
| 1,25(OH)2D | 54 ± 2 pg/mL | 15–60 pg/mL |
| Urinary calcium | 240 ± 11 mg/g creatinine | |
| Urine DPD | 17.6 ± 1.3 nmol/mmol creatinine | <14.6 nmol/mmol creatinine |
| Urine PYD | 46.8 ± 2.7 nmol/mmol creatinine | <51.8 nmol/mmol creatinine |
The Skeleton
Bone Markers
Both bone resorption and bone formation are stimulated by PTH. Markers of bone turnover, which reflect those dynamics, provide clues to the extent of skeletal involvement in PHPT.12 The study of bone markers in PHPT has been of considerable interest for several reasons. First, this inquiry sheds light on which markers accurately reflect skeletal activity in the patient with PHPT. Second, the evaluation of markers of bone turnover in PHPT has provided insight into the hyperparathyroid process in bone. Finally, clues to the extent of postoperative improvements in bone mineral density (BMD) might be provided by markers of bone turnover.
Bone Formation Markers
Bone formation is reflected by osteoblast products, including bone-specific alkaline phosphatase activity, osteocalcin, and type 1 procollagen peptide.12 Despite the availability of these sensitive measurements of bone formation, the total alkaline phosphatase activity—part of the multichannel biochemical screening profile—is still widely assessed in PHPT. In PHPT, levels can be mildly elevated, but in many patients, total alkaline phosphatase values are within normal limits.49,50 In a small study from our group,51 bone-specific alkaline phosphatase activity correlated with PTH levels and BMD at the lumbar spine and femoral neck. Osteocalcin is also generally increased in patients with PHPT.51–53 Osteocalcin correlates with other indices of bone formation. Assays for procollagen extension peptides reflect osteoblast activation and bone formation but have not been shown to have significant predictive or clinical utility in PHPT.53 In a small study of patients with PHPT, C-terminal propeptide of human type 1 procollagen (PICP) levels were higher than in control subjects,54 but distinct elevations were much less impressive than those seen for alkaline phosphatase, osteocalcin, or even hydroxyproline (see later).
Bone Resorption Markers
Markers of bone resorption include the osteoclast product, tartrate-resistant acid phosphatase (TRAP), and collagen breakdown products such as hydroxyproline, hydroxypyridinium cross-links of collagen, and N- and C-telopeptides of type 1 collagen.12 Urinary hydroxyproline, once the only available marker of bone resorption, no longer offers sufficient sensitivity or specificity to make it a useful tool in the assessment of patients with PHPT. Although urinary hydroxyproline was frankly elevated in patients with osteitis fibrosa cystica, in mild asymptomatic PHPT, it is now generally normal. Hydroxypyridinium cross-links of collagen, pyridinoline (PYD), and deoxypyridinoline (DPD), on the other hand, are often elevated in PHPT. They return to normal after parathyroidectomy.55 DPD and PYD both correlate positively with PTH concentrations. N- and C-terminal peptides of type I collagen (NTX and CTX) are likely to have utility, but they have not been studied extensively in PHPT. Other markers of bone resorption have also been limited in their application to bone turnover in PHPT. For example, studies of TRAP are limited, although levels have been shown to be elevated.47 In the case of the PYD cross-linked telopeptide domain of type I collagen (ICTP), pooled data from patients with high turnover diseases (i.e., PHPT as well as hyperthyroidism) suggest that this marker may reflect calcium kinetics and histomorphometric indices.12 Data specifically relevant to PHPT are not yet available. Bone sialoprotein, a phosphorylated glycoprotein which makes up approximately 5% to 10% of the noncollagenous bone protein, appears to reflect processes associated with both bone formation and bone resorption. In PHPT, bone sialoprotein levels are elevated and correlate with urinary PYD and DPD.55 Thus, sensitive assays of bone formation and bone resorption are both elevated in mild PHPT.
Longitudinal Bone Marker Studies
Studies of bone markers in the longitudinal follow-up of patients with PHPT are limited but indicate a reduction in these turnover markers following parathyroidectomy. Information from our group,55,56 Guo et al.,57 and Tanaka et al.58 all report declining levels of bone markers following surgery, although the choice of markers in the individual studies differed. Data are also emerging concerning the kinetics of change in bone resorption versus bone formation following parathyroidectomy. We have found that markers of bone resorption decline rapidly following cure of PHPT, while indices of bone formation follow a more gradual decrease.55 Urinary PYD and DPD decreased significantly as early as 2 weeks following parathyroidectomy, preceding reductions in alkaline phosphatase. Similar data were reported from Tanaka et al.,58 who demonstrated a discrepancy between changes in NTX (reflecting bone resorption) and osteocalcin (reflecting bone formation) following parathyroidectomy, and Minisola et al.,54 who reported a drop in bone resorptive markers and no significant change in alkaline phosphatase or osteocalcin. Short-term studies reported a brief increase in PICP immediately following parathyroidectomy, while bone resorptive markers fell promptly. The persistence of elevated bone formation markers coupled with rapid declines in bone resorption markers indicates a shift in the coupling between bone formation and bone resorption toward an anabolic buildup of bone mineral postoperatively. Increases in bone density postoperatively provide support for this idea.
Cytokines
IL-6 and TNF-α have been studied as possible mediators of bone resorption in PHPT. In vitro and in vivo data support an effect of PTH in stimulating production of IL-6, which in turn leads to increased osteoclastogenesis.59,60 Furthermore, antibodies to IL-6 prevent PTH-mediated bone resorption. In PHPT, serum levels of TNF-α and IL-6 are increased and fall following parathyroidectomy.61 Importantly, TNF and IL-6 concentrations correlate with the level of PTH in patients with PHPT. Bone turnover markers correlate with levels of these cytokines as well, supporting an important role of the cytokines in mediating the skeletal effects of PTH excess in PHPT.62,63 Furthermore, IL-6 soluble receptor has recently been reported to predict rates of bone loss in mild PHPT. Although cytokine levels did not correlate significantly with bone density measurements in the report of Grey et al.,61 it should be noted that bone density was not assessed at the site containing mostly cortical bone (the radius) where the catabolic effects of excess PTH would be expected to be seen most clearly. More information is needed to confirm these observations, especially with appropriate control subjects, and to test for potential involvement of other bone-resorptive cytokines in PHPT.
The involvement of other factors in the anabolic effect of PHPT on bone is also being studied. Levels of IGF-1, which is well documented to be a direct mediator of PTH action in bone, and insulin-like growth factor–binding protein 3 (IGFBP-3), the major binding protein for IGF-1, change following parathyroidectomy in PHPT.64 The alteration in the ratio of IGF-1 to IGFBP-3 supports enhanced delivery of IGF-1 to tissues following surgery, an increase inversely proportional to the observed rise in lumbar spine and femoral neck bone density. It is not known whether the anabolic properties of PTH at cancellous sites (e.g., lumbar spine) can be explained by IGF-1 prior to surgery.
Bone Densometry
The advent of bone mineral densitometry as a major diagnostic tool for osteoporosis occurred at a time when the clinical profile of PHPT was changing from a symptomatic to an asymptomatic disease. This fortuitous timing allowed questions about skeletal involvement in PHPT to be addressed when specific radiologic features of PHPT had all but disappeared. Bone mass measurements could provide information about the actual state of bone mineral with great accuracy and precision. The known physiologic proclivity of PTH to be catabolic at sites of cortical bone make a cortical site essential to any complete densitometric study of PHPT. By convention, the distal third of the radius is the site used. The early densitometric studies in PHPT also revealed another physiologic property of PTH, namely, to be anabolic at cancellous sites. The lumbar spine became an important site to measure not only because it is predominantly cancellous bone, but also because postmenopausal women are at risk for cancellous bone loss. In PHPT, bone density at the distal third of the radius is diminished.65,66 Bone density at the lumbar spine is only minimally reduced. The hip region, containing a relatively equal mixture of cortical and cancellous elements, shows bone density intermediate between the cortical and cancellous sites (Fig. 7-2). The results support not only the notion that PTH is catabolic for cortical bone but also the view that PTH is anabolic in cancellous bone.67–69 In postmenopausal women, the same pattern was observed.66 Postmenopausal women with PHPT, therefore, show a reversal of the pattern typically associated with postmenopausal bone loss: preferential loss of cancellous bone. The reduced bone density at the distal radius (cortical bone) and preserved density at the lumbar spine (cancellous bone) suggest that PHPT helps protect postmenopausal women from bone loss due to estrogen deficiency.
FIGURE 7-2 Bone densitometry in primary hyperparathyroidism. Data are shown in comparison to age- and sex-matched normal subjects. Divergence from expected values is different at each site (P < 0.0001). (Data from Silverberg SJ, Shane E, de la Cruz L et al: Skeletal disease in primary hyperparathyroidism, J Bone Miner Res 4:283–291, 1989.)
Observations of skeletal health in PHPT made by bone densitometry have established the importance of this technology in the evaluation of all patients with PHPT. Without this information, the data set leading to surgical recommendations is incomplete. The Consensus Development Conference on Asymptomatic Primary Hyperparathyroidism in 1990 implicitly acknowledged this point when bone mineral densitometry was included as a separate criterion for clinical decision making.70 Since that time, bone densitometry has become an indispensable component of both evaluating the patient and establishing clinical guidelines for management and monitoring.
The bone density profile in which there is relative preservation of skeletal mass at the vertebrae and diminution at the more cortical distal radius is not always seen in PHPT. While this pattern is evident in the vast majority of patients, a small group of patients have evidence of vertebral osteopenia at the time of presentation. In our natural history study, approximately 15% of patients had a lumbar spine Z score of less than −1.5 at the time of diagnosis.72 Only half of these patients were postmenopausal women, so not all vetebral bone loss could be attributed entirely to estrogen deficiency. These patients are of interest with regard to changes in bone density following parathyroidectomy and are discussed in further detail later. The extent of vertebral bone involvement will vary as a function of disease severity. In the typical mild form of the disease, the pattern described earlier is seen. When PHPT is more advanced, there will be more generalized involvement, and the lumbar spine will not appear to be protected. When PHPT is severe or more symptomatic, all bones can be extensively involved.
Bone Histomorphometry
Analyses of percutaneous bone biopsies from patients with PHPT have provided direct information that could only be indirectly surmised by bone densitometry and by bone markers. Both static and dynamic parameters present a picture of cortical thinning, maintenance of cancellous bone volume (Fig. 7-3), and a very dynamic process associated with high turnover and accelerated bone remodeling.
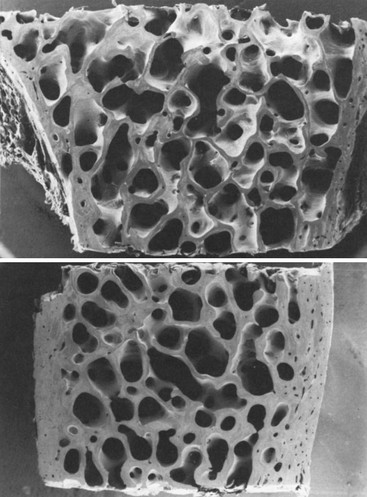
FIGURE 7-3 Scanning electron micrograph of bone biopsy specimens in a normal subject (top) and an age- and sex-matched patient with primary hyperparathyroidism (bottom). The cortices of the hyperparathyroid sample are markedly thinned, but cancellous bone and trabecular connectivity appear to be well preserved. (Magnification ×31.25.) (From Parisien MV, Silverberg SJ, Shane E et al: Bone disease in primary hyperparathyroidism, Endocrinol Metab Clin North Am 19:19–34, 1990.)
Cortical thinning, inferred by bone mineral densitometry, is clearly documented in a quantitative manner by iliac crest bone biopsy.72–74 Van Doorn et al.75 demonstrated a positive correlation between PTH levels and cortical porosity. These findings are consistent with the known effect of PTH to be catabolic at endocortical surfaces of bone. Osteoclasts are thought to erode more deeply along the corticomedullary junction under the influence of PTH.
Histomorphometric studies have contributed most by elucidating the nature of cancellous bone preservation in PHPT.75 Again, as suggested by bone densitometry, cancellous bone volume is clearly well preserved in PHPT. This is seen as well among postmenopausal women with PHPT. Several studies have shown that cancellous bone is actually increased in PHPT as compared to normal subjects.76,77 When cancellous bone volume is compared among age- and sex-matched subjects with PHPT or postmenopausal osteoporosis, a dramatic difference is evident (Fig. 7-4). Whereas postmenopausal women with osteoporosis have reduced cancellous bone volume, women with PHPT have higher cancellous bone volume.77 This observation suggests that while PHPT is said to be a risk factor for postmenopausal osteoporosis, it is a syndrome of bone loss. The region(s) of bone loss in PHPT is directed toward the cortical bone compartment, with good maintenance of cancellous bone volume unless the PHPT is unusually active.
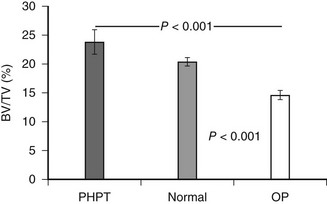
FIGURE 7-4 Cancellous bone volume in primary hyperparathyroidism. Cancellous bone volume was analyzed from bone biopsy specimens of the iliac crest. Comparisons are between 16 women with primary hyperparathyroidism, 17 women with postmenopausal osteoporosis, and 31 women with no known disorder of bone metabolism. Subjects were matched for age and other indices. (Data from Parisien M, Cosman F, Mellish RW et al: Bone structure in postmenopausal hyperparathyroid, osteoporotic and normal women, J Bone Miner Res 10:1393–1399, 1995.)
Preservation of cancellous bone volume even extends to comparisons with the expected losses associated with the effects of aging on cancellous bone physiology. In a study of 27 patients with PHPT (10 men and 17 women), static parameters of bone turnover (osteoid surface, osteoid volume, and eroded surface) were increased, as expected, in patients relative to control subjects.78 However, in control subjects, trabecular number varied inversely with age, while trabecular separation increased with advancing age. Both of these observations are expected concomitants of aging. In marked contrast, in the patients with PHPT, no such age dependency was seen. There was no relationship between trabecular number or separation and age in PHPT, suggesting that the actual plates and their connections were being maintained over time more effectively than one would have expected through the aging process. Thus, PHPT seems to retard the normal age-related processes associated with trabecular loss.
In PHPT, indices of trabecular connectivity are greater than expected, while indices of disconnectivity are decreased. When three matched groups of postmenopausal women were assessed (a normal group, a group with postmenopausal osteoporosis, and a group with PHPT), women with PHPT were shown to have trabeculae with less evidence of disconnectivity compared with normals, despite increased levels of bone turnover,76,78 so cancellous bone is preserved in PHPT through the maintenance of well-connected trabecular plates. In order to determine the mechanism of cancellous bone preservation in PHPT, static and dynamic histomorphometric indices were compared between normal and hyperparathyroid postmenopausal women. In normal postmenopausal women, there is an imbalance in bone formation and resorption which favors excess bone resorption. In postmenopausal women with PHPT, on the other hand, the adjusted apposition rate is increased. Bone formation, thus favored, may explain the efficacy of PTH at cancellous sites in patients with osteoporosis.67,79–81 Assessment of bone remodeling variables in patients with PHPT shows increases in the active bone-formation period77 (Table 7-4). The increased bone formation rate and total formation period may explain the preservation of cancellous bone seen in this disease.
Table 7-4
Wall Width and Remodeling Variables in Primary Hyperparathyroidism (PHPT) and Control Groups (Mean ± SEM)
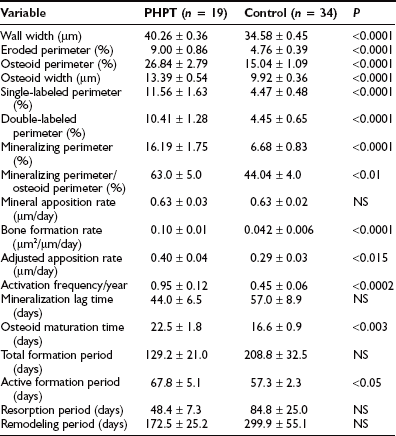
Modified from Dempster DW, Parisien M, Silverberg SJ et al: On the mechanism of cancellous bone preservation in postmenopausal women with mild primary hyperparathyroidism, J Clin Endocrinol Metab 84:1562–1566, 1999.
Recently, further analysis of trabecular microarchitecture has taken advantage of newer technologies that have largely been confirmatory. In a three-dimensional analysis of transiliac bone biopsies using microCT technology, a highly significant correlation was observed with the conventional histomorphometry82 described earlier. In comparison to age-matched control subjects without PHPT, postmenopausal women with PHPT had higher bone volume (BV/TV), higher bone surface area (BS/TV), higher connectivity density (Conn.D), and lower trabecular separation (Tb.Sp.). There were also less marked age-related declines in BV/TV and Conn.D as compared to controls, with no decline in BS/TV. Using the technique of backscattered electron imaging (qBEI) to evaluate trabecular BMD distribution (BMDD) in iliac crest bone biopsies, Roschger et al.83 showed reduced average mineralization density and increase in the heterogeneity of the degree of mineralization, consistent with reduced mean age of bone tissue. Studies of collagen maturity using Fourier Transform Infrared Spectroscopy provide further support for these observations.84 Bone strength, therefore, in PHPT has to take into account a number of factors related to skeletal properties of bone besides BMD.85
Fractures
Fractures were an integral element of classic PHPT, but their importance in modern-day disease is unclear. In a case-control study published in 1975, Dauphine et al.86 suggested that back pain and vertebral crush fractures might be part of the presenting clinical profile of PHPT. Since that time, reports on fracture incidence have been conflicting. A retrospective review of lateral chest radiographs of patients who underwent parathyroidectomy showed an increased incidence of vertebral fractures in one study,87 while Wilson et al.88 found no increase in such fractures in a cohort of 174 consecutive patients who had mild asymptomatic PHPT.
In a study that focused on hip fracture, a population-based prospective analysis (mean of 17 years’ duration; 23,341 person years) showed women with PHPT in Sweden not to be at increased risk.89 In a much smaller study (46 patients, 44 controls), fractures at any site were increased in hyperparathyroidism.90 This study is flawed not only by its small sample size but also by the unusually high fracture incidence in both patients (48%) and control subjects (28%) and by the use of thyroid medication in a significantly greater number of patients (28%) relative to control subjects.
The Mayo Clinic experience with PHPT and risk of fracture reviewed 407 cases of PHPT recognized during the 28-year period between 1965 and 1992.91 Fracture risk was assessed by comparing fractures at a number of sites with numbers of fractures expected on the basis of sex and age from the general population. The clinical presentation of these patients with PHPT was typical of the mild form of the disease, with the serum calcium being only modestly elevated at 10.9 ± 0.6 mg/dL. The data from this retrospective epidemiologic study indicate that overall fracture risk was significantly increased at many sites such as the vertebral spine, the distal forearm, the ribs, and the pelvis. There was no increase in hip fractures. After multivariate analysis, age and female sex remained significant independent predictors of fracture risk. These data, however, are subject to potential ascertainment bias. Patients with PHPT are typically followed more conscientiously, and thus fractures at some of these sites may have been recognized by greater surveillance. This may certainly be true of the vertebral spine and the ribs but unlikely in the case of fractures of the forearm. One might expect to see an increased incidence of distal forearm fractures, since the hyperparathyroid process tends to lead to reduction of cortical bone (distal forearm) in preference to cancellous bone (vertebral spine). Unfortunately, there were no densitometric data provided in this study, so one could not relate bone density to fracture incidence. It is difficult to know whether this study in fact confirms an expectation of preferential distal forearm fractures in PHPT or whether some other process is at work conferring universally greater fracture risk in these patients. In a more recent study, Vignali et al.92 studied the incidence of vertebral fractures in PHPT as determined by DXA-based vertebral fracture assessment (VFA). In this case control study, 150 consecutive patients and 300 healthy women matched for age and menopausal age were studied. Vertebral fractures were detected in more subjects with PHPT (24.6%) than the control subjects (4.0%; P < 0.001). Among asymptomatic PHPT patients, only those who met surgical guidelines showed a higher incidence of vertebral fractures compared with controls. We still lack prospective, controlled studies to determine fracture incidence in PHPT.
Even the expectation of an increased fracture risk at a cortical site, like the forearm, in PHPT is fraught with uncertainties. The expectation is based upon bone-density data and the presumption that in this disease, it is as predictive of fracture as it is in postmenopausal women who do not have PHPT. By analogy with osteoporosis, it is reasonable to consider bone density as a risk factor for fracture in PHPT, but other issues could lead to a different relationship between bone density and fracture risk in PHPT. It is now known that bone density is only one of several important qualities of bone. These other qualities are influential in the overall assessment of fracture risk. Bone size, for example, influences fracture risk. From the clinical trials of parathyroid hormone in the treatment of osteoporosis, as well as in observations of PHPT per se, it is likely that bone size is affected by PTH. Cortical thinning through PTH-mediated endosteal resorption is compensated for by PTH-mediated periosteal apposition, leading to bone that may be increased in cross-sectional diameter. This increase in bone size provides biomechanical protection for the skeleton. In PHPT, an interesting paradigm is set up: cortical thinning tends to increase fracture risk, whereas increased bone size tends to reduce fracture risk. In addition, as noted, microarchitecture of bone does not show the same kind of deterioration that is commonly seen in postmenopausal osteoporosis. The relative preservation of microarchitecture in PHPT is another factor that may protect bone. More recent studies in which hyperparathyroid bone has been studied with regard to bone mineralization density83 and collagen quality84 may also be relevant to this discussion (see earlier). These considerations emphasize the need for prospective studies of site-specific fracture incidence in PHPT.85,92–94
Nephrolithiasis
In the past, classic clinical descriptions of PHPT emphasized skeletal involvement and kidney stones as principal complications of the disease.95 The cause of nephrolithiasis in PHPT is probably multifactorial. An increase in the amount of calcium filtered at the glomerulus due to the hypercalcemia of hyperparathyroidism may lead to hypercalciuria despite the physiologic actions of PTH to facilitate calcium reabsorption. A component of absorptive hypercalciuria exists in this disorder. The enhanced intestinal calcium absorption is believed to be due to increased production of 1,25(OH)2D, a consequence of another physiologic action of PTH, namely to increase the synthesis of this active metabolite.96,97 Urinary calcium excretion is correlated with 1,25(OH)2D levels.97,98 In addition, increased intestinal calcium absorption seen in nephrolithiasis99 may also occur in PHPT. The skeleton provides yet another possible source for the increased levels of calcium in the glomerular filtrate. Hyperparathyroid bone resorption might contribute to hypercalciuria, and subsequently to nephrolithiasis, even though there is no convincing evidence to support this hypothesis.100 Finally, alteration in local urinary factors, such as a reduction in inhibitor activity or an increase in stone-promoting factors, may predispose some patients with PHPT to nephrolithiasis.100,101 It remains unclear whether the urine of patients with hyperparathyroid stone disease is different in this regard from that of other stone formers.
Studies in the 1970s and 1980s documented a higher incidence of renal stone disease than do reports of more recent experience. With the decreased incidence of osteitis fibrosa cystica, studies in the modern era have tended to focus on patients with kidney stones. Conflicting results have emerged, with one group providing evidence that 1,25(OH)2D3 plays an etiologic role in the development of nephrolithiasis in PHPT and other groups unable to document differences in 1,25(OH)2D3 levels between those with and without renal stone disease.95,100–102
Although the incidence of nephrolithiasis is much less common than the incidence in the classic, older presentation of PHPT, kidney stones remain the most common manifestation of symptomatic PHPT (see Table 7-1). Estimates in recent studies place the incidence of kidney stones at 15% to 20% of all patients.103 Other renal manifestations of PHPT include hypercalciuria, which is seen in approximately 40% of patients, and nephrocalcinosis, the frequency of which is unknown. It is important to note that in patients with PHPT who do not have renal stone disease, there is no relationship between extent of hypercalciuria and the development of kidney stones.104
In the 1930s, it was generally accepted that bone and stone disease did not coexist in the same patient with classic PHPT.1,6 Albright and Reifenstein1 postulated that low dietary calcium intake would lead to bone disease, while adequate or high dietary calcium levels would be associated with stone disease. Dent et al.,105 who provided convincing evidence against this construct, proposed the existence of two forms of circulating PTH, one causing renal stones and the other causing bone disease. A host of mechanisms, including differences in dietary calcium, calcium absorption, forms of circulating PTH, and levels of 1,25(OH)2D3, were proposed to account for the clinical distinction between bone and stone disease in PHPT.100,105 Today, there is no clear evidence for two distinct subtypes of PHPT. In our patients with PHPT, we could not identify a distinctive set of biochemical data for patients with stone disease.95 Furthermore, although our population did not include patients with classic hyperparathyroid bone disease, we found no evidence to support the notion that the processes affecting the skeleton and kidneys in hyperparathyroidism occur in different subsets of patients. Urinary calcium excretion per gram of creatinine, levels of 1,25(OH)2D, and BMD at all sites were indistinguishable among patients with and without nephrolithiasis. Cortical bone demineralization is as common and as extensive in those with and without nephrolithiasis.95,100
Other Organ Involvement
Neurocognitive and Neuropsychological Features
Over the years, PHPT has been associated with complaints referable to many different organ systems. Perhaps the most common complaints have been those of weakness and easy fatigability.31 Classic PHPT used to be associated with a distinct neuromuscular syndrome characterized by type II muscle cell atrophy.106,107 Originally described by Vicale in 1949,108 the syndrome consisted of easy fatigability, symmetric proximal muscle weakness, and muscle atrophy. Both the clinical and electromyographic features of this disorder were reversible after parathyroid surgery.109,110 In the milder, less symptomatic form of the disease that is common today, this disorder is rarely seen.111 In a group of 42 patients with mild disease, none had complaints consistent with the classic neuromuscular dysfunction described above. Although over half of all patients had nonspecific complaints of paresthesias and muscle cramps, electromyographic studies did not confirm the picture of past observations.
The “psychic groans” described by early observers of patients with classic PHPT remain a source of controversy today. Patients with PHPT often report some degree of behavioral and/or psychiatric symptomatology. A retrospective look at patients with more severe disease revealed a 23% incidence of psychiatric symptomatology (n = 441).112 More recent studies have shown some abnormalities on psychological tests preoperatively, with no improvement after surgery.113,114 The study of Solomon et al.115 studied psychiatric rather than neuropsychological symptoms.115 Using the SCL-90-R rating scale, they observed a constellation of abnormalities, most of which improved after successful surgery. The control group, who underwent neck surgery for nodular thyroid disease, had similar postoperative improvement. This study documented psychiatric symptoms in the patients with PHPT but could not distinguish the improvement in symptomatology to cure of hyperparathyroidism or to the effects of a successful surgical procedure.
The surgical literature provides further data supporting postoperative improvements. Data from Clark et al.116 on 152 patients (thyroid surgery control subjects) found 40% of patients reporting less fatigue after cure. Similar findings were reported by Pasieka and Parsons117 and Burney et al.118
The subject of neurocognitive and neuropsychological impairment was reviewed in detail by Silverberg et al.119 The more recent literature continues to be unclear on whether the symptomatology is specific to PHPT and whether it is improved following successful parathyroid surgery.119–124 Silverberg et al.119 have pointed out key limitations in experimental design in many of the published studies. The lack of adequate controls and the value of some of the quantitative instruments have been problematic. There are now three randomized, prospective trials in which this issue has been addressed.125–127 Despite the rigorous experimental design, with control built into these more recent studies, there was great variability in which features of PHPT if any were improved or not following successful parathyroid surgery. Some of the randomized clinical trials are not yet complete. It is clear that the issue remains unsettled.
Cardiovascular System
Interest in the effect of PHPT on cardiovascular function is rooted in pathophysiologic observations of the hypercalcemic state. Hypercalcemia has been associated with increases in blood pressure, left ventricular hypertrophy, heart muscle hypercontractility, and arrhythmias.128–130 Furthermore, evidence of calcium deposition has been documented in the form of calcifications in the myocardium, heart valves, and coronary arteries. The association of overt cardiovascular symptomatology with modern-day PHPT is unclear. Hypertension, a common feature of PHPT when it is part of a MEN syndrome with pheochromocytoma or hyperaldosteronism, has also been reported to be more prevalent in sporadic, asymptomatic PHPT than in appropriately matched control groups. The mechanism of this association is unknown, and the condition does not clearly remit following cure of the hyperparathyroid state.131,132
Cardiovascular Mortality
There is little doubt that in very active PHPT, cardiovascular mortality is increased.133–136 Of some interest are the postoperative observations in which the higher cardiovascular mortality rate persists for years after cure.137 These observations differ markedly from those in which asymptomatic PHPT have been studied. Although limited, the studies have not shown any increase in mortality.138–139 The Mayo Clinic studies help to bring these observations together. In the mildly hypercalcemic individuals, overall and cardiovascular mortality was reduced, but in those whose serum calcium was in the highest quartile, cardiovascular mortality was increased.139 The idea that the more common asymptomatic form of PHPT is not associated with increased mortality is supported by data from Nilsson et al.140 and by other studies141–142 in which more recently enrolled subjects had better survival than those who had been entered earlier and presumably had more active disease.
Hypertension
This discussion excludes subjects with MEN syndromes in whom a pheochromocytoma may be causative. But hypertension is also a frequent observation among those with mild disease. Most but not all studies have not shown that hypertension is reversed or ameliorated after successful parathyroid surgery for PHPT.143–146
Cardiac Manifestations of Primary Hyperparathyroidism
Coronary Artery Disease: Both calcium and PTH have independently been shown to be associated with coronary heart disease.147–148 Aside from autopsy studies such as those of Roberts and Waller,149 in which coronary atherosclerosis was seen in hyperparathyroid subjects with levels of calcium that would these days be occasioned with alarm (16.8 to 27.4 mg/dL), the more recent literature has been controversial. Even more recent studies, however, have tended to agree with the idea that the incidence of coronary artery disease in PHPT is more likely to be present as a function of the serum calcium level.142 Some studies have actually found that in mild PHPT, there is better exercise tolerance as determined by the electrocardiogram.150 If valvular and myocardial calcifications are regarded separately, the level of the serum calcium again seems to be the determinant.151–152 Given these differing views, it is also noteworthy that the risk of cardiovascular death in PHPT seems to be related more to traditional risk factors for cardiovascular disease than to the hyperparathyroidism per se.
Left Ventricular Hypertrophy: Left ventricular hypertrophy (LVH) is considered separately because it is itself a strong predictor of cardiovascular disease and mortality. Moreover, different from the parameters described above, in which involvement seems to be a function of the serum calcium level, LVH has been seen across a wide range of calcium levels.153–154 The idea has been advanced that LVH is more a function of the PTH level than it is the serum calcium.152,155–156 If it could be shown that when PTH is returned to normal after successful parathyroidectomy LVH is reversible, this observation could have important management implications. Unfortunately, the literature does not give a clear view on this point, with a few but by no means all studies suggesting regression of LVH following parathyroidectomy.152–153,155–157
Electrocardiographic Manifestations: Classically, marked hypercalcemia is associated with a reduced QT interval.158 In most patients with mild hypercalcemia, however, abnormalities on the electrocardiogram are not seen. Moreover, no other conduction abnormalities or arrhythmogenic potential are observed.159–160
Vascular Manifestations of Primary Hyperparathyroidism
Carotid Plaque: Calcium has been reported in a recent population-based study of Rubin et al.161 to be associated with carotid plaque thickness. Consistent abnormalities in carotid intima-medial thickness (IMT), a strong predictor of systemic atherosclerosis and cerebrovascular events, have not been confirmed in mild hyperparathyroidism.162–165
Vascular Function: The evidence implicating vascular dysfunction in PHPT has been focused upon those with more severe disease than we see now.162,166,167 However, in those with lower calcium levels, Baykan et al.168 also found impaired flow-mediated (endothelial) dilation that negatively correlated with calcium levels. There is a preliminary report on endothelial dysfunction in PHPT169 and two studies that have reported increased vascular stiffness.169–170
Gastrointestinal Manifestations
Primary hyperparathyroidism has long been thought to be associated with an increased incidence of peptic ulcer disease. Most recent studies suggest that the incidence of peptic ulcer disease in PHPT is approximately 10%, a figure similar to its percentage in the general population. An increased incidence of peptic ulcer disease is seen in patients with PHPT due to MEN1, in which approximately 40% of patients have clinically apparent gastrinomas (Zollinger-Ellison syndrome). In those patients, PHPT is associated with increased clinical severity of gastrinoma, and treatment of the associated PHPT has been reported to benefit patients with Zollinger-Ellison syndrome.171 Despite this, current recommendations (Consensus Conference Guidelines for Therapy of MEN1) state that the coexistence of Zollinger-Ellison syndrome does not represent sufficient indication for parathyroidectomy, since medical therapy is so successful.172
Although hypercalcemia can underlie pancreatitis, most large series have not reported an increased incidence of pancreatitis in patients with PHPT with serum calcium levels under 12 mg/dL. The Mayo Clinic experience from 1950 to 1975 found that only 1.5% of those with PHPT had coexisting pancreatitis, and alternative explanations for pancreatitis were found for several patients. Regarding pancreatitis in pregnancy in patients with PHPT, these conditions may coexist, but there is no evidence for a causal relationship between the disorders.173
Other Systemic Involvement
Vitamin D Deficiency and Symptomatic Primary Hyperparathyroidism
Much of this chapter has focused on asymptomatic PHPT as the predominating clinical profile in the modern era. Certainly in countries where biochemical screening tests are routinely employed, this description is accurate. However, reports from other countries have revisited the older, more classic descriptions of PHPT as a disease of “stones, bones, and groans.”174–176 The lack of routine screening tests does not explain completely this older form of the disease in the 1990s in these other countries. Rather, patients who have been described from China, Brazil, India, and Saudi Arabia have a common underlying vitamin D deficiency. Years ago, PHPT and vitamin D deficiency were described as a potent negative combination by Lumb and Stanbury.177 Even in mild, asymptomatic PHPT, we have shown that indices of disease activity are generally higher among those whose 25(OH)D levels are low.47 Mechanisms to explain this clinical observation are speculative, but it is intriguing to consider vitamin D–PTH gene interactions. An endogenous regulator of PTH gene function is 1,25(OH)2D.178 When vitamin D deficiency is present in PHPT, it is possible that the abnormal PTH cells are stimulated further to produce PTH.
Natural History
Over the past 25 years, new knowledge of the natural history of PHPT with or without surgery has been very helpful in guiding decisions regarding surgery in patients with asymptomatic PHPT. The longest prospective observational trial has been conducted by the authors and their colleagues.179–180 This project began in 1984 in an effort to define the natural history of asymptomatic PHPT. The study included detailed analyses of pathophysiologic, densitometric, histomorphometric, and other skeletal features of PHPT.179–180 Much of the information gleaned from that study have been summarized already in earlier sections of this chapter. The 15-year follow-up of this study constitutes the longest natural history study of this disorder and was recently reported.180
Recommendations for surgery or observation were made on the basis of the 1990 set of NIH guidelines, but both groups included patients who did or did not meet surgical guidelines. This is because some patients opted for surgery even if they didn’t meet the guidelines, while others opted for a conservative approach even if they did meet guidelines for surgery. As will be described in the following, this imperfect design was followed by three studies that were truly randomized but were of much shorter duration.125–127 The results with regard to natural history from all studies are remarkably concordant.
Natural History With Surgery
Parathyroidectomy resulted in normalization of the serum calcium and PTH levels permanently. Postoperatively, there was a marked improvement in BMD at all sites (lumbar spine, femoral neck, and distal one-third radius) amounting to gains above 10%. The improvement was most rapid at the lumbar spine, but all sites showed persistent gains for the 15 years of follow-up (Fig. 7-5). The improvements were seen in those who met and did not meet surgical criteria at study entry, confirming the salutary effect of parathyroidectomy in this regard on all patients.
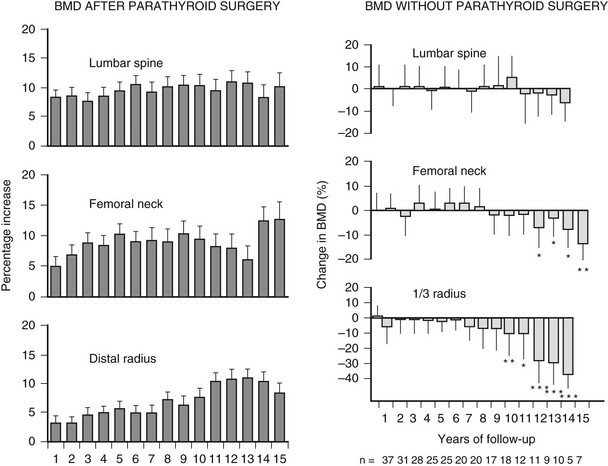
FIGURE 7-5 Longitudinal course of bone density in primary hyperparathyroidism. Data are presented as percentage change from preoperative baseline bone density measurement by site following parathyroidectomy (left) or in patients followed with no intervention (right). (Data from Rubin MR, Bilezikian JP, McMahon DJ et al: The natural history of primary hyperparathyroidism with or without parathyroid surgery after 15 years, J Clin Endocrinol Metab 93:3462–3470, 2008.)
Randomized Studies of The Natural History of Asymptomatic Primary Hyperparathyroidism
Although the long natural history study of asymptomatic PHPT has added much to our knowledge about this disease over time, randomized clinical trials have added data with a more rigorous design that have confirmed the observational data. The three trials are limited by their short duration. In 2004, Rao et al.125 reported on their randomized controlled trial of parathyroidectomy versus no surgery. The study was not completely enrolled and thus included only 53 subjects, assigned either to parathyroid surgery (n = 25) or to no surgery (n = 28). The follow-up was for at least 2 years. BMD significantly increased at the femoral neck and total hip, along with normalization of the serum calcium and PTH. In those who did not undergo parathyroid surgery, there were no changes in the lumbar spine or femoral neck bone density, but total hip density significantly declined. Forearm BMD declined, an oddity considering the vulnerability of this site to the catabolic actions of PTH. Biochemical indices were all stable.
In 2007, Bollerslev et al.126 reported interim results of their randomized trial of parathyroidectomy versus no surgery. This study from three Scandinavian countries was larger, with 191 patients who were randomized to medical observation or to surgery. After surgery, biochemical indices normalized and BMD increased. In the group that did not undergo parathyroid surgery, BMD did not change.
Also in 2007, Ambrogini et al.127 reported the results of their randomized controlled trial of parathyroidectomy versus observation. Surgery was associated with a significant increase in BMD of the lumbar spine and hip after 1 year.
Guidelines To Therapy
Parathyroidectomy remains the only currently available option for cure of PHPT. As the disease profile has changed, questions have been raised concerning the advisability of surgery in asymptomatic patients. If asymptomatic patients have a benign natural history, the surgical alternative is not an attractive one. On the other hand, asymptomatic patients may display levels of hypercalcemia or hypercalciuria that cause concern for the future. Similarly, bone-mass measurements can be frankly low at cortical or (less commonly) cancellous sites. In an effort to address such issues, two consensus development conferences (in 1991 and 2002) on the management of asymptomatic PHPT70,181 were followed in 2009 by the Third International Workshop on the Management of Asymptomatic Primary Hyperthyroidism, the proceedings of which have been published.182–187 The 2009 Workshop convened a panel of international experts who reviewed the evidence on aspects of the clinical profile of asymptomatic PHPT with a few towards revising the guidelines for surgery. The most recent guidelines that emerged from that conference should be helpful to the clinician faced with the asymptomatic hyperparathyroid patient: All symptomatic patients are advised to undergo parathyroidectomy. Surgery is advised in asymptomatic patients with (1) serum calcium greater than 1 mg/dL above the upper limits of normal; (2) reduction in creatinine clearance to less than 60 mL/min; (3) reduced bone density (T-score < −2.5 at any site or the presence of a fragility fracture); and (4) age younger than 50 years. The most recent guidelines are shown in Table 7-5. It is noteworthy that urinary calcium excretion is no longer regarded as a guideline for surgery, because urinary calcium excretion in subjects with PHPT who have not had a kidney stone is not predictive of the risk for subsequent nephrolithiasis (see earlier discussion). A second change in the guidelines reflects the fact that a fragility fracture is now included as a guideline for surgery. Since two-thirds of vertebral compression fractures are asymptomatic, this brings up the question of whether all patients with asymptomatic PHPT should have a vertebral x-ray or other imaging study (i.e., vertebral fracture assessment) to rule out this possibility. The 2009 Workshop did not address this question.

Surgery
A large percentage of those patients who meet the surgical guidelines listed in Table 7-5 are asymptomatic. Some asymptomatic patients who meet surgical guidelines elect not to have surgery. Among the reasons why surgery is not sought are personal choice, intercurrent medical conditions, and previous unsuccessful parathyroid surgery. Conversely, there are patients who meet none of the NIH guidelines for parathyroidectomy but opt for surgery nevertheless. Physician and patient input remain very important factors in the decision regarding parathyroid surgery.
Preoperative Localization of Hyperfunctioning Parathyroid Tissue
Noninvasive Imaging
Noninvasive parathyroid imaging studies include technetium (Tc)-99m sestamibi scintigraphy, ultrasound, computed tomography (CT) scanning, magnetic resonance imaging (MRI), and positron emission tomography (PET) scanning (see also Chapter 9). Tc-99m sestamibi is generally regarded to be the most sensitive and specific imaging modality, especially when it is combined with single-photon emission CT (SPECT). For the single parathyroid adenoma, sensitivity has ranged from 80% to 100%, with a 5% to 10% false-positive rate. On the other hand, sestamibi scintigraphy and the other localization tests have a relatively poor record in the context of multiglandular disease.188 The success of ultrasonography is highly operator dependent.189 In centers where there is great expertise, this noninvasive approach is most attractive. Abnormalities identified by ultrasound as possible parathyroid tissue may prove to be a thyroid nodule or lymph node, which underscores the importance of the skill and experience of the ultrasonographer. Rapid spiral thin-slice CT scanning of the neck and mediastinum with evaluation of axial, coronal, and sagittal views can add much to the search for elusive parathyroid tissue.190 MRI can also identify abnormal parathyroid tissue, but it is time consuming and expensive. It is also less sensitive than the other noninvasive modalities. It can nonetheless be useful when the search with these other noninvasive approaches has been unsuccessful. PET with or without simultaneous CT scan (PET/CT) can be used, but like MRI, it is expensive and does not have the kind of experiential basis that make it attractive. There are also specificity issues because FDG, the scanning agent, accumulates in the thyroid, making differentiation between parathyroid adenoma and thyroid nodules difficult.
Invasive Imaging
Parathyroid Fine-Needle Aspiration: Fine-needle aspiration (FNA) of a parathyroid gland, identified by any of the aforementioned modalities, can be performed and the aspirate analyzed for PTH. This technique is not recommended for routine de novo cases.191 A theoretic concern with this approach is the possibility that parathyroid cells could be deposited outside the parathyroid gland in the course of the aspiration. Autoseeding of parathyroid tissue would be an unwanted consequence of this procedure if it were to occur.
Arteriography and Selective Venous Sampling for Parathyroid Hormone
In situations where the gland has not been identified by any of the techniques described, the combination of arteriography and selective venous sampling can provide both anatomic and functional localization of abnormal parathyroid tissue. This approach, however, is costly and requires an experienced interventional radiologist. It is also performed in only a few centers in the United States. This approach is reserved now only for those individuals who have had previous unsuccessful parathyroid surgery in whom all other localization techniques have failed.192
Surgical Approach
In the hands of an expert parathyroid surgeon, parathyroidectomy is a highly successful procedure with infrequent complications. A standard surgical approach is the four-gland parathyroid gland exploration under general or local anesthesia, with or without preoperative localization. This approach has been reported to lead to surgical cure in over 95% of cases (see Chapter 9).193
Several alternative approaches have emerged that focus upon the single gland and not the total four-gland neck exploration that used to be routinely employed. Unilateral approaches are appealing in a disease in which only a single gland is involved in approximately 85% of cases. These procedures include a unilateral operation in which the gland on the same side that harbors the adenoma is ascertained to be normal. Since multiple parathyroid adenomas are unusual, a normal parathyroid gland is considered by some to be sufficient evidence for single-gland disease. Another limited surgical approach that has emerged in many centers as the approach of choice is the minimally invasive parathyroidectomy (MIP).194–195 Preoperative parathyroid imaging is necessary, and the procedure is directed only to the site where the abnormal parathyroid gland has been visualized.196 Preoperative blood is obtained for comparison of the PTH concentration with an intraoperative sample obtained after removal of the “abnormal” parathyroid gland. The availability of a rapid PTH assay in or near the operating room is necessary for this procedure. If the level falls by more than 50% following resection, into the normal range, the gland that has been removed is considered to be the sole source of overactive parathyroid tissue, and the operation is terminated. If the PTH level does not fall by more than 50%, into the normal range, the operation is extended to a more traditional one in a search for other overactive parathyroid tissue. There is a risk (albeit small) that the minimally invasive procedure may miss other overactive gland(s) that are suppressed in the presence of a dominant gland.
In Europe, MIP is being performed with an endoscopic camera.197–198 Yet another variation on this theme is the use of preoperative sestamibi scanning with an intraoperative gamma probe to help locate enlarged parathyroid glands. The MIP procedure seems to be as successful as more standard approaches, in the range of 95% to 98%.199–200
Immediate Postoperative Course
After surgery, biochemical indices return rapidly to normal.201,202 Serum calcium and PTH levels normalize, and urinary calcium excretion falls by as much as 50%. The serum calcium no longer tends to become abnormally low, a situation characteristic of an earlier time when PHPT was a symptomatic disease with overt skeletal involvement. The acute reversal of hyperparathyroidism was associated with a robust deposition of calcium into the skeleton at a pace that could not be compensated for by supplemental calcium. Thus, postoperative hypocalcemia was routine and sometimes a serious short-term complication (“hungry bone syndrome”). Occasionally, postoperative hypocalcemia still occurs, especially if preoperative bone turnover markers are elevated. More typically, however, the early postoperative course is not complicated by symptomatic hypocalcemia.
Postoperative course after successful parathyroid surgery leads to normalization of the biochemical indices of the disease and improvements in BMD, as mentioned (Fig. 7-6). The capacity of the skeleton to restore itself is seen dramatically in young patients with severe PHPT. Kulak et al.203 reported two patients with osteitis fibrosa cystica who experienced increases in bone density that ranged from 260% to 430% 3 to 4 years following surgery. Similar observations have been made by Tritos and Hartzband204 and by DiGregorio.205
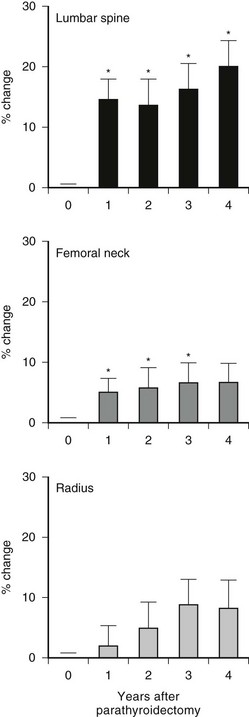
FIGURE 7-6 Bone mineral density following parathyroidectomy in 14 primary hyperparathyroid patients with vertebral osteopenia. The cumulative percentage change from preoperative baseline (year 0) is shown for each site. Asterisk denotes change from baseline at P < 0.05. (Data from Silverberg SJ, Locke FG, Bilezikian JP: Vertebral osteopenia: A new indication for surgery in primary hyperparathyroidism, J Clin Endocrinol Metab 81:4007–4012, 1996.)
Medical Management
Surgery is generally not recommended in patients who do not meet any surgical guidelines. Among typical cohorts in the United States, about 50% of patients with PHPT will fit into this category. The most recent guidelines for management of asymptomatic PHPT restated the position that it is reasonable to pursue a nonsurgical course of management in those who do not meet criteria for surgery. In those patients who are not going to have parathyroid surgery, the Workshop183 suggested a set of monitoring steps which are summarized in Table 7-6. This includes annual measurements of the serum calcium concentration, a calculated creatinine clearance, and regular monitoring of BMD.
Table 7-6
Comparison of New and Old Management Guidelines for Patients With Asymptomatic Primary Hyperparathyroidism Who Do Not Undergo Parathyroid Surgery
| Measurement | Older Guidelines | Newer Guidelines |
| Serum calcium | Semiannually | Annually |
| 24-hour urinary calcium | Annually | Not recommended |
| Creatinine clearance | Annually | Not recommended |
| Serum creatinine | Annually | Annually |
| Bone density | Annually | Annually or biannually |
| Abdominal x-ray | Annually | Not recommended |
From Bilezikian JP, Khan AA, Potts JT Jr on behalf of the Third International Workshop on the Management of Asymptomatic Primary Hyperthyroidism: Guidelines for the management of asymptomatic primary hyperparathyroidism: Summary statement from the Third International Workshop, J Clin Endocrinol Metab 94:335–339, 2009.
Diet
Dietary management of PHPT has long been an area of controversy. Many patients are advised to limit their dietary calcium intake because of the hypercalcemia. However, it is well known that low dietary calcium can lead to increased PTH levels in normal individuals.206–208 In patients with PHPT, even though the abnormal PTH tissue is not as sensitive to slight perturbations in the circulating calcium concentration, it is still possible that PTH levels will rise when dietary calcium is tightly restricted. Conversely, diets enriched in calcium could suppress PTH levels in PHPT, as shown by Insogna et al.209 Dietary calcium could also be variably influenced by ambient levels of 1,25(OH)2D. In patients with normal levels of 1,25(OH)2D3, Locker et al.210 noted no difference in urinary calcium excretion between those on high (1000 mg/day) and low (500 mg/day) calcium intake diets. On the other hand, in those with elevated levels of 1,25(OH)2D3, high calcium diets were associated with worsening hypercalciuria. This observation suggests that dietary calcium intake in patients can be liberalized to 1000 mg/day if 1,25(OH)2D3 levels are not increased but should be more tightly controlled if 1,25(OH)2D3 levels are elevated.
Pharmaceuticals
Oral phosphate can lower the serum calcium by up to 1 mg/dL.211–212 A complex interplay of mechanisms leads to this moderating effect of oral phosphate. First, calcium absorption falls in the presence of intestinal phosphorus. Second, concomitant increases in serum phosphorus will tend to reduce circulating 1,25(OH)2D3 levels. Third, phosphate can be an antiresorptive agent. Finally, increased serum phosphorus reciprocally lowers serum calcium. Problems with oral phosphate include limited gastrointestinal tolerance, possible further increase in PTH levels, and the possibility of soft-tissue calcifications after long-term use. It is essentially not used anymore in the management of PHPT.
Bisphosphonates
Bisphosphonates are conceptually attractive in PHPT because they are antiresorptive agents with an overall effect to reduce bone turnover. Although they do not affect PTH secretion directly, bisphosphonates could reduce serum and urinary calcium levels. Early studies with the first-generation bisphosphonates were disappointing. Etidronate has no effect.213 Clodronate use was associated in several studies with a reduction in serum and urinary calcium,214 but the effect was transient.
Alendronate has been studied most extensively in PHPT. Studies by Rossini et al.215 and Hassani et al.216 were followed by those of Chow et al.,217 Parker et al.,218 and Kahn et al.219 These studies were all characterized by a randomized, controlled design. Typically, BMD of the lumbar spine and hip regions increases along with reductions in bone turnover markers. Except for the study of Chow et al.,217 serum calcium was unchanged. These results suggest that a bisphosphonate-like alendronate might be useful in patients with low bone density in whom parathyroid surgery is not to be performed.
Estrogens and Selective Estrogen-Receptor Modulators
The earliest studies on the use of estrogen replacement therapy in PHPT date back to the early 1970s. A 0.5 to 1.0 mg/dL reduction in total serum calcium levels in postmenopausal women with PHPT who receive estrogen replacement therapy is generally seen. Gallagher and Nordin220 first reported a calcium-lowering effect in 10 postmenopausal women given ethinyl estradiol. A prompt lowering of both serum and urinary calcium excretion was noted after 1 week of therapy, with continued reductions at 4 weeks (mean serum calcium [normal range, 8.9 to 10.2 mg/dL]: 12.0 to 11.6 to 11.3 mg/dL; P < 0.0025; and mean urinary calcium, 402 to 291 to 283 mg/g creatinine; P < 0.0005). Subsequent studies have reported similar declines in serum and urinary calcium in response to both ethinyl estradiol and conjugated equine estrogens.221 Although levels of PTH were not measured in the earlier studies, Marcus et al.221 and Selby and Peacock222 reported no change in PTH as measured by a C-terminal radioimmunoassay. Grey et al.223 also found no changes in intact PTH levels.
Various approaches to skeletal dynamics have been used in studies of metabolic and skeletal responses to estrogen replacement therapy in women with PHPT. Gallagher and Wilkinson224 demonstrated normalization of calcium balance, with a decrease in output relative to intake. Studies of BMD in estrogen-treated patients with PHPT have documented a salutary effect of treatment on BMD at the femoral neck and lumbar spine.223
Raloxifene, a selective estrogen-receptor modulator, has been studied in PHPT, but the data are sparse. In a short-term (8-week) trial of 18 postmenopausal women, raloxifene (60 mg/day) was associated with a statistically significant although small (0.5 mg/dL) reduction in the serum calcium concentration and in markers of bone turnover.225 No long-term data or data on bone density are available.
Inhibition of Parathyroid Hormone
The most specific pharmacologic approach to primary PHPT is to inhibit the synthesis and secretion of PTH from the parathyroid glands. Interest is now focused upon compounds that act on the parathyroid cell calcium-sensing receptor. This G protein–coupled receptor recognizes calcium as its cognate ligand.226–228 When activated by increased extracellular calcium, the calcium-sensing receptor signals the cell via a G protein–transducing pathway to raise the intracellular calcium concentration, which inhibits PTH secretion. Molecules that mimic the effect of extracellular calcium by altering the affinity of calcium for the receptor could activate this receptor and inhibit parathyroid cell function. The phenylalkylamine (R)-N(3-methoxy-α-phenylethyl)-3-(2-chlorophenyl)-1-propylamine (R-568) is one such calcimimetic compound. R-568 was found to increase cytoplasmic calcium and reduce PTH secretion in vitro, as well as in normal postmenopausal women.229–230 This drug also was shown to inhibit PTH secretion in postmenopausal women with PHPT.231 A second-generation ligand, cinacalcet, has been the subject of recent more extensive investigation in PHPT. Studies conducted by the authors and their colleagues232–234 indicate that this drug can reduce the serum calcium concentration to normal in PHPT (Fig. 7-7), but despite normalization of the serum calcium concentration, PTH levels do not return to normal; they do fall by 35% to 50% after administration of drug. Urinary calcium excretion does not change; serum phosphorus levels increase but are maintained in the lower range of normal; and 1,25(OH)2D3 levels do not change. The average BMD does not change, even after 3 years of administration of cinacalcet. Marcocci235 has recently shown that cinacalcet is effective in subjects with intractable PHPT, Silverberg et al.236 have shown that cinacalcet reduces calcium levels effectively in inoperable parathyroid carcinoma.
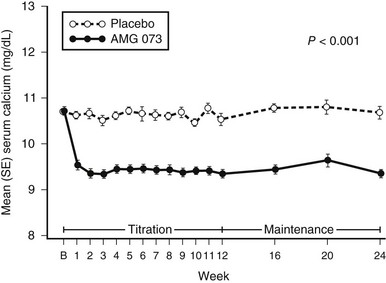
FIGURE 7-7 Changes in serum calcium concentrations with administration of the calcimimetic cinacalcet (solid line) or placebo (broken line) in patients with primary hyperparathyroidism. (Data from Shoback DM, Bilezikian JP, Turner SA et al: The calcimimetic cinacalcet normalizes serum calcium in subjects with primary hyperparathyroidism, J Clin Endocrinol Metab 88:5644–5649, 2003.)
Unusual Presentations
Neonatal Primary Hyperparathyroidism
Neonatal PHPT is a rare form of the disorder caused by homozygous inactivation of the calcium-sensing receptor.237 When present in a heterozygous form, it is a benign hypercalcemic state known as familial hypercalciuric hypercalcemia (FHH). However, in the homozygous neonatal form, hypercalcemia is severe and the outcome fatal unless recognized early. The treatment of choice is early subtotal parathyroidectomy to remove the majority of hyperplastic parathyroid tissue.
Primary Hyperparathyroidism In Pregnancy
Primary hyperparathyroidism in pregnancy is primarily of concern for its potential effect on the fetus and neonate.238–239 Complications of PHPT in pregnancy include spontaneous abortion, low birth weight, supravalvular aortic stenosis, and neonatal tetany. The last condition is a result of fetal parathyroid gland suppression by high levels of maternal calcium, which readily crosses the placenta during pregnancy. These infants, used to hypercalcemia in utero, have functional hypoparathyroidism after birth, and can develop hypocalcemia and tetany in the first few days of life. Today, with most patients (pregnant or not) presenting with a mild form of PHPT, an individualized approach to the management of the pregnant patient with PHPT is advised. Many of those with very mild disease can be followed safely, with successful neonatal outcomes without surgery. However, parathyroidectomy during the second trimester remains the traditional recommendation for this condition.
Acute Primary Hyperparathyroidism
Acute PHPT is known variously as parathyroid crisis, parathyroid poisoning, parathyroid intoxication, and parathyroid storm. Acute PHPT describes an episode of life-threatening hypercalcemia of sudden onset in a patient with PHPT.240–243 Clinical manifestations of acute PHPT are mainly those associated with severe hypercalcemia. Nephrocalcinosis or nephrolithiasis is frequently seen. Radiologic evidence of subperiosteal bone resorption is also commonly present. Laboratory evaluation is remarkable not only for very high serum calcium levels but for extreme elevations in PTH to approximately 20 times normal.242 In this way, acute PHPT resembles parathyroid carcinoma. A history of persistent mild hypercalcemia has been reported in 25% of patients. However, given the rarity of this condition, the risk of developing acute PHPT in a patient with mild asymptomatic PHPT is very low. Intercurrent medical illness with immobilization may precipitate acute PHPT. Early diagnosis, with aggressive medical management followed by surgical cure, is essential for a successful outcome.
Parathyroid Cancer
An indolent yet potentially fatal disease, parathyroid carcinoma accounts for less than 0.5% of cases of PHPT.244 The cause of the disease is unknown, and no clear risk factors have been identified. There is no evidence to support the malignant degeneration of previously benign parathyroid adenomas. Manifestations of hypercalcemia are the primary effects of parathyroid cancer. The disease tends not to have a bulk tumor effect, spreading slowly in the neck. Metastatic disease is a late finding, with lung (40%), liver (10%), and lymph node (30%) involvement seen most commonly.
The clinical profile of parathyroid cancer differs from that of benign PHPT in several important ways.244–245 First, no female predominance is seen among patients with carcinoma. Second, elevations in serum calcium and PTH are far greater. As a consequence, the hyperparathyroid disease tends to be much more severe, with the classic targets of PTH excess involved in most cases. Nephrolithiasis or nephrocalcinosis is seen in up to 60% of patients; overt radiologic evidence of skeletal involvement is seen in 35% to 90% of patients. A palpable neck mass, distinctly unusual in benign PHPT, has been reported in 30% to 76% of patients with parathyroid cancer.246
Grossly, malignant glands are large, often exceeding 12 g. They tend to be adherent to adjacent structures. Microscopically, the trabecular arrangement of the tumor cells is divided by thick, fibrous bands. Capsular and blood vessel invasion is common by these cells that often contain mitotic figures.246
Parathyroid carcinoma has also been reported in hereditary syndromes of hyperparathyroidism,247–250 particularly in hyperparathyroidism-jaw tumor (HPT-JT) syndrome,251 a rare autosomal disorder in which as many as 15% of patients will have malignant parathyroid disease. Because cystic changes are common, this disorder has also been referred to as cystic parathyroid adenomatosis.252 In HPT-JT, ossifying fibromas of the maxilla and mandible are seen in 30% of cases. Less commonly, kidney lesions, including cysts, polycystic disease, hamartomas, or Wilms tumors can be present.253 Parathyroid carcinoma has also been reported in familial isolated hyperparathyroidism.248,254 Recently, parathyroid carcinoma, as defined pathologically, has been reported in MEN1 syndrome and with somatic MEN1 mutations.255–257 However, recurrent parathyroid disease in MEN1 may mimic but not actually be due to malignancy. Only one case of parathyroid carcinoma has been reported in the MEN2A syndrome.258
Loss of the retinoblastoma tumor suppressor gene used to be considered a marker for parathyroid cancer,210 but more recent studies do not unequivocally support this impression.259 Work by Shattuck et al.260–261 have provided new insights into the molecular pathogenesis of parathyroid cancer. Parathyroid carcinomas from 10 of 15 patients with sporadic parathyroid cancer carried a mutation in the HRPT2 gene. The HRPT2 gene that encodes for the parafibromin protein has been shown to be mutated in a substantial number of patients with parathyroid cancer. Marcocci et al.244 have recently reviewed this topic, pointing out a potential role for parafibromin in parathyroid cancer. In 3 of 15 patients with parathyroid cancer, Shattuck et al.261 showed that the mutation was in the germline. The presence of the mutation in the germline suggests that this disease might be related in some way to the HPT-JT syndrome, in which this gene has been implicated.261 In addition, there is an increased risk of parathyroid cancer in the HPT-JT syndrome. In fact, in patients with a germline mutation, certain clinical features in them and their relatives are indicative of the HPT-JT syndrome or phenotypic variants.262–264
Chemotherapy has had a very limited role in this disease. Bradwell and Harvey265 have attempted an immunotherapeutic approach by injecting a patient who had severe hypercalcemia due to parathyroid cancer with immunogenic PTH. Coincident with a rise in antibody titer to PTH, previous refractory hypercalcemia fell impressively. A more recent report266 provided evidence of antitumor effect in a single case of PTH immunization in metastatic parathyroid cancer. Recent attention has been focused instead on control of hypercalcemia in this devastating disease. The intravenous bisphosphonates have been used in their capacity as agents that treat severe hypercalcemia. Although efficacious in that regard, they do not have an enduring effect. The calcimimetic agents hold promise for offering calcium-lowering effects on an outpatient basis. Our group267 reported on a single patient treated with the calcimimetic, R-568; despite widely metastatic disease, the patient had serum calcium levels maintained in a range that allowed him to return to normal functioning for nearly 2 years. A wider experience by Silverberg et al.236 gives even more evidence that cinacalcet has utility in the management of parathyroid cancer. The U.S. Food and Drug Administration approved this calcimimetic for the treatment of hypercalcemia in patients with parathyroid cancer. Based upon extensive studies,268 cinacalcet has also been approved for use in patients with secondary hyperparathyroidism on hemodialysis. Use of this agent in parathyroid cancer led to improvement in serum calcium levels and a decrease in symptoms of nausea, vomiting, and mental lethargy, which are common concomitants of marked hypercalcemia. There are no data on the effect of cinacalcet on tumor growth in parathyroid cancer. Although many questions remain, this agent offers an option for control of intractable hypercalcemia when surgical removal of cancerous tissue is no longer an option.
References
1. Albright, F, Reifenstein, EC. The Parathyroid Glands and Metabolic Bone Disease. Baltimore: Williams & Wilkins; 1948.
2. Cope, O. The story of hyperparathyroidism at the Massachusetts General Hospital. N Engl J Med. 1966;21:1174–1182.
3. Bauer, W. Hyperparathyroidism: Distinct disease entity. J Bone Joint Surg. 1933;15:135–141.
4. Bauer, W, Federman, DD. Hyperparathyroidism epitomized: Case of Captain Charles E. Martell. Metabolism. 1962;11:21–22.
5. Mandl, F. Therapeutische Versuch bei Ostitis fibrosa generalisata mittels Extirpation eines Epithelkšrperchentumors. Wien Klin Wochenschr. 1925;50:1343–1344.
6. Albright, F, Aub, JC, Bauer, W. Hyperparathyroidism: A common and polymorphic condition as illustrated by seventeen proven cases from one clinic. JAMA. 1934;102:1276–1287.
7. Silverberg, SJ, Bilezikian, JP. Clinical presentation of primary hyperparathyroidism in the United States. In: Marcus R, Levine MA, eds. The Parathyroids. New York: Academic Press; 2001:349–360.
8. Heath, H, Hodgson, SF, Kennedy, MA. Primary hyperparathyroidism: Incidence, morbidity, and economic impact in a community. N Engl J Med. 1980;302:189–193.
9. Mundy, GR, Cove, DH, Fisken, R. Primary hyperparathyroidism: Changes in the pattern of clinical presentation. Lancet. 1980;1:1317–1320.
10. Scholz, DA, Purnell, DC. Asymptomatic primary hyperparathyroidism. Mayo Clin Proc. 1981;56:473–478.
11. Bilezikian, JP, Primary hyperparathyroidism. DeGroot, L. Dartmouth: MDTEXT.COM Inc; 2009. www.ENDOTEXT.org
12. Silverberg, SJ, Bilezikian, JP. Primary hyperparathyroidism. In: Seibel MJ, Robins SP, Bilezikian JP, eds. Dynamics of Bone and Cartilage Metabolism. San Diego: Elsevier; 2006:767–778.
13. Silverberg, SJ, Bilezikian, JP. The diagnosis and management of asymptomatic primary hyperparathyroidism. Nat Clin Pract Endocrinol Metab. 2006;2:494–503.
14. Bilezikian, JP, Silverberg, SJ. Primary hyperparathyroidism. In: Rosen C, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. ed 7. Washington, DC: American Society for Bone and Mineral Research; 2008:302–306.
15. Golden, SH, Robinson, KA, Saldanha, I, Anton, B, Ladenson, PW. Prevalence and incidence of endocrine and metabolic disorders in the United States: A comprehensive review. J Clin Endocrinol Metab. 2009;94:1853–1878.
16. Rao, SD, Frame, B, Miller, MJ, et al. Hyperparathyroidism following head and neck irradiation. Arch Intern Med. 1980;140:205–207.
17. Nordenstrom, J, Strigard, K, Perbeck, L, et al. Hyperparathyroidism associated with treatment of manic-depressive disorders by lithium. Eur J Surg. 1992;158:207–211.
18. McHenry, CR, Rosen, IB, Rotstein, LE, et al. Lithiumogenic disorders of the thyroid and parathyroid glands as surgical disease. Surgery. 1992;108:1001–1005.
19. Attie, JN, Bock, G, Auguste, L. Multiple parathyroid adenomas: Report of 33 cases. Surgery. 1990;108:1014–1019.
20. Nudelman, IL, Deutsch, AA, Reiss, R. Primary hyperparathyroidism due to mediastinal parathyroid adenoma. Int Surg. 1987;72:104–108.
21. Sloane, JA. Parathyroid adenoma in submucosa of esophagus. Arch Pathol Lab Med. 1978;102:242–243.
22. Joseph, MP, Nadol, JB, Pilch, BZ, Goodman, ML. Ectopic parathyroid tissue in the hypopharyngeal mucosa (pyriform sinus). Head Neck Surg. 1982;5:70–74.
23. Gilmour, JR. Some developmental abnormalities of the thymus and parathyroids. J Pathol Bacteriol. 1941;52:213–218.
24. Wermers, RA, Khosla, S, Atkinson, EJ, et al. The rise and fall of primary hyperparathyroidism. Ann Intern Med. 1997;126:433–440.
25. Melton, LJ, III. The epidemiology of primary hyperparathyroidism in North America. J Bone Miner Res. 2002;17(Suppl 2):N12–N17.
26. Ringe, JD. Reversible hypertension in primary hyperparathyroidism: Pre- and postoperative blood pressure in 75 cases. Klin Wochenschr. 1984;62:465–469.
27. Broulik, PD, Horky, K, Pacovsky, V. Blood pressure in patients with primary hyperparathyroidism before and after parathyroidectomy. Exp Clin Endocrinol. 1985;86:346–352.
28. Rapado, A. Arterial hypertension and primary hyperparathyroidism. Am J Nephrol. 1986;6(suppl 1):49–50.
29. Bilezikian, JP, Aurbach, GD, Connor, TB, et al. Pseudogout following parathyroidectomy. Lancet. 1973;1:445–447.
30. Geelhoed, GW, Kelly, TR. Pseudogout as a clue and complication in primary hyperparathyroidism. Surgery. 1989;106:1036–1041.
31. Silverberg, SJ. Non-classical target organs in primary hyperparathyroidism. J Bone Min Res. 2003;17(Suppl 2):N117–N125.
32. Jacobs, TP, Bilezikian, JP. Rare causes of hypercalcemia. J Clin Endocrinol Metab. 2005;90:6316–6322.
33. Lepage, R, Roy, L, Brossard, JH, et al. A non-(1–84) circulating parathyroid hormone (PTH) fragment interferes significantly with intact PTH commercial assay measurements in uremic samples. Clin Chem. 1998;44:805–809.
34. Quarles, LD, Lobough, B, Murphy, G. Intact parathyroid hormone over-estimates the presence and severity of parathyroid-mediated osseous abnormalities in uremia. J Clin Endocrinol Metab. 1992;75:145–150.
35. John, MR, Goodman, WG, Gao, P, Cantor, T, Salusky, IB, Jueppiner, H. A novel immunoradiometric assay detects full-length human PTH but not amino-terminally truncated fragments: Implications for PTH measurement in renal failure. J Clin Endocrinol Metab. 1999;84:4287–4290.
36. Gao, P, Scheibel, S, D’Amour, P, et al. Development of a novel immunoradiometric assay exclusively for biologically active whole parathyroid hormone 1–84. Implications for improvement of accurate assessment of parathyroid function. J Bone Miner Res. 2001;16(4):605–614.
37. Slatopolsky, E, Finch, JL, Clay, P, et al. A novel mechanism for skeletal resistance in uremia. Kidney Int. 2000;58:753–761.
38. Silverberg, SJ, Brown, I, LoGerfo, P, Gao, P, Cantor, T, Bilezikian, JP. Clinical utility of an immunoradiometric assay for whole PTH (1–84) in primary hyperparathyroidism. J Clin Endocrinol Metab. 2003;88:4725–4730.
39. Souberbielle, JC, Cormier, C, Kindermans, C, et al. Vitamin D status and redefining serum parathyroid hormone reference range in the elderly. J Clin Endocrinol Metab. 2001;86:3086–3090.
40. D’Amour, P, Brossard, JH, Rousseau, L, et al. Amino-terminal form of parathyroid hormone (PTH) with immunologic similarities to hPTH(1–84) is overproduced in primary and secondary hyperparathyroidism. Clin Chem. 2003;49:2037–2044.
41. Rubin, MR, Silverberg, SJ, D’Amour, P, et al. An N-terminal molecular form of parathyroid hormone (PTH) distinct from hPTH(1–84) is overproduced in parathyroid carcinoma. Clin Chem. 2007;53:1470–1476.
42. Rao, DS, Wilson, RJ, Kleerekoper, M, Parfitt, AM. Lack of biochemical progression or continuation of accelerated bone loss in mild asymptomatic primary hyperparathyroidism. J Clin Endocrinol Metab. 1988;67:1294–1298.
43. Silverberg, SJ, Gartenberg, F, Jacobs, TP, et al. Longitudinal measurements of bone density and biochemical indices in untreated primary hyperparathyroidism. J Clin Endocrinol Metab. 1995;80:723–728.
44. Hagag, P, Revet-Zak, I, Hod, N, Horne, T, Rapoport, MJ, Weiss, M. Diagnosis of normocalcemic hyperparathyroidism by oral calcium loading test. J Endocrinol Invest. 2003;26:327–332.
45. Silverberg, SJ, Bilezikian, JP. “Incipient” primary hyperparathyroidism: a “forme fruste” of an old disease. J Clin Endocrinol Metab. 2003;88:5348–5352.
46. Lowe, H, McMahon, DJ, Rubin, MR, Bilezikian, JP, Silverberg, SJ. Normocalcemic primary hyperparathyroidism: further characterization of a new clinical phenotype. J Clin Endocrinol Metab. 2007;92:3001–3005.
47. Silverberg, SJ, Shane, E, Dempster, DW, Bilezikian, JP. Vitamin D deficiency in primary hyperparathyroidism. Am J Med. 1999;107:561–567.
48. Vieth, R, Bayley, TA, Walfish, PG, et al. Relevance of vitamin D metabolite concentrations in supporting the diagnosis of primary hyperparathyroidism. Surgery. 1991;110:1043–1046.
49. Silverberg, SJ, Gartenberg, F, Jacobs, TP, et al. Longitudinal measurements of bone density and biochemical indices in untreated primary hyperparathyroidism. J Clin Endocrinol Metab. 1995;80:723–728.
50. Seibel, MJ. Molecular Markers of bone metabolism in primary hyperparathyroidism. In: Bilezikian JP, ed. The Parathyroids: Basic and Clinical Concepts. New York: Academic Press; 2001:399–410.
51. Price, PA, Parthemore, JG, Deftos, LJ. New biochemical marker for bone metabolism. Measurement by radioimmunoassay of bone Gla-protein in the plasma of normal subjects and patients with bone disease. J Clin Invest. 1980;66:878–883.
52. Deftos, LJ, Parthemore, JG, Price, PA. Changes in plasma bone Gla-protein during treatment of bone disease. Calcif Tissue Int. 1982;34:121–124.
53. Ebeling, PR, Peterson, JM, Riggs, BL. Utility of type 1 procollagen propeptide assays for assessing abnormalities in metabolic bone diseases. J Bone Miner Res. 1992;7:1243–1250.
54. Minisola, S, Romagnoli, E, Scarnecchia, L, et al. Serum CITP in patients with primary hyperparathyroidism: Studies in basal conditions and after parathyroid surgery. Eur J Endocrinol. 1994;130:587–591.
55. Seibel, MJ, Gartenberg, F, Silverberg, SJ, et al. Urinary hydroxypyridinium cross-links of collagen in primary hyperparathyroidism. J Clin Endocrinol Metab. 1992;74:481–486.
56. Seibel, MJ, Woitge, HW, Pecherstorfer, M, et al. Serum immunoreactive bone sialoprotein as a new marker of bone turnover in metabolic and malignant bone disease. J Clin Endocrinol Metab. 1996;81:3289–3294.
57. Guo, CY, Thomas, WER, Al-Dehaimi, AW, et al. Longitudinal changes in bone mineral density and bone turnover in women with primary hyperparathyroidism. J Clin Endocrinol Metab. 1996;81:3487–3491.
58. Tanaka, Y, Funahashi, H, Imai, T, et al. Parathyroid function and bone metabolic markers in primary and secondary hyperparathyroidism. Semin Surg Oncol. 1997;13:125–133.
59. Greenfield, EM, Shaw, SM, Gornik, SA, Banks, MA. Adenyl cyclase and interleukin 6 are downstream effectors of PTH resulting in stimulation of bone resorption. J Clin Invest. 1995;96:1238–1244.
60. Pollock, JH, Blaha, MJ, Lavish, SA, et al. In vivo demonstration that PTH and PTHrP stimulate expression by osteoblasts of IL-6 and LIF. J Bone Miner Res. 1996;11:754–759.
61. Grey, A, Mitnick, M, Shapses, S, et al. Circulating levels of IL-6 and TNF-alpha are elevated in primary hyperparathyroidism and correlate with markers of bone resorption. J Clin Endocrinol Metab. 1996;81:3450–3454.
62. Insogna, K, Mitnick, M, Pascarella, J, et al. Role of the interleukin-6 soluble receptor cytokine system in mediating increased skeletal sensitivity to parathyroid hormone in perimenopausal women. J Bone Miner Res. 2002;17:N108–N116.
63. Grey, A, Mitnick, MA, Shapses, S, Ellison, A, Gundberg, C, Insogna, K. Circulating levels of interleukin-6 and tumor necrosis factor-alpha are elevated in PHPT and correlate with markers of bone resorption: A clinical research center study. J Clin Endocrinol Metab. 1996;81:3450–3454.
64. Rosen, CJ, Bing-you, R, Silverberg, SJ, Bilezikian, JP. Enhancement of cancellous bone mass after parathyroidectomy is associated with changes in circulating insulin-like growth factor binding proteins. J Bone Miner Res. 1994;9(suppl I):C424.
65. Silverberg, SJ, Shane, E, DeLaCruz, L, et al. Skeletal disease in primary hyperparathyroidism. J Bone Miner Res. 1989;4:283–291.
66. Bilezikian, JP, Silverberg, SJ, Shane, E, et al. Characterization and evaluation of asymptomatic primary hyperparathyroidism. J Bone Miner Res. 1991;6(suppl I):585–589.
67. Dempster, DW, Cosman, F, Parisien, M, et al. Anabolic actions of parathyroid hormone on bone. Endocr Rev. 1993;14:690–709.
68. Bilezikian, JP, Rubin, MR, Finkelstein, J. Parathyroid hormone as an anabolic therapy for women and men. J Endocrinol Invest. 2005;28(Suppl 7):41–49.
69. Canalis, E, Giustina, A, Bilezikian, JP. Mechanisms of anabolic therapies for osteoporosis. N Engl J Med. 2007;357:905–916.
70. National Institutes of Health. Consensus development conference statement on primary hyperparathyroidism. J Bone Miner Res. 1991;6:s9–s13.
71. Silverberg, SJ, Locker, FG, Bilezikian, JP. Vertebral osteopenia: A new indication for surgery in primary hyperparathyroidism. J Clin Endocrinol Metab. 1996;81:4007–4012.
72. Parisien, M, Silverberg, SJ, Shane, E, et al. The histomorphometry of bone in primary hyperparathyroidism: Preservation of cancellous bone structure. J Clin Endocrinol Metab. 1990;70:930–938.
73. Parfitt, AM. Accelerated cortical bone loss: Primary and secondary hyperparathyroidism. In: Uhthoff H, Stahl E, eds. Current Concepts of Bone Fragility. Berlin: Springer-Verlag; 1986:279–285.
74. Parfitt, AM. Surface specific bone remodeling in health and disease. In: Kleerekoper M, ed. Clinical Disorders of Bone and Mineral Metabolism. New York: Mary Ann Liebert; 1989:7–14.
75. Van Doorn, L, Lips, P, Netelenbos, JC, Hackengt, WHL. Bone histomorphometry and serum intact PTH (I-84) in hyperparathyroid patients. Calcif Tissue Int. 1989;44S:N36.
76. Parisien, M, Cosman, F, Mellish, RWE, et al. Bone structure in postmenopausal hyperparathyroid, osteoporotic and normal women. J Bone Miner Res. 1995;10:1393–1399.
77. Dempster, DW, Parisien, M, Silverberg, SJ, et al. On the mechanism of cancellous bone preservation in postmenopausal women with mild primary hyperparathyroidism. J Clin Endocrinol Metab. 1999;84:1562–1566.
78. Parisien, M, Mellish, RWE, Silverberg, SJ, et al. Maintenance of cancellous bone connectivity in primary hyperparathyroidism: Trabecular and strut analysis. J Bone Miner Res. 1992;7:913–920.
79. Lindsay, R, Nieves, J, Formica, C, et al. Randomised controlled study of effect of parathyroid hormone on vertebral-bone mass and fracture incidence among postmenopausal women on oestrogen with osteoporosis. Lancet. 1997;350:550–555.
80. Dempster, DW, Cosman, F, Kurland, ES, et al. Effects of daily treatment with parathyroid hormone on bone microarchitecture and turnover in patients with osteoporosis: A paired biopsy study. J Bone Miner Res. 2001;16:1846–1853.
81. Neer, RM, Arnaud, CD, Zanchetta, JR, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–1441.
82. Dempster, DW, Müller, R, Zhou, H, et al. Preserved three-dimensional cancellous bone structure in mild primary hyperparathyroidism. Bone. 2007;41:19–24.
83. Roschger, P, Dempster, DW, Zhou, H, et al. New observations on bone quality in mild primary hyperparathyroidism as determined by quantitative backscattered electron imaging. J Bone Miner Res. 2007;22:717–723.
84. Zoehrer, R, Dempster, DW, Bilezikian, JP, et al. Bone quality determined by Fourier transform infrared imaging analysis in mild primary hyperparathyroidism. J Clin Endocrinol Metab. 2008;93:3484–3489.
85. Bilezikian, JP. Bone strength in primary hyperparathyroidism. Osteoporos Int. 2003;14(Suppl 5):5113–5117.
86. Dauphine, RT, Riggs, BL, Scholz, DA. Back pain and vertebral crush fractures: An unemphasized mode of presentation for primary hyperparathyroidism. Ann Intern Med. 1975;83:365–367.
87. Kochesberger, G, Buckley, NJ, Leight, GS, et al. What is the clinical significance of bone loss in primary hyperparathyroidism. Arch Intern Med. 1987;147:1951–1953.
88. Wilson, RJ, Rao, S, Ellis, B, et al. Mild asymptomatic primary hyperparathyroidism is not a risk factor for vertebral fractures. Ann Intern Med. 1988;109:959–962.
89. Larsson, K, Ljunghall, S, Krusemo, UB, et al. The risk of hip fractures in patients with primary hyperparathyroidism: A population-based cohort study with a follow-up of 19 years. J Intern Med. 1993;234:585–593.
90. Kenny, AM, MacGillivray, DC, Pilbeam, CC, et al. Fracture incidence in postmenopausal women with primary hyperparathyroidism. Surgery. 1995;118:109–114.
91. Khosla, S, Melton, LJ, Wermers, RA, et al. Primary hyperparathyroidism and the risk of fracture: A population-based study. J Bone Miner Res. 1999;14:1700–1707.
92. Vignali, E, Viccica, C, Diacinti, D, et al. Morphometic vertebral fractures in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab. April 28, 2009.
93. Parfitt, AM. Parathyroid hormone and periosteal bone expansion. J Bone Min Res. 2002;17:1741–1743.
94. Mosekilde, L. Primary hyperparathyroidism and the skeleton. Clin Endocrinol (Oxf). 2008;69:1–19.
95. Silverberg, SJ, Shane, E, Jacobs, TP, et al. Nephrolithiasis and bone involvement in primary hyperparathyroidism, 1985–1990. Am J Med. 1990;89:327–334.
96. Pak, CYC, Ohata, M, Lawrence, EC, Snyder, W. The hypercalciurias: Causes, parathyroid functions and diagnostic criteria. J Clin Invest. 1974;54:387–391.
97. Kaplan, RA, Haussler, MR, Deftos, LJ, et al. The role of 1,25(OH)2D in the mediation of intestinal hyperabsorption of calcium in primary hyperparathyroidism and absorptive hypercalciuria. J Clin Invest. 1977;59:756–760.
98. Broadus, AE, Horst, RL, Lang, R, et al. The importance of circulating 1,25(OH)2D in the pathogenesis of hypercalciuria and renal stone formation in primary hyperparathyroidism. N Engl J Med. 1980;302:421–426.
99. Pak, CYC, Holt, K. Nucleation and growth of brushite and calcium oxalate in urine of stone formers. Metabolism. 1976;25:665–673.
100. Pak, CYC, Nicar, MJ, Peterson, R, et al. Lack of unique pathophysiologic background for nephrolithiasis in primary hyperparathyroidism. J Clin Endocrinol Metab. 1981;53:536–542.
101. Pak, CYC. Effect of parathyroidectomy on crystallization of calcium salts in urine of patients with primary hyperparathyroidism. Invest Urol. 1979;17:146–151.
102. Berger, AD, Wu, W, Eisner, BH, Cooperberg, MR, Duh, QY, Stoller, ML. Patients with primary hyperparathyroidism-why do some form stones? J Urol. 2009;181:2141–2145.
103. Klugman, VA, Favus, M, Pak, CYC. Nephrolithiasis in primary hyperparathyroidism. In: Bilezikian JP, ed. The Parathyroids: Basic and Clinical Concepts. New York: Academic Press; 2001:437–450.
104. Peacock, M. Primary hyperparathyroidism and the kidney: Biochemical and clinical spectrum. J Bone Miner Res. 2002;17(Suppl 2):N87–N94.
105. Dent, CE, Hartland, BV, Hicks, J, Sykes, ED. Calcium intake in patients with primary hyperparathyroidism. Lancet. 1961;2:336–342.
106. Aurbach, GD, Mallette, LE, Patten, BM, et al. Hyperparathyroidism: Recent studies. Ann Intern Med. 1973;79:566–581.
107. Patten, BM, Bilezikian, JP, Mallette, LE, et al. The neuromuscular disease of hyperparathyroidism. Ann Intern Med. 1974;80:182–194.
108. Vicale, CT. Diagnostic features of muscular syndrome resulting from hyperparathyroidism, osteomalacia owing to renal tubular acidosis and perhaps to related disorders of calcium metabolism. Trans Am Neurol Assoc. 1949;74:143–147.
109. Frame, B, Heinze, EG, Block, MA, Manson, AGA. Myopathy in primary hyperparathyroidism: Observations in three patients. Ann Intern Med. 1968;68:1022–1027.
110. Rollinson, RD, Gilligan, BS. Primary hyperparathyroidism presenting as a proximal myopathy. Aust N Z J Med. 1977;7:420–421.
111. Turken, SA, Cafferty, M, Silverberg, SJ, et al. Neuromuscular involvement in mild, asymptomatic primary hyperparathyroidism. Am J Med. 1989;87:553–557.
112. Joborn, C, Hetta, J, Johansson, H, et al. Psychiatric morbidity in primary hyperparathyroidism. World J Surg. 1998;2:476–481.
113. Brown, GG, Preisman, RC, Kleerekoper, MD. Neurobehavioral symptoms in mild hyperparathyroidism: related to hypercalcemia but not improved by parathyroidectomy. Henry Ford Med J. 1987;35:211–215.
114. Cogan, MG, Covey, CM, Arieff, AI, Clark, OH. Central nervous system manifestations of hyperparathyroidism. Am J Med. 1978;65:963–970.
115. Solomon, BL, Schaaf, M, Smallridge, RC. Psychologic symptoms before and after parathyroid surgery. Am J Med. 1994;96:101–106.
116. Clark, OH. Presidential address: Asymptomatic primary hyperparathyroidism: Is parathyroidectomy indicated? Surgery. 1994;116:947.
117. Pasieka, JL, Parsons, L. Prospective surgical outcome study of relief of symptoms following surgery in patients with primary hyperparathyroidism. World J Surg. 1998;22:513–519.
118. Burney, RE, Jones, KR, Christy, B, Thompson, NW. Health status improvement after surgical correction of primary hyperparathyroidism in patients with high and low preoperative calcium levels. Surgery. 1999;125:608–614.
119. Silverberg, SJ, Lewiecki, EM, Mosekilde, L, Peacock, M, Rubin, MR. Presentation of asymptomatic primary hyperparathyroidism: Proceedings of the Third International Workshop. J Clin Endocrinol Metab. 2009;94:351–365.
120. Caillard, C, Sebag, F, Mathonnet, M, et al. Prospective evaluation of quality of life (SF-36v2) and nonspecific symptoms before and after cure of primary hyperparathyroidism (1-year follow-up). Surgery. 2007;141:153–159.
121. Dotzenrath, CM, Kaetsch, AK, Pfingsten, H. Neuropsychiatric and cognitive changes after surgery for primary hyperparathyroidism. World J Surg. 2006;5:680–685.
122. Eigelberger, MS, Cheah, WK, Ituarte, PH, et al. The NIH criteria for parathyroidectomy in asymptomatic primary hyperparathyroidism: Are they too limited? Ann Surg. 2004;239:528–535.
123. Roman, SA, Sosa, JA, Mayes, L, et al. Parathyroidectomy improves neurocognitive deficits in patients with primary hyperparathyroidism. Surgery. 2005;138:1121–1128.
124. Walker, MD, McMahon, DJ, Inabnet, WB, et al. Neuropsychological features of primary hyperparathyroidism: A prospective study. J Clin Endocrinol Metab. 2009;94:1951–1959.
125. Rao, DS, Phillips, ER, Divine, GW, Talpos, GB. Randomized, controlled clinical trial of surgery vs no surgery in mild asymptomatic primary hyperparathyroidism. J Clin Endocrinol Metab. 2004;89:5415–5422.
126. Bollerslev, J, Jansson, S, Mollerup, CL, et al. Medical observation compared with parathyroidectomy, for asymptomatic primary hyperparathyroidism: A prospective, randomized trial. J Clin Endocrinol Metab. 2007;92:1687–1692.
127. Ambrogini, E, Cetani, F, Cianferotti, L, et al. Surgery or no surgery for mild asymptomatic primary hyperparathyroidism: A prospective, randomized clinical trial. J Clin Endocrinol Metab. 2007;92:3114–3121.
128. Symons, C, Fortune, F, Greenbaum, RA, Dandona, P. Cardiac hypertrophy, hypertrophic cardiomyopathy and hyperparathyroidism—an association. Br Heart J. 1985;54:539–542.
129. Diamond, TH, Kawalski, DL, van der Merwe, TL, Myburgh, DP. Hypercalcemia due to parathyroid adenoma and hypertrophic cardiomyopathy. S Afr Med J. 1987;71:448–449.
130. Stefenelli, T, Mayr, H, Bergler-Klein, J, et al. Primary hyperparathyroidism: Incidence of cardiac abnormalities and partial reversibility after successful parathyroidectomy. Am J Med. 1993;95:197–202.
131. Bradley, EL, III., Wells, JO. Primary hyperparathyroidism and hypertension. Am Surg. 1983;49:569–570.
132. Dominiczak, AF, Lyall, F, Morton, JJ. Blood pressure, left ventricular mass and intracellular calcium in primary hyperparathyroidism. Clin Sci. 1990;78:127–132.
133. Palmer, M, Adami, HO, Bergstrom, R, et al. Mortality after surgery for primary hyperparathyroidism: A follow-up of 441 patients operated on from 1956 to 1979. Surgery. 1987;102:1–7.
134. Ronni-Sivula, H. Causes of death in patients previously operated on for primary hyperparathyroidism. Ann Chir Gynaecol. 1985;74:13–18.
135. Hedback, G, Tisell, LE, Bengtsson, BA, et al. Premature death in patients operated on for primary hyperparathyroidism. World J Surg. 1990;14:829–835.
136. Ljunghall, S, Jakobsson, S, Joborn, C, Palmer, M, Rastad, J, Akerstrom, G. Longitudinal studies of mild primary hyperparathyroidism. J Bone Miner Res. 1991;6(Suppl 2):S111–S116.
137. Hedback, G, Oden, A, Tisell, LE. The influence of surgery on the risk of death in patients with primary hyperparathyroidism. World J Surg. 1991;15:399–405.
138. Soreide, JA, van Heerden, JA, Grant, CS, Yau Lo, C, Schleck, C, Ilstrup, DM. Survival after surgical treatment for primary hyperparathyroidism. Surgery. 1997;122:1117–1123.
139. Wermers, RA, Khosla, S, Atkinson, EJ, et al. Survival after the diagnosis of hyperparathyroidism: a population-based study. Am J Med. 1998;104:115–122.
140. Nilsson, IL, Yin, L, Lundgren, E, Rastad, J, Ekbom, A. Clinical presentation of primary hyperparathyroidism in Europe: nationwide cohort analysis on mortality from nonmalignant causes. J Bone Miner Res. 2002;17(Suppl 2):N68–74.
141. Hedbäck, G, Odén, A. Increased risk of death from primary hyperparathyroidism: an update. Eur J Clin Invest. 1998;28:271–276.
142. Vestergaard, P, Mollerup, CL, Frokjaer, VG, et al. Cardiovascular events before and after surgery for primary hyperparathyroidism. World J Surg. 2003;27:216–222.
143. Nainby-Luxmoore, JC, Langford, HG, Nelson, NC, et al. A case-comparison study of hypertension and hyperparathyroidism. J Clin Endocrinol Metab. 1982;55:303–306.
144. Ringe, JD. Reversible hypertension in primary hyperparathyroidism: pre- and postoperative blood pressure in 75 cases. Klin Wochenschr. 1984;62:465–469.
145. Rapado, A. Arterial hypertension and primary hyperparathyroidism. Incidence and follow-up after parathyroidectomy. Am J Nephrol. 1986;6(Suppl 1):49–50.
146. Lind, L, Jacobsson, S, Palmer, M, Lithell, H, Wengle, B, Ljunghall, S. Cardiovascular risk factors in primary hyperparathyroidism: a 15-year follow-up of operated and unoperated cases. J Intern Med. 1991;230:29–35.
147. Lind, L, Skarfors, E, Berglund, L, et al. Serum calcium: A new, independent, prospective risk factor for myocardial infarction in middle-aged men followed for 18 years. J Clin Epidemiol. 1997;50:967–973.
148. Kamycheva, E, Sundsfjord, J, Jorde, R. Serum parathyroid hormone levels predict coronary heart disease: The Tromso Study. Eur J Cardiovasc Prev Rehabil. 2004;11:69–74.
149. Roberts, WC, Waller, BF. Effect of chronic hypercalcemia on the heart. An analysis of 18 necropsy patients. Am J Med. 1981;71:371–384.
150. Nilsson, IL, Aberg, J, Rastad, J, Lind, L. Maintained normalization of cardiovascular dysfunction 5 years after parathyroidectomy in primary hyperparathyroidism. Surgery. 2005;137:632–638.
151. Stefenelli, T, Mayr, H, Bergler-Klein, J, et al. Primary hyperparathyroidism: incidence of cardiac abnormalities and partial reversibility after successful parathyroidectomy. Am J Med. 1993;95:197–202.
152. Dalberg, K, Brodin, LA, Juhlin-Dannfelt, A, Farnebo, LO. Cardiac function in primary hyperparathyroidism before and after operation. An echocardiographic study. Eur J Surg. 1996;162:171–176.
153. Nuzzo, V, Tauchmanova, L, Fonderico, F, et al. Increased intima-media thickness of the carotid artery wall, normal blood pressure profile and normal left ventricular mass in subjects with primary hyperparathyroidism. Eur J Endocrinol. 2002;147:453–459.
154. Nilsson, IL, Aberg, J, Rastad, J, Lind, L. Left ventricular systolic and diastolic function and exercise testing in primary hyperparathyroidism-effects of parathyroidectomy. Surgery. 2000;128:895–902.
155. Piovesan, A, Molineri, N, Casasso, F, et al. Left ventricular hypertrophy in primary hyperparathyroidism. Effects of successful parathyroidectomy. Clin Endocrinol (Oxf). 1999;50:321–328.
156. Almqvist, EG, Bondeson, AG, Bondeson, L, et al. Cardiac dysfunction in mild primary hyperparathyroidism assessed by radionuclide angiography and echocardiography before and after parathyroidectomy. Surgery. 2002;132:1126–1132.
157. Stefenelli, T, Abela, C, Frank, H, et al. Cardiac abnormalities in patients with primary hyperparathyroidism: implications for follow-up. J Clin Endocrinol Metab. 1997;82(1):106–112.
158. Lind, L, Ridefelt, P, Rastad, J, Akerstrom, G, Ljunghall, S. Cytoplasmic calcium regulation and the electrocardiogram in patients with primary hyperparathyroidism. Clin Physiol. 1994;14(1):103–110.
159. Rosenqvist, M, Nordenstrom, J, Andersson, M, Edhag, OK. Cardiac conduction in patients with hypercalcaemia due to primary hyperparathyroidism. Clin Endocrinol (Oxf). 1992;37(1):29–33.
160. Barletta, G, De Feo, ML, Del Bene, R, et al. Cardiovascular effects of parathyroid hormone: a study in healthy subjects and normotensive patients with mild primary hyperparathyroidism. J Clin Endocrinol Metab. 2000;85(5):1815–1821.
161. Baykan, M, Erem, C, Erdogan, T, et al. Assessment of left ventricular diastolic function and the Tei index by tissue Doppler imaging in patients with primary hyperparathyroidism. Clin Endocrinol (Oxf). 2007;66(4):483–488.
162. Nilsson, IL, Aberg, J, Rastad, J, Lind, L. Endothelial vasodilatory dysfunction in primary hyperparathyroidism is reversed after parathyroidectomy. Surgery. 1999;126(6):1049–1055.
163. Kosch, M, Hausberg, M, Vormbrock, K, Kisters, K, Rahn, KH, Barenbrock, M. Studies on flow-mediated vasodilation and intima-media thickness of the brachial artery in patients with primary hyperparathyroidism. Am J Hypertens. 2000;13(7):759–764.
164. Lumachi, F, Ermani, M, Frego, M, et al. Intima-media thickness measurement of the carotid artery in patients with primary hyperparathyroidism. A prospective case-control study and long-term follow-up. In Vivo. 2006;20(6B):887–890.
165. Fallo, F, Camporese, G, Capitelli, E, Andreozzi, GM, Mantero, F, Lumachi, F. Ultrasound evaluation of carotid artery in primary hyperparathyroidism. J Clin Endocrinol Metab. 2003;88(5):2096–2099.
166. Neunteufl, T, Katzenschlager, R, Abela, C, et al. Impairment of endothelium-independent vasodilation in patients with hypercalcemia. Cardiovasc Res. 1998;40(2):396–401.
167. Kosch, M, Hausberg, M, Vormbrock, K, et al. Impaired flow-mediated vasodilation of the brachial artery in patients with primary hyperparathyroidism improves after parathyroidectomy. Cardiovasc Res. 2000;47(4):813–818.
168. Baykan, M, Erem, C, Erdogan, T, et al. Impairment of flow mediated vasodilatation of brachial artery in patients with primary hyperparathyroidism. Int J Cardiovasc Imaging. 2007;23(3):323–328.
169. Smith, JC, Page, MD, John, R, et al. Augmentation of central arterial pressure in mild primary hyperparathyroidism. J Clin Endocrinol Metab. 2000;85(10):3515–3519.
170. Rubin, MR, Maurer, MS, McMahon, DJ, Bilezikian, JP, Silverberg, SJ. Arterial stiffness in mild primary hyperparathyroidism. J Clin Endocrinol Metab. 2005;90(6):3326–3330.
171. Marx, S. Multiple endocrine neoplasia type 1. In: Bilezikian JP, ed. The Parathyroids. New York: Academic Press; 2001:535–584.
172. Brandi, ML, Gagel, RF, Angeli, A. Consensus Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86:5658–5671.
173. Khoo, TK, Vege, SS, Abu-Lebdeh, HS, et al. Acute pancreatitis in primary hyperparathyroidism: a population-based study. J Clin Endocrinol Metab. 2009;94:2115–2118.
174. Harinarayan, DV, Gupta, N, Kochupillai, N. Vitamin D status in primary hyperparathyroidism in India. Clin Endocrinol. 1995;43:351–358.
175. Meng, XW, Xing, XP, Liu, SQ, Shan, ZW. The diagnosis of primary hyperparathyroidism—analysis of 134 cases. Ann Acad Med Singapore. 1994;16:116–122.
176. Luong, KVQ, Nguyen, LTH. Coexisting hyperthyroidism and hyperparathyroidism with vitamin D deficient osteomalacia in a Vietnamese immigrant. Endocr Pract. 1996;2:250–254.
177. Lumb, GA, Stanbury, SW. Parathyroid function in vitamin D deficiency in primary hyperparathyroidism. Am J Med. 1974;54:833–839.
178. Feldman, D. Vitamin D, parathyroid hormone and calcium: A complex regulatory network. Am J Med. 1999;107:637–639.
179. Silverberg, SJ, Shane, E, Jacobs, TP, Siris, E, Bilezikian, JP. A 10-year prospective study of primary hyperparathyroidism with or without parathyroid surgery. New Engl J Med. 1999;341:1249–1255.
180. Rubin, MR, Bilezikian, JP, McMahon, DJ, et al. The natural history of primary hyperparathyroidism with or without parathyroid surgery after 15 years. J Clin Endocrinol Metab. 2008;93:3462–3470.
181. Bilezikian, JP, Potts, JT, Jr., El-Hajj Fuleihan, G, et al. Summary statement from a workshop on asymptomatic primary hyperparathyroidism: a perspective for the 21st century. J Clin Endocrinol Metab. 2002;87:5353–5361.
182. Khan, AA, Bilezikian, JP, Potts, JT, Jr. on behalf of the Third International Workshop on Asymptomatic Primary Hyperparathyroidism the diagnosis and management of asymptomatic primary hyperparathyroidism revisited. J Clin Endocrinol Metab. 2009;94:333–334.
183. Bilezikian, JP, Khan, AA, Potts, JT, Jr. on behalf of the Third International Workshop on Asymptomatic Primary Hyperthyroidism guidelines for the management of asymptomatic primary hyperparathyroidism: Summary statement from the Third International Workshop. J Clin Endocrinol Metab. 2009;94:335–339.
184. Eastell, R, Arnold, A, Brandi, ML, et al. Diagnosis of Asymptomatic Primary Hyperparathyroidism: proceedings of the Third International Workshop. J Clin Endocrinol Metab. 2009;94:340–350.
185. Silverberg, SJ, Lewiecki, EM, Mosekilde, L, Peacock, M, Rubin, MR. Presentation of Asymptomatic Primary Hyperparathyroidism: proceedings of the Third International Workshop. J Clin Endocrinol Metab. 2009;94:351–365.
186. Udelsman, R, Pasieka, JL, Sturgeon, C, Young, JEM, Clark, OH. Surgery for asymptomatic primary yyperparathyroidism: proceedings of the Third International Workshop. J Clin Endocrinol Metab. 2009;94:366–372.
187. Khan, A, Grey, A, Shoback, D. Medical management of asymptomatic primary hyperparathyroidism: proceedings of the Third International Workshop. J Clin Endocrinol Metab. 2009;94:373–381.
188. Civelek, A, Ozalp, E, Donovan, P, Udelsman, R. Prospective evaluation of delayed technetium-99M sestamibi SPECT scintigraphy for preoperative localization of primary hyperparathyroidism. Surgery. 2002;131:149–157.
189. Van Husen, R, Kim, LT. Accuracy of surgeon-performed ultrasound in parathyroid localization. World J Surg. 2004:1122–1126.
190. Mortenson, ME, Evans, DB, Hunter, GJ, et al. Parathyroid exploration in the reoperative neck: improved preoperative localization with 4D-computer tomography. J Am Coll Surg. 2008;206:888–895.
191. Maser, C, Donovan, P, Satos, F, et al. Sonographically guided fine needle aspiration with rapid parathyroid hormone assay. Ann Surg Oncol. 2006;13:1690–1695.
192. Udelsman, R, Donovan, PI. Remedial parathyroid surgery: changing trends in 130 consecutive cases. Ann Surg. 2006;244:471–479.
193. Clark, OH. How should patients with primary hyperparathyroidism be treated? J Clin Endocrinol Metab. 2003;88:3011–3014.
194. Udelsman, R. Six hundred fifty-six consecutive explorations for primary hyperparathyroidism. Ann Surgery. 2002;235(5):665–672.
195. Irvin, CL, Solorzano, CC, Carneiro, DM. Quick intraoperative parathyroid hormone assay: Surgical adjunct to allow limited parathyroidectomy, improved success rate and predict outcome. World J Surg. 2004;28:1287–1292.
196. Udelsman, R, Donovan, POI, Sokoll, LT. One hundred consecutive minimally invasive parathyroid explorations. Ann Surg. 2000;232:331–339.
197. Miccoli, P, Berti, P, Materazzi, G, Ambrosini, CE, Fregoli, L, Donatini, G. Endoscopic bilateral neck exploration versus quick intraoperative parathormone assay (qPTHa) during endoscopic parathyroidectomy: A prospective randomized trial. Surg Endosc. 2008;22:398–400.
198. Henry, JF, Sebag, F, Tamagnini, P, Forman, C, Silaghi, H. Endoscopic parathyroid surgery: results of 365 consecutive procedures. World J Surg. 2004;28:1219–1223.
199. Westerdahl, J, Bergenfelz, A. Unilateral versus bilateral neck exploration for primary hyperparathyroidism: Five-year follow-up of a randomized controlled trial. Ann Surg. 2007;246(6):976–981.
200. Russell, CF, Dolan, SJ, Laird, JD. Randomized clinical trial comparing scan-directed unilateral versus bilateral cervical exploration for primary hyperparathyroidism due to solitary adenoma. Br J Surg. 2006;93(4):418–421.
201. Silverberg, SJ, Gartenberg, F, Jacobs, TP, et al. Increased bone mineral density following parathyroidectomy in primary hyperparathyroidism. J Clin Endocrinol Metab. 1995;80:729–734.
202. Silverberg, SJ, Shane, E, Jacobs, TP, et al. Primary hyperparathyroidism: 10-year course with or without parathyroid surgery. N Engl J Med. 1999;341:1249–1255.
203. Kulak, CAM, Bandeira, C, Voss, D, et al. Marked improvement in bone mass after parathyroidectomy in osteitis fibrosa cystica. J Clin Endocrinol Metab. 1998;83:732–735.
204. Tritos, NA, Hartzband, P. Rapid improvement of osteoporosis following parathyroidectomy in a premenopausal woman with acute primary hyperparathyroidism. Arch Intern Med. 1999;139:1498.
205. DiGregorio, S. Hiperparatiroidismo primario: Dramatico incremento de la masa ostea post paratiroidectomia. Diagn Osteol. 1999;1:11–15.
206. Dawson-Hughes, B, Stern, DT, Shipp, CC, Rasmussen, HM. Effect of lowering dietary calcium intake on fractional whole body calcium retention. J Clin Endocrinol Metab. 1998;67:62–68.
207. Barger-Lux, MJ, Heaney, RP. Effects of calcium restriction on metabolic characteristics of premenopausal women. J Clin Endocrinol Metab. 1993;76:103–107.
208. Calvo, MS, Kumar, R, Heath, H. Persistently elevated parathyroid hormone secretion and action in young women after four weeks of ingesting high phosphorus, low calcium diets. J Clin Endocrinol Metab. 1990;70:1334–1340.
209. Insogna, KL, Mitnick, ME, Stewart, AF, et al. Sensitivity of the parathyroid hormone-1, 25-dihydroxyvitamin D axis to variations in calcium intake in patients with primary hyperparathyroidism. N Engl J Med. 1985;313:1126–1130.
210. Locker, FG, Silverberg, SJ, Bilezikian, JP. Optimal dietary calcium intake in primary hyperparathyroidism. Am J Med. 1997;102:543–550.
211. Purnell, DC, Scholz, DA, Smith, LM, et al. Treatment of primary hyperparathyroidism. Am J Med. 1984;56:800–809.
212. Broadus, AE, Magee, JS, Mallette, LE, et al. A detailed evaluation of oral phosphate therapy in selected patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 1983;56:953–961.
213. Kaplan, RA, Geho, WB, Poindexter, C, et al. Metabolic effects of diphosphonate in primary hyperparathyroidism. J Clin Pharmacol. 1977;17:410–419.
214. Shane, E, Baquiran, DC, Bilezikian, JP. Effects of dichloromethylene diphosphonate on serum and urinary calcium in primary hyperparathyroidism. Ann Intern Med. 1981;95:23–27.
215. Rossini, M, Gatti, D, Isaia, G, Sartori, L, Braga, V, Adami, S. Effects of oral alendronate in elderly patients with osteoporosis and mild primary hyperparathyroidism. J Bone Miner Res. 2001;16:113–119.
216. Hassani, S, Braunstein, GD, Seibel, MJ, et al. Alendronate therapy of primary hyperparathyroidism. Endocrinologist. 2001;11:459–464.
217. Chow, CC, Chan, WB, Li, JKY, et al. Oral alendronate increases bone mineral density in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab. 2003;88:581–587.
218. Parker, CR, Blackwell, PJ, Fairbairn, KJ, Hosking, DJ. Hyperparathyroid-related osteoporosis: A 2-year study. J Clin Endocrinol Metab. 2002;87:4482–4489.
219. Kahn, AA, Bilezikian, JP, Kung, AW, et al. Alendronate in primary hyperparathyroidism: A double-blind, randomized, placebo-controlled trial. J Clin Endocrinol Metab. 2004;89:3319–3325.
220. Gallagher, JC, Nordin, BEC. Treatment with oestrogens of primary hyperparathyroidism in post-menopausal women. Lancet. 1972;1:503–507.
221. Marcus, R, Madvig, P, Crim, M, et al. Conjugated estrogens in the treatment of postmenopausal women with hyperparathyroidism. Ann Intern Med. 1984;100:633–640.
222. Selby, PL, Peacock, M. Ethinyl estradiol and norethindrone in the treatment of primary hyperparathyroidism in postmenopausal women. N Engl J Med. 1986;314:1481–1485.
223. Grey, AB, Stapleton, JP, Evans, MC, et al. Effect of hormone replacement therapy on BMD in post-menopausal women with primary hyperparathyroidism. Ann Intern Med. 1996;125:360–368.
224. Gallagher, JC, Wilkinson, R. The effect of ethinyl estradiol on calcium and phosphorus metabolism of post-menopausal women with primary hyperparathyroidism. Clin Sci Mol Med. 1973;45:785–802.
225. Rubin, MA, Lee, KH, McMahon, DJ, Silverberg, SJ. Raloxifene lowers serum calcium and markers of bone turnover in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab. 2003;88:1174–1178.
226. Nemeth, EF, Scarpa, A. Rapid mobilization of cellular calcium in bovine parathyroid cells evoked by extracellular divalent cations. Evidence for a cell surface calcium receptor. J Biol Chem. 1987;262:5188–5196.
227. Brown, EM, Gamba, G, Riccardi, D, et al. Cloning and characterization of an extracellular Ca2-sensing receptor from bovine parathyroid. Nature. 1993;366:575–580.
228. Brown, EM, Pollak, M, Seidman, CE. Calcium ion sensing cell surface receptors. N Engl J Med. 1995;333:234–240.
229. Fox, J, Hadfield, S, Petty, BA, Nemeth, EF. A first generation calcimimetic compound (NPS R-568) that acts on the parathyroid cell calcium receptor: A novel therapeutic approach for hyperparathyroidism. J Bone Miner Res. 1993;8:S181.
230. Heath, H, III., Sanguinetti, EL, Oglseby, S, Marriott, TB. Inhibition of human parathyroid hormone secretion in vivo by NPS R-568, a calcimimetic drug that targets the parathyroid cell surface calcium receptor. Bone. 1995;16:85S.
231. Silverberg, SJ, Marriott, TB, Bone, HG, III., et al. Short term inhibition of parathyroid hormone secretion by a calcium receptor agonist in primary hyperparathyroidism. N Engl J Med. 1997;307:1506–1510.
232. Shoback, DM, Bilezikian, JP, Turner, SA, McCary, LC, Guo, MD, Peacock, M. The calcimimetic AMG 073 normalizes serum calcium in patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 2003;88:5644–5649.
233. Peacock, M, Bilezikian, JP, Klassen, PS, Guo, MD, Turner, SA, Shoback, DM. Cinacalcet hydrochloride maintains long-term normocalcemia in patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 2005;90:135–141.
234. Wüthrich, RP, Martin, D, Bilezikian, JP. The role of calcimimetics in the treatment of hyperparathyroidism. Eur J Clin Invest. 2007;37:915–922.
235. Marcocci, C, Chanson, P, Shoback, D, et al. Cinacalcet reduces serum calcium concentrations in patients with intractable primary hyperparathyroidism. J Clin Endocrinol Metab. 2009 June 2.
236. Silverberg, SJ, Rubin, MR, Faiman, C, et al. Cinacalcet HCI reduces the serum calcium concentration in inoperable parathyroid carcinoma. J Clin Endocrinol Metab. 2007;92:3803–3808.
237. Marx, SJ, Fraser, D, Rapoport, A. Familial hypocalciuric hypercalcemia: Mild expression of the gene in heterozygotes and severe expression in homozygotes. Am J Med. 1985;78:15–22.
238. Kristoffersson, A, Dahlgren, S, Lithner, F, Jarhult, J. Primary hyperparathyroidism in pregnancy. Surgery. 1985;97:326–330.
239. Lowe, DK, Orwoll, ES, McClung, MR, et al. Hyperparathyroidism and pregnancy. Am J Surg. 1983;145:611–619.
240. Fitzpatrick, LA, Bilezikian, JP. Acute primary hyperparathyroidism. Am J Med. 1987;82:275–282.
241. Bayat-Mokhtari, F, Palmieri, GMA, Moinuddin, M, et al. Parathyroid storm. Arch Intern Med. 1980;140:1092–1095.
242. Fitzpatrick, LA. Acute primary hyperparathyroidism. In: Bilezikian JP, ed. The Parathyroids: Basic and Clinical Concepts. New York: Academic Press; 2001:527–534.
243. Shane, E, Bilezikian, JP. Parathyroid carcinoma: A review of 62 patients. Endocr Rev. 1982;3:218–226.
244. Marcocci, C, Cetani, F, Rubin, MR, Silverberg, SJ, Pinchera, A, Bilezikian, JP. Parathyroid carcinoma. J Bone Min Res. 2008;23:1869–1880.
245. Shane, E. Parathyroid carcinoma. J Clin Endocrinol Metab. 2001;86:485–493.
246. LiVolsi, V. Morphology of the parathyroid glands. In Becker KL, ed.: Principles and Practice of Endocrinology and Metabolism, ed 3, Philadelphia: Lippincott Williams & Wilkins, 2000.
247. Streeten, EA, Weinstein, LS, Norton, JA, et al. Studies in a kindred with parathyroid carcinoma. J Clin Endocrinol Metab. 1992;75:362–366.
248. Wassif, WS, Moniz, CF, Friedman, E, et al. Familial isolated hyperparathyroidism: a distinct genetic entity with an increased risk of parathyroid cancer. J Clin Endocrinol Metab. 1993;77:1485–1489.
249. Yoshimoto, K, Endo, H, Tsuyuguchi, M, et al. Itakura Familial isolated primary hyperparathyroidism with parathyroid carcinomas: clinical and molecular features. Clin Endocrinol (Oxf). 1998;48:67–72.
250. Marx, SJ, Simonds, WF, Agarwal, SK, et al. Hyperparathyroidism in hereditary syndromes: special expressions and special managements. J Bone Miner Res. 2002;17(Suppl 2):N37–43.
251. Chen, JD, Morrison, C, Zhang, C, Kahnoski, K, Carpten, JD, Teh, BT. Hyperparathyroidism-jaw tumour syndrome. J Intern Med. 2003;253:634–642.
252. Mallette, LE, Malini, S, Rappaport, MP, Kirkland, JL. Familial cystic parathyroid adenomatosis. Ann Intern Med. 1987;107:54–60.
253. Carpten, JD, Robbins, CM, Villablanca, A, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumors syndrome. Nat Genet. 2002;32:676–680.
254. Simonds, WF, James-Newton, LA, Agarwal, SK, et al. Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine (Baltimore). 2002;81:1–26.
255. Dionisi, S, Minisola, S, Pepe, J, et al. Concurrent parathyroid adenomas and carcinoma in the setting of multiple endocrine neoplasia type 1: presentation as hypercalcemic crisis. Mayo Clin Proc. 2002;77:866–869.
256. Agha, A, Carpenter, R, Bhattacharya, S, Edmonson, SJ, Carlsen, E, Monson, JP. Parathyroid carcinoma in multiple endocrine neoplasia type 1 (MEN1) syndrome: Two case reports of an unrecognized entity. J Endocrinol Invest. 2007;30:145–149.
257. Haven, CJ, van Puijenbroek, M, Tan, MH, et al. Identification of MEN1 and HRPT2 somatic mutations in paraffin-embedded (sporadic) parathyroid carcinomas. Clin Endocrinol (Oxf). 2007;67:370–376.
258. Jenkins, PJ, Satta, MA, Simmgen, M, et al. Metastatic parathyroid carcinoma in the MEN2A syndrome. Clin Endocrinol (Oxf). 1997;47:747–751.
259. Dotzenrath, C, Teh, BT, Farnebo, F, et al. Allelic loss of the retinoblastoma tumor suppressor gene: A marker for aggressive parathyroid tumors? J Clin Endocrinol Metab. 1996;81:3194.
260. Shattuck, TM, Valimaki, S, Abara, T, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Eng J Med. 2003;349:1722–1929.
261. Weinstein, LS, Simonds, WF. HRPT2, a marker of parathyroid cancer. N Eng J Med. 2003;349:1691–1692.
262. Carpten, JD, Robbins, CM, Villablanca, A, et al. HRPT2, encoding parafibromin is mutated in hyperparathyroidism-jw tumor sydrome. Nat Genet. 2002;32:676–680.
263. Marx, SJ, Simonds, WF, Agarwal, SK, et al. Hyperparathyroidism in hereditary syndromes: special expressions and special managements. J Bone Miner Res. 2002;17(Suppl 2):N37–N43.
264. Simonds, WF, James-Newton, LA, Agarwal, SK, et al. Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine (Baltimore). 2002;81:1–26.
265. Bradwell, AR, Harvey, TC. Control of hypercalcemia of parathyroid carcinoma by immunisation. Lancet. 1999;353:370–373.
266. Betea, D, Bradwell, AR, Harvey, TC, et al. Hormonal and biochemical normalization and tumor shrinkage induced by anti-parathyroid hormone immunotherapy in a patient with metastatic parathyroid carcinoma. J Clin Endocrinol Metab. 2004;89:3413–3420.
267. Collins, MT, Skarulis, MC, Bilezikian, JP, et al. Treatment of hypercalcemia secondary to parathyroid carcinoma with a novel calcimimetic agent. J Clin Endocrinol Metab. 1998;83:1083–1088.
268. Block, GA, Martin, KJ, deFrancisco, ALM, et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med. 2004;350:1516–1525.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 7
Primary Hyperparathyroidism
Natural History Without Surgery
Randomized Studies of the Natural History of Asymptomatic Primary Hyperparathyroidism
Primary hyperparathyroidism (PHPT) is characterized by hypercalcemia and elevated parathyroid hormone (PTH) levels. The disease today bears little resemblance to the severe disorder of “stones, bones, and groans” described by Fuller Albright and others in the 1930s.1–6 Osteitis fibrosa cystica was the hallmark of classic PHPT. Radiography of the skeleton showed brown tumors of the long bones, subperiosteal bone resorption, distal tapering of the clavicles and phalanges, and “salt-and-pepper”–appearing erosions of the skull on radiograph (Fig. 7-1).7 Nephrocalcinosis was present in 80% of patients, and neuromuscular dysfunction with muscle weakness was also common. With the advent of the automated serum chemistry autoanalyzer in the 1970s, the diagnosis of PHPT became much more common, with a four- to fivefold increase in incidence.8–10 Classic symptomatology, concomitantly, became much less common. In the United States and elsewhere in the developed world, symptomatic PHPT is now the exception rather than the rule, with more than three-fourths of patients having no symptoms attributable to their disease, making PHPT a disease which has “evolved” from its classic presentation (Table 7-1).11–14 Nephrolithiasis is still seen, although much less frequently than in the past. Now, radiologically evident bone disease is rare, but bone involvement is readily detected by bone mass measurement. This chapter describes the clinical picture of PHPT as it presents today, how it can be differentiated from other causes of hypercalcemia, and its clinical course. Issues in management, many of which are still unresolved, are also addressed. A detailed discussion of our current understanding of the etiology of this disease can be found in Chapter 8.
FIGURE 7-1 Radiologic representation of osteitis fibrosa cystica in classic primary hyperparathyroidism. A, Salt-and-pepper skull. B, Cystic bone disease of the clavicle. C, Subperiosteal bone resorption of the digits. D, Cortical erosions.
Pathology
By far the most common lesion found in patients with PHPT is the solitary parathyroid adenoma, occurring in 80% of patients.7,14 Several risk factors have been identified in the development of PHPT. These include a history of neck irradiation16 and prolonged use of lithium therapy for affective disorders.17–18 While in most cases a single adenoma is found, multiple parathyroid adenomas have been reported in 2% to 4% of cases.19 These may be familial or sporadic. Parathyroid adenomas can be discovered in many unexpected anatomic locations (see Chapter 5). Embryonal migration patterns of parathyroid tissue account for a plethora of possible sites of ectopic parathyroid adenomas. The most common sites are within the thyroid gland, the superior mediastinum, and within the thymus.20 Occasionally, the adenoma may ultimately be identified in the retroesophageal space, the pharynx, the lateral neck, and even the alimentary submucosa of the esophagus.21–23 On histologic examination, most parathyroid adenomas are encapsulated and composed of parathyroid chief cells. Adenomas containing mainly oxyphilic or oncocytic cells are rare but can give rise to clinical PHPT.
Clinical Presentation
The incidence of PHPT has changed dramatically.8,9,24,25 Prior to the advent of the multichannel autoanalyzer in the early 1970s, Heath et al.8 reported an incidence of 7.8 cases per 100,000 persons in Rochester, Minnesota. With the introduction of routine calcium measurements in the mid-1970s, this rate rose precipitously to 51.1 cases per 100,000 in the same community. Once prevalent cases were diagnosed (the “sweeping” effect), the incidence declined to approximately 27 per 100,000 persons per year. A report from Rochester, Minnesota, suggested that newly diagnosed cases of PHPT have been declining continuously since the mid-1970s.24 The decline in incidence is not explained by a change in the use of multichannel chemical screening because, in the United States, it is only in the very late 1990s that use of this technique became limited. Moreover, such declines in incidence are not apparent at other medical centers. It is possible that the special demographics of Rochester, Minnesota, combined with the rather complete discovery of PHPT in a population that receives virtually all of its care in one system (ideal epidemiologic surveillance) would naturally be associated with declining numbers for years thereafter. The analogy here might be to overfishing a small pond. Unrelated to the Rochester, Minnesota, experience, the United States may soon experience a decline in new cases of PHPT because multichannel screening is now more limited. On the other hand, greater appreciation of the potential of parathyroid hormone to be a catabolic force in postmenopausal women with osteoporosis has led to measurement of PTH even in subjects who do not have hypercalcemia. This trend has led to the emergence of a new entity, normocalcemic PHPT. A number of factors and forces are thus likely to influence the incidence of PHPT in the future (see later discussion).
Clinical Features
PHPT occurs predominantly in individuals in their middle years, with a peak incidence between ages 50 and 60 years. However, the disease is seen at all ages. Women are affected more frequently than men, in a ratio of approximately 3 : 1. At the time of diagnosis, most patients with PHPT do not have classic symptoms or signs associated with disease. Kidney stones are uncommon, and fractures are rare.14 Diseases associated epidemiologically with PHPT have included hypertension,26–28 peptic ulcer disease, gout, or pseudogout.29,30 Some concomitant disorders such as hypertension are commonly seen, but it is not established that any of these associated disorders are etiologically linked to the disease. The only exception is the MEN syndromes, in which MEN1 is often seen with peptic ulcer disease, and MEN2 may be associated with a pheochromocytoma. Constitutional complaints such as weakness, easy fatigability, depression, and intellectual weariness are seen with some regularity (see later discussion).31
Differential Diagnosis
The diagnosis of PHPT is made when hypercalcemia and elevated PTH levels are present. The other major cause of hypercalcemia, malignancy, is readily distinguished from PHPT. Patients with hypercalcemia of malignancy typically have advanced disease that has already been diagnosed. An exception is multiple myeloma, in which hypercalcemia can be the initial manifestation. These diseases can be easily distinguished; PTH levels are suppressed in multiple myeloma. The differential diagnosis of hypercalcemia, however, includes a number of other etiologies, including rare ones.32
Improved testing for PTH, especially immunoradiometric (IRMA) and immunochemiluminometric (ICMA) assay, has facilitated the distinction between PHPT and hypercalcemia of malignancy. In recent years, it has become clear that the “intact” immunoradiometric assay for PTH (“intact” IRMA) may significantly overestimate the concentration of biologically active parathyroid hormone. In 1998, Lepage et al.33 demonstrated a large non-(1–84) PTH fragment that comigrated with a large amino-terminally truncated fragment (PTH[7–84]) and had substantial cross-reactivity in commercially available IRMAs. This large, inactive moiety constituted as much as 50% (20% to 90%) of immunoreactivity by IRMA for PTH in individuals with chronic renal failure.34 Recognition of this molecule led to the development of a new IRMA utilizing affinity purified polyclonal antibodies to PTH(39–84) and to the extreme N-terminal amino acid regions, PTH(1–4).35,36 This assay detects only the full-length PTH molecule, PTH(1–84). It does not detect the large inactive fragment the normally circulates This assay has clear utility in uremic patients, in whom the “intact” IRMA has been shown to considerably overestimate elevations in biologically active hormone concentration.33,37,38 In PHPT, it is less clear that this assay will aid in the diagnostic evaluation.
A small percentage of patients with PHPT have PTH levels that are within the normal reference range as measured either by the classic IRMA or the newer PTH(1–84) assay. In these patients, levels tend to be in the upper range of normal. In PHPT, such values, although within the normal range, are clearly abnormal in a hypercalcemic setting. This is even more evident in patients younger than 45 years of age. Because PTH levels normally rise with age, the broad normal range for the older IRMA (10 to 65 pg/mL) reflects values for the entire population. In the younger-than-45 individual, one expects a narrower, lower normal range (10 to 45 pg/mL), so hypercalcemia and a PTH level of 50 pg/mL is distinctly abnormal. Occasionally, in either a younger or older patient, the PTH level as measured by the established IRMA will not even be in the upper end of the normal range but as low as 30 pg/mL. Such unusual examples generally require a more careful consideration of other causes of hypercalcemia. But in the end, these individuals are likely to have PHPT also because non–PTH dependent hypercalcemia should suppress the PTH concentration to levels that are either undetectable or at the lower limits of the reference range. Souberbielle et al.39 have illustrated that the normal range is very much a function of whether or not the reference population is or is not vitamin D deficient. When vitamin D deficient individuals were excluded, the upper limit of the PTH reference interval decreased from 65 to 46 pg/mL. When vitamin D–deficient individuals were excluded from the subjects used to establish a reference interval for “whole PTH,” the upper limit decreased from 44 to 34 ng/L.
Ninety percent of patients with hypercalcemia will be shown either to have PHPT or malignancy. Although there are many other causes of hypercalcemia (Table 7-2), they constitute only approximately 10% of the hypercalcemic population. Here also, the PTH assay is useful. With the exception of lithium and thiazide use and familial hypocalciuric hypercalcemia (FHH), virtually all other causes of hypercalcemia are associated with suppressed levels of PTH. If the patient can be safely withdrawn from lithium or thiazide, this should be attempted. Serum calcium and PTH levels are then reassessed 3 months later. If the serum calcium and PTH levels continue to be elevated, the diagnosis of PHPT is made. While patients can generally be readily withdrawn from a thiazide diuretic, patient safety must be the first consideration in any decision to withdraw lithium therapy. The recent emergence of a number of alternative therapeutic approaches may make this option realistic. FHH is differentiated from PHPT by (1) family history, (2) markedly lowered urinary calcium excretion, and (3) the specific gene abnormality (see Chapter 8).
Table 7-2
Rarely, a patient with malignancy will be shown to have elevated PTH levels due to ectopic secretion of native PTH from the tumor itself.32 Much more commonly, the malignancy is associated with the secretion of parathyroid hormone–related protein (PTHrP), a molecule that does not cross-react in the IRMA and ICMA assay. Finally, it is possible that a malignancy is present in association with PHPT. When the PTH level is elevated in someone with a malignancy, this is more likely to be the case than a true ectopic PTH syndrome.
Using the third-generation assay for PTH(1–84), a second molecular form of PTH(1–84) that is immunologically intact at both extremes has been identified. This molecule reacts only poorly in second-generation PTH assays. This molecular species represents less than 10% of the immunoreactivity in normal individuals and up to 15% in renal failure patients. It has been shown to be overexpressed, however, in a limited number of patients with a severe form of PHPT or with parathyroid cancer.41
With these points in mind, patients with elevated PTH levels and normal calcium levels have been reported. These subjects do not have serum calcium levels that extend occasionally into the abnormal range nor do they have any of the known secondary causes for an elevated PTH level. An explanation for why this entity is being seen today may reside in the fact that endocrinologists and other osteoporosis specialists currently evaluate the skeletal status of women at risk for osteoporosis not only with determination of bone density but also with calciotropic hormone measurements. These patients may represent the earliest manifestations of PHPT, a “forme fruste” of the disease. That these patients should exist is not surprising, insofar as clinical manifestations of PHPT are already present when the disorder is commonly diagnosed with hypercalcemia.42,43 One would expect the earliest phase of this disease to be characterized by elevated PTH levels in the absence of hypercalcemia. During this clinically silent period, the patient would not come to medical attention because the serum calcium is normal. However, if these patients were to have PTH levels measured, one might expect to discover them. Several reports describing these individuals have recently been reported, with several patients progressing to overt hypercalcemia while under observation.44–46 Frankly low or low-normal serum calcium concentrations suggest an adaptive response to hypocalcemia with high PTH levels. Secondary hyperparathyroidism can be seen in patients with renal insufficiency, malabsorption, or any of the other vitamin D–deficiency states. Rarely, patients with PHPT and coexisting vitamin D deficiency will present with low calcium concentration. PTH levels are high. In such patients, correction of the vitamin D deficiency is associated with a rise in serum calcium concentration into the hypercalcemic range.47
Other Biochemical Features
In PHPT, serum phosphorus tends to be in the lower range of normal, but frank hypophosphatemia is present in less than a fourth of patients. The hypophosphatemia, when present, represents the phosphaturic actions of PTH. Average total urinary calcium excretion is at the upper end of the normal range, with about 40% of all patients having hypercalciuria. Serum 25(OH)D levels tend to be in the lower end of the normal range. While mean values of 1,25(OH)2D3 are in the high-normal range, approximately a third of patients have frankly elevated levels of 1,25(OH)2D3.48 This pattern is due to the actions of PTH to facilitate the conversion of 25(OH)D to 1,25(OH)2D. A mild hyperchloremia is seen occasionally, due to the effect of PTH on renal acid-base balance. A typical biochemical profile is shown in Table 7-3.
Table 7-3
Biochemical Profile in Primary Hyperparathyroidism (n = 137)
| Patients (Mean ± SEM) | Normal Range | |
| Serum calcium | 10.7 ± 0.1 mg/dL | 8.2–10.2 mg/dL |
| Serum phosphorus | 2.8 ± 0.1 mg/dL | 2.5–4.5 mg/dL |
| Total alkaline phosphatase | 114 ± 5 IU/L | <100 IU/L |
| Serum magnesium | 2.0 ± 0.1 mg/dL | 1.8–2.4 mg/dL |
| PTH (IRMA) | 119 ± 7 pg/mL | 10–65 pg/mL |
| 25(OH)D | 19 ± 1 ng/mL | 30–100 ng/mL |
| 1,25(OH)2D | 54 ± 2 pg/mL | 15–60 pg/mL |
| Urinary calcium | 240 ± 11 mg/g creatinine | |
| Urine DPD | 17.6 ± 1.3 nmol/mmol creatinine | <14.6 nmol/mmol creatinine |
| Urine PYD | 46.8 ± 2.7 nmol/mmol creatinine | <51.8 nmol/mmol creatinine |
The Skeleton
Bone Markers
Both bone resorption and bone formation are stimulated by PTH. Markers of bone turnover, which reflect those dynamics, provide clues to the extent of skeletal involvement in PHPT.12 The study of bone markers in PHPT has been of considerable interest for several reasons. First, this inquiry sheds light on which markers accurately reflect skeletal activity in the patient with PHPT. Second, the evaluation of markers of bone turnover in PHPT has provided insight into the hyperparathyroid process in bone. Finally, clues to the extent of postoperative improvements in bone mineral density (BMD) might be provided by markers of bone turnover.
Bone Formation Markers
Bone formation is reflected by osteoblast products, including bone-specific alkaline phosphatase activity, osteocalcin, and type 1 procollagen peptide.12 Despite the availability of these sensitive measurements of bone formation, the total alkaline phosphatase activity—part of the multichannel biochemical screening profile—is still widely assessed in PHPT. In PHPT, levels can be mildly elevated, but in many patients, total alkaline phosphatase values are within normal limits.49,50 In a small study from our group,51 bone-specific alkaline phosphatase activity correlated with PTH levels and BMD at the lumbar spine and femoral neck. Osteocalcin is also generally increased in patients with PHPT.51–53 Osteocalcin correlates with other indices of bone formation. Assays for procollagen extension peptides reflect osteoblast activation and bone formation but have not been shown to have significant predictive or clinical utility in PHPT.53 In a small study of patients with PHPT, C-terminal propeptide of human type 1 procollagen (PICP) levels were higher than in control subjects,54 but distinct elevations were much less impressive than those seen for alkaline phosphatase, osteocalcin, or even hydroxyproline (see later).
Bone Resorption Markers
Markers of bone resorption include the osteoclast product, tartrate-resistant acid phosphatase (TRAP), and collagen breakdown products such as hydroxyproline, hydroxypyridinium cross-links of collagen, and N- and C-telopeptides of type 1 collagen.12 Urinary hydroxyproline, once the only available marker of bone resorption, no longer offers sufficient sensitivity or specificity to make it a useful tool in the assessment of patients with PHPT. Although urinary hydroxyproline was frankly elevated in patients with osteitis fibrosa cystica, in mild asymptomatic PHPT, it is now generally normal. Hydroxypyridinium cross-links of collagen, pyridinoline (PYD), and deoxypyridinoline (DPD), on the other hand, are often elevated in PHPT. They return to normal after parathyroidectomy.55 DPD and PYD both correlate positively with PTH concentrations. N- and C-terminal peptides of type I collagen (NTX and CTX) are likely to have utility, but they have not been studied extensively in PHPT. Other markers of bone resorption have also been limited in their application to bone turnover in PHPT. For example, studies of TRAP are limited, although levels have been shown to be elevated.47 In the case of the PYD cross-linked telopeptide domain of type I collagen (ICTP), pooled data from patients with high turnover diseases (i.e., PHPT as well as hyperthyroidism) suggest that this marker may reflect calcium kinetics and histomorphometric indices.12 Data specifically relevant to PHPT are not yet available. Bone sialoprotein, a phosphorylated glycoprotein which makes up approximately 5% to 10% of the noncollagenous bone protein, appears to reflect processes associated with both bone formation and bone resorption. In PHPT, bone sialoprotein levels are elevated and correlate with urinary PYD and DPD.55 Thus, sensitive assays of bone formation and bone resorption are both elevated in mild PHPT.
Longitudinal Bone Marker Studies
Studies of bone markers in the longitudinal follow-up of patients with PHPT are limited but indicate a reduction in these turnover markers following parathyroidectomy. Information from our group,55,56 Guo et al.,57 and Tanaka et al.58 all report declining levels of bone markers following surgery, although the choice of markers in the individual studies differed. Data are also emerging concerning the kinetics of change in bone resorption versus bone formation following parathyroidectomy. We have found that markers of bone resorption decline rapidly following cure of PHPT, while indices of bone formation follow a more gradual decrease.55 Urinary PYD and DPD decreased significantly as early as 2 weeks following parathyroidectomy, preceding reductions in alkaline phosphatase. Similar data were reported from Tanaka et al.,58 who demonstrated a discrepancy between changes in NTX (reflecting bone resorption) and osteocalcin (reflecting bone formation) following parathyroidectomy, and Minisola et al.,54 who reported a drop in bone resorptive markers and no significant change in alkaline phosphatase or osteocalcin. Short-term studies reported a brief increase in PICP immediately following parathyroidectomy, while bone resorptive markers fell promptly. The persistence of elevated bone formation markers coupled with rapid declines in bone resorption markers indicates a shift in the coupling between bone formation and bone resorption toward an anabolic buildup of bone mineral postoperatively. Increases in bone density postoperatively provide support for this idea.
Cytokines
IL-6 and TNF-α have been studied as possible mediators of bone resorption in PHPT. In vitro and in vivo data support an effect of PTH in stimulating production of IL-6, which in turn leads to increased osteoclastogenesis.59,60 Furthermore, antibodies to IL-6 prevent PTH-mediated bone resorption. In PHPT, serum levels of TNF-α and IL-6 are increased and fall following parathyroidectomy.61 Importantly, TNF and IL-6 concentrations correlate with the level of PTH in patients with PHPT. Bone turnover markers correlate with levels of these cytokines as well, supporting an important role of the cytokines in mediating the skeletal effects of PTH excess in PHPT.62,63 Furthermore, IL-6 soluble receptor has recently been reported to predict rates of bone loss in mild PHPT. Although cytokine levels did not correlate significantly with bone density measurements in the report of Grey et al.,61 it should be noted that bone density was not assessed at the site containing mostly cortical bone (the radius) where the catabolic effects of excess PTH would be expected to be seen most clearly. More information is needed to confirm these observations, especially with appropriate control subjects, and to test for potential involvement of other bone-resorptive cytokines in PHPT.
The involvement of other factors in the anabolic effect of PHPT on bone is also being studied. Levels of IGF-1, which is well documented to be a direct mediator of PTH action in bone, and insulin-like growth factor–binding protein 3 (IGFBP-3), the major binding protein for IGF-1, change following parathyroidectomy in PHPT.64 The alteration in the ratio of IGF-1 to IGFBP-3 supports enhanced delivery of IGF-1 to tissues following surgery, an increase inversely proportional to the observed rise in lumbar spine and femoral neck bone density. It is not known whether the anabolic properties of PTH at cancellous sites (e.g., lumbar spine) can be explained by IGF-1 prior to surgery.
Bone Densometry
The advent of bone mineral densitometry as a major diagnostic tool for osteoporosis occurred at a time when the clinical profile of PHPT was changing from a symptomatic to an asymptomatic disease. This fortuitous timing allowed questions about skeletal involvement in PHPT to be addressed when specific radiologic features of PHPT had all but disappeared. Bone mass measurements could provide information about the actual state of bone mineral with great accuracy and precision. The known physiologic proclivity of PTH to be catabolic at sites of cortical bone make a cortical site essential to any complete densitometric study of PHPT. By convention, the distal third of the radius is the site used. The early densitometric studies in PHPT also revealed another physiologic property of PTH, namely, to be anabolic at cancellous sites. The lumbar spine became an important site to measure not only because it is predominantly cancellous bone, but also because postmenopausal women are at risk for cancellous bone loss. In PHPT, bone density at the distal third of the radius is diminished.65,66 Bone density at the lumbar spine is only minimally reduced. The hip region, containing a relatively equal mixture of cortical and cancellous elements, shows bone density intermediate between the cortical and cancellous sites (Fig. 7-2). The results support not only the notion that PTH is catabolic for cortical bone but also the view that PTH is anabolic in cancellous bone.67–69 In postmenopausal women, the same pattern was observed.66 Postmenopausal women with PHPT, therefore, show a reversal of the pattern typically associated with postmenopausal bone loss: preferential loss of cancellous bone. The reduced bone density at the distal radius (cortical bone) and preserved density at the lumbar spine (cancellous bone) suggest that PHPT helps protect postmenopausal women from bone loss due to estrogen deficiency.
FIGURE 7-2 Bone densitometry in primary hyperparathyroidism. Data are shown in comparison to age- and sex-matched normal subjects. Divergence from expected values is different at each site (P < 0.0001). (Data from Silverberg SJ, Shane E, de la Cruz L et al: Skeletal disease in primary hyperparathyroidism, J Bone Miner Res 4:283–291, 1989.)
Observations of skeletal health in PHPT made by bone densitometry have established the importance of this technology in the evaluation of all patients with PHPT. Without this information, the data set leading to surgical recommendations is incomplete. The Consensus Development Conference on Asymptomatic Primary Hyperparathyroidism in 1990 implicitly acknowledged this point when bone mineral densitometry was included as a separate criterion for clinical decision making.70 Since that time, bone densitometry has become an indispensable component of both evaluating the patient and establishing clinical guidelines for management and monitoring.
The bone density profile in which there is relative preservation of skeletal mass at the vertebrae and diminution at the more cortical distal radius is not always seen in PHPT. While this pattern is evident in the vast majority of patients, a small group of patients have evidence of vertebral osteopenia at the time of presentation. In our natural history study, approximately 15% of patients had a lumbar spine Z score of less than −1.5 at the time of diagnosis.72 Only half of these patients were postmenopausal women, so not all vetebral bone loss could be attributed entirely to estrogen deficiency. These patients are of interest with regard to changes in bone density following parathyroidectomy and are discussed in further detail later. The extent of vertebral bone involvement will vary as a function of disease severity. In the typical mild form of the disease, the pattern described earlier is seen. When PHPT is more advanced, there will be more generalized involvement, and the lumbar spine will not appear to be protected. When PHPT is severe or more symptomatic, all bones can be extensively involved.
Bone Histomorphometry
Analyses of percutaneous bone biopsies from patients with PHPT have provided direct information that could only be indirectly surmised by bone densitometry and by bone markers. Both static and dynamic parameters present a picture of cortical thinning, maintenance of cancellous bone volume (Fig. 7-3), and a very dynamic process associated with high turnover and accelerated bone remodeling.
FIGURE 7-3 Scanning electron micrograph of bone biopsy specimens in a normal subject (top) and an age- and sex-matched patient with primary hyperparathyroidism (bottom). The cortices of the hyperparathyroid sample are markedly thinned, but cancellous bone and trabecular connectivity appear to be well preserved. (Magnification ×31.25.) (From Parisien MV, Silverberg SJ, Shane E et al: Bone disease in primary hyperparathyroidism, Endocrinol Metab Clin North Am 19:19–34, 1990.)
Cortical thinning, inferred by bone mineral densitometry, is clearly documented in a quantitative manner by iliac crest bone biopsy.72–74 Van Doorn et al.75 demonstrated a positive correlation between PTH levels and cortical porosity. These findings are consistent with the known effect of PTH to be catabolic at endocortical surfaces of bone. Osteoclasts are thought to erode more deeply along the corticomedullary junction under the influence of PTH.
Histomorphometric studies have contributed most by elucidating the nature of cancellous bone preservation in PHPT.75 Again, as suggested by bone densitometry, cancellous bone volume is clearly well preserved in PHPT. This is seen as well among postmenopausal women with PHPT. Several studies have shown that cancellous bone is actually increased in PHPT as compared to normal subjects.76,77 When cancellous bone volume is compared among age- and sex-matched subjects with PHPT or postmenopausal osteoporosis, a dramatic difference is evident (Fig. 7-4). Whereas postmenopausal women with osteoporosis have reduced cancellous bone volume, women with PHPT have higher cancellous bone volume.77 This observation suggests that while PHPT is said to be a risk factor for postmenopausal osteoporosis, it is a syndrome of bone loss. The region(s) of bone loss in PHPT is directed toward the cortical bone compartment, with good maintenance of cancellous bone volume unless the PHPT is unusually active.
FIGURE 7-4 Cancellous bone volume in primary hyperparathyroidism. Cancellous bone volume was analyzed from bone biopsy specimens of the iliac crest. Comparisons are between 16 women with primary hyperparathyroidism, 17 women with postmenopausal osteoporosis, and 31 women with no known disorder of bone metabolism. Subjects were matched for age and other indices. (Data from Parisien M, Cosman F, Mellish RW et al: Bone structure in postmenopausal hyperparathyroid, osteoporotic and normal women, J Bone Miner Res 10:1393–1399, 1995.)
Preservation of cancellous bone volume even extends to comparisons with the expected losses associated with the effects of aging on cancellous bone physiology. In a study of 27 patients with PHPT (10 men and 17 women), static parameters of bone turnover (osteoid surface, osteoid volume, and eroded surface) were increased, as expected, in patients relative to control subjects.78 However, in control subjects, trabecular number varied inversely with age, while trabecular separation increased with advancing age. Both of these observations are expected concomitants of aging. In marked contrast, in the patients with PHPT, no such age dependency was seen. There was no relationship between trabecular number or separation and age in PHPT, suggesting that the actual plates and their connections were being maintained over time more effectively than one would have expected through the aging process. Thus, PHPT seems to retard the normal age-related processes associated with trabecular loss.
In PHPT, indices of trabecular connectivity are greater than expected, while indices of disconnectivity are decreased. When three matched groups of postmenopausal women were assessed (a normal group, a group with postmenopausal osteoporosis, and a group with PHPT), women with PHPT were shown to have trabeculae with less evidence of disconnectivity compared with normals, despite increased levels of bone turnover,76,78 so cancellous bone is preserved in PHPT through the maintenance of well-connected trabecular plates. In order to determine the mechanism of cancellous bone preservation in PHPT, static and dynamic histomorphometric indices were compared between normal and hyperparathyroid postmenopausal women. In normal postmenopausal women, there is an imbalance in bone formation and resorption which favors excess bone resorption. In postmenopausal women with PHPT, on the other hand, the adjusted apposition rate is increased. Bone formation, thus favored, may explain the efficacy of PTH at cancellous sites in patients with osteoporosis.67,79–81 Assessment of bone remodeling variables in patients with PHPT shows increases in the active bone-formation period77 (Table 7-4). The increased bone formation rate and total formation period may explain the preservation of cancellous bone seen in this disease.
Table 7-4
Wall Width and Remodeling Variables in Primary Hyperparathyroidism (PHPT) and Control Groups (Mean ± SEM)
Modified from Dempster DW, Parisien M, Silverberg SJ et al: On the mechanism of cancellous bone preservation in postmenopausal women with mild primary hyperparathyroidism, J Clin Endocrinol Metab 84:1562–1566, 1999.
Recently, further analysis of trabecular microarchitecture has taken advantage of newer technologies that have largely been confirmatory. In a three-dimensional analysis of transiliac bone biopsies using microCT technology, a highly significant correlation was observed with the conventional histomorphometry82 described earlier. In comparison to age-matched control subjects without PHPT, postmenopausal women with PHPT had higher bone volume (BV/TV), higher bone surface area (BS/TV), higher connectivity density (Conn.D), and lower trabecular separation (Tb.Sp.). There were also less marked age-related declines in BV/TV and Conn.D as compared to controls, with no decline in BS/TV. Using the technique of backscattered electron imaging (qBEI) to evaluate trabecular BMD distribution (BMDD) in iliac crest bone biopsies, Roschger et al.83 showed reduced average mineralization density and increase in the heterogeneity of the degree of mineralization, consistent with reduced mean age of bone tissue. Studies of collagen maturity using Fourier Transform Infrared Spectroscopy provide further support for these observations.84 Bone strength, therefore, in PHPT has to take into account a number of factors related to skeletal properties of bone besides BMD.85
Fractures
Fractures were an integral element of classic PHPT, but their importance in modern-day disease is unclear. In a case-control study published in 1975, Dauphine et al.86 suggested that back pain and vertebral crush fractures might be part of the presenting clinical profile of PHPT. Since that time, reports on fracture incidence have been conflicting. A retrospective review of lateral chest radiographs of patients who underwent parathyroidectomy showed an increased incidence of vertebral fractures in one study,87 while Wilson et al.88 found no increase in such fractures in a cohort of 174 consecutive patients who had mild asymptomatic PHPT.
In a study that focused on hip fracture, a population-based prospective analysis (mean of 17 years’ duration; 23,341 person years) showed women with PHPT in Sweden not to be at increased risk.89 In a much smaller study (46 patients, 44 controls), fractures at any site were increased in hyperparathyroidism.90 This study is flawed not only by its small sample size but also by the unusually high fracture incidence in both patients (48%) and control subjects (28%) and by the use of thyroid medication in a significantly greater number of patients (28%) relative to control subjects.
The Mayo Clinic experience with PHPT and risk of fracture reviewed 407 cases of PHPT recognized during the 28-year period between 1965 and 1992.91 Fracture risk was assessed by comparing fractures at a number of sites with numbers of fractures expected on the basis of sex and age from the general population. The clinical presentation of these patients with PHPT was typical of the mild form of the disease, with the serum calcium being only modestly elevated at 10.9 ± 0.6 mg/dL. The data from this retrospective epidemiologic study indicate that overall fracture risk was significantly increased at many sites such as the vertebral spine, the distal forearm, the ribs, and the pelvis. There was no increase in hip fractures. After multivariate analysis, age and female sex remained significant independent predictors of fracture risk. These data, however, are subject to potential ascertainment bias. Patients with PHPT are typically followed more conscientiously, and thus fractures at some of these sites may have been recognized by greater surveillance. This may certainly be true of the vertebral spine and the ribs but unlikely in the case of fractures of the forearm. One might expect to see an increased incidence of distal forearm fractures, since the hyperparathyroid process tends to lead to reduction of cortical bone (distal forearm) in preference to cancellous bone (vertebral spine). Unfortunately, there were no densitometric data provided in this study, so one could not relate bone density to fracture incidence. It is difficult to know whether this study in fact confirms an expectation of preferential distal forearm fractures in PHPT or whether some other process is at work conferring universally greater fracture risk in these patients. In a more recent study, Vignali et al.92 studied the incidence of vertebral fractures in PHPT as determined by DXA-based vertebral fracture assessment (VFA). In this case control study, 150 consecutive patients and 300 healthy women matched for age and menopausal age were studied. Vertebral fractures were detected in more subjects with PHPT (24.6%) than the control subjects (4.0%; P < 0.001). Among asymptomatic PHPT patients, only those who met surgical guidelines showed a higher incidence of vertebral fractures compared with controls. We still lack prospective, controlled studies to determine fracture incidence in PHPT.
Even the expectation of an increased fracture risk at a cortical site, like the forearm, in PHPT is fraught with uncertainties. The expectation is based upon bone-density data and the presumption that in this disease, it is as predictive of fracture as it is in postmenopausal women who do not have PHPT. By analogy with osteoporosis, it is reasonable to consider bone density as a risk factor for fracture in PHPT, but other issues could lead to a different relationship between bone density and fracture risk in PHPT. It is now known that bone density is only one of several important qualities of bone. These other qualities are influential in the overall assessment of fracture risk. Bone size, for example, influences fracture risk. From the clinical trials of parathyroid hormone in the treatment of osteoporosis, as well as in observations of PHPT per se, it is likely that bone size is affected by PTH. Cortical thinning through PTH-mediated endosteal resorption is compensated for by PTH-mediated periosteal apposition, leading to bone that may be increased in cross-sectional diameter. This increase in bone size provides biomechanical protection for the skeleton. In PHPT, an interesting paradigm is set up: cortical thinning tends to increase fracture risk, whereas increased bone size tends to reduce fracture risk. In addition, as noted, microarchitecture of bone does not show the same kind of deterioration that is commonly seen in postmenopausal osteoporosis. The relative preservation of microarchitecture in PHPT is another factor that may protect bone. More recent studies in which hyperparathyroid bone has been studied with regard to bone mineralization density83 and collagen quality84 may also be relevant to this discussion (see earlier). These considerations emphasize the need for prospective studies of site-specific fracture incidence in PHPT.85,92–94
Nephrolithiasis
In the past, classic clinical descriptions of PHPT emphasized skeletal involvement and kidney stones as principal complications of the disease.95 The cause of nephrolithiasis in PHPT is probably multifactorial. An increase in the amount of calcium filtered at the glomerulus due to the hypercalcemia of hyperparathyroidism may lead to hypercalciuria despite the physiologic actions of PTH to facilitate calcium reabsorption. A component of absorptive hypercalciuria exists in this disorder. The enhanced intestinal calcium absorption is believed to be due to increased production of 1,25(OH)2D, a consequence of another physiologic action of PTH, namely to increase the synthesis of this active metabolite.96,97 Urinary calcium excretion is correlated with 1,25(OH)2D levels.97,98 In addition, increased intestinal calcium absorption seen in nephrolithiasis99 may also occur in PHPT. The skeleton provides yet another possible source for the increased levels of calcium in the glomerular filtrate. Hyperparathyroid bone resorption might contribute to hypercalciuria, and subsequently to nephrolithiasis, even though there is no convincing evidence to support this hypothesis.100 Finally, alteration in local urinary factors, such as a reduction in inhibitor activity or an increase in stone-promoting factors, may predispose some patients with PHPT to nephrolithiasis.100,101 It remains unclear whether the urine of patients with hyperparathyroid stone disease is different in this regard from that of other stone formers.
Studies in the 1970s and 1980s documented a higher incidence of renal stone disease than do reports of more recent experience. With the decreased incidence of osteitis fibrosa cystica, studies in the modern era have tended to focus on patients with kidney stones. Conflicting results have emerged, with one group providing evidence that 1,25(OH)2D3 plays an etiologic role in the development of nephrolithiasis in PHPT and other groups unable to document differences in 1,25(OH)2D3 levels between those with and without renal stone disease.95,100–102
Although the incidence of nephrolithiasis is much less common than the incidence in the classic, older presentation of PHPT, kidney stones remain the most common manifestation of symptomatic PHPT (see Table 7-1). Estimates in recent studies place the incidence of kidney stones at 15% to 20% of all patients.103 Other renal manifestations of PHPT include hypercalciuria, which is seen in approximately 40% of patients, and nephrocalcinosis, the frequency of which is unknown. It is important to note that in patients with PHPT who do not have renal stone disease, there is no relationship between extent of hypercalciuria and the development of kidney stones.104
In the 1930s, it was generally accepted that bone and stone disease did not coexist in the same patient with classic PHPT.1,6 Albright and Reifenstein1 postulated that low dietary calcium intake would lead to bone disease, while adequate or high dietary calcium levels would be associated with stone disease. Dent et al.,105 who provided convincing evidence against this construct, proposed the existence of two forms of circulating PTH, one causing renal stones and the other causing bone disease. A host of mechanisms, including differences in dietary calcium, calcium absorption, forms of circulating PTH, and levels of 1,25(OH)2D3, were proposed to account for the clinical distinction between bone and stone disease in PHPT.100,105 Today, there is no clear evidence for two distinct subtypes of PHPT. In our patients with PHPT, we could not identify a distinctive set of biochemical data for patients with stone disease.95 Furthermore, although our population did not include patients with classic hyperparathyroid bone disease, we found no evidence to support the notion that the processes affecting the skeleton and kidneys in hyperparathyroidism occur in different subsets of patients. Urinary calcium excretion per gram of creatinine, levels of 1,25(OH)2D, and BMD at all sites were indistinguishable among patients with and without nephrolithiasis. Cortical bone demineralization is as common and as extensive in those with and without nephrolithiasis.95,100
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
