33
Prenatal diagnosis
The spectrum of congenital abnormality
Screening for fetal abnormalities
Introduction
The identification of some ‘congenital abnormalities’ in pregnancy transforms what was previously an exciting and joyous event into a worrying and distressing time. Accuracy of information giving, tact, understanding and reassurance (if appropriate) are paramount. The very greatest of care should be taken in explaining any aberrant findings to parents. The advice given to parents is of such importance that it will frequently be necessary to involve senior members of the obstetric team (usually with subspecialty training), as well as members of other specialties, particularly paediatricians, and clinical geneticists and radiologists.
The aims of prenatal diagnosis are four-fold:
 the identification at an early gestation of congenital abnormalities incompatible with survival, or likely to result in significant handicap, in order to prepare parents, alert other specialist clinicians and offer the option of termination of pregnancy (TOP) (if appropriate)
the identification at an early gestation of congenital abnormalities incompatible with survival, or likely to result in significant handicap, in order to prepare parents, alert other specialist clinicians and offer the option of termination of pregnancy (TOP) (if appropriate)
 the identification of conditions which may influence the timing, site or mode of delivery
the identification of conditions which may influence the timing, site or mode of delivery
 the identification of fetuses who would benefit from early neonatal paediatric intervention
the identification of fetuses who would benefit from early neonatal paediatric intervention
 the identification of fetuses who may benefit from in-utero treatment (relatively rare).
the identification of fetuses who may benefit from in-utero treatment (relatively rare).
Discussion and non-directional counselling are mandtory. It should not be assumed that all parents are going to request TOP, even in the presence of lethal abnormality. Many couples have opted to continue pregnancies in the face of severe defects which have resulted in either intrauterine or early neonatal death, and have expressed the view that they found it easier to cope with their grief having held their child. It is essential that palliative care is coordinated by a multidisciplinary team and organized by specialist teams. Other parents, after careful and informed consideration, make the difficult decision to opt for TOP. More controversial still are the problems of chronic disease processes associated with a high risk of long-term handicap and potential suffering for both the child and the parents. The parents themselves must decide what action they wish to take – it is they who will have to live with the decisions they make. It is our role to advise, guide and respect their final wishes, irrespective of our own personal views.
Non-directive counselling
When parents are found to have an ‘abnormal baby’ they often know little or nothing about the congenital anomaly, its impact on pregnancy outcome, the long-term prognosis and the options for care. Parents know even less about termination or about recurrence risks of the condition. Parents need accurate information, and it is the role of obstetricians and genetic counsellors to inform accurately and in language that is clear to understand. Often parents ask the doctor what he or she would do, but it remains important to encourage the couple towards their own decision. It is often appropriate to involve other professional groups (i.e. paediatricians) in the discussions either jointly or at separate consultations.
Non-directive counselling has to be truly non-directive. The sentence ‘the risk of handicap is 5%’ sounds worse than ‘the baby has a 95% chance of being normal’. Expressions like ‘high risk’ or ‘severe handicap’ imply a value judgement and should be avoided if possible. ‘Common’ may be interpreted as anything from 1% to 99%, depending on the context and the listener.
It is preferable to give information in writing, to reinforce discussions. Parents need time to take in information, and it is often important that they take time to reflect on information and to consider any decisions they may make carefully. It can be extremely useful to arrange a follow-up appointment a day or two after the initial appointment.
The spectrum of congenital abnormality
Although most congenital abnormalities are individually rare, together they cause an enormous burden of suffering. Approximately 5% of fetuses have congenital malformations and 2% of newborn babies have a serious abnormality detectable at, or soon after, birth. The main ones are listed in Box 33.1.
Box 33.1
Selected congenital abnormalities
 Down syndrome (trisomy 21)
Down syndrome (trisomy 21)
 Edwards syndrome (trisomy 18)
Edwards syndrome (trisomy 18)
 Patau syndrome (trisomy 13)
Patau syndrome (trisomy 13)
 Triploidy
Triploidy
 Sex chromosome abnormalities
Sex chromosome abnormalities
 XO (Turner syndrome)
XO (Turner syndrome)
 XXY (Klinefelter syndrome)
XXY (Klinefelter syndrome)
 XYY
XYY
 XXX
XXX
 Apparently balanced rearrangements (translocations or inversions)
Apparently balanced rearrangements (translocations or inversions)
 Unbalanced chromosomal structural abnormalities
Unbalanced chromosomal structural abnormalities
 Gene disorders (e.g. fragile X syndrome, Huntington’s chorea, Tay–Sachs disease)
Gene disorders (e.g. fragile X syndrome, Huntington’s chorea, Tay–Sachs disease)
 Congenital heart disease
Congenital heart disease
 Neural tube defects (e.g. anencephaly, encephalocele, spina bifida)
Neural tube defects (e.g. anencephaly, encephalocele, spina bifida)
 Abdominal wall defects (e.g. exomphalos, gastroschisis)
Abdominal wall defects (e.g. exomphalos, gastroschisis)
 Genitourinary abnormalities (e.g. renal dysplasia, polycystic kidney disease, pyelectasis, posterior urethral valves, Potter syndrome)
Genitourinary abnormalities (e.g. renal dysplasia, polycystic kidney disease, pyelectasis, posterior urethral valves, Potter syndrome)
 Lung disorders (e.g. pulmonary hypoplasia, diaphragmatic herniae, cystic fibrosis)
Lung disorders (e.g. pulmonary hypoplasia, diaphragmatic herniae, cystic fibrosis)
 Toxoplasmosis
Toxoplasmosis
 Rubella
Rubella
 Cytomegalovirus
Cytomegalovirus
 Herpes simplex virus
Herpes simplex virus
 Chickenpox
Chickenpox
 Erythrovirus
Erythrovirus
 Hepatitis
Hepatitis
 Listeria monocytogenes
Listeria monocytogenes
 Syphilis
Syphilis
 Beta-haemolytic streptococci – group B
Beta-haemolytic streptococci – group B
Assessing the risk
In general, the risk for congenital abnormalities is very small, for most couples. The risk of autosomal trisomies, particularly trisomy 21 (Down syndrome) increases with increasing maternal age. In some instances, however, there may be a family history of an inherited condition, for example, Duchenne muscular dystrophy, cystic fibrosis, sickle cell disease or myotonic dystrophy. Consanguinity increases the risk of single gene anomalies, particularly in relation to autosomal recessive conditions. Structural abnormalities are also usually slightly more likely to occur in those with a family history and the risk is also higher for certain anomalies if the parents have had a previous child with an anomaly; for example, a woman who has had a child with spina bifida has an approximately 2% risk of a recurrence compared with the background risk of about 0.2%.
It is important to consider medical conditions when assessing prenatal risk of congenital anomalies. Women with pre-existing diabetes have an increased risk of cardiac and neural tube defects, and those with seizures are also at increased risk of fetal structural anomalies, particularly if taking potentially teratogenic anticonvulsants (e.g. congenital heart disease). The majority of structural and chromosomal problems, however, occur de novo, in those who have no predisposing history or recognized risk factors, and screening tests are offered to those at apparently low risk.
Screening for fetal abnormalities
The decision to screen for fetal abnormality rests with each individual couple. It is appropriate to offer testing to all couples: some wish no screening tests at all; others may be keen to consider all of the options (Table 33.1).
Table 33.1
Overview of potential screening programmes for chromosomal and structural abnormalities
| Programme | Advantages | Disadvantages |
| NT and serum at 11–14 weeks | Good detection of Down syndrome at early gestation (11–14 weeks) | Use of CVS may increase miscarriage rate Minimal detection of structural abnormalities |
| NT and serum at 11–14 weeks and FDS at 18 weeks | Good detection of Down syndrome at early gestation (11–14 weeks) Good detection of structural abnormalities |
Use of CVS may increase miscarriage rate |
| FDS at 18 weeks | Good detection of structural abnormalities | Minimal detection of Down syndrome |
| Serum screening alone at 16 weeks | Good detection of Down syndrome Amniocentesis may be safer than CVS Reasonable detection of open lesions, particularly neural tube defects |
Minimal detection of other structural abnormalities |
| Serum screening at 16 weeks with FDS at 18 weeks | Good detection of Down syndrome Amniocentesis may be safer than CVS Good detection of structural abnormalities |
Down syndrome not identified until relatively late (17–18 weeks) |
CVS, chorionic villus sampling; FDS, fetal detailed scan; NT, nuchal translucency.
Ultrasound scanning
Structural anomalies may be visualized on ultrasound scan and it is recommended that all women should be offered a detailed ultrasound at around 18–21 weeks’ gestation. This has the advantage that those with major or lethal anomalies (e.g. anencephaly, lethal skeletal dysplasias or renal agenesis) can be offered termination, and it also allows planned deliveries of those conditions which may require early neonatal intervention (e.g. gastroschisis or transposition of the great arteries).
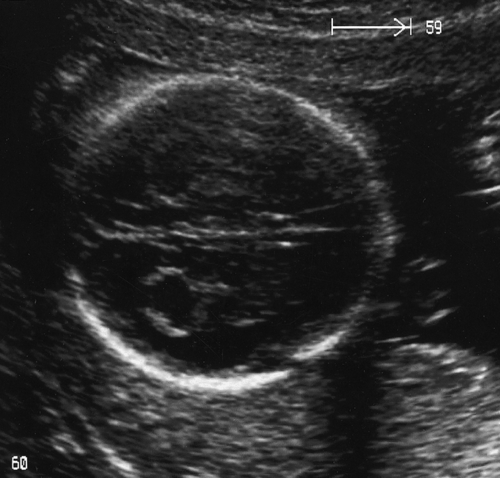
Ultrasound scanning has the limitation, however, that some anomalies may not be identified. It is likely, for example, that less than 50% of cardiac defects are recognized and the false reassurance provided by a scan may become a source of parental resentment. Furthermore, a ‘soft marker’ may be uncovered, the significance of which is often unclear. These soft markers are ultrasound appearances, which in themselves are not an abnormality, but which may point to other problems, particularly chromosomal abnormalities. They are found in approximately 5% of all pregnancies at a second-trimester scan and cause a lot of parental anxiety. Such markers include choroid plexus cysts (Fig. 33.1), mild renal pelvic dilatation, echogenic cardiac foci (Fig. 33.2) and mild cerebral ventricular dilatation. If the soft marker is isolated, the risk of chromosomal problems is low, but if more than one is found, or if there are any other structural defects, the risk of a chromosomal problem is very much higher. Ultrasound markers of chromosome anomaly are not sensitive or specific enough to be used for screening of aneuploidy.
Unlike structural abnormalities, chromosomal abnormalities can be much more difficult to identify on ultrasound scan. Around two-thirds of fetuses with Down syndrome (trisomy 21) will have a normal appearance at 18 weeks, and the remaining third may demonstrate only minor defects which are not pathognomonic of the condition. Most fetuses with the less common trisomies, e.g. Edwards syndrome (trisomy 18) or Patau syndrome (trisomy 13), do show some abnormality, although the abnormality is again often neither specific nor diagnostic. Since trisomy 18 and 13 are usually lethal in the perinatal period, there are fewer lifelong implications than for trisomy 21. Much of the screening work has therefore been directed at trisomy 21, in particular measuring specific markers in the maternal blood (serological screening), or measuring the thickness of nuchal fluid behind the fetal neck (nuchal translucency assessment).
Nuchal translucency: in the first trimester combined screening test
Screening for fetal chromosomal anomalies (principally trisomy 21) is also possible by measuring the fetal nuchal translucency (NT) on first-trimester ultrasound scanning (Fig. 33.3). The risk of Down syndrome increases with larger NT measurements. NT measurement is combined with first trimester biochemistry (measuring free β-HCG and PAPP-A) in order to provide an estimate of risk of Down syndrome. In the first trimester, it has been recommended that a ‘cut-off’ of high risk is taken as 1 in 150 at term. Using such a threshold, the first trimester combined screening test can detect up to a 90% of babies with trisomy 21, with a 2.5% risk of being offered an invasive, prenatal procedure. The sensitivity of the test may further be improved by the visualization of a fetal ‘nasal bone’ and exclusion of right sided cardiac ‘tricuspid regurgitation’. These latter tests are not routine in their use.
Parents then may be offered a prenatal diagnostic test. Chorionic villous sampling (CVS) may be used to establish a diagnosis at an earlier gestational age than with amniocentesis (see below), thereby allowing the option of an early surgical termination of pregnancy rather than a late medical termination. There is, however, evidence to suggest that parental psychological morbidity is independent of whether a diagnosis is made in the first or second trimester, and indeed medical termination may carry less psychological morbidity than surgical (even if medical complications are higher).
Increased nuchal translucency is also a marker for structural defects, particularly cardiac anomalies (when used in combination with tricuspid blood flow measurements to exclude regurgitation), renal, abdominal wall disease, as well as diaphragmatic herniae.
This may also be used in multiple pregnancies. In dichorionic twins, each twin has its ‘individualized risk’ and in monochorionic twins, the risk is the ‘average’ of that calculated for the two fetuses.
Serological screening
This is used almost exclusively to detect two abnormalities: spina bifida and chromosomal anomalies. Alpha-fetoprotein (αFP) is an alpha-globulin of similar molecular weight to albumin which is synthesized by the fetal liver. If there is a break in the fetal skin (for example with spina bifida), αFP escapes into the maternal circulation and the maternal serum level becomes elevated.
Normal serum αFP levels rise with advancing gestation and most laboratories report results as multiples of the median (MoM) for unaffected pregnancies at the gestation of sampling. A level of 1.0 is normal, and for screening purposes levels raised to more than 2.0–2.5 MoM indicate the need for detailed scanning to look for neural tube defects, multiple pregnancy, gastroschisis or intrauterine death. As there is a large overlap between normal and affected pregnancies, a raised level of maternal serum αFP is therefore only a screening test and not a diagnostic test. An ultrasound scan provides the final diagnosis.
It has also been observed that the level of maternal serum αFP is lower than expected when the fetus has Down syndrome. The reason for this remains unclear but the result can be combined with maternal age to give an estimated risk of the fetus being affected. The screening test with the greatest sensitivity and specificity for abnormal fetal chromosomal anomalies is the first trimester combined screening test. The Quadruple Test is an optional blood test offered to pregnant women who have booked too late for a first trimester combined screening test, or where a nuchal scan was attempted but the measurement was not obtained. This mostly happens due to the baby’s position when scanned. The Quadruple Test is offered and performed between 14 + 2–20 + 0 weeks of pregnancy to help identify pregnancies that have a higher chance of being affected with trisomy 21. Such serological testing may detect chromosomal anomalies (such as trisomy 21) in up to 85% of cases (with a false positive rate of between 3–5%). For this reason, within the NHS, first trimester combined screening is the ‘chromosomal screening test’ of choice.
Diagnosis of chromosomal abnormalities
Diagnostic tests are offered if screening tests suggest that the mother is above a certain risk level of carrying a baby with a chromosomal abnormality. A risk threshold of 1 in 250 at term or higher is commonly used for second trimester screening tests, and 1 in 150 at term in the first trimester.
Amniocentesis
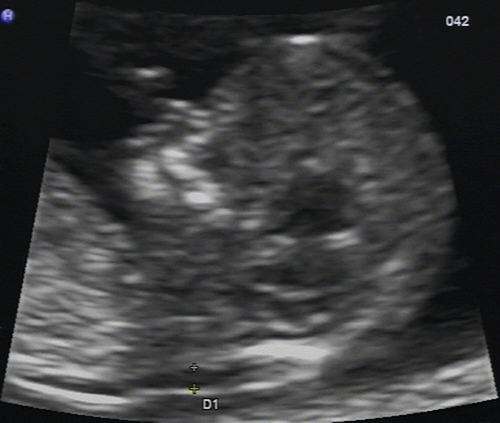
Diagnostic amniocentesis may be performed after 15 weeks’ gestation. A 22-gauge needle is inserted into the amniotic cavity under continuous ultrasound guidance and 10–15 mL of amniotic fluid are drawn off (Fig. 33.4). Rhesus-negative women are given 250 IU of anti-D immunoglobulin to prevent immunization. The risk of miscarriage is around 1%.
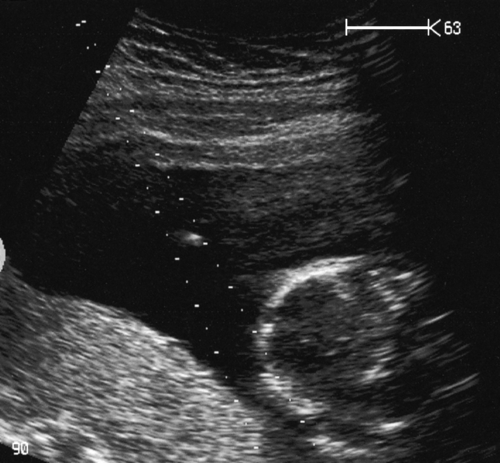
Fig. 33.4Amniocentesis is carried out under direct ultrasound guidance.
The tip of the needle can be seen between the dotted guidelines 2 cm above the fetal head.
Karyotype results are usually available within 3 weeks, but rapid FISH (fluorescence in-situ hybridization) or quantitative PCR (polymerase chain reaction) techniques may be used to exclude the commoner aneuploidies; the result is available within 3–4 working days.
Chorionic villus sampling
CVS or placental biopsy may be performed after 10 weeks’ gestation. Either a flexible cannula is passed through the cervix, or (more commonly) a needle (usually 18 to 20 gauge) is passed transabdominally – both under continuous, ultrasound guidance (Fig. 33.5). Results are usually available within 72 h, and the full karyotype within 12–15 days. CVS may carry the same procedure-related miscarriage rate as an amniocentesis, but due to a higher ‘background loss rate’, the procedure carries a higher overall risk of miscarriage (1.5–2%).
Here, the needle follows the on-screen needle guide dots.
Structural and chromosomal abnormalities
Down syndrome (trisomy 21)
The overall incidence is 1:650 live births, but the incidence increases with increasing maternal age:
 20 years 1:2000
20 years 1:2000
 30 years 1:900
30 years 1:900
 35 years 1:350
35 years 1:350
 36 years 1:240
36 years 1:240
 38 years 1:180
38 years 1:180
 40 years 1:100
40 years 1:100
 44 years 1:40.
44 years 1:40.
Most children born with trisomy 21, however, are born to younger mothers, as there are overall many more younger mothers than older ones. Although walking, language and self-care skills are usually attained, independent life is variable. Children with trisomy 21 often have a degree of neurodevelopmental delay (with a mean IQ around 50) and an association with congenital heart disease (in 20% of cases). Gastrointestinal atresia is more common, as a prenatal diagnosis and there is an association with early-onset dementia with similarities to Alzheimer disease. Overall, 20% die before the age of 1 year and 45% by the age of 60 years.
Of the cases, 95% are due to chromosomal non-dysjunction, with translocation 14:21 accounting for 2%; other translocations accounting for 2% and mosaicism 1%. Half of the translocations occur de novo. The recurrence risk for non-dysjunction is about 1:100 if the mother is < 35 years old and approximately four times the baseline risk if she is > 35 years old. If there is a 14:21 translocation in the mother, the recurrence risk is 1:10, and if in the father, 1:50.
Edwards syndrome (trisomy 18)
The incidence is around 1:2500 live births and most are due to non-dysjunction involving chromosome 18. The baby often is associated with early-onset fetal growth restriction (FGR), a small elongated head (strawberry-shaped on ultrasound), rocker bottom feet and an increased incidence of gastrointestinal and renal anomalies. Virtually all have congenital heart disease, often complex anomalies. Many babies are stillborn but of survivors, over 50% die before the age of 2 months and 90% before the end of the first year.
Patau syndrome (trisomy 13)
This is rare, at around 1:5000 live births. The fetus often has severe growth restriction, and an increased incidence of cleft palate, gastrointestinal atresias and holoprosencephaly. Again, most have complex congenital heart disease. The majority of babies are stillborn but with survivors, the children die before the age of 3 months and survival is rare after the age of 1 year.
Triploidy
These children rarely survive to birth and there is no survival beyond the neonatal period. Prenatally, the fetus is often severely growth restricted with numerous associated anomalies. Placental enlargement is a common association. In up to 5% of cases, there is trophoblastic disease and therefore, the pregnancy requires registration and follow-up (see Chapter 21).
Turner syndrome or monosomy X (45,X)
This occurs in around 1:3000 live births: 60% are pure monosomy X; just over 15% are mosaics (usually 45,X/46,XX); and the minority are complex deletions, rings or isochromosomes of Xq or Xp. The incidence is increased with increasing paternal age and decreased with increasing maternal age. Antenatally, they may be associated with a cystic hygroma ± generalized oedema and cardiac defects (Fig. 33.6). In over 50% of 45,X the association with hydrops fetalis leads to a fatal prenatal outcome. Live born children may have short stature, cubitus valgus, coarctation of the aorta, a bicuspid aortic valve, streak gonads and only occasionally, a lowered IQ. A small proportion may have fertility (particularly mosaics and deletions), although the incidence of premature ovarian failure is high.
XXX
The incidence of 1:1000 live births is doubled or tripled when the maternal age is more than 40 years. The phenotype and fertility are normal and the abnormality frequently goes unnoticed. There is, however, an increased risk of sex chromosome abnormalities (~ 4%) and premature menopause in the offspring. XXX + (i.e. more than three X chromosomes) is rare. Dysmorphism and mental handicap in this group are common, as is menstrual dysfunction. The individual may be fertile.
Klinefelter syndrome (XXY)
This is uncommon, at around 1:700–2000 live births. The individual is phenotypically a tall male, with occasionally a reduced IQ, sparse facial hair and gynaecomastia. It is the commonest single cause of male hypogonadism and is usually diagnosed in the investigation of male infertility. There is an association with hypothyroidism, diabetes and asthma. Azoospermia is usual.
XYY
Again, this is uncommon, at around 1:700 live births, and there is no association with maternal age. The IQ and fertility are usually normal and the suggestion of increased impulsive behaviour may be biased by the population sampled. Individuals are usually tall. The risk of sex chromosome abnormalities in offspring is approximately 4%.
Apparently balanced rearrangements (translocations or inversions)
If ‘apparently balanced rearrangements’ are found at prenatal diagnostic testing (CVS or amniocentesis), it is essential to check the karyotype of both parents. If one parent has the translocation and is phenotypically normal, it is likely that the fetus will be phenotypically normal as well. There is a chance that other offspring (or offspring of the fetus) will have an unbalanced translocation, and counselling ± karyotyping should be offered. In general, the smaller the section of chromosome involved, the greater the likelihood of a fetus surviving to term with an unbalanced translocation. Offspring may, of course, also have normal karyotypes without the translocation. If the translocation has occurred de novo, the overall risk of phenotypic abnormality is in the order of 10%, but as some chromosomal rearrangements are normal population variants, genetic advice should always be sought. Such chromosomal rearrangements are more commonly diagnosed by using prenatal microarray testing.
Unbalanced chromosomal structural abnormalities
Many chromosomal structural abnormalities are well characterized, but it is often difficult to be specific. Parental karyotyping is required and genetic advice should be sought. Mental impairment is common, and physical abnormality is possible. Again, specific clinical genetics input is mandatory.
Cystic hygroma
Cystic hygromas (see Fig. 33.6) are fluid-filled swellings at the back of the fetal neck, which probably develop because of a defect in the formation of lymphatic vessels. There is some ‘debate’ as to whether such an anomaly is a spectrum of disease with ‘enlarged nuchal translucency’. It is likely that the lymphatic and venous systems fail to connect and lymph fluid accumulates in the jugular lymph sacs. Cystic hygromas are more frequently divided by septae than nuchal translucency, and may be associated with skin oedema, ascites, pleural and pericardial effusions, and cardiac and renal abnormalities. There is also an association with common aneuploidies and it is appropriate to offer prenatal karyotyping. If generalized fetal hydrops is present, the prognosis is bleak. ‘Isolated cystic hygromas’, commonly associated with congenital anomalies of the nuchal lymphatic system may be surgically corrected postnatally and have a good prognosis. Only rarely are they so large as to result in problems with labour.
Congenital heart disease
This is the commonest congenital malformation in children and affects about 5–8:1000 live births. Of defects diagnosed antenatally, about 15% are associated with aneuploidy, most commonly trisomies 18 and 21.
The introduction of first trimester combined screening, with the formal examination of the four chamber view and Doppler insonation of right-sided cardiac A–V valve flow, has led to an increase in suspected diagnosis in the late first trimester.
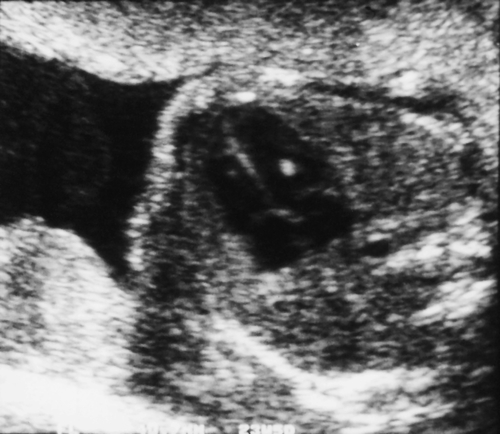
In the second trimester, the four-chamber view of the heart can also be used as a screening test (Fig. 33.2) and will identify 25–40% of all major abnormalities, particularly ventriculoseptal defect, ventricular hypoplasia (Fig. 33.7), valvular incompetence and arrhythmias. Moving above the four-chamber view allows the aorta and pulmonary artery outflow tracts to be visualized (Fig. 33.8). These are the recommended fetal cardiac views in the second trimester. In addition, many centres also use the visualization of the aortic arch as a mandatory cardiac view. This increases the sensitivity to identify approximately 75% of cardiac defects by screening. At 18 weeks, most of these major connections can be seen, but high-risk pregnancies (e.g. those with diabetes, or women taking anticonvulsants, or who have a personal or family history of congenital heart disease) should be re-scanned at 20–22 weeks for more minor defects.
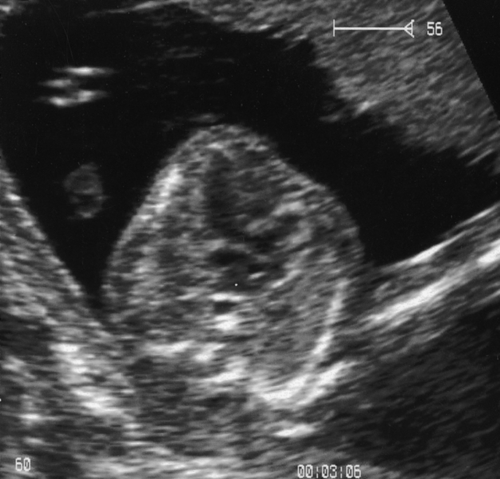
Fig. 33.7Hypoplastic right heart.
The normal four chamber view is not obtained. The baby died in the early neonatal period.
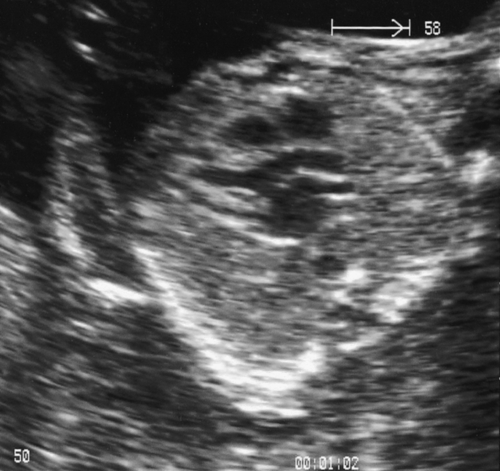
The aorta in this normal heart is seen to be arising exclusively from the left ventricle, excluding the diagnosis of tetralogy of Fallot.
Neural tube defects
The neural tube is formed from the closing of the neural folds, with both anterior and posterior neuropores closed by 6 weeks’ gestation (Fig. 33.9). Failure of closure of the anterior neuropore results in anencephaly or an encephalocele, and failure of posterior closure results in spina bifida. The European incidence of neural tube defects is around 1:1000 pregnancies.
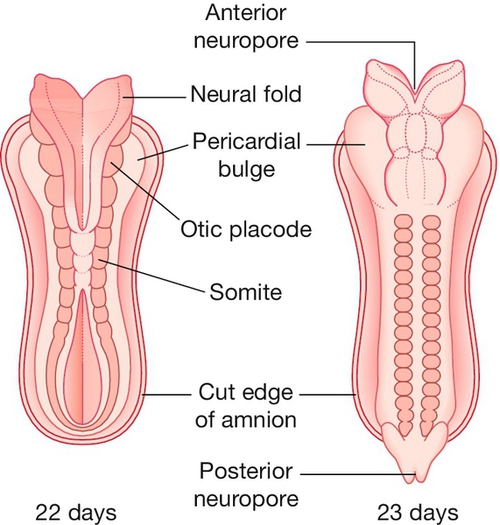
Anencephaly
The skull vault and cerebral cortex are absent. The infant is either stillborn or, if live born, will usually die shortly after birth.
Encephalocele
There is a bony defect in the cranial vault through which a dura mater sac (± brain tissue) protrudes (Fig. 33.10). This may be occipital or frontal. Small isolated encephaloceles carry a good prognosis, whereas those with microcephaly secondary to brain herniation carry a very poor prognosis.
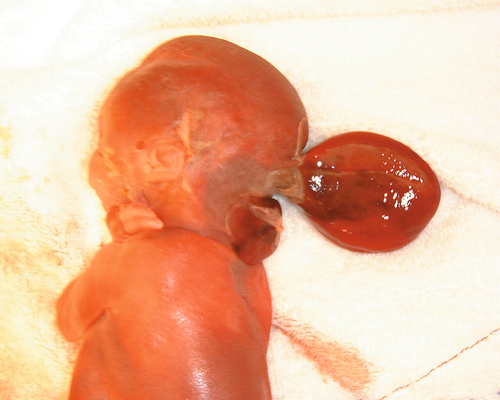
There is a defect in the posterior aspect of the skull, allowing brain tissue to herniate into the sac.
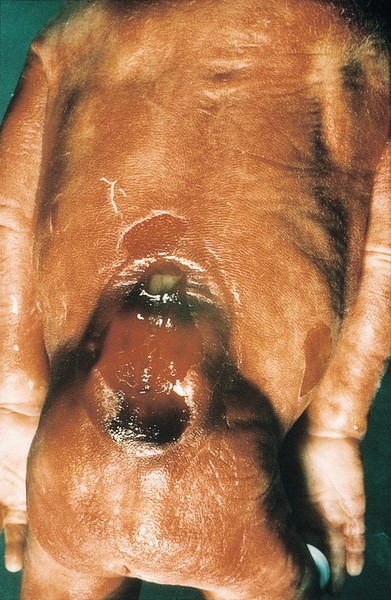
Spina bifida
Spina bifida (Figs 33.11 and 33.12) may take the form of a meningocele or a myelomeningocele. In a meningocele, the meninges of the neural tissue bulge through a posterior spinal wall defect, whereas in a myelomeningocele, the central canal of the cord is also exposed. Those with spinal meningoceles usually have normal lower limb neurology and 20% have hydrocephalus. Those with myelomeningoceles usually have abnormal lower limb neurology and many have hydrocephalus. In addition to immobility and neurodevelopmental delay, there may be problems with urinary tract infection, bladder dysfunction and bowel dysfunction.
There is a large lumbosacral defect, with the sac of the myelomeningocele clearly visible.
Such neural tube anomalies are now almost exclusively diagnosed at a first or second trimester ultrasound examination.
Daily folic acid taken from before conception reduces the recurrence risk of neural tube defects in those who have had a previously affected child. A pre-conceptual prophylactic dose for all women who are planning a pregnancy probably also offers some protection.
Abdominal wall anomalies
Exomphalos (or omphalocele)
This occurs following failure of the bowel to return to the abdominal cavity at 8 weeks’ gestation, after physiological ‘rotation’ and results in a defect through which the peritoneal sac protrudes (Fig. 33.13). The sac may contain both intestines and liver. There are chromosomal abnormalities in up to 30% (especially trisomy 18) and 10–50% have other associated malformations, particularly cardiac and renal anomalies. There is also a relatively rare association with ectopia vesicae and ectopia cardia (midline bladder and cardiac herniae). If the exomphalos is isolated (i.e. no other structural abnormalities), the chromosomes are normal and there is no bowel atresia or infarction, the prognosis is good (> 80% long-term survival). The sac rarely ruptures at vaginal delivery.
Gastroschisis
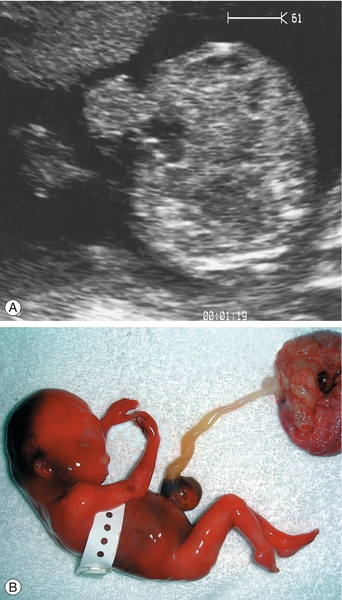
There is an abdominal wall defect usually to the right and below the insertion of the umbilical cord (Fig. 33.14). Small bowel (without a peritoneal covering) protrudes and floats free in the peritoneal fluid. Bowel atresias and cardiac lesions occur in up to 20% but the association with chromosomal abnormality is very small (< 1%). The prognosis is good if the bowel is viable, although 10% end in stillbirth despite apparently normal growth. Bowel dilatation may be associated with bowel obstruction or ischaemia. A small proportion of these babies have major bowel resection and associated long-term total parenteral nutrition requirements. This may adversely affect prognosis in this small number of babies (< 2%). These babies are usually small-for-gestational-age (SGA), have an increased risk of stillbirth (between 32–38 weeks) and require very close surveillance. The recurrence risk is less than 1%.
Genitourinary abnormalities
Multicystic dysplastic kidneys
The kidneys have large discrete non-communicating cysts with a central, more solid core and are thought to follow early developmental failure (Fig. 33.15). The inheritance is sporadic but may occasionally be associated with syndromes such as VATER/VACTERL. If the cysts affect only one kidney, the other is normal and there is adequate liquor, the prognosis is good. If the cysts are bilateral with associated severe oligohydramnios, the prognosis is very poor with high perinatal mortality.
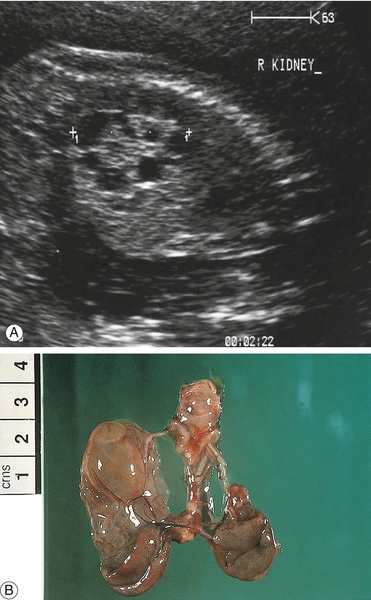
Fig. 33.15Dysplastic renal scan.
Note the enlarged kidney containing fluid-filled cysts: (A) ultrasound; (B) post-mortem specimen.
Polycystic kidney disease
Adult polycystic kidney disease has an autosomal dominant inheritance and is relatively benign, often not producing symptoms until the fifth decade of life. Many individuals have ultrasonically normal kidneys at birth.
Infantile polycystic kidney disease has an autosomal recessive inheritance. There is a wide range of expression, with the size of cysts ranging from microscopic to several millimetres across. Both kidneys are affected, and there may also be cysts present in the liver and pancreas. Ultrasound features of oligohydramnios, empty bladder and large symmetrical bright kidneys (Fig. 33.16) may not develop until later in pregnancy. They may be associated with other anomalies (central nervous system and abnormalities of the extremities, in Meckel–Gruber syndrome). If there is survival beyond the neonatal period, there may be later problems with raised blood pressure and progressive renal failure. Long-term survival is rare.
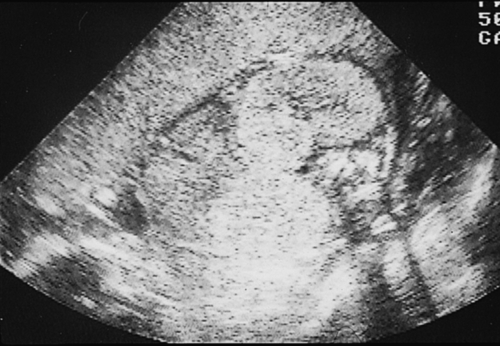
Fig. 33.16Infantile renal cystic scan.
Note anhydramnios and bright renal echoes from the microscopically small cysts.
Pyelectasis
Renal pelvic dilatation may be unilateral (79–90%) or bilateral. It is probably caused by a neuromuscular defect at the junction of the ureter and the renal pelvis, and presents with increasing pelvic dilatation in the presence of a normal ureter (Fig. 33.17). As there is an association with postnatal urinary tract infections and reflux nephropathy, it is reasonable to start all affected neonates on prophylactic antibiotics and arrange postnatal radiological follow-up. Even in those with mild dilatation (≥ 5 mm and < 10 mm), there is vesicoureteric reflux in 10–20%, although only a small proportion require surgery.
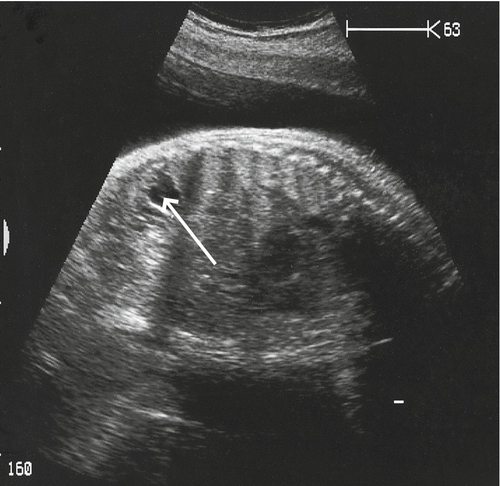
The renal pelvis is markedly dilated, although the renal cortex looks well preserved.
Posterior urethral valves (and congenital bladder neck obstruction)
In this condition, folds of mucosa at the bladder neck prevent urine leaving the bladder. The fetus is usually male, there is often oligohydramnios and on ultrasound, there are varying degrees of renal dysplasia. There is a chromosomal abnormality in 7% of isolated defects, and in one-third of those with other abnormalities. It may be possible to insert a ‘pigtail’ shunt between the bladder and amniotic cavity to relieve the obstruction, but the long-term prognosis is still often poor as the renal damage may not be reversible. Anomalies such as urethral atresia carry a worse prognosis overall.
Potter syndrome
Bilateral renal agenesis (Potter syndrome) is associated with extreme oligohydramnios, which leads to the Potter sequence of pulmonary hypoplasia (see below) and limb deformity (due to fetal compression). The condition is lethal. The recurrence risk is approximately 3%, although autosomal dominant forms with variable penetrance have been described.
Lung disorders
Pulmonary hypoplasia
Liquor is important for alveolar maturation, particularly in the second trimester. Without liquor (especially between 16–24 weeks), there will be a high rate of associated pulmonary hypoplasia (> 95%). Severe oligohydramnios occurs if there is very pre-term pre-labour membrane rupture or Potter syndrome (see above). Pulmonary hypoplasia also occurs with some congenital anomalies (such as diaphragmatic herniae and type III congenital cystic lung lesions).
Diaphragmatic hernia
Stomach, colon and even spleen can enter the chest through a defect in the diaphragm, most commonly on the left. The heart is pushed to the right and the lungs may become hypoplastic. The incidence of aneuploidy is 15–30% and there is an association with neural tube defects, congenital heart disease and renal and skeletal abnormalities. The overall survival for left-sided diaphragmatic herniae (which accounts for about 80% of cases) diagnosed antenatally in North America and Europe is up to 70%; the prognosis for right-sided herniae is relatively poor and may be associated with hydrops fetalis. Current research is looking at the best ways to predict those that have a higher mortality and morbidity, and to consider fetal therapy in this group. Polyhydramnios, mediastinal shift and left ventricular compression are poor antenatal prognostic factors. Postnatal surgery aims to reduce the hernia and close the diaphragmatic defect.
Single gene disorders
There is a very large number of single gene disorders, and many of these are amenable to prenatal diagnosis. A few of these are described below to illustrate some of the diagnostic issues.
Cystic fibrosis
The UK gene frequency for cystic fibrosis is 1:20 (i.e. heterozygote frequency) and the estimated overall couple risk for a live birth is around 1:2500 (there is probably an increased miscarriage rate in homozygotes). Clinically, there is respiratory, gastrointestinal, liver and pancreatic dysfunction and azoospermia is usual in males. The prognosis is very variable, although death around the age of 20–30 still occurs, the prognosis is improving and many affected individuals now live considerably longer. The health of an affected sibling is not a prognostic guide to the health of other siblings. Four mutant alleles account for 85% of the gene defects in the UK (the commonest being ΔF508), and antenatal screening for these is possible using saliva specimens, with CVS offered if both parents are gene carriers.
Fragile X syndrome
This is the commonest cause of moderate neurodevelopmental delay after trisomy 21 and the commonest form of inherited mental handicap. It is X-linked. Males are usually more severely affected than females. Speech delay is common and there is an associated behavioural phenotype with gaze aversion. The condition is caused by the expansion of a CGG triplet repeat on the X chromosome. Normal individuals have an average of 29 repeats, but for an unexplained reason, this may increase to a pre-mutation of 50–200 repeats. Those with a pre-mutation are phenotypically normal, but the pre-mutation is unstable during female meiosis and can expand to a full mutation of more than 200 repeats. There is an approximately 10% chance of this occurring (in the absence of a full mutation in that generation already). This causes the fragile X phenotype in 99% of males and around 30–50% of females. Parental screening is possible and CVS may be used to identify the degree of amplification of the CGG repeats in potential offspring.
Huntington’s chorea
The onset of this autosomal dominant condition is usually after the age of 30, although it may present as early as 10–15 years of age. There is dementia, mood change (usually depression) and choreoathetosis, progressing to death in approximately 15 years. It is caused by a CAG trinucleotide expansion on chromosome 4p and this allows accurate carrier and prenatal testing. There are major ethical issues around testing both mothers and their offspring.
Tay–Sachs disease
The gene frequency is 1:30 in Ashkenazi Jews, but is rare in other groups. There is a build-up of gangliosides within the CNS, leading to neurodevelopmental delay, paralysis and blindness. By the age of 4 years, the child is usually dead or in a vegetative state. Carriers may be screened by measuring the level of hexosaminidase A in leucocytes.
Prenatal congenital infection
Infections in pregnancy are especially important because of potential risks to the fetus. A number of agents are known to be teratogenic, particularly in the first and early second trimesters (e.g. rubella and cytomegalovirus). Others carry the risk of miscarriage, premature labour, severe neonatal sepsis or long-term carrier states.
Risk factors
Farm workers are at risk of chlamydial infection (which causes abortion in sheep) and listerial infection, both of which can cause abortion in humans. Care is required particularly at lambing time. Toxoplasma may also be acquired from cows, sheep and domestic cats. Certain foods have also been implicated in congenital infection (Table 33.2).
Table 33.2
Foods that carry potential infection risks in pregnancy
| Soft cheeses | Unpasteurized milk and its products may contain listeria. Those made from pasteurized milk are safe |
| Raw eggs | Must be avoided, as there is a risk of salmonella (including puddings) |
| Meat or pâté | Undercooked meat may transmit toxoplasma or rarely, listeria |
| Fruit | This should always be washed before eating as it may be contaminated with salmonella, toxoplasma or one of several intestinal parasites |
Specific infections
Infections (see also Table 33.3) in general, raise the maternal serological levels of immunoglobulins of both the IgG and IgM variety. Maternal IgG crosses the placenta, while IgM, a much larger molecule, does not. The fetus does not make IgM until beyond 20 weeks’ gestation and its presence in fetal or early neonatal blood implies infection. Infection does not necessarily mean that the infection has caused a problem, and absence of fetal or neonatal IgM at sampling does not completely exclude intrauterine infection. In high risk cases, the detection of pathogenic DNA in fetal blood or amniotic fluid by PCR has revolutionized the specificity of infective detection.
Chickenpox at term
Severe and even fatal cases of chickenpox can occur in neonates whose mothers develop chickenpox just before delivery, as the baby is born before maternal IgG production has increased sufficiently to allow passive transplacental protection. If maternal infection occurs 1–4 weeks before delivery, up to 50% of babies are infected and approximately a quarter of these develop clinical varicella. Severe infection is most likely to occur if the infant is born within 7 days of onset of the mother’s rash, when cord blood IgG is low.
If delivery occurs within 5 days of maternal infection, or if the mother develops chickenpox within 2 days of giving birth, the neonate should be given passive varicella zoster immunoglobulin and the infant should be monitored for around 2 weeks. If neonatal infection occurs, it should be treated with aciclovir. (See Table 33.3 for chickenpox in early pregnancy).
Hepatitis
Hepatitis A has not been associated with significant complications in pregnancy. All mothers should be screened antenatally for hepatitis B virus. The initial serological response to infection is with HBsAg, followed by HBeAg, a marker of high infectivity. Vertical transmission to the fetus is most likely to occur with acute infection (especially third trimester) or in the presence of HBeAg, and may lead to neonatal infection or long-term carriage. The baby should be given passive hepatitis B immunoglobulin at birth as well as an active hepatitis B immunization.
With hepatitis C, vertical transmission is related to viral load but is unlikely in the absence of detectable RNA. There is no evidence that treatment during pregnancy reduces the chance of transmission, and ribavirin is probably teratogenic. Hepatitis E infection in pregnancy, while uncommon, carries a 30% maternal mortality rate and possible risk of fetal loss.
Herpes simplex virus
An acute attack of primary genital herpes shortly before delivery may lead to a localized or systemic neonatal infection, including encephalitis. The risk of infection is greatest with a primary infection, but can occur with recurrence, although this risk decreases with time from the first attack. Screening is of no proven value, but caesarean section may be indicated in the presence of a primary infection.
Rubella
Rubella infection is outlined in Table 33.3 but its importance lies in the potential for prevention through vaccination. Immunity from natural infection is life-long. Seroconversion and life-long immunity occur in about 95% of vaccinated individuals and, as the benefits of herd immunity have been clearly demonstrated, many countries now immunize all pre-school children. Rubella antibodies are commonly checked at the first antenatal visit and postnatal vaccination is offered to those with low titres.
In common with other prenatal infections, it can be difficult to predict which fetuses are going to be infected, and which of these infected fetuses will be affected. Furthermore, the very process of sampling fetal blood in utero for such tests may encourage transplacental transmission.
Erythrovirus (previously called Human Parvovirus B19)
Erythrovirus infection in the first half of pregnancy can cause aplastic anaemia in the fetus, leading to high output cardiac failure and fetal demise (Fig. 33.18). Many babies though present with ‘hydrops fetalis’. The use of fetal middle cerebral artery Doppler peak systolic velocity measurements in this situation has been extremely valuable since the Doppler velocity correlates well with the degree of anaemia and allows an accurate determination of those who require in-utero transfusion. Transfusion is usually very successful and the outlook for babies receiving transfusion is good (up to 85% survival but prognosis is less good than that of rhesus disease).
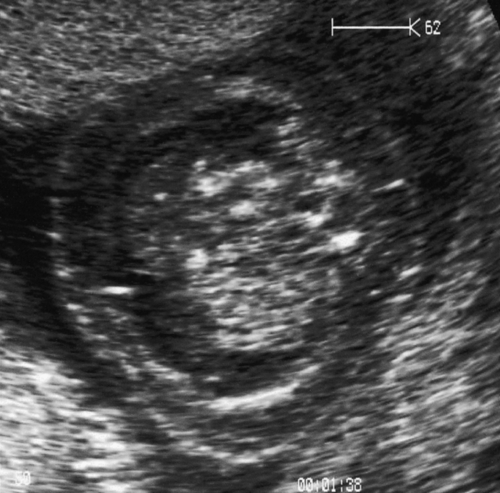
Fig. 33.18Hydrops fetalis caused by infection with erythrovirus.
There are ascites and marked skin oedema in this ultrasound image of a cross-section of the fetal abdomen.
Listeria monocytogenes
This is a rare bacterial infection transmitted by food, usually soft ripe cheeses, pâté, cooked–chilled meals and ready-to-eat foods that have not been thoroughly cooked. Following an initial gastroenteritis, which may be fleeting, bacteraemia results in bacilli crossing the placenta and causing amnionitis, pre-term labour (which may result in stillbirth) or spontaneous miscarriage. There may be meconium, neonatal jaundice, conjunctivitis or meningoencephalitis. Diagnosis is made by blood culture or by culture of liquor or placenta. Treatment is with high-dose amoxicillin or erythromycin.
Beta-haemolytic streptococci – group B
Between 5% and 20% of women carry this organism in the vagina. It is associated with pre-term rupture of the membranes. About 50% of babies become colonized at delivery but only about 1% of these develop infection. The neonatal mortality from infection may be as high as 80%, with 50% of those surviving meningitis having subsequent neurological impairment. Antenatal screening is not recommended in the UK (initial screen positives may become negative and vice versa) but those with known colonization should receive intrapartum penicillin. There is no evidence to support antenatal treatment of asymptomatic carriers, as carriage is rapidly re-established following treatment.
Syphilis
Congenital syphilis is rare in the UK but more common worldwide. Its prevalence is increasing. Severe early-onset FGR with or without congenital malformations (and hydrops) is often the presenting ultrasound sign. Those identified antenatally with positive serology should be treated with penicillin.
Termination of pregnancy for fetal abnormality
Prenatal diagnoses are often made after 14 weeks of pregnancy and termination is medical, rather than by surgical means. Termination of pregnancy under Clause E of the Abortion Act (1967) allows the offer of termination of pregnancy for fetal anomaly if two registered practitioners agree that a fetal anomaly carries significant risk of serious handicap. Such a consultation requires sensitivity. Not all couples will opt for prenatal termination of pregnancy and it is important that neonatal palliation as an alternative is discussed in selective cases. This requires multidisciplinary counselling and discussion. If parents opt for TOP, then after 22 weeks’ gestation, the obstetrician has a duty of care to be certain that the baby does not suffer and that it is not born alive. This requires the discussion of feticide in late termination of pregnancy. Parents may initially be reluctant to see the baby after delivery, but should be offered the opportunity. There is a multidisciplinary bereavement team that helps the management of this difficult period post-delivery. Many mothers are later comforted that they underwent a delivery and saw and named the baby. Photographs of the baby can also be taken for the parents.
Postmortem examination should be discussed and encouraged if the cause of the anomaly is unknown. If parents decline, they may accept a limited postmortem investigation with X-rays, clinical photographs and specimens for karyotype studies instead. Follow-up to discuss the results and their implications for subsequent pregnancy is important.