Malignancy-Associated Hypercalcemia and Medical Management
The History of Malignancy-Associated Hypercalcemia
Malignancy is the second most common cause of hypercalcemia in the general population and by far the most common cause among inpatients. Hypercalcemia was first reported in patients with cancer in the 1920s.1 The first large series of patients with malignancy-associated hypercalcemia (MAHC) was reported in 1936 by Gutman and colleagues.2 This group of patients primarily had multiple myeloma and breast cancer; skeletal invasion by tumor was extensive radiologically. The authors inferred that the cause of hypercalcemia in these patients was skeletal invasion by the malignancy.
This mechanism was assumed to be operative in all instances of MAHC until 1941, when Albright3 described a hypercalcemic patient with renal carcinoma and a solitary skeletal metastasis. He reasoned that a single bone metastasis was inadequate to cause hypercalcemia. Furthermore, he noted that the patient was hypophosphatemic, not hyperphosphatemic, as would be expected from the combination of rapid dissolution of skeletal phosphate-containing hydroxyapatite and parathyroid suppression induced by the hypercalcemia. Albright suggested that the hypercalcemia in this patient was etiologically distinct from that in the previously described patients with breast cancer and multiple myeloma and proposed that hypercalcemia resulted from secretion by the renal carcinoma of parathyroid hormone (PTH) or another humoral factor that resembled PTH. Support for Albright’s humoral hypothesis was presented in 1956, when two groups reported that surgical or other eradication of tumor reversed hypercalcemia in patients with carcinoma unaccompanied by skeletal involvement.4,5 After additional reports supporting the “humoral hypothesis,” Lafferty6 in 1966 reviewed 50 patients with humorally mediated hypercalcemia. By definition, these patients had no detectable skeletal metastases on radiographs or manifested disappearance of the hypercalcemia with tumor ablation or both. Histologically, the patients proved to have predominantly squamous (particularly lung), renal, bladder, and gynecologic malignancies.
Thus, by the end of the 1960s, two broad mechanistic categories of MAHC had clearly been demonstrated.7–9 In one group of patients, hypercalcemia develops through skeletal invasion and destruction by tumor, a condition we have referred to as local osteolytic hypercalcemia (LOH)2,7–10 (Table 8-1). In the other group of patients, hypercalcemia develops predominantly through a humoral mechanism, a condition we have referred to as humoral hypercalcemia of malignancy (HHM).11–13 Subsequently, two additional mechanistic subtypes were well described. These include authentic ectopic hyperparathyroidism and 1,25(OH)2-vitamin D–induced hypercalcemia in patients with lymphomas. These four types are summarized in Table 8-1 and are discussed in detail later.
Table 8-1
Tumor Histology in Malignancy-Associated Hypercalcemia
Tumors COMMONLY Associated With Local Osteolytic Hypercalcemia
Tumors COMMONLY Associated With Humoral Hypercalcemia of Malignancy
Squamous carcinoma (lung, head and neck, esophagus, cervix, vulva, skin)
Endocrine tumors (pheochromocytoma, adrenocortical carcinoma, islet carcinoma, carcinoids)
Tumors Associated With 1,25(OH)2D Overproduction
Tumors Associated With Authentic Ectopic PTH Secretion
Common Tumors Rarely Associated With Hypercalcemia
*Has been reported with many different histologic types of lymphoma.
Clinical Features of Malignancy-Associated Hypercalcemia
Hypercalcemia occurring in a patient with cancer indicates that the overall prognosis for the patient in question is very poor. For example, in a study by Ralson and collaborators,14 the onset of hypercalcemia was associated with a 30-day survival rate of only 50%.
The clinical manifestations of the hypercalcemia that accompanies cancer are no different from those that accompany other hypercalcemic disorders. Polyuria, polydipsia, dehydration, renal compromise, constipation, and varying degrees of neurologic dysfunction ranging from lethargy or confusion to coma are common. The electrocardiogram may show shortening of the QTc interval.15,16 The correlation between the degree of hypercalcemia and a given patient’s neurologic function is poor. Other factors (such as the rate of development of hypercalcemia, the presence of underlying central nervous system dysfunction, the presence of other metabolic disorders, and the use of a variety of medications) may profoundly influence the effects of a given degree of hypercalcemia. Patients with skeletal metastases may report skeletal pain or pathologic fractures or both. Sometimes the onset of hypercalcemia can be ascribed to factors other than tumor progression (e.g., recent immobilization, addition of a thiazide diuretic, prerenal or renal azotemia leading to inadequate renal calcium clearance, hypophosphatemia resulting from inadequate oral intake, gastrointestinal fluid losses, medications, parenteral calcium administration in the form of hyperalimentation). These events should be specifically sought because their correction may reverse a given patient’s hypercalcemia.
No study has precisely defined the relation of tumor size to the presence or absence of hypercalcemia. Nonetheless, to the extent that tumor size has been studied, it would seem clear that small, occult tumors rarely cause MAHC.2–12,17,18 A corollary of this statement is that when a patient has MAHC, the tumor that is responsible usually is readily apparent after only a modestly rigorous search; conversely, if a tumor has not been found after 2 or 3 days of evaluation in a hospitalized, hypercalcemic patient, it is unlikely that a malignancy is the cause of the hypercalcemia. Thus, a careful history and physical examination with attention to the skin, oropharynx, esophagus, pulmonary system, liver, genitourinary tract, hematopoietic system, and breasts and a limited laboratory and radiologic investigation focused on the hematologic system, esophagus, kidneys, bladder, gynecologic structures, and skeleton will almost invariably lead to rapid definition of the responsible tumor. Occasionally, retroperitoneal tumors (renal carcinomas, lymphomas, pancreatic tumors) may be difficult to demonstrate. Finally, endocrine tumors (e.g., islet carcinomas, pheochromocytomas, ovarian carcinoids) may lead to hypercalcemia and yet may be small and difficult or impossible to localize.
In approaching the evaluation and treatment of a patient with hypercalcemia and cancer, it is important to bear in mind that hypercalcemia may occur in patients with cancer for all the same reasons that it occurs in patients without cancer. For example, in a series of 133 patients with cancer and hypercalcemia encountered between 1978 and 1984, we identified 8 patients with cancer in whom hypercalcemia ultimately proved to result not from cancer but from coexisting primary hyperparathyroidism.12 Similarly, we have observed hypercalcemia resulting from tuberculosis, sarcoidosis, immobilization, vitamin D intoxication, hyperthyroidism, thiazide use, Addison’s disease, and other causes in patients initially perceived as having MAHC. Thus, the entire differential diagnosis of hypercalcemia should be entertained in every patient in whom hypercalcemia is identified, even if it appears at the outset that cancer will prove to be the ultimate cause. This approach is particularly important for the following reasons: (1) in contrast to the poor ultimate prognosis in MAHC, most causes of hypercalcemia other than cancer are readily treatable; (2) identifying a treatable, nonmalignant cause of hypercalcemia in a patient with cancer may dramatically change the overall perception of a case by the patient’s physicians; and (3) treatment approaches may vary.
It is important to say a word about the tumor histologies associated with hypercalcemia (see Table 8-1). Virtually all tumor types have been reported to cause hypercalcemia, but, as will be described in the sections on LOH and HHM, certain tumor types are particularly common causes of hypercalcemia. Conversely, certain other common tumor types (prostate, colon, oat cell, thyroid, and gastric carcinomas, and primary central nervous system malignancies are examples) almost never cause hypercalcemia2–12,17,18 (see Table 8-1). When these tumors are identified in a patient with hypercalcemia, other tumors or other nonmalignant causes of hypercalcemia should be sought.
Local Osteolytic Hypercalcemia
Hypercalcemia can result from direct skeletal involvement by a primary hematologic neoplasm or by skeletal metastases from a nonhematologic neoplasm. Patients with LOH account for approximately 20% of patients in a series of patients with MAHC.2–12,17–22 The malignancies that most commonly lead to LOH are multiple myeloma, leukemia, lymphoma, and breast cancer2–12,17–22 (see Table 8-1). This list is not exhaustive, for many other tumor types occasionally have been reported to cause hypercalcemia through skeletal metastasis.2–12,17–22
Hypercalcemia that occurs in patients with multiple myeloma and breast cancer was initially attributed to the direct physical destruction of bone by malignant cells. This concept is now seen as naive, for it is clear that simply having malignant tumor cells in the bone marrow compartment is insufficient to cause hypercalcemia. First, bone resorption surrounding malignant cells in bone marrow is always accomplished by osteoclasts, not tumor cells, indicating that osteoclast recruitment and activation by tumors are required. Second, although some tumor metastases in bone are commonly associated with LOH (breast cancer and myeloma are examples), certain other tumor types typically associated with destructive skeletal metastases (e.g., small cell and prostate carcinomas)23,24 only rarely cause hypercalcemia. Moreover, one large study reported an inverse correlation between the number of bone metastases and the serum calcium concentration in a series of hypercalcemic patients with breast cancer.25 Thus, the pathophysiology of the hypercalcemia in LOH is based on paracrine factors or cytokines that are capable of activating osteoclasts, and these are produced by only certain types of malignant cells in the bone marrow. It is these factors that produce hypercalcemia in LOH.
A search for these so-called osteoclast-activating factors, or OAFs, began in the 1970s with reports by Mundy and others26,27 that short-term cultures of bone marrow aspirates from patients with myeloma or lymphoma contain a bone-resorbing factor or family of bone-resorbing factors that are capable of stimulating osteoclasts in vitro. Despite work over the past four decades, the precise array of cytokines that compose OAFs remains incompletely resolved. At the time of this writing, the most attractive candidates include receptor activator of nuclear factor (NF)-κB ligand (RANK-L), macrophage inflammatory protein-1α (MIP-1α), interleukin-1α, interleukin-6, parathyroid hormone–related protein (PTHrP, see later), and tumor necrosis factor-α (TNF-α).28–31 In patients with lymphoma, studies by Dewhirst and others30 have suggested a role for interleukin-1. Thus, it seems likely that the generic term, OAF, actually constitutes a group of such factors that can mediate hypercalcemia in a tumor-specific fashion.
The discovery and elucidation of the RANK-RANK-L system in the late 1990s provided a particularly exciting advance and recently has been reviewed in detail as it relates to bone metastasis and myeloma.31,32 In brief, osteoclast precursors (macrophages) and mature osteoclasts express the receptor, RANK. Osteoblasts and marrow stromal cells normally express the ligand (RANK-L) and upregulate RANK-L in response to agents that recruit osteoclast precursors and induce them to form osteoclasts and initiate and enhance bone resorption. Thus, for example, PTH and PTHrP act via the PTH receptor on marrow stromal cells and osteoblasts and induce them to produce more RANK-L. This then stimulates osteoclast precursors to form more osteoclasts and induces mature osteoclasts to become more active as well. It is interesting to note that osteoblasts and marrow stromal cells also produce a soluble or secreted decoy RANK receptor called osteoprotegerin (OPG). OPG serves to balance the bone resorption induced by the RANK-RANK-L pathway. Increases in OPG production by marrow stromal cells and osteoblasts can completely prevent osteoclast recruitment and bone resorption. Thus, osteoclast number and activity ultimately depend on the balance between OPG and RANK-L in the marrow microenvironment. This, in turn, depends on how much OPG and RANK-L are produced by marrow stromal cells and osteoblasts, and, in the case of skeletal metastases or myeloma cells, whether they activate or serve as surrogates for the native RANK-RANK-L system. To be more specific, credible evidence now suggests that myeloma cells themselves may produce large amounts of RANK-L, and, through direct interaction with RANK on osteoclast precursors, may activate osteoclast recruitment and further activate bone resorption, leading to the massive osteolysis characteristic of multiple myeloma.28,31 These observations have not only pathophysiologic implications but potentially therapeutic ones as well, as is discussed later.
Another characteristic feature of myeloma bone disease is that bone formation by osteoblasts is absent. Thus, the skeletal demineralization of myeloma can be seen as being due in part to dramatic increases in bone resorption, as described earlier, acting in concert with dramatic reductions in osteoblastic bore formation. This marked uncoupling of bone formation from resorption is not well understood, but it is believed that cytokines or other paracrine factors produced by malignant plasma cells may be responsible for this suppression of bone formation in myeloma. In support of this possibility, Tian and colleagues33 recently reported that myeloma cells greatly overproduce DKK-1, an antagonist of the wnt signaling pathway that is known to be critical for osteoblast function. More recently, the Roodman group34 has reported that interleukin-3 (IL-3) secreted by malignant plasma cells can also act to suppress bone formation. DKK and IL-3 findings also suggest novel therapeutic strategies for inducing bone formation in patients with myeloma. It is possible that the marked infiltration of malignant plasma cells characteristic of myeloma replaces marrow stromal cells that might otherwise serve as a source of osteoblast precursors, and this lack of osteoblast precursors thus results in inadequate bone formation.
Progress also has been made in our understanding of the cellular mechanisms responsible for bone resorption by skeletal metastasis in breast cancer. An increasing number of reports suggest a role for PTHrP as a local or intraskeletal mediator of osteoclast activation in women with breast cancer bone metastases.29,35–38 Immunohistochemical analysis by Southby and associates37 demonstrated that 12 (92%) of 13 breast cancer skeletal metastases contained PTHrP, whereas only 3 (17%) of 18 nonskeletal breast cancer metastases contained PTHrP. These findings have been confirmed at the in situ hybridization level for PTHrP messenger RNA (mRNA) in breast cancers metastatic to bone or soft tissue.38 In addition to suggesting a role for PTHrP as a local bone-resorbing factor, these studies suggest that PTHrP somehow may serve to favor metastasis to the skeleton, as well as tumor growth, in patients with breast cancer. Guise and collaborators36 showed this concept to be true: In human breast cancer cell lines bioengineered to express PTHrP at high or low levels, those producing large quantities of PTHrP were more likely to lead to bone metastasis than were those expressing low levels. Moreover, after the development of bone metastases, a local skeletal vicious cycle appeared to develop in which PTHrP induces osteoclastic bone resorption, which in turn leads to the local release of TGF-β from resorbed bone. This locally released TGF-β further induces tumor-derived PTHrP production and accelerated bone resorption.36
Hypercalcemia will develop in approximately one third of women with breast cancer and bone metastases treated with estrogen or antiestrogens such as tamoxifen.39,40 The mechanisms responsible for this “estrogen flare” in breast cancer remain undefined. Frequently, the hypercalcemia will resolve spontaneously if hypercalcemia can be controlled over the short term and endocrine therapy can be continued.40 It has been suggested that the tamoxifen-induced hypercalcemic flare predicts a favorable tumor response. Valentin-Opran and colleagues41 suggested, after work with cultured breast cancer cell lines, that estrogen exposure enhances the production of undefined bone-resorbing factors.
From a clinical and biochemical standpoint, LOH is associated with accelerated bone resorption11–13,35,42 (Fig. 8-1), hypercalcemia, and appropriate suppression of PTH11,12 (Fig. 8-2), nephrogenous cyclic adenosine monophosphate (cAMP) (Fig. 8-3), and 1,25(OH)2-vitamin D (1,25[OH]2D) (Fig. 8-4).11,12 PTHrP is not detectable in the circulation19–22 (Fig. 8-5). As a result of hypercalcemia and suppressed PTH, fractional calcium excretion is increased11 (Fig. 8-6). With the presence of bone resorption, which delivers a phosphorus load into the extracellular fluid, together with suppression of PTH (limiting phosphorus excretion), one might expect the serum phosphorus concentration to be elevated in patients with LOH. Conversely, one might expect the serum phosphorus concentration to be low as a reflection of poor dietary intake and the phosphaturic effects of hypercalcemia. In fact, it is usually normal11,12 (Fig. 8-7), as is the tubular maximum for phosphorus (TmP/glomerular filtration rate [GFR])11,12 (see Fig. 8-7), which presumably reflects a balance between these opposing forces. Bone radionuclide scans typically display widespread metastases in breast cancer associated with LOH. In contrast, bone scans may be entirely negative in patients with multiple myeloma. This difference reflects the uptake of radionuclide in areas of bone formation (e.g., blastic metastases in breast cancer) but not in areas of osteoclastic activity. Scans may be positive in patients with myeloma in whom fractures and fracture callous formation have developed. As noted earlier, bone biopsy discloses markedly increased osteoclastic bone resorption in both breast cancer and myeloma associated with LOH (see Fig. 8-1), a finding reflected by increases in biochemical markers of bone resorption such as deoxypyridinoline and N-telopeptide excretion.42
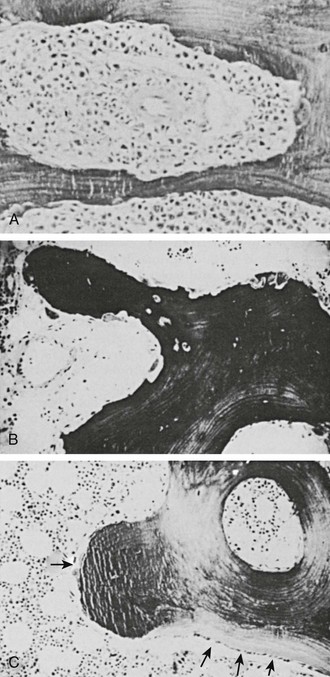
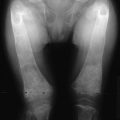
FIGURE 8-1 Bone biopsy photomicrographs. A, Local osteolytic hypercalcemia secondary to leukemia. Note the presence of leukemic cells in the marrow space and numerous osteoclasts lining the trabecular surface. B, Humoral hypercalcemia of malignancy caused by squamous carcinoma. Note the absence of tumor in the marrow space but the presence of numerous active osteoclasts on the trabecular surface. Also note the absence of osteoblasts and osteoid. C, Hyperparathyroidism. Note the abundant osteoclasts, osteoblasts, and osteoid. Osteoblasts are indicated by small arrows, and osteoblasts by large arrows. (B and C from Stewart AF, Vignery A, Silverate A, et al: Quantitative bone histomorphometry in humoral hypercalcemia of malignancy: uncoupling of bone cell activity, J Clin Endocrinol Metab 55:219–227, 1982.)
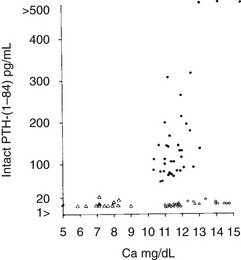
FIGURE 8-2 Immunoreactive parathyroid hormone in the serum of patients with primary hyperparathyroidism (solid circles), hypoparathyroidism (triangles), and hypercalcemia of malignancy (open circles) as measured with a two-site immunoradiometric assay for parathyroid hormone (1-84). Note the clear separation of patients with hyperparathyroidism from those with malignancy-associated hypercalcemia. (Data from Nussbaum SR, Zahradnik RJ, Lavigne JR, et al: Highly sensitive two-site immunoradiometric assay of parathyrin and its clinical utility in evaluating patients with hypercalcemia, Clin Chem 33:1364–1366, 1987.)
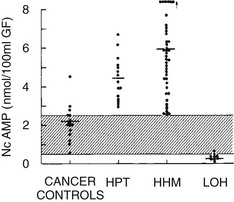
FIGURE 8-3 Nephrogenous cyclic adenosine monophosphate excretion in patients with normocalcemia and cancer (cancer controls), primary hyperparathyroidism (HPT), humoral hypercalcemia of malignancy (HHM), and local osteolytic hypercalcemia (LOH). (Data from Stewart AF, Horst R, Deftos LJ, et al: Biochemical evaluation of patients with cancer-associated hypercalcemia: evidence for humoral and non-humoral groups, N Engl J Med 303:1377–1383, 1980.)
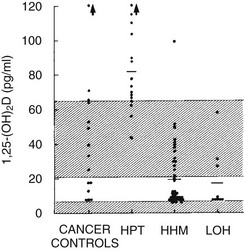
FIGURE 8-4 Plasma 1,25-dihydroxyvitamin D concentration in the four groups described in Figure 8-3. (Data from Stewart AF, Horst R, Deftos LJ, et al: Biochemical evaluation of patients with cancer-associated hypercalcemia: evidence for humoral and non-humoral groups, N Engl J Med 303:1377–1383, 1980.)
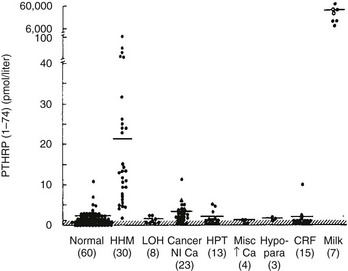
FIGURE 8-5 Immunoreactive parathyroid hormone–related protein (PTHrP) in plasma from patients with the clinical syndromes listed below the x-axis. Note that patients with humoral hypercalcemia of malignancy (HHM) have elevated circulating PTHrP concentrations. (Data from Burtis WJ, Brady TG, Orloff JJ, et al: Immunochemical characterization of circulating parathyroid hormone-related protein in patients with humoral hypercalcemia of malignancy, N Engl J Med 322:1106–1112, 1990.)
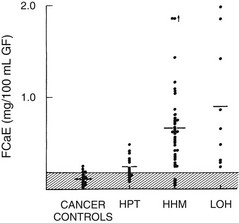
FIGURE 8-6 Fasting calcium excretion in the four groups described in Figure 8-3. Note that on average, patients with humoral hypercalcemia of malignancy (HHM) and local osteolytic hypercalcemia (LOH) appear to be more calciuric than do patients with primary hyperparathyroidism (HPT). Compare these findings with those in Figure 8-8. (Data from Stewart AF, Horst R, Deftos LJ, et al: Biochemical evaluation of patients with cancer-associated hypercalcemia: evidence for humoral and non-humoral groups, N Engl J Med 303:1377–1383, 1980.)
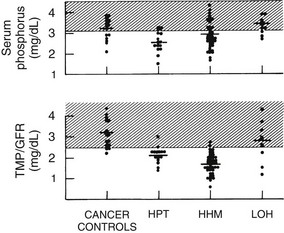
FIGURE 8-7 Serum phosphorus and renal phosphorus threshold in the four groups described in Figure 8-3. (Data from Stewart AF, Horst R, Deftos LJ, et al: Biochemical evaluation of patients with cancer-associated hypercalcemia: evidence for humoral and non-humoral groups, N Engl J Med 303:1377–1383, 1980.)
Humoral Hypercalcemia of Malignancy
Humoral hypercalcemia of malignancy (HHM) accounts for approximately 80% of patients in unselected series of individuals with MAHC.2–12,17–22 Approximately 50% of patients with HHM have an underlying squamous carcinoma of the lung, cervix, esophagus, larynx, oropharynx, vulva, skin, or other site2–12,17–22 (see Table 8-1). Carcinomas of the kidney, ovary, and bladder also are very common.2–14,19–22 It is interesting to note that breast cancer not only may cause MAHC through LOH and skeletal metastases but also may cause hypercalcemia in the absence of skeletal metastases in a classic HHM scenario43,44 (see Table 8-1). Human T cell lymphoma/leukemia virus-I lymphomas, 90% of which are associated with hypercalcemia, also operate through this mechanism.45,46 Finally, hypercalcemia resulting from endocrine tumors such as pheochromocytomas47,48 and islet cell carcinomas49,50 may cause hypercalcemia through this mechanism. As is the case with LOH, virtually every tumor type has been reported on occasion to cause HHM. It is interesting to note that not every tumor that causes hypercalcemia is malignant. Examples of systemic secretion of PTHrP by benign neoplasms (mammary hypertrophy and uterine leiomyomas are examples) have been reported to cause “humoral hypercalcemia of benignancy.”51
Since the advent of Albright’s humoral theory of hypercalcemia of malignancy in the 1940s,3 several substances have been proposed as candidates for the humoral mediator responsible for HHM. In the 1960s, Gordan and coworkers52 suggested that elevated circulating levels of four phytosterols (plant-derived vitamin D analogues) were present in patients with breast carcinoma. Subsequent studies showed, however, that these same phytosterols were present in equivalent concentrations in normal and lactating women, and that the potency of these analogues was inadequate to cause hypercalcemia.53 The phytosterol theory thus lost support.
With the discovery in the 1970s that prostaglandin E2 (PGE2) was a potent stimulator of bone resorption both in tissue culture54 and in experimental animals in vivo,55 the possibility arose that the hypercalcemia associated with HHM was due to systemic PGE2 secretion by tumors. Seyberth and colleagues56 and others reported that urinary metabolites of PGE2 were elevated in patients with MAHC, and that therapy with prostaglandin synthesis inhibitors (aspirin, indomethacin) reversed the hypercalcemia in several patients. However, subsequent, more extensive studies have not shown frequent responses to indomethacin.57 It is the current view of most investigators that PGE2 does not act as a systemic mediator of bone resorption in most cases of HHM, and that therapy with prostaglandin synthesis inhibitors is usually ineffective. It should be clear, however, that these observations do not exclude a role for PGE2 or other arachidonate metabolites in HHM at the local level within the skeleton.
As was noted earlier, Albright3 initially had suggested that PTH was the responsible humoral factor. This concept subsequently gained wide acceptance, as evidenced by the entrance into common usage in the 1960s and 1970s of the terms “ectopic hyperparathyroidism” and “pseudohyperparathyroidism.”6 Evidence in support of the “ectopic PTH” thesis included (1) the humoral nature of the syndrome4,5,10; (2) the hypophosphatemia and renal phosphate–wasting characteristic of the syndrome; and (3) the apparent failure of suppression of PTH observed in the early generations of PTH radioimmunoassays.58,59 It is now clear, as is described later, that although bona fide ectopic hyperparathyroidism does indeed exist (see later), it is extremely rare and fails to account for most instances of HHM.
Today, it is widely accepted that the vast majority of cases of HHM are due to the secretion of PTHrP by tumors. Evidence for this statement can be summarized as follows: (1) tumors associated with HHM produce and secrete PTHrP, which leads to elevated circulating concentrations of PTHrP19–22 (see Fig. 8-5); (2) PTHrP infusion into laboratory animals and humans reproduces the key features of the HHM syndrome in vivo60–65; and (3) infusion of neutralizing antisera against PTHrP reverses the HHM syndrome in laboratory animal models.66,67
The pathophysiology of HHM is now quite clear. Under normal circumstances, PTHrP is widely expressed among essentially all tissues and serves as a local paracrine or autocrine factor, but it does not enter the systemic circulation.68,69 However, in malignant and occasionally benign tumors, PTHrP gene expression may be significantly upregulated, and PTHrP now enters the circulation. Because of the structural homology between PTHrP and PTH, PTHrP is able to bind to and activate the common PTH/PTHrP receptor in bone and kidney, with the result that patients with HHM mimic many of the cardinal features of primary hyperparathyroidism. As is summarized later, hypercalcemia in HHM results from a combination of accelerated osteoclastic bone resorption and reduced ability of the kidney to clear calcium. Biochemically, patients with HHM display increases in circulating concentrations of PTHrP19–22 (see Fig. 8-5), and bone biopsies display a marked increase in osteoclastic resorption, accompanied by a decrease in osteoblastic bone formation13 (see Fig. 8-1). These histologic results have been corroborated more recently by the use of biochemical markers of bone turnover such as bone-specific alkaline phosphatase, osteocalcin, N-telopeptide of collagen, and deoxypyridinoline cross-links.42 This quantitatively striking uncoupling of bone resorption from formation leads to a large calcium flux from the skeleton into the extracellular fluid and accounts primarily for the hypercalcemia observed in HHM. The cellular basis for this dramatic uncoupling is not known. Circulating PTH concentrations are reduced in patients with HHM19–22,70 (see Fig. 8-2), but nephrogenous cyclic adenosine monophosphate (NcAMP) excretion is increased (see Fig. 8-3),11,12 a reflection of the increases in circulating PTHrP. Although the increase in NcAMP excretion is of little diagnostic importance today, the elevation in NcAMP and the ability of PTHrP to stimulate adenylyl cyclase in the kidney in vitro led to the identification and purification of PTHrP.69
HHM is associated with suppression of plasma 1,25(OH)2D11,12 (see Fig. 8-4). The mechanisms responsible for the reduction in 1,25(OH)2D in HHM as compared with primary hyperparathyroidism have recently begun to be elucidated. First, Horwitz et al.64 have demonstrated that chronic infusion of PTHrP in humans serves as a poor agonist of 1,25(OH)2D production, in contrast to infusion of PTH, which leads to robust, dose-related increases in 1,25(OH)2D. Second, Dean et al71 have recently demonstrated that whereas PTH and PTHrP may associate with the common human PTH-PTHrP receptor equivalently, their dissociation rates differ markedly, with PTH remaining bound to the receptor and activating signal transduction, while PTHrP dissociates rapidly, inactivating downstream signaling. Because of the low 1,25(OH)2D concentrations in patients with HHM, one might predict that intestinal calcium absorption does not contribute to their hypercalcemia, in contrast to those with primary hyperparathyroidism, in whom dietary calcium intake increases serum calcium. This inability to absorb dietary calcium in humans with malignancy-associated hypercalcemia has been directly documented using intestinal 45Ca absorption.72
Fractional calcium excretion by the kidney has been reported to be elevated10,11 (see Fig. 8-5) or reduced73,74 in patients with HHM. These conflicting reports result from the fact that accurate measurements of the GFR, on which calcium excretion measurement is based, are not possible in patients with advanced cancer, rapidly declining renal function, and markedly reduced muscle mass (and therefore creatinine release) who are undergoing aggressive hydration and diuretic therapy. To address this issue more directly, Horwitz and others62–64 recently demonstrated in normal healthy volunteers infused with PTH or PTHrP that both peptides have potent and equivalent effects in stimulating renal tubular (presumably distal) reabsorption of calcium (Fig. 8-8). Thus, in addition to the bone-resorbing effects of PTHrP, the anticalciuric effects of PTHrP contribute to hypercalcemia in HHM.
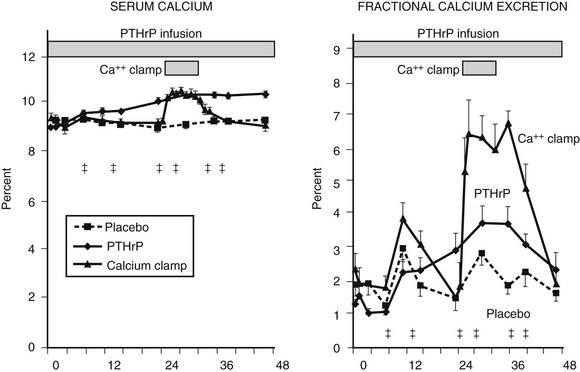
FIGURE 8-8 Fractional calcium excretion in normal subjects infused with calcium chloride or parathyroid hormone–related protein (PTHrP). Left, Serum calcium concentrations are shown in three groups of normal healthy volunteers infused with vehicle alone, with PTHrP to achieve a serum calcium of 10.3 mg/dL, or with calcium chloride (calcium clamp) to achieve an identical serum calcium of 10.3 mg/dL. Right, Response in fractional calcium excretion in the three groups. In the control (vehicle) group, fractional calcium excretion remains stable at approximately 2%. In the calcium clamp group, the fractional calcium excretion increases dramatically to approximately 6.5%, reflecting a combination of the increase in the filtered load of calcium from the calcium infusion and the suppression of endogenous PTH secretion. In the group receiving PTHrP, despite identical serum calcium concentration (and therefore an identical filtered load of calcium) to the calcium clamp group, fractional calcium excretion remains low, indicating that PTHrP is a potent stimulator of distal tubular calcium reabsorption. Studies comparing PTH with PTHrP indicate that the two peptides have equivalent effects in human renal calcium handling.62–64 (Data from Syed MA, Horwitz MJ, Tedesco MB, et al: Parathyroid hormone-related protein [1-36] stimulates renal tubular calcium reabsorption in normal human volunteers: implications for the pathogenesis of humoral hypercalcemia of malignancy, J Clin Endocrinol Metab 86:1525–1531, 2001.)
Finally, similar to PTH, PTHrP inhibits proximal tubular phosphorus reabsorption.60–64 Thus, in HHM, the serum phosphorus concentration is characteristically reduced11,12 (see Fig. 8-7) as long as renal function remains normal, a phenomenon that is reflected in a reduction in the TmP/GFR11,12 (see Fig. 8-7).
Bone radionuclide scans characteristically display a complete absence of skeletal metastases or the presence of only a few skeletal metastases,2–12,17–22 which is a reflection of the primarily humoral nature of the syndrome. It is important to note that the syndrome reverses with successful eradication of the tumor in question, thus underscoring the humoral nature of the syndrome.2–12,17,18 Unfortunately, this outcome is not common.
Authentic Ectopic Hyperparathyroidism
As noted earlier, from the 1940s through the 1970s, ectopic hyperparathyroidism was believed to be a common cause of paraneoplastic hypercalcemia. By the 1980s, the term had fallen into disuse, and the existence of authentic ectopic hyperparathyroidism was in doubt; most authors believed that all cases of what had previously been considered to be ectopic hyperparathyroidism were in fact cases of HHM. With the advent of sensitive and specific immunoassays and molecular probes for PTH and PTHrP, the situation has changed. Now, approximately 15 case reports describe patients with diverse types of cancer (e.g., small cell carcinomas of the lung, squamous carcinoma of the lung, hepatoma, thymoma, neuroendocrine tumors, clear cell adenocarcinoma of the ovary, thyroid papillary carcinoma) who also displayed elevations in immunoreactive PTH in plasma with the use of modern two-site PTH immunoassays or expressed PTH mRNA in their tumor, or both.75–82
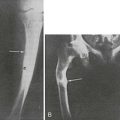
In one thoroughly studied case, reported by Nussbaum and associates,75 immunoreactive PTH values were found to be elevated in plasma by a sensitive and specific two-site PTH immunoradiometric assay. At surgery, a fivefold gradient of PTH was demonstrated across the ovarian tumor. PTH values decreased precipitately after tumor resection, and the serum calcium concentration normalized (Fig. 8-9). Neck exploration before the ovarian surgery had revealed four normal parathyroid glands, and resection of three-and-a-half parathyroid glands had no effect on the serum calcium concentration. PTH mRNA was abundantly present in the tumor, whereas PTHrP mRNA was undetectable, as was PTHrP in plasma.
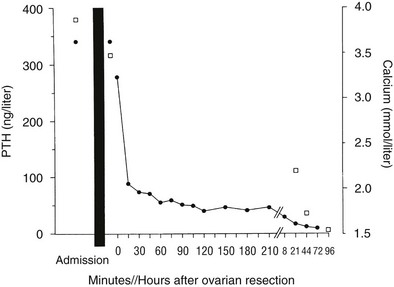
FIGURE 8-9 Serum calcium (open squares) and immunoreactive parathyroid hormone (PTH) (solid circles) in a patient with an ovarian carcinoma before and after oophorectomy, indicated by the black bar. The PTH immunoassay is that shown in Figure 8-2. Prior parathyroidectomy of 3.5 glands failed to influence her hypercalcemia or her elevated PTH concentration. (Data from Nussbaum SR, Gaz RD, Arnold A: Hypercalcemia and ectopic secretion of PTH by an ovarian carcinoma with rearrangement of the gene for PTH, N Engl J Med 323:1324–1328, 1990.)
The basis of PTH gene overexpression in this tumor was found to be twofold. First, a clonal rearrangement in the upstream region of one copy of the PTH gene in the ovarian carcinoma apparently served to abolish a silencer in this region of the gene or included a promoter region of a normal ovarian gene. Second, the PTH gene was amplified in the tumor. In contrast, in the report by Yoshimoto and colleagues76 describing ectopic hyperparathyroidism caused by a pulmonary small cell carcinoma, no such gene rearrangement or amplification events were identified, and the cause of the PTH expression was unexplained. In a recent report by VanHouten et al., ectopic production of PTH was documented from a pancreatic neuroendocrine tumor both in vivo and in vitro, and this was shown to be due not to a gene rearrangement or amplification, but to transcriptional activation of the PTH gene within the malignant neuroendocrine cells.82
Unusual Causes of Malignancy-Associated Hypercalcemia
It should be clear, however, that other humoral mediators undoubtedly exist in unusual patients. For example, a small number of patients with elevated PGE2 levels or clear responsiveness to prostaglandin synthesis inhibitors or both have been described,56,83 which suggests that in rare instances, PGE2 may act as a humoral, systemic agent.
In addition, more than 40 patients with a variety of types of lymphoma have been described in whom hypercalcemia occurred in the absence of bone metastases and with reduced urinary cAMP or NcAMP excretion but elevated circulating levels of 1,25(OH)2D84–86 (Fig. 8-10). These observations suggest that tumor-derived 1,25(OH)2D may have induced intestinal hyperabsorption of calcium or may have led to osteoclastic stimulation, or both. More recently, in patients harboring ovarian dysgerminomas, several groups have reported production and systemic elevations of 1,25(OH)2D leading to hypercalcemia.87,88
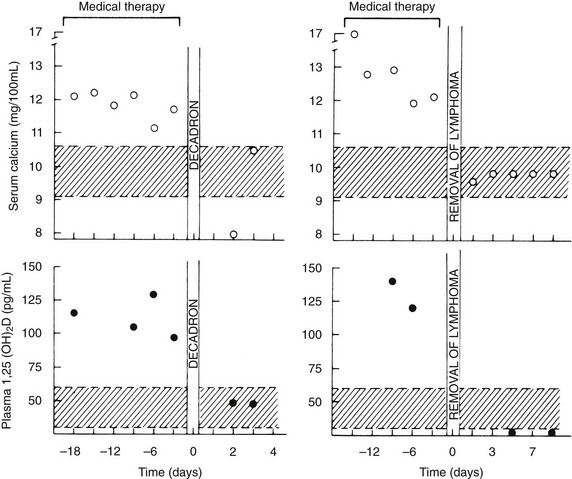
FIGURE 8-10 Serum calcium and plasma 1,25-dihydroxyvitamin (1,25[OH]2D) concentrations in patients with the 1,25(OH)2D-induced lymphoma syndrome before and after therapy. The patient shown in the two left panels was treated with dexamethasone (decadron) for systemic lymphoma. The patient in the two right panels had a splenectomy for a solitary splenic lymphoma. Note the decrease in plasma 1,25(OH)2D concentration after therapy in both patients and the normalization of serum calcium. Also compare the plasma 1,25(OH)2D concentrations in these patients versus those in patients with humoral hypercalcemia of malignancy and local osteolytic hypercalcemia shown in Figure 8-4. (Data from Rosenthal ND, Insogna KL, Godsall JW, et al: Elevations in circulating 1,25-dihydroxyvitamin D in three patients with lymphoma-associated hypercalcemia, J Clin Endocrinol Metab 60:29–33, 1985.)
Further, a patient with an ovarian dysgerminoma, no skeletal involvement, hypercalcemia in the presence of an elevated renal phosphorus threshold, and reduced levels of NcAMP and 1,25(OH)2D has been described.89 All these abnormalities promptly normalized after eradication of the patient’s tumor, which suggests that the tumor produced a humoral agent distinct from PTH, PTHrP, or 1,25(OH)2D.
Finally, Bringhurst and others90,91 described patients with hypercalcemia in the setting of metastatic malignant melanoma and bladder carcinoma. Excision of the tumor reversed the patients’ hypercalcemia, thus indicating a humoral mechanism. In culture, the tumors were shown to produce a bone-resorbing protein devoid of adenylate cyclase–stimulating activity or PTH-like bioactivity. In the aggregate, these examples collectively indicate that humoral forms of MAHC can occur through the production of factors other than 1,25(OH)2D, PTHrP, or PTH; however, these examples appear to be rare.
Treatment of Hypercalcemia
This section summarizes treatment of hypercalcemia of any cause but with particular reference to that due to cancer. The treatment of hypercalcemia caused by cancer has received complete attention in several recent reviews.92–94 As was noted earlier, the mean life expectancy in patients following a diagnosis of MAHC is 30 days.14 The most effective long-term therapy for MAHC is successful antitumor therapy. This point should be borne in mind and acted on early in the management of any given patient. Therapies aimed at treatment of hypercalcemia in patients with MAHC per se are rarely effective over the long term. Hence, long-term control of hypercalcemia depends on tumor eradication or debulking through surgery, chemotherapy, or radiotherapy. It is therefore essential and urgent that a plan for chemotherapy, surgery, or radiotherapy be developed and acted upon very early on in the course of treatment for MAHC. Said another way, therapy aimed at correcting hypercalcemia is largely short term and palliative: it should be used only to temporize while a more concrete and effective long-term treatment plan is formulated.
Rehydration
Thus, step one in treating hypercalcemia is to initiate a rehydration program.92–96 This is done after careful review of the patient’s clinical hydration status (lack of edema, neck vein distention, reduced skin turgor, and clinical and laboratory measures that might point to the presence of congestive heart failure or renal failure that might prevent aggressive rehydration). If no contraindications exist, intravenous hydration can be initiated with normal saline at a rate of between 200 and 1000 mL/hr, depending on the degree of dehydration and hypercalcemia and the ability to monitor the patient for congestive heart failure or other clinical syndromes of volume overload (with central venous pressure or pulmonary capillary wedge pressures as needed).
Diuresis
Administration of intravenous saline will initiate or enhance calciuresis.92–96 This is a result, in part, of an increase in glomerular blood flow and thus an increase in the filtered load of calcium. In addition, saline infusion inhibits calcium reabsorption in the proximal convoluted tubule. Renal calcium excretion can be further enhanced by the addition of a loop diuretic such as furosemide or ethacrynic acid, both of which inhibit calcium reabsorption in the thick ascending loop of Henle. However, because loop diuretics are also natriuretics and diuretics, they will exacerbate dehydration if they are initiated prematurely. Thus, loop diuretics should be initiated only after clinical evidence of adequate rehydration has accrued. Typically, therapy is initiated with furosemide at a dose of 20 mg IV every 2 to 12 hours, depending on the clinical circumstances, with careful monitoring of clinical volume status fluid balance, as well as potassium, magnesium, and electrolyte balance.
Antiresorptive Drugs
The Bisphosphonates
Bisphosphonates are structural analogues of pyrophosphate in which the two phosphate groups are joined by a carbon atom rather than an oxygen atom97–101 (Fig. 8-11). The P-C-P bond is thus analogous to the P-O-P in pyrophosphate, a natural metabolite that can inhibit calcification but is rapidly metabolized. Because the bisphosphonates are resistant to cleavage by pyrophosphatases, they are stable in the body. The bisphosphonates were developed in the search for more stable bone-seeking compounds that could imitate pyrophosphate action.97–101 The P-C-P bond is key for bone uptake; a hydroxyl group in the R1 position (see Fig. 8-11) increases affinity for hydroxyapatite in bone. It is now established that the R2 position is key to potency among the various bisphosphonates that have been developed. Once absorbed onto bone surface by the basic bisphosphonate pharmacophore, the nature of the substituent at the R2 position determines antiosteoclast potency and inhibition of bone resorption (Fig. 8-12). Substituents that contain nitrogen generally are more potent, with the most potent being those with a nitrogen within a heterocyclic ring (see Fig. 8-12).
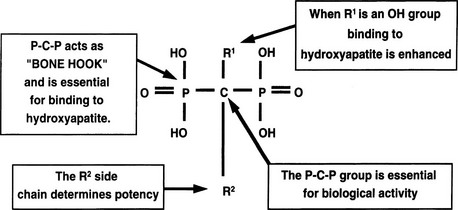
FIGURE 8-11 Structure of a bone-active bisphosphonate to show functional domains. (From Russell RG, Rogers MJ: Bisphosphonates: From the laboratory to the clinic and back again, Bone 25:97–106, 1999.)
Bisphosphonates are concentrated in areas of high bone turnover, are taken up by osteoclasts, and are potent inhibitors of bone destruction.97–101 Since their introduction almost 40 years ago, the precise cellular and biochemical mechanisms whereby the drugs act have been clarified, and successive generations of more potent compounds have been introduced.97–101 The bisphosphonate cellular target is principally the osteoclast, which seems selectively to take up the compounds during the process of bone resorption. The cellular actions are probably multiple, with evidence of impaired differentiation of osteoclast precursors, reduced osteoclast function, and accelerated osteoclast apoptosis, the net result being reduction in bone resorption. Studies in vitro have established two distinct cellular pathways by which bisphosphonates act on osteoclasts. Nitrogen-containing bisphosphonates interfere with enzymes in the mevalonate pathway that catalyze the formation of geranyl diphosphate and farnesyl diphosphate, which are key to prenylation of signaling proteins, especially those that accelerate GTP hydrolysis (GTPases). Without normal prenylation, these small proteins cannot attach to the osteoclast cell membrane, thus interfering with osteoclast function. Impairment of protein prenylation may also promote osteoclast apoptosis. Farnesyl or geranylgeranyl pyrophosphate added in vitro can reverse the blockade caused by the nitrogen-containing bisphosphonates.
Although there has been speculation about a cellular receptor for bisphosphonate, more recent evidence favors a still unidentified specific cellular uptake channel.102 In a series of experiments using osteoclasts and a related macrophage cell line, competition between several bisphosphonates for cellular uptake and biological effects was analyzed with varying doses of the two compounds administered to test for synergy at suboptimal doses of either, because the two bisphosphonates act by distinctive mechanisms. The overall results were interpreted as demonstrating competition for uptake through a common transport channel.102
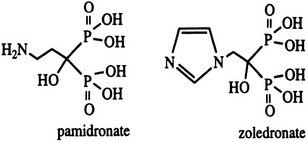
At present, the two bisphosphonates most widely used for treatment of MAHC hypercalcemia are pamidronate (aminohydroxy-propylidene bisphosphonate) and zoledronate (zolendronic acid) (see Fig. 8-11).97–101,103,104 Zoledronate is more potent on a milligram or molar basis and can be given more rapidly, thus facilitating inpatient and outpatient delivery. However, pamidronate is less expensive and displays similar efficacy.103 In general, bisphosphonates of all types are absorbed from the gastrointestinal tract with extremely poor efficiency (less than 1% to 2%).97–101 For this reason, zoledronate and pamidronate are administered intravenously.
Pamidronate, an aminobisphosphonate, is usually administered as a single intravenous infusion in a dosage of 60 to 90 mg over a 2-hour period intravenously, depending on the severity of the hypercalcemia.97–101,103,105 Treatment must be repeated when hypercalcemia recurs, but efficacy may wane with repeated dosing.32,35,36 The serum calcium normalizes in most patients with MAHC.97–101,103,105 It is critical to remember that this response typically requires 3 to 7 days.97–101,103,105 This means that once an initial dose is administered, a second dose generally should not be administered earlier than 3 to 4 days after the initial dose. The likelihood that the serum calcium level will normalize is related to both the dose of pamidronate administered and the severity of the initial hypercalcemia.
The most common side effect of intravenous pamidronate therapy is fever, which occurs in approximately 20% of patients and may be related to release of cytokines from osteoclasts, monocytes, and macrophages.97–101,103,105,106 Fever is generally mild (<2°C) and typically occurs only once in a patient’s lifetime, even if treatment is discontinued and restarted later. Other side effects of pamidronate include hypocalcemia (rare in patients with MAHC), hypophosphatemia, hypomagnesemia, and possibly reversible hepatotoxicity.97–101,103,105 A recently reported complication of pamidronate therapy is glomerulosclerosis with renal insufficiency and nephrotic syndrome, which occurred in seven patients with cancer treated at higher than usual doses (180 or 360 mg/mo rather than the usual 90 mg/mo).107 The interpretation was complicated by the use of other potent anticancer drugs, although high-dose pamidronate was the only common factor.107
Zoledronate generally is administered at a dose of 4 mg intravenously over a 15-minute period.97–101,103,104 It may be slightly more effective than pamidronate for the treatment of hypercalcemia.103 A randomized, double-blind study comparing zoledronic acid (at the recommended doses of 4 mg/5 minute infusion as well as 8 mg/5 minute infusion) with pamidronate (at the recommended 90 mg/2 hour infusion) in 250 patients with MAHC.103 Both doses of zoledronic acid normalized serum calcium levels slightly faster with more sustained responses than pamidronate (32 or 43 days for 4 vs. 8 mg of zoledronic acid, respectively, as compared with 18 days for pamidronate). On the other hand, although these differences were statistically significant, they were quantitatively small, leaving one to wonder whether the added expense of zoledronate as compared with pamidronate is merited. Zoledronic acid has an adverse event profile similar to pamidronate with pyrexia and occasional reports of renal toxicity presumed to be secondary to precipitation of calcium bisphosphonate complexes.108
Mithramycin
Mithramycin (also called plicamycin) is a chemotherapeutic agent that has been used in the treatment of neuroendocrine tumors and testicular tumors.109,110 It was found serendipitously to cause hypocalcemia as a result of its action to induce death in osteoclasts. Thus, for several decades it was used as a front-line treatment for MAHC, parathyroid carcinoma, and Paget’s disease. However, its use, particularly in large and repeated doses, was associated with azotemia, hepatitis, and coagulation defects. It has been eclipsed in clinical practice by the intravenous bisphosphonates, because of their efficacy and safety. On the other hand, it is a particularly effective drug, and its use should be considered in special circumstances.
Calcitonin
Calcitonin is a naturally occurring peptide made by the parafollicular cells within the thyroid gland in humans; it inhibits bone resorption and decreases renal tubular calcium reabsorption.111–115 Calcitonin has a rapid onset of action, but its calcium-lowering effect is limited because of rapid tachyphylaxis.111–115 For example, one early study reported that calcitonin lowered serum calcium levels by an average of 2 mg/dL with a peak effect in 6 hours.114 In another report, calcitonin lowered serum calcium levels in 75% of subjects within 2 hours, half of whom became normocalcemic in several hours.115 The calcium-lowering effect of calcitonin is clearly short-lived, however, with an increase in serum calcium levels often occurring within 48 to 96 hours despite continued calcitonin therapy.111–115 Because of this rapid tachyphylaxis, calcitonin has limited utility beyond 1 to 2 days as a single agent for the treatment of hypercalcemia, and it should no longer be widely used. It may be useful in rare patients with severe hypercalcemia in whom the clinical situation cannot wait an extra 24 hours for the hypocalcemic effects of pamidronate or zoledronate. The usual dosage of salmon calcitonin is 4 U/kg every 12 hours, although doses as high as 8 U/kg have been given every 6 hours.111–115 The most common side effects of calcitonin therapy are nausea and vomiting, which occur in approximately 10% to 15% of patients.111–115 Other side effects include flushing and abdominal cramps.111–115
Gallium Nitrate
Gallium nitrate inhibits the release of 45Ca from fetal rat bones in response to PTH and lymphokines and inhibits the ability of cultured osteoclasts to resorb cortical bone in vitro. It interacts with both hydroxyapatite and the cellular components of bone and accumulates preferentially in areas of active bone formation. Gallium nitrate has been shown to reduce the serum calcium in patients with MAHC,116 but it must be given continuously intravenously to achieve these effects. Thus, its use is limited for practical reasons. Given their efficacy and superior convenience of use, bisphosphonates remain the standard therapy in most patients.
Glucocorticoids
Several decades ago, before the development of mithramycin, calcitonin, and the intravenous bisphosphonates, glucocorticoids were a therapeutic agent of choice for patients with MAHC and sometimes were used in vitamin D intoxication.117,118 In large doses, glucocorticoids increase renal calcium clearance and urinary calcium excretion, directly decrease intestinal absorption of calcium, and inhibit tumor activity of 1-alpha hydroxylase, the enzyme that leads to production of 1,25(OH)2D. They also may inhibit production of bone-resorbing cytokines by tumors. In some tumors, particularly hematologic tumors, glucocorticoids have antitumor effects as well. These effects of glucocorticoids on the tumor, the kidney, and the intestine lower serum calcium levels. Thus, they still have a place in the management of occasional patients with MAHC due to lymphomas, leukemias, and/or plasma cell dyscrasias, particularly if they are included as part of an antitumor chemotherapeutic regimen or protocol. Outside of this context, however, they are rarely used.
Dialysis
Dialysis may seem a heroic therapeutic measure in patients with advanced cancer, and in most cases, it is not appropriate clinically, economically, or perhaps ethically. On the other hand, there are exceptions to this rule, such as a recently diagnosed patient with estrogen receptor–positive, epidermal growth factor receptor–positive breast cancer who has not yet undergone chemotherapy, but who presents with hypercalcemia and in renal failure because of ureteral obstruction or nephrotoxic antibiotic use. Another example might be a patient with multiple myeloma who presents with light chain nephropathy and hypercalcemia. In these rare patients, the prognostic outlook is positive, but standard measures to lower calcium (hydration, loop diuretics, and IV bisphosphonates) may be ineffective or contraindicated because of the renal failure. Hemodialysis or peritoneal dialysis against a low calcium bath can lower serum calcium levels rapidly and dramatically in such patients,119–121 and this can buy time for renal function to recover while an effective chemotherapeutic regimen is initiated. Dialysis membranes are quite permeable to calcium, and as much as 600 mg of calcium can be cleared per hour with hemodialysis. In a retrospective analysis of 33 patients with severe hypercalcemia of varying causes, calcium-free hemodialysis reduced serum calcium levels by an average of 6.9 mg/dL (1.71 mmol/L), but a partial rebound occurred in most patients within the first 24 hours.119 Transient hypotension was noted in more than one third of patients but resolved with fluid replacement.120 In another study of six patients with hypercalcemia of malignancy and renal failure, calcium-free hemodialysis reduced serum calcium levels without any adverse effects.121 Peritoneal dialysis can remove 500 to 2000 mg of calcium and can lower the serum calcium level by 3 to 12 mg/dL in 24 to 48 hours if a calcium-free dialysis solution is used.122–124
Phosphorus Replacement
On the other hand, overzealous phosphate replacement can be hazardous in the extreme in patients with hypercalcemia, because it may be associated with abrupt and severe hypocalcemia and seizures, as well as with acute onset of renal failure due to nephrocalcinosis.125 Thus, cautious phosphate replacement may be reasonable in subjects with serum phosphorus concentrations below 3.0 mg/dL. The authors’ practice is to use oral phosphorus replacement, in an initial dose in the range of 250 mg four times per day.93 Careful attention to both serum phosphorus and creatinine is required, as is withholding phosphorus replacement if the serum phosphorus rises above 3.0 mg/dL, or the serum creatinine rises above baseline values. In very rare patients who cannot receive oral medications but who have severe hypophosphatemia, it may be necessary to administer intravenous phosphorus as potassium phosphate. Small doses of phosphorus (e.g., 600 mg of phosphorus slowly over 24 hours) should be given, and extreme care should be taken to avoid hyperphosphatemia and increases in serum creatinine.
Future Therapies
The therapeutic armamentarium for treating hypercalcemia of malignancy is currently so effective and inexpensive that there may be little need for additional treatment modalities. On the other hand, at least a few potential agents may become available over the coming several years. These might include more potent or effective bisphosphonates, monoclonal antibodies directed against PTHrP,126 and monoclonal antibodies directed against cellular pathways that lead to recruitment, differentiation, and activation of osteoclasts, such as the RANK-RANK-ligand system, or recombinant osteoprotegerin,127 the natural decoy receptor for RANK-ligand. Among these, the most advanced in clinical trials is a monoclonal antibody, named denosumab, which binds to and inactivates RANK-ligand.128–130 Denosumab has been shown to be effective in neutralizing RANK-ligand in vivo in humans and thereby markedly blocking osteoclastic bone resorption in patients with osteoporosis. Through this antiresorptive mechanism, denosumab markedly reduces fractures and increases bone mineral density in postmenopausal women with osteoporosis, and in animal studies, reverses HHM.128–130 Because denosumab is at least as effective as bisphosphonates in osteoporosis, and because it is effective in animal models of MAHC,130 it would seem to follow that denosumab may be similarly or more effective in treating hypercalcemia in patients with cancer. Time will tell whether this is true.
References
1. Zondek, H, Petow, H, Siebert, W. Die bedeutung der calciumbestimmung im blute fur die diagnose der niereninsuffzientz. Z Klin Med. 1923;99:129–138.
2. Gutman, AB, Tyson, TL, Gutman, EB. Serum calcium, inorganic phosphorus and phosphatase activity in hyperparathyroidism, Paget’s disease, multiple myeloma and neoplastic disease of the bones. Arch Intern Med. 1936;57:379–413.
3. Case records of the Massachusetts General Hospital (case 27461). N Engl J Med. 1941;225:789–791.
4. Plimpton, CH, Gelhorn, A. Hypercalcemia in malignant disease without evidence of bone destruction. Am J Med. 1956;21:750–759.
5. Connors, TB, Howard, JF. The etiology of hypercalcemia associated with lung carcinoma. J Clin Invest. 1956;35:697–698.
6. Lafferty, FW. Pseudohyperparathyroidism. Medicine (Baltimore). 1966;45:247–260.
7. Rodman, JS, Sherwood, LM, Disorders of mineral metabolism in malignancy. Metabolic Bone Disease. Avioli, LV, Krane, SM, eds. Metabolic Bone Disease. New York: Academic Press; 1978;vol 2:555–631.
8. Myers, WPL. Hypercalcemia in neoplastic disease. Arch Surg. 1960;80:308.
9. Besarab, A, Caro, JF. Mechanisms of hypercalcemia in malignancy. Cancer. 1978;41:2276–2285.
10. Powell, D, Singer, FR, Murray, TM, et al. Nonparathyroid humoral hypercalcemia in patients with neoplastic diseases. N Engl J Med. 1973;289:176–181.
11. Stewart, AF, Horst, R, Deftos, LJ, et al. Biochemical evaluation of patients with cancer-associated hypercalcemia: evidence for humoral and non-humoral groups. N Engl J Med. 1980;303:1377–1383.
12. Godsall, JW, Burtis, WJ, Insogna, KL, et al. Nephrogenous cyclic AMP, adenylate cyclase–stimulating activity, and the humoral hypercalcemia of malignancy. Recent Prog Horm Res. 1986;40:705–750.
13. Stewart, AF, Vignery, A, Silvergate, A, et al. Quantitative bone histomorphometry in humoral hypercalcemia of malignancy: uncoupling of bone cell activity. J Clin Metab. 1982;55:219–227.
14. Ralson, SH, Gallagher, SJ, Patel, U, et al. Cancer-associated hypercalcemia: morbidity and mortality. Ann Intern Med. 1990;112:499–504.
15. Nierenberg, DW, Ransil, BJ. Q-aTc interval as a clinical indicator of hypercalcemia. Am J Cardiol. 1979;44:243–248.
16. Davis, TME, Singh, B, Choo, KE, et al. Dynamic assessment of the electrocardiographic QT intervals during citrate infusion in healthy volunteers. Br Heart J. 1995;75:523–526.
17. Omenn, GS, Roth, SI, Baker, WH. Hyperparathyroidism associated with malignant tumors of non-parathyroid origin. Cancer. 1969;24:1004–1012.
18. Skrabanek, P, McPartlin, J, Powell, DM. Tumor hypercalcemia and ectopic hyperparathyroidism. Medicine (Baltimore). 1980;59:262–282.
19. Burtis, WJ, Brady, TG, Orloff, JJ, et al. Immunochemical characterization of circulating parathyroid hormone–related protein in patients with humoral hypercalcemia of malignancy. N Engl J Med. 1990;322:1106–1112.
20. Budayr, AR, Nissenson, RA, Klein, RF, et al. Increased serum levels of PTH-like protein in malignancy-associated hypercalcemia. Ann Intern Med. 1989;111:807–810.
21. Blind, E, Raue, F, Gotzman, J, et al. Circulating levels of mid-regional parathyroid hormone-related protein in hypercalcemia of malignancy. Clin Endocrinol. 1992;37:290–297.
22. Pandian, MR, Morgan, CH, Carlton, E, et al. Modified immunoradiometric assay of parathyroid hormone-related protein: clinical application in the differential diagnosis of hypercalcemia. Clin Chem. 1992;38:282–288.
23. Bender, RA, Hansen, H. Hypercalcemia in bronchogenic carcinoma: a prospective study of 200 patients. Ann Intern Med. 1974;80:205–208.
24. Mahadevia, PS, Pamaswamy, A, Greenwald, ES, et al. Hypercalcemia in prostatic carcinoma. Arch Intern Med. 1983;143:1339–1342.
25. Ralston, S, Fogelman, I, Gardner, MD, et al. Hypercalcemia and metastatic bone disease: is there a link? Lancet. 1982;2:903–905.
26. Mundy, GR, Raisz, LG, Cooper, RA, et al. Evidence for the secretion of an osteoclast stimulating factor in myeloma. N Engl J Med. 1974;290:1041–1046.
27. Mundy, GR, Luben, RA, Raisz, LG, et al. Bone-resorbing activity in supernatants from lymphoid cell lines. N Engl J Med. 1974;290:867–871.
28. Callander, NS, Roodman, GD. Myeloma bone disease. Semin Hematol. 2001;38:276–285.
29. Horwitz, MJ, Stewart, AF. Malignancy-Associated Hypercalcemia. In: Favus M, ed. The American Society for Bone and Mineral Research Primer on Metabolic Bone Diseases and Disorders of Mineral Metabolism. ed 6. Washington D.C.: American Society for Bone and Mineral Research; 2006:195–199.
30. Dewhirst, FE, Stashenko, PP, Mole, JE, et al. Purification and partial sequence of human osteoclast activating factor: identity with human interleukin lB. J Immunol. 1985;135:2562.
31. Roodman, GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–1664.
32. Ross, FP. Osteoclast Biology and Bone Resorption. In: Favus M, ed. The American Society for Bone and Mineral Research Primer on Metabolic Bone Diseases and Disorders of Mineral Metabolism. ed 6. Washington D.C.: American Society for Bone and Mineral Research; 2006:30–35.
33. Tian, E, Zhan, F, Walker, R, et al. The role of the Wnt/beta-catenin signaling antagonist DKK-1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349:2483–2494.
34. Erlich, LA, Chung, HY, Ghobrial, I, et al. IL-3 is apotent inhibitor of osteoblast differentiation in multiple myeloma. Blood. 2005;106:1407–1414.
35. Galasko, CSB. Mechanisms of bone destruction in the development of skeletal metastases. Nature. 1976;263:507.
36. Guise, TA, Yin, JJ, Taylor, SD, et al. Evidence for a causal role of parathyroid hormone–related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest. 1996;98:1544.
37. Southby, J, Kissin, MW, Danks, JA, et al. Immunohistochemical localization of PTHrP in human breast cancer. Cancer Res. 1990;50:7710–7716.
38. Vargas, SJ, Gillespie, MT, Powell, GJ, et al. Localization of PTHrP mRNA expression in breast cancer and metastatic lesions by in situ hybridization. J Bone Miner Res. 1992;7:971–979.
39. Sztern, M, Barkan, A, Rakowsky, E, et al. Hypercalcemia in carcinoma of the breast without evidence of bone destruction. Cancer. 1981;48:2383–2385.
40. Legha, SS, Powell, K, Buzdar, AU, et al. Tamoxifen-induced hypercalcemia in breast cancer. Cancer. 1981;47:2803–2806.
41. Valentin-Opran, A, Eilon, G, Saez, S, et al. Estrogens stimulate release of bone-resorbing activity in cultured human breast cancer cells. J Clin Invest. 1985;72:726–731.
42. Nakayama, K, Fukumoto, S, Takeda, S, et al. Differences in bone and vitamin D metabolism between primary hyperparathyroidism and malignancy-associated hypercalcemia. J Clin Endocrinol Metab. 1996;81:607–611.
43. Grill, V, Ho, P, Body, JJ, et al. PTH-related protein: elevated levels in both humoral hypercalcemia of malignancy and hypercalcemia complicating metastatic breast cancer. J Clin Endocrinol. 1991;73:1309–1315.
44. Isales, C, Carcangiu, ML, Stewart, AF. Hypercalcemia in breast cancer: a reassessment of the mechanism. Am J Med. 1987;82:1143–1147.
45. Fukumoto, S, Matsumoto, T, Ikeda, T, et al. Clinical evaluation of adult T-cell lymphoma. Arch Intern Med. 1988;148:921–925.
46. Wantabe, T, Yamaguchi, K, Tatasuki, K, et al. Constitutive expression of PTHrP gene in HTLV-1 carriers and adult T-cell leukemia patients that can be trans-activated by HTLV-1 tax gene. J Exp Med. 1990;172:759–765.
47. Mune, T, Katakami, H, Kato, Y, et al. Production and secretion of parathyroid hormone-related protein in pheochromocytoma: participation of an α-adrenergic mechanism. J Clin Endocrinol Metab. 1993;76:757–762.
48. Stewart, AF, Hoecker, J, Segre, GV, et al. Hypercalcemia in pheochromocytoma: evidence for a novel mechanism. Ann Intern Med. 1985;102:776–779.
49. Mao, C, Carter, P, Schaefer, P, et al. Malignant islet cell tumor associated with hypercalcemia. Surgery. 1997;117:37–40.
50. Wu, T-J, Lin, C-L, Taylor, RL, et al. Increased parathyroid hormone-related peptide in patients with hypercalcemia associated with islet cell carcinoma. Mayo Clin Proc. 1997;72:1111–1115.
51. Dagdelen, S, Kalan, I, Gurlek, A. Humoral hypercalcemia of benignancy secondary to parathyroid hormone-related protein secreting uterine leiomyoma. Am J Med Sci. 2008;335:407–408.
52. Gordan, GS, Fitzpatrick, ME, Lubich, WP. Identification of osteolytic sterols in human breast cancer. Trans Assoc Am Physicians. 1967;80:183–189.
53. Haddad, JG, Couranz, SJ, Avioli, LV. Circulating phytosterols in normal females, lactating mothers, and breast cancer patients. J Clin Endocrinol Metab. 1970;30:174–180.
54. Klein, DC, Raisz, LG. Prostaglandins: stimulation of bone resorption in tissue culture. Endocrinology. 1970;86:1436–1440.
55. Tashjian, AH. Role of prostaglandins in the production of hypercalcemia by tumors. Cancer Res. 1978;38:4138–4141.
56. Seyberth, WJ, Segre, GV, Morgan, JL, et al. Prostaglandins as mediators of hypercalcemia associated with certain types of cancer. N Engl J Med. 1975;293:1278–1283.
57. Brenner, D, Harvey, HA, Lipton, A, et al. A study of prostaglandin E2 parathormone, and response to indomethacin in patients with hypercalcemia and malignancy. Cancer. 1982;49:556–561.
58. Benson, RC, Riggs, BL, Pickard, BM, et al. Radioimmunoassay of parathyroid hormone in hypercalcemic patients with malignant disease. Am J Med. 1974;56:821–826.
59. Riggs, BL, Arnaud, CD, Reynolds, JC, et al. Immunologic differentiation of primary hyperparathyroidism from hyperparathyroidism due to nonparathyroid cancer. J Clin Invest. 1971;50:2079–2083.
60. Everhart-Caye, M, Inzucchi, SE, Guinness-Henry, J, et al. Parathyroid hormone-related protein(1–36) is equipotent with parathyroid hormone(1–34) in humans. J Clin Endocrinol Metab. 1996;81:199.
61. Henry, JG, Mitnick, MA, Dann, PR, et al. Parathyroid hormone-related protein(1–36) is biologically active when administered subcutaneously to humans. J Clin Endocrinol Metab. 1997;82:900–906.
62. Syed, MA, Horwitz, MJ, Tedesco, MB, et al. Parathyroid hormone-related protein (1–36) stimulates renal tubular calcium reabsorption in normal human volunteers: implications for the pathogenesis of humoral hypercalcemia of malignancy. J Clin Endocrinol Metab. 2001;86:1525–1531.
63. Horwitz, MJ, Tedesco, MB, Sereika, S, et al. Direct comparison of sustained infusion of hPTHrP(1-36) versus hPTH(1-34) on serum calcium, plasma 1,25(OH)2 vitamin D concentrations and fractional calcium excretion in healthy human volunteers. J Clin Endocrinol Metab. 2003;88:1603–1609.
64. Horwitz, MJ, Tedesco, MB, Sereika, SM, et al. Continuous infusion of parathyroid hormone versus parathyroid hormone-related protein in humans: discordant effects on 1,25(OH)2 vitamin D and prolonged suppression of bone formation. J Bone Miner Res. 2005;20:1792–1803.
65. Stewart, A, Mangin, M, Wu, T, et al. A synthetic human parathyroid hormone-like protein stimulates bone resorption and causes hypercalcemia in rats. J Clin Invest. 1988;81:596–600.
66. Henderson, J, Bernier, S, D’Amour, P, et al. Effects of passive immunization against PTH-like peptide in hypercalcemic tumor-bearing rats and normocalcemic controls. Endocrinology. 1990;127:1310–1316.
67. Kukreja, SC, Shevrin, DH, Wimbiscus, SA, et al. Antibodies to PTH-related protein lower serum calcium in athymic mouse models of malignancy associated hypercalcemia due to human tumors. J Clin Invest. 1988;82:1798–1802.
68. Philbrick, WM, Wysolmerski, JJ, Galbraith, S, et al. Defining the physiologic roles of parathyroid hormone-related protein in normal physiology. Physiol Rev. 1996;76:127–173.
69. Strewler, GJ. The physiology of parathyroid hormone-related protein. N Engl J Med. 2000;342:177–185.
70. Nussbaum, SR, Zahradnik, RJ, Lavigne, JR, et al. Highly sensitive two-site immunoradiometric assay of parathyrin and its clinical utility in evaluating patients with hypercalcemia. Clin Chem. 1987;33:1364–1367.
71. Dean, T, Vilardaga, J-P, Potts, JT, et al. Altered selectivity of parathyroid hormone (PTH) and PTH-related protein (PTHrP) for distinct conformations of the PTH/PTHrP receptor. Mol Endocrinol. 2008;22:156–166.
72. Coombes, RC, Ward, MK, Greenberg, PB, et al. Calcium metabolism in cancer. Studies using calcium isotopes and immunoassay for parathyroid hormone and calcitonin. Cancer. 1976;38:2111–2120.
73. Ralston, SH, Fogelman, I, Gardner, MD, et al. Hypercalcemia of malignancy: evidence for a non-parathyroid humoral agent with an effect on renal tubular handling of calcium. Clin Sci. 1984;66:187–194.
74. Bonjour, J-P, Phillipe, J, Guelpa, G, et al. Bone and renal components in hypercalcemia of malignancy and response to a single infusion of clodronate. Bone. 1988;9:123–130.
75. Nussbaum, SR, Gaz, RD, Arnold, A. Hypercalcemia and ectopic secretion of PTH by an ovarian carcinoma with rearrangement of the gene for PTH. N Engl J Med. 1990;323:1324–1328.
76. Yoshimoto, K, Yamasaki, R, Sakai, H, et al. Ectopic production of PTH by small cell lung cancer in a patient with hypercalcemia. J Clin Endocrinol Metab. 1989;68:976–981.
77. Strewler, GJ, Budayr, AA, Clark, OH, et al. Production of parathyroid hormone by a malignant nonparathyroid tumor in a hypercalcemic patient. J Clin Endocrinol Metab. 1993;76:1373–1375.
78. Iguchi, H, Miyagi, C, Tomita, K, et al. Hypercalcemia caused by ectopic production of parathyroid hormone in a patient with papillary adenocarcinoma of the thyroid gland. J Clin Endocrinol Metab. 1998;83:2653–2657.
79. Nielsen, PK, Rasmussen, AK, Feldt-Rasmussen, U, et al. Ectopic production of intact parathyroid hormone by a squamous cell lung carcinoma in vivo and in vitro. J Clin Endocrinol Metab. 1996;81:3793–3796.
80. Rizzoli, R, Pache, J-C, Didierjean, L, et al. A thymoma as a cause of true ectopic hyperparathyroidism. J Clin Endocrinol Metab. 1994;79:912–915.
81. Schmeltzer, HJ, Hesch, RD, Mayer, H. Parathyroid hormone and PTH mRNA in a human small cell lung cancer. Recent Results Cancer Res. 1985;99:88–93.
82. VanHouten, JN, Yu, NY, Rimm, D, et al. Hypercalcemia of malignancy due to ectopic transactivation of the parathyroid hormone gene. J Clin Endocrinol Metab. 2006;91:580–583.
83. Metz, SA, McRae, JR, Robertson, RP. Prostaglandins as mediators of paraneoplastic syndromes: review and update. Metabolism. 1981;30:299–316.
84. Rosenthal, N, Insogna, KL, Godsall, JW, et al. Elevations in circulating 1,25 dihydroxyvitamin D in three patients with lymphoma-associated hypercalcemia. J Clin Endocrinol Metab. 1985;60:29–33.
85. Breslau, NA, McGuire, JL, Zerwekh, JE, et al. Hypercalcemia associated with increased serum 1,25 dihydroxyvitamin D in three patients with lymphoma. Ann Intern Med. 1984;100:1.
86. Seymour, JF, Gagel, RF, Hagemeister, FB, et al. Calcitriol production in hypercalcemia and normocalcemia patients with non-Hodgkin lymphoma. Ann Intern Med. 1994;121:633–640.
87. Evans, KN, Taylor, H, Zehnder, D, et al. Increased expression of 25-hydroxyvitamin D-1-alpha-hydroxylase in dysgerminomas: a novel form of humoral hypercalcemia of malignancy. Am J Pathol. 2004;165:807–813.
88. Masahito, H, Hara, F, Tomishige, H, et al. 1,25-Dihydroxyvitamin D–mediated hypercalcemia in ovarian dysgerminoma. Pediatr Hematol Oncol. 2008;25:73–78.
89. Stewart, AF, Broadus, AE, Schwartz, PE, et al. Hypercalcemia in gynecologic neoplasms. Cancer. 1982;49:2389–2394.
90. Bringhurst, FR, Bierer, BE, Godeau, F, et al. Humoral hypercalcemia of malignancy. J Clin Invest. 1986;77:456–464.
91. Bringhurst, FR, Varner, V, Segre, GV. Cancer-associated hypercalcemia: characterization of a new bone-resorbing factor [Abstract]. Clin Res. 1982;30:386.
92. Bilezikian, JP. Management of acute hypercalcemia. N Engl J Med. 1992;326:1196–1203.
93. Stewart, AF. Hypercalcemia associated with cancer. N Engl J Med. 2005;352:373–379.
94. Horwitz, MJ, Hodak, S, Stewart, AF. Non-Parathyroid Hypercalcemia. In Rosen CJ, ed.: The American Society for Bone and Mineral Research Primer on Metabolic Bone Diseases and Disorders of Mineral Metabolism, ed 7, Washington D.C.: American Society for Bone and Mineral Research, 2008.
95. Suki, WN, Yium, JJ, Von Minden, M, et al. Acute treatment of hypercalcemia with furosemide. N Engl J Med. 1970;283:836–840.
96. Hosking, DJ, Cowley, A, Bucknall, CA. Rehydration in the treatment of severe hypercalcaemia. Q J Med. 1981;50:473–481.
97. Fleisch, H. Bisphosphonates: mechanism of action. Endocr Rev. 1998;19:80–100.
98. Russell, RG, Rogers, MJ. Bisphosphonates: from the laboratory to the clinic and back again. Bone. 1999;25:97–106.
99. Black, D, Rosen, CJ. Bisphosphonates for the Prevention and Treatment of Osteoporosis. In: Favus M, ed. The American Society for Bone and Mineral Research Primer on Metabolic Bone Diseases and Disorders of Mineral Metabolism. ed 6. Washington D.C.: American Society for Bone and Mineral Research; 2006:30–35.
100. Rodan, GA, Fleisch, HA. Bisphosphonates: mechanisms of action. J Clin Invest. 1996;97:2692–2696.
101. Cheer, SM, Noble, S. Zolendronic acid. Drugs. 2001;61:799–805.
102. Frith, JC, Rogers, MJ. Antagonistic effects of different classes of bisphosphonates in osteoclasts and macrophages in vitro. J Bone Miner Res. 2003;18:204–212.
103. Major, P, Lortholary, A, Hon, J, et al. Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: A pooled analysis of two randomized controlled clinical trials. J Clin Oncol. 2001;19:558–567.
104. Body, JJ, Lortholary, A, Romieu, G, et al. A dose-finding study of zoledronate in hypercalcemic cancer patients. J Bone Miner Res. 1999;14:1557–1561.
105. Nussbaum, S, Younger, J, VandePol, C, et al. Single dose intravenous therapy with pamidronate for the treatment of hypercalcemia of malignancy: comparison of 30-, 60-, and 90-mg dosages. Am J Med. 1993;95:297–304.
106. Adami, S, Bhalla, AK, Dorizzi, R, et al. The acute-phase response after bisphosphonate administration. Calcif Tissue Int. 1987;41:326–331.
107. Markowitz, GS, Appel, GB, Fine, PL, et al. Collapsing focal segmental glomerulosclerosis following treatment with high-dose pamidronate. J Am Soc Nephrol. 2001;12:1164–1172.
108. Rosen, LS, Gordon, D, Kaminski, M, et al. Long-term efficacy and safety of zoledonic acid compared with pamidronate disodium in the treatment of skeletal complications in patients with advanced multiple myeloma or breast carcinoma: a randomized, double-blind, multicenter, comparative trial. Cancer. 2003;98(8):1735–1744.
109. Perlia, CP, Gubisch, NJ, Wolter, J, et al. Mithramycin treatment of hypercalcemia. Cancer. 1970;25:389–394.
110. Kennedy, BJ. Metabolic and toxic effects of mithramycin during tumor therapy. Am J Med. 1970;49:494–503.
111. Hosking, DJ, Gilson, D. Comparison of the renal and skeletal actions of calcitonin in the treatment of severe hypercalcaemia of malignancy. Q J Med. 1984;53:359–368.
112. Cochran, M, Peacock, M, Sachs, G, et al. Renal effects of calcitonin. BMJ. 1970;1:135–137.
113. Silva, OL, Becker, KL. Salmon calcitonin in the treatment of hypercalcemia. Arch Intern Med. 1973;132:337–339.
114. Kammerman, S, Canfield, RE. Effect of porcine calcitonin on hypercalcemia in man. J Clin Endocrinol Metab. 1970;31:70–75.
115. Wisneski, LA, Croom, WP, Silva, OL, et al. Salmon calcitonin in hypercalcemia. Clin Pharmacol Ther. 1978;24:219–222.
116. Warrell, RP, Jr., Israel, R, Frisone, M, et al. Gallium nitrate for acute treatment of cancer-related hypercalcemia. A randomized, double-blind comparison to calcitonin. Ann Intern Med. 1988;108:669–674.
117. Fulmer, DH, Dimich, AB, Rothschild, EO, et al. Treatment of hypercalcemia, comparison of intravenously administered phosphate, sulfate, and hydrocortisone. Arch Intern Med. 1972;129:923–930.
118. Percival, RC, Yates, AJ, Gray, RE, et al. Role of glucocorticoids in management of malignant hypercalcaemia. BMJ. 1984;289:287.
119. Camus, C, Charasse, C, Jouannic-Montier, I, et al. Calcium free hemodialysis: Experience in the treatment of 33 patients with severe hypercalcemia. Intensive Care Med. 1996;22:116–121.
120. Koo, WS, Jeon, DS, Ahn, SJ, et al. Calcium-free hemodialysis for the management of hypercalcemia. Nephron. 1996;72:424–428.
121. Cardella, CJ, Birkin, BL, Rapoport, A. Role of dialysis in the treatment of severe hypercalcemia: report of two cases successfully treated with hemodialysis and review of the literature. Clin Nephrol. 1979;12:285–290.
122. Nolph, KD, Stoltz, M, Maher, JF. Calcium free peritoneal dialysis, treatment of vitamin D intoxication. Arch Intern Med. 1971;128:809–814.
123. Hamilton, JW, Lasrich, M, Hirszel, P. Peritoneal dialysis in the treatment of severe hypercalcemia. J Dial. 1980;4:129–138.
124. Miach, PJ, Dawborn, JK, Martin, TJ, et al. Management of the hypercalcaemia of malignancy by peritoneal dialysis. Med J Aust. 1975;1:782–784.
125. Goldsmith, RS, Ingbar, SH. Inorganic phosphate treatment of hypercalcemia of diverse etiologies. N Engl J Med. 1966;274:1–7.
126. Sato, K, Onuma, E, Yocum, RC, et al. Treatment of malignancy-associated hypercalcemia and cachexia with humanized anti-parathyroid hormone-related protein antibody. Semin Oncol. 2003;30:167–173.
127. Body, J-J, Greipp, P, Coleman, RE, et al. A phase I study of AMGN-00007, a recombinant osteoprotegerin construct, in patients with multiple myeloma of breast cancer related bone metastases. Cancer. 2003;97:887–892.
128. McClung, MR, Leweicki, EM, Cohen, SB, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006;354:821–831.
129. Bone, HG, Bolognese, MA, Yuen, CK, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab. 2008;93:2149–2157.
130. Moroney, S, Warmington, K, Adamu, S, et al. The inhibition of RANKL causes greater suppression of bone resorption and hypercalcemia compared with bisphosphonates in two models of humoral hypercalcemia of malignancy. Endocrinology. 2005;146:3235.