[level-membership-for-hematology-oncology-and-palliative-medicine-category]
9 Primary CNS Lymphoma
Introduction
Primary CNS lymphomas (PCNSL) are extranodal malignant lymphomas arising within the brain, eyes, leptomeninges, or spinal cord in the absence of systemic lymphoma at the time of diagnosis. The incidence of PCNSL in western countries is 5 per 1 million person-years. Currently PCNSL are estimated to account for up to 1% of non-Hodgkin lymphomas (NHL) and 3% to 5% of all primary brain tumors. After a continual increase over the past two decades,1 epidemiologic data suggest a recent decrease in the incidence of PCNSL, particularly among young patients suffering from acquired immunodeficiency syndrome, and probably associated with the development of new active antiviral drugs. In contrast, the incidence remains high among older patients (over 60 years), most of whom are immunocompetent.2 The reason for the rising incidence of PCNSL among the immunocompetent population is obscure. PCNSL is also of interest because it is one of the only primary malignant brain tumors whose prognosis has improved considerably over the past two decades due to advances in treatment strategy. Although the prognosis remains poor for the majority of the patients, a substantial minority, representing approximately 20% to 30% of cases, can hope to be cured. Because long-term survivors are at increased risk of developing severe delayed cognitive dysfunction, future treatment should be aimed at improving the efficacy of treatment while limiting the risk of neurotoxicity. This review focuses on PCNSL in the immunocompetent population.
Pathology and Pathogenesis
In immunocompetent patients, all but 5% of PCNSLs are diffuse large B-cell lymphomas (DLBCL).3 Since they are morphologically indistinguishable from systemic DLBCL, the WHO classification of tumors of hematopoietic and lymphoid tissues does not recognize PCNSL as a separate entity.4 The remaining cases of PCNSL are T-cell lymphomas (2% to 5%)5 or, in rare instances, low-grade B-cell lymphomas of the lymphoplasmocytic (Waldenström macroglobulinemia), fol-licular, or mucosa-associated lymphoid tissue (MALT) type.6 Little is known about the tumorigenesis of PCNSL. In contrast to immunocompromised patients, the Epstein-Barr virus (EBV) does not appear to be involved in the pathogenesis of PCNSL in immunocompetent patients. The site of origin of lymphoma cells, the biological mechanisms involved in the neoplastic transformation of the lymphoid cells, and their intriguing confinement within the CNS during the course of the disease have yet to be elucidated. Indeed, the CNS does not contain resident lymphocytes under normal circumstances and lacks lymphatic vessels. However, recent evidence suggests that T and B cells enter the CNS under physiological conditions, and it has been hypothesized that PCNSL may originate from B cells derived from systemic lymphoid tissues and normally trafficking in and out of the CNS.7 PCNSL could derive from a benign CNS inflammatory process through a monoclonal proliferation of B cells. Another hypothesis is that PCNSL might represent the metastasis of an occult systemic lymphoma, eradicated by an intact immune system but escaping within the immune-privileged CNS. Analysis of clonally rearranged immunoglobulin heavy chain (IgH) genes revealed identical dominant PCR products in bone marrow aspirates, blood samples and tumor specimens from some PCNSL patients, suggesting that subclinical systemic disease can be detected at initial diagnosis, which supports a systemic origin of the tumor in these cases.8 In addition, B cell tropism for CNS might be acquired (before or after the oncogenic events) through specific interactions between selective homing receptors and their ligands expressed on CNS endothelial cells, as suggested by the distinctive angiotropism of CNS lymphoma cells.9,10
It also remains unclear whether the dismal outcome of PCNSL compared to systemic DLBCL is attributable to the immune-privileged cerebral location or reflects a specific aggressive intrinsic biologic behavior. Recently, expression profiling and genomic screening have provided new insights into understanding the poor prognosis of PCNSL. Based on lymphochip cDNA microarrays, two distinct gene expression profiles have been identified among systemic DLBCLs, indicative of different stages of B-cell differentiation. One subgroup expressed genes characteristic of germinal center B cells (GCB subgroup) whereas the other expressed genes normally induced during in vitro activation of peripheral blood B cells (ABC subgroup). Interestingly, patients with the GCB signature had a significantly better outcome than those with the ABC profile.11 PCNSLs have been shown to frequently expresses BCL612 and to carry an extremely high load of somatic mutations of immunoglobulin genes and in several oncogenes demonstrating aberrant ongoing hypermutation.13–15 Since such an ongoing hypermutation and BCL6 expression are considered as germinal center markers, it has been postulated that the cell of origin of PCNSL passed through the GC microenvironment and that these neoplasms correspond to the GCB subgroup as defined for DLBCL. However, recent immunoprofiling16 and gene expression studies using cDNA microarrays17,18 demonstrated that PCNSL may exhibit characteristics associated with both ABC and GCB subtypes. Thus, PCNSL may correspond to an overlapping B-cell differentiation time slot, i.e., late GC/early post-GC stage.
Pangenomic analyses of chromosomal imbalances by comparative genomic hybridization (CGH) have shown frequent chromosome 6 q loss (60% to 75%) in PCNSL.19–21 Nakamura et al.22 refined the candidate region suspected to contain a lymphoma-related tumor suppressor gene in the 6q22–23 locus by a fine LOH deletion mapping of 6 q in PCNSL. The PTPRK gene seems a relevant candidate gene, since it is involved in the regulation of cell contact and adhesion, and a loss of protein expression was observed in most (76%) of the PCNSLs tested. It belongs to the protein tyrosine phosphatase superfamily of enzymes. Further studies to identify gene mutations and/or rearrangements are needed to ascertain the involvement of PTPRK in PCNSL tumorigenesis. Interestingly, the authors showed that chromosome 6 q loss was found at a significantly higher rate in PCNSL than in systemic DLBCL and was correlated with shorter survival.22 An independent study of 75 newly diagnosed HIV-negative PCNSLs investigated by interphase FISH analysis has confirmed the frequent 6q22 chromosome deletion, with a prevalence of 45%, and its negative impact on overall survival.23 Chromosome 6 q loss would therefore represent a prognostic marker in PCNSL. Otherwise, the BCL6 gene has been found mutated and its chromosomal locus (3q27) recurrently involved by translocations (20%), suggesting that BCL6 activation through genomic rearrangements may play a role in PCNSL pathogenesis.23–25 BCL6 is frequently expressed in PCNSL, but there are conflicting data on its prognostic value as an immunohistochemical marker.16,26–29 These results provide evidence for an alternative pathogenesis in PCNSL compared to DLBCL explaining, in part, differences in clinical behavior and prognosis.
Diagnosis and Workup
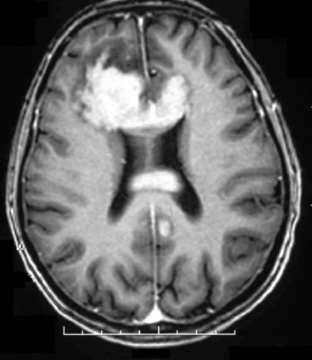
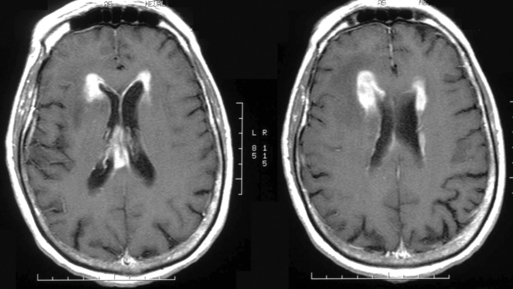
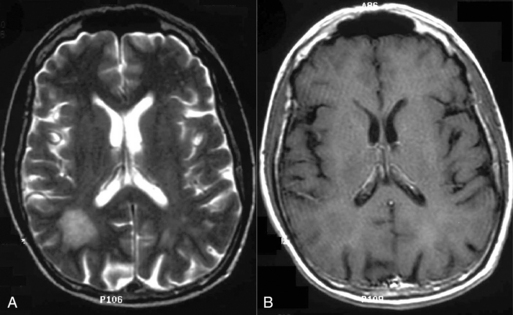
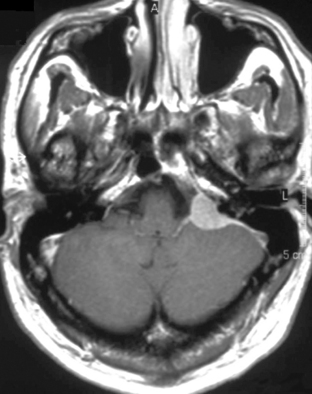
The clinical presentation of PCNSL includes focal symptoms and raised intracranial pressure. Changes in behavior and personality as well as confusion frequently occur in the elderly. The deep location of PCNSL explains why seizures are less frequent than in other brain tumors.30,31 CT scans and MRI typically show solitary (two thirds of cases) or multiple (one third of cases) periventricular, homogeneously-enhancing lesions (Figures 9-1 and 9-2).32–34 PCNSL is potentially associated with a large spectrum of radiological presentations and can simulate inflammatory (sarcoidosis, multiple sclerosis) or postinfectious diseases (ADEM) or other brain tumors (meningiomas, malignant gliomas, gliomatosis cerebri, brain metastases).35–37 The diagnosis may be difficult to establish, especially with the presence of nonenhancing infiltrating lesions (occurring in about 10% of cases) (Figure 9-3),38,39 or dural-based masses (Figure 9-4)40,41; perfusion MRI shows a much lower relative cerebral blood volume than that seen in malignant gliomas, and distinct signal-time intensity curves reflecting rapid leakage of contrast medium into the interstitial space.42 Similarly magnetic resonance spectroscopy at short echo times show massive elevations of lipid resonances in PCNSL, not usually seen in high-grade gliomas.43,44 Steroid-induced response, observed in approximately 40% of cases, is classic but not the rule and should not be used as a diagnostic test since it may prevent a pathological confirmation of the diagnosis. Rarely, spontaneous disappearance of lesions has been reported, hence the term “ghost tumors.”45,46 Ring enhancement, frequently seen in immunodeficient patients, is a pattern rarely seen in immunocompetent patients. However, none of these signs are specific, and diagnosis relies on the pathological study of a tumor sample. Cerebral biopsy can be avoided when lymphomatous cells are discovered in the cerebrospinal fluid (10% to 30% of patients) or in a vitreous-body biopsy (uveitis, sometimes asymptomatic, is present in 10% to 20% of cases at diagnosis). The current recommended staging evaluation for PCNSL includes body CT scans and bone marrow biopsy.47 In fact, systemic involvement is so rare at onset that extensive staging is not recommended by some authors, the only required examinations being HIV testing, chest radiography, analysis of cerebrospinal fluid, ocular slit-lamp examination, and a careful clinical assessment. However, in a recent retrospective study, 7% of patients were found to have systemic NHL by staging FDG body PET when body CT scans and bone marrow biopsies were negative.48 This higher incidence of systemic lymphoma than reported in prior series suggests that occult lymphoma may be more common than recognized previously. These findings warrant prospective validation, since the identification of a systemic site of the lymphoma has important implications in the management of PCNSL.
Treatment Options
TREATMENT OF NEWLY DIAGNOSED PCNSL
Age and performance status are the main independent prognostic factors consistently identified in a large number of studies.49–51 As PCNSL is a highly radiosensitive and chemosensitive infiltrative tumor, surgery is therefore restricted to diagnostic biopsy. Although the tumor appears on MRI as a unique contrast-enhancing lesion in the majority of the immunocompetent patients, whole-brain radiotherapy (WBRT) is recommended based on the microscopically diffuse nature of PCNSL. Despite a high rate of response, RT alone has shown a limited survival benefit in PCNSL with a median overall survival (OS) duration of 10 to 18 months and a 5-year survival rate of less than 5%.52 The only phase II trial of radiotherapy (conducted by the RTOG) delivered a total dose of 40 Gy with an additional 20-Gy boost to contrast-enhancing lesions and reported an overall survival of 11.6 months.53 Interestingly, most of the relapses occurred in sites that had received the maximum RT dose. These disappointing results have led to the use of chemotherapy in combination with WBRT. Since the 1990s, numerous convergent phase II studies have shown that the addition of high-dose methotrexate (MTX)-based chemotherapy to RT substantially improves survival compared with RT alone (median survival: 2 to 4 years; 5-year survival rate: 20% to 40%).54–69 In contrast, adding the CHOP regimen (standard chemotherapy for systemic lymphoma) to RT did not appear to improve survival compared with RT alone. This may reflect the poor central nervous system penetration of the chemotherapy agents included in these regimens. The optimal dose of irradiation postchemotherapy has never been prospectively investigated. Doses between 20 and 50 Gy to the whole brain (WB) with or without a tumor bed boost are currently used, with most of the protocols using a total dose of 40-45 Gys without a boost. For patients who achieve a complete response after high-dose MTX-based chemotherapy, it still remains unclear whether consolidation with WBRT improves disease control or survival. A subset analysis from a phase II trial that included 25 patients aged less than 60 years who achieved CR after initial chemotherapy and received either 45 Gy or 30.6 Gy as consolidation treatment showed a significantly higher recurrence and lower overall survival rate in the reduced dose RT group.61 Other studies did not share this observation. In two multicenter retrospective analyses, no differences were noted regarding disease-free or overall survival between patients in CR receiving WBRT as consolidation treatment or at the time of relapse.70,71 In the Memorial Sloan Kettering Cancer Center (MSKCC) experience, consolidation treatment with WBRT improved failure-free survival but not overall survival in complete-responding patients after MTX-based therapy, but increased the rates of neurotoxicity.72
In order to reduce neurotoxicity, several groups have explored the efficacy of various high-dose MTX-based chemotherapy regimens as initial treatment for newly diagnosed PCNSL. These studies converge to suggest that chemotherapy alone and deferred radiotherapy strategy allows comparable results in terms of survival to those reported for combined chemoradiotherapy, with better neurocognitive and quality of life outcomes.73–79 This supports the comparison of these approaches in a prospective randomized trial. While high-dose MTX is undoubtedly a key drug for PCNSL chemotherapy, interest in adding other agents to MTX exists in the scientific community. Two prospective phase II trials have investigated the efficacy of high-dose MTX (8 g/m2) as a single drug therapy,77,78 and similarly demonstrated a clearly shorter PFS (13 months) than that achieved with a polychemotherapy regimen. This suggests that other cytotoxic agents should be used with high-dose MTX in a chemotherapy-alone approach. However the optimal combination remains to be established. Alkylating agents able to effectively penetrate the CNS would be preferred. Concerning the optimal “high dose” of MTX to deliver, although there is no clear evidence of a dose response, a dose of greater than 3 g/m2 in a rapid infusion is recommended.80 In addition, this dose generally yields cytotoxic levels in the cerebral spinal fluid, thus theoretically avoiding the need to administer intrathecal chemotherapy for leptomeningeal coverage. In practice, when intravenous high dose MTX (>3 g/m2) is used, several authors recommend adding intrathecal CT only in cases of positive CSF cytology and withholding it in the absence of detectable subarachnoid disease.81
DELAYED NEUROTOXICITY
WBRT, high-dose MTX chemotherapy, and the combination of both treatments expose the patients to delayed neurotoxicity. This complication can occur as early as 3 months after the treatment and is characterized by attention deficit, memory impairment, ataxia, and urinary incontinence, potentially ultimately leading to dementia. Imaging shows confluent diffuse white matter changes and later cortical-subcortical atrophy. The physiopathology of this complication remains poorly understood; loss of oligodendrocyte progenitors and oxidative stress have been suggested as potential mechanisms. Lai et al.82 reported a well-documented series of five autopsied cases who died of treatment-related leukoencephalopathy. All had combined treatment and were in tumor remission. In addition to white matter rarefaction and spongiosis, fibrotic thickening of small vessels in the deep white matter and atherosclerosis of intracranial large vessels were systematically found, suggesting that a vascular process may be also an important component of this white matter injury. The risk of neurotoxicity increases sharply with patient age. In the elderly population (patients over age 60), virtually all long-term survivors will develop delayed neurotoxicity with its devastating consequences on quality of life and mortality.83 In the younger population (patients under age 60), the exact incidence of this complication is more difficult to determine. Cognitive dysfunctions are usually less severe and occur later than in the elderly population, although they may interfere with quality of life.64 An update of the MSKCC experience providing long-term data (median follow-up of 115 months) reported a 26% rate of neurotoxicity in surviving patients younger than 60 years (versus 75% in the elderly).68 However, this should be regarded as a minimum estimate in the absence of a psychometric evaluation. A series of 19 consecutive young patients (median age: 44 years) treated in a European clinical trial and in complete remission for a mean time of 24 months after combined therapy were investigated by an extensive neuropsychological evaluation.84 Cognitive impairments were found in 63% of patients (including 21% with severe cognitive deficits); only 42% of the patients had resumed work, and 67% had white matter abnormalities and cortical atrophy detectable by MRI. Although this study suffers from the absence of available baseline data at the completion of the treatment to assess the potential contribution of the tumor to cognitive dysfunction, it suggests that the incidence of delayed neurotoxicity after combined therapy is largely underestimated in young patients, albeit less severe than in the elderly. These results contrast sharply with a prospective neuropsychological study performed by Fliessbach et al.84 in a series of 23 patients successfully treated with a high-dose MTX-based chemotherapy without radiotherapy. Comparison between baseline evaluation at completion of the treatment and at the last follow-up (median: 44 months) showed a good preservation of cognitive functions, although one third of the patients demonstrated some degree of white matter changes at the MRI. This latter point confirmed some previously published reports showing that MTX-related leukoencephalopathy is a frequent but not necessarily universal accompaniment to deterioration of cognitive performance.86 The significantly better preservation of neurocognitive functions and quality of life observed in patients treated with chemotherapy alone as compared with those who received combined treatment was also supported by Correa et al.87 in a retrospective comparative neuropsychometric analysis of 28 patients from a single institution. The incorporation of systematic psychometric and quality of life evaluations with an appropriate standardized test battery is recommended for all future prospective trials.88 Interestingly, some functional polymorphisms interfering with the methionine metabolism might influence MTX neurotoxicity.89,90
TREATMENT IN THE ELDERLY
Elderly patients (i.e., ≥ 60 years of age), who experience a very poor prognosis and a high vulnerability to the delayed neurotoxicity, represent an important subgroup, accounting for approximately half of all cases of PCNSL. However, prospective trials specifically devoted to older patients are scarce. Most of the available data come from retrospective studies. In the elderly, PCNSLs exhibit a low radiosensitivity; the RTOG Phase II trial reported a short median survival of 7.6 months with RT alone.53 The high risk of neurotoxicity observed with the combined chemo-radiotherapy approach (see above) prompted several authors to defer radiotherapy in this population. The only multicenter phase II focusing on patients older than 60 and evaluating chemotherapy alone as initial treatment was conducted by the EORTC. The regimen consisted of high-dose MTX (1 g/m2) plus lomustine (CCNU), procarbazine, and intrathecal chemotherapy (MTX, cytarabine). The intent-to-treat response rate and median survival were 48% and 14.3 months respectively.98 Although this study showed less favorable results than those reported by other published studies (MS ranging from 18 to 34 months),68,74,75,95–100 it led, nevertheless, to the same conclusions and confirmed that chemotherapy alone is a valuable approach for treating elderly patients with PCNSL. Since median PFS was similar to the other studies, the lower OS may be explained by the salvage therapy. Hence, in the EORTC trial, only a small minority of patients were treated by WBRT at relapse. Altogether, chemotherapy alone appears to be more effective than RT alone and considerably reduces the risk of neurotoxicity (up to 8% of cases) as compared with that expected with combined treatment, allowing a substantial proportion of patients to reach prolonged remission without the need for consolidation radiotherapy and preserving their quality of life. Future protocols for the elderly should focus on defining the optimal chemotherapy regimen.
INTENSIVE CHEMOTHERAPY (ICT) WITH AUTOLOGOUS STEM-CELL TRANSPLANTATION (ASCT)
Intensive chemotherapy (ICT) with autologous stem-cell transplantation (ASCT) is the standard treatment for chemosensitive relapsing systemic NHL. Because ICT is expected to improve the BBB crossing, allowing cytotoxic agents to reach the brain at higher doses, this strategy has been evaluated for PCNSL. This procedure was first evaluated in refractory and recurrent cerebral and intraocular lymphoma with promising results in a single institutional pilot study.101 The protocol consisted of an induction cytarabine-etoposide combination (CYVE regimen) followed by high-dose chemotherapy with thiotepa, busulfan, and cyclophosphamide (TBC regimen). These results have been recently confirmed in a multicenter phase II trial using the same regimen including 43 patients.102 Twenty-seven patients (62% by intention-to-treat analysis) completed the full ICT-ASCT procedure, including 15 patients responsive and 12 nonresponsive to CYVE-induction salvage chemotherapy. Twenty-six of 27 patients achieved a complete response with prolonged remission; the median PFS and overall survival were 41 and 58 months respectively. Interestingly, all but one patient (in whom the disease was refractory to the salvage chemotherapy) achieved a complete response after ICT-ASCT (Figure 9-2). The intent-to-treat median PFS and overall survival of the whole population of this trial were 11 and 18 months. Altogether, these results compare favorably with those yet reported by other salvage treatments, including second line conventional chemotherapy regimens103,104 and radiotherapy alone.105,106 The favorable impact of ICT-ASCT on survival, regardless of the chemosensitivity status before IC, contrasts with the situation in relapsing systemic NH lymphomas, suggesting that ICT-ASCT might overcome resistance mediated by the BBB.
Several studies have evaluated ICT-ASCT as first-line treatment in newly-diagnosed PCNSL. BEAM protocol (BCNU, etoposide, cytarabine, and melphalan) or thiotepa-based chemotherapy were used as conditioning regimens, and increased the rate of complete remission after high-dose MTX-based induction chemotherapy. However, three of these trials included whole brain radiotherapy at the end of the procedure, making the analysis of the specific contribution of IC-ASCT to the encouraging survival results questionable. Subsequently, in order to minimize the risk of neurotoxicity, Illerhaus et al.112 modified their initial protocol by increasing the number of chemotherapy cycles and augmenting the thiotepa dose within the conditioning regimen while restricting WBRT only to patients not in complete response after finishing chemotherapy. In addition, they delivered ICT-ASCT to all of their patients, irrespective of their response to high-dose MTX. The preliminary results of this pilot study support the hypothesis that WBRT may not be necessary to cure many patients in CR after ICT-ASCT. The only trial that did not combine radiotherapy with ICT-ASCT used an induction high-dose MTX-cytarabine chemotherapy followed by an intensive BEAM regimen.107 The results were disappointing, with a short median event-free survival (9.3 months). This suggests that the drugs were used at suboptimal doses and that more aggressive regimens, including agents such as thiotepa and busulfan that penetrate the CNS, rather than standard lymphoma regimens may be warranted. This may be illustrated by encouraging results reported by Cheng et al.108 using the TBC pretransplant conditioning regimen without WBRT in a small series of 7 patients, with a median event-free survival that had not been reached at 24 months.
The evaluation of the neurocognitive tolerance of this approach is an important issue. Soussain et al.101,102 observed a 10% to 30% rate of neurotoxicity in relapsing patients treated by ICT-ASCT as salvage therapy, especially in the older and preirradiated patients. In the studies using a combined approach, i.e., ICT-ASCT followed by WBRT, for newly diagnosed PCNSL, the reported rates of severe neurotoxicity range from 0 to 20%.110–112 In contrast, this was not reported in the two studies using ICT-ASCT without WBRT as primary treatment.107,108 Further prolonged neurocognitive followup with psychometric evaluation is clearly needed. Neurotoxicity seems influenced by age of the patient, prior treatment (especially radiotherapy), and the CNS safety profile of the drug used. It remains to be determined whether ICT-ASCT can represent an interesting alternative option to radiotherapy as consolidation treatment.
SALVAGE TREATMENT
Although combined treatment has considerably improved the prognosis of PCNSL, one should not forget that about one third of the patients are refractory to initial treatment and that the majority of the patients who have achieved a complete remission will subsequently relapse. As discussed above, the most promising results have been reported with ICT-ASCT. Conventional second-line chemotherapy, such as temozolomide (TMZ),104 topotecan,113 intraarterial carboplatin,114 and high dose cytarabine combined with etoposide and ifosfamide103 have been also shown to be potentially active in relapsed PCNSL. These latter treatments achieved objective response rates (26% to 37%), 1-year PFS (13% to 22%) and 1-year overall survival (25% to 41%). Although the activity of TMZ and topotecan as single agents is modest in relapsed tumors, their role as part of a first-line MTX-based combination merits further investigation,99 particularly because of their relatively good safety profile. MTX reinduction may also yield to new remission in some patients who have previously achieved a prolonged remission with high-dose MTX-based chemotherapy.115 Two studies recently evaluated the activity and tolerance of WBRT delivered in relapsing PCNSL previously treated with high dose MTX-based CT alone as initial treatment.105,106 Interestingly, the response rate was high (70%) and the median survival from relapse ranged from 11 to 16 months, quite similar to what we would expect with WBRT as initial treatment.53 This suggests a preservation of radiation sensitivity at recurrence after high-dose MTX. Delayed neurotoxicity occurred in 15% to 22%, raising the possibility that when RT is deferred after high-dose MTX, the risk of neurotoxicity compared to immediate postchemotherapy irradiation is significantly lower.
IMMUNOTHERAPY BY ANTI-CD20 ANTIBODIES
Because most of PCNSLs are neoplastic B cells expressing the CD20 surface antigen, the chimeric monoclonal antibody rituximab is a potentially active treatment for this disease. It has been successfully used in systemic diffuse large B-cell lymphomas, in association with the CHOP regimen. However, the potential efficacy of rituximab in CNS tumors when delivered intravenously is limited by its high molecular weight, which prevents its penetration into the CNS through an intact blood-brain barrier. Pharmacokinetic studies have estimated that the CSF levels of rituximab are approximately 0.1% of matched serum levels after intravenous administration.116 Schulz et al.117 reported their experience using direct intraventricular/intrathecal administration of rituximab (10 to 40 mg), allowing them to reach a higher continuous concentration in the CSF, in a series of six patients. The only relevant toxicity was an acute reversible paraparesis associated with back pain related to a rapid tumor cell lysis in the CSF. An objective response was observed in all four patients with leptomeningeal disease, while no response was obtained in the two patients suffering from parenchymal tumor mass. Rubenstein et al.118 conducted a phase I study in recurrent CNS lymphoma and found that intraventricular rituximab monotherapy (10 to 25 mg) was feasible and effective. They reported a cytologic response in six of nine patients with lymphomatous meningitis. Interestingly, two out of three patients with concurrent intraocular lymphoma and one out of five with brain parenchymal lymphoma exhibited an objective response. These preliminary results suggest that intraventricular/intrathecal rituximab can be safely delivered and may have a role in the management of leptomeningeal and ocular disease, rather than in parenchymal tumors of PCNSL.
Intravenous rituximab has been used in combination with a high-dose MTX-based chemotherapy regimen (MPVA) as initial treatment before WBRT for newly diagnosed PCNSL,69 and with temozolomide as salvage treatment for recurrent parenchymal CNS lymphomas.119,120 Both combinations were well-tolerated except for a higher rate of neutropenia seen when rituximab was added to MPVA, and were associated with improved survival. However, the specific contribution of intravenous rituximab on these results remains speculative.
1. J.E. Olson, C.A. Janney, R.D. Rao, et al. The continuing increase in the incidence of primary central nervous system non-Hodgkin lymphoma: a surveillance, epidemiology, and end results analysis. Cancer. 2002;95:1504-1510.
2. N.S. Kadan-Lottick, M.C. Skluzacek, J.G. Gurney. Decreasing incidence rates of primary central nervous system lymphoma. Cancer. 2002;95:193-202.
3. S. Camilleri-Broët, A. Martin, A. Moreau, et al. Primary central nervous system lymphomas in 72 immunocompetent patients: pathologic findings and clinical correlations. Groupe Ouest Est d’étude des Leucémies et Autres Maladies du sang (GOELAMS). Am J Clin Pathol. 1998;110:607-612.
4. S.H. Swerdlow, E. Campo, N.L. Harris, et al. World Health Organization Classification of Tumours. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. In: WHO press, editors. WHO classification of tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon:IARC. 2008.
5. T.N. Shenkier. Unusual variants of primary central nervous system lymphoma. Hematol Oncol Clin North Am. 2005;19:651-664. vi
6. P.H. Tu, C. Giannini, A.R. Judkins, et al. Clinicopathologic and genetic profile of intracranial marginal zone lymphoma: a primary low-grade CNS lymphoma that mimics meningioma. J Clin Oncol. 2005;23:5718-5727.
7. A. Uccelli, F. Aloisi, V. Pistoia. Unveiling the enigma of the CNS as a B-cell fostering environment. Trends Immunol. 2005;26:254-259.
8. K. Jahnke, M. Hummel, A. Korfel, et al. Detection of subclinical systemic disease in primary CNS lymphoma by polymerase chain reaction of the rearranged immunoglobulin heavy-chain genes. J Clin Oncol. 2006;24:4754-4757.
9. A. Alter, M. Duddy, S. Hebert, et al. Determinants of human B cell migration across brain endothelial cells. J Immunol. 2003;170:4497-4505.
10. J.R. Smith, R.M. Braziel, S. Paoletti, et al. Expression of B-cell-attracting chemokine 1 (CXCL13) by malignant lymphocytes and vascular endothelium in primary central nervous system lymphoma. Blood. 2003;101:815-821.
11. A.A. Alizadeh, M.B. Elsen, R.E. Davis, et al. Distinct types of diffuse large B cell lymphoma identified by gene expression profiling. Nature. 2000;403:503-511.
12. L.M. Larocca, D. Capello, A. Rinelli, et al. The molecular and phenotypic profile of primary central nervous system lymphoma identifies distinct categories of the disease and is consistent with histogenetic derivation from germinal center-related B cells. Blood. 1998:1011-1019.
13. A.R. Thompsett, D.W. Ellison, F.K. Stevenson, et al. VH gene sequences from primary central nervous system lymphomas indicate derivation from highly mutated germinal center B cells with ongoing mutational activity. Blood. 1999;5:1738-1746.
14. M. Montesinos-Rongen, R. Küppers, D. Schlüler, et al. Primary central nervous system lymphomas are derived from germinal-center B cells and show a preferential usage of the V4–34 gene segment. Am J Pathol. 1999;155:2077-2086.
15. M. Montesinos-Rongen, D. Van Roost, C. Schaller, et al. Primary diffuse large B-cell lymphomas of the central nervous system are targeted by aberrant somatic hypermutation. Blood. 2004;103:1869-1875.
16. S. Camilleri-Broët, E. Crinière, P. Broët, et al. A uniform activated B-cell-like immunophenotype might explain the poor prognosis of primary central nervous system lymhpomas: analysis of 83 cases. Blood. 2006;107:190-196.
17. J.L. Rubinstein, J. Fridlyand, A. Shen, et al. Gene expression and angiotropism in primary CNS lymphoma. Blood. 2006;107:3716-3723.
18. M. Montesinos-Rongen, A. Brunn, S. Bentink, et al. Gene expression profiling suggests primary central nervous system lymphomas to be derived from a late germinal center B cell. Leukemia. 2008;22:400-405.
19. T. Weber, R.G. Weber, K. Kaulich, et al. Characteristic chromosomal imbalances in primary central nervous system lymphomas of the diffuse large B-cell type. Brain Pathol. 2000;10:73-84.
20. K. Harada, T. Nishizaki, H. Kubota, et al. Distinct primary central nervous system lymphoma defined by comparative genomic hybridization and laser scanning cytometry. Cancer Genet Cytogenet. 2001;125:147-150.
21. R. Boonstra, A. Koning, M. Mastik, et al. Analysis of chromosomal copy number changes and oncoprotein expression in primary central nervous system lymphomas: frequent loss of chromosome arm 6 q. Virchows Arch. 2003;443:164-169.
22. M. Nakamura, M. Kishi, T. Sakaki, et al. Novel tumor suppressor loci on 6q22–23 in primary central nervous system lymphomas. Cancer Res. 2003;63:737-741.
23. F.M. Cady, B.P. O’Neill, M.E. Law, et al. Del(6)(q22) and BCL6 rearrangements in primary CNS lymphoma are indicators of an aggressive clinical course. J Clin Oncol. 2008;26:4814-4819.
24. M. Montesinos-Rongen, R. Zühlke-Jenisch, S. Gesk, et al. Interphase cytogenetic analysis of lymphoma-associated chromosomal breakpoints in primary diffuse large B-cell lymphomas of the central nervous system. J Neuropathol Exp Neurol. 2002;61:926-933.
25. H. Schwindt, T. Akasaka, R. Zühlke-Jenisch, et al. Chromosomal translocations fusing the BCL6 gene to different partner loci are recurrent in primary central nervous lymphoma and may be associated with aberrant somatic hypermutation or defective class switch recombination. J Neuropathol Exp Neurol. 2006;65:776-782.
26. C.C. Chang, B. Kampalath, C. Schultz, et al. Expression of p53, c-Myc, or Bcl-6 suggests a poor prognosis in primary central nervous system diffuse large B-cell lymphoma among immunocompetent individuals. Arch Pathol Lab Med. 2003;127:208-212.
27. K.M. Braaten, R.A. Betensky, L. de Leval, et al. BCL-6 expression predicts improved survival in patients with primary central nervous system lymphoma. Clin Cancer Res. 2003;9:1063-1069.
28. C.H. Lin, K.T. Kuo, S.S. Chuang, et al. Comparison of the expression and prognostic significance of differentiation markers between diffuse large B-cell lymphoma of central nervous system origin and peripheral nodal origin. Clin Cancer Res. 2006;12:1152-1156.
29. O. Levy, L.M. Deangelis, D.A. Filippa, et al. Bcl-6 predicts improved prognosis in primary central nervous system lymphoma. Cancer. 2008;112:151-156.
30. B. Bataille, V. Delwail, E. Menet, et al. Primary intracerebral malignant lymphoma: report of 248 cases. J Neurosurg. 2000;92:261-266.
31. A.J.M. Ferreri, M. Reni. Primary central nervous system lymphoma. Crit Rev Oncol Hematol. 2007;63:257-268.
32. R.J. Clifford, D.F. Reese, B.W. Scheithauer. Radiographic findings in 32 cases of primary CNS lymphoma. AJNR. 1986;146:271-276.
33. K.K. Koeller, J.G. Smirniotopoulos, R.V. Jones. Primary central nervous system lymphoma: radiologic-pathologic correlation. Radiographics. 1997;17:1497-1526.
34. U. Bühring, U. Herrlinger, T. Krings, et al. MRI features of primary central nervous system lymphomas at presentation. Neurology. 2001;57:393-396.
35. L.M. DeAngelis. Primary central nervous system lymphoma imitates multiple sclerosis. J Neurooncol. 1990;9:177-181.
36. U. Herrlinger, M. Schabet, M. Bitzer, et al. Primary central nervous system lymphoma: from clinical presentation to diagnosis. J Neurooncol. 1999;43:219-226.
37. L. Ayuso-Peralta, M. Ortí-Pareja, M. Zurdo-Hernández, et al. Cerebral lymphoma presenting as a leukoencephalopathy. J Neurol Neurosurg Psychiatry. 2001;71:243-246.
38. L.M. De Angelis. Cerebral Lymphoma Presenting as a Nonenhancing Lesion on Computed Tomographic/Magnetic Resonance Scan. Ann Neurol. 1993;33:308-311.
39. R. Kanai, M. Shibuya, T. Hata, et al. A case of ‘lymphomatosis cerebri’ diagnosed in an early phase and treated by whole brain radiation: case report and literature review. J Neurooncol. 2008;86:83-88.
40. A. Benouaich, J.P. Delord, M. Danjou, et al. Primary dural lymphoma: a report of two cases with review of the literature. Rev Neurol. 2003;159:652-658.
41. I. Bódi, A. Hussain, R.W. Gullan, et al. January 2003: 56-year-old female with right frontal tumor of the dura. Brain Pathol. 2003;13:417-418.
42. M. Hartmann, S. Heiland, I. Harting, et al. Distinguishing of primary cerebral lymphoma from high-grade glioma with perfusion-weighted magnetic resonance imaging. Neuroscience Lett. 2003;338:119-122.
43. I. Harting, M. Hartmann, G. Jost, et al. Differentiating primary central nervous system lymphoma from glioma in humans using localised proton magnetic resonance spectroscopy. Neurosci Lett. 2003;342:163-166.
44. S. Taillibert, R. Guillevin, C. Menuel, et al. Brain lymphoma: usefulness of the magnetic resonance spectroscopy. Neuro Oncol. 2008;86:225-229.
45. M. Weller. Glucocorticoid treatment of primary CNS lymphoma. J Neurooncol. 1999;43:237-239.
46. B.S. Mathew, K.A. Carson, S.A. Grossman. Initial response to glucocorticoids. A potentially important prognosis factor in patients with Primary CNS lymphoma. Cancer. 2006;106:383-387.
47. L.E. Abrey, T. Batchelor, A.J.M. Ferreri, et al. Report of an International Workshop to Standardize baseline Evaluation and Response Criteria for Primary CNS Lymphoma. J Clin Oncol. 2005;23:5034-5043.
48. N.A. Mohile, L.S. DeAngelis, L.E. Abrey. The utility of body FDG PET in staging primary central nervous system lymphoma. Neuro Oncol. 2008;10:223-228.
49. A.J. Ferreri, J.Y. Blay, M. Reni, et al. Prognostic scoring system for primary CNS lymphomas: the International Extranodal Lymphoma Study Group experience. J Clin Oncol. 2003;21:266-272.
50. E.M. Bessell, F. Graus, A. Lopez-Guillermo, et al. Primary non-Hodgkin’s lymphoma of the CNS treated with CHOD/BVAM or BVAM chemotherapy before radiotherapy: long-term survival and prognostic factors. Int J Radiat Oncol Biol Phys. 2004;59:501-508.
51. L.E. Abrey, L. Ben-Porat, K.S. Panageas, et al. Primary central nervous system lymphoma: the Memorial Sloan-Kettering Cancer Center prognostic model. J Clin Oncol. 2006;24:5711-5715.
52. E.M. Bessell, K. Hoang-Xuan, A.J. Ferreri, et al. Primary central nervous system lymphoma: biological aspects and controversies in management. Eur J Cancer. 2007;43:1141-1152.
53. D.F. Nelson, K.L. Martz, H. Bonner, et al. Non-Hodgkin’s lymphoma of the brain: can high dose, large volumen radiation therapy improve survival? Report on a prospective trial by the Radiation Therapy Oncology Group (RTOG) 1992. Int J Radiat Oncol Biol Phys. 1992;23:9-17.
54. A.A. Gabbai, F.H. Hochberg, R.M. Linggood, et al. High-dose methotrexate for non-AIDS primary central nervous system lymphoma. Report of 13 cases. J Neurosurg. 1989;70:190-194.
55. L.M. De Angelis, J. Yahalom, H.T. Thaler, et al. Combined modality therapy for primary CNS lymphoma. J Clin Oncol. 1992;10:635-643.
56. J. Glass, M.L. Gruber, L. Cher, et al. Preirradiation methotrexate chemotherapy of primary central nervous system lymphoma: long-term outcome. J Neurosurg. 1994;81:188-195.
57. J.Y. Blay, D. Bouhour, C. Carrie, et al. The C5R protocol: a regimen of high-dose chemotherapy and radiotherapy in primary cerebral non-Hodgkin’s lymphoma of patients with no known cause of immunosuppression. Blood. 1995;15(86):2922-2929.
58. J. Glass, C. Shustik, F.H. Hochberg, et al. Therapy of primary central nervous system lymphoma with preirradiation methotrexate, cyclophosphamide, doxorubicin, vincristine, and dexamethasone (MCHOD). J Neurooncol. 1996;30:257-265.
59. M. Brada, D. Hjiyiannakis, F. Hines, et al. Short intensive primary chemotherapy and radiotherapy in sporadic primary CNS lymphoma (PCL). Int J Radiat Oncol Biol Phys. 1998;40:1157-1162.
60. P. O’Brien, D. Roos, G. Pratt, et al. Phase II multicenter study of brief single-agent methotrexate followed by irradiation in primary CNS lymphoma. J Clin Oncol. 2000;18:519-526.
61. E.M. Bessell, A. López-Guillermo, S. Villá, et al. Importance of radiotherapy in the outcome of patients with primary CNS lymphoma: an analysis of the CHOD/BVAM regimen followed by two different radiotherapy treatments. J Clin Oncol. 2002;20:231-236.
62. L.M. DeAngelis, W. Seiferheld, S.C. Schold, et al. Combination chemotherapy and radiotherapy for primary central nervous system lymphoma: Radiation Therapy Oncology Group Study 93–10. J Clin Oncol. 2002;20:4643-4648.
63. P.M. Poortmans, H.C. Kluin-Nelemans, H. Haaxma-Reiche, et al. High-dose methotrexate-based chemotherapy followed by consolidating radiotherapy in non-AIDS-related primary central nervous system lymphoma: European Organization for Research and Treatment of Cancer Lymphoma Group Phase II Trial 20962. J Clin Oncol. 2003;21:4483-4488.
64. A.M. Omuro, L.M. DeAngelis, J. Yahalom, et al. Chemoradiotherapy for primary CNS lymphoma: an intent-to-treat analysis with complete follow-up. Neurology. 2005;64:69-74.
65. A. Korfel, P. Martus, M.R. Nowrousian, et al. Response to chemotherapy and treating institution predict survival in primary central nervous system lymphoma. Br J Haematol. 2005;128:177-183.
66. A.J. Ferreri, S. Dell’Oro, M. Foppoli, et al. MATILDE regimen followed by radiotherapy is an active strategy against primary CNS lymphomas. Neurology. 2006;66:1435-1438.
67. L.E. Abrey, J. Yahalom, L. De Angelis. Treatment of primary CNS lymphoma: the next step. J Clin Oncol. 2000;18:3144-3150.
68. I.T. Gavrilovic, A. Hormigo, J. Yahalom, et al. Long-term follow-up of high-dose methotrexate-based therapy with and without whole brain irradiation for newly diagnosed primary CNS lymphoma. J Clin Oncol. 2006;24:4570-4574.
69. G.D. Shah, J. Yahalom, D.D. Correa, et al. Combined immunochemotherapy with reduced whole-brain radiotherapy for newly diagnosed primary CNS lymphoma. J Clin Oncol. 2007;25(30):4730-4735.
70. M. Reni, A.J. Ferreri. Therapeutic management of refractory or relapsed primary central nervous system lymphomas. Ann Hematol. 2001;80(Suppl. 3):B113-B117.
71. A.J. Ferreri, M. Reni, F. Pasini, et al. Multicenter study of treatment of primary CNS lymphoma. Neurology. 2002;58:1513-1520.
72. M. Ekenel, F.M. Iwamoto, L.S. Ben-Porat, et al. Primary central nervous system lymphoma: the role of consolidation treatment after a complete response to high-dose methotrexate-based chemotherapy. Cancer. 2008;113:1025-1031.
73. E.A. Neuwelt, D.L. Goldman, S.A. Dahlborg, et al. Primary CNS lymphoma treated with osmotic blood-brain barrier disruption: prolonged survival and preservation of cognitive function. J Clin Oncol. 1991;9:1580-1590.
74. L.D. McAllister, N.D. Doolittle, P.E. Guastadisegni, et al. Cognitive outcomes and long-term follow-up results after enhanced chemotherapy delivery for primary central nervous system lymphoma. Neurosurgery. 2000;46:51-60.
75. H. Pels, I.G. Schmidt-Wolf, A. Glasmacher, et al. Primary central nervous system lymphoma: results of a pilot and phase II study of systemic and intraventricular chemotherapy with deferred radiotherapy. J Clin Oncol. 2003;21:4489-4495.
76. V. Sandor, V. Stark-Vancs, D. Pearson, et al. Phase II trial of chemotherapy alone for primary CNS and intraocular lymphoma. J Clin Oncol. 1998;16:3000-3006.
77. T. Batchelor, K. Carson, A. O’Neill, et al. Treatment of primary CNS lymphoma with methotrexate and deferred radiotherapy: a report of NABTT 96–07. J Clin Oncol. 2003;21:1044-1049.
78. U. Herrlinger, W. Küker, M. Uhl, et al. NOA-03 trial of high-dose methotrexate in primary central nervous system lymphoma: final report. Ann Neurol. 2005;57:843-847.
79. A.M. Omuro, L. Taillandier, O. Chinot, et al. Methotrexate (MTX), procarbazine and CCNU for primary central nervous system lymphoma (PCNSL) in patients younger than 60: Can radiotherapy (RT) be deferred? J Clin Oncol. 24, 2006. abstract 1551
80. S. Hiraga, N. Arita, T. Ohnishi, et al. Rapid infusion of high-dose methotrexate resulting in enhanced penetration into cerebrospinal fluid and intensified tumor response in primary central nervous system lymphomas. Neurosurgery. 1999;91:221-230.
81. R.B. Khan, W. Shi, H.T. Thaler. Is intrathecal methotrexate necessary in the treatment of primary CNS lymphoma? J Neurooncol. 2002;58:175-178.
82. R. Lai, L.E. Abrey, M.K. Rosenblum, et al. Treatment-induced leukoencephalopathy in primary CNS lymphoma: a clinical and autopsy study. Neurology. 2004;62:451-456.
83. L.E. Abrey, L.M. DeAngelis, J. Yahalom. Long term survival in primary CNS lymphoma. J Clin Oncol. 1998;16:859-863.
84. H. Harder, H. Holtel, J.E. Bromberg, et al. Cognitive status and quality of life after treatment for primary CNS lymphoma. Neurology. 2004;62:544-547.
86. K. Fliessbach, H. Urbach, C. Helmstaedter, et al. Cognitive performance and magnetic resonance imaging findings after high-dose systemic and intraventricular chemotherapy for primary central nervous system lymphoma. Arch Neurol. 2003;60:563-568.
87. D.D. Correa, L.M. DeAngelis, W. Shi, et al. Cognitive functions in survivors of primary central nervous system lymphoma. Neurology. 2004;62:548-555.
88. D.D. Correa, L. Maron, H. Harder, et al. Cognitive functions in primary central nervous system lymphoma: literature review and assessment guidelines. Ann Oncol. 2007;18:1145-1151.
89. M. Linnebank, H. Pels, N. Kleczar, et al. MTX-induced white matter changes are associated with polymorphisms of methionine metabolism. Neurology. 2005;64:912-913.
90. M. Linnebank, S. Moskau, A. Jurgens, et al. Association of genetic variants of methionine metabolism with MTX-induced CNS white matter changes in patients with primary central nervous system lymphoma. Neuro Oncol. 2008. Sep 22
95. R.J. Freilich, J.Y. Delattre, A. Monjour, et al. Chemotherapy without radiation therapy as initial treatment for primary CNS lymphoma in older patients. Neurology. 1996;46:435-439.
96. S. Ng, M.A. Rosenthal, D. Ashley, et al. High-dose methotrexate for primary CNS lymphoma in the elderly. Neuro Oncol. 2000;2:40-44.
97. A. Juergens, H. Pels, U. Schlegel, et al. A Primary central nervous system lymphoma: results of a pilot and phase II study of systemic and intraventricular chemotherapy with deferred radiotherapy – Final report. J Neurol. 253(II/23–II24), 2006. abstract O95
98. K. Hoang-Xuan, L. Taillandier, O. Chinot, et al. Chemotherapy alone as initial treatment for primary CNS lymphoma in patients older than 60 years: a multicenter phase II study (26952) of the European Organization for Research and Treatment of Cancer Brain Tumor Group. J Clin Oncol. 2003;21:2726-2731.
99. A.M. Omuro, L. Taillandier, O. Chinot, et al. Temozolomide and methotrexate for primary central nervous system lymphoma in the elderly. J Neurooncol. 2007;85:207-211.
100. E.R. Gerstner, J.J. Zhu, D.A. Engler, et al. High dose methotrexate for elderly patients with primary central nervous system lymphoma. Neuro Oncol. 2008. Aug 29
101. C. Soussain, F. Suzan, K. Hoang-Xuan, et al. Results of intensive chemotherapy followed by hematopoietic stem-cell rescue in 22 patients with refractory or recurrent primary CNS lymphoma or intraocular lymphoma. J Clin Oncol. 2001;19:742-749.
102. C. Soussain, K. Hoang-Xuan, L. Taillandier, et al. Intensive chemotherapy followed by hematopoietic stem-cell rescue for refractory and recurrent primary CNS and intraocular lymphoma: Société Française de Greffe de Moëlle Osseuse-Thérapie Cellulaire. J Clin Oncol. 2008;26:2512-2518.
103. E. Arellano-Rodrigo, A. López-Guillermo, E.M. Bessell, et al. Salvage treatment with etoposide (VP-16), ifosfamide and cytarabine (Ara-C) for patients with recurrent primary central nervous system lymphoma. Eur J Haematol. 2003;70:219-224.
104. M. Reni, W. Mason, F. Zaja, et al. Salvage chemotherapy with temozolomide in primary CNS lymphomas: preliminary results of a phase II trial. Eur J Cancer. 2004;40:1682-1688.
105. P.L. Nguyen, A. Chakravarti, D.M. Finkelstein, et al. Results of whole-brain radiation as salvage of methotrexate failure for immunocompetent patients with primary CNS lymphoma. J Clin Oncol. 2005;23:1507-1513.
106. A.F. Hottinger, L.M. DeAngelis, J. Yahalom, et al. Salvage whole brain radiotherapy for recurrent or refractory primary CNS lymphoma. Neurology. 2007;69:1178-1182.
107. L.E. Abrey, C.H. Moskowitz, W.P. Mason, et al. Intensive methotrexate and cytarabine followed by high-dose chemotherapy with autologous stem-cell rescue in patients with newly diagnosed primary CNS lymphoma: an intent-to-treat analysis. J Clin Oncol. 2003;21:4151-4156.
108. T. Cheng, P. Forsyth, A. Chaudhry, et al. High-dose thiotepa, busulfan, cyclophosphamide and ASCT without whole-brain radiotherapy for poor prognosis primary CNS lymphoma. Bone Marrow Transplant. 2003;31:679-685.
110. M. Montemurro, T. Kiefer, F. Schüler, et al. Primary central nervous system lymphoma treated with high-dose methotrexate, high-dose busulfan/thiotepa, autologous stem-cell transplantation and response-adapted whole-brain radiotherapy: results of the multicenter Ostdeutsche Studiengruppe Hamato-Onkologie OSHO-53 phase II study. Ann Oncol. 2007;18:665-671.
111. G. Illerhaus, R. Marks, G. Ihorst, et al. High-dose chemotherapy with autologous stem-cell transplantation and hyperfractionated radiotherapy as first-line treatment of primary CNS lymphoma. J Clin Oncol. 2006;24:3865-3870.
112. G. Illerhaus, F. Müller, F. Feuerhake, et al. High-dose chemotherapy and autologous stem-cell transplantation without consolidating radiotherapy as first-line treatment for primary lymphoma of the central nervous system. Haematologica. 2008;93:147-148.
113. L. Fischer, E. Thiel, H.A. Klasen, et al. Response of relapsed or refractory primary central nervous system lymphoma (PCNSL) to topotecan. Neurology. 2004;62:1885-1887.
114. R.M. Tyson, T. Siegal, N.D. Doolittle, et al. Current status and future of relapsed primary central nervous system lymphoma (PCNSL). Leuk Lymphoma. 2003;44:627-633.
115. S.R. Plotkin, R.A. Betensky, F.H. Hochberg, et al. Treatment of relapsed central nervous system lymphoma with high-dose methotrexate. Clin Cancer Res. 2004;10:5643-5646.
116. J.L. Rubenstein, D. Combs, J. Rosenberg, et al. Rituximab therapy for CNS lymphomas: targeting the leptomeningeal compartment. Blood. 2003;101:466-468.
117. H. Schulz, H. Pels, I. Schmidt-Wolf, et al. Intraventricular treatment of relapsed central nervous system lymphoma with the anti-CD20 antibody rituximab. Haematologica. 2004;89:753-754.
118. J.L. Rubenstein, J. Fridlyand, L. Abrey, et al. Phase I study of intraventricular administration of rituximab in patients with recurrent CNS and intraocular lymphoma. J Clin Oncol. 2007;25:1350-1356.
119. E.T. Wong, R. Tishler, L. Barron, et al. Immunochemotherapy with rituximab and temozolomide for central nervous system lymphomas. Cancer. 2004;101:139-145.
120. R.H. Enting, A. Demopoulos, L.M. DeAngelis, et al. Salvage therapy for primary CNS lymphoma with a combination of rituximab and temozolomide. Neurology. 2004;63:901-903.
85. K. Fliessbach, C. Helmstaedter, H. Urbach, et al. Neuropsychological outcome after chemotherapy for primary CNS lymphoma: a prospective study. Neurology. 2005;64:1184-1188.
91. C. Schultz, C. Scott, W. Sherman, et al. Preirradiation chemotherapy with Cyclophosphamide, doxorubicin, vincristine, and Dexamethasone (CHOD) for PCNSL: Initial report of Radiation Therapy Oncology Group (RTOG) protocol 88–06. J Clin Oncol. 1996;14:556-564.
92. B.P. O’Neill, J.R. O’Fallon, J.D. Earle, et al. Primary central nervous system non-Hodgkin’s lymphoma: survival advantages with combined initial therapy? Int J Radiat Oncol Biol Phys. 1995;33:663-673.
93. B. Desablens, M. Gardembas, C. Haie-Meder, Primary CNS lymphoma. Long-term results of the GOELAMS LCP88 trial with focus on neurological complications among 152 patients. Ann Oncol. 1999;10(Suppl. 3):14.
94. E.M. Bessell, F. Graus, A. Lopez-Guillermo, et al. CHOD/BVAM regimen plus radiotherapy in patients with primary CNS non-Hodgkin’s lymphoma. Int J Radiat Oncol Biol Phys. 2001;50:457-464.
109. P. Colombat, A. Lemevel, P. Bertrand, et al. High-dose chemotherapy with autologous stem cell transplantation as first-line therapy for primary CNS lymphoma in patients younger than 60 years: a multicenter phase II study of the GOELAMS group. Bone Marrow Transplant. 2006;38:417-420.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
9 Primary CNS Lymphoma
Introduction
Primary CNS lymphomas (PCNSL) are extranodal malignant lymphomas arising within the brain, eyes, leptomeninges, or spinal cord in the absence of systemic lymphoma at the time of diagnosis. The incidence of PCNSL in western countries is 5 per 1 million person-years. Currently PCNSL are estimated to account for up to 1% of non-Hodgkin lymphomas (NHL) and 3% to 5% of all primary brain tumors. After a continual increase over the past two decades,1 epidemiologic data suggest a recent decrease in the incidence of PCNSL, particularly among young patients suffering from acquired immunodeficiency syndrome, and probably associated with the development of new active antiviral drugs. In contrast, the incidence remains high among older patients (over 60 years), most of whom are immunocompetent.2 The reason for the rising incidence of PCNSL among the immunocompetent population is obscure. PCNSL is also of interest because it is one of the only primary malignant brain tumors whose prognosis has improved considerably over the past two decades due to advances in treatment strategy. Although the prognosis remains poor for the majority of the patients, a substantial minority, representing approximately 20% to 30% of cases, can hope to be cured. Because long-term survivors are at increased risk of developing severe delayed cognitive dysfunction, future treatment should be aimed at improving the efficacy of treatment while limiting the risk of neurotoxicity. This review focuses on PCNSL in the immunocompetent population.
Pathology and Pathogenesis
In immunocompetent patients, all but 5% of PCNSLs are diffuse large B-cell lymphomas (DLBCL).3 Since they are morphologically indistinguishable from systemic DLBCL, the WHO classification of tumors of hematopoietic and lymphoid tissues does not recognize PCNSL as a separate entity.4 The remaining cases of PCNSL are T-cell lymphomas (2% to 5%)5 or, in rare instances, low-grade B-cell lymphomas of the lymphoplasmocytic (Waldenström macroglobulinemia), fol-licular, or mucosa-associated lymphoid tissue (MALT) type.6 Little is known about the tumorigenesis of PCNSL. In contrast to immunocompromised patients, the Epstein-Barr virus (EBV) does not appear to be involved in the pathogenesis of PCNSL in immunocompetent patients. The site of origin of lymphoma cells, the biological mechanisms involved in the neoplastic transformation of the lymphoid cells, and their intriguing confinement within the CNS during the course of the disease have yet to be elucidated. Indeed, the CNS does not contain resident lymphocytes under normal circumstances and lacks lymphatic vessels. However, recent evidence suggests that T and B cells enter the CNS under physiological conditions, and it has been hypothesized that PCNSL may originate from B cells derived from systemic lymphoid tissues and normally trafficking in and out of the CNS.7 PCNSL could derive from a benign CNS inflammatory process through a monoclonal proliferation of B cells. Another hypothesis is that PCNSL might represent the metastasis of an occult systemic lymphoma, eradicated by an intact immune system but escaping within the immune-privileged CNS. Analysis of clonally rearranged immunoglobulin heavy chain (IgH) genes revealed identical dominant PCR products in bone marrow aspirates, blood samples and tumor specimens from some PCNSL patients, suggesting that subclinical systemic disease can be detected at initial diagnosis, which supports a systemic origin of the tumor in these cases.8 In addition, B cell tropism for CNS might be acquired (before or after the oncogenic events) through specific interactions between selective homing receptors and their ligands expressed on CNS endothelial cells, as suggested by the distinctive angiotropism of CNS lymphoma cells.9,10
It also remains unclear whether the dismal outcome of PCNSL compared to systemic DLBCL is attributable to the immune-privileged cerebral location or reflects a specific aggressive intrinsic biologic behavior. Recently, expression profiling and genomic screening have provided new insights into understanding the poor prognosis of PCNSL. Based on lymphochip cDNA microarrays, two distinct gene expression profiles have been identified among systemic DLBCLs, indicative of different stages of B-cell differentiation. One subgroup expressed genes characteristic of germinal center B cells (GCB subgroup) whereas the other expressed genes normally induced during in vitro activation of peripheral blood B cells (ABC subgroup). Interestingly, patients with the GCB signature had a significantly better outcome than those with the ABC profile.11 PCNSLs have been shown to frequently expresses BCL612 and to carry an extremely high load of somatic mutations of immunoglobulin genes and in several oncogenes demonstrating aberrant ongoing hypermutation.13–15 Since such an ongoing hypermutation and BCL6 expression are considered as germinal center markers, it has been postulated that the cell of origin of PCNSL passed through the GC microenvironment and that these neoplasms correspond to the GCB subgroup as defined for DLBCL. However, recent immunoprofiling16 and gene expression studies using cDNA microarrays17,18 demonstrated that PCNSL may exhibit characteristics associated with both ABC and GCB subtypes. Thus, PCNSL may correspond to an overlapping B-cell differentiation time slot, i.e., late GC/early post-GC stage.
Pangenomic analyses of chromosomal imbalances by comparative genomic hybridization (CGH) have shown frequent chromosome 6 q loss (60% to 75%) in PCNSL.19–21 Nakamura et al.22 refined the candidate region suspected to contain a lymphoma-related tumor suppressor gene in the 6q22–23 locus by a fine LOH deletion mapping of 6 q in PCNSL. The PTPRK gene seems a relevant candidate gene, since it is involved in the regulation of cell contact and adhesion, and a loss of protein expression was observed in most (76%) of the PCNSLs tested. It belongs to the protein tyrosine phosphatase superfamily of enzymes. Further studies to identify gene mutations and/or rearrangements are needed to ascertain the involvement of PTPRK in PCNSL tumorigenesis. Interestingly, the authors showed that chromosome 6 q loss was found at a significantly higher rate in PCNSL than in systemic DLBCL and was correlated with shorter survival.22 An independent study of 75 newly diagnosed HIV-negative PCNSLs investigated by interphase FISH analysis has confirmed the frequent 6q22 chromosome deletion, with a prevalence of 45%, and its negative impact on overall survival.23 Chromosome 6 q loss would therefore represent a prognostic marker in PCNSL. Otherwise, the BCL6 gene has been found mutated and its chromosomal locus (3q27) recurrently involved by translocations (20%), suggesting that BCL6 activation through genomic rearrangements may play a role in PCNSL pathogenesis.23–25 BCL6 is frequently expressed in PCNSL, but there are conflicting data on its prognostic value as an immunohistochemical marker.16,26–29 These results provide evidence for an alternative pathogenesis in PCNSL compared to DLBCL explaining, in part, differences in clinical behavior and prognosis.
Diagnosis and Workup
The clinical presentation of PCNSL includes focal symptoms and raised intracranial pressure. Changes in behavior and personality as well as confusion frequently occur in the elderly. The deep location of PCNSL explains why seizures are less frequent than in other brain tumors.30,31 CT scans and MRI typically show solitary (two thirds of cases) or multiple (one third of cases) periventricular, homogeneously-enhancing lesions (Figures 9-1 and 9-2).32–34 PCNSL is potentially associated with a large spectrum of radiological presentations and can simulate inflammatory (sarcoidosis, multiple sclerosis) or postinfectious diseases (ADEM) or other brain tumors (meningiomas, malignant gliomas, gliomatosis cerebri, brain metastases).35–37 The diagnosis may be difficult to establish, especially with the presence of nonenhancing infiltrating lesions (occurring in about 10% of cases) (Figure 9-3),38,39 or dural-based masses (Figure 9-4)40,41; perfusion MRI shows a much lower relative cerebral blood volume than that seen in malignant gliomas, and distinct signal-time intensity curves reflecting rapid leakage of contrast medium into the interstitial space.42 Similarly magnetic resonance spectroscopy at short echo times show massive elevations of lipid resonances in PCNSL, not usually seen in high-grade gliomas.43,44 Steroid-induced response, observed in approximately 40% of cases, is classic but not the rule and should not be used as a diagnostic test since it may prevent a pathological confirmation of the diagnosis. Rarely, spontaneous disappearance of lesions has been reported, hence the term “ghost tumors.”45,46 Ring enhancement, frequently seen in immunodeficient patients, is a pattern rarely seen in immunocompetent patients. However, none of these signs are specific, and diagnosis relies on the pathological study of a tumor sample. Cerebral biopsy can be avoided when lymphomatous cells are discovered in the cerebrospinal fluid (10% to 30% of patients) or in a vitreous-body biopsy (uveitis, sometimes asymptomatic, is present in 10% to 20% of cases at diagnosis). The current recommended staging evaluation for PCNSL includes body CT scans and bone marrow biopsy.47 In fact, systemic involvement is so rare at onset that extensive staging is not recommended by some authors, the only required examinations being HIV testing, chest radiography, analysis of cerebrospinal fluid, ocular slit-lamp examination, and a careful clinical assessment. However, in a recent retrospective study, 7% of patients were found to have systemic NHL by staging FDG body PET when body CT scans and bone marrow biopsies were negative.48 This higher incidence of systemic lymphoma than reported in prior series suggests that occult lymphoma may be more common than recognized previously. These findings warrant prospective validation, since the identification of a systemic site of the lymphoma has important implications in the management of PCNSL.
Treatment Options
TREATMENT OF NEWLY DIAGNOSED PCNSL
Age and performance status are the main independent prognostic factors consistently identified in a large number of studies.49–51 As PCNSL is a highly radiosensitive and chemosensitive infiltrative tumor, surgery is therefore restricted to diagnostic biopsy. Although the tumor appears on MRI as a unique contrast-enhancing lesion in the majority of the immunocompetent patients, whole-brain radiotherapy (WBRT) is recommended based on the microscopically diffuse nature of PCNSL. Despite a high rate of response, RT alone has shown a limited survival benefit in PCNSL with a median overall survival (OS) duration of 10 to 18 months and a 5-year survival rate of less than 5%.52 The only phase II trial of radiotherapy (conducted by the RTOG) delivered a total dose of 40 Gy with an additional 20-Gy boost to contrast-enhancing lesions and reported an overall survival of 11.6 months.53 Interestingly, most of the relapses occurred in sites that had received the maximum RT dose. These disappointing results have led to the use of chemotherapy in combination with WBRT. Since the 1990s, numerous convergent phase II studies have shown that the addition of high-dose methotrexate (MTX)-based chemotherapy to RT substantially improves survival compared with RT alone (median survival: 2 to 4 years; 5-year survival rate: 20% to 40%).54–69 In contrast, adding the CHOP regimen (standard chemotherapy for systemic lymphoma) to RT did not appear to improve survival compared with RT alone. This may reflect the poor central nervous system penetration of the chemotherapy agents included in these regimens. The optimal dose of irradiation postchemotherapy has never been prospectively investigated. Doses between 20 and 50 Gy to the whole brain (WB) with or without a tumor bed boost are currently used, with most of the protocols using a total dose of 40-45 Gys without a boost. For patients who achieve a complete response after high-dose MTX-based chemotherapy, it still remains unclear whether consolidation with WBRT improves disease control or survival. A subset analysis from a phase II trial that included 25 patients aged less than 60 years who achieved CR after initial chemotherapy and received either 45 Gy or 30.6 Gy as consolidation treatment showed a significantly higher recurrence and lower overall survival rate in the reduced dose RT group.61 Other studies did not share this observation. In two multicenter retrospective analyses, no differences were noted regarding disease-free or overall survival between patients in CR receiving WBRT as consolidation treatment or at the time of relapse.70,71 In the Memorial Sloan Kettering Cancer Center (MSKCC) experience, consolidation treatment with WBRT improved failure-free survival but not overall survival in complete-responding patients after MTX-based therapy, but increased the rates of neurotoxicity.72
In order to reduce neurotoxicity, several groups have explored the efficacy of various high-dose MTX-based chemotherapy regimens as initial treatment for newly diagnosed PCNSL. These studies converge to suggest that chemotherapy alone and deferred radiotherapy strategy allows comparable results in terms of survival to those reported for combined chemoradiotherapy, with better neurocognitive and quality of life outcomes.73–79 This supports the comparison of these approaches in a prospective randomized trial. While high-dose MTX is undoubtedly a key drug for PCNSL chemotherapy, interest in adding other agents to MTX exists in the scientific community. Two prospective phase II trials have investigated the efficacy of high-dose MTX (8 g/m2) as a single drug therapy,77,78 and similarly demonstrated a clearly shorter PFS (13 months) than that achieved with a polychemotherapy regimen. This suggests that other cytotoxic agents should be used with high-dose MTX in a chemotherapy-alone approach. However the optimal combination remains to be established. Alkylating agents able to effectively penetrate the CNS would be preferred. Concerning the optimal “high dose” of MTX to deliver, although there is no clear evidence of a dose response, a dose of greater than 3 g/m2 in a rapid infusion is recommended.80 In addition, this dose generally yields cytotoxic levels in the cerebral spinal fluid, thus theoretically avoiding the need to administer intrathecal chemotherapy for leptomeningeal coverage. In practice, when intravenous high dose MTX (>3 g/m2) is used, several authors recommend adding intrathecal CT only in cases of positive CSF cytology and withholding it in the absence of detectable subarachnoid disease.81
DELAYED NEUROTOXICITY
WBRT, high-dose MTX chemotherapy, and the combination of both treatments expose the patients to delayed neurotoxicity. This complication can occur as early as 3 months after the treatment and is characterized by attention deficit, memory impairment, ataxia, and urinary incontinence, potentially ultimately leading to dementia. Imaging shows confluent diffuse white matter changes and later cortical-subcortical atrophy. The physiopathology of this complication remains poorly understood; loss of oligodendrocyte progenitors and oxidative stress have been suggested as potential mechanisms. Lai et al.82 reported a well-documented series of five autopsied cases who died of treatment-related leukoencephalopathy. All had combined treatment and were in tumor remission. In addition to white matter rarefaction and spongiosis, fibrotic thickening of small vessels in the deep white matter and atherosclerosis of intracranial large vessels were systematically found, suggesting that a vascular process may be also an important component of this white matter injury. The risk of neurotoxicity increases sharply with patient age. In the elderly population (patients over age 60), virtually all long-term survivors will develop delayed neurotoxicity with its devastating consequences on quality of life and mortality.83 In the younger population (patients under age 60), the exact incidence of this complication is more difficult to determine. Cognitive dysfunctions are usually less severe and occur later than in the elderly population, although they may interfere with quality of life.64 An update of the MSKCC experience providing long-term data (median follow-up of 115 months) reported a 26% rate of neurotoxicity in surviving patients younger than 60 years (versus 75% in the elderly).68 However, this should be regarded as a minimum estimate in the absence of a psychometric evaluation. A series of 19 consecutive young patients (median age: 44 years) treated in a European clinical trial and in complete remission for a mean time of 24 months after combined therapy were investigated by an extensive neuropsychological evaluation.84 Cognitive impairments were found in 63% of patients (including 21% with severe cognitive deficits); only 42% of the patients had resumed work, and 67% had white matter abnormalities and cortical atrophy detectable by MRI. Although this study suffers from the absence of available baseline data at the completion of the treatment to assess the potential contribution of the tumor to cognitive dysfunction, it suggests that the incidence of delayed neurotoxicity after combined therapy is largely underestimated in young patients, albeit less severe than in the elderly. These results contrast sharply with a prospective neuropsychological study performed by Fliessbach et al.84 in a series of 23 patients successfully treated with a high-dose MTX-based chemotherapy without radiotherapy. Comparison between baseline evaluation at completion of the treatment and at the last follow-up (median: 44 months) showed a good preservation of cognitive functions, although one third of the patients demonstrated some degree of white matter changes at the MRI. This latter point confirmed some previously published reports showing that MTX-related leukoencephalopathy is a frequent but not necessarily universal accompaniment to deterioration of cognitive performance.86
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
