Figure 23.1 Schematic diagram of classification of PHT: prehepatic, hepatic and posthepatic factors.
These are derived from a dilated venous plexus in the lamina propria of the lower oesophagus, usually supplied from the left gastric vein and draining through perforating veins into the azygos venous system. There are a number of thin-walled parallel vessels that run within the mucosal folds in a longitudinal manner (‘palisade zones’) which are superficial and related to the distal 3–5 cm of the oesophagus with poor connective tissue support and constitute the majority of those which bleed. These can make up almost 30% of the cross-sectional area in this zone.
BOX 23.2:Gastric varices: the Sarin (1992) classification [3]
Gastroesophageal
• Type 1: Lesser curve varices (75%) extending to oesophagogastric junction.
• Type 2: Greater curvature. These veins meet the upper end of the cardia of the stomach and drain into the left gastric and short gastric veins.
Isolated
• Type 1: Fundal varices with a high (78%) incidence of bleeding.
• Type 2: Distal stomach or proximal duodenum.
Extrahepatic symptoms, other than PHT, appear to be rare (at least clinically) in the absence of cirrhosis. However, in an interesting study of 12 children referred for consideration of meso-Rex bypass (MRB) surgery, minimal hepatic encephalopathy was detected by a battery of psychometric testing, and was reversed by restoration of portal blood flow [7]. A more recent study looking for features of minimal hepatic encephalopathy using serum ammonia levels, electroencephalography (EEG) and psychometric testing also found suggestive features in about half of a group of 13 children [8]. A larger study involving 70 children from India identified the same phenomenon in a proportion but was able to reverse all testing abnormalities simply with oral lactulose [9].
Dilated periumbilical veins (the famous caput medusae* with hepatofugal blood flow may be uncommonly seen in long-standing PHT (Figure 23.2). This recanalisation of the umbilical veins when combined with a distinct bruit has been labelled the Cruveilhier–Baumgarten* syndrome. Dilated abdominal wall veins with blood flowing towards the thorax might suggest caval occlusion and the possibility of Budd–Chiari syndrome (B-CS). Other features, such as telangiectasia, palmer erythema and jaundice, are more associated with chronic liver disease than PHT per se.
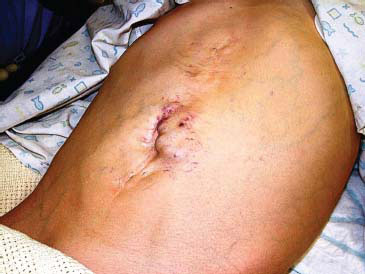
Figure 23.2 Umbilical varices in a child with complete thrombosis of his portal venous system due to complicated neonatal surgery.
23.2.1.1 PORTAL BILIOPATHY
Formation of choledochal varices in long-standing PVO was first described in 1965 [10], and when they impact upon the bile duct lumen, leading to obstruction and changes in liver biochemistry, they can be termed portal biliopathy. This is relatively common in adults but has a low incidence in children, with Maksoud-Filho et al. reporting an incidence of only 3% in their series from Brazil [6]. There is some suggestion that it is more frequently found in those with PVO rather than cirrhosis, although this may simply be related to the duration of PHT and accompanying venous collateral formation.
Shin et al. [11] divided biliary abnormalities in portal biliopathy into three types: varicoid, fibrotic and mixed. Varicoid irregularities were smooth indentations suggested to be caused by paracholedochal plexus dilatation (cavernous transformation). In the fibrotic type, smooth localised strictures may be seen at the level of the CBD and are possibly ischaemic in nature. Symptoms may include intermittent jaundice, cholangitis, haemobilia and even stone formation [12], and this then indicates the need for treatment. Endoscopic ultrasound (US) may also have a particular role in diagnosis by trying to determine the contribution of varices to luminal obstruction. Most experience has been acquired in adults with this condition [13,14], and endoscopic biliary stenting ± removal of stones is the first-line therapy, although resistant cases may warrant MRB (in those due to PVO) or portosystemic shunting to reduce portal venous pressure and allow more direct biliary surgery.
23.2.1.2 PORTAL HYPERTENSIVE GASTROPATHY
This is initially recognised endoscopically as a fine, mottled punctate rash on the gastric mucosa, progressing to reddening affecting the crests of gastric folds and leading ultimately to a ‘snakeskin’ appearance due to mucosal oedema (Figure 23.3). Contact bleeding is common in established cases, and it may be a cause of symptomatic gastrointestinal bleeding leading to anaemia or even melaena, but this is fairly uncommon. Gastropathy is usually an issue in older children with PVO who have already undergone a programme of variceal obliteration, which may indeed be the cause. Helicobacter pylori seems to have no role in aetiology, and biopsy is not necessary. Medical treatment aimed at lowering PHT (e.g. propranolol) and reducing gastric acid output (e.g. ranitidine) may help.
23.2.2 Portal vein thrombosis
This is the most common single cause of PHT in children, having many distinguishing features from cirrhotic liver disease in terms of aetiology, natural history and clinical management, and which deserve special attention as possibly the most important cause of PHT. The classic paper on the subject is Shelia Sherlock’s† description of 97 adults and children (55%) with PVO published in 1979 [15]. In this series, one of the more common causes appeared to be abdominal infection (e.g. appendicitis) and septicaemia, but in about half the cause was entirely obscure. The mortality was high (24%) and the use of surgery extensive – 64 patients underwent 114 operations for variceal haemorrhage.
23.2.2.1 PATHOPHYSIOLOGY
Acute PVO is surprisingly well tolerated and frequently and surprisingly asymptomatic because of two phenomena:
1. Arterial rescue – Rapid compensatory increase in arterial flow reducing the effects of hepatocyte ischaemia.
2. Venous rescue – This starts within a few days and involves neoformation of venous collaterals, with completion around 3–5 weeks.
There may be a hyperkinetic circulation (low systemic vascular resistance and a high cardiac output) evident during this compensatory phase.
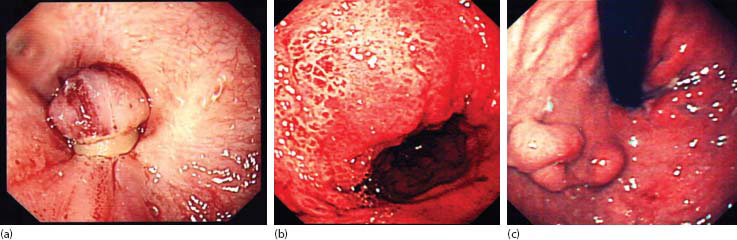
Figure 23.3 Varices: (a) banded oesophageal varices, (b) severe portal hypertensive gastropathy and (c) gastric varices.
Chronic PVO, arbitrarily defined if lasting >60 days or if a cavernoma is evident, may be the cause of further detrimental effects:
1. PHT – As discussed above, this leads principally to variceal formation and splenomegaly. How long it takes to develop actual varices is a moot point. The first point of contact with most of these children will be their index bleed, and for those with PVO, the median age is just more than 4 years of age [16].
2. Diminution in liver mass – The effects of this are less easy to discern and, aside from mild elevation in the international normalised ratio (INR) (indicative of diminished production of coagulation factors), require sophisticated investigation.
23.2.2.2 EPIDEMIOLOGY
PVO is surprisingly common in the population, with some early postmortem studies suggesting that about 1% of the population are affected [17]. However, more recent studies using noninvasive imaging suggest that the true incidence may be even higher, at 5%–10% of an otherwise normal population. There is a single national study estimating the incidence of PVT. Thus, from a series of 32 children (<15 years) in Israel, the incidence was 0.72 cases per million children [18].
23.2.2.3 AETIOLOGY AND RISK FACTORS
The cause is not clear in most cases; however, certain observations can be made (Table 23.1) [19–21]. About 20% will have other extra-abdominal malformations, suggesting that the PVO is also a congenital anomaly [16]. There may be a particular relationship with skeletal (e.g. absent radius), cardiac and craniofacial anomalies. In other cases, the onset of PVO appears to be in the neonatal period, most obviously in relation to umbilical vein catheterisation (12% in our series), but also episodes of neonatal sepsis and dehydration (7%) appear more frequently in this cohort [16].
Later during childhood, a focus of sepsis such as appendicitis may prompt septic emboli via the portal vein (portal pyaemia), which in turn may cause portal vein thrombosis and consequent PHT. Compared with older series [15], this is much less frequent, accounting for perhaps 8% of our current series [16]. Occasionally, obstruction and infiltration of the portal vein by adjacent abscess or tumour (e.g. hepatoblastoma) occurs, but this too remains an unusual cause of PHT in childhood [21,22].
23.2.2.3.1 Umbilical vein sepsis and catheterisation
Catheterisation of the umbilical vein for neonatal venous access has been around since the 1960s and resulted in a greater number of clinically evident PVO. The actual incidence of clinically significant PVT following use of a umbilical vein catheter (UVC), however, is not known. Older observational studies suggested an incidence of 1%–2% [23]. In a more recent well-controlled study of 200 infants with varying durations of placement of UVC [24], the incidence of thrombus formation at the catheter tip was 4%–7% detected by echocardiography, although this may not mean much, as the thrombus needs to propagate proximally to involve the portal vein proper.
Furthermore, a diagnosis of PVT does not necessarily imply the development of PHT. Indeed, in a Toronto series of 133 infants with PVT, only about 25% actually went on to develop significant PHT [25]. About 75% had had placement of a UVC during the neonatal period as the presumed cause.
23.2.2.3.2 Postsplenectomy
PVO may also be seen as a complication after splenectomy due to propagation of a clot along the splenic vein. Older series using retrospective techniques estimated the risk at <5% for open splenectomy, with increased risk for those requiring surgery for haematological or malignant reasons. However, there appears to be a greater risk when the splenectomy is accomplished laparoscopically [26,27]. A comparative Japanese study using contrast computed tomography (CT) scan to detect portal vein or splenic thrombosis showed thrombosis in 55% of the laparoscopic group compared with 19% of the open group [26]. Similar findings were also shown in a prospective study of 40 patients [28]. Nine (22%) developed splenic vein thrombosis, progressing to the portal vein in five (12%).
Table 23.1 Cause of portal vein occlusion: adults and children compared
|
Adultsa |
Childrenb |
|
|
Congenital (presumed) |
n/a |
20%–30% |
|
Neonatal acquired |
n/a |
UVC |
|
Neonatal sepsis/dehydration |
||
|
Neoplasia |
Myeloproliferative disorders |
Hepatoblastoma |
|
HCC |
HCC |
|
|
Pancreatic cancer |
||
|
Intra-abdominal sepsis |
Appendicitis |
Appendicitis |
|
Diverticular disease |
||
|
Crohn’s disease |
||
|
Inflammatory bowel disease |
||
|
Thombogenesis |
Protein C/S deficiencies (4%–12% and 3%–4%) |
Protein C/S deficiencies (<5% and <2%) |
|
Factor V Leiden (<2%) |
Factor V Leiden (5%–16%) |
|
|
Antithrombin III deficiency (<5%) |
Antithrombin III deficiency (<3%) |
|
|
MTHFR mutations (12%–20%) |
MTHFR mutations (3%–50%) |
|
|
Paroxysmal nocturnal haemoglobinuria |
||
|
Antiphospholipid syndrome |
||
|
Pregnancy |
||
|
Oral contraceptive pill |
||
|
Myeloproliferative disorders |
Polycythaemia rubra vera |
|
|
Myelofibrosis |
||
|
Trauma |
Postsplenectomy |
Postsplenectomy |
a After El-Hamid N, et al., Journal of Pediatric Gastroenterology & Nutrition 2008, 47: 630–634; Janssen HL, et al., Gut2001, 49: 720–724; Bhattacharyya M, et al., American Journal of Clinical Pathology 2004, 121: 844–847.
b After Heller C, et al., British Journal of Haematology 2000, 111: 534–539.
23.2.2.3.3 Thombophilia
Hepatic vein thrombosis (B-CS) is a much more likely consequence of disturbed thrombogenesis than PVO. To illustrate this, of 30 within our series of children with PVO who had screening for thrombophilia [21], not one had a consistent genetic thrombophilia defect (including screening for JAK2 V617F and factor V Leiden). Notwithstanding this, a number of inherited defects of thrombolysis might indicate a genetic predisposition. These include mutations in factor V Leiden and methyltetrahydrofolate reductase (MTHFR) deficiency, protein C (and possibly protein S) deficiencies and antithrombin III deficiency. One genetic study reported MTHFR mutations in 16 (heterozygote in 12, homozygote in 4) of 31 children with PVO [28], although 23% of controls also showed such mutations. This raises the question of whether the MTHFR gene polymorphism (without hyperhomocysteinaemia) actually contributes to thrombophilia.
Caution in interpretation should also be reserved for some children with apparently low levels in protein C levels (up to 30%), as the diminished synthetic function seen in some cases of PVO may be the cause, rather than an inherited defect [21,29–32]. This is not seen with protein S, as it is partially synthesised outside of the liver.
23.2.2.3.4 Patterns of thrombosis
The portal venous system can be variably occluded, and there does seem to be a relationship with aetiology [33]. These can be broadly classified into (1) occlusion limited to the portal vein, (2) occlusion of the portal vein extending to involve the superior mesenteric vein, and (3) as for number 2, but also involving the splenic vein (Figure 23.4).
23.2.2.4 HEPATOPORTAL SCLEROSIS (AKA OBLITERATIVE PORTAL VENOPATHY)
Hepatoportal sclerosis, named by Mikkelsen in 1965 [34], is a condition defined by the presence of a normal portal vein and major intrahepatic branches, but thickening and sclerosis of the small intrahepatic portal veins, leading to PHT. Occasionally, changes in the liver itself can be seen, such as surface nodularity, caudate lobe hypertrophy and right hepatic lobe atrophy [35]. Histological features typically include fibrosis confined to the portal triads, mural thickening of portal venules (phlebosclerosis) and megasinusoids.
Aetiology is unclear in most children, but some studies in adults have incriminated exposure to toxic substances (e.g. arsenic or vinyl chloride) or sometimes drugs such as vitamin A or 6-mercaptopurine. There also appears to be a relationship to human immunodeficiency virus (HIV), or at least the highly active antiretroviral therapy used to treat it.
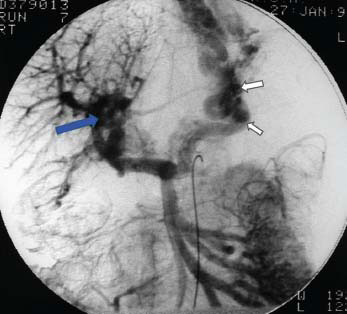
Figure 23.4 Venous phase of angiogram showing discrete portal vein block (blue arrow) and collateral formation with obvious oesophagogastric varices (white arrows).
There also appears to be a marked geographical disparity, with most of the larger series being reported from India [36] and China. In some of these areas, the cause appears to be drinking water or traditional medicines (e.g. Fowler’s solution) contaminated by arsenic.
The diagnosis can be suggested by US and CT scan but should be defined by indirect venography or MRV, where a ‘withered tree’ appearance can be seen [35,37], and a generous liver biopsy.
There is no defined treatment for the pathology itself, but the resultant PHT should be treated along conventional lines. Long-standing cases can end in liver failure, and transplantation has been used in adults.
23.2.3 Budd*–Chiari† syndrome and venoocclusive disease
There are a multitude of posthepatic or postsinusoidal causes of PHT, including those where the level of obstruction is at a microscopic level involving the hepatic venules (usually referred to as veno-occlusive disease [VOD]), or the hepatic vein confluence either alone or by extension to involve the hepatic segment of the vena cava itself (Figure 23.5). Historically, chronic constrictive pericarditis typically due to TB was also considered a cause.
23.2.3.1 AETIOLOGY
Congenital webs impeding venous return have been described but are rare in practice, although one of the larger series is from South Africa describing nine cases, with eight being from a particular region of Namibia [39]. Most cases elsewhere seem to be acquired in the some way, and typically B-CS is related to an underlying malignant or thrombogenic disorder. Table 23.2 lists some predisposing causes presenting during childhood. Intravascular tumour infiltration may arise from a Wilms’ tumour and obstruct the cava and then hepatic venous ostia on its way to the right atrium. Alternatively, hepatoblastoma or hepatocellular carcinoma (HCC) may also directly invade the large hepatic veins or induce adjacent thrombosis.
Hepatic venous occlusion and consequent B-CS are also possible complications post liver transplantation and are considered in detail elsewhere.
VOD, also termed sinusoidal obstruction syndrome when associated with chemotherapy or bone marrow transplant, and is usually drug or toxin induced. It was originally recognised as a complication of pyrozolidine alkaloid poisoning found in some Jamaican bush teas and herbal remedies (Table 23.2) [39]. Nowadays, it is much more prevalent in association with more aggressive haematological rescue therapies following myeloablative therapy. For instance, almost 20% of stem cell transplants are complicated by VOD.
23.2.3.2 CLINICAL FEATURES
The hallmark of these is hepatomegaly due to venous congestion, certainly in the acute phase, together with relative sparing of the caudate lobe, which then usually hypertrophies due to its independent venous drainage [40,41]. Cases can be divided into fulminant, acute and chronic according to where they are in the natural history, with increasing fibrosis causing parenchymal necrosis and ultimately cirrhosis. Ascites is usually prominent from the outset, and there may be lower limb oedema, particularly if there is caval obstruction as well. PHT causes the development of varices and splenomegaly as before.
Jaundice may be seen at the disease onset as it probably inflammatory, but by the time the disease has entered its chronic phase, most children are anicteric and usually relatively well.
VOD has similar clinical features to B-CS, although a much higher incidence of involvement of other organs, particularly the kidney. The so-called Baltimore criteria for diagnosis of VOD list jaundice (bilirubin > 34 µmol/L) and the presence of at least two of the following symptoms: hepatomegaly, ascites and weight gain [42].
23.2.3.3 DIAGNOSIS AND MANAGEMENT
The key diagnostic tool is detailed Doppler ultrasonography. Normally, hepatic venous flow is triphasic on Doppler US and is altered markedly in B-CS. There may also be visible thrombus within the veins, evidence of a structural web or stenosis and, if chronic, evidence of intrahepatic collateral vessel formation. These are seen as small, usually subcapsular spiderweb collaterals. Flow in the portal vein proper may be reversed and hepatofugal.

Figure 23.5 B-CS. George Budd (1808–1882) and Hans Chiari (1851–1916).
Table 23.2 Causes of B-CS and VOD
|
B-CS |
VOD (sinusoidal occlusion syndrome) |
Comments |
|
|
Increased Thrombogenicity |
|||
|
Paroxysmal nocturnal haemoglobinuria (PNH) [39] |
n/a |
Causes pancytopenia and haemolysis due to increased membrane susceptibility to complement; usually X-linked |
|
|
Factor V Leiden, protein C/S deficiency |
n/a |
||
|
Janus tyrosine kinase 2 (JAK2) mutation (V617F) |
n/a |
||
|
Behçet’s disease |
n/a |
Autoimmune, multisystemic disorder; usually of Turkish/Kurdish origin |
|
|
Drugs |
|||
|
n/a |
Actinomycin D, cytosine arabinoside, mithramycin, 6-thioguanine, anti-CD33 antibody gemtuzumab ozogamicin |
||
|
n/a |
Stem cell rescue following myeloablation |
||
|
Toxins |
|||
|
n/a |
Pyrozolidine alkaloids, e.g. Senecio (ragwort) and Jamaican ‘bush tea’ |
||
|
n/a |
Arsenic |
CT or magnetic resonance (MR) scanning should show hepatomegaly and maybe heterogeneous enhancement (so-called nutmeg liver). The latter may also show nodular regenerative hyperplasia in chronic B-CS. These are seen as large nodules with increased signal intensity on T1-weighted images and low to intermediate signal intensity on T2-weighted images.
Liver biopsy is warranted in B-CS and will show signs of venous congestion, liver cell loss and fibrosis predominantly located in the centrilobular area. VOD will show nonthrombotic obstruction of hepatic venules by subendothelial swelling due to injury of the sinusoidal wall in Zone III.
Transjugular or transfemoral selective hepatic venography is definitive in most cases of B-CS and then may go on to be therapeutic, with a wide range of options, from angioplasty to stent placement [43].
Treatment depends largely on the degree of PHT or ascites either condition has caused. Anticoagulation and medical therapy should be considered the first-line treatment but is effective only early in the disease’s course. Currently, transjugular intrahepatic portosystemic shunt (TIPS) and angioplasty or stent placement techniques have replaced direct surgical exposure or bypass procedures such as portocaval, mesocaval or mesoatrial shunting. Radiological demonstration of a ‘congenital’ web usually mandated surgical exploration and venous reconstruction with total hepatic vascular isolation. For those with chronic B-CS, liver transplantation may be indicated as a rescue therapy, with the possibility of lifelong anticoagulation afterwards [44].
Table 23.3 Causes of arterioportal hypertension
|
Cause |
Characteristics |
Treatment |
|
|
Congenital |
Unknown |
Failure to thrive, GI bleeding, splenomegaly |
Embolisation, resection, arterial ligation, portal diversion |
|
Trauma • Blunt • Penetrating (e.g. knife, gunshot and liver biopsy) |
Formation and rupture of pseudoaneurysm |
Intraperitoneal bleeding, GI bleeding |
Conservative embolisation |
|
Hereditary haemorrhagic Telangectasia |
Autosomal dominant; mutations in three genes related to angiogenic biology |
Typically presents with epistaxis; widespread vascular malformations, including intrahepatic; usually AV but maybe AP |
Medical treatment available (bevacizumab – anti-VEGF) |
|
Tumours (particularly HCC) |
Abnormal angiogenesis |
Resection, transplantation |
|
|
Cirrhosis |
Abnormal angiogenesis |
Transplantation |
Note: VEGF, vascular endothelial growth factor; AV, arteriovenous; AP, arterioportal; GI, gastrointestinal.
The treatment of VOD is still evolving and is primarily supportive, but there appears to be an increasing role for defibrotide in both prophylaxis and treatment, particularly in those related to stem cell transplant [44,45]. This is a single-stranded polydeoxyribonucleotide derived from porcine tissue with anticoagulant and antifibrotic properties, and trials in adults and children have suggested reduction in morbidity and mortality due to VOD. Established VOD may present long after the acute presentation and early treatment phase with variceal complications. In such cases, variceal management should proceed along conventional lines.
23.2.4 Arterioportal hypertension
Arterioportal hypertension due to arterioportal fistula (APF) was first reported by Gryboski et al. almost 50 years ago [46]. In itself, it is an unusual cause of PHT caused by arterial blood leaking into the portal venous system. It does have enough characteristics to differentiate it from the usual causes, which are usually due to elevated resistance to flow rather than arterial supplementation of venous flow.
23.2.4.1 AETIOLOGY
Table 23.3 illustrates the usual classification. In paediatric practice, most are usually of congenital origin, but overall, the most common cause is due to some kind of penetrating trauma – even the biopsy needle of the liver physician or, in our reported case, a partial hepatectomy 6 years previously [47]. It may also be more widespread than appreciated in generalised liver pathology, such as cirrhosis.
23.2.4.2 CLINICAL FEATURES
Congenital APF usually presents in infancy with failure to thrive and splenomegaly [48,49]. If sustained, then varicieal formation may occur and therefore predispose to gastrointestinal bleeding and rarely to ascites. There may be an unusual distribution of varices (e.g. gastric rather than oesophageal) due to the partial nature of the arterial leak. Rarely in children but described in adults is an arterial ‘steal’ phenomenon with abdominal pain and diarrhoea.
Site and distribution can be variable. In some, it is isolated and unilobar, whereas in others, there are multiple fistulas. Sometimes, these declare themselves and open up only after ‘effective’ treatment by embolisation and so forth.
There is a predisposition to congenital vascular anomalies in children with Down’s syndrome.* Within this spectrum are some with APF (Figure 23.6). These appear to be somewhat different in that there is usually an aneurysmal dilatation involving the left portal vein and fed by numerous arteries.
23.2.4.3 DIAGNOSIS AND MANAGEMENT
US supplemented with colour-flow Doppler is the usual investigation which raises the possibility of an APF and shows reversed pulsatile flow in the portal vein.
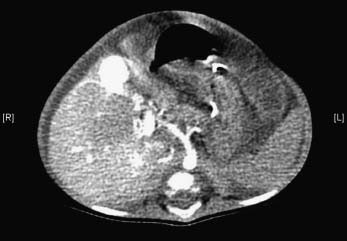
Figure 23.6 Arterioportal hypertension. Four-month-old child with Down’s syndrome and born with duodenal atresia. A CT angiogram shows a hypertrophied hepatic artery feeding an aneurysm in an umbilical fissure and early filling of the left portal vein.
Contrast-enhanced CT scans will confirm the site of the arterial leak and early visualisation of the portal vein.
Transcatheter embolisation should be the first line of management, as it is precise and relatively nontraumatic. A number of agents are actually used, including detachable coils, Gelfoam™ and glue. Nonetheless, its longer-term efficacy remains undecided, particularly in congenital APF. Precise appreciation of the anatomy of the leak is essential in planning surgical intervention, but the possibilities include liver resection and ligation of the feeding hepatic artery. For those with bilobar or recurrent fistulas, transplantation remains an option.
We reported a less intuitive alternative as palliation for one child who had recurrence of postembolisation of multiple intrahepatic APFs of congenital origin [50]. This involved division of his portal vein and end-to-side portocaval anastomosis. This allowed his mesenteric venous system to decompress while the recurrent APFs maintained his liver blood flow.
23.2.5 Investigation of portal hypertension
These fall broadly into
1. Definition of degree of portal vein thrombosis. The diagnosis should be suggested by US and Doppler flow studies usually showing cavernomatous transformation (aka cavernoma) of the portal vein. The degree of flow within the intrahepatic portal venous system should be estimated, together with any evidence of nodular transformation. Further imaging usually relies on contrast venography, which is currently achieved by contrast CT or MR imaging. Any question of surgical intervention mandates a full appreciation of the relevant venous anatomy, including patency of splenic and superior mesenteric veins from the splanchnic side and patency of the left renal vein and vena cava from the systemic side.
2. Assessment of predisposing factors. In the absence of clinically obvious predisposing factors such as UVC use during neonatal life, a search should be made for evidence of abnormal coagulation and tendency to increased thrombogenesis. Slight prolongation of the INR is typical of those with PVO and need have no prognostic significance. Similarly, levels of antithrombin III and proteins C and S should be assessed critically. The definition of a hypercoagulable condition may preclude surgery or alternatively warrant long-term anticoagulation.
3. Assessment of effects of PHT
a. US should allow assessment of spleen size and the presence of any ascites. The principal consequence of PVO is the development of varices, and therefore full upper gastrointestinal endoscopy to the level of the third part of the duodenum should show the presence and grade of varices in the stomach and oesophagus, together with any stigmata suggesting imminent risk of bleeding. The rectum and distal sigmoid should also be endoscopically assessed for the presence of rectal varices and haemorrhoids.
b. Hypersplenism is suggested by thrombocytopenia (<100 × 109/L), leucopenia and anaemia.
23.2.5 MANAGEMENT
There is a role for anticoagulation (low-molecular-weight heparin followed by warfarin) in acute portal (or mesenteric) vein thrombosis, and complete recanalisation can be expected in about 50% following 6 months of therapy. Success is related to the period of delay following onset, but ideally it should be before 1 week [51].
There are many ways to manage the effects of chronic PVO, and there is considerable controversy over some of the choices. Much of the problem concerns the very poor evidence base and the lack of comparative trials in children. Thus, there have been no trials of endoscopic variceal management versus bypass or shunt surgery in children, with each option having the potential for problems and complications. In practice, as there is a range of effects of PVO, there should be a range of available options to allow a tailored approach to each patient. The advent of MRB in the surgical repertoire during the 1990s has reignited the debate as to the role of surgery early on in the course of this disease. Its key proponent and in fact its originator, Jean De Ville de Goyet, argues strongly that most cases of PVO are suitable for and should undergo MRB, perhaps even before any actual bleeding episodes [52,53]. However, this ignores the long history of successful endoscopic management in perhaps 90% of children without the need for surgical intervention (Table 23.4) and minimises the problems associated with MRB. For instance, in the recently published Paris series of surgically treated PVO, 43 children underwent an attempted MRB with a success rate of only 60% in the long term [58]. Conventional portosystemic shunts had a much better long-term patency rate, but of course always have the spectre of encephalopathy. Box 23.3 outlines the endoscopy versus surgery debate.
Table 23-4 Conservatively managed portal vein occlusion in children
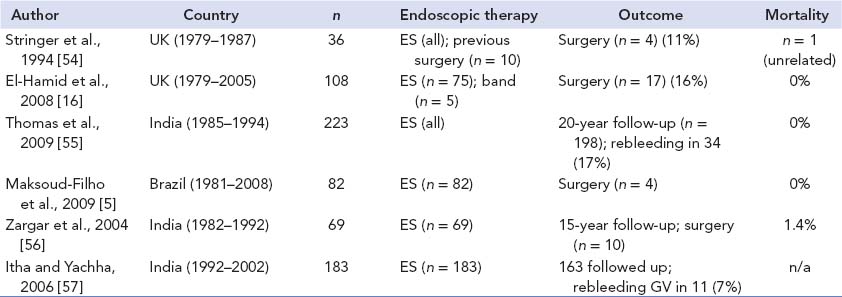
Note: ES, endoscopic sclerotherapy; band, variceal banding; GV, gastric varices.
23.2.5.1 BLEEDING OESOPHAGOGASTRIC VARICES
Clinical history and examination should indicate whether there is an underlying chronic parenchymal liver disease (postbiliary atresia, congenital hepatic fibrosis, cystic fibrosis, etc.) or PVO – the two most likely reasons for de novo bleeding. Children with the latter condition are much more robust to bleeding episodes, while the latter may decompen-sate quite quickly.
The emergency management of bleeding varices or, much more usually, recently bled varices is to restore circulating blood volume (aiming for haemoglobin = 8–10 g/dL; haematocrit > 24%) and correct any apparent coagulopathy (intravenous [IV] vitamin K, fresh frozen plasma and cryoprecipitate, if INR > 2) and thrombocytopenia (if <50 × 109/L). A recent trial of recombinant factor VIIa in adult cirrhotics failed to show any advantage over standard therapy [59].
IV antibiotics (e.g. ciprofloxacin and third-generation cephalosporin) are also mandatory if cirrhosis is the underlying cause. Acute pharmacological control of PHT (see Table 23.5) can be initiated in most hospital environments and should precede acute endoscopy to allow a modicum of control. Endoscopic visualisation of actively bleeding varix within a stomach full of blood is by no means easy.
23.2.5.1.1 Sengstaken–Blakemore tube
Various devices were invented in the 1950s designed to apply pressure and tamponade bleeding oesophagogastric varices. The template is the Sengstaken*–Blackmore† (S-B) tube, which has inflatable gastric and oesophageal balloons, together with two aspiratable lumens, one proximal for saliva in the pharynx and proximal oesophagus and the other for gastric aspiration (Figure 23.7).
* Robert William Sengstaken Jr. (1923–1978), – American neuro-surgeon.
In our practice, all S-B tubes are placed under general anaesthesia with endotracheal intubation. Although children do not have the same high risks of parallel bleeding episodes from peptic ulcers that cirrhotic adults do, preliminary endoscopy should confirm that it is varices which are the cause of the bleeding.
The tube is inserted through the mouth until clearly in the stomach; the gastric balloon is inflated with diluted contrast (to check position) and pulled back to impact the oesophagogastric junction (OGJ), thus exerting variceal compression. It is usually not necessary to inflate the oesophageal balloon in children. OGJ pressure is usually maintained by bringing the tube to the side of the mouth and taping it to the cheek. A 24-hour period of traction is maintained to allow correction of any coagulopathy and haemodynamic instability. Typically, traction is relaxed every 4–5 h for a 10-min period to prevent mucosal ischaemia. Box 23.4 illustrates the series from King’s College Hospital of its use and outcomes during the period 1997–2012 [60].
The main problems encountered usually involve acute control in infants and small children because currently available S-B tubes are really designed for the longer and wider oesophagus of older children. Sometimes, in the emergency situation it may be necessary to use a more appropriately sized urethral catheter and its inflated balloon to achieve the necessary traction control.
23.2.5.2 MEDICAL MANAGEMENT (TABLE 23.5)
Vasopressin is the most potent splanchnic vasoconstrictor which leads to a decrease in portal venous inflow and decline in portal pressure. Its clinical application is severely limited by multiple side effects caused by vasospasm in other organs, including cardiac, mesenteric and peripheral ischaemia; arrhythmias; and hypertension. Terlipressin, an analogue of vasopressin, is used in clinical practice, although not commonly in children. There is some evidence of superiority over the usual alternative octreotide, so that in a recent Cochrane review it had a significant effect on mortality in adult cirrhotics. Certainly, its effects are exerted over a much longer period of up to 3 h following a single IV bolus, and unlike its parent molecule, there are no untoward effects on the coronary circulation. It may also reverse the hepatorenal syndrome, if present.
[level-membership-for-pediatrics-category]
VARICEAL OBLITERATION
Pro
• Safe, quick, easy
• Obliteration achieved in >95%
• Decreased risk of complications
Con
• Multiple GAs
• Accepts low platelet count/splenomegaly
• Accepts risk of minimal hepatic encephalopathy
• Recurrent, potentially unmanageable bleeding (10%) later
MESO-REX BYPASS SURGERY
Pro
• Liver revascularisation
• Increased platelet count
• Effect on spleen
CON
• Anatomically unsuitability (20%)
• Potential for PS shunt
• Thrombosis rate (10%–20%)
• Cosmetically obvious scars (all)
• Complications of laparotomy (e.g. chylous ascites)
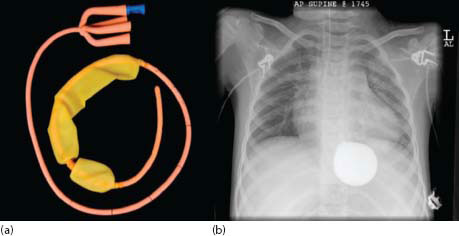
Figure 23.7 (a) Sengstaken tube and (b) X-ray of tube in situ with balloon inflated in correct position. (Reproduced with permission from Jayakumar S, et al., Journal of Pediatric Surgery 2015, 50: 1142–1146.)
***
Nineteen children required an S-B tube to control torrential haematemesis in a 16-year period. Most had underlying intrahepatic disease (e.g. biliary atresia, n = 8) or PVO (n = 5). One was later found to be bleeding from an aortooesophageal fistula, not varices.
• Survived to transplant n = 5
• Survived to portosystemic shunt n = 2
• Survived to complete endoscopic eradication n = 5
• Long-term mortality was high n = 6 (31%).
Table 23.5 Drug therapy for PHT
|
Dose (adult) |
Dose (child) |
Comments |
|
|
Acute |
|||
|
Somatostatin |
250 μg→250 μg/h |
n/a |
|
|
Octreotide |
IV 20–50 μg bolus→25–50 μg/h |
IV 1–2 μg/kg bolus→1–2 μg/kg/h |
|
|
Vapreotide (Sanvar™) |
IV 50 μg bolus→50 μg/h |
n/a |
|
|
Vasopressin (No Longer Used) Analogues |
|||
|
Terlipressin (Glypressin™) |
2 mg bolus IV, repeated, then 1 mg q 4 hour IV |
n/a |
No effect on coronary perfusion |
|
Chronic |
|||
|
Non-Selective ß-Blockade |
|||
|
Propranolol |
20 mg b.d. (initial) |
2–8 mg/kg/day |
Aim to achieve 25% reduction in baseline heart rate |
|
Nadolol |
40 mg o.d. (initial) |
n/a |
Decreased side effects; c.f. propranolol |
|
Depot Somatostatin Analogues |
|||
|
Lanreotide (Somatuline LA™) |
30 mg IM q week |
n/a |
|
|
Octreotide long-acting repeatable (OCT- LAR™) |
n/a |
2.5–20 mg IM/monthly |
A more selective approach was signposted by Bosch et al. [61], who published their landmark study in 1981 showing that an IV somatostatin bolus of 250 μg could reduce wedged hepatic venous pressure by 28% and estimated hepatic blood flow by 17% in cirrhotic adults. Later studies confirmed similar desired effects in both cirrhotics and noncirrhotics and also perhaps more relevant reductions in azygous blood flow and actual variceal pressure. The somatostatin analogue octreotide has a much longer half-life than its parent (50- to 100-fold) and has a high affinity only to somatostatin receptors subtype 2 (sst2). Both agents have a multiplicity of mechanisms, including vasoconstriction of the mesenteric arterial bed and reduction in intrahepatic resistance (in cirrhosis) by sinusoidal dilatation, possibly by an effect on the hepatic stellate cells, and by inhibition of glucagon-induced postprandial splanchnic hyperaemia. The effects of octreotide, however, seem to be more subject to individual variation and may wear off with repetition (tachyphylaxis).
Long-term pharmacological control of PHT is possible and, when used as secondary prophylaxis, reduces rebleeding episodes. Oral propranolol, the most common agent, decreases portal flow by reduction of cardiac output (via β1 receptor antagonism) and diminishes intrahepatic and splanchnic vasoconstriction (via β2 receptor antagonism). of course, there are no randomised controlled trials (RCTs) in children, and sometimes, it can be difficult to attain the usually recommended heart rate reduction of 20%–25% [62].
Long-acting depot preparations of octreotide (e.g. OCTLAR™) are now available, and we have recently reported their use in nine children (2.5–20 mg intramuscularly monthly) who were unsuitable for more definitive surgical procedures [63]. Remarkably, complete cessation of chronic bleeding was achieved in seven without side effects.
23.2.5.3 ENDOSCOPIC MANAGEMENT OF OESOPHAGEAL AND GASTRIC VARICES
IS causing thrombosis and obliteration of variceal columns in the oesophagus has been used successfully in children since the 1980s [6,54,64]. Its primary role has been therapeutic following the onset of bleeding, although it can have a more debatable role in primary prophylaxis.
The nature of the sclerosant used has varied, and there has been little by way of comparative trials. Table 23.4 [65–69] illustrates the composition of some reported in the literature. The current technique can be divided into a purposeful intravariceal injection or one where the aim is paravariceal. The latter is less effective, but with the intention of building up paravariceal inflammatory tissue and inducing neighbouring thrombosis in thin-walled veins over a period of time. IS of oesophageal varices is nowadays restricted to infants where banding is not possible due to size constraints.
Table 23.6 Endoscopic sclerotherapy: sclerosants
|
Sclerosant |
Dosea |
Comments |
References |
|
Oesophageal Varices |
|||
|
Fatty Acid Derivatives |
|||
|
Ethanolamine oleate (2%–5%) |
0.5–2 mL/injection |
Most popular in children, decreased complications |
60–64 |
|
Sodium morrhuate (5%) |
0.5–2 mL/injection (maximum 15 mL) |
Derived from cod liver oil |
|
|
Synthetic |
|||
|
Sodium tetradecyl sulphate (STD) (1% and 3%) (Thrombovar™) |
0.5–1 mL/injection (adult maximum dose is 10 mL per session) |
Popular in North America |
62, 65 |
|
Polidocanolb(0.5%–3% solutions) |
1–2 mL/injection (adult maximum dose is 10 mL) |
Used mainly in Europe and Asia |
|
|
Others |
|||
|
Ethanol (absolute)b |
|||
|
0.5–1 mL (adult maximum dose is 4 mL) |
Increased complications due to tissue destruction |
65 |
|
|
Gastric Varices |
|||
|
Lipiodol and n-butyl cyanoacrylate |
0.5 mL – superglue mixed with 1.5 mL base (usually single only) |
Caution with retrieval of needle through endoscope |
66, 67 |
a There have been no dosage studies in paediatric practice, and most published figures are by comparison with adult studies.
b Not licensed in the United States.
Endoscopic treatment of gastric varices is also possible, typically for those converging on the OGJ from lesser curve (Type 1) or fundus (Type 2) (Box 23.2). IS using most of the sclerosants listed in Table 23.6 is not recommended for gastric varices, as the resultant ulcers are deeper, bleed more and are resistant to treatment. This requires the use of a tissue adhesive and a change in technique [70,71]. Our choice is to use a mixture of Lipiodol™ vehicle (1.5 mL) and n-butyl cyanoacrylate (0.5 mL) (Histoacryl™) in aliquots, with the radio-opaque agent delaying premature hardening by increasing the polymerisation time. The actual injection technique involves intravariceal puncture initially with saline, then the Histoacryl mixture and then saline again to avoid blockage of the endoscope lumen. Tissue adhesives do not actually initiate an inflammatory or thrombotic reaction but physically occlude the variceal lumen and immediately control any bleeding.
The first reports of variceal banding in children involved bands applied individually [72] and were fairly laborious. Today, there are plenty of multiband devices where six bands, at least, are available without replacing the endoscope [73]. Banding usually begins on the right side at the OGJ, targeting those with obvious stigmata first and then moving spirally to target more proximal columns (Figure 23.3a). The suction should be the maximum available and the bander angulated into the varix and wall. Suction should lead to a ‘red-out’ on the screen and then the tripwire fired. Successful band ligation should resemble a ‘mushroom’ with the band securely around the stalk. There will be spontaneous necrosis and passing of the band in perhaps 5–7 days. The resulting mucosal ulcer might bleed, but this is unusual.
There is little comparative data [74] and few RCTs comparing the two procedures in children. An Indian RCT in 49 children with PVO showed equivalent variceal obliteration rates, but achieved in fewer endoscopic sessions (four vs. six) with less in the way of rebleeding rate and major complications (25% vs. 4%) [75].
Prophylactic IS or banding is still a controversial area in children [68,76]. Both reduce the overall incidence of bleeding from oesophageal varices that were eradicated, but IS has a higher rate of complications than banding and is probably not warranted (unless in infants [68]). In addition, both may increase the development of congestive hypertensive gastropathy (although probably not gastric varices). The key predictive endoscopic findings are stigmata (red spots, wales* and ‘varices on varices’) and large size.
23.2.5.3.1 Complications
IS can cause chest pain, mediastinitis, pleural effusion and bacteraemia. Ulceration, particularly if the injection trespassed onto the stomach, can be slow to heal and then be complicated by stenosis. In older series in children, this was the most common complication occurring in up to 40% of the series [16,64]. Cerebral stroke, pulmonary embolism and portal vein embolisation have all been reported in adults following injection of tissue glue into varices [70]. Long-term effects of the tissue glue itself have not been determined, but bucrylate-induced peritoneal sarcomas have also been described in a rat model [77]. Currently, however, this particular tissue adhesive is not available in the United States or Japan.
Although frequently prescribed, H2 receptor antagonists (e.g. ranitidine), omeprazole and sucralfate have had limited efficacy in adult trials [78,79]. By contrast, prophylactic antibiotics at the time of IS does reduce bacteraemia [80,81], although that is a much less common event in children.
Specific complications of banding include inadvertent mucosal tears, induced variceal rupture due to the suction and incomplete band control and suction of the entire circumference into the banding chamber, leading to oesophageal stenosis. Similarly, if too many bands are placed around the circumference, this will also lead to stenosis. Oesophageal perforation is the most dangerous complication, a risk which is exacerbated in the younger child. Still, the overall incidence of these complications is substantially less than that associated with variceal IS, rendering the latter almost obsolete for the older child.
23.2.5.4 RADIOLOGICAL MANAGEMENT OF OESOPHAGEAL AND GASTRIC VARICES
The TIPS procedure is a well-established tool to manage severe complications of PHT due to cirrhosis or fibrosis in adults, but there is very little real experience in children, and its use is confined to only the larger centres. Di Giorgio et al., from a large North Italian centre [82], recently reported a large series of 11 children (ranging in ages from 2.2 to 18 years) with PHT who underwent TIPS using a polytetrafluoroethylene (PTFE)-covered stent. There was effective diminution in the portosystemic gradient and resolution of complications in all but one. Interestingly, no patient developed overt hepatic encephalopathy. A recent series from Seattle, Washington, described TIPS in 12 predominantly adolescents with 8–10 mm PTFE shunts [83]. All were successful, with none needing revision in a median follow-up period of more than 20 months. Clearly, size is an issue in children, but even the smallest can still be considered for TIPS. The youngest appears to be an 8-month-old child with trisomy 21 born with biliary atresia and suffering recurrent bleeding episodes from varices. She underwent successful TIPS with a 6 mm bare stent, requiring a larger exchange some months later [84].
Other percutaneous techniques sometimes used in adults, such as embolisation of varices, are little used in children. One exception may be Burke et al.’s report [85] of a creation of a percutaneous mesocaval shunt to control variceal bleeding in a child, although this relied upon an unusual anatomical approximation of cava and retroperitoneal vessel.
1. Balfour GW, Stewart TG. Case of enlarged spleen complicated with ascitis and both varicose dilatation and thrombosis of portal vein. Edinburgh Medical Journal 1869; 14: 55.
2. Crafoord C, Frenckner P. New surgical treatment of varicose veins of the oesophagus. Acta Otolaryngologica (Stockholm) 1939; 27: 422–429.
3. Sarin SK, Lahoti D, Saxena SP, et al. Prevalence, classification and natural history of gastric varices: a long-term follow-up study in 568 portal hypertension patients. Hepatology 1992; 16: 1343–1349.
4. Lautz TB, Sundaram SS, Whitington PF, et al. Growth impairment in children with extrahepatic portal vein obstruction is improved by mesenterico-left portal vein bypass. Journal of Pediatric Surgery 2009; 44: 2067–2070.
5. Mehrotra RN, Bhatia V, Dabadghao P, et al. Extrahepatic portal vein obstruction in children: anthropometry, growth hormone, and insulin-like growth factor I. Journal of Pediatric Gastroenterology & Nutrition 1997; 25: 520–523.
6. Maksoud-Filho JG, Gonçalves ME, Cardoso SR, et al. Long-term follow-up of children with extrahepatic portal vein obstruction: impact of an endoscopic sclerotherapy program on bleeding episodes, hepatic function, hypersplenism, and mortality. Journal of Pediatric Surgery 2009; 44: 1877–1883.
7. Mack CL, Zelko FA, Lokar J, et al. Surgically restoring portal blood flow to the liver in children with primary extra-hepatic portal vein thrombosis improves fluid neurocognitive ability. Pediatrics 2006; 117: e405–e412.
8. D’Antiga L, Dacchille P, Boniver C, et al. Clues for minimal hepatic encephalopathy in children with noncirrhotic portal hypertension. Journal of Pediatric Gastroenterology & Nutrition 2014; 59: 689–694.
9. Sharma P, Sharma BC. Lactulose for minimal hepatic encephalopathy in patients with extrahepatic portal vein obstruction. Saudi Journal of Gastroenterology 2012; 18: 168–172.
10. Hunt AH. Compression of the common bile duct by an enlarging collateral vein in case of portal hypertension. British Journal of Surgery 1965; 52: 636–637.
11. Shin SM, Kim S, Lee JW, et al. Biliary abnormalities associated with portal biliopathy: evaluation on MR cholangiography. AJR American Journal of Roentgenology 2007; 188: W341–W347.
12. Chiu B, Superina R. Extrahepatic portal vein thrombosis is associated with an increased incidence of cholelithiasis. Journal of Pediatric Surgery 2004; 39: 1059–1061.
13. Perlemuter G, Béjanin H, Fritsch J, et al. Biliary obstruction caused by portal cavernoma: a study of 8 cases. Journal of Hepatology 1996; 25: 58–63.
14. Dhiman RK, Puri P, Chawla Y, et al. Biliary changes in extrahepatic portal venous obstruction: compression by collaterals or ischemic? Gastrointestinal Endoscopy 1999; 50: 646–652.
15. Webb LJ, Sherlock S. The aetiology, presentation and natural history of extra-hepatic portal venous obstruction. Quarterly Journal of Medicine 1979; 48: 627–639.
16. El-Hamid N, Taylor RM, Marinello D, et al. Aetiology and management of extrahepatic portal vein obstruction in children: King’s College Hospital experience. Journal of Pediatric Gastroenterology & Nutrition 2008; 47: 630–634.
17. Ogren M, Bergqvist D, Bjorck M, et al. Portal vein thrombosis: prevalence, patient characteristics and lifetime risk: a population study based on 23,796 consecutive autopsies. World Journal of Gastroenterology 2006; 12: 2115–2119.
18. Weiss B, Shteyer E, Vivante A, et al. Etiology and long-term outcome of extrahepatic portal vein obstruction in children. World Journal of Gastroenterology 2010; 16: 4968–4972.
19. Janssen HL, Wijnhoud A, Haagsma EB, et al. Extrahepatic portal vein thrombosis: aetiology and determinants of survival. Gut 2001; 49: 720–724.
20. Bhattacharyya M, Makharia G, Kannan M, et al. Inherited prothrombotic defects in Budd-Chiari syndrome and portal vein thrombosis: a study from North India. American Journal of Clinical Pathology 2004; 121: 844–847.
21. Heller C, Schobess R, Kurnik K, et al. Abdominal venous thrombosis in neonates and infants: role of prothrombotic risk factors – a multicentre case-control study. For the Childhood Thrombophilia Study Group. British Journal of Haematology 2000; 111: 534–539.
22. Kanazawa H, Sakamoto S, Matsunami M, et al. Technical refinement in living-donor liver transplantation for hepatoblastoma with main portal vein tumor thrombosis – a pullout technique. Pediatric Transplantation 2014; 18: E266–E269.
23. Schwartz DS, Gettner PA, Konstantino MM, et al. Umbilical venous catheterization and the risk of portal vein thrombosis. Journal of Pediatrics 1997; 13: 760–762.
24. Butler-O’Hara M, Buzzard CJ, Reubens L, et al. A randomized trial comparing long-term and short-term use of umbilical venous catheters in premature infants with birth weights of less than 1251 grams. Pediatrics 2006; 118: e25–e35.
25. Morag I, Epelman M, Daneman A, et al. Portal vein thrombosis in the neonate: risk factors, course, and outcome. Journal of Pediatrics 2006; 148: 735–739.
26. Rossi E, Michelini ME, Pignatti CB, et al. A case of portal vein thrombosis after laparoscopy-assisted splenectomy and cholecystectomy in a child. Journal of Pediatric Surgery 2007; 42: 1449–1451.
27. Ikeda M, Sekimoto M, Takiguchi S, et al. High incidence of thrombosis of the portal venous system after laparoscopic splenectomy: a prospective study with contrast-enhanced CT scan. Annals of Surgery 2005; 241: 208–216.
28. Pietrabissa A, Moretto C, Antonelli G, et al. Thrombosis in the portal venous system after elective laparoscopic splenectomy. Surgical Endoscopy 2004; 18: 1140–1143.
29. Pietrobattista A, Luciani M, Abraldes JG, et al. Extrahepatic portal vein thrombosis in children and adolescents: influence of genetic thrombophilic disorders. World Journal of Gastroenterology 2010; 16: 6123–6127.
30. Dubuisson C, Boyer-Neumann C, Wolf M, et al. Protein C, protein S and antithrombin III in children with portal vein obstruction. Journal of Hepatology 1997; 27: 132–135.
31. Pinto RB, Silveira TR, Bandinelli E, Röhsig L. Portal vein thrombosis in children and adolescents: the low prevalence of hereditary thrombophilic disorders. Journal of Pediatric Surgery 2004; 39: 1356–1361.
32. Fisher NC, Wilde JT, Roper J, Elias E. Deficiency of natural anticoagulant proteins C, S, and antithrombin in portal vein thrombosis: a secondary phenomenon? Gut 2000; 46: 534–539.
33. Stringer MD, Heaton ND, Karani J, et al. Patterns of portal vein occlusion and their aetiological significance. British Journal of Surgery 1994; 81: 1328–1331.
34. Mikkelsen WP, Edmondson HA, Peters RL, et al. Extra-and intrahepatic portal hypertension without cirrhosis (hepatoportal sclerosis). Annals of Surgery 1965; 162: 602–620.
35. Krishnan P, Fiel MI, Rosenkrantz AB, et al. Hepatoportal sclerosis: CT and MRI appearance with histopathologic correlation. AJR American Journal of Roentgenology 2012; 198: 370–376.
36. Dhiman RK, Chawla Y, Vasishta RK, et al. Noncirrhotic portal fibrosis (idiopathic portal hypertension): experience with 151 patients and a review of the literature. Journal of Gastroenterology & Hepatology 2002; 17: 6–16.
37. Carson JA, Tunell WP, Barnes P, Altshuler G. Hepatoportal sclerosis in childhood: a mimic of extrahepatic portal vein obstruction. Journal of Pediatric Surgery 1981; 16: 291–296.
38. Hoffman HD, Stockland B, von der Heyden U. Membranous obstruction of the inferior vena cava with the Budd-Chiari syndrome in children: a report of nine cases. Journal of Pediatric Gastroenterology & Nutrition 1987; 6: 878–884.
39. Wyatt HA, Mowat AP, Layton M. Paroxysmal nocturnal haemoglobinuria and Budd-Chiari syndrome. Archives of Disease in Childhood 1995; 72: 241–242.
40. Gentil-Kocher S, Bernard O, Brunelle F, et al. Budd-Chiari syndrome in children: report of 22 cases. Journal of Pediatrics 1988; 113 (1 Pt 1): 30–38.
41. Cauchi JA, Oliff S, Baumann U, et al. The Budd-Chiari syndrome in children: the spectrum of management. Journal of Pediatric Surgery 2006; 41: 1919–1923.
42. Bulley SR, Strahm B, Doyle J, Dupuis LL. Defibrotide for the treatment of hepatic veno-occlusive disease in children. Pediatric Blood Cancer 2007; 48: 700–704.
43. Bozorgmanesh A, Selvam DA, Caridi JG. Budd-Chiari syndrome: hepatic venous web outflow obstruction treated by percutaneous placement of hepatic vein stent. Seminars in Interventional Radiology 2007; 24: 100–105.
44. Srinivasan P, Rela M, Prachalias A, et al. Liver transplantation for Budd-Chiari syndrome. Transplantation 2002; 73: 973–977.
45. Keating GM. Defibrotide: a review of its use in severe hepatic veno-occlusive disease following haematopoietic stem cell transplantation. Clinical Drug Investigation 2014; 34: 895–904.
46. Gryboski JD, Clemett A. Congenital hepatic artery aneurysm with superior mesenteric artery insufficiency: a steal syndrome. Pediatrics 1967; 39: 344–347.
47. Davenport M, Redkar R, Howard ER, Karani J. Arterioportal hypertension: a rare complication of partial hepatectomy. Pediatric Surgery International 1999; 15: 543–545.
48. Heaton ND, Davenport M, Karani J, Mowat AP, Howard ER. Congenital hepatoportal arteriovenous fistula. Surgery 1995; 117: 170–174.
49. Vauthey JN, Tomczak RJ, Helmberger T, et al. The arterioportal fistula syndrome: clinicopathologic features, diagnosis, and therapy. Gastroenterology 1997; 113: 1390–1401.
50. Sutcliffe R, Mieli-Vergani G, Dhawan A, et al. A novel treatment of congenital hepatoportal arteriovenous fistula. Journal of Pediatric Surgery 2008; 43: 571–573.
51. Francoz C, Belghiti J, Vilgrain V, et al. Splanchnic vein thrombosis in candidates for liver transplantation: usefulness of screening and anticoagulation. Gut 2005; 54: 691–697.
52. de Ville de Goyet J, D’Ambrosio G, Grimaldi C. Surgical management of portal hypertension in children. Seminars in Pediatric Surgery 2012; 21: 219–232.
53. Sharif K, McKiernan P, de Ville de Goyet J. Mesoportal bypass for extrahepatic portal vein obstruction in children: close to a cure for most! Journal of Pediatric Surgery 2010; 45: 272–276.
54. Stringer MD, Howard ER. Long-term outcome after injection sclerotherapy for oesophageal varices in children with extrahepatic portal hypertension. Gut 1994; 35: 257–259.
55. Thomas V, Jose T, Kumar S. Natural history of bleeding after esophageal variceal eradication in patients with extrahepatic portal venous obstruction; a 20-year follow-up. Indian Journal of Gastroenterology 2009; 28: 206–211.
56. Zargar SA, Yattoo GN, Javid G, et al. Fifteen-year follow up of endoscopic injection sclerotherapy in children with extrahepatic portal venous obstruction. Journal of Gastroenterology & Hepatology 2004; 19: 139–145.
57. Itha S, Yachha SK. Endoscopic outcome beyond esophageal variceal eradication in children with extrahepatic portal venous obstruction. Journal of Pediatric Gastroenterology & Nutrition 2006; 42: 196–200.
58. Guérin F, Bidault V, Gonzales E, et al. Meso-Rex bypass for extrahepatic portal vein obstruction in children. British Journal of Surgery 2013; 100: 1606–1613.
59. Bosch J, Thabut D, Bendtsen F, et al. Recombinant factor VIIa for upper gastrointestinal bleeding in patients with cirrhosis: a randomized, double-blind trial. Gastroenterology 2004; 127: 1123–1130.
60. Jayakumar S, Patel S, Davenport M, Ade-Ajayi N. Surviving Sengstaken. Journal of Pediatric Surgery 2015; 50: 1142–1146.
61. Bosch J, Kravetz D, Rodes J. Effects of somatostatin on hepatic and systemic hemodynamics in patients with cirrhosis of the liver: comparison with vasopressin. Gastroenterology 1981; 80: 518–525.
62. Ozsoylu S, Kocak N, Yuce A. Propranolol therapy for portal hypertension in children. Journal of Pediatrics 1985; 106: 317–321.
63. O’Meara M, Cicalese MP, Bordugo A, et al. Successful use of long-acting octreotide for intractable chronic gastrointestinal bleeding in children. Journal of Pediatric Gastroenterology & Nutrition 2015; 60: 48–53.
64. Stringer MD, Howard ER. Long-term outcome after injection sclerotherapy for esophageal varices in children with extrahepatic portal hypertension. Gut 1994; 35: 257–259.
65. Kitano S, Wada H, Yamaga H, et al. Comparative effects of 5% ethanolamine oleate versus 5% sodium morrhuate for sclerotherapy of oesophageal varices. Journal of Gastroenterology & Hepatology 1991; 6: 476–480.
66. Kitano S, Iso Y, Yamaga H, et al. Trial of sclerosing agents in patients with oesophageal varices. British Journal of Surgery 1988; 75: 751–753.
67. Gonçalves ME, Cardoso SR, Maksoud JG. Prophylactic sclerotherapy in children with esophageal varices: long-term results of a controlled prospective randomized trial. Journal of Pediatric Surgery 2000; 35: 401–405.
68. Duché M, Habès D, Roulleau P, et al. Prophylactic endoscopic sclerotherapy of large esophagogastric varices in infants with biliary atresia. Gastrointestinal Endoscopy 2008; 67: 732–737.
69. Sarin SK, Kumar A. Sclerosants for variceal sclerotherapy: a critical appraisal. American Journal of Gastroenterology 1990; 85: 641–649.
70. Thakeb F, Salama Z, Salama H, et al. The value of combined use of N-butyl-2-cyanoacrylate and ethanolamine oleate in the management of bleeding esophagogastric varices. Endoscopy 1995; 27: 358–364.
71. Kang EJ, Jeong SW, Jang JY, et al. Long-term result of endoscopic Histoacryl (N-butyl-2-cyanoacrylate) injection for treatment of gastric varices. World Journal of Gastroenterology 2011; 17: 1494–1500.
72. Price MR, Sartorelli KH, Karrer FM, et al. Management of esophageal varices in children by endoscopic variceal ligation. Journal of Pediatric Surgery 1996; 31: 1056–1059.
73. McKiernan PJ, Beath SV, Davison SM. A prospective study of endoscopic esophageal variceal ligation using a multiband ligator. Journal of Pediatric Gastroenterology & Nutrition 2002; 34: 207–211.
74. Kim SJ, Oh SH, Jo JM, et al. Experiences with endoscopic interventions for variceal bleeding in children with portal hypertension: a single center study. Journal of Pediatric Gastroenterology, Hepatology & Nutrition 2013; 16: 248–253.
75. Zargar SA, Javid G, Khan BA, et al. Endoscopic ligation compared with sclerotherapy for bleeding esophageal varices in children with extrahepatic portal venous obstruction. Hepatology 2002; 36: 666–672.
76. Duché M, Ducot B, Ackermann O, et al. Experience with endoscopic management of high-risk gastroesophageal varices, with and without bleeding, in children with biliary atresia. Gastroenterology 2013; 145: 801–807.
77. Binmoeller KF, Soehendra N. ‘Superglue’: the answer to variceal bleeding and fundal varices? Endoscopy 1995; 27: 392–396.
78. Garg PK, Sidhu SS, Bhargava DK. Role of omeprazole in prevention and treatment of postendoscopic variceal sclerotherapy esophageal complications: double-blind randomized study. Digestive Diseases Science 1995; 40: 1569–1574.
79. Paquet KJ, Koussouris P, Keinath R, et al. Comparison of sucralfate with placebo in the treatment of esophageal ulcers following therapeutic sclerotherapy of esophageal varices: a prospective controlled randomized trial. American Journal of Medicine 1991; 91: S147S–S150.
80. Rolando N, Gimson A, Philpott-Howard J, et al. Infectious sequelae after endoscopic sclerotherapy of oesophageal varices: role of antibiotic prophylaxis. Journal of Hepatology 1993; 18: 290–294.
81. Selby WS, Norton ID, Pokorny CS, Benn RAV. Bacteremia and bacterascites after endoscopic sclerotherapy for bleeding esophageal varices and prevention by intravenous cefotaxime: a randomized trial. Gastrointestinal Endoscopy 1994; 40: 680–684.
82. Di Giorgio A, Agazzi R, Alberti D, et al. Feasibility and efficacy of transjugular intrahepatic portosystemic shunt (Tips) in children. Journal of Pediatric Gastroenterology, Hepatology & Nutrition 2012; 54: 594–600.
83. Vo NJ, Shivaram G, Andrews RT, et al. Midterm follow-up of transjugular intrahepatic portosystemic shunts using polytetrafluoroethylene endografts in children. Journal of Vascular Interventional Radiology 2012; 23: 919–924.
84. Chlapoutaki CE, Franchi-Abella S, Habes D, Pariente D. Custom-made covered transjugular intrahepatic portosystemic shunt (TIPS) in an infant with trisomy 22 and biliary atresia. Pediatric Radiology 2009; 39: 739–742.
85. Burke C, Taylor AG, Ring EJ, Kerlan RK Jr. Creation of a percutaneous mesocaval shunt to control variceal bleeding in a child. Pediatric Radiology 2013; 43: 1218–1220.
* Jean Cruveilhier (1791–1874), French anatomist who also described features of multiple sclerosis, and Paul Clemens von Baumgarten (1848–1928), German pathologist who along with Robert Kock identified the tuberculosis bacillus.
† Dame Sheila Sherlock (1918–2001), Doyenne of British hepatology, worked at Royal Free Hospital as first female professor of medicine in the United Kingdom.
† Hans Chiari (1851–1916), Austrian pathologist; also famous for naming the Arnold–Chiari malformation of the brainstem, and later (1899) contributed the histological and postmortem features.
† Arthur Blakemore (1897–1970), American vascular surgeon.
* Wales – not the country. A term used to describe the ridges in the fabric corduroy.
* Caput medusae–head of Medusa, one of a trio of mythical Gorgons with snakes for hair, which if sighted turned the viewer to stone.
* George Budd (1808–1882), English physician who worked at King’s College Hospital, London, and described three cases related to abscess formation and phlebitis in 1845.
* John Langdon Down (1828–1896), English physician. He characterised this group of children using the term Mongolism in a descriptive sense in 1866. Down’s syndrome became the preferred term in 1961, not least due to pressure from the Republic of Mongolia.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
[/not-level-membership-for-pediatrics-category]
