Chapter 55 Posterior Fossa Tumors in the Pediatric Population
Multidisciplinary Management
Primary brain tumors are the most common solid tumors in the pediatric population, comprising 20% to 25% of all childhood cancers. About 60% to 70% of all pediatric brain tumors originate in the posterior fossa.1–4 The reason that pediatric brain tumors have a propensity to occur in the posterior fossa has not yet been elucidated. By far, the most common posterior fossa tumors of childhood are medulloblastomas, ependymomas, and astrocytomas. As for uncommon tumors, these include atypical teratoid/rhabdoid tumors (AT/RTs), teratoma, hemangioblastoma, dermoid, and epidermoid. Each tumor may have a vastly different prognosis and response to treatment depending on the pathologies. Recent advancements have increased our knowledge of the cell biology, facilitated earlier diagnosis and improved neurosurgical resections and adjuvant treatment. These in turn have not only improved the survival of children but also their quality of life. Although much progress has been made in the diagnosis and treatment of posterior fossa tumors, they still cause the most cancer-related deaths in this age group.5,6 This review focuses on the multidisciplinary management of the major types of pediatric posterior fossa tumors. Current advances in tumor diagnosis and surgery, along with adjuvant therapeutic modalities, will be discussed.
Symptoms and Signs
Posterior fossa tumors often cause hydrocephalus by blocking cerebrospinal (CSF) outflow pathways, with resulting signs and symptoms of raised intracranial pressure (ICP).7 Symptoms typically begin with intermittent headache, often worse in the morning followed by vomiting and eventually gait disturbance. Funduscopy must be performed in children with suspected posterior fossa or supratentorial tumors because papilledema is a common finding. Sixth nerve palsies resulting from intracranial hypertension and ataxia from a combination of cerebellar compression and hydrocephalus are also common.
The presenting signs and symptoms are dependent on the growth rate of tumors, the age of the patient and the location of the tumor. Most children with medulloblastomas present with a short clinical history, for less than 1.5 months in approximately 50% of patients and less than 3 months in approximately 75% of patients.7 Ependymomas are relatively fast-growing tumors and the median duration of symptoms is 2 to 3 months.8 As for cerebellar astrocytoma, the symptomatic period ranges from 5 to 9 months and is usually significantly longer than other posterior fossa tumors.9 Rapid, sudden deterioration in neurologic status may result from acute obstructive hydrocephalus or hemorrhage into either the tumor or the subarachnoid space.10–13 Older children may complain of headache, neck stiffness, dizziness or diplopia. On neurologic examination, they will demonstrate truncal or appendicular ataxia, dysmetria, nystagmus, or cranial nerve palsies. In comparison with older children, the presentation in infants and young children is often more insidious, with gradual progression.14 The diagnosis is suspected in the setting of irritability, loss of appetite, weight loss and failure to thrive. In addition, they may display signs of increased ICP, including lethargy, drowsiness, vomiting, sun-setting, a full fontanelle, or an increasing head circumference. Midline tumors of the cerebellum usually cause truncal ataxia, whereas hemispheric lesions cause ipsilateral dysmetria. Tumor in the obex and area postrema causes vomiting even if the tumor is small and there is no hydrocephalus. Cranial neuropathies such as facial weakness, hearing loss and swallowing dysfunction may present in tumors that exit the foramen of Luschka from the cerebellopontine angle or in tumors demonstrating brain stem invasion.
Head-tilt, signifying descent of the cerebellar tonsils into the foramen magnum with compression of the C1 or C2 nerve roots, may be observed. Development of a stiff neck or head tilt usually suggests tonsillar involvement by the tumor or signs of impending herniation.15–17 Rarely, children initially present with symptoms of spinal dissemination, such as back and leg pain or paraparesis.18 The transmission of increased pressure to the upper cervical spinal cord may cause syringomyelia and related symptoms in patients with cerebellar astrocytomas.19
Radiologic Findings
Medulloblastoma
Approximately two thirds of medulloblastomas are located in the midline arising from the vermis or inferior medullary velum and widening the space between the cerebellar tonsils. Isolated involvement of the hemisphere is seen in older children and young adults.20,21
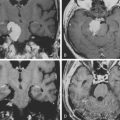
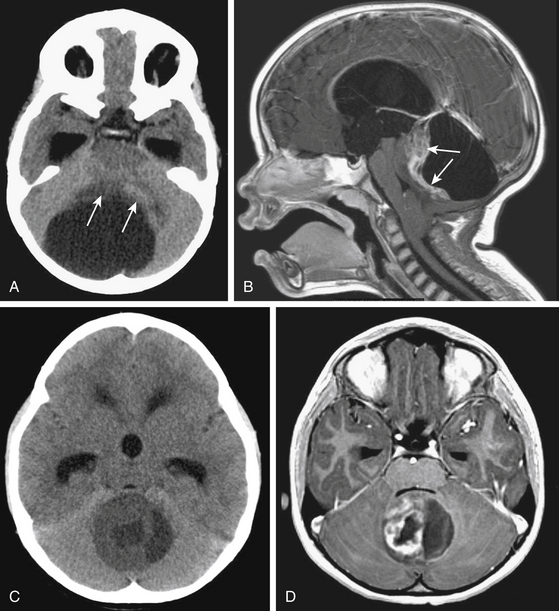
Due to the increased nuclear to cytoplasmic ratio of the tumor cells, medulloblastoma typically appears as a homogenous iso- to hyper-dense midline mass within the posterior fossa on non-contrast CT scan (Fig. 55-1A), with variable amounts of peritumoral edema and hydrocephalus whereas the solid portion of cerebellar astrocytomas are hypodense on precontrast studies.22 By CT, medulloblastoma can also contain calcification, necrosis, cystic degeneration, and hemorrhage and can invade the brain stem. Following contrast administration, this tumor demonstrates homogenous enhancement (Fig. 55-1B).
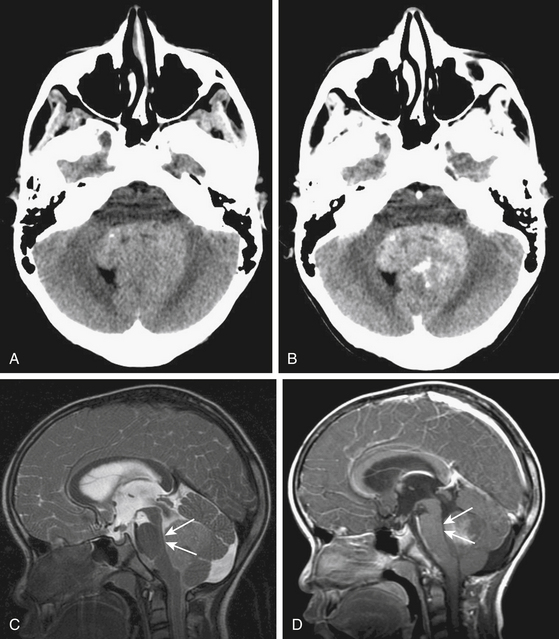
On MRI, medulloblastoma typically appears as a hypo- to iso-intense mass on T1-weighted images (T1WI), and iso- to hyper-intense on T2-weighted images (T2WI)22,23 (Fig. 55-1C). Diffusion-weighted images (DWI) demonstrate restricted diffusion.24 The enhancement pattern is variable: enhancement may be uniform or patchy22,23 (Fig. 55-1D). The heterogeneity probably results from cysts and calcification. Intracranial subarachnoid or intraventricular dissemination should be carefully identified because the perimesencephalic cisterns, tentorial surface and suprasellar cisterns are frequent sites for dissemination at the time of tumor diagnosis.
Ependymoma
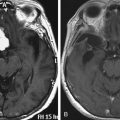
On CT scan, a posterior fossa ependymoma is an iso- or hyper-dense midline cerebellar mass with calcifications, small cysts, and heterogeneous enhancement after intravenous contrast administration25 (Fig. 55-2A). Hemorrhage is present in up to 13% and calcifications are frequent in 25% to 50%. MR imaging shows marked heterogeneity of the tumor due to small cysts as well as areas of old hemorrhage. Precontrast T1- and T2-weighted images commonly reveal an iso- to hyper-intense signal intensity. Most tumors show heterogeneous and irregular enhancement following gadolinium administration26 (Fig. 55-2B).
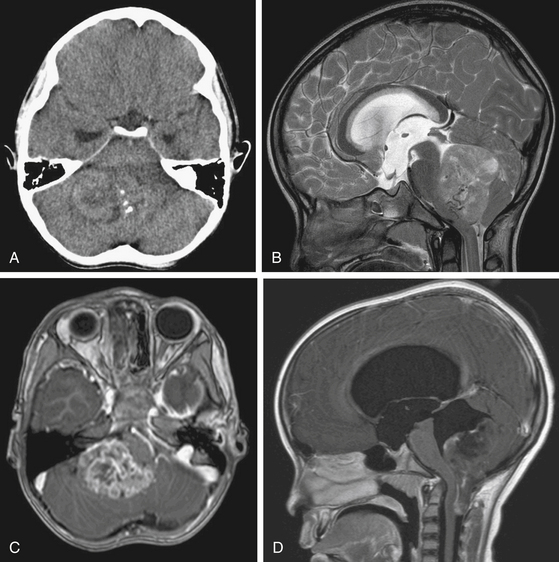
The most characteristic appearance supporting the diagnosis of ependymoma is of a mass arising from the lower brain stem and projecting into the fourth ventricle; in half or more tumors, extension through the foramen of Luschka into the cerebellopontine angle or through the foramen magnum into the cervical spinal canal is found (Fig. 55-2C and D). The vertebral arteries and posterior inferior cerebellar arteries may be displaced or encased.
Cerebellar Astrocytoma
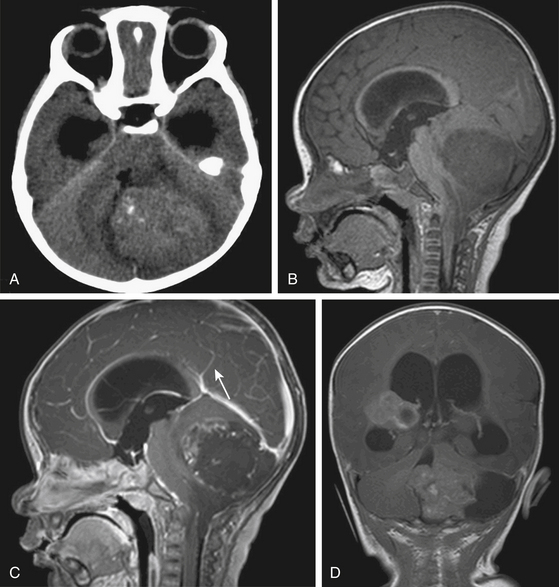
Cerebellar astrocytomas can be either vermian or hemispheric in location. They can be predominantly cystic with a solid mass, the so-called mural nodule, in 30% of cases; they can be, solid with cystic areas in 40% to 50%; and wholly solid in 20% to 30%.9,27,28 Cystic tumors tend to be located within the hemispheres and solid tumors in the midline alone or in combination with extension into one or both hemispheres.29,30
On noncontrast CT, the solid portion of the tumor is usually iso- to hypodense and the cyst is hypodense but denser than CSF because of the high protein concentration (Fig. 55-3A and C). Calcification can be seen in up to 20%, and cysts in 70%.31,32 On MRI, solid parts of the tumor are generally iso- or hypo-intense signal intensity masses on T1-weighted sequences and hyperintense masses on T2-weighted and FLAIR sequences.33 Contrast enhancement is usually irregular caused by cysts and tumor necrosis22 (Fig. 55-3B and D). Tumors of pilocytic pathology show intense enhancement of their solid portions.34 Nonenhancing tumors are almost never of pilocytic histology. Frequently, the infiltrative fibrillary astrocytomas do not show much gadolinium enhancement.35 Both pathologies can invade the cerebellar peduncle and brain stem.
Atypical Teratoid/Rhabdoid Tumors
AT/RTs usually originate in the cerebellar hemispheres instead of the midline and have a predilection for the cerebello-pontine angle.36,37 Their pattern of growth is aggressive with significant mass effect on the adjacent fourth ventricle and brain stem, and early CSF dissemination. AT/RTs may also arise in the spinal cord, pineal gland and suprasellar region.38,39
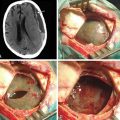
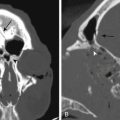
On noncontrast CT, the solid portions of the tumor demonstrate hyperdensity, presumably due to the high cellularity of the tumor36,38 (Fig. 55-4A). On MRI, solid portions of the tumor are low signal intensity on T1-weighted images and isointense or decreased signal on T2-weighted images.36,38,40 In addition, frequent necrosis, cysts, and sometimes hemorrhage are identified, giving an appearance of striking heterogeneity.36,37,41 Enhancement can be variable (Fig. 55-4B to D).
Invasion of the Brain Stem and Cerebellar Peduncle
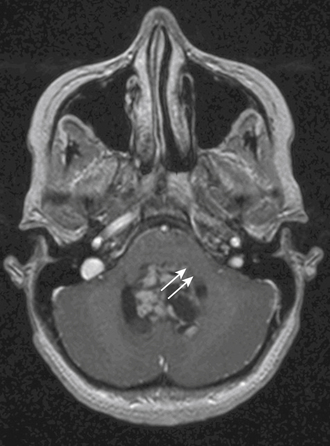
Invasion of the brain stem and cerebellar peduncle is found in up to 40% of newly diagnosed medulloblastoma.4,42 In another study, brain stem or peduncle involvement is identified in 34% of cerebellar pilocytic and diffuse astrocytomas.43 Cerebellar peduncle involvement is suggested by an ill-defined border on the affected side on postcontrast MRI (Fig. 55-5). On MR midsagittal images without/with contrast infusion, a cerebrospinal fluid space between the tumor and the floor of the fourth ventricle may be found, which can indicate that there is no invasion of the floor of the fourth ventricle (Fig. 55-1C and D). However, absence of a cerebrospinal fluid space does not always indicate the invasion of tumor. As such, it is difficult to affirm unequivocally that the tumor is compressing or invading the floor of the fourth ventricle by imaging studies alone.
Work-Up for Tumor Dissemination
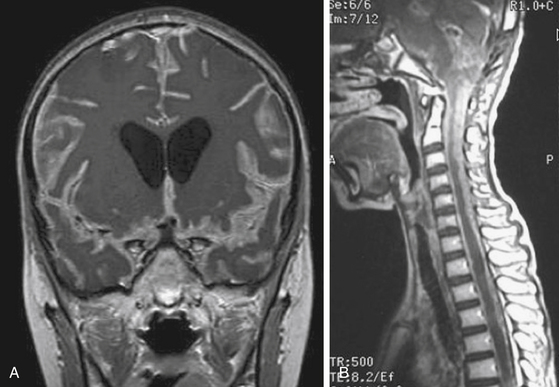
At diagnosis, patients rarely present with symptoms of tumor dissemination. However, spinal metastases can be found in about 20% to 50% of medulloblastoma, 11% to 17% of ependymoma, 25% to 34% of AT/RT at the time of diagnosis.38,39,44–49 The current standard of care should be to obtain pre- and post-contrast scans of the brain and spine when the presumptive diagnosis of a malignant posterior fossa tumor is made. In the first few weeks after posterior fossa surgery, artifacts from blood products can be very difficult to differentiate from CSF spread of tumor.50,51 These problems are avoided by performing preoperative staging MR studies of the spine. Postsurgical MRI should be performed within 48 to 72 hours of surgery or at least 2 weeks later to avoid the difficulty of distinguishing between postoperative changes and tumor dissemination or residual tumor. By MRI scans, leptomeningeal dissemination appears as diffuse enhancement of the meninges and/or enhancing clumps along the spinal cord and cauda equina on T1-weighted images (Fig. 55-6).
In addition to spinal MRI, CSF cytologic examination for malignant cells has been used for the diagnosis of leptomeningeal disease pre- or post-operatively. However, spinal MR imaging has a greater diagnostic accuracy than CSF cytology in the early detection of disseminated disease.52,53 CSF cytologic analysis performed more than 2 to 3 weeks after surgery can reduce false-positive results. Negative CSF cytologic results do not always exclude the tumor dissemination.54,55
Surgery
Management of Hydrocephalus
Obstructive hydrocephalus is reported in 70% to 80% of children with posterior fossa tumors and is frequently the cause of clinical deterioration at the time of diagnosis.56,57 Depending on the patient’s symptoms and the severity of the associated hydrocephalus, a decision must be made whether to treat the hydrocephalus up front or at the time of tumor resection. In the 1960s, when a child presented in a poor clinical state as a result of a delayed diagnosis, the routine placement of a preoperative shunt significantly reduced the overall morbidity and mortality rate.58,59 However, neurosurgeons have questioned the need for a routine shunt because only 15% to 30% of patients ultimately require permanent CSF diversion following resection of the tumor.57,60 In addition to the usual complications associated with ventriculoperitoneal (VP) shunts such as infection and blockage, rare complications such as upward herniation, tumor hemorrhage, and peritoneal seeding of the intracranial tumor may occur.61 Accordingly, most centers are using a combination of corticosteroids, early surgery, and external ventricular drainage rather than VP shunting.62,63
Postoperative hydrocephalus occurs within the first few days to months after surgery. Rarely, it will develop several months or years after surgery, or at the time of a local recurrence or of subarachnoid spread.64,65 Factors requiring placement of postoperative shunt include more severe hydrocephalus at diagnosis, young age, large preoperative tumor size, midline localization and incomplete tumor removal.56,60,66,67 Patients with medulloblastoma, as opposed to other posterior fossa tumors, also appear to be at higher risk for postoperative hydrocephalus requiring a VP shunt.56,68
The effectiveness of endoscopic third ventriculostomy (ETV) has been evaluated in the management of preoperative and postoperative hydrocephalus. ETV, when performed prior to posterior fossa surgery, reduces the incidence of hydrocephalus because preoperative normalization of CSF hydrodynamics decreases the risk of permanent postoperative impairment of the CSF circulation.67 However, the routine application of preoperative ETV is not indicated because of the small number of patients requiring definitive treatment for hydrocephalus.57,68 ETV may be used for persistent or progressive hydrocephalus following tumor removal.57
Patient Positioning
Several options for patient positioning are available for patients with posterior fossa tumors: prone, Concorde, lateral decubitus, “park-bench” and the sitting position. Pediatric patients are typically placed in the prone position. For lesions extending superiorly through the aqueduct, the head is tilted slightly to the patient’s right, away from the surgeon and flexed far forward (Concorde position) so that the rostral extent of the tumor can be removed with the surgeon sitting behind the patient’s shoulder. The lateral decubitus positioning can be used for tumors that occur in the cerebellopontine angle or lateral cerebellum. The sitting position has several distinct advantages including limited use of retraction, and a clear operative field. However, it is difficult to position infants and young children in the sitting position. Dangers include cardiovascular instability, hypotension, air embolism, pneumocephalus, subdural hematoma, and the rapid escape of cerebrospinal fluid from the ventricular system.
Opening and Closure
If the lesion is located in the vermis or paravermian region, a midline skin incision suffices. If the lesion occupies one hemisphere without a contiguous attachment to the midline, a paramedian incision is preferred and a hockey stick incision can be used to allow for a wider craniectomy. In the midline posterior fossa exposure, the incision is extended from slightly rostral to the inion down to the C1–C2 spinal process. Skin flaps are mobilized by undermining the subcutaneous plane to prepare a fascial flap for closure and the fascia is incised using a Y- or T-shaped incision for reapproximating tightly at the superior nuchal line. The cervical musculature is mobilized laterally off the occiput and cervical laminae in the midline avascular plane. The foramen magnum is exposed, and the dura is identified up to the posterior arch of C1. A posterior fossa craniotomy is then performed (Fig. 55-7). Bilateral burr holes are made just below the transverse sinus from the midline. The dura is separated from the inner table of the skull, which is typically not adherent in children. At the foramen magnum, the junction of the pericranium and the dura is sharply dissected with caution because of the occipital sinus in the midline near the foramen magnum. If the tumor extends into the upper cervical spinal canal, a laminectomy of the upper vertebrae may be performed. Monopolar cautery should be used carefully when dissecting the superolateral surface of C1 to prevent injury to an aberrant vertebral artery. C1 can be bifid and is often cartilaginous in infants and young children. After extending a laminectomy below C2, young children have an increased risk of swan neck deformity.69 Therefore, it is prudent to remove the smallest amount of bone possible.
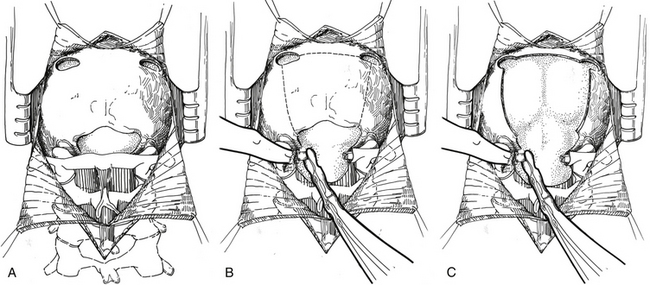
The dura is then opened in a Y-fashion with care taken to obtain hemostasis of the occipital sinuses (Fig. 55-8A). When the vertical incision extends to the foramen magnum, it may extend below the falx cerebella. If there is significant bleeding from the midline occipital sinus and circular dural sinuses, it should be controlled with obliquely placed hemostatic clips or suture ligatures. If clips are used, they can be removed as the dura is sutured because metal clips may result in artifact on postoperative MRI studies. Intraoperative ultrasonography and neuronavigation may be used to define the location of tumors. The surgeon should inspect the surface of the exposed cerebellum and spinal cord for evidence of leptomeningeal seeding. After the cisterna magna has been exposed, cerebrospinal fluid is aspirated for cytology and drained to decrease ICP.
After tumor removal, meticulous hemostasis is achieved with judicious use of bipolar coagulation and gentle pressure, and may be confirmed with a Valsalva maneuver. The dura is always closed in a watertight fashion, followed by replacement of the bone flap and multilayer closure of the superficial tissues. It is important to close the fascial layer tightly near the inion to prevent postoperative leak of cerebrospinal fluid and the formation of a pseudomeningocele.
Tumor Removal
Transvermian and Telovelar Approaches to Midline Cerebellar Tumors
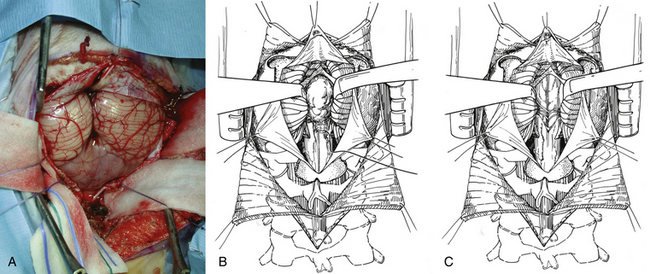
The two most common surgical approaches to midline cerebellar tumors adjacent to the fourth ventricle are the transvermian and telovelar approaches. The transvermian approach involves incising the inferior vermis of the cerebellum and retracting the two halves of the vermis in opposite lateral directions to remove the tumor. The telovelar approach uses the dissection of the cerebellomedullary fissure, which is a natural cleft between the cerebellum and medulla oblongata, and the dissection of the tela choroidea and inferior medullary velum along the natural avascular planes.70–74 The greatest advantage of the telovelar approach is the superolateral exposure of the fourth ventricle and the complete exposure of the foramen of Luschka, which can be accessed without removal of cerebellum or adjustment of the retractors.75,76 However, a limitation of this exposure can be expected when approaching the rostral portion of the fourth ventricle. The transvermian approach provides a greater working angle in this area and a better visualization of the midline inferior portion of the superior medullary velum and fastigium. Nevertheless, the main disadvantages of this approach are a limited lateral exposure and the potential for complications due to iatrogenic injury from excessive vermian dissection and retraction.
In the transvermian approach, the cerebellar tonsils are separated in the midline and it is sometimes helpful to retract the inferior vermis rostrally or incise the caudal vermis to prevent injury to the inferior vermian veins. The posterior inferior cerebellar arteries are usually displaced laterally. The distal portion of the vermis is divided to expose the dorsal surface of tumor (Fig. 55-8B). Cottonoid patties are used to help to protect the normal neural structures and to dissect along the tumor–cerebellar interface. Once planes around the tumor are developed, the tumor is internally debulked, with suction or with the Cavitron. Specimens are now taken for pathologic assessment and tumor banking. As the tumor is debulked, the lateral tumor–cerebellum interface continues to be developed. The uninvolved side of the cerebellar peduncle is first dissected from the surrounding cerebellar tissue, the floor of the fourth ventricle and the aqueduct are then identified, and tumor invading the involved cerebellar peduncle is resected (Fig. 55-8C). The dentate nucleus is located just above the ipsilateral tonsil, and one should take meticulous care to avoid trauma to it during resection of the tumor from the upper pole of the tonsil.
The telovelar approach is performed by opening the cerebellomedullary fissure, which includes dissection of the tonsillomedullary and uvulotonsillar space. The two cerebellar tonsils are then retracted laterally to expose the floor of the fissure, which includes the inferior medullary velum and tela choroidea. The tela choroidea, which forms the caudal part of the roof of the fourth ventricle, is incised from the foramen of Magendie and then followed laterally to the foramen of Luschka on both sides. In large tumors, the uvulotonsillar cleft is stretched, and the anatomy may be distorted. Aggressive dissection in this area before decompression can result in breaching the pial plane and entering either the vermis or tonsil leading to neurologic deficit, such as ataxia and cerebellar mutism. Placing the retractor on the superomedial part of the tonsil to visualize the superolateral corner of the fourth ventricle can injure the dentate nucleus. Dissection and decompression should be done simultaneously to minimize retraction and reduce associated injuries in large-sized tumors.77,78
Techniques When Operating Near the Brain Stem
Care must be taken to identify areas where tumor may invade the floor of the fourth ventricle. One of the most important factors of successful tumor resection is the identification of the floor of the fourth ventricle. If the tumor is not adherent to the floor of the fourth ventricle, one can place Cottonoid patties along the ventricular floor as the dissection proceeds. If the tumor adheres to or invades the floor of the fourth ventricle, the intact floor of the fourth ventricle next to the invasive tumor is identified and the last portion of the tumor is resected at the plane of the floor of the fourth ventricle. The sylvian aqueduct should be covered with Cottonoid patties to prevent blood from entering the third ventricle. The tumor should not be chased into the brain stem as this will lead to serious neurologic morbidity. Hemorrhage from the floor of the fourth ventricle should not be cauterized, because this may result in unexpected cranial nerve damage. Placement of a Gelfoam pack and gentle compression should result in the hemostasis needed.
Intraoperative Neuromonitoring
Intraoperative monitoring of the cranial nerves and the brain stem is frequently used for posterior fossa tumors. Monitoring may be helpful to decrease morbidity and to allow a more aggressive resection. Brain stem auditory-evoked potentials (BAEPs) are resistant to most anesthetic agents. Somatosensory-evoked potentials (SSEPs) are affected by the inhalation anesthetics, although the effects to nitrous oxide are minimal.79 Intravenous anesthetics affect SSEPs significantly less than do inhalation anesthetics. Propofol with nitrous oxide has minimal latency shift and amplitude reduction effects on SSEPs and offers promise as an effective adjunct to neuroanesthesia.80,81
Operative Complications
The most common complication is cerebellar ataxia. This may be transient and often rapidly resolves over several weeks. The portion of the tumor involving the unilateral cerebellar peduncle can be aggressively resected, and gross total resection is possible with minimal neurologic deficits.82 However, if bilateral cerebellar peduncles are involved, radical surgical manipulation should be avoided and may result in severe permanent cerebellar ataxia. When the dentate nucleus is damaged, the disequilibrium and intentional tremor during voluntary movement of the extremities may be found.
Cerebellar mutism is a unique postoperative complication seen in 5% to 30% of children following the resection of a posterior fossa mass lesion.71,83,84 Vermian incision and lateral retraction of the dentate nuclei may explain the development of the cerebellar mutism syndrome particularly in children. Cerebellar mutism is a transient complication that may appear after removal of midline cerebellar tumors involving the vermis in children. It typically arises 1 to 2 days following surgery with patients initially displaying diminished speech progressing to mutism and typically resolves during the ensuing weeks to months. Cerebellar mutism is characterized by a lack of speech output in awake patients with intact speech comprehension, and is sometimes associated with oropharyngeal apraxia.
Cranial nerve dysfunction, such as abducens palsy and facial weakness, internuclear ophthalmoplegia, horizontal gaze palsy, swallowing difficulty, and vocal cord palsy are related to irritation of the brain stem or lower cranial nerve. Deficits with vocal cord apposition and swallowing may cause complications such as aspiration pneumonia and respiratory distress. Swallowing problems related to lower cranial nerve manipulation can generally be expected to recover function over time.3,85,86 On the contrary, the symptoms with nuclear involvement of the brain stem may take longer to recover or may never recover completely.
Postoperative pseudomeningoceles or subsequent CSF leak affect 23% to 28% of all children with posterior fossa tumors.87,88 The mechanisms leading to pseudomeningocele formation are unclear, but probably include inadequate dural and wound closures. Small pseudomeningoceles may respond well to pressure bandages, needle aspiration, and lumbar CSF drainage. However, pseudomeningoceles may be a manifestation of hydrocephalus and in some cases may require CSF diversion to control.
Specific Tumors
Medulloblastoma
Epidemiology and Genetics
Medulloblastomas are the most common malignant brain tumor in children comprising 20% to 25% of all pediatric brain tumors and are usually found in the posterior fossa.89 The majority (85%) arise from the cerebellar vermis and in the minority, they arise laterally from the cerebellar hemisphere. The median age at diagnosis is approximately 6 to 9 years.20,90 There is a slight male predominance (male:female ratio 2:1).91,92 Up to 30% of cases occur in adulthood.93
Several cancer predisposition syndromes are associated with medulloblastoma including Gorlin’s syndrome, Turcot’s syndrome, Li-Fraumeni syndrome, and Rubenstein-Taybi syndrome.94 A frequent genetic abnormality is amplification of the MYC family of oncogenes, which is seen in up to 10% of cases.95–97 Molecular subgroups, characterized by Wnt/wingless signaling, sonic hedgehog signaling (SHH), expression of neuronal differentiation genes or photoreceptor genes have been identified.97–99 Based on these signaling pathways, targeted therapies have been suggested using agents such as cyclopamine and HhAnTag (small molecule inhibitor of the SHH).100–102 Common chromosomal copy number changes include gain of chromosomes 1q and 7, as well as loss of chromosomes 22, 11, 10q, and 17p.95,103,104 Loss of 17p is observed in up to 50% of cases, and may occur in the context of an isochromosome 17q.95,103
Pathology
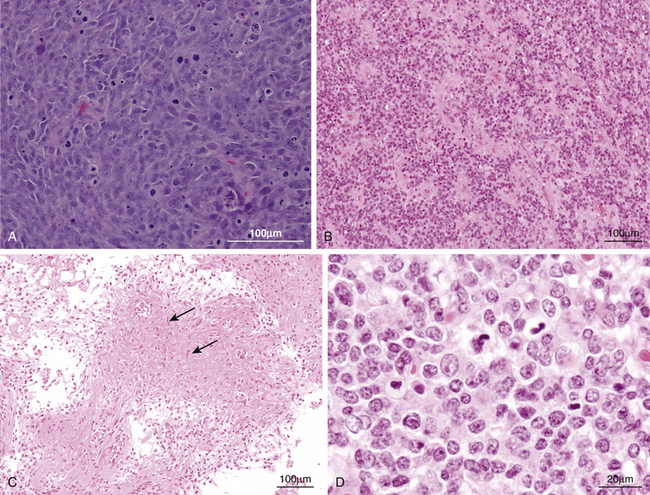
Medulloblastomas are considered WHO grade IV tumors.104 Histologic variants include the classic, desmoplastic/nodular, medulloblastoma with extensive nodularity, anaplastic, and large-cell subtypes. The classic medulloblastomas (70% to 80% of cases) appear as a dense sheet of small, basophilic cells with little cytoplasm, round-to-oval hyperchromatic nuclei and frequent mitoses (Fig. 55-9A). Homer-Wright rosettes are commonly seen.104 Evidence of differentiation along neuronal or glial lineage is seen in up to 50% of cases.105 The desmoplastic/nodular subtype accounts for approximately 15% to 20% of cases and is characterized by pale, reticulin-free nodules surrounded by reticulin-positive collagen fibers.106 These nodules represent regions of more advanced neuronal differentiation. The large-cell and anaplastic subtypes have a greater percentage of poorly differentiated, anaplastic cellular regions. Medulloblastoma with extensive nodularity typically occurs in patients under 3 years of age, and shows the presence of lobular, grapelike nodules as a result of expansion of the reticulin-free zones.
Staging and Prognostic Factors
Careful pre- and postoperative staging is crucial in order to direct therapy and provide an estimate of patient prognosis. The staging system proposed by Chang et al. classified patient lesions based on two parameters: tumor and metastasis stage.107 Currently, high-risk patients are defined as children with more than 1.5 cm2 postoperative tumor residual, presenting at 3 years old or younger and/or the presence of metastases. All other patients are considered standard or average-risk.
One of the factors most consistently associated with prognosis is age at diagnosis.44,108 Poor outcomes in younger patients may be a reflection of differences in treatment patterns, such as the delay or avoidance of adjuvant radiotherapy. Absence of metastatic disease at diagnosis and maximal safe surgical resection have been associated with improved survival. With respect to histologic subtypes, medulloblastoma with extensive nodularity has a very good prognosis and the desmoplastic subtype may have a better prognosis.109–113 The large-cell and anaplastic subtypes demonstrate the worst prognosis.114,115
As for molecular markers, over-expression of TrkC (neurotrophin-3 receptor) and nuclear positivity for β-catenin (a marker of Wnt pathway activation) have been associated with a better prognosis.116–119 High expression of the oncogenes erbB2 and myc are associated with worse outcomes.120–122 Metastatic medulloblastoma has been associated with increased expression of genes involved with mitogen signaling including platelet-derived growth factor-alpha (PDFGR-α) and RAS/MAPK signaling components.123
Radiation Therapy
Craniospinal radiation (CSI) is an essential adjuvant treatment because medulloblastomas have a propensity for leptomeningeal spread. The standard postoperative CSI regimen is 36 Gy to the entire neuraxis, with a boost to the posterior fossa to a total dose of 54 Gy.124,125 In recent years, a risk-adapted therapeutic strategy has been employed due to adverse sequelae of full dose radiation. High-risk patients older than age 3 years typically receive the standard-dose CSI plus adjuvant chemotherapy. In average-risk patients, reduced-dose CSI has been examined. The average-risk patients receiving reduced-dose CSI without adjuvant chemotherapy demonstrated a worse outcome compared with conventional dose CSI.126–128 However, the reduced-dose CSI (23.4 Gy) in combined with adjuvant chemotherapy offered survival and disease control rates comparable to those achieved with standard dose CSI.129–131 In a phase III study, the 5-year overall survival and event-free survival rates were 86% and 81%, respectively.130 Another method being investigated for maximizing target radiation dose and reducing toxicity to adjacent normal brain includes the use of conformal boost radiotherapy limited to the tumor bed alone. The limited radiotherapy boost to the tumor bed produced disease control rates comparable to studies in which the entire posterior fossa is targeted.131–134 For average-risk patients, 5-year disease-free survival and posterior fossa control rate were 86% and 94%, respectively.134
CSI can be associated with long-term CNS toxicities. Acute postradiation effects occur in the first week and consist of temporary drowsiness, nausea, and headaches. Subacute effects occur after 6 to 10 weeks and are reversible lethargy and fatigue. Late changes include cognitive impairment, growth abnormalities, hypopituitarism, severe sensorineural hearing loss, moyamoya syndrome, and secondary neoplasms (gliomas, meningiomas).135
Chemotherapy
Several studies reported a survival benefit for average and high-risk patients treated with adjuvant chemotherapy following surgical resection and CSI.52,129,136,137 After CSI plus adjuvant chemotherapy, the 5-year, progression-free survival rate was 67% for patients with metastatic disease at the time of diagnosis.52 Neoadjuvant chemotherapy has received attention but showed no clear benefit over conventional therapeutic strategies.127,138–140 In the CCG921 trial, high-risk patients treated with postradiation chemotherapy had better 5-year progression-free survival (63%) compared with a neoadjuvant chemotherapy plus postradiation chemotherapy (45%).140 Neoadjuvant chemotherapy was also associated with subsequent radiotherapy-induced myelotoxicity, leading to higher rates of treatment interruptions.139 The strategy of high-dose, myeloablative chemotherapy followed by hematopoietic stem cell rescue has been tried as an option for recurrent disease, high-risk patients, disseminated disease, or children less than 36 months of age, in the avoidance of radiotherapy.136,141–145 Overall, treatment-related mortality rates between 10% to 20% are seen with this strategy. For children less than 3 years old without metastasis, the 5-year event-free and overall survival rates for patients with gross total resection and residual resection were 64% and 79%, and 29% and 57%, respectively.144 In average- and high-risk patients, 5-year overall survival rates of 85% and 70% have been reported following postoperative risk-adapted CSI followed by high-dose chemotherapy and stem cell rescue, respectively.136
Chemotherapy has been associated with a variety of adverse events such as fatigue, nausea, vomiting, loss of appetite, stomatitis, myelosuppression, and infection. Some studies have found greater hematologic toxicity when chemotherapy was combined with radiation therapy compared with radiotherapy alone.139 Less common side effects include nephrotoxicity, hepatotoxicity, cardiomyopathy, urinary bladder, sensorineural hearing loss, acute myelogenous leukemia, or pulmonary fibrosis depending on the agent used.
Follow-up and Recurrence
Long-term follow-up imaging has become the standard of care for medulloblastoma patients, with most receiving repeat brain and spine MRI every 3 to 6 months in the first 2 years following treatment.
Collin’s law states that tumor relapse should occur within a period equal to the patient’s age at diagnosis plus 9 months. This usually holds true for medulloblastoma in spite of several exceptions. The most common site of recurrent disease is the primary tumor site. However, up to 60% of patients will display evidence of disseminated disease at relapse.90,146 Metastasis outside the nervous system is uncommon.146 Bone lesions are the most common and lymph nodes are the second most common site. Early detection of recurrence offers the opportunity to test novel therapies in the setting of minimal disease burden. However, there is a controversy to the proven benefit because of the high percentage of dissemination at recurrence.
Ependymoma
Epidemiology and Genetics
Ependymomas account for 5% to 10% of pediatric CNS tumors and can be located throughout the CNS.147 They are the third most common brain tumors in children, following astrocytoma and medulloblastoma. More than 70% of ependymomas occur in the posterior fossa.148 The mean age at the time of diagnosis is between 3 and 6 years, with more than 25% found in children under the age of 3 years. Males are 1.4 times more likely to develop ependymomas than females are.
The genetic alterations observed are loss of heterozygosity in chromosome 22q, monosomy 17, and loss of a region on 6q.149–151 The 22q region is of special interest because it contains the neurofibromatosis type 2 (NF-2) tumor suppressor gene and an increased incidence of ependymomas is seen in NF-2. The intriguing finding of SV40-like DNA sequences raises the question of a possible viral role in tumor pathogenesis.152 Although ependymomas from the supratentorial space, posterior fossa, and spinal cord have very similar histology, they are biologically distinct diseases with their own transcriptional profiles and distinct sets of genetic abnormalities.153
Pathology
Ependymomas are glial neoplasms arising from ependymal cell layers adjacent to the ventricular system or central spinal canal.154 According to the WHO criteria, the two-tiered classification of ependymoma (ependymoma as grade II and anaplastic ependymoma as grade III) has been used. The cells are usually uniform, cuboidal to elongated with oval or round nuclei. The key pathologic feature is perivascular pseudorosettes, in which tumor cells are arranged radially around vessels (Fig. 55-9B). True ependymal rosettes may be present in a minority of cases. The histological differences between ependymoma and anaplastic ependymoma include the presence of nuclear atypia, marked mitotic activity, high cellularity, and often prominent vascular proliferation. Numerous studies concerning the correlation between histopathologic features and clinical outcome have yielded conflicting, inconsistent results.46,47,155,156
Prognostic Factors
Surgical resection is the best prognostic factor. A number of studies have shown that complete surgical resection offers the best hope of cure.47,48,157,158 Surgery and radiation therapy yield 5-year progression-free survival ranging from 60% to 87% after complete resection to 0% to 33% after incomplete surgical resection.47,148,159–165 Postoperative imaging is essential. In patients where postoperative imaging shows residual disease that is surgically accessible, there may be a role for early second look surgery or perhaps delayed surgery for resection of residual disease after chemotherapy and radiation. These tumors are relatively well-demarcated and distinct from adjacent brain parenchyma. However, depending on location, tumors arising from the roof of the fourth ventricle are the easiest to totally remove. Lateral tumors with a large cerebellopontine angle component are the most difficult to totally remove as they are often adherent to cranial nerves and vascular structures.166 Morbidity remains high secondary to brain stem and cranial nerve injury. The complete removal rates of the mid-floor type (origin of fourth ventricular floor), lateral type (extension to cerebellopontine angle), and roof type (origin of fourth ventricular roof) tumors are approximately 23%, 0%, and 100%, respectively.167
The incidence of dissemination of ependymoma is only 11% to 17%.47,48 Nonetheless, it is important to demonstrate its presence or absence, because disseminated disease is a strong adverse prognostic factor. Perioperative disease staging with craniospinal MRI and CSF cytology is recommended. Controversial results have been published regarding the efficacy of age, histologic composition, radiotherapy, and chemotherapy on progression-free survival.147,165,168
Treatment
Intracranial ependymomas are relatively radiosensitive and chemoresistant. This further supports the need to completely extirpate primary ependymoma when feasible. Radiation therapy is the adjuvant therapy of choice. It is generally recommended that even after a gross total tumor resection, patients with localized disease undergo local tumor bed irradiation. The dose of radiation for the treatment of ependymoma has traditionally been in the range of 4500 to 5600 cGy. Craniospinal irradiation (CSI) is indicated for patients with disseminated disease and anaplastic histology does not appear to be an indication for CSI because most of subtypes are prone to local treatment failure. There was no difference in the distant failure rate for those receiving CSI versus posterior fossa only, for ependymoma (5%) versus anaplastic ependymoma (8.7%).156,169–171 With the advent of stereotactic conformal radiotherapy, treatment can be highly focused on the tumor bed while sparing adjacent normal brain or brain stem in order to minimize radiation-associated toxicity.172
Adjuvant chemotherapy combined with radiation therapy has not yielded a significant improvement in survival for pediatric infratentorial ependymoma.170,173,174 However, chemotherapy is efficacious to defer irradiation at the time of relapse and provide improved survivals in very young patients.161,175–178 High-dose chemotherapy followed by autologous bone marrow transplantation has been tried for recurrent disease, but ependymoma does not appear to be as sensitive as medulloblastoma.179,180 These data are not sufficient to justify the use of chemotherapy except in younger children to substitute for or delay radiation therapy, or patients in whom surgery and radiation therapy have failed to control tumor growth.
Tumors recur predominantly at the primary tumor site, suggesting that they arise from residual ependymoma cells.46,160,162,181,182 Most instances of spinal metastases follow failure in the posterior fossa. It is very unusual to see isolated recurrence in the spine. After recurrence, repeated surgeries can be performed. Because the primary problem with ependymomas is local tumor control, brachytherapy with 125iodine and radiosurgery have been reported to be an option in patients with recurrent disease. In current treatment paradigms, brachytherapy has been replaced with radiosurgery.165,170,183
Cerebellar Astrocytoma
Epidemiology
The cerebellar astrocytoma represents over 10% of pediatric brain tumors and over 25% of posterior fossa tumors, making it one of the most common intracranial neoplasms of childhood. Over 70% of cerebellar astrocytomas are seen in children.184 A mean age at presentation of 6 to 8 years is found, and these tumors are rarely found in children under 1 year of age or in adults over the age of 40.9 There is no gender predilection.
Pathology
Most cerebellar astrocytomas are low grade neoplasms, especially in childhood. These are divided into two pathologic entities. Pilocytic astrocytomas are WHO grade I lesions and account for 65% to 85% of cerebellar astrocytoma.9,27,28 Diffuse astrocytomas are considered as WHO grade II and account for 15% to 35%. Pilocytic astrocytomas exhibit a biphasic appearance, with compact areas composed mainly of bipolar cells with hair-like projections alternating with areas of loosely aggregated astrocytes containing stellate astrocytes and microcysts (Fig. 55-9C). In this pathology, the presence of endothelial proliferation does not imply a higher grade. Diffuse astrocytomas are composed of fibrillary neoplastic astrocytes on the background of loosely structured tumor matrix. Malignant tumors account for less than 5% of all pediatric cerebellar astrocytomas.185
Prognostic Factors
Long-term prognosis is dependent on the extent of resection, presence of brain stem invasion and histological features of malignancy.9,27,28,185–188 Total resection is associated with considerably better outcome than is subtotal or partial resection. Total resection overall carries a 5-year survival of 90%, compared to subtotal resection with 48.5%.27 There is no difference between diffuse and pilocytic astrocytoma in outcome for totally resected tumors. The diffuse type has been considered more prone to recurrence but the histologic type is not as definitive a predictor of recurrence as is extent of removal. Brain stem involvement is a significant adverse prognostic factor for cerebellar astrocytoma and the poor prognosis of solid tumors is related with the increased likelihood of brain stem involvement. There is a tendency for some tumors to invade the subarachnoid space and grow along the surface of the cerebellum, but this is not ominous and does not usually portend a negative prognosis. However, patients with malignant pathology will have poor clinical courses.
Treatment
The surgical goal is gross total resection of all contrast-enhancing tumor tissue, which virtually guarantees the patient a cure. Total tumor resection is limited by attachment or invasion of the tumor into the brain stem and both pilocytic and diffuse astrocytomas can invade the brain stem. Tumors of the cyst-mural nodule form are more often amenable to total gross removal because of their discrete margins and lateral placement. Tumors of the solid form may also be clearly demarcated from the surrounding cerebellum. However, with or without visible cysts, these may show an increased involvement of the brain stem, which in turn may make complete neurosurgical resection more difficult to achieve.27,32,189
All patients should have postoperative brain imaging with and without contrast enhancement to evaluate the extent of resection. The postoperative imaging is more accurate than the surgeon’s assessment in determining whether a total removal has been accomplished.28 If surgically accessible, bulky disease is seen on the postoperative images, early second-look surgery and resection of tumor may be advisable in some cases. If there is unexpected postoperative enhancement (with small residual tumor) in the patients in whom a total resection was thought to have been accomplished, the neurosurgeon can safely follow these patients with serial imaging studies rather than pursue a second operation. The growth potential of these tumors is low and the dedifferentiation to a more malignant type is rare.
There is no role for adjuvant radiation or chemotherapy once a cerebellar astrocytoma has been completely removed with confirmation by postoperative imaging. The value of postoperative radiotherapy as initial adjuvant treatment following subtotal removal has been debated and does not significantly improve survival.27,190 However, it may be useful for the treatment of progression following a subtotal surgical excision where there is obvious infiltration of the brain stem or cerebellar peduncles.191 These tumors may violate Collins’ law by recurring late.192
Atypical Teratoid/Rhabdoid Tumors
Epidemiology and Genetics
AT/RTs of the CNS in infants and children are unique histologic entities with an extremely aggressive natural history. AT/RTs form 1% to 2% of all brain tumors in the pediatric population.38,39,49,193,194 Two thirds of AT/RTs will take origin in the posterior fossa. Three quarters of patients are age 3 years or younger at the time of diagnosis and the median age at diagnosis ranges from 16 to 32 months. There is a 3:2 male predominance.
Monosomy 22 is the principal cytogenetic abnormality, and was initially demonstrated in 1990.195 Subsequently, truncating mutations of hSNF5/INI1 on chromosome 22q11 were found in a series of cell lines derived from renal rhabdoid tumors. Somatic mutations of hSNF5/INI1 were then identified in a series of CNS AT/RT.196,197 These studies suggest that mutations in the tumor suppressor gene INI1 predispose children to the development of AT/RTs.
Pathology
Histologically, AT/RTs resemble the more common and less aggressive primitive neuroectodermal tumor/medulloblastoma (PNET/MB) with which they have been misdiagnosed in the past. It is important to distinguish AT/RTs from PNET/MBs because AT/RTs are aggressive and usually fatal malignancies, whereas PNET/MBs have well-defined treatment protocols leading to decent 5-year survival rates.
The microscopic characteristics of AT/RT may be variable, but they almost always contain visible rhabdoid cells. The typical rhabdoid cells have an eccentric round nucleus with a prominent nucleolus and a plump cell body (Fig. 55-9D). Careful study of these tumors disclosed fields of rhabdoid cells with or without areas of primitive neuroepithelial cells, which are composed entirely (13%) or partly (77%) of rhabdoid cells.38 In a quarter to a third of tumors, mesenchymal and/or epithelial elements are seen as well. Mitotic figures are typically abundant, and necrosis and hemorrhage are also common. In immunohistochemical studies, there are three antibodies whose epitopes are almost always expressed: epithelial membrane antigen (EMA), vimentin, and smooth-muscle actin. Today, immunohistochemistry for INI1 protein typically shows absence of expression in AT/RTs, but not in PNET/MBs.
Treatment
The initial treatment for most children with AT/RT is maximal safe neurosurgical resection of tumor. The interface of the AT/RT and cerebellum may be infiltrative and ill-defined, and tumors primarily in the cerebello-pontine angle may incorporate cranial nerve roots in the vicinity.39 Total or near total resection of the tumor is feasible in about 30% of patients.38 Postoperative cranio-spinal imaging is essential. Spinal imaging at initial presentation reveals evidence of disease in 25% to 34% patients.38,39,49 Patterns of relapse show local disease alone in 31%, leptomeningeal dissemination alone in 11% and both in 58%.38 The prognosis for children with AT/RT remains poor. The median time to progression is 4.5 months and the median reported survivals range from 6 to 11 months.38,39,49 The few patients who underwent surgery with our without radiation therapy but had no chemotherapy experienced a median survival of 2.5 months.
As children with AT/RT frequently present with leptomeningeal dissemination, or develop it at the time of relapse, it is desirable to administer CSI after primary tumor resection for children greater than age 3 years and chemotherapy. While radiation therapy does not seem to alter the progression of disease in children with AT/RT, most children will receive radiation at some point in the course of their disease.38,39 Patients with AT/RT respond poorly to chemotherapy with or without radiation therapy, and only 6 in 36 children had a greater than 50% reduction in tumor mass. The longest lasting response to chemotherapy was 10 months.38 However, there are a few reports with prolonged survival after intensified chemotherapy. One study showed that after intensive therapy including intrathecal chemotherapy, two of three children were alive without evidence of disease 36 and 89 months after diagnosis.194 Three of four children in another study treated with high dose chemotherapy with stem cell rescue survived more than 12 months and one is alive with no evidence of disease 46 months from diagnosis.49
Abdollahzadeh M., Hoffman H.J., Blazer S.I., et al. Benign cerebellar astrocytoma in childhood: experience at the Hospital for Sick Children 1980-1992. Childs Nerv Syst. 1994;10:380-383.
Culley D.J., Berger M.S., Shaw D., et al. An analysis of factors determining the need for ventriculoperitoneal shunts after posterior fossa tumor surgery in children. Neurosurgery. 1994;34:402-407.
David K.M., Casey A.T., Hayward R.D., et al. Medulloblastoma: is the 5-year survival rate improving? A review of 80 cases from a single institution. J Neurosurg. 1997;86:13-21.
Hayostek C.J., Shaw E.G., Scheithauer B., et al. Astrocytomas of the cerebellum: a comparative clinicopathologic study of pilocytic and diffuse astrocytomas. Cancer. 1993;72:856-869.
Koral K., Gargan L., Bowers D.C., et al. Imaging characteristics of atypical teratoid-rhabdoid tumor in children compared with medulloblastoma. AJR Am J Roentgenol. 2008;190:809-814.
Lee Y.Y., Van Tassel P., Bruner J.M., et al. Juvenile pilocytic astrocytomas: CT and MR characteristics. AJR Am J Roentgenol. 1989;152:1263-1270.
Louis D.N., Ohgaki H., Wiestler O.D., Cavenee W.K. World Health Organization Classification of Tumours of the Central Nervous System. Lyon: IARC; 2007.
Meyers S.P., Kemp S.S., Tarr R.W. MR imaging features of medulloblastomas. AJR Am J Roentgenol. 1992;158:859-865.
Meyers S.P., Wildenhain S.L., Chang J.K., et al. Postoperative evaluation for disseminated medulloblastoma involving the spine: contrast-enhanced MR findings, CSF cytologic analysis, timing of disease occurrence, and patient outcomes. AJNR Am J Neuroradiol. 2000;21:1757-1765.
Miller J.P., Cohen A.R. Surgical management of tumors of the fourth ventricle. In: Schmidek H.H., Roberts D.W. Schmidek and Sweet operative neurosurgical techniques: indications, methods, and results. 5th ed. Philadelphia: Elsevier; 2006:881-909.
Mussi A.C., Rhoton A.L.Jr. Telovelar approach to the fourth ventricle: microsurgical anatomy. J Neurosurg. 2000;92:812-823.
Nazar G.B., Hoffman H.J., Becker J.E., et al. Infratentorial ependymomas in childhood: prognostic factors and treatment. J Neurosurg. 1990;72:408-417.
Nejat F., El Khashab M., Rutka J.T. Initial management of childhood brain tumors: neurosurgical considerations. J Child Neurol. 2008;23:1136-1148.
Park T.S., Hoffman H.J., Hendrick E.B., et al. Medulloblastoma: clinical presentation and management. Experience at the hospital for sick children, Toronto, 1950-1980. J Neurosurg. 1983;58:543-552.
Rorke L.B., Packer R.J., Biegel J.A. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. 1996;85:56-65.
Rutka J.T. Medulloblastoma. Clin Neurosurg. 1997;44:571-585.
Rutka J.T., Kuo J.S. Pediatric surgical neuro-oncology: current best care practices and strategies. J Neurooncol. 2004;69:139-150.
Rutka J.T., Kuo J.S., Carter M., et al. Advances in the treatment of pediatric brain tumors. Expert Rev Neurother. 2004;4:879-893.
Sainte-Rose C., Cinalli G., Roux F.E., et al. Management of hydrocephalus in pediatric patients with posterior fossa tumors: the role of endoscopic third ventriculostomy. J Neurosurg. 2001;95:791-797.
Schneider J.H.Jr., Raffel C., McComb J.G. Benign cerebellar astrocytomas of childhood. Neurosurgery. 1992;30:58-62.
Shemie S., Jay V., Rutka J., et al. Acute obstructive hydrocephalus and sudden death in children. Ann Emerg Med. 1997;29:524-528.
Tanriover N., Ulm A.J., Rhoton A.L.Jr., et al. Comparison of the transvermian and telovelar approaches to the fourth ventricle. J Neurosurg. 2004;101:484-498.
Taylor M.D., Mainprize T.G., Rutka J.T. Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery. 2000;47:888-901.
Tortori-Donati P., Fondelli M.P., Cama A., et al. Ependymomas of the posterior cranial fossa: CT and MRI findings. Neuroradiology. 1995;37:238-243.
Undjian S., Marinov M., Georgiev K. Long-term follow-up after surgical treatment of cerebellar astrocytomas in 100 children. Childs Nerv Syst. 1989;5:99-101.
1. Ross J.A., Severson R.K., Pollock B.H., et al. Childhood cancer in the United States. A geographical analysis of cases from the Pediatric Cooperative Clinical Trials groups. Cancer. 1996;77:201-207.
2. Ullrich N.J., Pomeroy S.L. Pediatric brain tumors. Neurol Clin. 2003;21:897-913.
3. Newman L.A., Boop F.A., Sanford R.A., et al. Postoperative swallowing function after posterior fossa tumor resection in pediatric patients. Childs Nerv Syst. 2006;22:1296-1300.
4. Nejat F., El Khashab M., Rutka J.T. Initial management of childhood brain tumors: neurosurgical considerations. J Child Neurol. 2008;23:1136-1148.
5. Rutka J.T., Kuo J.S., Carter M., et al. Advances in the treatment of pediatric brain tumors. Expert Rev Neurother. 2004;4:879-893.
6. Rutka J.T., Kuo J.S. Pediatric surgical neuro-oncology: current best care practices and strategies. J Neurooncol. 2004;69:139-150.
7. Park T.S., Hoffman H.J., Hendrick E.B., et al. Medulloblastoma: clinical presentation and management. Experience at the hospital for sick children, Toronto, 1950-1980. J Neurosurg. 1983;58:543-552.
8. Miller J.P., Cohen A.R. Surgical management of tumors of the fourth ventricle. In: Schmidek H.H., Roberts D.W. Schmidek and Sweet operative neurosurgical techniques: indications, methods, and results. 5th ed. Philadelphia: Elsevier; 2006:881-909.
9. Abdollahzadeh M., Hoffman H.J., Blazer S.I., et al. Benign cerebellar astrocytoma in childhood: experience at the Hospital for Sick Children 1980-1992. Childs Nerv Syst. 1994;10:380-383.
10. Shemie S., Jay V., Rutka J., et al. Acute obstructive hydrocephalus and sudden death in children. Ann Emerg Med. 1997;29:524-528.
11. Byard R.W., Bourne A.J., Hanieh A. Sudden and unexpected death due to hemorrhage from occult central nervous system lesions. A pediatric autopsy study. Pediatr Neurosurg. 1991-1992;17:88-94.
12. Yokota A., Kajiwara H., Matsuoka S., et al. Subarachonid hemorrhage from brain tumors in childhood. Childs Nerv Syst. 1987;3:65-69.
13. Laurent J.P., Bruce D.A., Schut L. Hemorrhagic brain tumors in pediatric patients. Childs Brain. 1981;8:263-270.
14. Kayama T., Yoshimoto T., Shimizu H., et al. Neonatal medulloblastoma. J Neurooncol. 1993;15:157-163.
15. Modha A., Vassilyadi M., George A., et al. Medulloblastoma in children—the Ottawa experience. Childs Nerv Syst. 2000;16:341-350.
16. Packer R.J., Cogen P., Vezina G., et al. Medulloblastoma: clinical and biologic aspects. Neuro Oncol. 1999;1:232-250.
17. Turgut M., Akalan N., Bertan V., et al. Acquired torticollis as the only presenting symptom in children with posterior fossa tumors. Childs Nerv Syst. 1995;11:86-88.
18. Pezeshkpour G.H., Henry J.M., Armbrustmacher V.W. Spinal metastases. A rare mode of presentation of brain tumors. Cancer. 1984;54:353-356.
19. Kumar C., Panagapoulos K., Kalbag R.M., et al. Cerebellar astrocytoma presenting as a syringomyelic syndrome. Surg Neurol. 1987;27:187-190.
20. Roberts R.O., Lynch C.F., Jones M.P., et al. Medulloblastoma: a population-based study of 532 cases. J Neuropathol Exp Neurol. 1991;50:134-144.
21. Maleci A., Cervoni L., Delfini R. Medulloblastoma in children and in adults: a comparative study. Acta Neurochir (Wien). 1992;119:62-67.
22. Vezina L.G., Packer R.J. Infratentorial brain tumors of childhood. Neuroimaging Clin North Am. 1994;4:423-436.
23. Meyers S.P., Kemp S.S., Tarr R.W. MR imaging features of medulloblastomas. AJR Am J Roentgenol. 1992;158:859-865.
24. Koral K., Gargan L., Bowers D.C., et al. Imaging characteristics of atypical teratoid-rhabdoid tumor in children compared with medulloblastoma. AJR Am J Roentgenol. 2008;190:809-814.
25. Naidich T.P., Zimmerman R.A. Primary brain tumors in children. Semin Roentgenol. 1984;19:100-114.
26. Tortori-Donati P., Fondelli M.P., Cama A., et al. Ependymomas of the posterior cranial fossa: CT and MRI findings. Neuroradiology. 1995;37:238-243.
27. Sgouros S., Fineron P.W., Hockley A.D. Cerebellar astrocytoma of childhood: long-term follow-up. Childs Nerv Syst. 1995;11:89-96.
28. Schneider J.H.Jr., Raffel C., McComb J.G. Benign cerebellar astrocytomas of childhood. Neurosurgery. 1992;30:58-62.
29. Geissinger J.D. Astrocytomas of the cerebellum in children. Long-term study. Arch Neurol. 1971;24:125-135.
30. Ilgren E.B., Stiller C.A. Cerebellar astrocytomas. Part I. Macroscopic and microscopic features. Clin Neuropathol. 1987;6:185-200.
31. Chang T., Teng M.M., Lirng J.F. Posterior cranial fossa tumours in childhood. Neuroradiology. 1993;35:274-278.
32. Undjian S., Marinov M., Georgiev K. Long-term follow-up after surgical treatment of cerebellar astrocytomas in 100 children. Childs Nerv Syst. 1989;5:99-101.
33. Kuroiwa T., Numaguchi Y., Rothman M.I., et al. Posterior fossa glioblastoma multiforme: MR findings. AJNR Am J Neuroradiol. 1995;16:583-589.
34. Lee Y.Y., Van Tassel P., Bruner J.M., et al. Juvenile pilocytic astrocytomas: CT and MR characteristics. AJR Am J Roentgenol. 1989;152:1263-1270.
35. Zimmerman R.A., Bilaniuk L.T., Rebsamen S. Magnetic resonance imaging of pediatric posterior fossa tumors. Pediatr Neurosurg. 1992;18:58-64.
36. Arslanoglu A., Aygun N., Tekhtani D., et al. Imaging findings of CNS atypical teratoid/rhabdoid tumors. AJNR Am J Neuroradiol. 2004;25:476-480.
37. Lee Y.K., Choi C.G., Lee J.H. Atypical teratoid/rhabdoid tumor of the cerebellum: report of two infantile cases. AJNR Am J Neuroradiol. 2004;25:481-483.
38. Rorke L.B., Packer R.J., Biegel J.A. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. 1996;85:56-65.
39. Burger P.C., Yu I.T., Tihan T., et al. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol. 1998;22:1083-1092.
40. Fenton L.Z., Foreman N.K. Atypical teratoid/rhabdoid tumor of the central nervous system in children: an atypical series and review. Pediatr Radiol. 2003;33:554-558.
41. Yoon C.S., Chuang S., Jay V. Primary malignant rhabdoid tumor of the brain: CT and MR findings. Yonsei Med J. 2000;41:8-16.
42. Gajjar A., Sanford R.A., Bhargava R., et al. Medulloblastoma with brain stem involvement: the impact of gross total resection on outcome. Pediatr Neurosurg. 1996;25:182-187.
43. Hayostek C.J., Shaw E.G., Scheithauer B., et al. Astrocytomas of the cerebellum: a comparative clinicopathologic study of pilocytic and diffuse astrocytomas. Cancer. 1993;72:856-869.
44. David K.M., Casey A.T., Hayward R.D., et al. Medulloblastoma: is the 5-year survival rate improving? A review of 80 cases from a single institution. J Neurosurg. 1997;86:13-21.
45. Bartels U., Shroff M., Sung L., et al. Role of spinal MRI in the follow-up of children treated for medulloblastoma. Cancer. 2006;107:1340-1347.
46. Nazar G.B., Hoffman H.J., Becker J.E., et al. Infratentorial ependymomas in childhood: prognostic factors and treatment. J Neurosurg. 1990;72:408-417.
47. Sutton L.N., Goldwein J., Perilongo G., et al. Prognostic factors in childhood ependymomas. Pediatr Neurosurg. 1990-1991;16:57-65.
48. Rezai A.R., Woo H.H., Lee M., et al. Disseminated ependymomas of the central nervous system. J Neurosurg. 1996;85:618-624.
49. Hilden J.M., Watterson J., Longee D.C., et al. Central nervous system atypical teratoid tumor/rhabdoid tumor: response to intensive therapy and review of the literature. J Neurooncol. 1998;40:265-275.
50. Shaw D.W., Weinberger E., Brewer D.K., et al. Spinal subdural enhancement after suboccipital craniectomy. AJNR Am J Neuroradiol. 1996;17:1373-1377.
51. Wiener M.D., Boyko O.B., Friedman H.S., et al. False-positive spinal MR findings for subarachnoid spread of primary CNS tumor in postoperative pediatric patients. AJNR Am J Neuroradiol. 1990;11:1100-1103.
52. Packer R.J., Sutton L.N., Elterman R., et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg. 1994;81:690-698.
53. Meyers S.P., Wildenhain S.L., Chang J.K., et al. Postoperative evaluation for disseminated medulloblastoma involving the spine: contrast-enhanced MR findings, CSF cytologic analysis, timing of disease occurrence, and patient outcomes. AJNR Am J Neuroradiol. 2000;21:1757-1765.
54. Balhuizen J.C., Bots G.T., Schaberg A., et al. Value of cerebrospinal fluid cytology for the diagnosis of malignancies in the central nervous system. J Neurosurg. 1978;48:747-753.
55. Fouladi M., Gajjar A., Boyett J.M., et al. Comparison of CSF cytology and spinal magnetic resonance imaging in the detection of leptomeningeal disease in pediatric medulloblastoma or primitive neuroectodermal tumors. J Clin Oncol. 1999;17:3234-3237.
56. Due-Tonnessen B.J., Helseth E. Management of hydrocephalus in children with posterior fossa tumors: role of tumor surgery. Pediatr Neurosurg. 2007;43:92-96.
57. Fritsch M.J., Doerner L., Kienke S., et al. Hydrocephalus in children with posterior fossa tumors: role of endoscopic third ventriculostomy. J Neurosurg. 2005;103:40-42.
58. Hekmatpanah J., Mullan S. Ventriculo-caval shunt in the management of posterior fossa tumors. J Neurosurg. 1967;26:609-613.
59. Abraham J., Chandy J. Ventriculo-atrial shunt in the management of posterior-fossa tumours: preliminary report. J Neurosurg. 1963;20:252-253.
60. Kombogiorgas D., Natarajan K., Sgouros S. Predictive value of preoperative ventricular volume on the need for permanent hydrocephalus treatment immediately after resection of posterior fossa medulloblastomas in children. J Neurosurg Pediatr. 2008;1:451-455.
61. McLaurin R.L. Disadvantages of the preoperative shunt in posterior fossa tumors. Clin Neurosurg. 1983;30:286-292.
62. Rappaport Z.H., Shalit M.N. Perioperative external ventricular drainage in obstructive hydrocephalus secondary to infratentorial brain tumours. Acta Neurochir (Wien). 1989;96:118-121.
63. Shalit M.N., Ben Ari Y., Eynan N. The management of obstructive hydrocephalus by the use of external continuous ventricular drainage. Acta Neurochir (Wien). 1979;47:161-172.
64. Culley D.J., Berger M.S., Shaw D., et al. An analysis of factors determining the need for ventriculoperitoneal shunts after posterior fossa tumor surgery in children. Neurosurgery. 1994;34:402-407.
65. Lee M., Wisoff J.H., Abbott R., et al. Management of hydrocephalus in children with medulloblastoma: prognostic factors for shunting. Pediatr Neurosurg. 1994;20:240-247.
66. Kombogiorgas D., Sgouros S., Walsh A.R., et al. Outcome of children with posterior fossa medulloblastoma: a single institution experience over the decade 1994-2003. Childs Nerv Syst. 2007;23:399-405.
67. Sainte-Rose C., Cinalli G., Roux F.E., et al. Management of hydrocephalus in pediatric patients with posterior fossa tumors: the role of endoscopic third ventriculostomy. J Neurosurg. 2001;95:791-797.
68. Morelli D., Pirotte B., Lubansu A., et al. Persistent hydrocephalus after early surgical management of posterior fossa tumors in children: is routine preoperative endoscopic third ventriculostomy justified? J Neurosurg. 2005;103:247-252.
69. Steinbok P., Boyd M., Cochrane D. Cervical spine deformity following craniotomy and upper cervical laminectomy for posterior fossa tumors in children. Childs Nerv Syst. 1989;5:25-28.
70. Matsushima T., Fukui M., Inoue T., et al. Microsurgical and magnetic resonance imaging anatomy of the cerebello-medullary fissure and its application during fourth ventricle surgery. Neurosurgery. 1992;30:325-330.
71. Dailey A.T., McKhann G.M.2nd, Berger M.S. The pathophysiology of oral pharyngeal apraxia and mutism following posterior fossa tumor resection in children. J Neurosurg. 1995;83:467-475.
72. Matsushima T., Inoue T., Inamura T., et al. Transcerebellomedullary fissure approach with special reference to methods of dissecting the fissure. J Neurosurg. 2001;94:257-264.
73. Mussi A.C., Rhoton A.L.Jr. Telovelar approach to the fourth ventricle: microsurgical anatomy. J Neurosurg. 2000;92:812-823.
74. Rhoton A.L.Jr. Cerebellum and fourth ventricle. Neurosurgery. 2000;47(suppl 3):S7-27.
75. Tanriover N., Ulm A.J., Rhoton A.L.Jr., et al. Comparison of the transvermian and telovelar approaches to the fourth ventricle. J Neurosurg. 2004;101:484-498.
76. Deshmukh V.R., Figueiredo E.G., Deshmukh P., et al. Quantification and comparison of telovelar and transvermian approaches to the fourth ventricle. Neurosurgery. 2006;58(4 suppl 2):ONS202-ONS206.
77. El-Bahy K. Telovelar approach to the fourth ventricle: operative findings and results in 16 cases. Acta Neurochir (Wien). 2005;147:137-142.
78. Rajesh B.J., Rao B.R., Menon G., et al. Telovelar approach: technical issues for large fourth ventricle tumors. Childs Nerv Syst. 2007;23:555-558.
79. Peterson D.O., Drummond J.C., Todd M.M. Effects of halothane, enflurane, isoflurane, and nitrous oxide on somatosensory evoked potentials in humans. Anesthesiology. 1986;65:35-40.
80. Jellinek D., Jewkes D., Symon L. Noninvasive intraoperative monitoring of motor evoked potentials under propofol anesthesia: effects of spinal surgery on the amplitude and latency of motor evoked potentials. Neurosurgery. 1991;28:551-557.
81. Taniguchi M., Nadstawek J., Pechstein U., et al. Total intravenous anesthesia for improvement of intraoperative monitoring of somatosensory evoked potentials during aneurysm surgery. Neurosurgery. 1992;30:891-897.
82. Tomita T. Surgical management of cerebellar peduncle lesions in children. Neurosurgery. 1986;18:568-575.
83. Pollack I.F., Polinko P., Albright A.L., et al. Mutism and pseudobulbar symptoms after resection of posterior fossa tumors in children: incidence and pathophysiology. Neurosurgery. 1995;37:885-893.
84. Robertson P.L., Muraszko K.M., Holmes E.J., et al. Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma: a prospective study by the Children’s Oncology Group. J Neurosurg. 2006;105:444-451.
85. Mukand J.A., Blackinton D.D., Crincoli M.G., et al. Incidence of neurologic deficits and rehabilitation of patients with brain tumors. Am J Phys Med Rehabil. 2001;80:346-350.
86. Shiminski-Maher T. Brainstem tumors in childhood: preparing patients and families for long- and short-term care. Pediatr Neurosurg. 1996;24:267-271.
87. Parizek J., Sercl M., Michl A., et al. Posterior fossa duraplasty in children: remarks on surgery and clinical and CT follow-up. Childs Nerv Syst. 1994;10:444-449.
88. Steinbok P., Singhal A., Mills J., et al. Cerebrospinal fluid (CSF) leak and pseudomeningocele formation after posterior fossa tumor resection in children: a retrospective analysis. Childs Nerv Syst. 2007;23:171-174.
89. Rutka J.T. Medulloblastoma. Clin Neurosurg. 1997;44:571-585.
90. Belza M.G., Donaldson S.S., Steinberg G.K., et al. Medulloblastoma: freedom from relapse longer than 8 years—a therapeutic cure? J Neurosurg. 1991;75:575-582.
91. Agerlin N., Gjerris F., Brincker H., et al. Childhood medulloblastoma in Denmark 1960-1984. A population-based retrospective study. Childs Nerv Syst. 1999;15:29-36.
92. Gurney J.G., Kadan-Lottick N. Brain and other central nervous system tumors: rates, trends, and epidemiology. Curr Opin Oncol. 2001;13:160-166.
93. Ang C., Hauerstock D., Guiot M.C., et al. Characteristics and outcomes of medulloblastoma in adults. Pediatr Blood Cancer. 2008;51:603-607.
94. Taylor M.D., Mainprize T.G., Rutka J.T. Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery. 2000;47:888-901.
95. Bayani J., Zielenska M., Marrano P., et al. Molecular cytogenetic analysis of medulloblastomas and supratentorial primitive neuroectodermal tumors by using conventional banding, comparative genomic hybridization, and spectral karyotyping. J Neurosurg. 2000;93:437-448.
96. Tong C.Y., Hui A.B., Yin X.L., et al. Detection of oncogene amplifications in medulloblastomas by comparative genomic hybridization and array-based comparative genomic hybridization. J Neurosurg. 2004;100:187-193.
97. Kool M., Koster J., Bunt J., et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3:e3088.
98. Clifford S.C., Lusher M.E., Lindsey J.C., et al. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle. 2006;5:2666-2670.
99. Thompson M.C., Fuller C., Hogg T.L., et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924-1931.
100. Taipale J., Chen J.K., Cooper M.K., et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005-1009.
101. Romer J.T., Kimura H., Magdaleno S., et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/-) p53(-/-) mice. Cancer Cell. 2004;6:229-240.
102. Romer J., Curran T. Targeting medulloblastoma: small-molecule inhibitors of the Sonic Hedgehog pathway as potential cancer therapeutics. Cancer Res. 2005;65:4975-4978.
103. Bigner S.H., McLendon R.E., Fuchs H., et al. Chromosomal characteristics of childhood brain tumors. Cancer Genet Cytogenet. 1997;97:125-134.
104. Louis D.N., Ohgaki H., Wiestler O.D., Cavenee W.K. World Health Organization Classification of Tumours of the Central Nervous System. Lyon: IARC; 2007.
105. Packer R.J., Sutton L.N., Rorke L.B., et al. Prognostic importance of cellular differentiation in medulloblastoma of childhood. J Neurosurg. 1984;61:296-301.
106. McManamy C.S., Pears J., Weston C.L., et al. Nodule formation and desmoplasia in medulloblastomas-defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol. 2007;17:151-164.
107. Chang C.H., Housepian E.M., Herbert C.Jr. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology. 1969;93:1351-1359.
108. Tomita T., McLone D.G. Medulloblastoma in childhood: results of radical resection and low-dose neuraxis radiation therapy. J Neurosurg. 1986;64:238-242.
109. Giangaspero F., Perilongo G., Fondelli M.P., et al. Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg. 1999;91:971-977.
110. Suresh T.N., Santosh V., Yasha T.C., et al. Medulloblastoma with extensive nodularity: a variant occurring in the very young-clinicopathological and immunohistochemical study of four cases. Childs Nerv Syst. 2004;20:55-60.
111. Chatty E.M., Earle K.M. Medulloblastoma. A report of 201 cases with emphasis on the relationship of histologic variants to survival. Cancer. 1971;28:977-983.
112. Eberhart C.G., Kepner J.L., Goldthwaite P.T., et al. Histopathologic grading of medulloblastomas: a Pediatric Oncology Group study. Cancer. 2002;94:552-560.
113. Garton G.R., Schomberg P.J., Scheithauer B.W., et al. Medulloblastoma—prognostic factors and outcome of treatment: review of the Mayo Clinic experience. Mayo Clin Proc. 1990;65:1077-1086.
114. Giangaspero F., Rigobello L., Badiali M., et al. Large-cell medulloblastomas. A distinct variant with highly aggressive behavior. Am J Surg Pathol. 1992;16:687-693.
115. Lamont J.M., McManamy C.S., Pearson A.D., et al. Combined histopathological and molecular cytogenetic stratification of medulloblastoma patients. Clin Cancer Res. 2004;10:5482-5493.
116. Segal R.A., Goumnerova L.C., Kwon Y.K., et al. Expression of the neurotrophin receptor TrkC is linked to a favorable outcome in medulloblastoma. Proc Natl Acad Sci USA. 1994;91:12867-12871.
117. Kim J.Y., Sutton M.E., Lu D.J., et al. Activation of neurotrophin-3 receptor TrkC induces apoptosis in medulloblastomas. Cancer Res. 1999;59:711-719.
118. Grotzer M.A., Janss A.J., Fung K., et al. TrkC expression predicts good clinical outcome in primitive neuroectodermal brain tumors. J Clin Oncol. 2000;18:1027-1035.
119. Ellison D.W., Onilude O.E., Lindsey J.C., et al. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005;23:7951-7957.
120. Aldosari N., Bigner S.H., Burger P.C., et al. MYCC and MYCN oncogene amplification in medulloblastoma. A fluorescence in situ hybridization study on paraffin sections from the Children’s Oncology Group. Arch Path Lab Med. 2002;126:540-544.
121. Gilbertson R.J., Pearson A.D., Perry R.H., et al. Prognostic significance of the c-erbB-2 oncogene product in childhood medulloblastoma. Br J Cancer. 1995;71:473-477.
122. Grotzer M.A., Hogarty M.D., Janss A.J., et al. MYC messenger RNA expression predicts survival outcome in childhood primitive neuroectodermal tumor/medulloblastoma. Clin Cancer Res. 2001;7:2425-2433.
123. MacDonald T.J., Brown K.M., LaFleur B., et al. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat Genet. 2001;29:143-152.
124. Paterson E., Farr R.F. Cerebellar medulloblastoma: treatment by irradiation of the whole central nervous system. Acta Radiol. 1953;39:323-336.
125. Bloom H.J., Wallace E.N., Henk J.M. The treatment and prognosis of medulloblastoma in children. A study of 82 verified cases. Am J Roentgenol Radium Ther Nucl Med. 1969;105:43-62.
126. Deutsch M., Thomas P.R., Krischer J., et al. Results of a prospective randomized trial comparing standard dose neuraxis irradiation (3,600 cGy/20) with reduced neuraxis irradiation (2,340 cGy/13) in patients with low-stage medulloblastoma. A Combined Children’s Cancer Group-Pediatric Oncology Group Study. Pediatr Neurosurg. 1996;24:167-176.
127. Bailey C.C., Gnekow A., Wellek S., et al. Prospective randomised trial of chemotherapy given before radiotherapy in childhood medulloblastoma. Med Pediatr Oncol. 1995;25:166-178. International Society of Paediatric Oncology (SIOP) and the (German) Society of Paediatric Oncology (GPO): SIOP II
128. Thomas P.R., Deutsch M., Kepner J.L., et al. Low-stage medulloblastoma: final analysis of trial comparing standard-dose with reduced-dose neuraxis irradiation. J Clin Oncol. 2000;18:3004-3011.
129. Packer R.J., Goldwein J., Nicholson H.S., et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: a Children’s Cancer Group Study. J Clin Oncol. 1999;17:2127-2136.
130. Packer R.J., Gajjar A., Vezina G., et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24:4202-4208.
131. Carrie C., Muracciole X., Gomez F., et al. Conformal radiotherapy, reduced boost volume, hyperfractionated radiotherapy, and online quality control in standard-risk medulloblastoma without chemotherapy: results of the French M-SFOP 98 protocol. Int J Radiat Oncol Biol Phys. 2005;63:711-716.
132. Merchant T.E., Kun L.E., Krasin M.J., et al. Multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys. 2008;70:782-787.
133. Wolden S.L., Dunkel I.J., Souweidane M.M., et al. Patterns of failure using a conformal radiation therapy tumor bed boost for medulloblastoma. J Clin Oncol. 2003;21:3079-3083.
134. Douglas J.G., Barker J.L., Ellenbogen R.G., et al. Concurrent chemotherapy and reduced-dose cranial spinal irradiation followed by conformal posterior fossa tumor bed boost for average-risk medulloblastoma: efficacy and patterns of failure. Int J Radiat Oncol Biol Phys. 2004;58:1161-1164.
135. Jenkin D. The radiation treatment of medulloblastoma. J Neurooncol. 1996;29:45-54.
136. Gajjar A., Chintagumpala M., Ashley D., et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7:813-820.
137. Packer R.J., Sutton L.N., Goldwein J.W., et al. Improved survival with the use of adjuvant chemotherapy in the treatment of medulloblastoma. J Neurosurg. 1991;74:433-440.
138. Kuhl J., Muller H.L., Berthold F., et al. Preradiation chemotherapy of children and young adults with malignant brain tumors: results of the German pilot trial HIT ’88/’89. Klin Padiatr. 1998;210:227-233.
139. Kortmann R.D., Kuhl J., Timmermann B., et al. Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: results of the German prospective randomized trial HIT ’91. Int J Radiat Oncol Biol Phys. 2000;46:269-279.
140. Zeltzer P.M., Boyett J.M., Finlay J.L., et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children’s Cancer Group 921 randomized phase III study. J Clin Oncol. 1999;17:832-845.
141. Graham M.L., Herndon J.E.2nd, Casey J.R., et al. High-dose chemotherapy with autologous stem-cell rescue in patients with recurrent and high-risk pediatric brain tumors. J Clin Oncol. 1997;15:1814-1823.
142. Strother D., Ashley D., Kellie S.J., et al. Feasibility of four consecutive high-dose chemotherapy cycles with stem-cell rescue for patients with newly diagnosed medulloblastoma or supratentorial primitive neuroectodermal tumor after craniospinal radiotherapy: results of a collaborative study. J Clin Oncol. 2001;19:2696-2704.
143. Chi S.N., Gardner S.L., Levy A.S., et al. Feasibility and response to induction chemotherapy intensified with high-dose methotrexate for young children with newly diagnosed high-risk disseminated medulloblastoma. J Clin Oncol. 2004;22:4881-4887.
144. Dhall G., Grodman H., Ji L., et al. Outcome of children less than three years old at diagnosis with non-metastatic medulloblastoma treated with chemotherapy on the “Head Start” I and II protocols. Pediatr Blood Cancer. 2008;50:1169-1175.
145. Guruangan S., Dunkel I.J., Goldman S., et al. Myeloablative chemotherapy with autologous bone marrow rescue in young children with recurrent malignant brain tumors. J Clin Oncol. 1998;16:2486-2493.
146. Taylor R.E., Bailey C.C., Robinson K., et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: the International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 Study. J Clin Oncol. 2003;21:1581-1591.
147. Shu H.K., Sall W.F., Maity A., et al. Childhood intracranial ependymoma: twenty-year experience from a single institution. Cancer. 2007;110:432-441.
148. Pollack I.F., Gerszten P.C., Martinez A.J., et al. Intracranial ependymomas of childhood: long-term outcome and prognostic factors. Neurosurgery. 1995;37:655-666.
149. Von Haken M.S., White E.C., Daneshvar-Shyesther L., et al. Molecular genetic analysis of chromosome arm 17p and chromosome arm 22q DNA sequences in sporadic pediatric ependymomas. Genes Chromosomes Cancer. 1996;17:37-44.
150. Rubio M.P., Correa K.M., Ramesh V., et al. Analysis of the neurofibromatosis 2 gene in human ependymomas and astrocytomas. Cancer Res. 1994;54:45-47.
151. Kramer D.L., Parmiter A.H., Rorke L.B., et al. Molecular cytogenetic studies of pediatric ependymomas. J Neurooncol. 1998;37:25-33.
152. Zhen H.N., Zhang X., Bu X.Y., et al. Expression of the simian virus 40 large tumor antigen (Tag) and formation of Tag-p53 and Tag-pRb complexes in human brain tumours. Cancer. 1999;86:2124-2132.
153. Taylor M.D., Poppleton H., Fuller C., et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323-335.
154. Hamilton R.L., Pollack I.F. The molecular biology of ependymomas. Brain Pathol. 1997;7:807-822.
155. Schiffer D., Giordana M.T. Prognosis of ependymoma. Childs Nerv Syst. 1998;14:357-361.
156. Teo C., Nakaji P., Symons P., et al. Ependymoma. Childs Nerv Syst. 2003;19:270-285.
157. Kovalic J.J., Flaris N., Grigsby P.W., et al. Intracranial ependymoma long term outcome, patterns of failure. J Neurooncol. 1993;15:125-131.
158. Rogers L., Pueschel J., Spetzler R., et al. Is gross-total resection sufficient treatment for posterior fossa ependymomas? J Neurosurg. 2005;102:629-636.
159. Awaad Y.M., Allen J.C., Miller D.C., et al. Deferring adjuvant therapy for totally resected intracranial ependymoma. Pediatr Neurol. 1996;14:216-219.
160. Chiu J.K., Woo S.Y., Ater J., et al. Intracranial ependymoma in children: analysis of prognostic factors. J Neurooncol. 1992;13:283-290.
161. Grill J., Le Deley M.C., Gambarelli D., et al. Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age: a multicenter trial of the French Society of Pediatric Oncology. J Clin Oncol. 2001;19:1288-1296.
162. Healey E.A., Barnes P.D., Kupsky W.J., et al. The prognostic significance of postoperative residual tumor in ependymoma. Neurosurgery. 1991;28:666-671.
163. Palma L., Celli P., Mariottini A., et al. The importance of surgery in supratentorial ependymomas. Long-term survival in a series of 23 cases. Childs Nerv Syst. 2000;16:170-175.
164. Vinchon M., Soto-Ares G., Riffaud L., et al. Supratentorial ependymoma in childhood. Pediatr Neurosurg. 2001;34:77-87.
165. Loefffler J.S., Rossitch E.Jr., Siddon R., et al. Role of stereotactic radiosurgery with a linear accelerator in treatment of intracranial arteriovenous malformations and tumors in children. Pediatrics. 1990;85:774-782.
166. Sanford R.A., Kun L.E., Heideman R.L., et al. Cerebellar pontine angle ependymoma in infants. Pediatr Neurosurg. 1997;27:84-91.
167. Ikezaki K., Matsushima T., Inoue T., et al. Correlation of microanatomical localization with postoperative survival in posterior fossa ependymomas. Neurosurgery. 1993;32:38-44.
168. Figarella-Branger D., Civatte M., Bouvier-Labit C., et al. Prognostic factors in intracranial ependymomas in children. J Neurosurg. 2000;93:605-613.
169. Salazar O.M. A better understanding of CNS seeding and a brighter outlook for postoperatively irradiated patients with ependymomas. Int J Radiat Oncol Biol Phys. 1983;9:1231-1234.
170. Sneed P.K., Russo C., Scharfen C.O., et al. Long-term follow-up after high-activity 125I brachytherapy for pediatric brain tumors. Pediatr Neurosurg. 1996;24:314-322.
171. Merchant T.E., Haida T., Wang M.H., et al. Anaplastic ependymoma: treatment of pediatric patients with or without craniospinal radiation therapy. J Neurosurg. 1997;86:943-949.
172. Merchant T.E., Mulhern R.K., Krasin M.J., et al. Preliminary results from a phase II trial of conformal radiation therapy and evaluation of radiation-related CNS effects for pediatric patients with localized ependymoma. J Clin Oncol. 2004;22:3156-3162.
173. Evans A.E., Anderson J.R., Lefkowitz-Boudreaux I.B., et al. Adjuvant chemotherapy of childhood posterior fossa ependymoma: cranio-spinal irradiation with or without adjuvant CCNU, vincristine and prednisone: a Children’s Cancer Group study. Med Pediatr Oncol. 1996;27:8-14.
174. Souweidane M.M., Bouffet E., Finlay J. The role of chemotherapy in newly diagnosed ependymoma of childhood. Pediatr Neurosurg. 1998;28:273-278.
175. Bouffet E., Foreman N. Chemotherapy for intracranial ependymomas. Childs Nerv Syst. 1999;15:563-570.
176. Duffner P.K., Krischer J.P., Sanford R.A., et al. Prognostic factors in infants and very young children with intracranial ependymomas. Pediatr Neurosurg. 1998;28:215-222.
177. Duffner P.K., Horowitz M.E., Krischer J.P., et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328:1725-1731.
178. Geyer J.R., Zeltzer P.M., Boyett J.M., et al. Survival of infants with primitive neuroectodermal tumors and malignant ependymomas of the CNS treated with eight drugs in 1 day: a report from the Childrens Cancer Group. J Clin Oncol. 1994;12:1607-1615.
179. Grill J., Kalifa C., Doz F., et al. A high-dose busulfan-thiotepa combination followed by autologous bone marrow transplantation in childhood recurrent ependymoma. A phase-II study. Pediatr Neurosurg. 1996;25:7-12.
180. Mason W.P., Goldman S., Yates A.J., et al. Survival following intensive chemotherapy with bone marrow reconstitution for children with recurrent intracranial ependymoma—a report of the Children’s Cancer Group. J Neurooncol. 1998;37:135-143.
181. Lyons M.K., Kelly P.J. Posterior fossa ependymomas: report of 30 cases and review of the literature. Neurosurgery. 1991;28:659-664.
182. Vanuytsel L.J., Bessell E.M., Ashley S.E., et al. Intracranial ependymoma: long-term results of a policy of surgery and radiotherapy. Int J Radiat Oncol Biol Phys. 1992;23:313-319.
183. Grabb P.A., Lunsford L.D., Albright A.L., et al. Stereotactic radiosurgery for glial neoplasms of childhood. Neurosurgery. 1996;38:696-701.
184. Ilgren E.B., Stiller C.A. Cerebellar astrocytomas. Clinical characteristics and prognostic indices. J Neurooncol. 1987;4:293-308.
185. Kulkarni A.V., Becker L.E., Jay V., et al. Primary cerebellar glioblastomas multiforme in children. Report of four cases. J Neurosurg. 1999;90:546-550.
186. Davis C.H., Joglekar V.M. Cerebellar astrocytomas in children and young adults. J Neurol Neurosurg Psychiatry. 1981;44:820-828.
187. Palma L., Russo A., Celli P. Prognosis of the so-called “diffuse” cerebellar astrocytoma. Neurosurgery. 1984;15:315-317.
188. Szenasy J., Slowik F. Prognosis of benign cerebellar astrocytomas in children. Childs Brain. 1983;10:39-47.
189. Klein D.M., McCullough D.C. Surgical staging of cerebellar astrocytomas in childhood. Cancer. 1985;56:1810-1811.
190. Garcia D.M., Marks J.E., Latifi H.R., et al. Childhoood cerebellar astrocytomas: is there a role for postoperative irradiation? Int J Radiat Oncol Biol Phys. 1990;18:815-818.
191. Lapras C., Patet J.D., Lapras C.Jr., et al. Cerebellar astrocytomas in childhood. Childs Nerv Syst. 1986;2:55-59.
192. Austin E.J., Alvord E.C.Jr. Recurrences of cerebellar astrocytomas: a violation of Collins’ law. J Neurosurg. 1988;68:41-47.
193. Rickert C.H., Paulus W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Childs Nerv Syst. 2001;17:503-511.
194. Olson T.A., Bayar E., Kosnik E., et al. Successful treatment of disseminated central nervous system malignant rhabdoid tumor. J Pediatr Hematol Oncol. 1995;17:71-75.
195. Biegel J.A., Rorke L.B., Packer R.J., et al. Monosomy 22 in rhabdoid or atypical tumors of the brain. J Neurosurg. 1990;73:710-714.
196. Versteege I., Sevenet N., Lange J., et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203-206.
197. Biegel J.A., Zhou J.Y., Rorke L.B., et al. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59:74-79.