[level-membership-for-dermatology-category]
Porphyria cutanea tarda
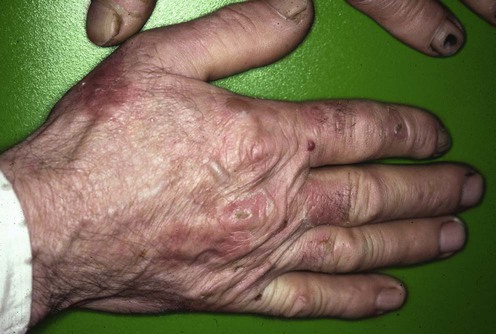
Specific investigations







First-line therapies
 Serial phlebotomies
Serial phlebotomies Chloroquine, hydroxychloroquine
Chloroquine, hydroxychloroquineSecond-line therapies
 Deferoxamine (desferrioxamine)
Deferoxamine (desferrioxamine) Erythropoietin
ErythropoietinThird-line therapies
 Antiretroviral therapy
Antiretroviral therapy Interferon-α
Interferon-α Vitamins E and C
Vitamins E and C Plasmapheresis, plasma exchange
Plasmapheresis, plasma exchange High-flux hemodialysis
High-flux hemodialysis Enteric sorbents (cholestyramine, activated charcoal)
Enteric sorbents (cholestyramine, activated charcoal) Metabolic alkalinization by oral sodium bicarbonate
Metabolic alkalinization by oral sodium bicarbonate Cimetidine
Cimetidine Photothermolysis
PhotothermolysisRemoval of plasma porphyrins with high-flux hemodialysis in porphyria cutanea tarda associated with end stage renal disease.
Carson RW, Dunnigan EJ, DuBose TD Jr, Goeger DE, Anderson KE. J Am Soc Nephrol 1992; 2: 1445–50.
High-flux hemodialysis may remove porphyrins more effectively than conventional hemodialysis.
[/level-membership-for-dermatology-category][not-level-membership-for-dermatology-category]
Porphyria cutanea tarda
