[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 4
Bone Development and Remodeling
Skeletal Development
Mechanisms of Bone Development
Intramembranous Ossification
The cranial and facial bones are derived largely from the neural crest through the process of intramembranous bone formation. In areas where bones are formed, mesenchymal precursor cells aggregate and form regions of high cell density, called condensations, that represent outlines of future skeletal elements. In intramembranous bones, the cells in these condensations then differentiate directly into osteoblasts that deposit “osteoid” or bone matrix rich in type I collagen, a process that occurs in close spatial interaction with vascular tissue. As the osteoblasts mature, the bone matrix becomes progressively mineralized. Terminally differentiated osteoblasts eventually become entrapped in the bone as osteocytes. Through modeling and remodeling (see later), the respective bones will reach their final shape and size. Membranous bones that derive as such from condensations that never form chondrocytes during development are restricted to the flat bones of the skull (calvarial bones and mandibles) and parts of the clavicles.1–5 The subperiosteal bone collar or provisional cortex of long bones can be considered to form also by intramembranous bone formation, because the osteoblasts in this region differentiate directly from the perichondrial/periosteal mesenchyme and form bone adjacent to, instead of directly upon, a cartilage-derived matrix (see later). Unlike the formation of intramembranous bone directly from condensations, the formation of this intramembranous bone is regulated directly by signals from the adjacent chondrocytes, so can be thought of developmentally as part of endochondral bone formation. Intramembranous bone formation also occurs during bone repair after fracture, particularly when the bones are stabilized (fixed).6–8
Endochondral Ossification
Endochondral ossification is the mechanism responsible for the formation of all long bones of the axial skeleton (vertebrae and ribs) and the appendicular skeleton (limbs). Most of the axial skeleton is derived from cells of the paraxial mesoderm that condense early in embryogenesis on both sides of the neural tube and the notochord. Portions of this mesoderm form segmented structures called somites, portions of which later become the sclerotomes that will give rise to the vertebral bodies. The appendicular skeleton arises from the lateral plate mesoderm. The mechanisms underlying the early condensation, segmentation, differentiation, and patterning events define the precise arrangement of the individual anatomic elements and their patterning along the proximal-distal, dorsal-ventral, and posterior-anterior body axes. These mechanisms involve actions and cross-talk of several morphogens, including fibroblast growth factors (FGFs), sonic hedgehog (Shh), bone morphogenetic proteins (BMPs), and Wnts, as well as control by Notch signaling and by transcription factors encoded by HOX, PAX1, and TBX genes.9
As in intramembranous ossification, the development of the long bones proper starts with mesenchymal progenitor cells forming condensations at the sites where the bones will be formed.10 Yet, in the mesenchymal condensations of endochondral bones, cells do not differentiate into osteoblasts but instead differentiate into chondrocytes that synthesize a characteristic extracellular matrix (ECM) rich in type II collagen and specific proteoglycans. As such, a cartilaginous model or anlage is established that prefigures the future bone. In mice, these differentiated cartilage structures appear around embryonic day 12, with the limb elements emerging in sequence along the proximodistal axis (i.e., hip to toes, shoulder to fingers). The sequential steps of the endochondral ossification process starting from this stage are illustrated in Fig. 4-1. Initially, the cartilage further enlarges through chondrocyte proliferation and matrix production. Chondrocytes in the midportion of the bone model then stop proliferating, undergo further maturation, and ultimately become hypertrophic. These large hypertrophic chondrocytes secrete a distinct matrix, containing type X collagen, which rapidly becomes calcified. Concomitantly, cells in the connective tissues surrounding the cartilage element called the perichondrium differentiate into osteoblasts that deposit mineralized bone matrix—the “bone collar”—around the cartilage template. This bone collar forms the initiation site of the cortical bone, the dense outer envelope of compact, lamellar bone that provides the long bone with most of its strength and rigidity (see Fig. 4-1).
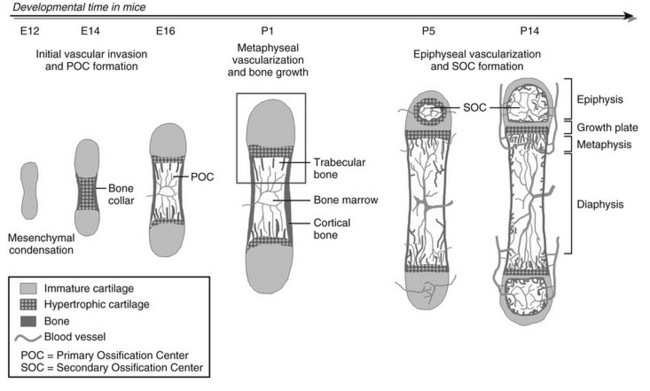
FIGURE 4-1 Stepwise schematic diagram of long bone development through endochondral ossification. Around embryonic day (E) 12 in mice, mesenchymal progenitor cells condense and differentiate into chondrocytes to form the cartilage anlagen that prefigure future long bones. Chondrocytes in the center become hypertrophic, while cells in the surrounding perichondrium differentiate into osteoblasts, forming a bone collar, the provisional cortical bone. The hypertrophic cartilage core subsequently is invaded by blood vessels and eroded and replaced by bone and marrow (primary ossification center [POC]). In the metaphysis, hypertrophic cartilage of the growth cartilage is continually replaced by trabecular bone, a process that relies on metaphyseal vascularization and mediates longitudinal bone growth. Around postnatal day (P) 5, epiphyseal vessels invade the avascular cartilage and initiate a secondary center of ossification (SOC). Discrete layers of residual chondrocytes form growth plates between the epiphyseal and metaphyseal bone centers to support further postnatal longitudinal bone growth. Ultimately (in humans), the growth plates close and growth stops. A detailed view of perinatal bone structure (boxed area) is provided in Fig. 4-2.
At this time in development (around embryonic day 14 to 15 in mice), the hypertrophic cartilage core becomes invaded by blood vessels. This process is accompanied by apoptosis of terminally differentiated hypertrophic chondrocytes, resorption of the calcified cartilage matrix by invading osteoclasts or related “chondroclasts,” and deposition of mineralized bone matrix on the remnants of calcified cartilage by perichondrium/periosteum-derived osteoblasts. The net result is replacement of the cartilage model by bone, vascular, and marrow elements: the primary ossification center (see Fig. 4-1). With the disappearance of the diaphyseal cartilage, the remaining chondrocytes, restricted to the opposing ends of the long bone, provide the engine for subsequent bone lengthening. This process is typified by precise temporal and spatial regulation of chondrocyte proliferation and differentiation, with the chondrocytes first flattening out and forming longitudinal columns of rapidly proliferating cells, and next, as they reach the ends of the columns closest to the center of the bones, maturing further to hypertrophic chondrocytes (Fig. 4-2). Finally, at the border with the metaphysis, the terminally differentiated chondrocytes undergo apoptosis, and the calcified hypertrophic cartilage matrix is replaced with cancellous or trabecular bone (primary spongiosa) in a process requiring progressive neovascularization by metaphyseal capillaries (see Fig. 4-1 and cellular details in Fig. 4-2). Thus, similar to the initial formation of the primary ossification center, endochondral bone formation at the growth cartilage involves rigorous coupling of vascular invasion with maturation and activity of chondrocytes, osteoclasts, and osteoblasts.
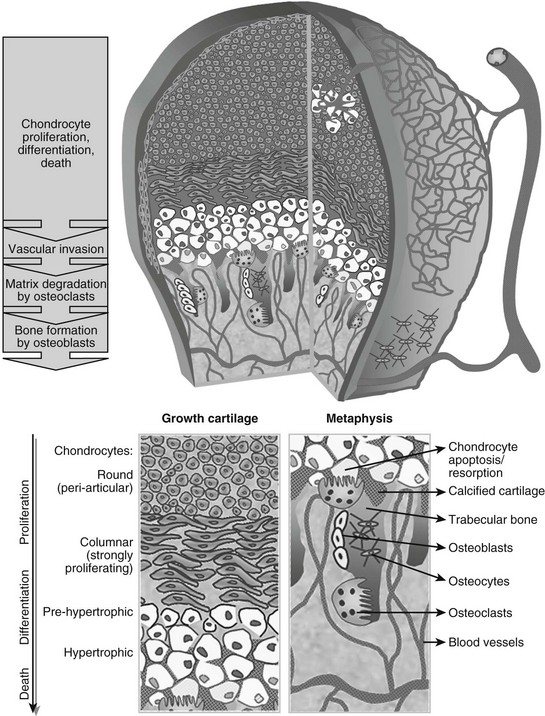
FIGURE 4-2 Schematic view of the cellular structure of developing long bones. The epiphysis is composed of chondrocytes, organized in layers of proliferation (round periarticular and flat columnar proliferating cells), progressive differentiation toward the metaphysis (pre-hypertrophic and hypertrophic chondrocytes), and cell death (apoptosis). The avascular cartilage is supplied by the epiphyseal blood vessel network that overlays its surface. In the metaphysis, blood vessels invading the terminal hypertrophic chondrocytes, osteoclasts resorbing the cartilage, and osteoblasts building bone on the cartilage remnants all act coordinately to replace the cartilage anlagen with bone and marrow.
The mechanisms of embryonic bone development described here are largely recapitulated in the adult upon repair of bone defects. In contrast to soft tissues, which repair predominantly through the production of fibrous scar tissue at the site of injury, the skeleton possesses an astounding capacity to regenerate upon damage. As such, bone defects heal by forming new bone that is indistinguishable from adjacent, uninjured bone tissue. It has been appreciated for a long time that fracture repair in the adult bears close resemblance to fetal skeletal tissue development, with both intramembranous and/or endochondral bone formation processes occurring depending on the type of fracture. In recent years, this close resemblance has been supported by genetic and molecular studies showing that similar signaling pathways (see later) are at work in both settings,8,11–13 although additionally, some signaling molecules that are dispensable for development have been found to play essential roles in fracture repair.14,15
Cellular and Molecular Control of Skeletal Development
Chondrogenesis
As outlined previously, a crucial step in endochondral bone development fundamentally entails the differentiation of cells in the mesenchymal condensations into chondrocytes. At least some of the mesenchymal cells are probably osteochondroprogenitor cells with the potential to differentiate into chondrocytes or osteoblasts. The decision to follow a given differentiation pathway is determined by the expression of key transcription factors, likely under the influence of canonical Wnt signaling. As will be discussed in greater detail later, Wnt signaling results in the stabilization of β-catenin, which then can act as a transcription factor and regulate the expression of downstream target genes. Recent studies have provided evidence that in the perichondrium, where cells are destined to become osteoblasts, Wnt signaling is high (at least in part induced by Shh),16 leading to high levels of β-catenin and inducing the expression of genes that mediate osteoblast differentiation (such as Runx2, a master regulator of osteoblastogenesis, see later), while inhibiting transcription of genes required for chondrocyte differentiation (such as Sox9). In the absence of β-catenin, these cells become chondrocytes instead of osteoblasts, as revealed via genetic modifications in mice. Conversely, in the inner region of the condensations, Wnt signaling must be low as these cells become chondrocytes.17–19 The initial conversion of mesenchymal progenitor cells into chondrocytes is driven by the transcription factor Sox9, whereas the combined action of Sox9, 5 and 6, is required to direct the subsequent differentiation of chondrocytes throughout all phases of the chondrocyte lineage.20–23
In the growth cartilage, the most extensively studied cartilage, differentiation of committed cells along the chondrocyte lineage characteristically gives rise to a stratified organization of small, round periarticular chondrocytes (also previously termed resting or reserve chondrocytes, because these cells proliferate only slowly after birth and can become columnar chondrocytes), flattened, columnar proliferating chondrocytes, and pre-hypertrophic and hypertrophic chondrocytes (see Figs. 4-2 and 4-3). Because progression of the chondrocytes through these stages is the driving force of actual bone development and growth, it is not surprising that the process is tightly controlled by a myriad of local signaling molecules, the best characterized being BMPs and transforming growth factor-β (TGFβ), FGFs, parathyroid hormone (PTH)-related protein (PTHrP), and Indian hedgehog (Ihh). The importance of these molecules is reflected by the fact that mutations in their receptors have been found to cause severe human dwarfing conditions, such as constitutive activating mutations of FGF receptor (FGFR)3 in human achondroplasia and thanatophoric dysplasia24–27 and PTH/PTHrP receptor mutations in Jansen and Blomstrand chondrodysplasia.28–30 Here, we will briefly review the basic mechanisms by which these signaling systems control the pace of proliferation and differentiation of growth chondrocytes.31–33
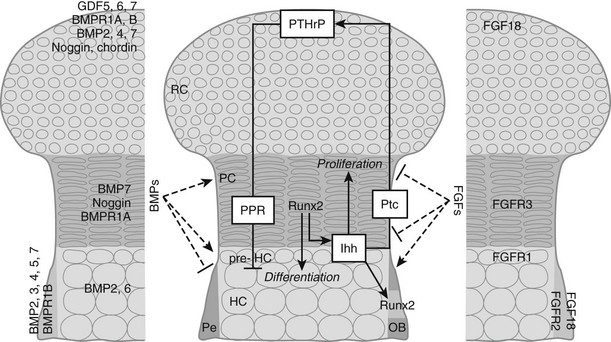
FIGURE 4-3 Regulation of chondrocyte proliferation and differentiation. Schematic representation of the epiphysis, with zones of periarticular round chondrocytes (RC), strongly proliferating columnar chondrocytes (PC), and (pre-)hypertrophic chondrocytes ([pre-]HC). Hypertrophic cartilage is surrounded by perichondrium (Pe), where osteoblasts (OB) develop. Centrally, the Indian hedgehog (Ihh)/parathyroid hormone-related protein (PTHrP)-negative feedback loop is indicated. Cells at the articular end of the bone secrete PTHrP, acting through the PTH/PTHrP receptor (PPR) on columnar chondrocytes to keep them in the proliferating pool and delay their differentiation. When the distance from the bone end becomes sufficiently large, cells escape the differentiation block and become Ihh-expressing pre-hypertrophic chondrocytes. In an as yet unknown manner, Ihh induces PTHrP expression. In addition, Ihh directly stimulates chondrocyte proliferation and converts perichondrial cells into OBs by inducing runt-related transcription factor 2 (Runx2), thereby determining the site of bone collar formation. Runx2 also favors Ihh expression and hypertrophic differentiation. Several bone morphogenetic protein (BMP) and fibroblast growth factor (FGF) family members are expressed at specific sites in the growth cartilage (left and right panels, respectively). BMPs and FGFs act antagonistically to modulate chondrocyte proliferation, Ihh expression, and terminal hypertrophic differentiation. GDF, Growth and differentiation factor; Ptc, patched; BMPR, BMP receptor; FGFR, FGF receptor.
BMPs, secreted proteins belonging to the TGFβ superfamily, transduce signals through serine/threonine kinase receptors, composed of type I and II subtypes, activating intracellular proteins of the Smad family that relay the BMP signal to target genes in the nucleus.34 BMPs were first discovered as agents in bone matrix capable of inducing ectopic formation of cartilage and bone after injection into subcutaneous tissues.35–38 Subsequent studies have shown that BMPs, some of which are also called growth and differentiation factors (GDFs) are, in fact, signaling proteins crucial for regulating development in almost all the principal organs and tissues.39,40 In the morphogenesis of the skeleton, BMPs have been implicated in early limb patterning, as well as in the subsequent processes of chondrogenesis and osteogenesis. Initially, the role of BMPs in the correct formation of mesenchymal condensations was highlighted by the mutation of the BMP5 gene in the mouse skeletal mutant short ear (se)41 and by abnormalities observed in brachypodism (bp) mice that were attributed to mutations in the GDF5 gene.42 In both se and bp mice, the abnormalities were traced back to altered size or shape already apparent in the mesenchymal condensations of the respective skeletal elements that were affected. More recently, studies in which two BMP type I receptors were ablated at the condensation stage demonstrated that BMP signaling is essential for converting condensing mesenchyme into chondrocytes; the mutant cells expressed undetectable amounts of the essential Sox9, L-Sox5, and Sox6 transcription factors.43 Similarly, when the potent BMP inhibitor noggin was introduced in early-stage mouse or chick limbs, mesenchymal condensation and chondrogenesis did not take place.44,45 BMPs also play critical roles at later stages of cartilage development. These functions have been difficult to document in vivo because in some cases, inactivation of specific BMPs led to severe early defects and lethality (e.g., BMP-2, BMP-4), precluding investigation of the later stages; in other cases, removal of individual family members (such as BMP-7) displayed no defects in skeletogenesis, presumably because of functional redundancy between the various BMP ligands and receptors (around 20 BMP family members have been identified to date). Further, various models have yielded superficially contradictory results, presumably reflecting the multiple actions of BMPs and the interactions of BMP signaling with other pathways. Recent and ongoing studies therefore employ conditional (site-specific) and/or combined (multiple targets) mutagenesis strategies. As such, it was shown in vivo that BMP signaling (through type I receptors on chondrocytes) positively regulates chondrocyte proliferation and survival, while concomitantly delaying the conversion to terminal hypertrophic differentiation,46 confirming previous limb culture results.47,48 These effects likely involve interactions between the BMP, FGF, and Ihh signaling pathways (see later); indeed BMP signaling seems to antagonize FGF actions and to promote Ihh expression. Studies in which BMP-2 and BMP-4 were ablated in limb development also revealed an essential role of BMP signaling in osteoblast differentiation (see later).49 It is not surprising that perichondrium, chondrocytes, and osteoblasts express multiple BMPs, BMP receptors, and BMP antagonists to regulate this essential pathway (see Fig. 4-3).
The FGF pathway similarly involves multiple ligands and receptors in regulating skeletal development.50 FGF1851,52 and FGF953 appear to be the most important FGF ligands in regulating chondrogenesis identified so far. These ligands activate FGFR3 expressed on proliferating chondrocytes. Activating mutations in the FGFR3 gene cause dominantly inherited human dwarfing chondrodystrophies due to impaired chondrocyte proliferation,24 and FGFR3 inactivation in mice increases chondrocyte proliferation and prolongs growth.54,55 This finding was surprising given that FGFs in most tissues act as potent mitogens; although this may pertain in early chondrogenesis, in which FGF18 stimulates proliferation,56 later in development, signaling through FGFR3 constitutes a master block on chondrocyte proliferation through activation of STAT1, which activates the cell cycle inhibitor p21Waf1/Cip1.57–59 In addition, although previous in vitro experiments had indicated that FGF signaling accelerates the late steps of chondrocyte hypertrophy,47 mouse models of achondroplasia and thanatophoric dysplasia have shown delayed hypertrophic differentiation of chondrocytes, strongly suggesting that FGFR3 signaling in vivo inhibits chondrocyte differentiation.25,60–63 This effect is relayed by activation of the mitogen-activated protein kinase (MAPK) pathway in chondrocytes, which affects longitudinal growth by regulating hypertrophic chondrocyte differentiation and matrix deposition.64–66 In vitro and in vivo experiments suggest that snail1 is required downstream of FGFR3 signaling for regulating its effects on chondrocyte proliferation and differentiation through activation of the Stat1/p21 and MAPK/Erks pathways, respectively.67 Yet, the effects of FGFR3 are regulated only in part by direct signaling in chondrocytes, and in part indirectly by modulating the expression of the Ihh/PTHrP/BMP signaling pathways. Indeed, mice harboring an activating mutation in FGFR3 have decreased expression of Ihh and its receptor and downstream target Patched (Ptc), and of BMP4, whereas in mice lacking FGFR3, Ihh, Ptc, and BMP4 expression are upregulated.60,68,69 During chondrocyte differentiation, the FGF and BMP pathways generally appear to antagonize each other,46 and both FGF signaling and BMP signaling regulate Ihh production (see Fig. 4-3). Thus, each of these pathways has multiple mechanisms for communicating with each other as they regulate chondrocyte and osteoblast differentiation.
A major regulatory system in growth chondrocytes is provided by Ihh, a member of the conserved family of hedgehog proteins, and PTHrP.32 The Ihh/PTHrP pathway forms a negative-feedback loop that regulates the onset of hypertrophic differentiation (see Fig. 4-3). Ihh is produced by prehypertrophic and early hypertrophic chondrocytes and signals through its receptor Ptc to stimulate expression of PTHrP by chondrocytes located near the periarticular ends of bone. Increased production of Ihh or production of Ihh closer to the cells making PTHrP leads to increased PTHrP production. PTHrP in turn signals back to its receptor—the PTH/PTHrP receptor that also responds to PTH in osteoblasts and kidney—that is expressed at low levels by proliferating chondrocytes and strongly by prehypertrophic cells. This signaling slows down chondrocyte differentiation and keeps chondrocytes in the proliferative state (see Fig. 4-3). This slowed differentiation consequently delays the generation of cells that can produce Ihh; therefore, the production of PTHrP is lowered. Thus, a negative-feedback signaling pathway allows PTHrP and Ihh to regulate each other and, in turn, to control the pace of chondrocyte development in the growth plate.32,70,71 It is important to note that the phenotype of the Ihh-/- mice72 revealed that Ihh has additional functions in endochondral bone formation, independent of regulation of PTHrP production. Indeed, these mutant mice also displayed a marked decrease in chondrocyte proliferation and absence of mature osteoblasts and bone collar formation.72 The regulation of chondrocyte proliferation represents a direct action of Ihh signaling to chondrocytes.73,74 Ihh also accelerates the conversion of round periarticular chondrocytes to flat columnar chondrocytes.75 Control of bone collar formation is mediated by Ihh-induced direct actions on perichondrial cells that are probably bipotential osteochondroprogenitors, and that are driven into the osteoblast lineage under the influence of Ihh stimulating the expression of Runx2. Runx2, an essential transcription factor in osteoblast development (see later), conversely also regulates Ihh expression and hypertrophic differentiation.72,76–79 From this, it is evident that Ihh is a master regulator of endochondral bone development, coordinating chondrocyte proliferation, chondrocyte maturation, and osteoblast differentiation (see Fig. 4-3). Recently, a continued role for Ihh in the postnatal growth plate has been revealed by employing inducible mutagenesis in mice.80
Aside from the above mentioned factors, chondrogenesis is affected by several other growth factors, cytokines, and hormones, including growth hormone (GH) and insulin-like growth factors (IGFs), C-natriuretic peptide, retinoids, thyroid hormone, estrogen, androgen, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], glucocorticoids, and others.32,81
Importantly, a main difference between mesenchymal condensations that differentiate toward the osteoblast lineage in and becoming intramembranous bones, and endochondral cartilaginous condensations is that the latter typically develop over a prolonged period in the absence of blood vessels. As an avascular tissue, cartilage consequently faces the challenge of hypoxia.82,83 The main mediator of cellular responses to hypoxia is the transcription factor hypoxia-inducible factor (HIF).84 Recently, HIF1α was shown to influence chondrocyte differentiation and to be required for survival of chondrocytes, because mice with conditional inactivation of HIF1α displayed aberrant apoptosis of chondrocytes located in the center of the growth cartilage, farthest from blood vessels.85,86 The precise mechanisms by which HIF1 prevents chondrocyte apoptosis have yet to be fully explained. Part of the explanation may include an indirect survival effect of HIF1, mediated through induction of vascular endothelial growth factor (VEGF), as deletion of VEGF from cartilage also resulted in the apoptotic phenotype.87,88 Alternatively, and not mutually exclusively, HIF1 may facilitate a vital switch in the metabolism of hypoxic chondrocytes by inducing the anaerobic glycolytic pathway,86,89–91 and thereby allow their further differentiation up to the hypertrophic stage, when the chondrocytes induce vascular invasion (see next).
Vascular Invasion of Cartilage
Both temporally and spatially, chondrocyte differentiation is followed by vascular invasion of the terminally differentiated, hypertrophic cartilage. This step is an absolute requirement for endochondral bone development and growth to proceed, as physical blockage of blood vessel invasion into the hypertrophic cartilage of fetal skeletal explants completely halts their development,92 and blocking the blood supply of bone in vivo results in reduced longitudinal growth.93,94 The importance of skeletal vascularization goes beyond endochondral bone development. In fact, as a rule, any type of bone formation occurs in close spatial and temporal association with vascularization of the ossified tissue. The reasons that the vascular system is crucial for bone development, maintenance, and repair obviously include its intrinsic function to supply oxygen, nutrients, and growth factors/hormones to bone cells as required for their specified activities. In addition, the blood vessels serve to bring in (precursors of) osteoclasts that will degrade the cartilage or bone extracellular matrix, to remove end products of the resorption processes, and to bring in progenitors of osteoblasts that will deposit bone.95–97
During endochondral ossification, the initially avascular cartilage template is replaced by highly vascularized bone and marrow tissue through three consecutive vascularization events (see Fig. 4-1). First, initial vascular invasion of the cartilage anlagen during embryonic development (sometimes called quiescent angiogenesis) involves endothelial cells invading from the perichondrial tissues and organizing into immature blood vessels in the primary center of ossification. Second, capillary invasion at the metaphyseal border of the growth cartilage mediates rapid bone lengthening. Third, vascularization of the cartilage ends initiates the formation of secondary ossification centers (see Fig. 4-1). Although the vessel systems associated with the different stages of cartilage vascularization are believed to be substantially different,98,99 a common feature is that invasion of cartilage at all times is preceded by chondrocyte hypertrophy. Immature chondrocytes are resistant to vascular invasion because of the production of angiogenic inhibitors, such as chondromodulin-I, troponin-I, and thrombospondins.100–103 In contrast, when chondrocytes become hypertrophic, they switch to production of angiogenic stimulators, thereby becoming a target for capillary invasion and angiogenesis.104,105 One exceptionally potent angiogenic stimulator, VEGF, has received much attention, as it is expressed at very high levels by hypertrophic cartilage, as well as by osteoblasts and osteoclasts. Although the regulation of VEGF expression is still largely unresolved and may vary depending on the developmental stage and specific location and cell type, several mechanisms have been implicated to date. Hypoxia seems to be a crucial trigger of VEGF expression in chondrocytes, osteoblasts, and possibly osteoclasts, through mechanisms that involve HIF both in vitro106–109 and in vivo.110 During early bone development, VEGF transcription may be induced by Runx2 (see later), given that expression of VEGF was impaired in Runx2-deficient mice and no blood vessel ingrowth into the cartilage occurred.111 In addition, several hormones [including PTH, GH, and 1,25(OH)2D3] and locally produced growth factors (e.g., FGFs, TGFβ, BMPs, IGFs, platelet-derived growth factor [PDGF]) have been demonstrated to be involved, at least in vitro, in the regulation of VEGF expression.112
Important insight into the crucial physiologic function of VEGF-mediated angiogenesis in bone was first provided by Gerber et al.,113 who inhibited VEGF action in juvenile mice through administration of a soluble VEGF receptor chimeric protein (sFlt-1). VEGF inhibition impaired vascular invasion of the growth plate, and concomitantly, trabecular bone formation and bone growth were reduced and the hypertrophic cartilage zone became enlarged, likely as the result of reduced osteoclast-mediated resorption (see later).113 Additional mouse genetic studies performed over the past decade have univocally demonstrated that VEGF is an essential mediator of all three key vascularization stages of endochondral bone development. Although a mouse with the VEGF gene ablated in all cells could not be employed owing to early lethality of even heterozygous VEGF knockout embryos, several alternative mutagenesis approaches have been and are being exploited to investigate the role of VEGF in bone. These models include Cre/LoxP-mediated conditional inactivation of the VEGF gene (VEGFa) in type II collagen–expressing chondrocytes, and expression of only one of the three major VEGF isoforms by genetically engineered mice.87,88,114–116 Altogether, these models have exposed multiple essential roles of VEGF and its isoforms in endochondral ossification, not only as a key inducer of vascularization but also as a direct modulator of bone development by affecting the various cell types involved. Perichondrial cells, osteoblasts, and osteoclasts are well documented to express several VEGF receptors and to respond to VEGF signaling through enhanced recruitment, differentiation, activity, and/or survival.112,117,118 These pleiotrophic actions of VEGF on various cells in the bone environment may contribute to the tight coordination of vascularization, ossification, and matrix resorption that is characteristically seen in endochondral ossification. Indeed, the current model is that VEGF secretion in the bone environment will attract blood vessels and stimulate endothelial cells to form new blood vessels, which will be indirectly associated with increased potential delivery of osteoblast and osteoclast progenitors.81 At the same time, VEGF signaling directly stimulates the recruitment and differentiation of osteoblasts to form bone.112,114,119,120 VEGF works as a chemoattractant stimulating osteoclast invasion of cartilage, and enhances osteoclast differentiation, survival, and resorptive activity.121–127 A positive-feedback system is likely established, as chondroclast/osteoclast-derived matrix metalloproteinase (MMP) 9 may release more matrix-bound VEGF from the cartilage that is being resorbed (Fig. 4-4).81,128
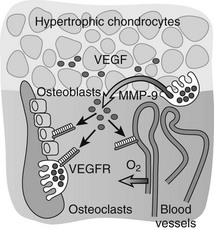
FIGURE 4-4 Vascular invasion and the role of vascular endothelial growth factor (VEGF). VEGF is produced at high levels by hypertrophic chondrocytes and is sequestered in part in the cartilage matrix upon its secretion. Trapped VEGF can be released from the matrix by proteases such as matrix metalloproteinase (MMP) 9 secreted by osteoclasts/chondroclasts during cartilage resorption. VEGF then can bind to its receptors (VEGFRs) on endothelial cells and stimulate the guided attraction of blood vessels to invade the terminal cartilage. Osteoblasts and osteoclasts also express VEGF receptors, and VEGF can affect their differentiation and function both directly and indirectly by enhancing metaphyseal vascularization, thus playing a coordinative role in the key events that mediate bone development and growth.
Osteoclastogenesis and Skeletal Tissue Resorption
At the time of vascular invasion of hypertrophic cartilage, the chondrocytes undergo apoptosis, and remaining cellular debris and matrix components become degraded and are digested by co-invading osteoclasts (or related cells termed chondroclasts that have been postulated in the context of resorption of cartilage128) through their proteolytic enzyme products.81 Beyond development, the osteoclast remains a key participant in the regulation of bone mass, as bone is constantly being remodeled throughout life (see later). Several pathologic conditions are due to an imbalance between bone formation and resorption, most frequently involving excessive osteoclast activity. Such skeletal diseases include osteoporosis, a common low bone mass disorder typically prevalent in postmenopausal women, as well as periodontal disease, rheumatoid arthritis, multiple myeloma, and metastatic cancer. On the other hand, osteopetrosis, a rare human disease characterized by increased bone mass and obliteration of the bone marrow cavity, is caused by impaired osteoclast differentiation and/or function. Many efforts have been made to dissect the regulatory pathways leading to functional osteoclasts.129–134
Osteoclastogenesis: Osteoclasts are giant multinucleated cells that have the unique capacity to efficiently degrade mineralized tissues. Osteoclasts are derived from hematopoietic precursor cells of the myelomonocytic lineage and share a common precursor with macrophages.131 Recognition of the intertwining control and close interactions of the immune and skeletal systems has led recently to the emergence of the novel integrating field of osteoimmunology.131,132 Early differentiation of the bipotential macrophage/osteoclast precursor cells is regulated by PU.1, a versatile hematopoietic cell-specific transcriptional regulator of the ETS family of transcription factors. The commitment to macrophages/osteoclasts is dependent on a high level of activity of PU.1131,135–137 (Fig. 4-5); PU.1 regulates the lineage fate decision of early progenitors by directly controlling expression of the c-Fms gene, which is a key determinant of differentiation into the macrophage/osteoclast lineage (see later).138,139 Consequently, PU.1-null mice lack both macrophages and osteoclasts, and are osteopetrotic.136,140,141 PU.1 also regulates the transcription of another key osteoclastogenesis control gene, encoding receptor activator of nuclear factor-κB (RANK) (see later) in myeloid progenitors.142 Although mature macrophages and osteoclasts have some cell surface markers in common, the latter express high levels of tartrate-resistant acid phosphatase (TRAP), cathepsin K, vitronectin, calcitonin receptor, and αvβ3 integrin, which typify the osteoclast lineage (see later).81
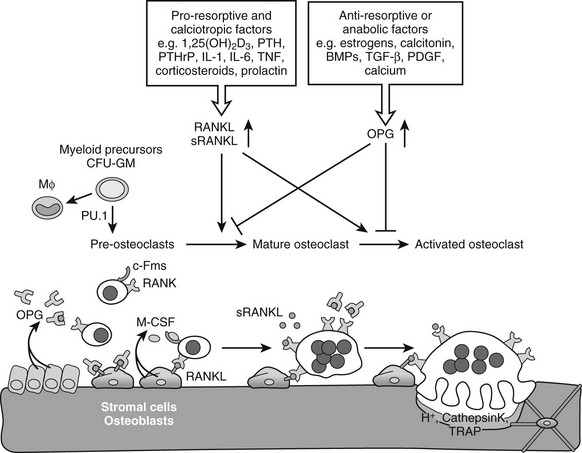
FIGURE 4-5 Osteoclastogenesis. The transcription factor PU.1 is indispensable for development of the osteoclast and macrophage (Mϕ) lineages from a common myeloid precursor. Macrophage-colony stimulating factor (M-CSF) acting upon the receptor c-Fms is required for the conversion of the progenitor to become a pre-osteoclast. Signaling through the membrane-bound receptor activator of nuclear factor-κB (RANK) promotes further differentiation of pre-osteoclasts to mature polykaryotic osteoclasts and their subsequent activation to bone-resorbing cells that secrete protons and lytic enzymes. RANK signaling is induced by receptor activator of nuclear factor-κB ligand (RANKL), present on osteoblasts and bone marrow stromal cells, as well as on lymphocytes and possibly in a soluble form in serum. The soluble antagonist osteoprotegerin (OPG) competes with RANK for RANKL binding, thereby functioning as a negative regulator of osteoclast differentiation, activation, and survival. RANKL expression is induced by pro-resorptive and calciotropic factors that stimulate osteoclastogenesis. Conversely, OPG is induced by factors that block bone catabolism and promote anabolic effects.
After the precursor cells have committed to the osteoclast lineage, they are subjected to a complex multistep process that culminates in the generation of mature, multinuclear, activated osteoclasts; these steps include proliferation, maturation, and fusion of differentiating precursor cells, and finally activation of resorption (see Fig. 4-5).81,130,132,134 Two critical cytokines are essential for osteoclastogenesis: macrophage-colony stimulating factor (M-CSF) and receptor activator of nuclear factor-κB ligand (RANKL) (also known previously as osteoprotegerin ligand [OPGL], osteoclast differentiation factor [ODF], or tumor necrosis factor [TNF]-related activation-induced cytokine [TRANCE]). Both of these signaling molecules are strongly expressed by bone marrow stromal cells (i.e., osteoblast progenitors) and osteoblasts; M-CSF is produced in both a soluble and a membrane-bound form, and RANKL is made exclusively as a membrane protein by osteoblasts. The process of osteoclastogenesis requires direct cell-to-cell interaction between stromal/osteoblastic cells and osteoclast precursors, presumably because of these key membrane-bound ligands (see Fig. 4-5). Both PTH and 1,25(OH)2D3 and several other osteotropic factors stimulating resorption increase the expression of RANKL on stromal/osteoblastic cells130,143,144 (see later). Moreover, before the identification of RANKL, in vitro investigation of osteoclastogenesis relied on contact-dependent co-culture of osteoclast progenitors with stromal/osteoblastic cells stimulated with PTH or 1,25(OH)2D3. Now we know that RANKL, together with M-CSF, is sufficient to induce in vitro osteoclast differentiation from spleen- or bone marrow–derived precursors in the complete absence of stromal/osteoblastic cells.145 Furthermore, not only osteoblasts but also chondrocytes146–148 and T cells149,150 synthesize and secrete RANKL and are able to support osteoclastogenesis. This may be important physiologically during osteoclast differentiation and invasion at the hypertrophic cartilage during endochondral bone development and growth, and it definitely plays a major role pathologically. For instance, production of RANKL by T cells has been implicated as an activator of osteoclastic resorption in inflammatory mediated bone and cartilage destruction, as is seen in several autoimmune disorders, including rheumatoid arthritis,81,130,151 likely working synergistically with TNFα.152,153
M-CSF and RANKL affect several steps of the osteoclastogenesis cascade by binding to their respective receptors, c-Fms and RANK, which are expressed by osteoclast progenitors and osteoclasts at all stages of differentiation. M-CSF signaling is essential early on in the lineage for proliferation, differentiation, and survival of osteoclast/macrophage precursors. The essential roles of M-CSF were illustrated through analysis of mice carrying the op/op mutation, an inactivating point mutation in the M-CSF gene; these mice have osteopetrosis with low numbers of macrophages and a complete lack of mature osteoclasts.154–157
RANKL is an essential inducer of multiple aspects of osteoclastogenesis, including osteoclast differentiation, fusion, activation of mature osteoclasts to resorb mineralized bone, and survival.158–161 Through these actions, RANKL potently stimulates bone resorption. RANKL acts by binding to its signal transducing receptor, RANK, a member of the TNF receptor family present on pre-osteoclasts and mature osteoclasts. Consistent herewith, an activating mutation of the human RANK gene was found in patients with familial expansile osteolysis, a disease of excess bone resorption.162 Recent genetic and cell biological studies have begun to elucidate the complex signaling cascade downstream of RANK/RANKL.130,163 Briefly stated and oversimplified, the binding of RANKL to RANK on the surface of osteoclast precursors recruits the adaptor protein TNF receptor–associated factor (TRAF) 6 to the cytoplasmic domain of RANK, leading to activation of NF-κB and its translocation to the nucleus. NF-κB increases c-Fos expression, and c-Fos, as a component of the activator protein (AP)-1 complex, interacts with the master transcription factor for osteoclastogenesis, nuclear factor of activated T cells c1 (NFATc1), to induce osteoclast-specific genes (such as the genes encoding TRAP and calcitonin receptor; see later).164–167
The actions of RANKL are regulated negatively by a secreted soluble decoy receptor termed osteoprotegerin (OPG) (previously also known as osteoclastogenesis inhibitory factor [OCIF]), also a member of the TNF receptor superfamily. OPG sequesters RANKL molecules and thereby blocks their binding to RANK.130,168 As such, OPG protects bone from excessive resorption; this conclusion is supported by the finding that certain homozygous deletions of OPG in humans can cause juvenile Paget’s disease, a disorder characterized by increased bone remodeling, osteopenia, and fractures.169 The relative concentrations of RANKL and OPG in bone thus are major determinants of bone mass and strength. OPG is widely expressed; it is not surprising that its expression by osteoblasts and stromal cells is positively regulated by bone anabolic or antiresorptive factors, such as estrogen and calcitonin.130
The essence of the RANK/RANKL/OPG cascade was further clarified by mouse genetic studies. Mice lacking RANKL170 or RANK171,172 and mice with increased circulating OPG by transgenic overexpression173 were severely osteopetrotic owing to a block in osteoclastogenesis. Conversely, targeted mutagenesis of OPG,174,175 overexpression of an sRANKL transgene,176 and administration of RANKL159,160 in mice all led to increased osteoclast formation, activation, and/or survival and resulted in an osteoporotic phenotype. In summary, RANKL and OPG act in an antagonistic fashion to regulate bone resorption, and their respective expression levels are under the control of proresorptive and antiresorptive factors, including several hormones, cytokines, and growth factors (see Fig. 4-5). Of note, RANK is broadly expressed, and these mouse studies indicate that the RANK/RANKL system functions in tissues beyond bone. For instance, RANK/RANKL regulates lymph node formation and lactational mammary gland development in mice.172,177 OPG has nonskeletal functions too, as it protects large arteries of mice from medial calcification.174
Other regulatory molecules have been implicated recently in the late stages of osteoclast differentiation, the fusion process, and activation of resorption. Co-stimulatory molecules acting in concert with M-CSF and RANKL to complete osteoclastogenesis include proteins containing an immunoreceptor tyrosine-based activation motif (ITAM) domain that is critical for the activation of calcium signaling and is found in adapter molecules like DNAX-activating protein (DAP)12 and the Fc receptor γ (FcRγ).178 The resultant increase in intracellular calcium is required for activation of NFATc1. The fusion of mononuclear osteoclast precursor cells into mature multinucleated osteoclasts is regulated by a membrane protein called dendritic cell-specific transmembrane protein (DC-STAMP). DC-STAMP–deficient cells failed to fuse, and these mononuclear osteoclasts had reduced resorptive efficiency in vitro. Consequently, DC-STAMP–deficient mice exhibited increased bone mass.179 These data may suggest that multinucleation and osteoclast enlargement could be associated with a higher degree of resorption efficiency.180,181 Pathologically huge osteoclasts, containing substantially more nuclei than normal osteoclasts do, are seen for instance in Paget’s disease of bone (up to 100 nuclei per cell; normal range is 3 to 20 nuclei per cell), in which localized excessive bone remodeling is initiated by increases in osteoclast-mediated bone resorption.182
Additional studies on the regulatory components of osteoclastogenesis not only will expand our basic understanding of the molecular mechanisms of osteoclast differentiation during bone development and remodeling but also offer opportunities to develop therapeutic means of intervention in osteoclast-related diseases. OPG and soluble recombinant RANK suppress osteoclastogenesis, while antibodies to RANK can stimulate osteoclast formation.183 From the clinical point of view, the RANKL signaling pathway thus holds great promise as a strategy for suppressing excessive osteoclast formation in a variety of bone diseases, including osteoporosis, autoimmune arthritis, periodontitis, Paget’s disease, and bone tumors/metastases.
Bone Resorption: Osteoclast Action and Proteolytic Enzymes: Bone resorption involves both dissolution of bone mineral and degradation of organic bone matrix. Osteoclasts are highly specialized to perform both of these functions. Upon activation of mature multinucleated osteoclasts, the cells attach themselves firmly to the bone surface, using specialized actin-rich podosomes (actin ring), through cytoskeleton reorganization and cellular polarization.184–187 Within these tightly sealed zones of adhesion to the mineralized matrix, the osteoclasts form convoluted, villus-like membranes called “ruffled borders,” which drastically increase the surface area of the cell membrane facing the resorption lacuna (Howship’s lacuna). Via these ruffled membranes, the osteoclasts secrete abundant hydrochloric acid (involving the vacuolar H+-ATPase proton pump), mediating acidification of the compartment between the cell and the bone surface, as well as a myriad of enzymes such as lysosomal cathepsins, the phosphatase TRAP, and proteolytic MMPs (see later). The acidity of the environment leads to dissolution of the mineral phase (crystalline hydroxyapatite), activation of lytic enzymes, and digestion of organic matrix compounds (see Fig. 4-5). The sealing mechanism allows localized dissolving and degrading of the mineralized bone matrix, while simultaneously protecting neighboring cells from harm.188,189 During the resorption process, dissolution of hydroxyapatite releases large amounts of soluble calcium, phosphate, and bicarbonate. Removal of these ions is needed (e.g., to maintain the acidic pH in the resorption lacuna) and apparently involves vesicular pathways and direct ion transport via different ion exchangers, channels, and pumps. The degradation products of the organic matrix after enzymatic digestion are transcytosed through the cell for secretion at the basolateral membrane.188,189
These complex processes of osteoclast recruitment, polarization on the bone surface, and export of acid and enzymes are orchestrated by many factors, including RANKL,190–192 as well as by integrin-mediated signaling from the bone matrix itself.193–195 The latter, which is particularly represented by the αvβ3 integrin in osteoclasts, was suggested to be important for osteoclast functioning based on the finding that inhibition of signaling through this αvβ3 integrin inhibited osteoclast-mediated bone resorption in vitro and in animal models of osteoporosis and malignant osteolysis (reviewed in refs. 196, 197). Integrins are heterodimeric cell-surface receptors, composed of an α and a β subunit, that mediate cell–matrix interactions and thus adhesion. The αvβ3 integrin, among various integrins the most highly expressed in osteoclasts, recognizes RGD(Arg-Gly-Asp)-containing matrix proteins such as vitronectin, osteopontin, and bone sialoprotein. Several components of the αvβ3 integrin signaling pathway localize to the sealing zone of actively resorbing osteoclasts, suggesting that they play a role in linking the matrix adhesion of osteoclasts to cytoskeletal organization, cell polarization, and activation for bone resorption. For these reasons, αvβ3 integrin appeared as a favorable candidate target for antiresorptive therapeutic interventions; in fact, small molecule inhibitors of the integrin are in clinical trial for the treatment of osteoporosis.194,198
Among the molecules that are important for the functional activity of osteoclasts per se, such as β3 integrin, but also c-Src, cathepsin K, carbonic anhydrase II, TRAP, and several ion channel proteins, many have been found to cause an osteopetrotic phenotype when deleted in mice or altered in humans. The absence of these genes does not affect the differentiation into morphologically normal osteoclasts; however, the osteoclasts are not functional, and they fail to resorb bone effectively.81 For instance, cathepsin K, the key enzyme in the digestion of bone matrix by its activity in degrading type I collagen, is highly expressed by activated osteoclasts and secreted in the resorption lacuna.199–201 Its deletion in mice led to osteopetrosis,201–203 and mutations in the human cathepsin K gene cause pycnodysostosis.204 Highly selective and potent cathepsin K inhibitors have been developed recently and have been shown to be useful antiresorptive agents for the treatment of osteoporosis, as well as promising therapeutic tools to reduce breast cancer–induced osteolysis and skeletal tumor burden.205–208
Besides cathepsin K, several proteolytic enzyme groups are involved in the degradation of organic components (collagens and proteoglycans) of bone and cartilage matrices after the mineral is dissolved.209–211 One of these is the MMP family, which constitutes over 25 members, including secreted collagenases, stromelysins, gelatinases, and membrane-type (MT)-MMPs.212,213 MMPs are synthesized as latent proenzymes that, upon proteolytic activation, can degrade numerous extracellular matrix components. As such, they are involved in the development, growth, and repair of tissues, but also in pathologic conditions associated with excessive matrix degradation, such as rheumatoid arthritis, osteoarthritis, and tumor metastasis.210,213–215 Several MMPs, including MMP9 and MMP14 (also known as MT1-MMP), are highly expressed in osteoclasts/chondroclasts,128,216,217 but they are produced by many other cell types as well. Both of these molecules play a role in the cartilage resorption process associated with invasion by osteoclasts during endochondral ossification.122,128,218–220 MMPs and proteolytic enzymes containing a disintegrin and metalloprotease domain (ADAMs) also possibly affect osteoclastogenesis per se, by modulating the bioavailability and presentation of RANKL through the proteolytic cleavage of its transmembrane form to soluble RANKL.221,222
Finally, after a limited period of resorptive activity, the osteoclast is thought to die via apoptosis (see later),223,224 and the resorbed area of cartilage or bone is, in conditions of development, growth, and bone health, efficiently replaced by newly formed bone through the action of osteoblasts.
Osteoblastogenesis
The final step required to rebuild cartilage to bone during development consists of the differentiation of osteoblasts and the deposition of mineralized bone tissue by this specialized cell type. Osteoblasts are mesenchymal cells thought to descend from mesenchymal stem cells that are pluripotent and have the capacity to differentiate into a variety of cell types, including muscle cells (myoblasts), fat cells (adipocytes), chondrocytes, and osteoblasts (Fig. 4-6).225–227 Differentiation of mesenchymal stem cells into distinct lineages is regulated by the expression of different transcription factors. MyoD transcription factors (MyoD, Myf5, and myogenin) regulate myogenic differentiation; C/EBP family (C/EBPβ, C/EBPδ, and C/EBPα) and PPARγ2 transcription factors regulate adipocyte differentiation (see Fig. 4-6).228 In certain settings, osteoblasts and chondrocytes seem to share a common precursor, termed osteochondroprogenitor. Specific transcription factors direct the osteochondroprogenitors into the chondrocyte (the Sox family transcription factors Sox9, Sox5, and Sox6) or the osteoblast lineages (β-catenin, Runx2, and osterix; discussed later) (see Fig. 4-6).31,229,230
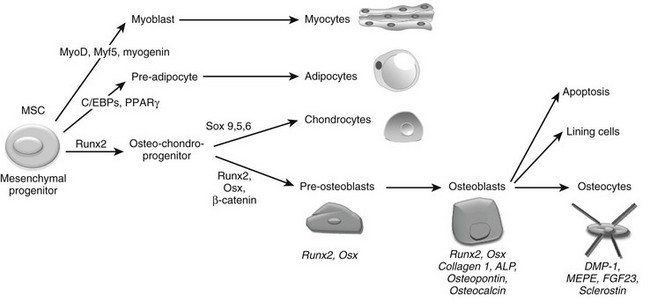
FIGURE 4-6 Osteoblast differentiation. Osteoblasts are derived from mesenchymal stem cells (MSCs) or progenitors that can become myocytes, adipocytes, or chondrocytes (driven by the indicated transcription factors) or can be directed into the osteoblast lineage, possibly through an osteochondroprogenitor intermediate. The transcription factors Runx2, Osx, and β-catenin mediate osteoblast differentiation and functioning. Progressive osteoblast differentiation is characterized by changes in gene expression typifying specific stages as indicated. See text for details.
Committed pre-osteoblasts further differentiate into mature osteoblasts that secrete large amounts of type I collagen and other matrix components, which together are called osteoid. Subsequently, osteoblasts direct the mineralization of the osteoid in a process that requires active alkaline phosphatase (ALP), expressed on the osteoblast membrane. These mature osteoblasts express characteristic genes, including the gene encoding osteocalcin.231 Ultimately, the cells die through apoptosis, or are converted to bone-lining cells, or become embedded within the bone matrix as osteocytes. Osteocytes are found dispersed throughout the bone matrix and have the potential to live as long as the organism itself. In fact, osteocytes are the most abundant cell type in bone, constituting over 90% of adult bone cells.232,233 As osteoblasts become encased by the mineralized bone matrix that they themselves synthesized, they dramatically change their shape, sending out numerous dendritic processes that run inside lacunar cannaliculi.234,235 These processes allow osteocytes to communicate with each other, with the vasculature, and with cells on the bone surface.233,236–239 Osteocytes are well positioned to sense and respond to mechanical forces, and a growing body of evidence documents that osteocytes mediate the actions of mechanical forces to increase bone mass (see later). Osteocytes synthesize a series of proteins unique to this most differentiated member of the osteoblast lineage. These include dentin matrix protein (DMP)-1, matrix extracellular phosphoglycoprotein (MEPE) and FGF23, secreted proteins involved in systemic phosphate homeostasis, and sclerostin (SOST), an inhibitor of Wnt action (see later).237,240–244 Despite their relative inactivity compared with osteoblasts, osteocytes play a central role in the determination and maintenance of bone structure (see later).
Wnt/β-Catenin Signaling: Wnts are a large family of secreted growth factors (19 different members in mouse and human genomes) that play essential roles in multiple developmental processes. Wnts are also required for adult tissue maintenance, and perturbations in Wnt signaling can lead to tumor formation and other diseases.17,245–248 In the skeletal system, mutations in Wnt signaling components lead to skeletal malformations and diseases such as osteoporosis-pseudoglioma249 and osteoarthritis.250
Wnts can transduce their signals through several different downstream signaling pathways. The best understood pathway is the canonical or Wnt/β-catenin pathway (Fig. 4-7).251 Central to this pathway is the regulation of the protein stability of β-catenin, which acts as a transcription factor in the canonical Wnt pathway and is also involved in cell adhesion by binding to cadherins. In the absence of Wnts, cytoplasmic β-catenin is constitutively degraded through its phosphorylation by glycogen synthase kinase 3-β (GSK3-β) in a large protein complex brought together by axin and adenomatous polyposis coli (APC).245,247,252,253 Phosphorylated β-catenin is recognized by a β-transducin repeat containing protein (β-TrCP) that targets it for proteasome-mediated degradation. Upon Wnt stimulation, the ligands bind to two synergistically acting families of Wnt (co)receptors, the Frizzled (Fz) receptor family members and low-density lipoprotein receptor-related proteins (LRP5 or LRP6), leading to the recruitment of axin to LRP5/6 in the plasma membrane. The sequestering of axin at the plasma membrane likely leads to disassembly of the β-catenin destruction complex, thus inhibiting the phosphorylation/degradation of β-catenin.254–259 Stabilized β-catenin protein then accumulates in the cytoplasm and translocates to the nucleus, where it interacts with members of the T cell factor/lymphoid enhancer factor (TCF/LEF) family of DNA-binding transcription factors to regulate the expression of downstream target genes (see Fig. 4-7).260–262
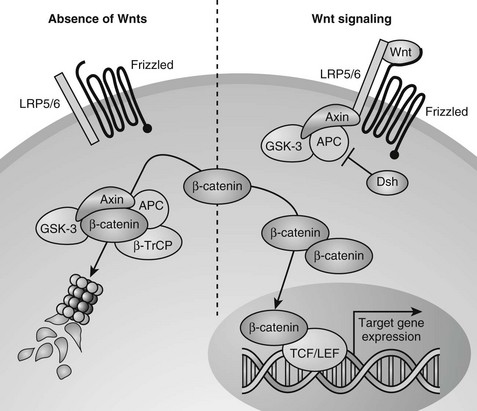
FIGURE 4-7 Canonical Wnt/β-catenin signaling pathway. In the absence of Wnt signals (left), β-catenin is associated with glycogen synthase kinase 3-β (GSK3-β) in a destruction complex that also contains Axin and adenomatous polyposis coli (APC). A hyperphosphorylated β-catenin is created by GSK3β and is bound by the β-transducin repeat containing protein (β-TrCP) component of the ubiquitin ligase complex, targeting β-catenin for proteasomal degradation. Wnt signaling through the Frizzled and LRP5/6 receptor complex (right) leads to inhibition of GSK3β through activation of Dishevelled (Dsh). Consequently, when Wnt signaling is active, β-catenin is stabilized, and the level of β-catenin in the cytoplasm becomes elevated. Accumulated β-catenin can translocate to the nucleus to interact with T cell factor/lymphoid enhancer factor (TCF/LEF) proteins and mediate transcription of target genes.
During the past decade, canonical Wnt signaling has been shown to play a significant role in the control of osteoblastogenesis and bone formation. Recent genetic studies analyzing conditional β-catenin loss- and gain-of-function mouse models provided compelling evidence that β-catenin is a crucial transcription factor determining the osteoblast lineage commitment of early osteochondroprogenitors, by inducing osteoblastic and suppressing chondrocytic differentiation.17,263 Indeed, inactivation of β-catenin in mesenchymal progenitor cells blocked osteoblast differentiation, and mesenchymal cells in the perichondrium and calvarium differentiated into chondrocytes instead.16,18,264 In addition, recent evidence showed that β-catenin plays an important role in the coupling between osteoblast and osteoclast activity by stimulating differentiated osteoblasts to produce OPG, an inhibitor of osteoclast formation (see earlier).265,266
In several clinical cases, mutations have been found in the Wnt receptor complexes that are associated with changes in bone mineral density and fractures. Loss-of-function mutations in LRP5 receptors cause osteoporosis-pseudoglioma syndrome,249 whereas gain-of-function mutations in the same group that render LRP5 with reduced affinities for DKK1 and sclerostin (SOST), secreted antagonists of canonical Wnt signaling, result in high bone mass phenotypes.267–271 These actions of LRP5 are indirect actions on bone: Activation of LRP5 in the duodenum suppresses serotonin secretion, and the lower blood levels of serotonin lead to an increase in bone mass through less activation of serotonin Htr1b receptors on osteoblasts.272
The protein inhibitors of Wnt signaling may have additional clinical relevance. For example, osteolytic bone lesions in patients with multiple myeloma are associated with the production of DKK1,273,274 and homozygous loss-of-function mutations in SOST have been identified in patients diagnosed with sclerosteosis, a disfiguring disease associated with high bone mass.275,276 Mutations affecting regulation of SOST transcription also cause van Buchem disease, a disease that resembles sclerosteosis.241,277 These findings highlight the important role of canonical Wnt signaling in regulating human bone mass. Because osteocytes are the major producer of SOST, inhibition of this Wnt antagonist may offer a promising strategy for preventing bone loss.17,278,279 Extensive research therefore is being conducted in this area based on the prospect that it may lead to the identification of targets of pharmacologic intervention useful in the management of osteoporosis.
Runx2 and Osx: Runx2, runt-related transcription factor 2 (previously known as core-binding factor a1 [Cbfa1], Osf2, or AML3), a transcription factor of the ancient runt family, has been demonstrated as absolutely essential for the induction of osteoblast differentiation and endochondral and intramembranous bone formation. Runx2-deficient mice completely lack osteoblasts and do not form bone at all; instead a completely cartilaginous skeleton develops without any true bone matrix.280,281 In heterozygous (haploinsufficient) Runx2 mutant mice, the defect in osteoblast differentiation is limited to intramembranous bones.280 The resulting phenotype in these mice closely resembles the cleidocranial dysplasia (CCD) syndrome in humans, a dominantly inherited developmental disorder of bone, and RUNX2 was found to be mutated in most CCD patients.282,283 Consistent with its function as an early transcriptional regulator of osteoblast differentiation, Runx2 is an early molecular marker of the osteoblast lineage, being highly expressed in perichondrial mesenchyme and in all osteoblasts.280,284 Hypertrophic chondrocytes also express Runx2, and Runx2 plays important roles in cartilage biology, as was mentioned earlier.
Runx2 can be sufficient to induce osteoblast differentiation in vitro.285 Moreover, Runx2-null calvarial cells spontaneously differentiate into adipocytes and differentiate into chondrocytes in the presence of BMP-2 in vitro, but they do not differentiate into osteoblasts.286 Thus, Runx2 is both sufficient and essential for differentiation of mesenchymal cells into osteoblasts, and it inhibits their differentiation into adipocytes and chondrocytes. Runx2 mediates osteoblast differentiation by inducing ALP activity, by regulating the expression of a variety of bone matrix protein genes, and by stimulating mineralization in immature mesenchymal cells and osteoblastic cells.287 Furthermore, Runx2 regulates the expression of RANKL and OPG in osteoblasts, thus affecting osteoclast differentiation (see earlier).288,289
The DNA-binding sites of Runx2 have been identified in major osteoblast-specific genes, including the genes that encode type I collagen, (Col1a1), osteopontin, osteonectin, bone sialoprotein, osteocalcin, and Runx2 itself, and Runx2 induced the expression of these genes or activated their promoters in vitro.287 It is puzzling that although Runx2 strongly induced osteocalcin expression in vitro,285,290 its overexpression in osteoblasts in vivo severely reduced osteocalcin expression.291 Yet, expression of a dominant-negative form of Runx2 in osteoblasts also led to virtual absence of osteocalcin expression and caused impaired postnatal bone formation.292 What is clear from these and other findings is that the regulation of different stages of osteoblast differentiation by Runx2 is very complex; Runx2 transcriptional regulation involves interactions with a myriad of transcriptional activators and repressors and other co-regulatory proteins that are under continued investigation. The current model is that Runx2 triggers the expression of major bone matrix protein genes and the acquisition of an osteoblastic phenotype at an early stage of osteoblast differentiation, while inhibiting the late osteoblast maturation stages and the transition into osteocytes. As such, Runx2 may play an important role in maintaining a supply of immature osteoblasts.287
Runx2 also regulates the expression of Osterix (encoded by the Osx or Sp7 gene), an SP family transcription factor with three zinc-finger motifs. Osterix is expressed in osteoblast progenitors, in osteoblasts, and at a lower level in (pre-)hypertrophic chondrocytes.293 Similar to Runx2-deficient mice, mice lacking Osterix showed complete lack of osteoblasts and absence of both intramembranous and endochondral bone formation.293 Thus, Osterix is a third transcription factor that is essential for osteoblast differentiation. Because Runx2 is expressed in the mesenchymal cells of Osx-null mice but Osterix is not expressed in Runx2-null mice, it can be concluded that Osterix acts downstream of Runx2.293 Furthermore, the Osx gene contains a consensus Runx2-binding site in its promoter region, suggesting that Osterix might be a direct target of Runx2.294 The transcriptional activity of Osterix involves its interaction with NFATc1, cooperatively forming a complex that binds to DNA and induces the expression of Col1a1.295 The subtleties of how Osterix regulates osteoblast differentiation and function and which osteoblastic genes are directly regulated by Osterix have not yet been elucidated. Expression of genes characteristic of mature osteoblasts (such as those encoding bone sialoprotein, osteopontin, osteonectin, and osteocalcin) was absent in cells surrounding chondrocytes in Osx-null mice, and instead these cells express genes characteristic of chondrocytes (Sox9, Sox5, Col2a1).293 Osterix has also been reported to inhibit chondrogenesis in vitro.296,297 Thus, Osterix may be important for directing precursor cells away from the chondrocyte lineage and toward the osteoblast lineage. Overall, it is currently thought that Runx2 has a crucial role in the earliest determination stage of the osteoblast lineage, driving mesenchymal progenitors to become pre-osteoblasts, while Osterix regulates at a later stage the differentiation of pre-osteoblasts to functional, bone-forming osteoblasts expressing high levels of osteoblast markers (see Fig. 4-6).
Other Factors and Signaling Molecules Involved in Osteoblastogenesis: Besides β-catenin, Runx2, and Osterix, various other (non–bone-specific) transcription factors are involved in osteoblast differentiation and function, albeit not to a similar critical extent, as their inactivation does not completely abrogate bone formation. These include ATF4,298–300 Msx1 and Msx2,301–304 Dlx5, Dlx6, and Dlx3,305–308 Twist,309,310 activator protein-1 (AP1) and its related molecules (Fos/Jun),311–315 and Schnurri-2 and -3.230,316–322 A myriad of morphogens and signaling molecules control the activity of the transcription factors just described. These molecules include Wnts, as discussed before, as well as locally produced BMPs and TGFβ, Hedgehogs (particularly Ihh), FGFs, Notch ligands, IGFs, PDGF, and systemic factors like PTH, GH, prostaglandins, estrogens, androgens, 1,25(OH)2D3 and glucocorticoids.81,97,323
Signaling pathways involving BMPs, Ihh, and FGFs have been mentioned previously in this chapter for their roles in chondrogenesis, but these signals also exert important functions in osteoblastogenesis and bone formation. Among BMP family members, BMP-2, BMP-4, and BMP-7 have been studied most extensively for their roles in osteogenesis during development, postnatal bone formation and remodeling, and fracture repair.15,40,49 The double knockout of BMP-2 and BMP-4 in the limb completely disrupted osteoblast differentiation, demonstrating the crucial roles of these two BMPs in osteoblast differentiation.49 Effects of BMP signaling in later stages of osteoblast differentiation are suggested by studies using BMP antagonists. Targeting of noggin overexpression to differentiated osteoblasts by the osteocalcin promoter results in osteopenia by 8 months of age.324 Likewise, overexpression of gremlin, another BMP antagonist, in differentiated osteoblasts results in reduced bone mineral density and fractures.325
BMP-2 has been shown to play a critical role in osteogenic differentiation: It promotes the commitment of pluripotent mesenchymal cells to the osteoblast lineage326–329 and has been demonstrated to induce the expression of both Runx2 and Osx during osteoblastogenesis.327,330–333 The precise mechanisms responsible for BMP-2-mediated gene regulation during osteoblast differentiation and bone formation are not well elucidated because of the considerable complexity and involvement of a myriad of interacting pathways. Nevertheless, the activity of BMP-2 as a potent inducer of bone formation is being applied to repair bone defects in humans.323,334,335 A recent human genetic study indicated that polymorphisms in the BMP-2 gene are linked to a high risk for osteoporosis.336
The Hedgehog family member Ihh is required for endochondral but not for intramembranous bone formation, by controlling osteoblast differentiation in the perichondrium of long bones. Ihh-null mice completely lack endochondral ossification because of the lack of osteoblasts.72,337 In these mice, Runx2 is expressed in chondrocytes but not in perichondrial cells. As well, nuclear β-catenin is absent in the perichondrial cells of Ihh-deficient mice.16 Thus, Ihh is required for inducing the initial activity of Runx2 and β-catenin in perichondrial cells and triggering them to become endochondral osteoblasts, thereby coupling chondrocyte maturation with osteoblast differentiation during endochondral ossification. Chondrocyte-derived Ihh remains crucial for sustaining trabecular bone in the postnatal skeleton.80 Skeletal abnormalities have been described in mutant mice lacking some of the intracellular mediators of Ihh signaling termed Gli proteins (three related transcription factors Gli1, Gli2, and Gli3)338–341; Gli2 mediates Ihh-induced osteoblast differentiation in mesenchymal cell lines by associating with Runx2 and stimulating its expression and function,342 as well as by inducing BMP-2 expression.343
FGF signaling has been implicated in the proliferation of immature osteoblasts and the anabolic function of mature osteoblasts in vivo.50–52,344–348 Whether and how osteoblast differentiation per se is affected by FGFs is currently elusive, but recent evidence indicates that FGF signaling induces BMP-2 expression and stimulates the expression and transcriptional activities of Runx2.349–354 Conversely, Runx2 has been demonstrated to form a complex with Lef1 or TCF that binds to the promoter region of the gene encoding FGF18, inducing its expression.355 These findings underscore once more how the various pathways that are essential for endochondral and intramembranous bone development physically and functionally converge. Moreover, the ultimate outcome relies on the complex integration of stimulatory and inhibitory signaling. For instance, Notch signaling suppresses osteoblast differentiation by diminishing Runx2 transcriptional activity via the Wnt/β-catenin pathway, actions that may be required to maintain a pool of undifferentiated mesenchymal progenitors in the bone marrow.356–359
Bone Remodeling and Skeletal Homeostasis
Throughout life, bone tissue maintains its integrity and responds to changes in functional demands by continuously turning over, a process termed bone remodeling.360,361 Bone remodeling is needed to remove older matrix and cells and stress-induced microcracks to ensure biomechanical stability and to regulate mineral homeostasis of the whole organism. Osteoclasts initiate bone remodeling and perform the actual removal of old bone matrix, while osteoblasts subsequently become activated to lay down new bone. Disturbances in the delicate balance between bone resorption and bone formation lead to disorders such as osteopetrosis (reduced function of the osteoclasts), osteosclerosis (increased function of the osteoblasts), and osteoporosis (low bone mass caused by greater bone resorption than bone formation). The two first disease states are characterized by a high bone mass; osteoporosis, the more frequent condition, is typified by a decrease in bone density, severe trabecular and cortical porosity, and a high incidence of fractures. Here we will briefly review the principles and main regulatory determinants of the bone remodeling process, and will outline some of the current insights into how osteoclasts and particularly osteoblasts function to sustain additional skeletal functions in a broader physiologic perspective.
Principles of Bone Remodeling
Bone is constantly being renewed by undergoing a process of self-destruction followed by a regenerative process. Depending on mechanical and physiologic needs, osteoclasts resorb particular pockets or trenches of existing trabecular and cortical mineralized bone tissue, whereas osteoblasts lay down new bone at appropriate sites to replace the lost bone (Fig. 4-8). Packets of bone that are renewed during remodeling are called bone-remodeling units (BRUs) or bone multicellular units (BMUs).360 It has been estimated that the adult human skeleton contains more than a million of these microscopic remodeling foci at any one time.361 The bone in an activated BRU is first removed by osteoclastic bone resorption through a process that takes a few weeks (see Fig. 4-8). Lost bone is replaced by osteoblastic bone formation, lasting 3 to 4 months for one packet. This discrepancy in the kinetics of bone resorption and formation explains in part how increased resorption, even when accompanied by coupled increased formation, can cause bone loss, for example, in estrogen deficiency or hyperparathyroidism. Bone resorption that starts the remodeling cycle may be initiated by osteoblast lineage cells perceiving and responding to a remodeling signal (see later). The signal responsible for the completion of resorption in a remodeling cycle has not been determined in vivo, but several factors that reduce osteoclast formation or activity could play a role (see later). In healthy bones, cessation of resorption is followed by bone formation. During the time lag that occurs between the end of resorption and the beginning of formation, called the reversal phase, small cells are seen on the resorbed surface, perhaps osteoblast precursors that are attracted to the BRU. At this stage of the remodeling cycle, plump cuboidal osteoblastic cells differentiate on the bone surface and deposit the organized matrix that then becomes mineralized. The bone formation period is completed after approximately 4 months, when the respective packet of bone, either on the surface of a trabecula or in the cortical haversian canal system, enters a quiescent period until the next remodeling cycle (see Fig. 4-8).360
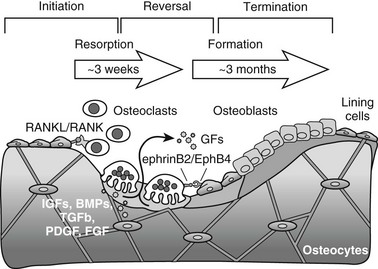
FIGURE 4-8 Three-phase model of bone remodeling. The skeleton is a metabolically active organ that undergoes continuous remodeling throughout life. Bone remodeling involves the removal of mineralized bone by osteoclasts, followed by the formation and subsequent mineralization of bone matrix by osteoblasts. Initiation starts with recruitment of hematopoietic precursors and their differentiation to osteoclasts, induced by osteoblast lineage cells that express osteoclastogenic ligands such as receptor activator of nuclear factor-κB ligand (RANKL). Osteoclasts become multinucleated and resorb bone. Transition is marked by switching from bone resorption to formation via coupling factors, possibly including diffusible growth factors (GFs) (e.g., hormones), membrane-bound molecules (e.g., ephrins), and factors embedded in bone matrix that become released upon osteoclastic bone resorption and can stimulate osteoblast recruitment, differentiation, and/or activity. During the termination phase, the resorbed lacuna is refilled through bone formation by osteoblasts that later flatten to form a layer of lining cells on the bone surface or become osteocytes connected by canaliculi within the bone.
Skeletal homeostasis remains intact as long as the activities of both osteoclasts and osteoblasts are balanced (coupled), and the net bone mass is maintained. This balance implies the existence of mechanisms that tightly coordinate the differentiation of osteoblasts and osteoclasts, as well as their migration to locations where they function. One prime aspect of the coupling principle is provided by the direct control of osteoclastogenesis by cells of the osteoblast lineage. Indeed, as was outlined previously in this chapter, osteoblasts and stromal cells control bone degradation by expressing M-CSF required for the proliferation of osteoclast precursors and RANKL, mediating the differentiation of hematopoietic osteoclast precursors toward mature multinucleated cells (osteoclastogenesis). Conversely, osteoclasts may reciprocally stimulate osteoblast differentiation and function to initiate the anabolic arm of the remodeling process. Osteoclastic bone resorption may well locally release the myriad of growth factors that are stored in the bone matrix, which can subsequently act as potent osteoblast stimuli. Growth factors like TGFβ, BMPs, IGFs, and PDGF are known to be incorporated into the bone matrix in high concentrations and to have the potency to attract osteoblastic cells and/or progenitors, and/or to stimulate proliferation and osteoblast differentiation and activity.362–364 Direct signaling by osteoclasts to cells of the osteoblast lineage has been demonstrated recently and may participate in coupled osteoblastogenesis and bone formation. For instance, the ephrin/Eph receptor system allows bidirectional signaling between osteoclasts and osteoblasts; osteoclasts express ephrin B2, and osteoblasts express its EphB4 receptor, both membrane-bound proteins.365 Signaling through EphB4 into osteoblasts (forward signaling) enhances osteogenic differentiation, whereas signaling through ephrin B2 into osteoclast precursors (reverse signaling) suppresses osteoclast differentiation.365 The overall outcome of such interaction thus is predicted to favor bone formation.364,366 It has also been reported that the v-ATPase V0 subunit D2 not only is involved in osteoclast fusion but also regulates the secretion by osteoclasts of still unidentified factors that inhibit the differentiation of osteoblast precursors into mature cells.367
Control of Bone Remodeling and Bone Mass
Bone remodeling and the resultant net bone mass are influenced by a myriad of signals (Fig. 4-9); mechanical stimuli, endocrine molecules, and central regulatory systems profoundly affect bone resorption and bone formation and hence the balance between the two processes, in addition to local factors (discussed previously).
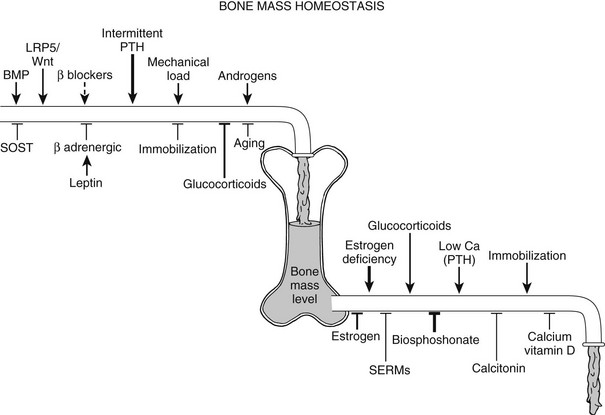
FIGURE 4-9 Control of skeletal homeostasis and bone mass. Schematic representation of the servo system that maintains bone mass at steady-state levels. Physiologic and pharmacologic stimulators and inhibitors of bone formation and resorption are listed. The relative impact, where known, is represented by the thickness of the arrows. Solid lines are current therapies, and dotted lines, putative ones. SERM, Selective estrogen receptor modulator. (Modified from Harada S, Rodan GA: Control of osteoblast function and regulation of bone mass, Nature 423:349–355, 2003.)
Mechanical Loading
Increasing evidence suggests that the main mechanosensing cell type of bone is the osteocyte (see earlier). The osteocyte is ideally situated within the lamellar bone to sense mechanical forces and may communicate signals to other osteocytes, osteoblasts, and osteoclasts on the bone surface through the connecting canalicular network.233,368–370 Osteocytes have been demonstrated to have the potential to stimulate bone resorption in vitro and in vivo.371,372 This modulation of bone remodeling has been suggested to be elicited by osteocyte apoptosis, which can be consequential to unloading.373 Conversely, mechanical stimulation is capable of maintaining osteocyte viability.374,375 Recent studies underscore the importance of osteocyte viability in maintenance of bone tissue health and the response to mechanical loading. Experimental destruction of osteocytes in murine bone, via targeted diphtheria toxin receptor expression under the control of the osteocyte-specific DMP-1 promoter, quickly led to a large-scale increase in bone resorption, decreased bone formation, and trabecular bone loss. Concomitantly, these mice were resistant to unloading-induced bone loss, indicating the requirement for osteocytes in the response to mechanical signals.376
The mechanisms by which a mechanical stimulus is transduced into biochemical signals in osteocytes and osteoblasts and the means whereby these cells then modulate bone remodeling activities have not been clearly identified. Influences that have been implicated in the process include shearing forces produced by fluid movement (e.g., in the canaliculi surrounding the osteocytic dendrites) and a variety of membrane proteins, including integrins, connexins, and stretch-sensitive ion channels.233,236,239 For instance, mechanical stimulation increases expression of connexins, membrane-spanning proteins that form regulated channels; this allows the direct exchange of small molecules with adjacent cells, resulting in intercellular communication between cells.239,377–379 Moreover, osteocytes respond in vitro and in vivo to increased load by altering their signaling. For example, in response to loading, osteocytes upregulate nitric oxide production, release prostaglandin (PG)E-2 and IGF-1, and decrease glutamate transporter expression.233 DMP-1 expression is also robustly increased upon mechanical stimulation.380 DMP-1 inactivation in mice is associated with a hypomineralized phenotype linked with elevated FGF-23 and defective osteocyte lacuna/canalicular network formation.381 MEPE production in osteocytes is mechanosensitive as well, showing delayed production after mechanical stimulation, quite unlike that of DMP-1.382 Targeted disruption of MEPE results in increased bone mass and confers a degree of resistance to age-related trabecular bone loss.383 Because both DMP-1 and MEPE can regulate phosphate metabolism and bone mass, these findings suggest a potential connection between exercise, local bone mineralization, phosphate homeostasis, and kidney function orchestrated via the osteocyte, which may be important toward an understanding of the full spectrum of consequences of the loss of osteocytes observed in aging bones.233,237,243,384
Of great interest also is the recent observation that mechanical loading changes the expression levels of SOST in osteocytes, resulting in a rapid decrease in sclerostin production.385 As was mentioned, sclerostin inhibits Wnt signaling through binding to LRP5/6240,241; SOST-null mice have a very high bone mass,386,387 whereas conversely transgenic mice overexpressing sclerostin in osteocytes suffer severe bone loss.388 Osteocytes thus appear to use the Wnt/β-catenin pathway to transmit signals of mechanical loading to cells on the bone surface.236,389
Systemic Factors Regulating Calcium Homeostasis
Bone remodeling also represents a major mechanism used by land vertebrates to regulate mineral homeostasis. The removal of calcium from bone, to maintain calcium homeostasis acutely, is facilitated by osteoclastic resorption of calcium-containing bone. The principal mediators in this process are PTH and its downstream effector 1,25(OH)2D3. Both these hormones act on osteoblastic cells to increase the production of RANKL and thereby potently activate osteoclastogenesis.143,144,390
The actions of PTH on bone are complex, with multiple direct actions on cells of the osteoblast lineage that vary, both with the state of differentiation of the cell and with its location in bone (periosteal vs. endosteal). The actions of PTH to increase osteoclast number and action dominate when levels of PTH are continuously elevated, but PTH also increases the rate of bone formation. In fact, when PTH is administered intermittently, this effect predominates and leads to a net increase in bone mass.391–394 PTH enhances Runx2 expression and activity when administered intermittently; however, Runx2 is also involved in the catabolic effect of PTH through the induction of RANKL.395,396 Intermittent PTH promotes osteoblast differentiation also in part by its ability to promote exit from the cell cycle, to activate Wnt signaling in osteoblasts, and to inhibit production of the Wnt antagonist sclerostin in osteocytes.394,397–399 Furthermore, intermittent PTH prolongs osteoblast survival.400 By controlling osteoblast function, PTH increases bone formation on cancellous, endocortical, and periosteal bone surfaces and has become a valuable clinical anabolic treatment strategy for osteoporotic conditions.401,402
Sex Steroids
Another significant endocrine input into bone remodeling and homeostasis is the pronounced influence of sex steroids, essential for maintenance of the adult skeleton (see Fig. 4-9). Skeletal preservation by estrogen in females may be related evolutionarily to the need for calcium stores for embryonic skeletal development. In mammalian adult males and females, including humans, estrogen has been identified as the major inhibitor of bone resorption by reducing osteoclast number. This explains the profound bone loss associated with estrogen deficiency, with postmenopausal osteoporosis being the most prominent type of bone pathology in humans.223,403,404 Treatment of women with hormone replacement therapy (estrogen alone or estrogen plus progesterone) has been shown to prevent this bone loss.405
Recent studies have shed light on the mechanism of estrogen action on osteoclasts: Osteoclasts express estrogen receptors (ERα and ERβ) and estrogen acts directly on osteoclasts to increase apoptosis.406,407 The regulation of osteoclast lifespan by estrogen involves induction of the Fas/FasL system causing apoptosis, a pathway that may be involved both in osteoclast cell-autonomous effects407 and in indirect mechanisms mediated via the osteoblast.408 Furthermore, estrogen can have nongenomic effects, or rapid signaling effects, thus inducing the phosphorylation of components of various signaling pathways (e.g., the MAPK pathway) or calcium regulation. Such effects may contribute to the induction of osteoclast apoptosis by estrogen without the need for direct binding of ERα to DNA.403,409 Besides affecting osteoclast lifespan, estrogen was reported to decrease the responsiveness of osteoclast progenitor cells to RANKL, thereby reducing osteoclastogenesis.410
A variety of other mechanisms contribute to the beneficial effects of estrogen on bone. A prime role is played by cytokines regulated by estrogen that indirectly lead to changes in osteoclast number: Estrogen suppresses the production of osteoclastogenic cytokines such as interleukin (IL)-1, IL-6, IL-7, and TNFα in T cells and osteoblasts.411–415 As well, a portion of the effects ascribed to estrogen deficiency were suggested recently to be in fact mediated by the resultant increase in pituitary gland–derived follicle-stimulating hormone (FSH) acting on osteoclasts.416,417
Androgens are important not only as a source of estrogen, through the action of aromatase, but also for their direct effect in stimulating bone formation.418,419 Testosterone, responsible for the male phenotype characterized by a larger skeleton, has complex effects on bone metabolism. As an anabolic steroid, it stimulates bone formation in both males and females. In addition, testosterone can inhibit bone resorption directly, acting through the androgen receptor and through conversion by aromatase to estrogens. Androgens also increase periosteal bone formation, leading to larger and therefore stronger bones. Loss of androgens in males from chemical or surgical castration or an age-associated decline in androgen levels has the same adverse effect on the skeleton as estrogen loss in women, albeit the loss of testosterone in aging men is not as universal or as abrupt as the loss of estrogen at the time of menopause in women.418–420
Other Systemic Regulators of Bone Remodeling
In contrast to sex steroids, glucocorticoid excess is catabolic to bone, as is illustrated by glucocorticoid-induced osteoporosis, a devastating consequence of long-term use of glucocorticoids.421 The mechanisms for glucocorticoid-induced bone loss are complex, including suppression of renal calcium reabsorption, reduction in intestinal calcium absorption, and hypogonadism, all of which lead to increased bone resorption and bone turnover. Perhaps more important, glucocorticoids have direct effects on the skeleton.421 Glucocorticoids impair the replication, differentiation, and function of osteoblasts and induce the apoptosis of mature osteoblasts and osteocytes, leading to dramatic suppression of bone formation.422–425 As well, glucocorticoids act directly on osteoclasts and prolong their lifespan; in addition, glucocorticoids may sensitize bone cells to regulators of bone remodeling and favor osteoclastogenesis, thus leading to overall increased bone resorption.426–428
Influences on bone remodeling by the GH/IGF axis,429 thyroid hormones,430 and prostaglandins431 are reviewed in the indicated recent references.
Central Control of Bone Remodeling
The central nervous system (CNS) coordinates the activities of multiple organs throughout the body. Given the likely importance of coordinating skeletal responses to a variety of systemic challenges, perhaps it is not surprising that recent work shows that signals from the CNS and the sympathetic nervous system (SNS) profoundly influence bone remodeling and resultant bone mass (Fig. 4-10).
FIGURE 4-10 Interactions between bone, brain, and energy metabolism. The skeleton affects cells involved in metabolic control, such as pancreatic β cells and fat cells, which signal to brain centers and peripheral tissues to regulate energy and metabolic homeostasis (left). Conversely, the central nervous system and brain neuropeptides regulate bone homeostasis (right). Osteoblasts express osteotesticular protein tyrosine phosphatase (OT-PTP), a product of the Esp gene, which regulates γ-carboxylation of the osteoblast-specific protein osteocalcin. Whereas carboxylated osteocalcin has high affinity to bind the bone mineral, uncarboxylated osteocalcin is secreted into the serum and acts in a hormone-like manner to influence energy metabolism by increasing β cell proliferation and insulin secretion in the pancreas, and by increasing adipocyte secretion of adiponectin, an insulin-sensitizing adipokine. As a result, insulin sensitivity is enhanced. Thus, the skeleton functions as an endocrine regulator of energy metabolism (left). Adipocytes also secrete the adipokine leptin, which binds to leptin receptors in the hypothalamus and regulates appetite, reproduction, and energy expenditure (right). Leptin also regulates bone mass (right). The leptin signal in the brain leads to stimulation of the sympathetic nervous system (SNS) and activation of the β2-adrenergic receptor (Adrβ2) on osteoblasts, which decreases osteoblast proliferation (mediated by molecular clock genes) and bone formation, while increasing bone resorption through stimulation of receptor activator of nuclear factor-κB ligand (RANKL) expression. Yet, leptin also has inhibitory effects on bone resorption by binding to its receptor on neurons in the arcuate nucleus of the hypothalamus and increasing expression of the anorexigenic peptides cocaine- and amphetamine-related transcript (CART). Via an unknown mechanism, CART inhibits RANKL expression. Other neuropeptides, such as neuromedin U (NMU) and neuropeptide Y (NPY), also influence bone metabolism and thus play a role in the central control of bone mass.
Leptin, an adipocyte-derived hormone mainly known for controlling appetite, energy metabolism, and reproduction, acts through the leptin receptor in the CNS (hypothalamus) and ultimately through the SNS to negatively regulate bone formation by osteoblasts.432,433 Sympathetic signaling via β2-adrenergic receptors (Adrβ2) in osteoblasts inhibits proliferation through transcriptional factors such as Period (per), which also regulate the circadian clock,434 and additionally favors bone resorption by increasing osteoblastic expression of RANKL435 (see Fig. 4-10). These conclusions were drawn from a series of studies using mutant mice with deficiencies or lesions in portions of the leptin-hypothalamic-sympathetic pathways432,433,435; all of these models displayed a similar bone phenotype characterized by increased bone mass due to vastly increased osteoblast activity and bone formation accompanied by a mild increase in osteoclastic bone resorption. Part of this outcome is explained further by another action of leptin through hypothalamic receptors, which is to inhibit bone resorption by stimulating the expression of cocaine- and amphetamine-regulated transcript (CART), leading—through as yet unknown mechanisms—to reduced RANKL expression.435,436 Evidence that leptin regulates bone turnover in part through a CNS/β-adrenergic system relay has driven attention toward the potential therapeutic benefits of β-adrenergic blockers in improving bone mass and strength in osteoporosis.437–439 It is interesting to note that whereas the downstream effectors of leptin action on bone remodeling seem to be largely independent of its functions in energy metabolism and reproduction, a reciprocal regulatory function on energy metabolism and adiposity is exerted by osteoblasts (as outlined in the next section). In addition to CART, leptin interacts with the hypothalamic neuropeptides neuropeptide Y and neuromedin U, which themselves have been implicated in the central control of bone remodeling440–442 (see Fig. 4-10). Furthermore, other neuroendocrine connections to bone remodeling have been made with the findings in mouse genetic and pharmacologic studies that both thyroid-stimulating hormone (TSH) and FSH act directly on bone cells to influence remodeling.416,443,444 For additional readings on these subjects, see references 437 through 439, and 445 through 448.
Additional Roles of Osteoblasts and Osteoclasts In Integrating Skeletal Physiologic Functions
Interplay Between Bone Cells and the Immune and Hematopoietic Systems
With the growing elucidation of the molecular control of osteoclastogenesis, links with the immune system have increasingly emerged and given rise to a novel discipline of bone-related studies termed osteoimmunology.131 This field encompasses the complex interactions and molecular and functional overlap of the immune system and bone cells. For instance, not only osteoblasts and bone marrow stromal cells, but also immune cells, express M-CSF and RANKL, pivotal regulators of osteoclastogenesis. Particularly certain subsets of T cells rapidly upregulate surface expression of RANKL on activation, and therefore are capable of regulating osteoclast differentiation and activity. Additional cytokines, either produced by or regulating T cells, are responsible for the upregulation of osteoclast formation observed in a variety of conditions such as inflammation (e.g., arthritic joint disease), hyperparathyroidism, and estrogen deficiency causing osteoporosis.411 These factors, which include TNFα, IL-1, and IL-6, function as co-stimulatory molecules for osteoclastogenesis. Another level of molecular overlap is provided by the fact that most osteoclastogenic cytokines, including M-CSF and RANKL, also regulate macrophages or dendritic cells that share myeloid precursor cells from the bone marrow during development.131,133
Although poorly understood at present, B cells also seem to modulate the bone remodeling balance.449 Recently, it was shown that B cells constitute an important source of OPG in vivo, and that mice deficient in B cells or with arrested B cell differentiation have reduced bone mass.450,451 B cell progenitors themselves are capable of differentiating into osteoclasts in vitro.452
The close interaction between immune progenitors and the skeleton is facilitated by their proximity in the bone marrow. Moreover, bones do not merely house the bone marrow but also provide the appropriate environment for physiologic and pathologic blood cell development. Recent data have highlighted the importance of osteoblasts and osteogenic stromal cells as key regulators of hematopoietic cell development, hematopoietic stem cell maintenance, and blood cell mobilization, by providing the appropriate microenvironment for the hematopoietic system.453–458 Accordingly, changes in bone that affect the bone marrow environment, in particular the osteoblast niche, can disrupt hematopoietic stem cells, leading to altered hematopoietic and immune cell development and/or potentially initiating myeloproliferative disease.454,459–461 It remains to be determined whether and to what extent bone remodeling may dynamically respond to the changing demands of hematopoiesis. As a suggestive example, osteoclast resorptive activity on the endosteal bone surface, where hematopoietic cells reside,456,462 was found to be capable of mediating the release of these hematopoietic cells to mobilize them into the circulation.463,464 Furthermore, human hematologic diseases such as myelofibrotic disease often are associated with secondary bone pathology, suggesting that dysregulated hematopoietic differentiation may affect bone formation and/or resorption processes.
Skeletal Control of Energy Metabolism
The concept of an adipocyte-derived hormone regulating bone, as outlined earlier (leptin; see Fig. 4-10), led Karsenty to postulate the existence of reciprocal endocrine regulation from the skeleton. In search of bone-derived signals secreted into the circulation and able to modulate energy metabolism and adiposity, his research group discovered that a form of osteocalcin, one of the few osteoblast-specific gene products, acts as a hormone that increases insulin-sensitive glucose utilization and pancreatic β cell mass and reduces visceral fat (see Fig. 4-10).465 Osteoblastic secretion of the hormone-like, undercarboxylated form of osteocalcin is controlled by a receptor-like protein tyrosine phosphatase, termed osteotesticular protein tyrosine phosphatase (OT-PTP), which is encoded by the Esp gene and expressed exclusively in osteoblasts. Mice lacking OT-PTP in osteoblasts have increased β cell activity that protects them from induced obesity and diabetes. These phenotypes are corrected by deleting one allele of osteocalcin. Osteocalcin deficiency in knockout mice leads to decreased insulin and adiponectin secretion, insulin resistance, higher serum glucose levels, and increased adiposity.465,466 These findings indicate that the skeleton plays a previously unrecognized role in the regulation of energy metabolism and thereby may contribute to the onset and severity of metabolic disorders. The full significance of these novel findings established through elegant mouse genetics has yet to be fully defined in humans.
References
1. Wilkie, AO, Morriss-Kay, GM. Genetics of craniofacial development and malformation. Nat Rev Genet. 2001;2:458–468.
2. Morriss-Kay, GM, Wilkie, AO. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat. 2005;207:637–653.
3. Tapadia, MD, Cordero, DR, Helms, JA. It’s all in your head: new insights into craniofacial development and deformation. J Anat. 2005;207:461–477.
4. Huang, LF, Fukai, N, Selby, PB, et al. Mouse clavicular development: analysis of wild-type and cleidocranial dysplasia mutant mice. Dev Dyn. 1997;210:33–40.
5. Matsuoka, T, Ahlberg, PE, Kessaris, N, et al. Neural crest origins of the neck and shoulder. Nature. 2005;436:347–355.
6. Le, AX, Miclau, T, Hu, D, et al. Molecular aspects of healing in stabilized and non-stabilized fractures. J Orthop Res. 2001;19:78–84.
7. Carter, DR, Beaupre, GS, Giori, NJ, et al. Mechanobiology of skeletal regeneration. Clin Orthop Relat Res. 1998;355(suppl):S41–S55.
8. Behonick, DJ, Xing, Z, Lieu, S, et al. Role of matrix metalloproteinase 13 in both endochondral and intramembranous ossification during skeletal regeneration. PLoS ONE. 2007;2:e1150.
9. Tabin, C, Wolpert, L. Rethinking the proximodistal axis of the vertebrate limb in the molecular era. Genes Dev. 2007;21:1433–1442.
10. Hall, BK, Miyake, T. All for one and one for all: condensations and the initiation of skeletal development. Bioessays. 2000;22:138–147.
11. Ferguson, CM, Miclau, T, Hu, D, et al. Common molecular pathways in skeletal morphogenesis and repair. Ann N Y Acad Sci. 1998;857:33–42.
12. Thompson, Z, Miclau, T, Hu, D, et al. A model for intramembranous ossification during fracture healing. J Orthop Res. 2002;20:1091–1098.
13. Gerstenfeld, LC, Cullinane, DM, Barnes, GL, et al. Fracture healing as a post-natal developmental process: molecular, spatial, and temporal aspects of its regulation. J Cell Biochem. 2003;88:873–884.
14. Maes, C, Coenegrachts, L, Stockmans, I, et al. Placental growth factor mediates mesenchymal cell development, cartilage turnover, and bone remodeling during fracture repair. J Clin Invest. 2006;116:1230–1242.
15. Tsuji, K, Bandyopadhyay, A, Harfe, BD, et al. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet. 2006;38:1424–1429.
16. Hu, H, Hilton, MJ, Tu, X, et al. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development. 2005;132:49–60.
17. Day, TF, Yang, Y. Wnt and hedgehog signaling pathways in bone development. J Bone Joint Surg Am. 2008;90(suppl 1):19–24.
18. Day, TF, Guo, X, Garrett-Beal, L, et al. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–750.
19. Hill, TP, Taketo, MM, Birchmeier, W, et al. Multiple roles of mesenchymal beta-catenin during murine limb patterning. Development. 2006;133:1219–1229.
20. Akiyama, H, Chaboissier, MC, Martin, JF, et al. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16:2813–2828.
21. Akiyama, H, Stadler, HS, Martin, JF, et al. Misexpression of Sox9 in mouse limb bud mesenchyme induces polydactyly and rescues hypodactyly mice. Matrix Biol. 2007;26:224–233.
22. Bi, W, Deng, JM, Zhang, Z, et al. Sox9 is required for cartilage formation. Nat Genet. 1999;22:85–89.
23. Smits, P, Li, P, Mandel, J, et al. The transcription factors L-Sox5 and Sox6 are essential for cartilage formation. Dev Cell. 2001;1:277–290.
24. Vajo, Z, Francomano, CA, Wilkin, DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocr Rev. 2000;21:23–39.
25. Naski, MC, Wang, Q, Xu, J, et al. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat Genet. 1996;13:233–237.
26. Bellus, GA, McIntosh, I, Smith, EA, et al. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat Genet. 1995;10:357–359.
27. Shiang, R, Thompson, LM, Zhu, YZ, et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78:335–342.
28. Schipani, E, Langman, CB, Parfitt, AM, et al. Constitutively activated receptors for parathyroid hormone and parathyroid hormone-related peptide in Jansen’s metaphyseal chondrodysplasia. N Engl J Med. 1996;335:708–714.
29. Schipani, E, Kruse, K, Juppner, H. A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science. 1995;268:98–100.
30. Karaplis, AC, He, B, Nguyen, MT, et al. Inactivating mutation in the human parathyroid hormone receptor type 1 gene in Blomstrand chondrodysplasia. Endocrinology. 1998;139:5255–5258.
31. Goldring, MB, Tsuchimochi, K, Ijiri, K. The control of chondrogenesis. J Cell Biochem. 2006;97:33–44.
32. Kronenberg, HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336.
33. Kappen, C, Neubuser, A, Balling, R, et al. Molecular basis for skeletal variation: insights from developmental genetic studies in mice. Birth Defects Res B Dev Reprod Toxicol. 2007;80:425–450.
34. Shi, Y, Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700.
35. Sampath, TK, Reddi, AH. Dissociative extraction and reconstitution of extracellular matrix components involved in local bone differentiation. Proc Natl Acad Sci U S A. 1981;78:7599–7603.
36. Urist, MR. Bone: formation by autoinduction. Science. 1965;150:893–899.
37. Wozney, JM, Rosen, V, Celeste, AJ, et al. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242:1528–1534.
38. Luyten, FP, Cunningham, NS, Ma, S, et al. Purification and partial amino acid sequence of osteogenin, a protein initiating bone differentiation. J Biol Chem. 1989;264:13377–13380.
39. Hogan, BL. Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev. 1996;10:1580–1594.
40. Chen, D, Zhao, M, Mundy, GR. Bone morphogenetic proteins. Growth Factors. 2004;22:233–241.
41. Kingsley, DM. What do BMPs do in mammals? Clues from the mouse short-ear mutation. Trends Genet. 1994;10:16–21.
42. Storm, EE, Huynh, TV, Copeland, NG, et al. Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature. 1994;368:639–643.
43. Yoon, BS, Ovchinnikov, DA, Yoshii, I, et al. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci U S A. 2005;102:5062–5067.
44. Pizette, S, Niswander, L. BMPs are required at two steps of limb chondrogenesis: formation of prechondrogenic condensations and their differentiation into chondrocytes. Dev Biol. 2000;219:237–249.
45. Tsumaki, N, Nakase, T, Miyaji, T, et al. Bone morphogenetic protein signals are required for cartilage formation and differently regulate joint development during skeletogenesis. J Bone Miner Res. 2002;17:898–906.
46. Yoon, BS, Pogue, R, Ovchinnikov, DA, et al. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development. 2006;133:4667–4678.
47. Minina, E, Kreschel, C, Naski, MC, et al. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev Cell. 2002;3:439–449.
48. Pathi, S, Rutenberg, JB, Johnson, RL, et al. Interaction of Ihh and BMP/Noggin signaling during cartilage differentiation. Dev Biol. 1999;209:239–253.
49. Bandyopadhyay, A, Tsuji, K, Cox, K, et al. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2:e216.
50. Ornitz, DM. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 2005;16:205–213.
51. Liu, Z, Xu, J, Colvin, JS, et al. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 2002;16:859–869.
52. Ohbayashi, N, Shibayama, M, Kurotaki, Y, et al. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 2002;16:870–879.
53. Hung, IH, Yu, K, Lavine, KJ, et al. FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev Biol. 2007;307:300–313.
54. Colvin, JS, Bohne, BA, Harding, GW, et al. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–397.
55. Deng, C, Wynshaw-Boris, A, Zhou, F, et al. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921.
56. Liu, Z, Lavine, KJ, Hung, IH, et al. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev Biol. 2007;302:80–91.
57. Sahni, M, Ambrosetti, DC, Mansukhani, A, et al. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 1999;13:1361–1366.
58. Su, WC, Kitagawa, M, Xue, N, et al. Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature. 1997;386:288–292.
59. Legeai-Mallet, L, Benoist-Lasselin, C, Munnich, A, et al. Overexpression of FGFR3, Stat1, Stat5 and p21Cip1 correlates with phenotypic severity and defective chondrocyte differentiation in FGFR3-related chondrodysplasias. Bone. 2004;34:26–36.
60. Naski, MC, Colvin, JS, Coffin, JD, et al. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development. 1998;125:4977–4988.
61. Segev, O, Chumakov, I, Nevo, Z, et al. Restrained chondrocyte proliferation and maturation with abnormal growth plate vascularization and ossification in human FGFR-3(G380R) transgenic mice. Hum Mol Genet. 2000;9:249–258.
62. Wang, Y, Spatz, MK, Kannan, K, et al. A mouse model for achondroplasia produced by targeting fibroblast growth factor receptor 3. Proc Natl Acad Sci U S A. 1999;96:4455–4460.
63. Iwata, T, Chen, L, Li, C, et al. A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum Mol Genet. 2000;9:1603–1613.
64. Murakami, S, Kan, M, McKeehan, WL, et al. Up-regulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proc Natl Acad Sci U S A. 2000;97:1113–1118.
65. Murakami, S, Balmes, G, McKinney, S, et al. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev. 2004;18:290–305.
66. Yasoda, A, Komatsu, Y, Chusho, H, et al. Overexpression of CNP in chondrocytes rescues achondroplasia through a MAPK-dependent pathway. Nat Med. 2004;10:80–86.
67. de Frutos, CA, Vega, S, Manzanares, M, et al. Snail1 is a transcriptional effector of FGFR3 signaling during chondrogenesis and achondroplasias. Dev Cell. 2007;13:872–883.
68. Li, C, Chen, L, Iwata, T, et al. A Lys644Glu substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by activation of STATs and ink4 cell cycle inhibitors. Hum Mol Genet. 1999;8:35–44.
69. Chen, L, Li, C, Qiao, W, et al. A Ser(365)→Cys mutation of fibroblast growth factor receptor 3 in mouse downregulates Ihh/PTHrP signals and causes severe achondroplasia. Hum Mol Genet. 2001;10:457–465.
70. Lanske, B, Karaplis, AC, Lee, K, et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273:663–666.
71. Vortkamp, A, Lee, K, Lanske, B, et al. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622.
72. St Jacques, B, Hammerschmidt, M, McMahon, AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–2086.
73. Karp, SJ, Schipani, E, St Jacques, B, et al. Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related protein dependent and independent pathways. Development. 2000;127:543–548.
74. Kobayashi, T, Chung, UI, Schipani, E, et al. PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development. 2002;129:2977–2986.
75. Kobayashi, T, Soegiarto, DW, Yang, Y, et al. Indian hedgehog stimulates periarticular chondrocyte differentiation to regulate growth plate length independently of PTHrP. J Clin Invest. 2005;115:1734–1742.
76. Inada, M, Yasui, T, Nomura, S, et al. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev Dyn. 1999;214:279–290.
77. Kim, IS, Otto, F, Zabel, B, et al. Regulation of chondrocyte differentiation by Cbfa1. Mech Dev. 1999;80:159–170.
78. Takeda, S, Bonnamy, JP, Owen, MJ, et al. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 2001;15:467–481.
79. Ueta, C, Iwamoto, M, Kanatani, N, et al. Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J Cell Biol. 2001;153:87–100.
80. Maeda, Y, Nakamura, E, Nguyen, MT, et al. Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc Natl Acad Sci U S A. 2007;104:6382–6387.
81. Karsenty, G, Wagner, EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2:389–406.
82. Provot, S, Schipani, E. Fetal growth plate: a developmental model of cellular adaptation to hypoxia. Ann N Y Acad Sci. 2007;1117:26–39.
83. Schipani, E. Hypoxia and HIF-1alpha in chondrogenesis. Ann N Y Acad Sci. 2006;1068:66–73.
84. Semenza, GL. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107:1–3.
85. Provot, S, Zinyk, D, Gunes, Y, et al. Hif-1alpha regulates differentiation of limb bud mesenchyme and joint development. J Cell Biol. 2007;177:451–464.
86. Schipani, E, Ryan, HE, Didrickson, S, et al. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15:2865–2876.
87. Maes, C, Stockmans, I, Moermans, K, et al. Soluble VEGF isoforms are essential for establishing epiphyseal vascularization and regulating chondrocyte development and survival. J Clin Invest. 2004;113:188–199.
88. Zelzer, E, Mamluk, R, Ferrara, N, et al. VEGFA is necessary for chondrocyte survival during bone development. Development. 2004;131:2161–2171.
89. Rajpurohit, R, Koch, CJ, Tao, Z, et al. Adaptation of chondrocytes to low oxygen tension: relationship between hypoxia and cellular metabolism. J Cell Physiol. 1996;168:424–432.
90. Mobasheri, A, Richardson, S, Mobasheri, R, et al. Hypoxia inducible factor-1 and facilitative glucose transporters GLUT1 and GLUT3: putative molecular components of the oxygen and glucose sensing apparatus in articular chondrocytes. Histol Histopathol. 2005;20:1327–1338.
91. Pfander, D, Cramer, T, Schipani, E, et al. HIF-1alpha controls extracellular matrix synthesis by epiphyseal chondrocytes. J Cell Sci. 2003;116:1819–1826.
92. Colnot, C, Lu, C, Hu, D, et al. Distinguishing the contributions of the perichondrium, cartilage, and vascular endothelium to skeletal development. Dev Biol. 2004;269:55–69.
93. Trueta, J, Amato, VP. The vascular contribution to osteogenesis. III. Changes in the growth cartilage caused by experimentally induced ischaemia. J Bone Joint Surg Br. 1960;42:571–587.
94. Brashear, HR, Jr. Epiphyseal avascular necrosis and its relation to longitudinal bone growth. J Bone Joint Surg Am. 1963;45:1423–1438.
95. Brandi, ML, Collin-Osdoby, P. Vascular biology and the skeleton. J Bone Miner Res. 2006;21:183–192.
96. Erlebacher, A, Filvaroff, EH, Gitelman, SE, et al. Toward a molecular understanding of skeletal development. Cell. 1995;80:371–378.
97. Karsenty, G. The complexities of skeletal biology. Nature. 2003;423:316–318.
98. Trueta, J, Morgan, JD. The vascular contribution to osteogenesis. I. Studies by the injection method. J Bone Joint Surg Br. 1960;42:97–109.
99. Brighton, CT. Structure and function of the growth plate. Clin Orthop Relat Res. 1978;136:22–32.
100. Moses, MA, Sudhalter, J, Langer, R. Identification of an inhibitor of neovascularization from cartilage. Science. 1990;248:1408–1410.
101. Hiraki, Y, Inoue, H, Iyama, K, et al. Identification of chondromodulin I as a novel endothelial cell growth inhibitor: purification and its localization in the avascular zone of epiphyseal cartilage. J Biol Chem. 1997;272:32419–32426.
102. Shukunami, C, Hiraki, Y. Chondromodulin-I and tenomodulin: the negative control of angiogenesis in connective tissue. Curr Pharm Des. 2007;13:2101–2112.
103. Moses, MA, Wiederschain, D, Wu, I, et al. Troponin I is present in human cartilage and inhibits angiogenesis. Proc Natl Acad Sci U S A. 1999;96:2645–2650.
104. Gerber, HP, Ferrara, N. Angiogenesis and bone growth. Trends Cardiovasc Med. 2000;10:223–228.
105. Descalzi, CF, Melchiori, A, Benelli, R, et al. Production of angiogenesis inhibitors and stimulators is modulated by cultured growth plate chondrocytes during in vitro differentiation: dependence on extracellular matrix assembly. Eur J Cell Biol. 1995;66:60–68.
106. Cramer, T, Schipani, E, Johnson, RS, et al. Expression of VEGF isoforms by epiphyseal chondrocytes during low-oxygen tension is HIF-1 alpha dependent. Osteoarthritis Cartilage. 2004;12:433–439.
107. Kim, HH, Lee, SE, Chung, WJ, et al. Stabilization of hypoxia-inducible factor-1alpha is involved in the hypoxic stimuli-induced expression of vascular endothelial growth factor in osteoblastic cells. Cytokine. 2002;17:14–27.
108. Akeno, N, Czyzyk-Krzeska, MF, Gross, TS, et al. Hypoxia induces vascular endothelial growth factor gene transcription in human osteoblast-like cells through the hypoxia-inducible factor-2alpha. Endocrinology. 2001;142:959–962.
109. Knowles, HJ, Athanasou, NA. Hypoxia-inducible factor is expressed in giant cell tumour of bone and mediates paracrine effects of hypoxia on monocyte-osteoclast differentiation via induction of VEGF. J Pathol. 2008;215:56–66.
110. Wang, Y, Wan, C, Deng, L, et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest. 2007;117:1616–1626.
111. Zelzer, E, Glotzer, DJ, Hartmann, C, et al. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech Dev. 2001;106:97–106.
112. Dai, J, Rabie, AB. VEGF: an essential mediator of both angiogenesis and endochondral ossification. J Dent Res. 2007;86:937–950.
113. Gerber, HP, Vu, TH, Ryan, AM, et al. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5:623–628.
114. Maes, C, Carmeliet, P, Moermans, K, et al. Impaired angiogenesis and endochondral bone formation in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Mech Dev. 2002;111:61–73.
115. Zelzer, E, McLean, W, Ng, YS, et al. Skeletal defects in VEGF(120/120) mice reveal multiple roles for VEGF in skeletogenesis. Development. 2002;129:1893–1904.
116. Haigh, JJ, Gerber, HP, Ferrara, N, et al. Conditional inactivation of VEGF-A in areas of collagen2a1 expression results in embryonic lethality in the heterozygous state. Development. 2000;127:1445–1453.
117. Maes, C, Carmeliet, G. Vascular and nonvascular roles of VEGF in bone development. In: Ruhrberg C, ed. VEGF in Development. Austin: Springer; 2008:79–90.
118. Zelzer, E, Olsen, BR. Multiple roles of vascular endothelial growth factor (VEGF) in skeletal development, growth, and repair. Curr Top Dev Biol. 2005;65:169–187.
119. Fiedler, J, Leucht, F, Waltenberger, J, et al. VEGF-A and PlGF-1 stimulate chemotactic migration of human mesenchymal progenitor cells. Biochem Biophys Res Commun. 2005;334:561–568.
120. Mayr-Wohlfart, U, Waltenberger, J, Hausser, H, et al. Vascular endothelial growth factor stimulates chemotactic migration of primary human osteoblasts. Bone. 2002;30:472–477.
121. Aldridge, SE, Lennard, TW, Williams, JR, et al. Vascular endothelial growth factor receptors in osteoclast differentiation and function. Biochem Biophys Res Commun. 2005;335:793–798.
122. Engsig, MT, Chen, QJ, Vu, TH, et al. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J Cell Biol. 2000;151:879–889.
123. Henriksen, K, Karsdal, M, Delaisse, JM, et al. RANKL and vascular endothelial growth factor (VEGF) induce osteoclast chemotaxis through an ERK1/2-dependent mechanism. J Biol Chem. 2003;278:48745–48753.
124. Nakagawa, M, Kaneda, T, Arakawa, T, et al. Vascular endothelial growth factor (VEGF) directly enhances osteoclastic bone resorption and survival of mature osteoclasts. FEBS Lett. 2000;473:161–164.
125. Niida, S, Kondo, T, Hiratsuka, S, et al. VEGF receptor 1 signaling is essential for osteoclast development and bone marrow formation in colony-stimulating factor 1-deficient mice. Proc Natl Acad Sci U S A. 2005;102:14016–14021.
126. Yang, Q, McHugh, KP, Patntirapong, S, et al. VEGF enhancement of osteoclast survival and bone resorption involves VEGF receptor-2 signaling and beta(3)-integrin. Matrix Biol. 2008;27:589–599.
127. Yao, S, Liu, D, Pan, F, et al. Effect of vascular endothelial growth factor on RANK gene expression in osteoclast precursors and on osteoclastogenesis. Arch Oral Biol. 2006;51:596–602.
128. Vu, TH, Shipley, JM, Bergers, G, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422.
129. Nakashima, T, Takayanagi, H. The dynamic interplay between osteoclasts and the immune system. Arch Biochem Biophys. 2008;473:166–171.
130. Boyle, WJ, Simonet, WS, Lacey, DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342.
131. Lorenzo, J, Horowitz, M, Choi, Y. Osteoimmunology: interactions of the bone and immune system. Endocr Rev. 2008;29:403–440.
132. Takayanagi, H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7:292–304.
133. Boyce, BF, Yao, Z, Zhang, Q, et al. New roles for osteoclasts in bone. Ann N Y Acad Sci. 2007;1116:245–254.
134. Xing, L, Schwarz, EM, Boyce, BF. Osteoclast precursors, RANKL/RANK, and immunology. Immunol Rev. 2005;208:19–29.
135. Scott, EW, Simon, MC, Anastasi, J, et al. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577.
136. DeKoter, RP, Singh, H. Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science. 2000;288:1439–1441.
137. Tondravi, MM, McKercher, SR, Anderson, K, et al. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature. 1997;386:81–84.
138. DeKoter, RP, Walsh, JC, Singh, H. PU.1 regulates both cytokine-dependent proliferation and differentiation of granulocyte/macrophage progenitors. EMBO J. 1998;17:4456–4468.
139. Zhang, DE, Hetherington, CJ, Chen, HM, et al. The macrophage transcription factor PU.1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol Cell Biol. 1994;14:373–381.
140. Tondravi, MM, McKercher, SR, Anderson, K, et al. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature. 1997;386:81–84.
141. McKercher, SR, Torbett, BE, Anderson, KL, et al. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996;15:5647–5658.
142. Kwon, OH, Lee, CK, Lee, YI, et al. The hematopoietic transcription factor PU.1 regulates RANK gene expression in myeloid progenitors. Biochem Biophys Res Commun. 2005;335:437–446.
143. Huang, JC, Sakata, T, Pfleger, LL, et al. PTH differentially regulates expression of RANKL and OPG. J Bone Miner Res. 2004;19:235–244.
144. Kitazawa, S, Kajimoto, K, Kondo, T, et al. Vitamin D3 supports osteoclastogenesis via functional vitamin D response element of human RANKL gene promoter. J Cell Biochem. 2003;89:771–777.
145. Quinn, JM, Elliott, J, Gillespie, MT, et al. A combination of osteoclast differentiation factor and macrophage-colony stimulating factor is sufficient for both human and mouse osteoclast formation in vitro. Endocrinology. 1998;139:4424–4427.
146. Kishimoto, K, Kitazawa, R, Kurosaka, M, et al. Expression profile of genes related to osteoclastogenesis in mouse growth plate and articular cartilage. Histochem Cell Biol. 2006;125:593–602.
147. Masuyama, R, Stockmans, I, Torrekens, S, et al. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest. 2006;116:3150–3159.
148. Usui, M, Xing, L, Drissi, H, et al. Murine and chicken chondrocytes regulate osteoclastogenesis by producing RANKL in response to BMP2. J Bone Miner Res. 2008;23:314–325.
149. Horwood, NJ, Kartsogiannis, V, Quinn, JM, et al. Activated T lymphocytes support osteoclast formation in vitro. Biochem Biophys Res Commun. 1999;265:144–150.
150. Weitzmann, MN, Cenci, S, Rifas, L, et al. T cell activation induces human osteoclast formation via receptor activator of nuclear factor kappaB ligand-dependent and -independent mechanisms. J Bone Miner Res. 2001;16:328–337.
151. Kong, YY, Feige, U, Sarosi, I, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309.
152. Lam, J, Takeshita, S, Barker, JE, et al. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106:1481–1488.
153. Fuller, K, Murphy, C, Kirstein, B, et al. TNFalpha potently activates osteoclasts, through a direct action independent of and strongly synergistic with RANKL. Endocrinology. 2002;143:1108–1118.
154. Felix, R, Cecchini, MG, Hofstetter, W, et al. Impairment of macrophage colony-stimulating factor production and lack of resident bone marrow macrophages in the osteopetrotic op/op mouse. J Bone Miner Res. 1990;5:781–789.
155. Kodama, H, Yamasaki, A, Abe, M, et al. Transient recruitment of osteoclasts and expression of their function in osteopetrotic (op/op) mice by a single injection of macrophage colony-stimulating factor. J Bone Miner Res. 1993;8:45–50.
156. Kodama, H, Yamasaki, A, Nose, M, et al. Congenital osteoclast deficiency in osteopetrotic (op/op) mice is cured by injections of macrophage colony-stimulating factor. J Exp Med. 1991;173:269–272.
157. Yoshida, H, Hayashi, S, Kunisada, T, et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444.
158. Armstrong, AP, Tometsko, ME, Glaccum, M, et al. A RANK/TRAF6-dependent signal transduction pathway is essential for osteoclast cytoskeletal organization and resorptive function. J Biol Chem. 2002;277:44347–44356.
159. Burgess, TL, Qian, Y, Kaufman, S, et al. The ligand for osteoprotegerin (OPGL) directly activates mature osteoclasts. J Cell Biol. 1999;145:527–538.
160. Lacey, DL, Timms, E, Tan, HL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176.
161. Lacey, DL, Tan, HL, Lu, J, et al. Osteoprotegerin ligand modulates murine osteoclast survival in vitro and in vivo. Am J Pathol. 2000;157:435–448.
162. Hughes, AE, Ralston, SH, Marken, J, et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet. 2000;24:45–48.
163. Takayanagi, H. Mechanistic insight into osteoclast differentiation in osteoimmunology. J Mol Med. 2005;83:170–179.
164. Takayanagi, H, Kim, S, Koga, T, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901.
165. Grigoriadis, AE, Wang, ZQ, Cecchini, MG, et al. c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994;266:443–448.
166. Iotsova, V, Caamano, J, Loy, J, et al. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997;3:1285–1289.
167. Lomaga, MA, Yeh, WC, Sarosi, I, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–1024.
168. Boyce, BF, Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. 2008;473:139–146.
169. Whyte, MP, Obrecht, SE, Finnegan, PM, et al. Osteoprotegerin deficiency and juvenile Paget’s disease. N Engl J Med. 2002;347:175–184.
170. Kong, YY, Yoshida, H, Sarosi, I, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323.
171. Li, J, Sarosi, I, Yan, XQ, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci U S A. 2000;97:1566–1571.
172. Dougall, WC, Glaccum, M, Charrier, K, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424.
173. Simonet, WS, Lacey, DL, Dunstan, CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319.
174. Bucay, N, Sarosi, I, Dunstan, CR, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268.
175. Mizuno, A, Amizuka, N, Irie, K, et al. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun. 1998;247:610–615.
176. Mizuno, A, Kanno, T, Hoshi, M, et al. Transgenic mice overexpressing soluble osteoclast differentiation factor (sODF) exhibit severe osteoporosis. J Bone Miner Metab. 2002;20:337–344.
177. Fata, JE, Kong, YY, Li, J, et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell. 2000;103:41–50.
178. Koga, T, Inui, M, Inoue, K, et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004;428:758–763.
179. Yagi, M, Miyamoto, T, Sawatani, Y, et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202:345–351.
180. Trebec, DP, Chandra, D, Gramoun, A, et al. Increased expression of activating factors in large osteoclasts could explain their excessive activity in osteolytic diseases. J Cell Biochem. 2007;101:205–220.
181. Bar-Shavit, Z. The osteoclast: a multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell. J Cell Biochem. 2007;102:1130–1139.
182. Roodman, GD, Windle, JJ. Paget disease of bone. J Clin Invest. 2005;115:200–208.
183. Hsu, H, Lacey, DL, Dunstan, CR, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci U S A. 1999;96:3540–3545.
184. Gil-Henn, H, Destaing, O, Sims, NA, et al. Defective microtubule-dependent podosome organization in osteoclasts leads to increased bone density in Pyk2(-/-) mice. J Cell Biol. 2007;178:1053–1064.
185. Jurdic, P, Saltel, F, Chabadel, A, et al. Podosome and sealing zone: specificity of the osteoclast model. Eur J Cell Biol. 2006;85:195–202.
186. Luxenburg, C, Geblinger, D, Klein, E, et al. The architecture of the adhesive apparatus of cultured osteoclasts: from podosome formation to sealing zone assembly. PLoS ONE. 2007;2:e179.
187. Luxenburg, C, Parsons, JT, Addadi, L, et al. Involvement of the Src-cortactin pathway in podosome formation and turnover during polarization of cultured osteoclasts. J Cell Sci. 2006;119:4878–4888.
188. Teitelbaum, SL. Osteoclasts: what do they do and how do they do it? Am J Pathol. 2007;170:427–435.
189. Vaananen, HK, Laitala-Leinonen, T. Osteoclast lineage and function. Arch Biochem Biophys. 2008;473:132–138.
190. Wittrant, Y, Theoleyre, S, Couillaud, S, et al. Regulation of osteoclast protease expression by RANKL. Biochem Biophys Res Commun. 2003;310:774–778.
191. Fujisaki, K, Tanabe, N, Suzuki, N, et al. Receptor activator of NF-kappaB ligand induces the expression of carbonic anhydrase II, cathepsin K, and matrix metalloproteinase-9 in osteoclast precursor RAW264.7 cells. Life Sci. 2007;80:1311–1318.
192. Sundaram, K, Nishimura, R, Senn, J, et al. RANK ligand signaling modulates the matrix metalloproteinase-9 gene expression during osteoclast differentiation. Exp Cell Res. 2007;313:168–178.
193. Nakamura, I, Pilkington, MF, Lakkakorpi, PT, et al. Role of alpha(v)beta(3) integrin in osteoclast migration and formation of the sealing zone. J Cell Sci. 1999;112(Pt 22):3985–3993.
194. Nakamura, I, Duong, IT, Rodan, SB, et al. Involvement of alpha(v)beta3 integrins in osteoclast function. J Bone Miner Metab. 2007;25:337–344.
195. Pfaff, M, Jurdic, P. Podosomes in osteoclast-like cells: structural analysis and cooperative roles of paxillin, proline-rich tyrosine kinase 2 (Pyk2) and integrin alphaVbeta3. J Cell Sci. 2001;114:2775–2786.
196. Nakamura, I, Duong, IT, Rodan, SB, et al. Involvement of alpha(v)beta3 integrins in osteoclast function. J Bone Miner Metab. 2007;25:337–344.
197. Rodan, SB, Rodan, GA. Integrin function in osteoclasts. J Endocrinol. 1997;154(suppl):S47–S56.
198. Rodan, SB, Rodan, GA. Integrin function in osteoclasts. J Endocrinol. 1997;154(suppl):S47–S56.
199. Bossard, MJ, Tomaszek, TA, Thompson, SK, et al. Proteolytic activity of human osteoclast cathepsin K: expression, purification, activation, and substrate identification. J Biol Chem. 1996;271:12517–12524.
200. Everts, V, Korper, W, Hoeben, KA, et al. Osteoclastic bone degradation and the role of different cysteine proteinases and matrix metalloproteinases: differences between calvaria and long bone. J Bone Miner Res. 2006;21:1399–1408.
201. Kamiya, T, Kobayashi, Y, Kanaoka, K, et al. Fluorescence microscopic demonstration of cathepsin K activity as the major lysosomal cysteine proteinase in osteoclasts. J Biochem. 1998;123:752–759.
202. Gowen, M, Lazner, F, Dodds, R, et al. Cathepsin K knockout mice develop osteopetrosis due to a deficit in matrix degradation but not demineralization. J Bone Miner Res. 1999;14:1654–1663.
203. Saftig, P, Hunziker, E, Wehmeyer, O, et al. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc Natl Acad Sci U S A. 1998;95:13453–13458.
204. Johnson, MR, Polymeropoulos, MH, Vos, HL, et al. A nonsense mutation in the cathepsin K gene observed in a family with pycnodysostosis. Genome Res. 1996;6:1050–1055.
205. Le Gall, C, Bellahcene, A, Bonnelye, E, et al. A cathepsin K inhibitor reduces breast cancer induced osteolysis and skeletal tumor burden. Cancer Res. 2007;67:9894–9902.
206. Le Gall, C, Bonnelye, E, Clezardin, P. Cathepsin K inhibitors as treatment of bone metastasis. Curr Opin Support Palliat Care. 2008;2:218–222.
207. Pearse, RN. New strategies for the treatment of metastatic bone disease. Clin Breast Cancer. 2007;8(suppl 1):S35–S45.
208. Stoch, SA, Wagner, JA. Cathepsin K inhibitors: a novel target for osteoporosis therapy. Clin Pharmacol Ther. 2008;83:172–176.
209. Delaisse, JM, Andersen, TL, Engsig, MT, et al. Matrix metalloproteinases (MMP) and cathepsin K contribute differently to osteoclastic activities. Microsc Res Tech. 2003;61:504–513.
210. Cawston, TE, Wilson, AJ. Understanding the role of tissue degrading enzymes and their inhibitors in development and disease. Best Pract Res Clin Rheumatol. 2006;20:983–1002.
211. Krane, SM, Inada, M. Matrix metalloproteinases and bone. Bone. 2008;43:7–18.
212. Ortega, N, Behonick, D, Stickens, D, et al. How proteases regulate bone morphogenesis. Ann N Y Acad Sci. 2003;995:109–116.
213. Sternlicht, MD, Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516.
214. Egeblad, M, Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174.
215. Martin, TJ, Moseley, JM. Mechanisms in the skeletal complications of breast cancer. Endocr Relat Cancer. 2000;7:271–284.
216. Sato, T, del Carmen, OM, Hou, P, et al. Identification of the membrane-type matrix metalloproteinase MT1-MMP in osteoclasts. J Cell Sci. 1997;110(Pt 5):589–596.
217. Tezuka, K, Nemoto, K, Tezuka, Y, et al. Identification of matrix metalloproteinase 9 in rabbit osteoclasts. J Biol Chem. 1994;269:15006–15009.
218. Colnot, C, Thompson, Z, Miclau, T, et al. Altered fracture repair in the absence of MMP9. Development. 2003;130:4123–4133.
219. Holmbeck, K, Bianco, P, Chrysovergis, K, et al. MT1-MMP-dependent, apoptotic remodeling of unmineralized cartilage: a critical process in skeletal growth. J Cell Biol. 2003;163:661–671.
220. Holmbeck, K, Bianco, P, Caterina, J, et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92.
221. Hikita, A, Yana, I, Wakeyama, H, et al. Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-kappaB ligand. J Biol Chem. 2006;281:36846–36855.
222. Lynch, CC, Hikosaka, A, Acuff, HB, et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell. 2005;7:485–496.
223. Manolagas, SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–137.
224. Hughes, DE, Dai, A, Tiffee, JC, et al. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nat Med. 1996;2:1132–1136.
225. Caplan, AI, Bruder, SP. Mesenchymal stem cells: building blocks for molecular medicine in the 21st century. Trends Mol Med. 2001;7:259–264.
226. Chen, Y, Shao, JZ, Xiang, LX, et al. Mesenchymal stem cells: a promising candidate in regenerative medicine. Int J Biochem Cell Biol. 2008;40:815–820.
227. Jiang, Y, Jahagirdar, BN, Reinhardt, RL, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418:41–49.
228. Nishimura, R, Hata, K, Ikeda, F, et al. Signal transduction and transcriptional regulation during mesenchymal cell differentiation. J Bone Miner Metab. 2008;26:203–212.
229. Harada, S, Rodan, GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–355.
230. Karsenty, G. Transcriptional control of skeletogenesis. Annu Rev Genomics Hum Genet. 2008;9:183–196.
231. Stein, GS, Lian, JB. Molecular mechanisms mediating proliferation/differentiation interrelationships during progressive development of the osteoblast phenotype. Endocr Rev. 1993;14:424–442.
232. Knothe Tate, ML, Adamson, JR, Tami, AE, et al. The osteocyte. Int J Biochem Cell Biol. 2004;36:1–8.
233. Noble, BS. The osteocyte lineage. Arch Biochem Biophys. 2008;473:106–111.
234. Kamioka, H, Honjo, T, Takano-Yamamoto, T. A three-dimensional distribution of osteocyte processes revealed by the combination of confocal laser scanning microscopy and differential interference contrast microscopy. Bone. 2001;28:145–149.
235. Sugawara, Y, Kamioka, H, Honjo, T, et al. Three-dimensional reconstruction of chick calvarial osteocytes and their cell processes using confocal microscopy. Bone. 2005;36:877–883.
236. Bonewald, LF, Johnson, ML. Osteocytes, mechanosensing and Wnt signaling. Bone. 2008;42:606–615.
237. Strom, TM, Juppner, H. PHEX, FGF23, DMP1 and beyond. Curr Opin Nephrol Hypertens. 2008;17:357–362.
238. Datta, HK, Ng, WF, Walker, JA, et al. The cell biology of bone metabolism. J Clin Pathol. 2008;61:577–587.
239. Civitelli, R. Cell-cell communication in the osteoblast/osteocyte lineage. Arch Biochem Biophys. 2008;473:188–192.
240. Poole, KE, van Bezooijen, RL, Loveridge, N, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19:1842–1844.
241. van Bezooijen, RL, Ten Dijke, P, Papapoulos, SE, et al. SOST/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev. 2005;16:319–327.
242. Toyosawa, S, Shintani, S, Fujiwara, T, et al. Dentin matrix protein 1 is predominantly expressed in chicken and rat osteocytes but not in osteoblasts. J Bone Miner Res. 2001;16:2017–2026.
243. Liu, S, Gupta, A, Quarles, LD. Emerging role of fibroblast growth factor 23 in a bone-kidney axis regulating systemic phosphate homeostasis and extracellular matrix mineralization. Curr Opin Nephrol Hypertens. 2007;16:329–335.
244. Nampei, A, Hashimoto, J, Hayashida, K, et al. Matrix extracellular phosphoglycoprotein (MEPE) is highly expressed in osteocytes in human bone. J Bone Miner Metab. 2004;22:176–184.
245. Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480.
246. Taipale, J, Beachy, PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–354.
247. Logan, CY, Nusse, R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810.
248. Klaus, A, Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398.
249. Gong, Y, Slee, RB, Fukai, N, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–523.
250. Loughlin, J, Dowling, B, Chapman, K, et al. Functional variants within the secreted frizzled-related protein 3 gene are associated with hip osteoarthritis in females. Proc Natl Acad Sci U S A. 2004;101:9757–9762.
251. Grigoryan, T, Wend, P, Klaus, A, et al. Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 2008;22:2308–2341.
252. Rubinfeld, B, Albert, I, Porfiri, E, et al. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026.
253. Behrens, J, Jerchow, BA, Wurtele, M, et al. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280:596–599.
254. Mao, J, Wang, J, Liu, B, et al. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell. 2001;7:801–809.
255. Pinson, KI, Brennan, J, Monkley, S, et al. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000;407:535–538.
256. Tamai, K, Semenov, M, Kato, Y, et al. LDL-receptor-related proteins in Wnt signal transduction. Nature. 2000;407:530–535.
257. Tamai, K, Zeng, X, Liu, C, et al. A mechanism for Wnt coreceptor activation. Mol Cell. 2004;13:149–156.
258. Zeng, X, Huang, H, Tamai, K, et al. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development. 2008;135:367–375.
259. Zeng, X, Tamai, K, Doble, B, et al. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature. 2005;438:873–877.
260. Behrens, J, von Kries, JP, Kuhl, M, et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642.
261. Moon, RT, Bowerman, B, Boutros, M, et al. The promise and perils of Wnt signaling through beta-catenin. Science. 2002;296:1644–1646.
262. Shitashige, M, Hirohashi, S, Yamada, T. Wnt signaling inside the nucleus. Cancer Sci. 2008;99:631–637.
263. Kolpakova, E, Olsen, BR. Wnt/beta-catenin—a canonical tale of cell-fate choice in the vertebrate skeleton. Dev Cell. 2005;8:626–627.
264. Hill, TP, Spater, D, Taketo, MM, et al. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8:727–738.
265. Glass, DA, Bialek, P, Ahn, JD, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–764.
266. Glass, DA, Karsenty, G. In vivo analysis of Wnt signaling in bone. Endocrinology. 2007;148:2630–2634.
267. Ai, M, Holmen, SL, Van Hul, W, et al. Reduced affinity to and inhibition by DKK1 form a common mechanism by which high bone mass-associated missense mutations in LRP5 affect canonical Wnt signaling. Mol Cell Biol. 2005;25:4946–4955.
268. Balemans, W, Devogelaer, JP, Cleiren, E, et al. Novel LRP5 missense mutation in a patient with a high bone mass phenotype results in decreased DKK1-mediated inhibition of Wnt signaling. J Bone Miner Res. 2007;22:708–716.
269. Semenov, MV, He, X. LRP5 mutations linked to high bone mass diseases cause reduced LRP5 binding and inhibition by SOST. J Biol Chem. 2006;281:38276–38284.
270. Boyden, LM, Mao, J, Belsky, J, et al. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346:1513–1521.
271. Little, RD, Carulli, JP, Del Mastro, RG, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70:11–19.
272. Yadav, VK, Ryu, JH, Suda, N, et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135:825–837.
273. Qiang, YW, Barlogie, B, Rudikoff, S, et al. Dkk1-induced inhibition of Wnt signaling in osteoblast differentiation is an underlying mechanism of bone loss in multiple myeloma. Bone. 2008;42:669–680.
274. Tian, E, Zhan, F, Walker, R, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349:2483–2494.
275. Balemans, W, Van Hul, W. Identification of the disease-causing gene in sclerosteosis—discovery of a novel bone anabolic target? J Musculoskelet Neuronal Interact. 2004;4:139–142.
276. Balemans, W, Cleiren, E, Siebers, U, et al. A generalized skeletal hyperostosis in two siblings caused by a novel mutation in the SOST gene. Bone. 2005;36:943–947.
277. Balemans, W, Patel, N, Ebeling, M, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–97.
278. Baron, R, Rawadi, G, Roman-Roman, S. Wnt signaling: a key regulator of bone mass. Curr Top Dev Biol. 2006;76:103–127.
279. Krishnan, V, Bryant, HU, MacDougald, OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202–1209.
280. Otto, F, Thornell, AP, Crompton, T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771.
281. Komori, T, Yagi, H, Nomura, S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764.
282. Lee, B, Thirunavukkarasu, K, Zhou, L, et al. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet. 1997;16:307–310.
283. Mundlos, S, Otto, F, Mundlos, C, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773–779.
284. Rabie, AB, Tang, GH, Hagg, U. Cbfa1 couples chondrocytes maturation and endochondral ossification in rat mandibular condylar cartilage. Arch Oral Biol. 2004;49:109–118.
285. Ducy, P, Zhang, R, Geoffroy, V, et al. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754.
286. Kobayashi, H, Gao, Y, Ueta, C, et al. Multilineage differentiation of Cbfa1-deficient calvarial cells in vitro. Biochem Biophys Res Commun. 2000;273:630–636.
287. Komori, T. Regulation of skeletal development by the Runx family of transcription factors. J Cell Biochem. 2005;95:445–453.
288. Enomoto, H, Shiojiri, S, Hoshi, K, et al. Induction of osteoclast differentiation by Runx2 through receptor activator of nuclear factor-kappa B ligand (RANKL) and osteoprotegerin regulation and partial rescue of osteoclastogenesis in Runx2-/- mice by RANKL transgene. J Biol Chem. 2003;278:23971–23977.
289. Thirunavukkarasu, K, Halladay, DL, Miles, RR, et al. The osteoblast-specific transcription factor Cbfa1 contributes to the expression of osteoprotegerin, a potent inhibitor of osteoclast differentiation and function. J Biol Chem. 2000;275:25163–25172.
290. Harada, H, Tagashira, S, Fujiwara, M, et al. Cbfa1 isoforms exert functional differences in osteoblast differentiation. J Biol Chem. 1999;274:6972–6978.
291. Liu, W, Toyosawa, S, Furuichi, T, et al. Overexpression of Cbfa1 in osteoblasts inhibits osteoblast maturation and causes osteopenia with multiple fractures. J Cell Biol. 2001;155:157–166.
292. Ducy, P, Starbuck, M, Priemel, M, et al. A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev. 1999;13:1025–1036.
293. Nakashima, K, Zhou, X, Kunkel, G, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29.
294. Nishio, Y, Dong, Y, Paris, M, et al. Runx2-mediated regulation of the zinc finger Osterix/Sp7 gene. Gene. 2006;372:62–70.
295. Koga, T, Matsui, Y, Asagiri, M, et al. NFAT and Osterix cooperatively regulate bone formation. Nat Med. 2005;11:880–885.
296. Kaback, LA, Soung, DY, Naik, A, et al. Osterix/Sp7 regulates mesenchymal stem cell mediated endochondral ossification. J Cell Physiol. 2008;214:173–182.
297. Tominaga, H, Maeda, S, Miyoshi, H, et al. Expression of osterix inhibits bone morphogenetic protein-induced chondrogenic differentiation of mesenchymal progenitor cells. J Bone Miner Metab. 2009;27:36–45.
298. Elefteriou, F, Benson, MD, Sowa, H, et al. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasia. Cell Metab. 2006;4:441–451.
299. Yang, X, Karsenty, G. ATF4, the osteoblast accumulation of which is determined post-translationally, can induce osteoblast-specific gene expression in non-osteoblastic cells. J Biol Chem. 2004;279:47109–47114.
300. Yang, X, Matsuda, K, Bialek, P, et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology: implication for Coffin-Lowry syndrome. Cell. 2004;117:387–398.
301. Liu, YH, Tang, Z, Kundu, RK, et al. Msx2 gene dosage influences the number of proliferative osteogenic cells in growth centers of the developing murine skull: a possible mechanism for MSX2-mediated craniosynostosis in humans. Dev Biol. 1999;205:260–274.
302. Liu, YH, Kundu, R, Wu, L, et al. Premature suture closure and ectopic cranial bone in mice expressing Msx2 transgenes in the developing skull. Proc Natl Acad Sci U S A. 1995;92:6137–6141.
303. Satokata, I, Ma, L, Ohshima, H, et al. Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat Genet. 2000;24:391–395.
304. Satokata, I, Maas, R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet. 1994;6:348–356.
305. Acampora, D, Merlo, GR, Paleari, L, et al. Craniofacial, vestibular and bone defects in mice lacking the Distal-less-related gene Dlx5. Development. 1999;126:3795–3809.
306. Holleville, N, Mateos, S, Bontoux, M, et al. Dlx5 drives Runx2 expression and osteogenic differentiation in developing cranial suture mesenchyme. Dev Biol. 2007;304:860–874.
307. Li, H, Marijanovic, I, Kronenberg, MS, et al. Expression and function of Dlx genes in the osteoblast lineage. Dev Biol. 2008;316:458–470.
308. Robledo, RF, Rajan, L, Li, X, et al. The Dlx5 and Dlx6 homeobox genes are essential for craniofacial, axial, and appendicular skeletal development. Genes Dev. 2002;16:1089–1101.
309. Bialek, P, Kern, B, Yang, X, et al. A twist code determines the onset of osteoblast differentiation. Dev Cell. 2004;6:423–435.
310. Lee, MS, Lowe, GN, Strong, DD, et al. TWIST, a basic helix-loop-helix transcription factor, can regulate the human osteogenic lineage. J Cell Biochem. 1999;75:566–577.
311. Jochum, W, David, JP, Elliott, C, et al. Increased bone formation and osteosclerosis in mice overexpressing the transcription factor Fra-1. Nat Med. 2000;6:980–984.
312. Sabatakos, G, Sims, NA, Chen, J, et al. Overexpression of DeltaFosB transcription factor(s) increases bone formation and inhibits adipogenesis. Nat Med. 2000;6:985–990.
313. Wagner, EF. Functions of AP1 (Fos/Jun) in bone development. Ann Rheum Dis. 2002;61(suppl 2):ii40–ii42.
314. Wagner, EF, Eferl, R. Fos/AP-1 proteins in bone and the immune system. Immunol Rev. 2005;208:126–140.
315. Kenner, L, Hoebertz, A, Beil, T, et al. Mice lacking JunB are osteopenic due to cell-autonomous osteoblast and osteoclast defects. J Cell Biol. 2004;164:613–623.
316. Glimcher, LH, Jones, DC, Wein, MN. Control of postnatal bone mass by the zinc finger adapter protein Schnurri-3. Ann N Y Acad Sci. 2007;1116:174–181.
317. Jones, DC, Wein, MN, Oukka, M, et al. Regulation of adult bone mass by the zinc finger adapter protein Schnurri-3. Science. 2006;312:1223–1227.
318. Saita, Y, Takagi, T, Kitahara, K, et al. Lack of Schnurri-2 expression associates with reduced bone remodeling and osteopenia. J Biol Chem. 2007;282:12907–12915.
319. Franceschi, RT, Ge, C, Xiao, G, et al. Transcriptional regulation of osteoblasts. Cells Tissues Organs. 2009;189:144–152.
320. Komori, T. Regulation of osteoblast differentiation by transcription factors. J Cell Biochem. 2006;99:1233–1239.
321. Marie, PJ. Transcription factors controlling osteoblastogenesis. Arch Biochem Biophys. 2008;473:98–105.
322. Lian, JB, Stein, GS, Javed, A, et al. Networks and hubs for the transcriptional control of osteoblastogenesis. Rev Endocr Metab Disord. 2006;7:1–16.
323. Yamaguchi, A, Komori, T, Suda, T. Regulation of osteoblast differentiation mediated by bone morphogenetic proteins, hedgehogs, and Cbfa1. Endocr Rev. 2000;21:393–411.
324. Wu, XB, Li, Y, Schneider, A, et al. Impaired osteoblastic differentiation, reduced bone formation, and severe osteoporosis in noggin-overexpressing mice. J Clin Invest. 2003;112:924–934.
325. Gazzerro, E, Pereira, RC, Jorgetti, V, et al. Skeletal overexpression of gremlin impairs bone formation and causes osteopenia. Endocrinology. 2005;146:655–665.
326. Chen, D, Ji, X, Harris, MA, et al. Differential roles for bone morphogenetic protein (BMP) receptor type IB and IA in differentiation and specification of mesenchymal precursor cells to osteoblast and adipocyte lineages. J Cell Biol. 1998;142:295–305.
327. Ryoo, HM, Lee, MH, Kim, YJ. Critical molecular switches involved in BMP-2-induced osteogenic differentiation of mesenchymal cells. Gene. 2006;366:51–57.
328. Katagiri, T, Yamaguchi, A, Komaki, M, et al. Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J Cell Biol. 1994;127:1755–1766.
329. Katagiri, T, Yamaguchi, A, Ikeda, T, et al. The non-osteogenic mouse pluripotent cell line, C3H10T1/2, is induced to differentiate into osteoblastic cells by recombinant human bone morphogenetic protein-2. Biochem Biophys Res Commun. 1990;172:295–299.
330. Matsubara, T, Kida, K, Yamaguchi, A, et al. BMP2 regulates Osterix through Msx2 and Runx2 during osteoblast differentiation. J Biol Chem. 2008;283:29119–29125.
331. Javed, A, Bae, JS, Afzal, F, et al. Structural coupling of Smad and Runx2 for execution of the BMP2 osteogenic signal. J Biol Chem. 2008;283:8412–8422.
332. Lee, KS, Kim, HJ, Li, QL, et al. Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol Cell Biol. 2000;20:8783–8792.
333. Celil, AB, Campbell, PG. BMP-2 and insulin-like growth factor-I mediate Osterix (Osx) expression in human mesenchymal stem cells via the MAPK and protein kinase D signaling pathways. J Biol Chem. 2005;280:31353–31359.
334. Govender, S, Csimma, C, Genant, HK, et al. Recombinant human bone morphogenetic protein-2 for treatment of open tibial fractures: a prospective, controlled, randomized study of four hundred and fifty patients. J Bone Joint Surg Am. 2002;84:2123–2134.
335. Kirker-Head, CA, Boudrieau, RJ, Kraus, KH. Use of bone morphogenetic proteins for augmentation of bone regeneration. J Am Vet Med Assoc. 2007;231:1039–1055.
336. Styrkarsdottir, U, Cazier, JB, Kong, A, et al. Linkage of osteoporosis to chromosome 20p12 and association to BMP2. PLoS Biol. 2003;1:E69.
337. Long, F, Chung, UI, Ohba, S, et al. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development. 2004;131:1309–1318.
338. Hilton, MJ, Tu, X, Cook, J, et al. Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development. 2005;132:4339–4351.
339. Hui, CC, Joyner, AL. A mouse model of Greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat Genet. 1993;3:241–246.
340. Miao, D, Liu, H, Plut, P, et al. Impaired endochondral bone development and osteopenia in Gli2-deficient mice. Exp Cell Res. 2004;294:210–222.
341. Mo, R, Freer, AM, Zinyk, DL, et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development. 1997;124:113–123.
342. Shimoyama, A, Wada, M, Ikeda, F, et al. Ihh/Gli2 signaling promotes osteoblast differentiation by regulating Runx2 expression and function. Mol Biol Cell. 2007;18:2411–2418.
343. Zhao, M, Qiao, M, Harris, SE, et al. The zinc finger transcription factor Gli2 mediates bone morphogenetic protein 2 expression in osteoblasts in response to hedgehog signaling. Mol Cell Biol. 2006;26:6197–6208.
344. Jacob, AL, Smith, C, Partanen, J, et al. Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev Biol. 2006;296:315–328.
345. Ornitz, DM, Marie, PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–1465.
346. Eswarakumar, VP, Horowitz, MC, Locklin, R, et al. A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proc Natl Acad Sci U S A. 2004;101:12555–12560.
347. Eswarakumar, VP, Monsonego-Ornan, E, Pines, M, et al. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development. 2002;129:3783–3793.
348. Yu, K, Xu, J, Liu, Z, et al. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074.
349. Naganawa, T, Xiao, L, Coffin, JD, et al. Reduced expression and function of bone morphogenetic protein-2 in bones of Fgf2 null mice. J Cell Biochem. 2008;103:1975–1988.
350. Choi, KY, Kim, HJ, Lee, MH, et al. Runx2 regulates FGF2-induced Bmp2 expression during cranial bone development. Dev Dyn. 2005;233:115–121.
351. Franceschi, RT, Xiao, G. Regulation of the osteoblast-specific transcription factor, Runx2: responsiveness to multiple signal transduction pathways. J Cell Biochem. 2003;88:446–454.
352. Kim, BG, Kim, HJ, Park, HJ, et al. Runx2 phosphorylation induced by fibroblast growth factor-2/protein kinase C pathways. Proteomics. 2006;6:1166–1174.
353. Kim, HJ, Kim, JH, Bae, SC, et al. The protein kinase C pathway plays a central role in the fibroblast growth factor-stimulated expression and transactivation activity of Runx2. J Biol Chem. 2003;278:319–326.
354. Xiao, G, Jiang, D, Gopalakrishnan, R, et al. Fibroblast growth factor 2 induction of the osteocalcin gene requires MAPK activity and phosphorylation of the osteoblast transcription factor, Cbfa1/Runx2. J Biol Chem. 2002;277:36181–36187.
355. Reinhold, MI, Naski, MC. Direct interactions of Runx2 and canonical Wnt signaling induce FGF18. J Biol Chem. 2007;282:3653–3663.
356. Engin, F, Yao, Z, Yang, T, et al. Dimorphic effects of Notch signaling in bone homeostasis. Nat Med. 2008;14:299–305.
357. Zanotti, S, Smerdel-Ramoya, A, Stadmeyer, L, et al. Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology. 2008;149:3890–3899.
358. Hilton, MJ, Tu, X, Wu, X, et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat Med. 2008;14:306–314.
359. Sciaudone, M, Gazzerro, E, Priest, L, et al. Notch 1 impairs osteoblastic cell differentiation. Endocrinology. 2003;144:5631–5639.
360. Martin, TJ, Seeman, E. Bone remodelling: its local regulation and the emergence of bone fragility. Best Pract Res Clin Endocrinol Metab. 2008;22:701–722.
361. Harada, S, Rodan, GA. Bone development and remodeling. In: Degroot LJ, Jameson JL, eds. Endocrinology. St Louis: Elsevier, 2005.
362. Martin, TJ, Sims, NA. Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med. 2005;11:76–81.
363. Sims, NA, Gooi, JH. Bone remodeling: multiple cellular interactions required for coupling of bone formation and resorption. Semin Cell Dev Biol. 2008;19:444–451.
364. Matsuo, K, Irie, N. Osteoclast-osteoblast communication. Arch Biochem Biophys. 2008;473:201–209.
365. Zhao, C, Irie, N, Takada, Y, et al. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006;4:111–121.
366. Edwards, CM, Mundy, GR. Eph receptors and ephrin signaling pathways: a role in bone homeostasis. Int J Med Sci. 2008;5:263–272.
367. Lee, SH, Rho, J, Jeong, D, et al. v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat Med. 2006;12:1403–1409.
368. Skerry, TM. The response of bone to mechanical loading and disuse: fundamental principles and influences on osteoblast/osteocyte homeostasis. Arch Biochem Biophys. 2008;473:117–123.
369. You, L, Temiyasathit, S, Lee, P, et al. Osteocytes as mechanosensors in the inhibition of bone resorption due to mechanical loading. Bone. 2008;42:172–179.
370. Han, Y, Cowin, SC, Schaffler, MB, et al. Mechanotransduction and strain amplification in osteocyte cell processes. Proc Natl Acad Sci U S A. 2004;101:16689–16694.
371. Kogianni, G, Mann, V, Noble, BS. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J Bone Miner Res. 2008;23:915–927.
372. Zhao, S, Zhang, YK, Harris, S, et al. MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J Bone Miner Res. 2002;17:2068–2079.
373. Aguirre, JI, Plotkin, LI, Stewart, SA, et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res. 2006;21:605–615.
374. Mann, V, Huber, C, Kogianni, G, et al. The influence of mechanical stimulation on osteocyte apoptosis and bone viability in human trabecular bone. J Musculoskelet Neuronal Interact. 2006;6:408–417.
375. Noble, BS, Peet, N, Stevens, HY, et al. Mechanical loading: biphasic osteocyte survival and targeting of osteoclasts for bone destruction in rat cortical bone. Am J Physiol Cell Physiol. 2003;284:C934–C943.
376. Tatsumi, S, Ishii, K, Amizuka, N, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–475.
377. Cherian, PP, Siller-Jackson, AJ, Gu, S, et al. Mechanical strain opens connexin 43 hemichannels in osteocytes: a novel mechanism for the release of prostaglandin. Mol Biol Cell. 2005;16:3100–3106.
378. Civitelli, R. Connexin43 modulation of osteoblast/osteocyte apoptosis: a potential therapeutic target? J Bone Miner Res. 2008;23:1709–1711.
379. Gluhak-Heinrich, J, Gu, S, Pavlin, D, et al. Mechanical loading stimulates expression of connexin 43 in alveolar bone cells in the tooth movement model. Cell Commun Adhes. 2006;13:115–125.
380. Yang, W, Lu, Y, Kalajzic, I, et al. Dentin matrix protein 1 gene cis-regulation: use in osteocytes to characterize local responses to mechanical loading in vitro and in vivo. J Biol Chem. 2005;280:20680–20690.
381. Feng, JQ, Ward, LM, Liu, S, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310–1315.
382. Gluhak-Heinrich, J, Pavlin, D, Yang, W, et al. MEPE expression in osteocytes during orthodontic tooth movement. Arch Oral Biol. 2007;52:684–690.
383. Gowen, LC, Petersen, DN, Mansolf, AL, et al. Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J Biol Chem. 2003;278:1998–2007.
384. Quarles, LD. Endocrine functions of bone in mineral metabolism regulation. J Clin Invest. 2008;118:3820–3828.
385. Robling, AG, Niziolek, PJ, Baldridge, LA, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866–5875.
386. Loots, GG, Kneissel, M, Keller, H, et al. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005;15:928–935.
387. Li, X, Ominsky, MS, Niu, QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–869.
388. Winkler, DG, Sutherland, MK, Geoghegan, JC, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22:6267–6276.
389. Robinson, JA, Chatterjee-Kishore, M, Yaworsky, PJ, et al. Wnt/beta-catenin signaling is a normal physiological response to mechanical loading in bone. J Biol Chem. 2006;281:31720–31728.
390. Horwood, NJ, Elliott, J, Martin, TJ, et al. Osteotropic agents regulate the expression of osteoclast differentiation factor and osteoprotegerin in osteoblastic stromal cells. Endocrinology. 1998;139:4743–4746.
391. Poole, KE, Reeve, J. Parathyroid hormone—a bone anabolic and catabolic agent. Curr Opin Pharmacol. 2005;5:612–617.
392. Martin, TJ, Quinn, JM, Gillespie, MT, et al. Mechanisms involved in skeletal anabolic therapies. Ann N Y Acad Sci. 2006;1068:458–470.
393. Potts, JT, Gardella, TJ. Progress, paradox, and potential: parathyroid hormone research over five decades. Ann N Y Acad Sci. 2007;1117:196–208.
394. Jilka, RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone. 2007;40:1434–1446.
395. Krishnan, V, Moore, TL, Ma, YL, et al. Parathyroid hormone bone anabolic action requires Cbfa1/Runx2-dependent signaling. Mol Endocrinol. 2003;17:423–435.
396. Fu, Q, Manolagas, SC, O’Brien, CA. Parathyroid hormone controls receptor activator of NF-kappaB ligand gene expression via a distant transcriptional enhancer. Mol Cell Biol. 2006;26:6453–6468.
397. Keller, H, Kneissel, M. SOST is a target gene for PTH in bone. Bone. 2005;37:148–158.
398. Leupin, O, Kramer, I, Collette, NM, et al. Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J Bone Miner Res. 2007;22:1957–1967.
399. O’Brien, CA, Plotkin, LI, Galli, C, et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE. 2008;3:e2942.
400. Bellido, T, Ali, AA, Plotkin, LI, et al. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts: a putative explanation for why intermittent administration is needed for bone anabolism. J Biol Chem. 2003;278:50259–50272.
401. Girotra, M, Rubin, MR, Bilezikian, JP. The use of parathyroid hormone in the treatment of osteoporosis. Rev Endocr Metab Disord. 2006;7:113–121.
402. Kousteni, S, Bilezikian, JP. The cell biology of parathyroid hormone in osteoblasts. Curr Osteoporos Rep. 2008;6:72–76.
403. Krum, SA, Brown, M. Unraveling estrogen action in osteoporosis. Cell Cycle. 2008;7:1348–1352.
404. Novack, DV. Estrogen and bone: osteoclasts take center stage. Cell Metab. 2007;6:254–256.
405. Rossouw, JE, Anderson, GL, Prentice, RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321–333.
406. Kameda, T, Mano, H, Yuasa, T, et al. Estrogen inhibits bone resorption by directly inducing apoptosis of the bone-resorbing osteoclasts. J Exp Med. 1997;186:489–495.
407. Nakamura, T, Imai, Y, Matsumoto, T, et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts 1. Cell. 2007;130:811–823.
408. Krum, SA, Miranda-Carboni, GA, Hauschka, PV, et al. Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J. 2008;27:535–545.
409. Kousteni, S, Chen, JR, Bellido, T, et al. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science. 2002;298:843–846.
410. Srivastava, S, Toraldo, G, Weitzmann, MN, et al. Estrogen decreases osteoclast formation by down-regulating receptor activator of NF-kappa B ligand (RANKL)-induced JNK activation. J Biol Chem. 2001;276:8836–8840.
411. Pacifici, R. Estrogen deficiency, T cells and bone loss. Cell Immunol. 2008;252:68–80.
412. Pacifici, R, Brown, C, Puscheck, E, et al. Effect of surgical menopause and estrogen replacement on cytokine release from human blood mononuclear cells. Proc Natl Acad Sci U S A. 1991;88:5134–5138.
413. Weitzmann, MN, Pacifici, R. Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest. 2006;116:1186–1194.
414. Weitzmann, MN, Roggia, C, Toraldo, G, et al. Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J Clin Invest. 2002;110:1643–1650.
415. Jilka, RL, Hangoc, G, Girasole, G, et al. Increased osteoclast development after estrogen loss: mediation by interleukin-6. Science. 1992;257:88–91.
416. Sun, L, Peng, Y, Sharrow, AC, et al. FSH directly regulates bone mass. Cell. 2006;125:247–260.
417. Martin, TJ, Gaddy, D. Bone loss goes beyond estrogen. Nat Med. 2006;12:612–613.
418. Vanderschueren, D, Gaytant, J, Boonen, S, et al. Androgens and bone. Curr Opin Endocrinol Diabetes Obes. 2008;15:250–254.
419. Vanderschueren, D, Vandenput, L, Boonen, S, et al. Androgens and bone. Endocr Rev. 2004;25:389–425.
420. Manolagas, SC, Kousteni, S, Jilka, RL. Sex steroids and bone. Recent Prog Horm Res. 2002;57:385–409.
421. Canalis, E, Mazziotti, G, Giustina, A, et al. Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos Int. 2007;18:1319–1328.
422. Migliaccio, S, Brama, M, Fornari, R, et al. Glucocorticoid-induced osteoporosis: an osteoblastic disease. Aging Clin Exp Res. 2007;19:5–10.
423. O’Brien, CA, Jia, D, Plotkin, LI, et al. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–1841.
424. Weinstein, RS, Jilka, RL, Parfitt, AM, et al. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids: potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–282.
425. Yun, SI, Yoon, HY, Jeong, SY, et al. Glucocorticoid induces apoptosis of osteoblast cells through the activation of glycogen synthase kinase 3beta. J Bone Miner Metab. 2009;27:140–148.
426. Jia, D, O’Brien, CA, Stewart, SA, et al. Glucocorticoids act directly on osteoclasts to increase their life span and reduce bone density. Endocrinology. 2006;147:5592–5599.
427. Kim, HJ, Zhao, H, Kitaura, H, et al. Glucocorticoids suppress bone formation via the osteoclast. J Clin Invest. 2006;116:2152–2160.
428. Kim, HJ, Zhao, H, Kitaura, H, et al. Glucocorticoids and the osteoclast. Ann N Y Acad Sci. 2007;1116:335–339.
429. Giustina, A, Mazziotti, G, Canalis, E. Growth hormone, insulin-like growth factors, and the skeleton. Endocr Rev. 2008;29:535–559.
430. Bassett, JH, Williams, GR. Critical role of the hypothalamic-pituitary-thyroid axis in bone. Bone. 2008;43:418–426.
431. Miller, SB. Prostaglandins in health and disease: an overview. Semin Arthritis Rheum. 2006;36:37–49.
432. Ducy, P, Amling, M, Takeda, S, et al. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100:197–207.
433. Takeda, S, Elefteriou, F, Levasseur, R, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111:305–317.
434. Fu, L, Patel, MS, Bradley, A, et al. The molecular clock mediates leptin-regulated bone formation. Cell. 2005;122:803–815.
435. Elefteriou, F, Ahn, JD, Takeda, S, et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 2005;434:514–520.
436. Ahn, JD, Dubern, B, Lubrano-Berthelier, C, et al. Cart overexpression is the only identifiable cause of high bone mass in melanocortin 4 receptor deficiency. Endocrinology. 2006;147:3196–3202.
437. Bonnet, N, Pierroz, DD, Ferrari, SL. Adrenergic control of bone remodeling and its implications for the treatment of osteoporosis. J Musculoskelet Neuronal Interact. 2008;8:94–104.
438. Takeda, S. Central control of bone remodelling. J Neuroendocrinol. 2008;20:802–807.
439. Karsenty, G, Ducy, P. The hypothalamic control of bone mass, implication for the treatment of osteoporosis. Ann Endocrinol (Paris). 2006;67:123.
440. Sato, S, Hanada, R, Kimura, A, et al. Central control of bone remodeling by neuromedin U. Nat Med. 2007;13:1234–1240.
441. Baldock, PA, Sainsbury, A, Couzens, M, et al. Hypothalamic Y2 receptors regulate bone formation. J Clin Invest. 2002;109:915–921.
442. Baldock, PA, Allison, SJ, Lundberg, P, et al. Novel role of Y1 receptors in the coordinated regulation of bone and energy homeostasis. J Biol Chem. 2007;282:19092–19102.
443. Abe, E, Marians, RC, Yu, W, et al. TSH is a negative regulator of skeletal remodeling. Cell. 2003;115:151–162.
444. Sun, L, Vukicevic, S, Baliram, R, et al. Intermittent recombinant TSH injections prevent ovariectomy-induced bone loss. Proc Natl Acad Sci U S A. 2008;105:4289–4294.
445. Wong, IP, Zengin, A, Herzog, H, et al. Central regulation of bone mass. Semin Cell Dev Biol. 2008;19:452–458.
446. Karsenty, G. Convergence between bone and energy homeostases: leptin regulation of bone mass. Cell Metab. 2006;4:341–348.
447. Cirmanova, V, Bayer, M, Starka, L, et al. The effect of leptin on bone: an evolving concept of action. Physiol Res. 2008;57(suppl 1):S143–S151.
448. Patel, MS, Elefteriou, F. The new field of neuroskeletal biology. Calcif Tissue Int. 2007;80:337–347.
449. Horowitz, MC, Bothwell, AL, Hesslein, DG, et al. B cells and osteoblast and osteoclast development. Immunol Rev. 2005;208:141–153.
450. Horowitz, MC, Xi, Y, Pflugh, DL, et al. Pax5-deficient mice exhibit early onset osteopenia with increased osteoclast progenitors. J Immunol. 2004;173:6583–6591.
451. Li, Y, Toraldo, G, Li, A, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood. 2007;109:3839–3848.
452. Sato, T, Shibata, T, Ikeda, K, et al. Generation of bone-resorbing osteoclasts from B220+ cells: its role in accelerated osteoclastogenesis due to estrogen deficiency. J Bone Miner Res. 2001;16:2215–2221.
453. Calvi, LM, Adams, GB, Weibrecht, KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846.
454. Visnjic, D, Kalajzic, Z, Rowe, DW, et al. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood. 2004;103:3258–3264.
455. Porter, RL, Calvi, LM. Communications between bone cells and hematopoietic stem cells. Arch Biochem Biophys. 2008;473:193–200.
456. Zhang, J, Niu, C, Ye, L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841.
457. Adams, GB, Martin, RP, Alley, IR, et al. Therapeutic targeting of a stem cell niche. Nat Biotechnol. 2007;25:238–243.
458. Wu, JY, Purton, LE, Rodda, SJ, et al. Osteoblastic regulation of B lymphopoiesis is mediated by Gsα-dependent signaling pathways. Proc Natl Acad Sci U S A. 2008;105:16976–16981.
459. Walkley, CR, Olsen, GH, Dworkin, S, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129:1097–1110.
460. Walkley, CR, Shea, JM, Sims, NA, et al. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007;129:1081–1095.
461. Larsson, J, Ohishi, M, Garrison, B, et al. Nf2/merlin regulates hematopoietic stem cell behavior by altering microenvironmental architecture. Cell Stem Cell. 2008;3:221–227.
462. Gong, JK. Endosteal marrow: a rich source of hematopoietic stem cells. Science. 1978;199:1443–1445.
463. Aicher, A, Kollet, O, Heeschen, C, et al. The Wnt antagonist Dickkopf-1 mobilizes vasculogenic progenitor cells via activation of the bone marrow endosteal stem cell niche. Circ Res. 2008;103:796–803.
464. Kollet, O, Dar, A, Shivtiel, S, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12:657–664.
465. Lee, NK, Sowa, H, Hinoi, E, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–469.
466. Lee, NK, Karsenty, G. Reciprocal regulation of bone and energy metabolism. Trends Endocrinol Metab. 2008;19:161–166.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 4
Bone Development and Remodeling
Skeletal Development
Mechanisms of Bone Development
Intramembranous Ossification
The cranial and facial bones are derived largely from the neural crest through the process of intramembranous bone formation. In areas where bones are formed, mesenchymal precursor cells aggregate and form regions of high cell density, called condensations, that represent outlines of future skeletal elements. In intramembranous bones, the cells in these condensations then differentiate directly into osteoblasts that deposit “osteoid” or bone matrix rich in type I collagen, a process that occurs in close spatial interaction with vascular tissue. As the osteoblasts mature, the bone matrix becomes progressively mineralized. Terminally differentiated osteoblasts eventually become entrapped in the bone as osteocytes. Through modeling and remodeling (see later), the respective bones will reach their final shape and size. Membranous bones that derive as such from condensations that never form chondrocytes during development are restricted to the flat bones of the skull (calvarial bones and mandibles) and parts of the clavicles.1–5 The subperiosteal bone collar or provisional cortex of long bones can be considered to form also by intramembranous bone formation, because the osteoblasts in this region differentiate directly from the perichondrial/periosteal mesenchyme and form bone adjacent to, instead of directly upon, a cartilage-derived matrix (see later). Unlike the formation of intramembranous bone directly from condensations, the formation of this intramembranous bone is regulated directly by signals from the adjacent chondrocytes, so can be thought of developmentally as part of endochondral bone formation. Intramembranous bone formation also occurs during bone repair after fracture, particularly when the bones are stabilized (fixed).6–8
Endochondral Ossification
Endochondral ossification is the mechanism responsible for the formation of all long bones of the axial skeleton (vertebrae and ribs) and the appendicular skeleton (limbs). Most of the axial skeleton is derived from cells of the paraxial mesoderm that condense early in embryogenesis on both sides of the neural tube and the notochord. Portions of this mesoderm form segmented structures called somites, portions of which later become the sclerotomes that will give rise to the vertebral bodies. The appendicular skeleton arises from the lateral plate mesoderm. The mechanisms underlying the early condensation, segmentation, differentiation, and patterning events define the precise arrangement of the individual anatomic elements and their patterning along the proximal-distal, dorsal-ventral, and posterior-anterior body axes. These mechanisms involve actions and cross-talk of several morphogens, including fibroblast growth factors (FGFs), sonic hedgehog (Shh), bone morphogenetic proteins (BMPs), and Wnts, as well as control by Notch signaling and by transcription factors encoded by HOX, PAX1, and TBX genes.9
As in intramembranous ossification, the development of the long bones proper starts with mesenchymal progenitor cells forming condensations at the sites where the bones will be formed.10 Yet, in the mesenchymal condensations of endochondral bones, cells do not differentiate into osteoblasts but instead differentiate into chondrocytes that synthesize a characteristic extracellular matrix (ECM) rich in type II collagen and specific proteoglycans. As such, a cartilaginous model or anlage is established that prefigures the future bone. In mice, these differentiated cartilage structures appear around embryonic day 12, with the limb elements emerging in sequence along the proximodistal axis (i.e., hip to toes, shoulder to fingers). The sequential steps of the endochondral ossification process starting from this stage are illustrated in Fig. 4-1. Initially, the cartilage further enlarges through chondrocyte proliferation and matrix production. Chondrocytes in the midportion of the bone model then stop proliferating, undergo further maturation, and ultimately become hypertrophic. These large hypertrophic chondrocytes secrete a distinct matrix, containing type X collagen, which rapidly becomes calcified. Concomitantly, cells in the connective tissues surrounding the cartilage element called the perichondrium differentiate into osteoblasts that deposit mineralized bone matrix—the “bone collar”—around the cartilage template. This bone collar forms the initiation site of the cortical bone, the dense outer envelope of compact, lamellar bone that provides the long bone with most of its strength and rigidity (see Fig. 4-1).
FIGURE 4-1 Stepwise schematic diagram of long bone development through endochondral ossification. Around embryonic day (E) 12 in mice, mesenchymal progenitor cells condense and differentiate into chondrocytes to form the cartilage anlagen that prefigure future long bones. Chondrocytes in the center become hypertrophic, while cells in the surrounding perichondrium differentiate into osteoblasts, forming a bone collar, the provisional cortical bone. The hypertrophic cartilage core subsequently is invaded by blood vessels and eroded and replaced by bone and marrow (primary ossification center [POC]). In the metaphysis, hypertrophic cartilage of the growth cartilage is continually replaced by trabecular bone, a process that relies on metaphyseal vascularization and mediates longitudinal bone growth. Around postnatal day (P) 5, epiphyseal vessels invade the avascular cartilage and initiate a secondary center of ossification (SOC). Discrete layers of residual chondrocytes form growth plates between the epiphyseal and metaphyseal bone centers to support further postnatal longitudinal bone growth. Ultimately (in humans), the growth plates close and growth stops. A detailed view of perinatal bone structure (boxed area) is provided in Fig. 4-2.
At this time in development (around embryonic day 14 to 15 in mice), the hypertrophic cartilage core becomes invaded by blood vessels. This process is accompanied by apoptosis of terminally differentiated hypertrophic chondrocytes, resorption of the calcified cartilage matrix by invading osteoclasts or related “chondroclasts,” and deposition of mineralized bone matrix on the remnants of calcified cartilage by perichondrium/periosteum-derived osteoblasts. The net result is replacement of the cartilage model by bone, vascular, and marrow elements: the primary ossification center (see Fig. 4-1). With the disappearance of the diaphyseal cartilage, the remaining chondrocytes, restricted to the opposing ends of the long bone, provide the engine for subsequent bone lengthening. This process is typified by precise temporal and spatial regulation of chondrocyte proliferation and differentiation, with the chondrocytes first flattening out and forming longitudinal columns of rapidly proliferating cells, and next, as they reach the ends of the columns closest to the center of the bones, maturing further to hypertrophic chondrocytes (Fig. 4-2). Finally, at the border with the metaphysis, the terminally differentiated chondrocytes undergo apoptosis, and the calcified hypertrophic cartilage matrix is replaced with cancellous or trabecular bone (primary spongiosa) in a process requiring progressive neovascularization by metaphyseal capillaries (see Fig. 4-1 and cellular details in Fig. 4-2). Thus, similar to the initial formation of the primary ossification center, endochondral bone formation at the growth cartilage involves rigorous coupling of vascular invasion with maturation and activity of chondrocytes, osteoclasts, and osteoblasts.
FIGURE 4-2 Schematic view of the cellular structure of developing long bones. The epiphysis is composed of chondrocytes, organized in layers of proliferation (round periarticular and flat columnar proliferating cells), progressive differentiation toward the metaphysis (pre-hypertrophic and hypertrophic chondrocytes), and cell death (apoptosis). The avascular cartilage is supplied by the epiphyseal blood vessel network that overlays its surface. In the metaphysis, blood vessels invading the terminal hypertrophic chondrocytes, osteoclasts resorbing the cartilage, and osteoblasts building bone on the cartilage remnants all act coordinately to replace the cartilage anlagen with bone and marrow.
The mechanisms of embryonic bone development described here are largely recapitulated in the adult upon repair of bone defects. In contrast to soft tissues, which repair predominantly through the production of fibrous scar tissue at the site of injury, the skeleton possesses an astounding capacity to regenerate upon damage. As such, bone defects heal by forming new bone that is indistinguishable from adjacent, uninjured bone tissue. It has been appreciated for a long time that fracture repair in the adult bears close resemblance to fetal skeletal tissue development, with both intramembranous and/or endochondral bone formation processes occurring depending on the type of fracture. In recent years, this close resemblance has been supported by genetic and molecular studies showing that similar signaling pathways (see later) are at work in both settings,8,11–13 although additionally, some signaling molecules that are dispensable for development have been found to play essential roles in fracture repair.14,15
Cellular and Molecular Control of Skeletal Development
Chondrogenesis
As outlined previously, a crucial step in endochondral bone development fundamentally entails the differentiation of cells in the mesenchymal condensations into chondrocytes. At least some of the mesenchymal cells are probably osteochondroprogenitor cells with the potential to differentiate into chondrocytes or osteoblasts. The decision to follow a given differentiation pathway is determined by the expression of key transcription factors, likely under the influence of canonical Wnt signaling. As will be discussed in greater detail later, Wnt signaling results in the stabilization of β-catenin, which then can act as a transcription factor and regulate the expression of downstream target genes. Recent studies have provided evidence that in the perichondrium, where cells are destined to become osteoblasts, Wnt signaling is high (at least in part induced by Shh),16 leading to high levels of β-catenin and inducing the expression of genes that mediate osteoblast differentiation (such as Runx2, a master regulator of osteoblastogenesis, see later), while inhibiting transcription of genes required for chondrocyte differentiation (such as Sox9). In the absence of β-catenin, these cells become chondrocytes instead of osteoblasts, as revealed via genetic modifications in mice. Conversely, in the inner region of the condensations, Wnt signaling must be low as these cells become chondrocytes.17–19 The initial conversion of mesenchymal progenitor cells into chondrocytes is driven by the transcription factor Sox9, whereas the combined action of Sox9, 5 and 6, is required to direct the subsequent differentiation of chondrocytes throughout all phases of the chondrocyte lineage.20–23
In the growth cartilage, the most extensively studied cartilage, differentiation of committed cells along the chondrocyte lineage characteristically gives rise to a stratified organization of small, round periarticular chondrocytes (also previously termed resting or reserve chondrocytes, because these cells proliferate only slowly after birth and can become columnar chondrocytes), flattened, columnar proliferating chondrocytes, and pre-hypertrophic and hypertrophic chondrocytes (see Figs. 4-2 and 4-3). Because progression of the chondrocytes through these stages is the driving force of actual bone development and growth, it is not surprising that the process is tightly controlled by a myriad of local signaling molecules, the best characterized being BMPs and transforming growth factor-β (TGFβ), FGFs, parathyroid hormone (PTH)-related protein (PTHrP), and Indian hedgehog (Ihh). The importance of these molecules is reflected by the fact that mutations in their receptors have been found to cause severe human dwarfing conditions, such as constitutive activating mutations of FGF receptor (FGFR)3 in human achondroplasia and thanatophoric dysplasia24–27 and PTH/PTHrP receptor mutations in Jansen and Blomstrand chondrodysplasia.28–30 Here, we will briefly review the basic mechanisms by which these signaling systems control the pace of proliferation and differentiation of growth chondrocytes.31–33
FIGURE 4-3 Regulation of chondrocyte proliferation and differentiation. Schematic representation of the epiphysis, with zones of periarticular round chondrocytes (RC), strongly proliferating columnar chondrocytes (PC), and (pre-)hypertrophic chondrocytes ([pre-]HC). Hypertrophic cartilage is surrounded by perichondrium (Pe), where osteoblasts (OB) develop. Centrally, the Indian hedgehog (Ihh)/parathyroid hormone-related protein (PTHrP)-negative feedback loop is indicated. Cells at the articular end of the bone secrete PTHrP, acting through the PTH/PTHrP receptor (PPR) on columnar chondrocytes to keep them in the proliferating pool and delay their differentiation. When the distance from the bone end becomes sufficiently large, cells escape the differentiation block and become Ihh-expressing pre-hypertrophic chondrocytes. In an as yet unknown manner, Ihh induces PTHrP expression. In addition, Ihh directly stimulates chondrocyte proliferation and converts perichondrial cells into OBs by inducing runt-related transcription factor 2 (Runx2), thereby determining the site of bone collar formation. Runx2 also favors Ihh expression and hypertrophic differentiation. Several bone morphogenetic protein (BMP) and fibroblast growth factor (FGF) family members are expressed at specific sites in the growth cartilage (left and right panels, respectively). BMPs and FGFs act antagonistically to modulate chondrocyte proliferation, Ihh expression, and terminal hypertrophic differentiation. GDF, Growth and differentiation factor; Ptc, patched; BMPR, BMP receptor; FGFR, FGF receptor.
BMPs, secreted proteins belonging to the TGFβ superfamily, transduce signals through serine/threonine kinase receptors, composed of type I and II subtypes, activating intracellular proteins of the Smad family that relay the BMP signal to target genes in the nucleus.34 BMPs were first discovered as agents in bone matrix capable of inducing ectopic formation of cartilage and bone after injection into subcutaneous tissues.35–38 Subsequent studies have shown that BMPs, some of which are also called growth and differentiation factors (GDFs) are, in fact, signaling proteins crucial for regulating development in almost all the principal organs and tissues.39,40 In the morphogenesis of the skeleton, BMPs have been implicated in early limb patterning, as well as in the subsequent processes of chondrogenesis and osteogenesis. Initially, the role of BMPs in the correct formation of mesenchymal condensations was highlighted by the mutation of the BMP5 gene in the mouse skeletal mutant short ear (se)41 and by abnormalities observed in brachypodism (bp) mice that were attributed to mutations in the GDF5 gene.42 In both se and bp mice, the abnormalities were traced back to altered size or shape already apparent in the mesenchymal condensations of the respective skeletal elements that were affected. More recently, studies in which two BMP type I receptors were ablated at the condensation stage demonstrated that BMP signaling is essential for converting condensing mesenchyme into chondrocytes; the mutant cells expressed undetectable amounts of the essential Sox9, L-Sox5, and Sox6 transcription factors.43 Similarly, when the potent BMP inhibitor noggin was introduced in early-stage mouse or chick limbs, mesenchymal condensation and chondrogenesis did not take place.44,45 BMPs also play critical roles at later stages of cartilage development. These functions have been difficult to document in vivo because in some cases, inactivation of specific BMPs led to severe early defects and lethality (e.g., BMP-2, BMP-4), precluding investigation of the later stages; in other cases, removal of individual family members (such as BMP-7) displayed no defects in skeletogenesis, presumably because of functional redundancy between the various BMP ligands and receptors (around 20 BMP family members have been identified to date). Further, various models have yielded superficially contradictory results, presumably reflecting the multiple actions of BMPs and the interactions of BMP signaling with other pathways. Recent and ongoing studies therefore employ conditional (site-specific) and/or combined (multiple targets) mutagenesis strategies. As such, it was shown in vivo that BMP signaling (through type I receptors on chondrocytes) positively regulates chondrocyte proliferation and survival, while concomitantly delaying the conversion to terminal hypertrophic differentiation,46 confirming previous limb culture results.47,48 These effects likely involve interactions between the BMP, FGF, and Ihh signaling pathways (see later); indeed BMP signaling seems to antagonize FGF actions and to promote Ihh expression. Studies in which BMP-2 and BMP-4 were ablated in limb development also revealed an essential role of BMP signaling in osteoblast differentiation (see later).49 It is not surprising that perichondrium, chondrocytes, and osteoblasts express multiple BMPs, BMP receptors, and BMP antagonists to regulate this essential pathway (see Fig. 4-3).
The FGF pathway similarly involves multiple ligands and receptors in regulating skeletal development.50 FGF1851,52 and FGF953 appear to be the most important FGF ligands in regulating chondrogenesis identified so far. These ligands activate FGFR3 expressed on proliferating chondrocytes. Activating mutations in the FGFR3 gene cause dominantly inherited human dwarfing chondrodystrophies due to impaired chondrocyte proliferation,24 and FGFR3 inactivation in mice increases chondrocyte proliferation and prolongs growth.54,55 This finding was surprising given that FGFs in most tissues act as potent mitogens; although this may pertain in early chondrogenesis, in which FGF18 stimulates proliferation,56 later in development, signaling through FGFR3 constitutes a master block on chondrocyte proliferation through activation of STAT1, which activates the cell cycle inhibitor p21Waf1/Cip1.57–59 In addition, although previous in vitro experiments had indicated that FGF signaling accelerates the late steps of chondrocyte hypertrophy,47 mouse models of achondroplasia and thanatophoric dysplasia have shown delayed hypertrophic differentiation of chondrocytes, strongly suggesting that FGFR3 signaling in vivo inhibits chondrocyte differentiation.25,60–63 This effect is relayed by activation of the mitogen-activated protein kinase (MAPK) pathway in chondrocytes, which affects longitudinal growth by regulating hypertrophic chondrocyte differentiation and matrix deposition.64–66 In vitro and in vivo experiments suggest that snail1 is required downstream of FGFR3 signaling for regulating its effects on chondrocyte proliferation and differentiation through activation of the Stat1/p21 and MAPK/Erks pathways, respectively.67 Yet, the effects of FGFR3 are regulated only in part by direct signaling in chondrocytes, and in part indirectly by modulating the expression of the Ihh/PTHrP/BMP signaling pathways. Indeed, mice harboring an activating mutation in FGFR3 have decreased expression of Ihh and its receptor and downstream target Patched (Ptc), and of BMP4, whereas in mice lacking FGFR3, Ihh, Ptc, and BMP4 expression are upregulated.60,68,69 During chondrocyte differentiation, the FGF and BMP pathways generally appear to antagonize each other,46 and both FGF signaling and BMP signaling regulate Ihh production (see Fig. 4-3). Thus, each of these pathways has multiple mechanisms for communicating with each other as they regulate chondrocyte and osteoblast differentiation.
A major regulatory system in growth chondrocytes is provided by Ihh, a member of the conserved family of hedgehog proteins, and PTHrP.32 The Ihh/PTHrP pathway forms a negative-feedback loop that regulates the onset of hypertrophic differentiation (see Fig. 4-3). Ihh is produced by prehypertrophic and early hypertrophic chondrocytes and signals through its receptor Ptc to stimulate expression of PTHrP by chondrocytes located near the periarticular ends of bone. Increased production of Ihh or production of Ihh closer to the cells making PTHrP leads to increased PTHrP production. PTHrP in turn signals back to its receptor—the PTH/PTHrP receptor that also responds to PTH in osteoblasts and kidney—that is expressed at low levels by proliferating chondrocytes and strongly by prehypertrophic cells. This signaling slows down chondrocyte differentiation and keeps chondrocytes in the proliferative state (see Fig. 4-3). This slowed differentiation consequently delays the generation of cells that can produce Ihh; therefore, the production of PTHrP is lowered. Thus, a negative-feedback signaling pathway allows PTHrP and Ihh to regulate each other and, in turn, to control the pace of chondrocyte development in the growth plate.32,70,71 It is important to note that the phenotype of the Ihh-/- mice72 revealed that Ihh has additional functions in endochondral bone formation, independent of regulation of PTHrP production. Indeed, these mutant mice also displayed a marked decrease in chondrocyte proliferation and absence of mature osteoblasts and bone collar formation.72 The regulation of chondrocyte proliferation represents a direct action of Ihh signaling to chondrocytes.73,74 Ihh also accelerates the conversion of round periarticular chondrocytes to flat columnar chondrocytes.75 Control of bone collar formation is mediated by Ihh-induced direct actions on perichondrial cells that are probably bipotential osteochondroprogenitors, and that are driven into the osteoblast lineage under the influence of Ihh stimulating the expression of Runx2. Runx2, an essential transcription factor in osteoblast development (see later), conversely also regulates Ihh expression and hypertrophic differentiation.72,76–79 From this, it is evident that Ihh is a master regulator of endochondral bone development, coordinating chondrocyte proliferation, chondrocyte maturation, and osteoblast differentiation (see Fig. 4-3). Recently, a continued role for Ihh in the postnatal growth plate has been revealed by employing inducible mutagenesis in mice.80
Aside from the above mentioned factors, chondrogenesis is affected by several other growth factors, cytokines, and hormones, including growth hormone (GH) and insulin-like growth factors (IGFs), C-natriuretic peptide, retinoids, thyroid hormone, estrogen, androgen, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3], glucocorticoids, and others.32,81
Importantly, a main difference between mesenchymal condensations that differentiate toward the osteoblast lineage in and becoming intramembranous bones, and endochondral cartilaginous condensations is that the latter typically develop over a prolonged period in the absence of blood vessels. As an avascular tissue, cartilage consequently faces the challenge of hypoxia.82,83 The main mediator of cellular responses to hypoxia is the transcription factor hypoxia-inducible factor (HIF).84 Recently, HIF1α was shown to influence chondrocyte differentiation and to be required for survival of chondrocytes, because mice with conditional inactivation of HIF1α displayed aberrant apoptosis of chondrocytes located in the center of the growth cartilage, farthest from blood vessels.85,86 The precise mechanisms by which HIF1 prevents chondrocyte apoptosis have yet to be fully explained. Part of the explanation may include an indirect survival effect of HIF1, mediated through induction of vascular endothelial growth factor (VEGF), as deletion of VEGF from cartilage also resulted in the apoptotic phenotype.87,88 Alternatively, and not mutually exclusively, HIF1 may facilitate a vital switch in the metabolism of hypoxic chondrocytes by inducing the anaerobic glycolytic pathway,86,89–91 and thereby allow their further differentiation up to the hypertrophic stage, when the chondrocytes induce vascular invasion (see next).
Vascular Invasion of Cartilage
Both temporally and spatially, chondrocyte differentiation is followed by vascular invasion of the terminally differentiated, hypertrophic cartilage. This step is an absolute requirement for endochondral bone development and growth to proceed, as physical blockage of blood vessel invasion into the hypertrophic cartilage of fetal skeletal explants completely halts their development,92 and blocking the blood supply of bone in vivo results in reduced longitudinal growth.93,94 The importance of skeletal vascularization goes beyond endochondral bone development. In fact, as a rule, any type of bone formation occurs in close spatial and temporal association with vascularization of the ossified tissue. The reasons that the vascular system is crucial for bone development, maintenance, and repair obviously include its intrinsic function to supply oxygen, nutrients, and growth factors/hormones to bone cells as required for their specified activities. In addition, the blood vessels serve to bring in (precursors of) osteoclasts that will degrade the cartilage or bone extracellular matrix, to remove end products of the resorption processes, and to bring in progenitors of osteoblasts that will deposit bone.95–97
During endochondral ossification, the initially avascular cartilage template is replaced by highly vascularized bone and marrow tissue through three consecutive vascularization events (see Fig. 4-1). First, initial vascular invasion of the cartilage anlagen during embryonic development (sometimes called quiescent angiogenesis) involves endothelial cells invading from the perichondrial tissues and organizing into immature blood vessels in the primary center of ossification. Second, capillary invasion at the metaphyseal border of the growth cartilage mediates rapid bone lengthening. Third, vascularization of the cartilage ends initiates the formation of secondary ossification centers (see Fig. 4-1). Although the vessel systems associated with the different stages of cartilage vascularization are believed to be substantially different,98,99 a common feature is that invasion of cartilage at all times is preceded by chondrocyte hypertrophy. Immature chondrocytes are resistant to vascular invasion because of the production of angiogenic inhibitors, such as chondromodulin-I, troponin-I, and thrombospondins.100–103 In contrast, when chondrocytes become hypertrophic, they switch to production of angiogenic stimulators, thereby becoming a target for capillary invasion and angiogenesis.104,105 One exceptionally potent angiogenic stimulator, VEGF, has received much attention, as it is expressed at very high levels by hypertrophic cartilage, as well as by osteoblasts and osteoclasts. Although the regulation of VEGF expression is still largely unresolved and may vary depending on the developmental stage and specific location and cell type, several mechanisms have been implicated to date. Hypoxia seems to be a crucial trigger of VEGF expression in chondrocytes, osteoblasts, and possibly osteoclasts, through mechanisms that involve HIF both in vitro106–109 and in vivo.110 During early bone development, VEGF transcription may be induced by Runx2 (see later), given that expression of VEGF was impaired in Runx2-deficient mice and no blood vessel ingrowth into the cartilage occurred.111 In addition, several hormones [including PTH, GH, and 1,25(OH)2D3] and locally produced growth factors (e.g., FGFs, TGFβ, BMPs, IGFs, platelet-derived growth factor [PDGF]) have been demonstrated to be involved, at least in vitro, in the regulation of VEGF expression.112
Important insight into the crucial physiologic function of VEGF-mediated angiogenesis in bone was first provided by Gerber et al.,113 who inhibited VEGF action in juvenile mice through administration of a soluble VEGF receptor chimeric protein (sFlt-1). VEGF inhibition impaired vascular invasion of the growth plate, and concomitantly, trabecular bone formation and bone growth were reduced and the hypertrophic cartilage zone became enlarged, likely as the result of reduced osteoclast-mediated resorption (see later).113 Additional mouse genetic studies performed over the past decade have univocally demonstrated that VEGF is an essential mediator of all three key vascularization stages of endochondral bone development. Although a mouse with the VEGF gene ablated in all cells could not be employed owing to early lethality of even heterozygous VEGF knockout embryos, several alternative mutagenesis approaches have been and are being exploited to investigate the role of VEGF in bone. These models include Cre/LoxP-mediated conditional inactivation of the VEGF gene (VEGFa) in type II collagen–expressing chondrocytes, and expression of only one of the three major VEGF isoforms by genetically engineered mice.87,88,114–116 Altogether, these models have exposed multiple essential roles of VEGF and its isoforms in endochondral ossification, not only as a key inducer of vascularization but also as a direct modulator of bone development by affecting the various cell types involved. Perichondrial cells, osteoblasts, and osteoclasts are well documented to express several VEGF receptors and to respond to VEGF signaling through enhanced recruitment, differentiation, activity, and/or survival.112,117,118 These pleiotrophic actions of VEGF on various cells in the bone environment may contribute to the tight coordination of vascularization, ossification, and matrix resorption that is characteristically seen in endochondral ossification. Indeed, the current model is that VEGF secretion in the bone environment will attract blood vessels and stimulate endothelial cells to form new blood vessels, which will be indirectly associated with increased potential delivery of osteoblast and osteoclast progenitors.81 At the same time, VEGF signaling directly stimulates the recruitment and differentiation of osteoblasts to form bone.112,114,119,120 VEGF works as a chemoattractant stimulating osteoclast invasion of cartilage, and enhances osteoclast differentiation, survival, and resorptive activity.121–127 A positive-feedback system is likely established, as chondroclast/osteoclast-derived matrix metalloproteinase (MMP) 9 may release more matrix-bound VEGF from the cartilage that is being resorbed (Fig. 4-4).81,128
FIGURE 4-4 Vascular invasion and the role of vascular endothelial growth factor (VEGF). VEGF is produced at high levels by hypertrophic chondrocytes and is sequestered in part in the cartilage matrix upon its secretion. Trapped VEGF can be released from the matrix by proteases such as matrix metalloproteinase (MMP) 9 secreted by osteoclasts/chondroclasts during cartilage resorption. VEGF then can bind to its receptors (VEGFRs) on endothelial cells and stimulate the guided attraction of blood vessels to invade the terminal cartilage. Osteoblasts and osteoclasts also express VEGF receptors, and VEGF can affect their differentiation and function both directly and indirectly by enhancing metaphyseal vascularization, thus playing a coordinative role in the key events that mediate bone development and growth.
Osteoclastogenesis and Skeletal Tissue Resorption
At the time of vascular invasion of hypertrophic cartilage, the chondrocytes undergo apoptosis, and remaining cellular debris and matrix components become degraded and are digested by co-invading osteoclasts (or related cells termed chondroclasts that have been postulated in the context of resorption of cartilage128) through their proteolytic enzyme products.81 Beyond development, the osteoclast remains a key participant in the regulation of bone mass, as bone is constantly being remodeled throughout life (see later). Several pathologic conditions are due to an imbalance between bone formation and resorption, most frequently involving excessive osteoclast activity. Such skeletal diseases include osteoporosis, a common low bone mass disorder typically prevalent in postmenopausal women, as well as periodontal disease, rheumatoid arthritis, multiple myeloma, and metastatic cancer. On the other hand, osteopetrosis, a rare human disease characterized by increased bone mass and obliteration of the bone marrow cavity, is caused by impaired osteoclast differentiation and/or function. Many efforts have been made to dissect the regulatory pathways leading to functional osteoclasts.129–134
Osteoclastogenesis: Osteoclasts are giant multinucleated cells that have the unique capacity to efficiently degrade mineralized tissues. Osteoclasts are derived from hematopoietic precursor cells of the myelomonocytic lineage and share a common precursor with macrophages.131 Recognition of the intertwining control and close interactions of the immune and skeletal systems has led recently to the emergence of the novel integrating field of osteoimmunology.131,132 Early differentiation of the bipotential macrophage/osteoclast precursor cells is regulated by PU.1, a versatile hematopoietic cell-specific transcriptional regulator of the ETS family of transcription factors. The commitment to macrophages/osteoclasts is dependent on a high level of activity of PU.1131,135–137 (Fig. 4-5); PU.1 regulates the lineage fate decision of early progenitors by directly controlling expression of the c-Fms gene, which is a key determinant of differentiation into the macrophage/osteoclast lineage (see later).138,139 Consequently, PU.1-null mice lack both macrophages and osteoclasts, and are osteopetrotic.136,140,141 PU.1 also regulates the transcription of another key osteoclastogenesis control gene, encoding receptor activator of nuclear factor-κB (RANK) (see later) in myeloid progenitors.142 Although mature macrophages and osteoclasts have some cell surface markers in common, the latter express high levels of tartrate-resistant acid phosphatase (TRAP), cathepsin K, vitronectin, calcitonin receptor, and αvβ3 integrin, which typify the osteoclast lineage (see later).81
FIGURE 4-5 Osteoclastogenesis. The transcription factor PU.1 is indispensable for development of the osteoclast and macrophage (Mϕ) lineages from a common myeloid precursor. Macrophage-colony stimulating factor (M-CSF) acting upon the receptor c-Fms is required for the conversion of the progenitor to become a pre-osteoclast. Signaling through the membrane-bound receptor activator of nuclear factor-κB (RANK) promotes further differentiation of pre-osteoclasts to mature polykaryotic osteoclasts and their subsequent activation to bone-resorbing cells that secrete protons and lytic enzymes. RANK signaling is induced by receptor activator of nuclear factor-κB ligand (RANKL), present on osteoblasts and bone marrow stromal cells, as well as on lymphocytes and possibly in a soluble form in serum. The soluble antagonist osteoprotegerin (OPG) competes with RANK for RANKL binding, thereby functioning as a negative regulator of osteoclast differentiation, activation, and survival. RANKL expression is induced by pro-resorptive and calciotropic factors that stimulate osteoclastogenesis. Conversely, OPG is induced by factors that block bone catabolism and promote anabolic effects.
After the precursor cells have committed to the osteoclast lineage, they are subjected to a complex multistep process that culminates in the generation of mature, multinuclear, activated osteoclasts; these steps include proliferation, maturation, and fusion of differentiating precursor cells, and finally activation of resorption (see Fig. 4-5).81,130,132,134 Two critical cytokines are essential for osteoclastogenesis: macrophage-colony stimulating factor (M-CSF) and receptor activator of nuclear factor-κB ligand (RANKL) (also known previously as osteoprotegerin ligand [OPGL], osteoclast differentiation factor [ODF], or tumor necrosis factor [TNF]-related activation-induced cytokine [TRANCE]). Both of these signaling molecules are strongly expressed by bone marrow stromal cells (i.e., osteoblast progenitors) and osteoblasts; M-CSF is produced in both a soluble and a membrane-bound form, and RANKL is made exclusively as a membrane protein by osteoblasts. The process of osteoclastogenesis requires direct cell-to-cell interaction between stromal/osteoblastic cells and osteoclast precursors, presumably because of these key membrane-bound ligands (see Fig. 4-5). Both PTH and 1,25(OH)2D3 and several other osteotropic factors stimulating resorption increase the expression of RANKL on stromal/osteoblastic cells130,143,144 (see later). Moreover, before the identification of RANKL, in vitro investigation of osteoclastogenesis relied on contact-dependent co-culture of osteoclast progenitors with stromal/osteoblastic cells stimulated with PTH or 1,25(OH)2D3. Now we know that RANKL, together with M-CSF, is sufficient to induce in vitro osteoclast differentiation from spleen- or bone marrow–derived precursors in the complete absence of stromal/osteoblastic cells.145 Furthermore, not only osteoblasts but also chondrocytes146–148 and T cells149,150 synthesize and secrete RANKL and are able to support osteoclastogenesis. This may be important physiologically during osteoclast differentiation and invasion at the hypertrophic cartilage during endochondral bone development and growth, and it definitely plays a major role pathologically. For instance, production of RANKL by T cells has been implicated as an activator of osteoclastic resorption in inflammatory mediated bone and cartilage destruction, as is seen in several autoimmune disorders, including rheumatoid arthritis,81,130,151 likely working synergistically with TNFα.152,153
M-CSF and RANKL affect several steps of the osteoclastogenesis cascade by binding to their respective receptors, c-Fms and RANK, which are expressed by osteoclast progenitors and osteoclasts at all stages of differentiation. M-CSF signaling is essential early on in the lineage for proliferation, differentiation, and survival of osteoclast/macrophage precursors. The essential roles of M-CSF were illustrated through analysis of mice carrying the op/op mutation, an inactivating point mutation in the M-CSF gene; these mice have osteopetrosis with low numbers of macrophages and a complete lack of mature osteoclasts.154–157
RANKL is an essential inducer of multiple aspects of osteoclastogenesis, including osteoclast differentiation, fusion, activation of mature osteoclasts to resorb mineralized bone, and survival.158–161 Through these actions, RANKL potently stimulates bone resorption. RANKL acts by binding to its signal transducing receptor, RANK, a member of the TNF receptor family present on pre-osteoclasts and mature osteoclasts. Consistent herewith, an activating mutation of the human RANK gene was found in patients with familial expansile osteolysis, a disease of excess bone resorption.162 Recent genetic and cell biological studies have begun to elucidate the complex signaling cascade downstream of RANK/RANKL.130,163 Briefly stated and oversimplified, the binding of RANKL to RANK on the surface of osteoclast precursors recruits the adaptor protein TNF receptor–associated factor (TRAF) 6 to the cytoplasmic domain of RANK, leading to activation of NF-κB and its translocation to the nucleus. NF-κB increases c-Fos expression, and c-Fos, as a component of the activator protein (AP)-1 complex, interacts with the master transcription factor for osteoclastogenesis, nuclear factor of activated T cells c1 (NFATc1), to induce osteoclast-specific genes (such as the genes encoding TRAP and calcitonin receptor; see later).164–167
The actions of RANKL are regulated negatively by a secreted soluble decoy receptor termed osteoprotegerin (OPG) (previously also known as osteoclastogenesis inhibitory factor [OCIF]), also a member of the TNF receptor superfamily. OPG sequesters RANKL molecules and thereby blocks their binding to RANK.130,168 As such, OPG protects bone from excessive resorption; this conclusion is supported by the finding that certain homozygous deletions of OPG in humans can cause juvenile Paget’s disease, a disorder characterized by increased bone remodeling, osteopenia, and fractures.169 The relative concentrations of RANKL and OPG in bone thus are major determinants of bone mass and strength. OPG is widely expressed; it is not surprising that its expression by osteoblasts and stromal cells is positively regulated by bone anabolic or antiresorptive factors, such as estrogen and calcitonin.130
The essence of the RANK/RANKL/OPG cascade was further clarified by mouse genetic studies. Mice lacking RANKL170 or RANK171,172 and mice with increased circulating OPG by transgenic overexpression173 were severely osteopetrotic owing to a block in osteoclastogenesis. Conversely, targeted mutagenesis of OPG,174,175 overexpression of an sRANKL transgene,176 and administration of RANKL159,160 in mice all led to increased osteoclast formation, activation, and/or survival and resulted in an osteoporotic phenotype. In summary, RANKL and OPG act in an antagonistic fashion to regulate bone resorption, and their respective expression levels are under the control of proresorptive and antiresorptive factors, including several hormones, cytokines, and growth factors (see Fig. 4-5). Of note, RANK is broadly expressed, and these mouse studies indicate that the RANK/RANKL system functions in tissues beyond bone. For instance, RANK/RANKL regulates lymph node formation and lactational mammary gland development in mice.172,177 OPG has nonskeletal functions too, as it protects large arteries of mice from medial calcification.174
Other regulatory molecules have been implicated recently in the late stages of osteoclast differentiation, the fusion process, and activation of resorption. Co-stimulatory molecules acting in concert with M-CSF and RANKL to complete osteoclastogenesis include proteins containing an immunoreceptor tyrosine-based activation motif (ITAM) domain that is critical for the activation of calcium signaling and is found in adapter molecules like DNAX-activating protein (DAP)12 and the Fc receptor γ (FcRγ).178 The resultant increase in intracellular calcium is required for activation of NFATc1. The fusion of mononuclear osteoclast precursor cells into mature multinucleated osteoclasts is regulated by a membrane protein called dendritic cell-specific transmembrane protein (DC-STAMP). DC-STAMP–deficient cells failed to fuse, and these mononuclear osteoclasts had reduced resorptive efficiency in vitro. Consequently, DC-STAMP–deficient mice exhibited increased bone mass.179 These data may suggest that multinucleation and osteoclast enlargement could be associated with a higher degree of resorption efficiency.180,181 Pathologically huge osteoclasts, containing substantially more nuclei than normal osteoclasts do, are seen for instance in Paget’s disease of bone (up to 100 nuclei per cell; normal range is 3 to 20 nuclei per cell), in which localized excessive bone remodeling is initiated by increases in osteoclast-mediated bone resorption.182
Additional studies on the regulatory components of osteoclastogenesis not only will expand our basic understanding of the molecular mechanisms of osteoclast differentiation during bone development and remodeling but also offer opportunities to develop therapeutic means of intervention in osteoclast-related diseases. OPG and soluble recombinant RANK suppress osteoclastogenesis, while antibodies to RANK can stimulate osteoclast formation.183 From the clinical point of view, the RANKL signaling pathway thus holds great promise as a strategy for suppressing excessive osteoclast formation in a variety of bone diseases, including osteoporosis, autoimmune arthritis, periodontitis, Paget’s disease, and bone tumors/metastases.
Bone Resorption: Osteoclast Action and Proteolytic Enzymes: Bone resorption involves both dissolution of bone mineral and degradation of organic bone matrix. Osteoclasts are highly specialized to perform both of these functions. Upon activation of mature multinucleated osteoclasts, the cells attach themselves firmly to the bone surface, using specialized actin-rich podosomes (actin ring), through cytoskeleton reorganization and cellular polarization.184–187 Within these tightly sealed zones of adhesion to the mineralized matrix, the osteoclasts form convoluted, villus-like membranes called “ruffled borders,” which drastically increase the surface area of the cell membrane facing the resorption lacuna (Howship’s lacuna). Via these ruffled membranes, the osteoclasts secrete abundant hydrochloric acid (involving the vacuolar H+-ATPase proton pump), mediating acidification of the compartment between the cell and the bone surface, as well as a myriad of enzymes such as lysosomal cathepsins, the phosphatase TRAP, and proteolytic MMPs (see later). The acidity of the environment leads to dissolution of the mineral phase (crystalline hydroxyapatite), activation of lytic enzymes, and digestion of organic matrix compounds (see Fig. 4-5). The sealing mechanism allows localized dissolving and degrading of the mineralized bone matrix, while simultaneously protecting neighboring cells from harm.188,189 During the resorption process, dissolution of hydroxyapatite releases large amounts of soluble calcium, phosphate, and bicarbonate. Removal of these ions is needed (e.g., to maintain the acidic pH in the resorption lacuna) and apparently involves vesicular pathways and direct ion transport via different ion exchangers, channels, and pumps. The degradation products of the organic matrix after enzymatic digestion are transcytosed through the cell for secretion at the basolateral membrane.188,189
These complex processes of osteoclast recruitment, polarization on the bone surface, and export of acid and enzymes are orchestrated by many factors, including RANKL,190–192 as well as by integrin-mediated signaling from the bone matrix itself.193–195 The latter, which is particularly represented by the αvβ3 integrin in osteoclasts, was suggested to be important for osteoclast functioning based on the finding that inhibition of signaling through this αvβ3 integrin inhibited osteoclast-mediated bone resorption in vitro and in animal models of osteoporosis and malignant osteolysis (reviewed in refs. 196, 197). Integrins are heterodimeric cell-surface receptors, composed of an α and a β subunit, that mediate cell–matrix interactions and thus adhesion. The αvβ3 integrin, among various integrins the most highly expressed in osteoclasts, recognizes RGD(Arg-Gly-Asp)-containing matrix proteins such as vitronectin, osteopontin, and bone sialoprotein. Several components of the αvβ3 integrin signaling pathway localize to the sealing zone of actively resorbing osteoclasts, suggesting that they play a role in linking the matrix adhesion of osteoclasts to cytoskeletal organization, cell polarization, and activation for bone resorption. For these reasons, αvβ3 integrin appeared as a favorable candidate target for antiresorptive therapeutic interventions; in fact, small molecule inhibitors of the integrin are in clinical trial for the treatment of osteoporosis.194,198
Among the molecules that are important for the functional activity of osteoclasts per se, such as β3 integrin, but also c-Src, cathepsin K, carbonic anhydrase II, TRAP, and several ion channel proteins, many have been found to cause an osteopetrotic phenotype when deleted in mice or altered in humans. The absence of these genes does not affect the differentiation into morphologically normal osteoclasts; however, the osteoclasts are not functional, and they fail to resorb bone effectively.81 For instance, cathepsin K, the key enzyme in the digestion of bone matrix by its activity in degrading type I collagen, is highly expressed by activated osteoclasts and secreted in the resorption lacuna.199–201 Its deletion in mice led to osteopetrosis,201–203 and mutations in the human cathepsin K gene cause pycnodysostosis.204 Highly selective and potent cathepsin K inhibitors have been developed recently and have been shown to be useful antiresorptive agents for the treatment of osteoporosis, as well as promising therapeutic tools to reduce breast cancer–induced osteolysis and skeletal tumor burden.205–208
Besides cathepsin K, several proteolytic enzyme groups are involved in the degradation of organic components (collagens and proteoglycans) of bone and cartilage matrices after the mineral is dissolved.209–211 One of these is the MMP family, which constitutes over 25 members, including secreted collagenases, stromelysins, gelatinases, and membrane-type (MT)-MMPs.212,213 MMPs are synthesized as latent proenzymes that, upon proteolytic activation, can degrade numerous extracellular matrix components. As such, they are involved in the development, growth, and repair of tissues, but also in pathologic conditions associated with excessive matrix degradation, such as rheumatoid arthritis, osteoarthritis, and tumor metastasis.210,213–215 Several MMPs, including MMP9 and MMP14 (also known as MT1-MMP), are highly expressed in osteoclasts/chondroclasts,128,216,217 but they are produced by many other cell types as well. Both of these molecules play a role in the cartilage resorption process associated with invasion by osteoclasts during endochondral ossification.122,128,218–220 MMPs and proteolytic enzymes containing a disintegrin and metalloprotease domain (ADAMs) also possibly affect osteoclastogenesis per se, by modulating the bioavailability and presentation of RANKL through the proteolytic cleavage of its transmembrane form to soluble RANKL.221,222
Finally, after a limited period of resorptive activity, the osteoclast is thought to die via apoptosis (see later),223,224 and the resorbed area of cartilage or bone is, in conditions of development, growth, and bone health, efficiently replaced by newly formed bone through the action of osteoblasts.
Osteoblastogenesis
The final step required to rebuild cartilage to bone during development consists of the differentiation of osteoblasts and the deposition of mineralized bone tissue by this specialized cell type. Osteoblasts are mesenchymal cells thought to descend from mesenchymal stem cells that are pluripotent and have the capacity to differentiate into a variety of cell types, including muscle cells (myoblasts), fat cells (adipocytes), chondrocytes, and osteoblasts (Fig. 4-6).225–227 Differentiation of mesenchymal stem cells into distinct lineages is regulated by the expression of different transcription factors. MyoD transcription factors (MyoD, Myf5, and myogenin) regulate myogenic differentiation; C/EBP family (C/EBPβ, C/EBPδ, and C/EBPα) and PPARγ2 transcription factors regulate adipocyte differentiation (see Fig. 4-6).228 In certain settings, osteoblasts and chondrocytes seem to share a common precursor, termed osteochondroprogenitor. Specific transcription factors direct the osteochondroprogenitors into the chondrocyte (the Sox family transcription factors Sox9, Sox5, and Sox6) or the osteoblast lineages (β-catenin, Runx2, and osterix; discussed later) (see Fig. 4-6).31,229,230
FIGURE 4-6 Osteoblast differentiation. Osteoblasts are derived from mesenchymal stem cells (MSCs) or progenitors that can become myocytes, adipocytes, or chondrocytes (driven by the indicated transcription factors) or can be directed into the osteoblast lineage, possibly through an osteochondroprogenitor intermediate. The transcription factors Runx2, Osx, and β-catenin mediate osteoblast differentiation and functioning. Progressive osteoblast differentiation is characterized by changes in gene expression typifying specific stages as indicated. See text for details.
Committed pre-osteoblasts further differentiate into mature osteoblasts that secrete large amounts of type I collagen and other matrix components, which together are called osteoid. Subsequently, osteoblasts direct the mineralization of the osteoid in a process that requires active alkaline phosphatase (ALP), expressed on the osteoblast membrane. These mature osteoblasts express characteristic genes, including the gene encoding osteocalcin.231 Ultimately, the cells die through apoptosis, or are converted to bone-lining cells, or become embedded within the bone matrix as osteocytes. Osteocytes are found dispersed throughout the bone matrix and have the potential to live as long as the organism itself. In fact, osteocytes are the most abundant cell type in bone, constituting over 90% of adult bone cells.232,233 As osteoblasts become encased by the mineralized bone matrix that they themselves synthesized, they dramatically change their shape, sending out numerous dendritic processes that run inside lacunar cannaliculi.234,235 These processes allow osteocytes to communicate with each other, with the vasculature, and with cells on the bone surface.233,236–239 Osteocytes are well positioned to sense and respond to mechanical forces, and a growing body of evidence documents that osteocytes mediate the actions of mechanical forces to increase bone mass (see later). Osteocytes synthesize a series of proteins unique to this most differentiated member of the osteoblast lineage. These include dentin matrix protein (DMP)-1, matrix extracellular phosphoglycoprotein (MEPE) and FGF23, secreted proteins involved in systemic phosphate homeostasis, and sclerostin (SOST), an inhibitor of Wnt action (see later).237,240–244 Despite their relative inactivity compared with osteoblasts, osteocytes play a central role in the determination and maintenance of bone structure (see later).
Wnt/β-Catenin Signaling: Wnts are a large family of secreted growth factors (19 different members in mouse and human genomes) that play essential roles in multiple developmental processes. Wnts are also required for adult tissue maintenance, and perturbations in Wnt signaling can lead to tumor formation and other diseases.17,245–248 In the skeletal system, mutations in Wnt signaling components lead to skeletal malformations and diseases such as osteoporosis-pseudoglioma249 and osteoarthritis.250
Wnts can transduce their signals through several different downstream signaling pathways. The best understood pathway is the canonical or Wnt/β-catenin pathway (Fig. 4-7).251 Central to this pathway is the regulation of the protein stability of β-catenin, which acts as a transcription factor in the canonical Wnt pathway and is also involved in cell adhesion by binding to cadherins. In the absence of Wnts, cytoplasmic β-catenin is constitutively degraded through its phosphorylation by glycogen synthase kinase 3-β (GSK3-β) in a large protein complex brought together by axin and adenomatous polyposis coli (APC).245,247,252,253 Phosphorylated β-catenin is recognized by a β-transducin repeat containing protein (β-TrCP) that targets it for proteasome-mediated degradation. Upon Wnt stimulation, the ligands bind to two synergistically acting families of Wnt (co)receptors, the Frizzled (Fz) receptor family members and low-density lipoprotein receptor-related proteins (LRP5 or LRP6), leading to the recruitment of axin to LRP5/6 in the plasma membrane. The sequestering of axin at the plasma membrane likely leads to disassembly of the β-catenin destruction complex, thus inhibiting the phosphorylation/degradation of β-catenin.254–259 Stabilized β-catenin protein then accumulates in the cytoplasm and translocates to the nucleus, where it interacts with members of the T cell factor/lymphoid enhancer factor (TCF/LEF) family of DNA-binding transcription factors to regulate the expression of downstream target genes (see Fig. 4-7).260–262
FIGURE 4-7 Canonical Wnt/β-catenin signaling pathway. In the absence of Wnt signals (left), β-catenin is associated with glycogen synthase kinase 3-β (GSK3-β) in a destruction complex that also contains Axin and adenomatous polyposis coli (APC). A hyperphosphorylated β-catenin is created by GSK3β and is bound by the β-transducin repeat containing protein (β-TrCP) component of the ubiquitin ligase complex, targeting β-catenin for proteasomal degradation. Wnt signaling through the Frizzled and LRP5/6 receptor complex (right) leads to inhibition of GSK3β through activation of Dishevelled (Dsh). Consequently, when Wnt signaling is active, β-catenin is stabilized, and the level of β-catenin in the cytoplasm becomes elevated. Accumulated β-catenin can translocate to the nucleus to interact with T cell factor/lymphoid enhancer factor (TCF/LEF) proteins and mediate transcription of target genes.
During the past decade, canonical Wnt signaling has been shown to play a significant role in the control of osteoblastogenesis and bone formation. Recent genetic studies analyzing conditional β-catenin loss- and gain-of-function mouse models provided compelling evidence that β-catenin is a crucial transcription factor determining the osteoblast lineage commitment of early osteochondroprogenitors, by inducing osteoblastic and suppressing chondrocytic differentiation.17,263 Indeed, inactivation of β-catenin in mesenchymal progenitor cells blocked osteoblast differentiation, and mesenchymal cells in the perichondrium and calvarium differentiated into chondrocytes instead.16,18,264 In addition, recent evidence showed that β-catenin plays an important role in the coupling between osteoblast and osteoclast activity by stimulating differentiated osteoblasts to produce OPG, an inhibitor of osteoclast formation (see earlier).265,266
In several clinical cases, mutations have been found in the Wnt receptor complexes that are associated with changes in bone mineral density and fractures. Loss-of-function mutations in LRP5 receptors cause osteoporosis-pseudoglioma syndrome,249 whereas gain-of-function mutations in the same group that render LRP5 with reduced affinities for DKK1 and sclerostin (SOST), secreted antagonists of canonical Wnt signaling, result in high bone mass phenotypes.267–271 These actions of LRP5 are indirect actions on bone: Activation of LRP5 in the duodenum suppresses serotonin secretion, and the lower blood levels of serotonin lead to an increase in bone mass through less activation of serotonin Htr1b receptors on osteoblasts.272
The protein inhibitors of Wnt signaling may have additional clinical relevance. For example, osteolytic bone lesions in patients with multiple myeloma are associated with the production of DKK1,273,274 and homozygous loss-of-function mutations in SOST have been identified in patients diagnosed with sclerosteosis, a disfiguring disease associated with high bone mass.275,276 Mutations affecting regulation of SOST transcription also cause van Buchem disease, a disease that resembles sclerosteosis.241,277
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
