Figure 3.1 Breakdown of haem moiety via biliverdin to bilirubin.
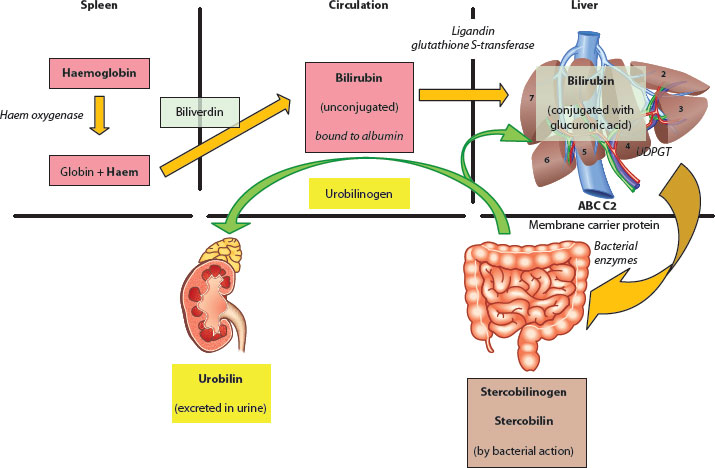
Figure 3.2 Overview of excretion of bilirubin and its metabolism.
It is the main by-product (via biliverdin) from the oxidation and breakdown of the haem moiety arising from red cell destruction (80%) and other proteins, such as myoglobin and catalase (20%), in the reticuloendothelial system tissue of the spleen, bone marrow and so forth. The rate-limiting step in this sequence is microsomal haem oxygenase, and about 250–400 mg/day of bilirubin can be produced in an adult (Figures 3.1 and 3.2).
Bilirubin is a fat-soluble compound which has to be transported attached to albumin on a high-affinity binding site (1:1) within the circulation to the sinusoids of the liver. There, albumin-bound bilirubin is able to pass through the fenestrations and, via an active transport process in the hepatocyte membrane (binding to glutathione S-transferase, aka ligandin), moves to the endoplasmic reticulum, where it undergoes conjugation. The aim is to alter its physiochemical properties into a polar, water-soluble molecule ready for excretion. The main carbohydrate conjugate is glucuronic acid, but glucose and xylose are also possible; the main enzyme involved is therefore uridine diphosphate glucuronosyl transferase (UDPGT). Gilbert* syndrome is caused by functional defects in this enzyme and leads to an essentially benign, recurrent unconjugated jaundice, particularly at times of stress or fasting.
Jaundice is clinically detectable in white skin and sclera when it is >50 μmol/L (≈3 mg/dL).
The classical method of measuring bilirubin in blood uses the diazo reaction and is known as the van den Bergh test, with two forms being identified. The direct reaction estimates the conjugated bilirubin fraction, and with the addition of a promoter (alcohol), the indirect reaction measures the unconjugated fraction.
The normal total bilirubin range is 5–20 μmol/L (≈0.3–1.2 mg/dL) (>95% unconjugated).
Up to 60% of newborn infants will exhibit so-called physiological jaundice, and in these, it is always unconjugated, appearing from 24 h of life and usually fading in the second week. Its origin is multifactorial and appears related to immature liver enzyme systems (such as glucuronyl transferase), a higher turnover of red cells (incorporating the transition from fetal to adult haemoglobin) and, occasionally, to the effects of breastfeeding.
Conjugated bilirubin (mostly as the diglucuronide form) is then secreted into the biliary canaliculus via a complex carrier-mediated mechanism. There are a number of inborn errors of metabolism that are characterised by a failure of this conjugation process, but the classical example is that of the Crigler–Najjar* syndrome [5], where there are high serum levels of unconjugated bilirubin due to defects in the UDPGT gene.
Bilirubin is further metabolised within the distal small intestine by bacterial β-glucuronidase action to urobilinogen compounds, and a proportion is reabsorbed in the distal ileum and recirculated enterohepatically. Further conversion in the colon occurs to stercobilin and stercobilinogen, both of which give faeces its characteristic colour and odour. Acquisition of such organisms is age dependent and not really complete until the second year of life [6] (Boxes 3.2 and 3.3).
3.3.3 Bile acids
Bile acids are synthesised in the hepatocyte by a complex multienzyme process (with at least 17 steps) from cholesterol. This, in itself, is an almost completely insoluble, hydrophobic compound, but with bile acids and phospholipids it is able to exist in a clear, single-phase solution in the bile. Bile acids have four main functions:
1. Main pathway for degradation and excretion of excess cholesterol
2. Maintaining solubility of cholesterol in bile
3. Increasing intestinal dietary lipid absorption (by formation of mixed micelles)
4. Facilitating fat-soluble vitamin absorption
There are two primary bile acids, cholic (~30% of total bile acids) and chenodeoxycholic (~45%) acid, and two secondary bile acids, deoxycholic and lithocholic acid. The latter compounds are the product of anaerobic colonic bacterial action and later portal venous reabsorption [7]. All bile acids are conjugated in the hepatocyte with the amino acids taurine and glycine, and secreted into the biliary canaliculus by a series of membrane-bound enzymes (e.g. bile salt export pump), rendering them impermeable to reabsorption across cell membranes (Figure 3.3). Intestinal deconjugation (again by bacterial action) occurs to allow distal ileal reabsorption and recirculation.
The enterohepatic circulation is a complex uptake mechanism in the ileal enterocyte and via the hepatocyte sinusoidal membrane that efficiently recycles bile acids [8]. Both the ileal Na+–bile acid transporter and the hepatocyte Na+-dependent–bile acid transporter are homologous but different proteins. Sinusoidal uptake from portal venous blood ensures a very low circulating level of total bile acids (<5 μmol/L).
Actual bile acid loss in faeces is <5% of the bile acid pool, but even this may have pathological consequences, as highly hydrophobic molecules such as deoxycholic acid are cytotoxic and may act a colon tumour promoter.
3.3.4 Hepatic (or canalicular) bile
Hepatic bile is the fluid secreted directly into the biliary canaliculus and is classically divided into two components, named because of the predominant organic solute:
1. Bile acid–dependent flow (BADF) (~50%): Actively transported into canaliculus with osmotic movement of water and electrolytes via paracellular junctions.
2. Bile acid–independent flow (BAIF): Related to secretion of osmotically active reduced glutathione and bicarbonate into the canaliculus. The former hydrolyses to glutamate, cysteine and glycine by the action of γ-glutamyl transpeptidase (γ-GT), which then appears in the bile.
There is then an active process moderated by the cholangiocytes of the biliary canaliculi which tends to dilute and alkalinise by the secretion of bicarbonate and chloride and reabsorption of glucose. This latter process is achieved by two glucose cotransporters, SGLT1 and GLUT1, on the apical membrane of the cholangiocyte and leads to very low levels of glucose in bile (<1 mmol/L). Cholangiocytes also have the cystic fibrosis transductance regulator (CFTR) on their membranes, and this too has a key role in the alkalinisation of bile. About one-half of the hepatic bile output enters the gallbladder, where it is then concentrated by a factor of 4–6 by removal of water and electrolytes.
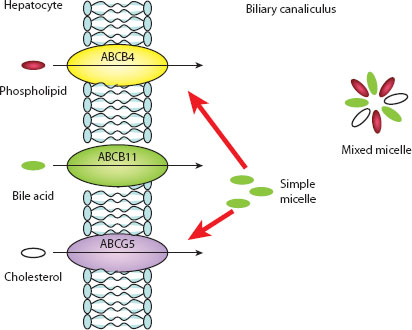
Figure 3.3 Excretion and formation of mixed micelles. (Reproduced by permission of author, Ms. E. Makin.)
3.3.5 Hormonal control of bile flow
There are a variety of foregut hormones which have an action on the flow of bile and produce a phasic response to the oral ingestion of food. This is largely triggered by the presence of acid and peptides on the S-cells in the duodenum and proximal jejunum.
1. Secretin * is a polypeptide of 27 amino acids that is secreted by the S-cells of the duodenal mucosa with stimulatory effects on canalicular bile flow by increasing bicarbonate secretion. One other mechanism involved is via its effect on aquaporin-1 water channels, present in the apical and basolateral membrane of cholangiocytes [9]. It also regulates the output of pancreatic juice, again by increasing bicarbonate secretion, and inhibits gastric acid production from the parietal cells, leading to an alkaline duodenal environment. Secretin is also now believed to be a neuropeptide, although its physiological role remains to be defined, but there is also some evidence of it playing a role in osmoregulation by its action on various extravisceral sites.
2. Bombesin (aka gastrin-releasing peptide) is a polypeptide of 14 amino acids which also stimulates biliary bicarbonate and fluid output [10].
3. Vasoactive intestinal polypeptide is believed to be the principal noncholinergic, nonadrenergic neurotransmitter in the gastrointestinal (GI) tract and has an effect on cholangiocytes to stimulate bicarbonate secretion and increase bile flow [11].
4. Cholecystokinin (CCK) is actually a family of hormones, with the most active being an octapeptide (CCK-8). It has structural similarities to gastrin and shares some of the same receptors. It is secreted by I-cells in the duodenal and small bowel mucosa in response to protein and fat, with its main effects on the liver, gallbladder, pancreas and, interestingly, central nervous system (CNS). CCK causes the gallbladder and, to a lesser extent, common hepatic duct contraction and relaxes the sphincter of Oddi† by a direct effect on smooth muscle which is independent of autonomic innervation.
In the pancreas, it stimulates increased enzyme output. CCK receptors are widely distributed in the CNS, and it can therefore act as a neurotransmitter, causing nausea, anxiety and perhaps inhibition of hunger (anorexigenic).
The physiological role of other GI hormones is still unclear, but a number appear to have stimulatory hepatobiliary actions. For instance, gastrin has a weak secretin-like effect, and glucagons can also induce BAIF choleresis. The range of inhibitory hormones is less but includes somatostatin, substance P and the distal gut hormone pancreatic peptide YY. Somatostatin, in particular, has been shown to reduce total hepatic bile output, reduce bile acid secretion and stimulate the sphincter of Oddi, impairing bile flow [12].
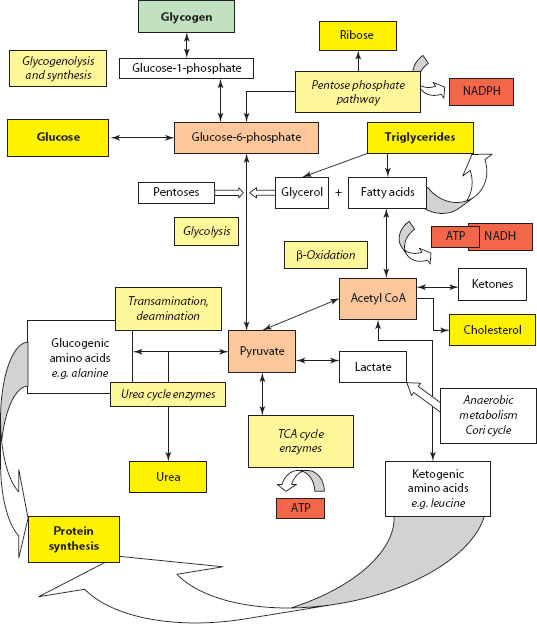
Figure 3.4 Schematic overview of metabolic pathways.
The CNS has an absolute requirement for glucose as a metabolic substrate, at least in the early part of the fasting period, and the liver plays a central and primary role in the control of its level in the blood (Figure 3.4). Control is achieved by a complex interplay between three principal metabolic pathways:
1. Glycogenesis – creation of storage carbohydrate polymer from glucose and controlled by the action of the enzyme complex, glycogen synthase, and regulated by c-AMP-dependent protein kinase, promoted by the action of insulin.
2. Glycogenolysis – creation and breakdown of storage carbohydrate polymer from glucose and controlled by the enzyme glycogen phosphorylase, which produces glucose-1-phosphate, isomerising to glucose-6-phosphate for glucolysis and energy release in muscle, and for circulating glucose by the action of glucose-6-phosphatase in liver. Hormonal control is by the action of glucagon, epinephrine (adrenaline) and cortisol.
3. Gluconeogenesis – creation of glucose from other molecules (e.g. amino acids).
Glycogen is found principally in the liver (making up ~10% of its mass) and muscle. However, because there is a greater overall muscle mass, most of the body’s glycogen is found in the latter.
About 40% of an oral glucose load is stored by glycogen synthesis, principally in the liver (~65%), although there is also significant storage by muscle (~25%) and kidney (~10%). About 60% of this liver glycogen arises as a result of the direct pathway described already, with the remainder indirectly via conversion from 3-carbon compounds such as lactate, pyruvate and alanine [13,14].
The CNS requirement for circulating glucose means that other mechanisms must exist following extinction of glycogen storage, that is gluconeogenesis. The process of gluconeogenesis is exclusive to the liver and the kidney, although there are substrate differences; for instance, alanine gluconeogenesis only occurs in the liver, while glutamine gluconeogenesis primarily occurs in the kidney [15]. The most important substrates are pyruvate, lactate, glycerol and glucogenic amino acids (especially alanine).
3.4.1 Ketone bodies (acetoacetate, β-hydroxybutyrate and acetone)
These circulate normally in low concentrations (<0.3 mM) and are perfectly capable of crossing the blood–brain barrier to be used as CNS fuel if given the chance (e.g. in the fasted state). They are also the preferential glucose-sparing fuel in the heart muscle and renal cortical tissue. Ketogenesis occurs in the hepatocyte mitochondrial matrix, where free fatty acids (FFAs) are first broken down to acetyl CoA via β-oxidation. The acetyl CoA is then used in ketogenesis. The primary regulator of ketone body synthesis is fatty acid availability; hence, increases in plasma FFA concentration (by high glucagon and low insulin levels) cause increased β-oxidation and high concentrations of acetyl CoA, resulting in ketone body synthesis (Box 3.4).
3.5 PROTEIN SYNTHESIS AND NITROGEN METABOLISM
About 25% of all protein synthesis occurs in the liver, with the major products being albumin, α1-globulins (e.g. high-density [HDL] and very high-density [VHDL] lipoproteins, glycoproteins and haptoglobulins), α2-globulins (e.g. ceruloplasmin, plasminogen and prothrombin), β-globulins (e.g. low-density [LDL] and very low-density [VLDL] lipo-proteins and transferrin) and coagulation factors.
3.5.1 Albumin
The main circulating plasma protein in fetal life is liver derived and termed α-fetoprotein (AFP), which in postnatal life is replaced by albumin. The normal range 35–40 g/dL, and it has a half-life of ~20 days.
Albumin consists of 584 amino acids with a molecular weight of ~66,500 and is synthesised in the rough endoplasmic reticulum of the hepatocyte and secreted via a series of intermediates (preproalbumin and prealbumin) into the circulation directly via the space of Disse or via hepatic lymphatics. There is a continual flow of albumin into the extravascular space (about 4% per hour – the so-called transcapillary escape rate).
It has three principal roles:
1. Maintenance of intravascular oncotic pressure (up to 80%)
2. Binding of drugs and transport (e.g. bilirubin)
3. Scavenging of free radicals
The site of albumin degradation is not known with precision but is probably the sinusoidal endothelial network in the liver. There is no storage form of albumin, and therefore changes in its level depend on its synthesis or its loss from the body.
Impaired albumin synthesis is seen in protein malnutrition and cirrhosis (either by a reduced liver cell mass per se or by nutrient diminution consequent on disordered vascularity). It is also under a degree of hormonal control and diminishes with low growth hormone levels and increases, at least in normal subjects, with exogenous administration [16]. Stress or inflammation tends to cause an initial fall in synthesis due to a rise in production of acute phase proteins, but later there is an actual increase in albumin synthesis. Low plasma albumin levels are often associated with fluid shifts rather than any actual diminution in synthesis.
Lactate is a product of anaerobic glycolysis, principally in muscle, which can be taken up and metabolised in various tissues (e.g. myocardium) to provide energy. Lactate can be a substrate for gluconeogenesis, but only in the liver and the kidney.
Glucose–lactate–glucose cycling between the tissues is known as the Cori * cycle. The total amount of lactate involved is large (up to 1500 mmol/day in adults), and if liver mitochondrial metabolism is not adequate, then the condition of lactic acidosis results.
The normal lactate concentration in blood is 0.5–2 mmol/L; >5 mmol/L implies severe lactic acidosis (with decreasing pH).
Lactic acidosis is divided by cause into
1. Type A – caused by poor tissue perfusion and increased production or decreased utilisation of lactate caused by liver disease, decreased gluconeogenesis and so forth
2. Type B – no clinical evidence of poor tissue perfusion or oxygenation exists, but related to systemic disease, diabetes or malignancy (B1); drug induced (e.g. metformin, isoniazid, halothane or salbutamol) (B2); or inborn error of metabolism (B3)
* Carl (1896–1984) and Gerty Cori (née Radnitz) (1896–1957), Czech physiologists who won the Nobel Prize for this work in 1947. Gerty was the first female recipient of the Nobel Prize for Medicine.
3.5.2 Nitrogen metabolism
The liver is the major site of nitrogen metabolism in the body. This arises either endogenously from amino acid and nucleic acid breakdown or exogenously as ingested dietary proteins which are metabolised by anaerobic and aerobic colonic bacteria to ammonia and carried in the portal venous system. Nitrogen, derived from excess amino acids, can be eliminated via transamination, deamination and urea formation, with the residual carbon skeleton being conserved as carbohydrate (gluconeogenesis) or synthesised to fatty acids.
Amino acids can therefore be categorised as either
1. Glucogenic: That is, those that produce pyruvate or tricarboxylic acid (TCA) cycle intermediates (e.g. α-ketoglutarate or oxaloacetate). Most amino acids are at least partly glucogenic.
2. Ketogenic: That is, those that metabolise directly to acetyl CoA or acetoacetyl CoA (e.g. lysine and leucine).
A smaller number of amino acids (isoleucine, phenylalanine, threonine, tryptophan and tyrosine) have both glucogenic and ketogenic potential (Box 3.5).
Dietary fat absorbed into the enterocyte can be
1. Repackaged into chylomicron or lipoprotein particles for distribution to the body tissues
2. Stored within the enterocyte in a lipid droplet
3. Partitioned into other lipids, including cholesteryl ester or phospholipid
4. Oxidised
Ammonia, as the end product of nitrogen metabolism, is excreted as water-soluble urea in the urea cycle (intermediates arginine, ornithine and citrulline).
Hyperammoniaemia (>40 mmol/L) occurs in a number of liver disorders:
1. Primary urea cycle enzyme defects (rarely)
2. Acute liver failure, cirrhosis and so forth
3. Portosystemic shunts
Elevated serum ammonia is related to the development of hepatic encephalopathy, although the actual mechanism is multifactorial and still debated. It includes changes in cerebral haemodynamics, production of ‘false neurotransmitters’, activation of central γ-aminobutyric acid–benzodiazepine receptors by endogenous ligands, zinc deficiency and alterations in cerebral metabolism [18,19].
Actual measurement of plasma ammonia is difficult because it is so unstable – ideally, it should be arterial, collected on ice, separated from red cells within 30 min and measured within 3 h.
There is an early peak in blood triglyceride levels about 20 min after a meal, triggered by a cephalic reflex to ingested fat, but consisting of lipids consumed and stored in an enterocyte storage pool from a previous meal [17]. This is then followed some hours later by a more prolonged triglyceride surge.
Lipid is transported away from the enterocytes in large chylomicron–triglyceride particles in the lacteal system. Conversion to and release of fatty acids occurs through the activity of lipoprotein lipase bound to capillary endothelium of most major organs. Tissue LPL is under the influence of the state of physical activity, insulin and glucose levels. Some nonesterified fatty acids (NEFAs) are released back into the blood – referred to as ‘spillover’.
About 10% of ingested fat is metabolised directly by the liver, and in addition, it is the main organ for synthesis of fatty acids and lipoproteins. Fatty acids are synthesised from acetyl CoA (via malonyl CoA) in the cytosol, a process promoted by insulin. Triglyceride synthesis also occurs from glycerol and fatty acids. Conversely, β-oxidation of fatty acids occurs in mitochondria. Briefly, this consists of a process involving at least 25 different enzymes with the initial production of acetyl CoA, NADH and FADH2. Acetyl CoA enters the TCA (Krebs cycle), and the others act as electron donors to the electron transport chain. This process appears to be the hepatocyte’s main source of ATP for its own energy supply.
Cholesterol, triglyceride and phospholipids are the three main classes of circulating lipids. As might be expected, they are all insoluble in water and are all carried in solution by complexing with lipoproteins. These were first described in the 1920s and subsequently named according to their ‘buoyant density’ as
1. Chylomicrons: Largest molecule and produced by enterocytes containing large amounts of triglyceride (>80%). They exhibit a short half-life (~30 min) and function as a transporting agent for dietary lipid to the lipid storage area in adipose tissue.
2. VLDLs: Main lipoproteins secreted by the liver containing synthesised triglyceride, often in response to increasing circulating fatty acids, and controlled by oestrogens, insulin and possibly thyroid hormones. Sent to adipose tissue.
3. LDLs: Arise as a result of degradation of VLDLs and carry cholesterol from liver to cells.
4. HDLs: Secreted from hepatocytes as bioconcave discs but change morphology by the action of a hepatic enzyme, lecithin cholesterol acyltransfersase (LCAT), into a more spherical object capable of transporting cholesterol within its core.
5. VHDLs: Contain mostly albumin and carry FFAs.
Abnormal accumulation of fat (triglyceride) in hepatocytes is termed steatosis and is apparent as membrane-bound liposomes within the cytoplasm. Most histological processing causes dissolution of these, and they appear as empty spaces but specific lipid stains (e.g. Sudan Red and osmium tetroxide) will visualise them. The clinical picture is termed nonalcoholic fatty liver disease (NAFLD) (defined as fat in >5% of hepatocytes) and may progress through the inflammatory phase of nonalcoholic steatohepatitis (NASH) to overt fibrosis and cirrhosis [20].
Esteller A. Physiology of bile secretion. World Journal of
Gastroenterology 2008; 14: 5641–5649.
Paumgartner G. Medical treatment of cholestatic liver diseases: from pathobiology to pharmacological targets. World Journal of Gastroenterology 2006; 12: 4445–4451.
1. Lautt WW. The 1995 Ciba-Geigy Award lecture. Intrinsic regulation of hepatic blood flow. Canadian Journal of Physiology and Pharmacology 1996; 74: 223–233.
2. Eipel C, Abshagen K, Vollmar B. Regulation of hepatic blood flow: the hepatic arterial buffer response revisited. World Journal of Gastroenterology 2010; 16: 6046–6057.
3. Oura T, Taniguchi M, Shimamura T, et al. Does the permanent portacaval shunt for a small-for-size graft in a living donor liver transplantation do more harm than good? American Journal of Transplantation 2008; 8: 250–252.
4. Jakimowicz J, Stultiëns G, Smulders F. Laparoscopic insufflation of the abdomen reduces portal venous flow. Surgical Endoscopy 1998; 12: 129–132.
5. Crigler JF, Najjar VA. Congenital familial nonhemolytic jaundice with kernicterus. Pediatrics 1952; 10: 169–179.
6. Norin KE, Gustafsson BE, Lindblad BS, Midtvedt T. The establishment of some microflora associated biochemical characteristics in feces from children during the first years of life. Acta Paediatrica Scandanavia 1985; 74: 207–212.
7. Aries V, Crowther JS, Drasar BS, Hill MJ. Degradation of bile salts by human intestinal bacteria. Gut 1969; 10: 575–576.
8. Redinger RN. The coming of age of our understanding of the enterohepatic circulation of bile salts. American Journal of Surgery 2003; 185: 168–172.
9. Cantor P, Mortensen PE, Myhre J, et al. The effect of the cholecystokinin receptor antagonist MK-329 on meal-stimulated pancreaticobiliary output in humans. Gastroenterology 1992; 102: 1742–1751.
10. Marinelli RA, Tietz PS, Pham LD, et al. Secretin induces the apical insertion of aquaporin-1 water channels in rat cholangiocytes. American Journal of Physiology: Gastrointestinal and Liver Physiology 1999; 276: G280–G286.
11. Thimister PW, Hopman WP, Tangerman A, et al. Effect of intraduodenal bile salt on pancreaticobiliary responses to bombesin and to cholecystokinin in humans. Hepatology 1998; 28: 1454–1460.
12. Nyberg B, Angelin B, Einarsson K. Somatostatin does not block the effect of vasoactive intestinal peptide on bile secretion in man. European Journal of Clinical Investigations 1992; 22: 60–66.
13. Woerle HJ, Meyer C, Dostou JM, et al. Pathways for glucose disposal after meal ingestion in humans. American Journal of Physiology Endocrinology and Metabolism 2003; 284: E716–E725.
14. Petersen KF, Cline GW, Gerard DP, et al. Contribution of net hepatic glycogen synthesis to disposal of an oral glucose load in humans. Metabolism 2001; 50: 598–601.
15. Stumvoll M, Meyer C, Kreider M, et al. Effects of glucagon on renal and hepatic glutamine gluconeogenesis in normal postabsorptive humans. Metabolism 1998; 47: 1227–1232.
16. Barle H, Rahlen L, Essen P, et al. Stimulation of human albumin synthesis and gene expression by growth hormone treatment. Clinical Nutrition 2001; 20: 59–67.
17. Lambert JE, Parks EJ. Postprandial metabolism of meal triglyceride in humans. Biochimica et Biophysica Acta 2012; 1821: 721–726. doi: 10.1016/j. bbalip.2012.01.006.
18. Riordan SM, Williams R. Treatment of hepatic encephalopathy. New England Journal of Medicine 1997; 337: 473–479.
19. Strauss GI, Knudsen GM, Kondrup J, et al. Cerebral metabolism of ammonia and amino acids in patients with fulminant hepatic failure. Gastroenterology 2001; 121: 1109–1119.
20. Liu Q, Bengmark S, Qu S. The role of hepatic fat accumulation in pathogenesis of non-alcoholic fatty liver disease (NAFLD). Lipids in Health and Disease 2010; 9: 42. doi: 10.1186/1476-511X-9-42.
* Joseph Disse (1852–1912), German anatomist.
* Augustin Nicolas Gilbert (1858–1927), French physician at Hôtel-Dieu, Paris.
* John Fielding Crigler (b. 1919) and Victor Assad Najjar (b. 1914), American paediatricians working in Boston.
* Secretin was the original ‘hormone’ recognised in 1902 by William Bayliss and Ernest Starling at University College, London.
† Ruggero Oddi (1864–1913). He described this while studying as a medical student in Perugia, Italy.