Chapter 100 Poisoning and Drug-Induced Neurologic Diseases
Introduction
Poisonings are common. In 2003, nearly 2.4 million cases of poisoning were reported to the USA’s poison control centers, 93 percent of which occurred at home [Watson et al., 2004]. Of these, 1.58 million involved persons younger than 20 years of age, and 1.24 million (or 52 percent of all poisoning victims) were children younger than 5 years [Watson et al., 2004]. Intoxication is the second most common injury (behind falls) in children younger than 48 months [Agran et al., 2003]. Data from other countries suggest similar demographics [Andiran and Sarikayalar, 2004].
Neurotoxins are among the most commonly encountered toxins, although they are the culprits less commonly in children younger than 6 years [Watson et al., 2004]. Poisonings and drug-induced neurologic disease may mimic infection, trauma, neoplasm, psychiatric illness, or metabolic disorders. Intoxications should be considered in the differential diagnosis of a child with an unexplained change in sensorium, seizures, ataxia, involuntary movements, muscle weakness, or autonomic dysfunction. Prompt recognition and management of intoxication reduce both mortality and morbidity in most cases.
Neurotoxins may have more long-term cognitive sequelae. Unfortunately, studies of low-level environmental exposures often suffer from imperfect control of confounding factors [Mink et al., 2004; Weiss et al., 2004]; this situation may limit the conclusions that may be drawn from these studies.
Most childhood poisonings are accidental [Nhachi and Kasilo, 1994; Singh et al., 1995; Watson et al., 2004]. The most common agents implicated in children younger than 6 years are cosmetics, personal care products, and household cleaning solutions. Important factors contributing to accidental poisonings include parental factors, such as improper medicinal or chemical storage, lapses in child monitoring, and ignorance of poison control methods. Likewise, children may be noncompliant, may mistake a potential toxin for food, may imitate parental medication-taking behavior, or simply may be curious. One unusual example was that of an 11-year-old male who ingested 35 percent hydrogen peroxide solution. Although H2O2 exposure may result in irreversible cerebral injury, his magnetic resonance imaging (MRI) test demonstrated bilateral posterior reversible diffusion restriction; he later made a full recovery [Cannon et al., 2003]. Failure of “childproof” containers was cited in 18 percent of poisonings in one study [Brayden et al., 1993].
Adolescents and preadolescents are at risk for recreational drug and solvent abuse. The rate of substance abuse by preteens in the United Kingdom approaches 5 percent [McArdle, 2004]. Aggressive attempts to identify substances in such intoxications may reveal unsuspected agents [Dresen et al., 2007]. Poisoning occurs deliberately in suicide attempts, child abuse, and other attempts to harm a child. In a 2002 Polish study, 14 percent of poisonings in children younger than 15 years were intentional (including self-inflicted) [Kotwica and Czerczak, 2002]. Two-thirds of suicide victims younger than 19 years in the United Kingdom use self-poisoning [Camidge et al., 2003]. In Hong Kong, poisoning is implicated in 15–20 percent of murders and suicides in the general population [Chan et al., 2003]. These high percentages of nonaccidental poisoning deaths, compared with the United States, may reflect differences in firearm regulation. Among the pediatric population, those at highest risk for abuse are infants and preschoolers; teenagers have the highest risk for suicide [McClure, 1994; McClure et al., 1996]. Abuse of alcohol and other recreational drugs may predict suicide attempts, although the method may involve neither [Hawton and Fagg, 1992; Hawton et al., 1993]. Common suicide methods identified recently in teenagers include hanging, exhaust inhalation, and overdose on medications, including paracetamol, benzodiazepines, and tricyclic antidepressants [Andiran and Sarikayalar, 2004; Ghazi-Khansari and Oreizi, 1995]. Pesticides, herbicides, and caustic agents may also be used; fatalities caused by ingestion of such agents may be the result of suicidal intent rather than accidental exposure more often in adolescents than in adults [Andiran and Sarikayalar, 2004; Klein-Schwartz and Smith, 1997; Thompson et al., 1995].
In the United States, with a preponderance of firearm-related deaths, a fatal outcome of Russian roulette is often classified as suicide. This may be a forensic determination rather than a reflection of intent. Although cause of death is a gunshot wound, a recent South Carolina report notes alcohol and or marijuana use in 6 of 8 victims. In these cases, the recreational agents likely are potentiating adolescent risk-taking behaviors with accidentally fatal outcomes [Collins, 2010].
Poisoning as a result of child abuse usually occurs in conjunction with a background of other, separate injuries. Still, poisoning may occur in isolation or within a more cryptic history of unexplained illnesses; this is especially evident in Munchausen’s syndrome by proxy [Chadwick, 1997]. A diagnosis of Munchausen’s syndrome by proxy suggests the secondary gain of medical contact as motive; it is sometimes difficult to differentiate Munchausen’s syndrome by proxy from other causes of nonaccidental injury. The incidence of nonaccidental poisoning in the British Isles is more than 2.8 per 100,000 children younger than 1 year each year, and more than 0.5 per 100,000 children younger than 16 years each year [McClure et al., 1996]. Methods reported include forced ingestion of antiepileptic drugs, opioids, and caustics, as well as various other agents [Gotschlich and Beltran, 1995; McClure et al., 1996]. Unfortunately, escalation of Munchausen’s syndrome by proxy often occurs, with suffocation a frequent terminal event [Chadwick, 1997; McClure et al., 1996]. One instructive case of Munchausen’s syndrome by proxy was that of an 11-year-old female with a history of cyclic vomiting. Investigation eventually revealed arsenic poisoning by her mother [Embry, 1987].
Conversely, some children and adults present with complaints of symptoms that are believed to be the result of a toxic exposure, but for which no toxin is identified. In these cases, symptoms often are vague; litigation is common (30 percent), and the patients or families do not easily accept refutation of the alleged exposure [Leikin et al., 2004].
Finally, chemical agents have been used as weapons against groups of people. Although their use in battle is not new, recent events have raised concern over covert use of chemical agents to attack civilian populations. In the 1994 Matsumoto nerve agent terrorist attack using sarin, 58 of 600 exposed people required hospital admission, and 7 died. The most common symptom was miosis, as the victims were exposed via vapors rather than direct contact with liquid nerve agent [Newmark, 2008]. Central nervous system (CNS) effects and transient cardiomyopathy also occurred [Okudera, 2002]. The U.S. Department of Health and Human Services has published a brief guide to toxidromes associated with likely chemical weapons used covertly [Patel et al., 2003].
Clinical trials, preclinical studies, anecdotal reports, and epidemiologic data have contributed to our understanding of neurotoxins [Erinoff, 1995; Fray and Robbins, 1996; Indulski and Lutz, 1996; Kurz et al., 1995]. Detailed discussion of antiepileptic drugs and immunization side effects is omitted here; these topics are covered in other chapters.
Emergency Evaluation and Management
Management of the poisoned child requires skilled immediate stabilization of the patient and appropriate corrective and supportive therapy. It requires also that the physician review the history and examine the child carefully for clues that may suggest poisoning or drug effects. Discovery of such evidence is not always easy, and a high index of suspicion is necessary. Three-quarters of all poisonings are by ingestion. Although cosmetics and household cleaning solutions are ingested, medications account for most deaths [Watson et al., 2004; Bronstein, et al., 2008]. Careful physical examination helps to establish the cause of the child’s distress and guide therapy. Neurologic findings may result from the drug or toxic agent itself, or from CNS hypoxia or ischemia caused by a generalized disturbance of circulation and respiration. Systemic abnormalities that occur after various types of intoxication may include cardiac dysrhythmias, gastrointestinal disturbances, and varying degrees of metabolic acidosis. Dysrhythmias, including marked degrees of bradycardia with cyanide or physostigmine intoxication, may be clues to the identity of the toxic agent and signal the need for emergency treatment. Cardiac conduction abnormalities, such as prolonged QT interval with phenothiazine overdose and widened QRS interval with tricyclic antidepressants, quinine, or quinidine overdose, may be present. Gastrointestinal complications, including severe diarrhea and vomiting, may occur with lithium, mercury, phosphorus, arsenic, mushroom, and organophosphate poisoning. Metabolic acidosis with a large anion gap may result from intoxication with cyanide, methyl alcohol, ethylene glycol, propylene glycol, and salicylates [Gardner et al., 2004]. Finally, steps should be taken to protect the child from future exposures. Appropriate general management steps are outlined in Box 100-1; details may be found in several references [Arena, 1985; Banner et al., 1994; Chan et al., 1993; Goetz, 1985; Gosselin et al., 1984; Haddad and Winchester, 1983; Osterhoudt et al., 2004; Leikin and Paloucek, 1995; POISINDEX, 2010; TOXNET, 2010; Zimmerman, 2003, Newmark, 2008]. Therapy for specific toxidromes is discussed with the individual agents.
Box 100-1 Suggested General Management of Suspected Intoxications and Poisoning
Testing
The value of routine toxicologic testing in cases of possible intoxications is debated in the literature. The low sensitivity of specific tests and the inability to test for all possible agents, medicolegal concerns, and cost-effectiveness have fueled arguments that routine testing should be limited [Bond, 1995]. Ideally, history and physical examination narrow the list of possible exposures and direct specific toxicologic testing. However, young children may not provide adequate history, and poisoned children sometimes present with unfamiliar or atypical clinical features [Lifshitz et al., 1997]. These situations may require a more extensive search for the toxic agent.
Toxicologic screening methods have other limitations as well. Most are not designed specifically for use in children, although modified panels or pediatric protocols are available [Badcock and Zoanetti, 1996]. Many physicians are unaware of the agents actually identified by the blood and urine panels available at their institutions. A standard screen for drugs of abuse may detect barbiturates, benzodiazepines, opioids, amphetamines, cocaine metabolites (benzoylecgonine), phencyclidine palmitate, and marijuana metabolites (tetrahydrocannabinol) by immunoassay, but other drugs, such as lysergic acid diethylamide (LSD), may remain undetected [Bond, 1995]. Other broad drug screens may detect phenothiazines, tricyclic antidepressants, ethanol or other volatile substances, and sympathomimetic amines. Urine screens for drugs of abuse may detect 2–9 agents (e.g., tetrahydrocannabinol, cocaine, opioids, amphetamines, barbiturates, benzodiazepines, methamphetamines, phencyclidine palmitate, tricyclic antidepressants in Diagnostix kits). Other commercially available screens include EMIT (Behring Diagnostics, San Jose, California), Abuscreen (Roche Diagnostic Systems, Basel, Switzerland), and others. Mach et al. compared on-site screening to gas chromatography/mass spectroscopy (GC/MS) and found roughly 20 percent discordance between the screening test and GC/MS [Mach, et al., 2007]. Thin-layer chromatography and ultraviolet spectroscopy are also popular, but older tests, including crude spot tests, are still available. The sensitivity of the screens and the need for subsequent confirmation (i.e., by high-performance liquid chromatography) depend on the methods used. Likewise, routine urine screens for heavy metals may identify only arsenic, mercury, and lead. Results from some screens may be obtained in less than 1 hour, but broad screens may require 24 hours for results; specific tests may require a week [Pathology and Laboratory Medicine, 1994]. Hair sample-based toxicologic testing has become increasingly popular and overcomes many limitations of other modalities. The test is minimally invasive, does not rely on dilution methods, and gives historical information. Use of a wide variety of substances can be determined from hair samples, including drugs of abuse, haloperidol, antidepressants, sympathomimetics, and heavy metals, including mercury [Hoffman and Nelson, 2001; Leikin and Paloucek, 1995; Schoeman et al., 2009, 2010]. The primary cocaine metabolite, benzoylecgonine, and acidic, polar drugs are difficult to identify reliably in hair samples, and the test is probably not appropriate for acute toxicity testing situations, although, in the case of cocaine, qualitative and semiquantitative analysis of more chronic use can be demonstrated [Katikaneni et al., 2002; Stephens et al., 2004].
In a prospective pediatric emergency department study, using urine GC/MS drug screens, the best clinical predictors of poisoning were odor on the child’s breath, symptoms and signs consistent with poisoning, and poison actually on the child’s clothing [Hwang et al., 2003]. Positive predictive values for these three variables were 100, 92, and 86 percent, respectively.
Other Ancillary Testing
Ancillary testing is directed by specific clinical findings. Electroencephalography (EEG) is important to exclude subtle or subclinical seizures in intoxicated patients who display altered mental status. Neuroimaging studies have at least three roles. First, in the acutely intoxicated child or teenager, a cranial computed tomography (CT) scan may be required if there is suspicion of concomitant trauma or hemorrhage. Second, brain MRI findings may explain a patient’s neurologic findings when clinical history and other laboratory studies do not. As neuroimaging of an increasing number of poisoned children and adults has occurred, a couple of recurring MRI patterns have become evident. Agents that cause hypoxic injury, including cellular respiratory poisons, may result in an MRI picture of bilateral symmetric cortical and subcortical gray-matter injury, although white-matter changes occur as well [Halavaara et al., 2002; Kim et al., 2003; Rachinger et al., 2002]. This includes methanol poisoning [Karayel et al., 2010]. Medications used for immunosuppression and those that cause hypertension or changes in blood–brain barrier permeability may contribute to a characteristic neuroradiographic pattern of bilateral posterior reversible leukoencephalopathy (so-called posterior reversible leukoencephalopathy syndrome) [Renard et al., 2004]. Third, the increasing use of diffusion-weighted MRI sequences and apparent diffusion coefficient calculations (apparent diffusion coefficient mapping) often provides a method to delineate early cerebral injury.
Neurologic Examination
Examination of a poisoned child includes initial and serial assessment of neurologic status; this assessment guides clinical management, predicts prognosis, and often identifies the offending agent. Assessment of mental status is important because several poisons, medications, and recreational drugs may be associated with affective symptoms, and may also cause acute changes in sensorium, such as irritability, other affective symptoms, delirium, coma, lethargy, and seizures. Although psychotropic or analgesic medications have the greatest potential to cause these effects, antihistamines and other routinely used pediatric medications are common culprits [Bassett et al., 1996]. Environmental and biologic toxins likewise may cause change in sensorium, although more often in younger children. More chronic effects include dementia, subtle learning difficulties, and apparent psychiatric illness. Agents that cause seizures or changes in sensorium are listed in Box 100-2.
Box 100-2 Selected Agents that Cause Changes in Sensorium or Seizures
Medications
















































































































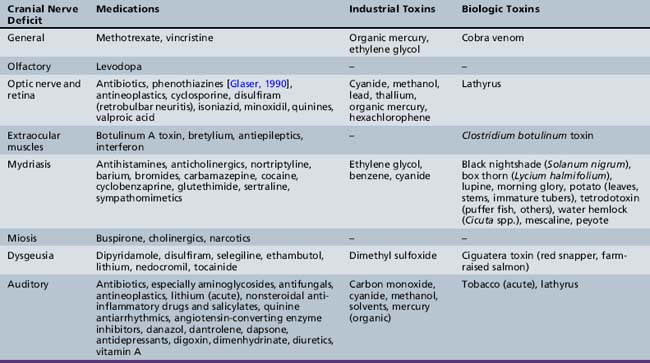
Cranial nerve examination may reveal specific toxin-induced syndromes. Decreased visual acuity or changes in color perception may suggest anticholinergic or cardiac glycoside toxicity, respectively. Papilledema reflects increased intracranial pressure and may suggest pseudotumor cerebri, or benign intracranial hypertension, caused by systemically administered steroids, excessive vitamin A intake, or use of outdated tetracyclines. Visual impairment and a macular cherry-red spot has been reported in association with dapsone poisoning. The patient had concomitant peripheral neuropathy, and the macular changes were considered secondary to toxic retinal damage [Abhayambika et al., 1990]. Pupillary dysfunction may be caused by medication use, abuse of street drugs, and exposure to various environmental or biologic toxins [Leikin and Paloucek, 1995; Slamovits and Glaser, 1990]. Nystagmus is common in intoxications, especially with antiepileptic agents; bradykinetic extraocular movements follow exposure to antidopaminergic medications, and extraocular muscle paresis is an important indicator of botulism poisoning [Glaser and Bachynski, 1990]. Loss of the blink reflex and facial paresis may occur as part of a generalized encephalopathy, such as in profound narcotic intoxications, or occur in isolation, such as in botulism or vincristine-induced neuropathy. Acute hearing loss resulting from poisoning is uncommon but may be caused by aminoglycoside use, toxicity, or overdose of nicotine or lithium. Vestibular dysfunction is one of the most common presenting symptoms of drug-induced neurologic syndromes, including those caused by exposure to antiepileptic drugs, antibiotics, and metals [Mount et al., 1995; Wood et al., 1996]. Vestibular dysfunction should be differentiated from vagally mediated “dizziness,” although both may result from intoxications. Isolated toxin-induced disorders of the pharyngeal and neck musculature (e.g., loss of ability to swallow in clostridial toxidromes) are rare, but dysgeusia is not; the latter occurs commonly with use of lithium-containing preparations. Table 100-1 lists toxins that cause cranial nerve deficits.
Motor weakness may be due to poisoning of the anterior horn cells, peripheral nerves, neuromuscular junction, or muscles. Proximal myopathies (e.g., caused by steroid use) or distal motor neuropathies (e.g., caused by metal exposure) should be differentiated from infectious, inflammatory, metabolic, and degenerative processes. Profound weakness may occur with use of depolarizing agents and neuromuscular blockers. Rigidity may result from either central or peripheral disinhibition (Boxes 100-3 to 100-5) [Katz et al., 1996; Reeves et al., 1996]. Extrapyramidal disorders may be caused by exposure to environmental toxins but also are well-recognized side effects of many medications (Box 100-6). Drugs implicated most frequently in movement disorders are antipsychotics and related agents, calcium channel antagonists, CNS stimulants, antidepressants, antiepileptic drugs, antiparkinsonian drugs, and lithium [Jimenez-Jimenez et al., 1997; Kerrick et al., 1995]. With severe intoxications, the cardiovascular effects of many of these medications overshadow the acute neurologic symptoms. Antibiotics, antidepressants, and some antineoplastic drugs may cause myoclonus (Box 100-7) [Chow et al., 2003].


































Box 100-4 Selected Agents Causing Peripheral Neuropathy
































(Additional references: Le Quintrec and Le Quintrec [1991]; Dyck and Thomas [1993]; Leikin and Paloucek [1995]; Patel et al. [2003]; REPROTOX [2010].)
Box 100-5 Selected Agents Associated with Paralysis and Muscular Rigidity


















Box 100-6 Selected Agents Associated with Parkinsonism and Other Acute Extrapyramidal Reactions



















































Ataxia may be due to cerebellar or peripheral nerve dysfunction (Box 100-8). Cerebellar dysfunction may also manifest as nystagmus, scanning speech, hypotonia with pendular reflexes, dyssynergy, dysmetria, or dysdiadochokinesis [DeJong, 1967; Findley, 1996]. Tremor is common in several intoxications (Box 100-9).































































Common Toxidromes
The neurologic examination may indicate poisonings and drug intoxications that fit clinically into one of several toxidromes resulting from a predominant neurotransmitter derangement or disruption of one arm of the autonomic nervous system [Babe and Serafin, 1996; Brown and Taylor, 1996; Haddad and Winchester, 1983; Hoffman and Lefkowitz, 1996; Leikin and Paloucek, 1995; Reisine and Pasternak, 1996; Sanders-Bush and Mayer, 1996]. The major toxidromes include the following: anticholinergic, cholinergic, sympathomimetic, serotonergic, antihistaminic, and narcotic/sedative-hypnotic syndromes.
Cholinergic toxidromes, which include both muscarinic and nicotinic effects, are produced by organophosphate insecticides, carbamate insecticides, nicotine, physostigmine, and their congeners. Consciousness may be extremely depressed, and the patient may be comatose with respiratory depression; pinpoint pupils; widespread fasciculations; salivation, lacrimation, urination, and defecation (SLUD syndrome); bronchoconstriction; pulmonary edema; hypotension or hypertension; and bradycardia. Seizures may occur. Young children, however, may not display typical peripheral cholinergic signs and symptoms [Lifshitz et al., 1997; Patel et al., 2003].
Sympathomimetic, or stimulant, toxidromes occur with ingestion (sometimes at subtherapeutic doses) of amphetamines, methylphenidate, xanthines (caffeine), ephedrine and related agents, nicotine, and cocaine. Signs range from restlessness, anorexia, and insomnia to euphoria and, in severe cases, seizures [Conway et al., 1990; Rivkin and Gilmore, 1989]. Mydriasis is present, and increased motor activity and tremor may also occur.
Although serotonergic side effects have been noted with several drugs (isoniazid, tranylcypromine, and l-tryptophan in combination with monoamine oxidase inhibitors), the proliferation of newer, relatively selective, serotonin reuptake inhibitors (SSRIs), used to treat depression, obsessive-compulsive disorder, other anxiety disorders, and headache, has fostered new interest in this toxidrome. A recent trend of using atypical antipsychotics adjunctively with SSRIs may further predispose patients to serotonergic toxicity [Rim and Gitlin, 2010]. Atypical antipsychotics alter serotonergic activity through complex interactions at multiple 5-hydroxytryptamine (5-HT) receptor subtypes. Recreational overdose of dextromethorphan may also result in a serotonergic toxidrome (Ganetsky, et al., 2007).
Onset of serotonergic toxicity is often sudden. Central manifestations of this syndrome include confusion, myoclonus, tremor, ataxia, hyperreflexia, and fever; peripheral findings consist of sweating, facial flushing, trismus, and diarrhea [Sternbach, 1991]. These symptoms may be distilled to a triad of mental status changes, autonomic instability, and neuromuscular excitation.
Antihistaminic agents can be divided into histamine 1 and histamine 2 blockers. Both may cause significant CNS side effects at therapeutic doses, although the degree to which they do so is agent-specific. Older histamine1 blockers, such as chlorpheniramine, act at both central and peripheral histamine 1 receptors, monoaminergic receptors, and muscarinic receptors. They cause sedation and delirium, the latter especially in children, and subtle cognitive deficits [Gengo, 1996]. They may also have significant toxicity, including movement disorders and autonomic dysfunction related to their antagonism of dopaminergic, β-adrenergic, serotonergic, and muscarinic receptors. With overdose, ataxia and seizures may supervene. Agents such as terfenadine and clemastine are less likely to cause CNS toxicity as a result of more histamine 1 selectivity and less CNS penetration [Babe and Serafin, 1996]. However, toxicity may be delayed by several days because of slower CNS penetration. Histamine 2 blockers, such as cimetidine, ranitidine, and newer agents, tend not to have significant antihistaminic side effects (except cimetidine) in children at therapeutic doses. At high doses, mental status changes predominate, especially with cimetidine.
The opioid-induced toxidrome, which is readily diagnosable and treatable by administration of naloxone, includes CNS depression with coma, pinpoint pupils, muscle flaccidity, respiratory depression, and bradycardia. All opioid-derived agents may cause this syndrome. Sedative-barbiturate toxidromes are manifested primarily by lethargy or more profound depression and respiratory depression, miotic pupils, and generalized hypotonia with areflexia and ataxia. Signs and symptoms may be progressive, especially in intoxication with longer-acting agents, such as phenobarbital. In the absence of administration of a specific antidote, such as naloxone for opioids or flumazenil for benzodiazepines, differentiation between causative agents may prove difficult on clinical grounds. Hillyard et al. and others have reported reversal of sedative hypnotic-induced coma, as well as that involving other lipophilic agents, using lipid emulsion. Further studies are needed to understand both the efficacy and side effects of this treatment modality [Hillyard et al., 2010].
Chronic Fatigue Syndrome and Related Disorders
Fatigue is a common complaint in both adult and pediatric medicine. It may take the form of simple “lack of energy,” or present as a more pervasive syndrome attended by an “infectious” prodrome and a host of somatic symptoms, including weakness, malaise, and sleep, appetite, and emotional or neuropsychiatric disturbances. This cluster of symptoms has been codified as chronic fatigue syndrome. The disorder is marked by extreme, disabling fatigability of longer than 6 months’ duration, lasting an average of 2–3 years, but usually with eventual full recovery. In a cohort of Chicago primary care adolescent patients, the rate of chronic fatigue syndrome was 4.4 percent [Mears et al., 2004]. Diagnostic criteria have been developed, although the disorder’s heterogeneity is recognized [Farrar et al., 1995].
Putative etiologies for chronic fatigue syndrome are numerous. Postinfectious and postviral causes have been explored most thoroughly, although there is little direct evidence of an association between infection and chronic fatigue syndrome. In one study, an apparent infection rate of 72 percent in chronic fatigue syndrome patients yielded a proven infection rate of only 7 percent [Salit, 1997]. Other contributing factors included trauma, surgery, antecedent stressful events, and allergies. Psychiatric factors also are suggested [Huibers et al., 2004]. Allergies have been implicated as a result of findings of possible but inconsistent immunologic alterations in chronic fatigue syndrome patients, a 65 percent rate of positive allergy history in chronic fatigue syndrome patients, and anecdotal reports of chronic fatigue syndrome onset after immunizations [Farrar et al., 1995; Salit, 1997]. Rigorous evaluation of the cohort of Salit revealed no correlation between chronic fatigue syndrome and exposure to animals, raw milk, meat, cigarette smoking, or foreign travel [Salit, 1997].
Neurobehavioral abnormalities in children are also ascribed frequently to allergies or “environmental toxins.” Some models for testing the causal relationship in adults exist but are applicable only in individual assessments, and the field is in its infancy [Weiss, 1994]. There are no large-scale studies of toxic causes of childhood behavioral disorders. A systematic approach to a child in whom there is concern for such a causal exposure begins with a careful history and physical examination to discern any evidence of exposure to known toxins. Temporal and spatial aspects of the history may be especially important. Other possible causes, including psychosocial factors, should be investigated as well. Results of this evaluation should direct any subsequent testing. Elimination of the putative offending agent (or removal of the child from the environment) should be an option, but disruption of the child’s life is a significant consideration in cases in which there is little evidence of environmental toxic exposure. Cognitive-behavioral therapy has been associated with improvement in chronic fatigue syndrome symptoms in adolescents and adults. Although relapse of some symptoms is common, functional outcome, including school attendance, improves [Stulemeijer et al., 2005].
Specific Agents
Poisons and Environmental Toxins
Biologic Toxins
Biologic toxins include snake, tick, insect, and fish toxins, botanical toxins, and bacterial toxins (e.g., diphtheria, tetanus, botulism) [Piradov et al., 1991; Stommel, 2008]. Botanical poisonings are especially common among children; whereas most are mild and result from ingestion of easily accessible houseplants, other botanical poisonings may have more serious consequences (see Table 100-1, Box 100-2, and Boxes 100-4 to 100-9) [Krenzelok et al., 1996]. Current parental concern over possible but unproven vaccination side effects may result in a resurgence of previously controlled neurotoxic pathogens. See also reviews by Hardin and Arena [1974], Kunkel [1983], Langford and Boor [1996], and Stommel [2008].
Snake venoms, which, in the United States, are primarily from rattlesnakes and copperheads, are strong neurotoxins. In addition to the local effects of envenomation, there are systemic symptoms of fever, nausea, vomiting, diarrhea, and tachycardia, as well as altered blood coagulation. Neurotoxins affect the neuromuscular junction, causing paralysis of the diaphragm and the intercostal and limb muscles and, more rarely, altered states of consciousness and convulsions. There is controversy concerning the use of local incisions, fasciotomy, application of ice packs, and steroids. Exercise hastens absorption of venom. Intravenous administration of specific antivenoms is recommended [Grant et al., 1997; Hawgood and Sir Joseph Fayrer, 1996; Podgorny, 1983]. The occurrence of the various envenomations depends partially on regional fauna. In New South Wales, Australia, the percentage of poisoning from envenomation ranges from 3.1 percent (females aged 15–19 years) to 84.1 percent (males aged 5–9 years) [Lam, 2003]. In the United States, young males comprise the largest risk group due to accidental or intentional exposure to potentially poisonous snakes [Stommel, 2008].
Under-appreciated in the USA, but a significant concern in Mexico and Central America, is scorpion envenomation [Stommel, 2008; Cheng et al., 2007]. Deaths from scorpion stings far exceed those from snake bites in these areas. Scorpion toxins have complex actions, but their neurotoxicity is due to massive excitation of both the parasympathetic and sympathetic nervous systems.
Tick paralysis is an infrequent but treatable cause of acute paralysis or ataxia caused by neurotoxins from Dermacentor andersoni (wood tick), Dermacentor variabilis (dog tick), Amblyomma americanum (Lone Star tick), Amblyomma maculatum, Ixodes scapularis (black-legged tick), and Ixodes pacificus (western black-legged tick) [Centers for Disease Control and Prevention, 1996a]. Although most prevalent in states west of the Rocky Mountains, tick paralysis also occurs in the southeastern and northeastern United States [Schaumburg and Albers, 2008]. Small children are most likely to be paralyzed. The scalp and neck at the hairline are the most common sites for attachment of the gravid tick, which produces a neurotoxin that prevents liberation of acetylcholine at the neuromuscular junction [Swift and Ignacio, 1975]. Within 5–6 days after tick attachment, the child develops restlessness and irritability, followed, within 24 hours, by progressive ataxia and difficulty walking, or by symmetric ascending flaccid paralysis with loss of deep tendon reflexes. Removal of the tick in the early stage of the disease leads to reversal of the clinical manifestations within 24 hours, whereas failure to remove the tick may result in progressive bulbar paralysis and death. Insect repellents, especially permethrin-containing products applied to children’s clothing, may be used as a preventive measure.
Botulinum toxin blocks presynaptic release of acetylcholine and causes neuromuscular junction blockade. Infection with Clostridium botulinum spores results primarily in three different clinical syndromes: childhood or adult food-borne, infantile, and wound botulism. Inhalation botulism has been reported. Food-borne botulism occurs after ingestion of food, which is frequently home-cooked and canned, that is contaminated with C. botulinum. Nonacidic canned vegetable products and soups are implicated frequently, and recent reports also implicate dairy products as a source of botulism poisoning [Aureli et al., 1996; Townes et al., 1996]. There are prominent gastrointestinal symptoms of diarrhea and vomiting, followed by rapidly developing progressive paralytic disease associated with visual blurring and diplopia, dysphagia, dysarthria, and subsequent weakness of extremities and intercostal muscles. Generally, onset is within 2 days; a prolonged incubation period may correlate with milder disease [Stommel, 2008]. Antitoxin is effective only if given early, and practical concerns regarding availabilty and administration preclude its use in most cases [Cherington, 1974; Newmark, 2008)]. Treatment otherwise is supportive and neuromuscular effects may linger, even after recovery.
Infantile botulism, which occurs almost exclusively in the first months of life, can be a severe paralytic disease affecting the limbs, trunk, bulbar musculature, and cranial nerves. It is preceded almost universally by a history of severe constipation without other gastrointestinal symptoms. This illness has been reported in clusters in both California and Pennsylvania, but it also has been observed in isolated instances throughout the United States and, in some instances, has been related to the ingestion of honey-containing botulinum spores [Glatman-Freedman, 1996; Johnson et al., 1979; Midura, 1996].
Wound botulism, the rarest form of botulism, is both a local infection of contaminated wounds and a systemic intoxication. Incubation period is longer than in the food-borne type. It can produce a paralytic illness that may be indistinguishable clinically from the disorder produced by food-borne botulism. The most common cause of wound botulism in the United States is secondary to intravenous drug abuse [Centers for Disease Control and Prevention, 1996b]. Rarely, wound botulism may result from small intranasal or septal abscesses in nasal cocaine abusers. Botulism mimicking Guillain-Barré syndrome has been reported [Griffin et al., 1997].
Mortality for wound botulism is approximately 15 percent. Food borne botulism carries a 5 percent mortality [Stommel, 2008].
Pediatric tetanus is extremely rare in the vaccination era. Neonatal tetanus is reported, resulting from infection of the umbilical stump. Some forms of culturally specific perinatal manipulation of the umbilical cord may increase this risk. Presentation is generalized rigidity and mortality is high, especially in neonates less than 10 days of age [Bennett et al., 1999].
Insecticides
Organophosphate and carbamate insecticides account for most childhood insecticide exposures. Fortunately, most childhood exposures do not result in poisoning. Fewer than 10 percent of the 8500 insecticide exposures reported to 16 regional poison control centers in 1983 were symptomatic or associated with more than minor symptoms [Veltri and Litovitz, 1984]. A later nine-state study from 1999 to 2002 confirmed that, even with active community spraying programs, symptomatic exposure was rare. In that study, 133 cases were reported among a population of 118 million people. Of these cases, 29 resulted from a single incident in which a mosquito-control truck inadvertently sprayed a malathion-containing product during a softball game. Only one person, who survived, had severe toxicity [Patel et al., 2003]. However, these agents are acetylcholinesterase inhibitors, which may result in serious cholinergic signs. Deaths have been reported in other series [Levy-Khademi et al., 2007]. Toxins can be absorbed through the skin and gastrointestinal tract, but most childhood poisonings are the result of ingestion. Signs of acute intoxication occur usually within 12–24 hours, but can develop in minutes after an acute exposure, indicating severe intoxication and a medical emergency. Salivation, lacrimation, bronchoconstriction, wheezing, and increased pulmonary secretions are all muscarinic manifestations. Peripheral nicotinic signs include muscle weakness, decreased respiratory effort, and muscle fasciculations. CNS signs are anxiety, restlessness, confusion, headache, slurred speech, ataxia, and generalized seizures. Opsoclonus has been reported with diazinon poisoning in an adult [Liang et al., 2003]. With low-dose organophosphate exposure, muscarinic signs predominate, but in more severe acute intoxication, the nicotinic and CNS signs appear. In the series of Levy-Khademi et al., 71 percent of poisoned childen presented with encephalopathy and/or convulsions [Levy-Khademi et al., 2007]. Nicotinic effects appear to mediate delayed peripheral toxicity. With longer-acting agents, such as chlorpyrifos (Dursban), recovery is protracted and possibly incomplete [Sherman, 1995]. Rarely, a myopathy may complicate the picture [Marrs, 1993; Rusyniak and Nanagas, 2004].
A scoring system called Acute Physiology and Chronic Health Evaluation II (APACHE II) has been applied to adults with organophosphate poisoning and has indicated high predictive value for intensive care unit admission and subsequent mortality [Lee and Tai, 2001]. Cholinesterase levels may be used to predict subsequent weaning off mechanical ventilation in surviving patients.
Treatment includes decontamination, general supportive therapy, and administration of specific antidote, including atropine and pralidoxime or obidoxime, the oximes being cholinesterase reactivators [Mortensen, 1986]. Prompt decontamination protects both the patient and attendant health-care workers [Stacey et al., 2004]. The dose of atropine for children younger than 12 years is 0.02–0.05 mg/kg, followed by maintenance doses of 0.02–0.05 mg/kg repeated every 10–30 minutes (maximum dose, 1–2 mg), until cholinergic signs are reversed. For children older than 12 years, the adult dose of atropine (1–2 mg) may be used. As pralidoxime does not cross the blood–brain barrier, atropine must be continued in addition to pralidoxime to reverse central and muscarinic receptor overstimulation. The response to antidote virtually confirms the diagnosis suggested by the history of exposure and the characteristic acute cholinergic symptoms, although intermediate (1–4 days after exposure) and delayed effects are not influenced by atropine or oxime therapy [Marrs, 1993]. These delayed effects are not seen generally in insecticides available in the United States and Europe, and it is thought that the delayed central effects are mediated, not by cholinergic mechanisms, but rather by direct phosphorylation of neuronal structures [Marrs, 1993]. A newer therapeutic agent, liposome encapsulating organophosphorus hydrolase, has demonstrated promise in the laboratory, raising the median lethal dose of paraoxon beyond that provided by pralidoxime and atropine combined [Petrikovics et al., 2004].
Endosulfan, a chlorinated hydrocarbon, may pose a chronic and endemic threat to children with repeated exposure. Acute intoxication causes respiratory distress, gagging, vomiting, diarrhea, agitation, convulsions, ataxia, and coma in severe cases. Mild cases may present with only anxiety and gastrointestinal symptoms [Durukan et al., 2009]. Fatal status epilepticus occurs, and endosulfan-related fatalities occurred in Sri Lanka in 1998 before the agent was banned that year [Roberts et al., 2003]. Dewan and associates reported recurrent convulsions and several deaths in a rural area of India due to wheat flour being stored in containers previously used for endosulfan storage [Dewan et al., 2004]. Amitraz, a formamidine pesticide, has side effects similar to clonidine overdose (CNS depression, miosis, bradycardia, hypotension, emesis, and hyperglycemia). With adequate support, death is rare [Aydin et al., 2002].
Nerve Agents
Nerve agents are related chemically to organophosphate insecticides but are far more potent [Newmark, 2008]. A single drop of VX on the skin may kill a person. Childhood exposure would likely be a result of deliberate attack with nerve agents. They are divided into two classes: G agents and V agents. Tabun (GA), Sarin (GB), and Soman (GD) are clear, tasteless, colorless, and mostly odorless. Tabun and Soman may have a slightly fruity odor, but this property is not clinically useful. VX is amber and oily, but also odorless and tasteless. All may exert their effects within seconds to minutes, depending on route of exposure. Onset after cutaneous exposure may be delayed up to a day. Presenting symptoms may depend on type of exposure. Vapor exposure may present with miosis and concomitant blurring and dimming of vision, followed by salivation. Transdermal exposure first causes local sweating and fasciculations, followed by systemic compromise. Pupillary changes may occur later in the course. Management, as with insecticide poisoning, rests on decontamination; airway, breathing, and cardiovascular stabilization; and specific antidotes, as described previously [Rotenberg and Newmark, 2003; Smythies and Golomb, 2004; Newmark, 2008]. Field treatment requires aggressive treatment with monitoring of respiratory status, rather than attention to peripheral signs and symptoms of cholinergic toxicity. Benzodiazepines are the drugs of choice to treat nerve agent-induced status epilepticus.
Lindane
Lindane (1,2,3,4,5,6-hexachlorocyclohexane) is a topical pediculicide used commonly throughout the world. Its use has been associated with significant neurotoxicity, especially in infants. Lindane passes easily into the CNS; brain levels may greatly exceed blood levels [Fischer, 1994].
Acute lindane neurotoxicity manifests usually as generalized seizures. Mental status changes and extrapyramidal movements may occur as well. Onset of symptoms after topical use occurs within a few hours, generally with complete resolution within 48 hours. Although topical lindane poisoning in young adults is reported, it most commonly results from inappropriate or excessive use of the medication in children [Boffa et al., 1995; Fischer, 1994; Forrester et al., 2004]. Occupational exposure may result in chronic lindane poisoning, manifesting as emotional changes, muscle jerking, and alteration of the EEG [Aks et al., 1995]. Accidental ingestion of lindane is usually reported in toddlers. This situation appears to result from parental misunderstanding of application instructions. High levels may be attained quickly, and vomiting and seizures result, although vomiting and ocular pain are common [Forrester et al., 2004]. Myonecrosis may occur with blood lindane levels exceeding 0.6 g/mL, whereas blood levels in excess of 1.2 g/mL may be fatal [Aks et al., 1995].
Treatment of acute topical exposure is supportive, with control of seizures necessary. Barbiturates or benzodiazepines are the antiepileptic drugs of choice; phenytoin should be avoided because it may exacerbate lindane-induced seizures [Aks et al., 1995]. Gastric lavage may be attempted within 30–60 minutes after ingestion; subsequently, activated charcoal or cholestyramine speeds gastrointestinal excretion of lindane. Prevention of lindane poisoning is easier than its treatment. Permethrin 5 percent preparations may be safer in infants and young toddlers. Repeated exposures within 7 days and increased topical absorption (e.g., with open wounds or after a hot bath) enhance toxicity. Finally, pediculicidal preparations available in the United States contain 1 percent lindane; lindane 0.3 percent preparations have been effective in other nations [Surber and Rufli, 1995].
Insect repellents
The most effective general insect repellent currently in use is diethyltoluamide (permethrin is more effective against ticks), a compound available in many topical preparations. Although increasing the diethyltoluamide concentration up to 75 percent increases its efficacy, preparations containing more than 20 percent diethyltoluamide are likely to cause neurotoxicity, and most products for children contain 8–10 percent diethyltoluamide [Brown and Herbert, 1997]. Children using compounds with as little as 20 percent diethyltoluamide may suffer seizures (8–48 hours after exposure), ataxia, and tremor [Oransky et al., 1989; Sudakin, 2003]. There is some evidence, in fact, that there is no absolutely safe level of diethyltoluamide exposure for children.
Metals
Lead
Acute lead poisoning is rare. The syndrome of acute lead encephalopathy in children with pica, who ate lead paint chips from peeling paint and plaster in older dwellings, consisted of listlessness, drowsiness, and irritability, followed by seizures and signs and symptoms of acutely increased intracranial pressure. Severe cerebral edema resulting from acute lead encephalopathy often resulted in significant neurologic sequelae. Blood levels in acute pediatric lead encephalopathy exceed 90 mcg/dL [Kumar, 2008]. Use of lead-based paints for indoor use was banned in the United States in 1978, with a consequent decrease in lead toxicity since.
Chronic lead exposure as a result of ingestion of lead paint, inhalation of automobile exhaust fumes, or inhalation of industrial pollutants may result in high levels of blood lead and is the more common form of intoxication today. Lead poisoning is defined as a blood lead level of greater than 10 mcg/dL, although this threshold is undergoing critical reappraisal as perhaps too liberal [Lidsky and Schneider, 2003; Wilkin et al., 2004; Murata et al., 2009]. Most children with lead poisoning are asymptomatic, but presentation with behavioral changes and gastrointestinal disturbance, initially thought secondary to a viral syndrome, has been reported [George et al., 2010]. The child in that case had autism and consumed lead paint. His lead level at diagnosis was 216 mcg/dL.
A lead concentration of 25 mcg/dL or higher in blood is reported to be associated with learning problems and neurobehavioral effects [Bellinger, 1995; Bellinger et al., 1991; Landrigan et al., 1975; LaPorte and Talbott, 1978; Winneke et al., 1990; Winneke, 1996]. Measurement of full-scale intelligence may be relatively insensitive in detecting the deleterious effects of chronic lead exposure in children; subtests measuring visual motor and reaction performance skills appear to be more sensitive. Some authorities recommend treatment with ethylenediaminetetraacidic acid (EDTA) for children who have even mildly elevated lead levels (i.e., greater than 25 mcg/dL) [American Academy of Pediatrics, 1995; Piomelli et al., 1984; Trachtenbarg, 1996]. A newer oral chelation agent, meso 2,3-dimercaptosuccinic acid, has been approved recently and used for children with blood lead levels greater than 45 mcg/dL [Berlin, 1997]. Acute lead encephalopathy at any blood lead level constitutes a life-threatening emergency and should be managed accordingly [American Academy of Pediatrics Committee on Drugs, 1995; Kumar 2008].
Mercury
Organic mercury poisoning has occurred as a result of ingestion of fish (contaminated by mercury-polluted water), grain (contaminated by mercurial fungicides), or pork (contaminated by feeding pigs mercury fungicide-treated grain) [Cranmer et al., 1996; Zepp et al., 1974]. Minamata disease occurred in people who ate fish from mercury-contaminated waters, and exemplified the danger of organic mercury bioamplification as it ascends the food chain [Bigham and Vandal, 1996]. The symptoms and signs of organic mercury poisoning include ataxia, peripheral neuropathy, choreoathetosis, visual loss, confusion, and coma [Patel et al., 2003]. Subclinical mercury neurotoxicity has been demonstrated as well in occupational exposures, using visual-evoked potentials and electromyography [Urban et al., 1996]. Dimercaprol and 2,3-dimercaptosuccinic acid effectively chelate and remove mercury from the tissues, but do not necessarily reverse the clinical manifestations. Heavy-metal poisoning may also cause a peripheral neuropathy, although this is rare in children. Ingestion of elemental mercury, such as from a broken thermometer, does not usually cause mercury poisoning; however, Koyun and colleagues reported serious mercury intoxication in three unrelated teenagers after exposure to a broken manometer [Koyun et al., 2004]. Despite chelation therapy, one patient died. Although other heavy-metal poisoning is usually associated with industrial exposure affecting workers, poisoning also may occur in children. Smelting operations may release large amounts of lead, zinc, cadmium, and arsenic into the surrounding environment [American Academy of Pediatrics Committee on Drugs, 1997].
Perhaps no topic concerning mercury has resulted in as much controversy as an alleged link between thimerosal and autism. In such debates, causal links are difficult to prove, and often even more difficult to disprove once suggested. Most peer-reviewed literature does not support an association beyond coincident timing of vaccines and manifestations of autism [Kingman et al., 2005; Kumar, 2008]. Furthermore, historical evidence (e.g., Minamata disease) reveals a different developmental neuropathology regarding mercury poisoning [Nelson and Bauman, 2003]. Please see Chapter 104 for further discussion.
Thallium
Thallium was once used as treatment for night sweats in tuberculosis and topically for ringworm in children (with mortality rates reported as high as 5 percent) [Dyck and Thomas, 1993]. The primary modern sources of thallium are rodenticides and insecticides. Ingestion may be accidental or intentional. Thallium poisoning presents with the clinical triad of abdominal pain, alopecia, and mixed peripheral neuropathy. Although there may be gastritis and hepatic injury, abdominal pain is usually secondary to peripheral neuropathy [Burnett, 1990]. Alopecia occurs suddenly after a 2- to 3-week delay, and is associated with an acneiform rash over the nose, cheeks, and nasolabial folds [Dyck and Thomas, 1993]. The neuropathy is both demyelinating and axonal. Other neurologic effects of thallium include cranial neuropathies involving optic and oculomotor nerves, CNS depression, dementia, delirium, and seizures. CNS levels of thallium may continue to rise for at least a few days after the initial exposure [Sharma et al., 2004]. Cardiovascular involvement includes hypertension and myocardial damage, with shock and death occurring in massive doses [Sharma et al., 2004]. In adults, acute exposure to as little as 1 g of thallium may be fatal; 5 mg/kg is toxic in children [Burnett, 1990; Patel et al., 2003]. Additional clues to diagnosis of thallium poisoning include the presence of painful distal glossitis, Mees’ lines, and black thallium deposits at the base of abnormally tapered hairs [Feldman and Levisohn, 1993]. Cerebrospinal fluid protein may be elevated as well. Thallium crosses the placenta and may affect the fetus.
Arsenic
Chronic arsenic poisoning is well recognized. Although intentional poisoning captures the imagination, accidental exposure through the workplace or environmental contamination poses a larger public health problem. As inorganic arsenic poses a greater threat of acute poisoning than does the less toxic organic arsenic, most severe accidental poisonings occur by contamination of water supply [Abernathy et al., 2003]. Inhalation of smoke from coal and industrial wastes may induce inorganic arsenic poisoning. Chromated copper arsenate-treated wood, still ubiquitous in construction and landscaping in the United States, can leach arsenic into nearby soil and release it when burned. Acute poisoning manifests with nausea and vomiting, abdominal pain, and diarrhea, together with cutaneous and neurologic findings [Ratnaike, 2003]. Severe inorganic arsenic poisoning also results in encephalopathy in older children [Patel et al., 2003]. A Guillain-Barré-like neuropathy may follow [Kumar, 2008]. Reports describe endemic chronic arsenic intoxication within entire families and towns, resulting from contamination of the local water supply or from chronic dust exposure [Gerr et al., 2000]. In these epidemics, children are affected with the typical keratoderma and distal peripheral neuropathy, and may also be mentally retarded [Das et al., 1995; Foy et al., 1992–1993;Mazumder et al., 1992]. Other acute cutaneous findings may include facial edema, transient flushing, conjunctival hemorrhage, and maculopapular rash in intertriginous areas. Mees’ lines may be found several months after exposure [Uede and Furukawa, 2003]. Unlike thallium, arsenic typically does not cause alopecia [Rusyniak et al., 2002].
Heavy-metal exposure, including cadmium, lead, arsenic, mercury, and thallium, may occur from intake of medicinals. Ernst and Coon provide a review of such metal toxicity in traditional Chinese medicines [Ernst and Coon, 2001].
Solvents
Isopropyl alcohol poisoning most commonly results from the ingestion of common household “rubbing alcohol.” Isopropyl alcohol vaporizes readily and is rapidly absorbed through the lungs. Alcohol sponging for fever reduction, especially in a poorly ventilated room, can result in intoxication and coma. Ingestion of as little as 20 mL can produce symptoms [McFadden and Haddow, 1969]. Peak serum levels are reached in about 1 hour, with adverse initial symptoms, primarily gastrointestinal complaints, occurring within 30 minutes after ingestion. CNS effects include headache, confusion, dizziness, ataxia, stupor, and deep coma, which may be caused partially by the formation of acetone during metabolism [Mydler et al., 1993]. Hypotension and hypothermia may occur. The diagnosis is suggested by history, characteristic odor on the patient’s breath, and the presence of large amounts of ketones in the urine without significant metabolic acidosis. Urine toxicology may be revealing [Mydler et al., 1993]. Treatment is primarily supportive. Intermittent or continuous gastric lavage has been suggested, but use of syrup of ipecac is contraindicated; charcoal may be beneficial [Leikin and Paloucek, 1995].
Intoxication has also been reported with benzyl alcohol, which was used as an antibacterial agent and a preservative in saline solution. This form of intoxication, called the gasping syndrome, was reported after the administration of saline solutions as intravenous flushes in small, preterm infants [American Academy of Pediatrics Committee on Drugs, 1997; Gershanik et al., 1982]. The infusion of small amounts of flush solutions containing 0.9 percent benzyl alcohol may cause severe metabolic acidosis, encephalopathy, respiratory depression with gasping, and death. Intoxication from other sources of benzyl alcohol (i.e., as a preservative in medications other than saline) is unlikely.
Children may suffer from clinically significant environmental exposure to solvents. This exposure may occur especially in areas with little child labor protection, resulting in occupational exposure [Saddik et al., 2003].
Other Nonpharmacologic Compounds
A patient presented in coma, after ingesting matchstick heads, of which potassium chlorate is the major component; his MRI indicated bilateral symmetric deep gray-matter and temporal lobe hyperintensity on T2-weighted images. He recovered after hyperbaric oxygen therapy and supportive care [Mutlu et al., 2003].
Drugs of Abuse
Cocaine
Cocaine abuse has reached epidemic proportions, especially among young adults. Lifetime incidence of cocaine use is 15 percent among high school seniors and more than 20 percent among college students. There is a suggestion of a decrease in cocaine use, although the same may not be true of crack cocaine. Almost 1 in 20 high-school seniors have tried crack cocaine [O’Malley et al., 1991].
Cocaine, a stimulant alkaloid, is derived from the leaves of the shrub Erythroxylum cocoa. Cocaine inhibits reuptake of norepinephrine, dopamine, and serotonin. Acute toxicity, manifested by tremors, diaphoresis, tachycardia, cardiac dysrhythmias, vasoconstriction, and hypertension, is probably the result of cocaine’s effect on norepinephrine reuptake. Behavioral effects are most likely caused by its actions on the dopaminergic system. Cocaine use results in an enhanced sense of well-being, increased alertness and excitement, and apparent enhancement of the reward response. These effects are followed by anxiety, depression, exhaustion, and sometimes paranoid psychosis. Addiction follows continued use. Stroke, intracranial hemorrhage, and seizures have been reported [Levine et al., 1990; Sloan et al., 1991; Walsh and Garg, 1997; Saleh et al., 2010]. Infants and children passively exposed to cocaine may have similar complications; however, some have disputed these effects [Wurtzel et al., 1988]. Increased cerebral blood flow velocity has been found in infants exposed to cocaine [Van de Bor et al., 1990].
Narcotics
Data regarding numbers of established narcotic users in childhood are scanty; a great percentage of narcotic users never come to the attention of poison control centers. High-school students are among the population at risk, as are children younger than 5 years of age who reside with adults who are narcotic abusers. Narcotics such as morphine, heroin, hydromorphone (Dilaudid), levorphanol (Dromoran), meperidine (Demerol), and methadone share the toxic properties of the opiate group, but the onset and duration of toxic signs vary after ingestion. Methodone intoxication, both accidental and intentional, has been described even in infants [Glatstein et al., 2009]. Opiate users may also be exposed simultaneously to multiple other toxic agents, which are either adulterants of the opiates or are taken concurrently. The signs and symptoms of acute opiate poisoning most commonly follow rapid intravenous administration, and include apnea, circulatory collapse, convulsions, and cardiopulmonary arrest. The less severely poisoned patient experiences varying degrees of CNS depression, ranging from drowsiness to profound coma, miotic pupils, and respiratory depression.
Narcotic poisoning in younger children more commonly results from ingestion of various analgesics, such as meperidine, codeine, oxycodone (Percodan), and pentazocine (Talwin). Ingestion of a large dose, whether accidental or iatrogenic, of the antidiarrheal medication diphenoxylate (Lomotil), which also contains atropine, may result in signs of opiate toxicity with CNS and respiratory depression, as well as signs of atropine overdose, such as facial flushing, tachycardia, dry mouth, hyperpyrexia, and agitation [Brunton, 1996; POISINDEX, 2010].
Nanan and associates reported the case of a 14-year-old female with intentional morphine sulfate overdose and leukoencephalopathy involving the corpus callosum, centrum semiovale, and cerebellar white matter [Nanan et al., 2000]. The MRI findings were not explained adequately by hypoxic-ischemic injury, and a drug-induced neurotoxicity was suggested. Other cases of severe neurotoxicity resulting from polypharmacy are reported [Nebelsieck, 2004]. Injected heroin also has been reported to produce a spongiform leukoencephalopathy [Robertson et al., 2001].
Cannabis
Cannabis usually is taken recreationally by inhalation (smoking marijuana cigarettes) or ingestion. The active compound is tetrahydrocannabinol. Tetrahydrocannabinol intoxication produces psychomotor slowing acutely, short-term memory loss, analgesia, and appetite stimulation. Coma has been reported in children ingesting cannabis-laced cookies. Cannabis ingestion should be considered in cases of coma, especially when associated with conjunctival hyperemia, pupillary dilatation, and tachycardia, and when there is no other obvious cause [Boros et al., 1996].
Tetrahydrocannabinol acts through the CB1 receptor (cannabinoid receptor type 1) [Iverson, 2003]. Interestingly, anandamide, an endogenous ligand for the CB1 receptor, has been implicated in the “runner’s high” [Sparling et al., 2003]. Long-term biologic sequelae of cannabis use are ill defined. Dependency may occur, as may apparently reversible mild cognitive impairment. Psychosocial consequences may be more severe.
Gamma-Hydroxybutyrate
Gamma-hydroxybutyrate is a schedule I sedative-hypnotic drug. Although it is used volitionally as a recreational drug, its most infamous use is when it is given surreptitiously as a “date-rape” drug. There is a suggestion in the literature that its sedative effects may be reversed by physostigmine, but no conclusive data exist [Caldicott and Kuhn, 2001; Traub et al., 2002]. As discussed previously, it may be found in addition to other agents in screens of people suspected of substance abuse.
Volatile Solvents and Propellants
Hydrocarbon-based solvents frequently are used by adolescents for their euphoria-producing effects. The compounds commonly in use include various glues, rubber and model airplane cements, typewriter correction fluids, gasoline, lighter fluids, and aerosols [Doring et al., 2002]. Neurologic effects develop within minutes and last several hours, with an initial excitatory stage of euphoria, mental confusion, ataxia, and dysarthria, followed by lethargy, sleep, and coma. Cerebellar dysfunction and sensorimotor neuropathy from chronic gasoline sniffing have been reported [Prockop, 1979; Prockop et al., 1974; Young et al., 1977]. Permanent cognitive impairment and CT evidence of diffuse atrophy of the cerebral hemispheres, cerebellum, and brainstem have also been documented [Hormes et al., 1986]. More sequelae occur with abuse of leaded gasoline [Cairney et al., 2004]. One MRI series demonstrated cerebral atrophy, white-matter hyperintensity on T2-weighted images, and hypointensity involving the basal ganglia and thalami on T2-weighted images, as well as focal enhancement. Although a correlation was found between the degree of neurologic impairment and extent of white-matter disease, no correlation was seen between radiographic evidence of damage to subcortical gray structures and clinical findings [Caldemeyer et al., 1996]. Late sequelae of recreational hydrocarbon inhalation may also include peripheral neuropathy [Kumar, 2008].
Nitrous Oxide
Nitrous oxide abuse is governed by access and therefore is rare in children, although it has been reported in college students [Kumar, 2008]. Neurotoxicity is related to nitrous oxide’s ability to interfere with the cobalamin co-factor of methionine synthetase. Nitrous oxide irreversibly oxidizes the cobalt atom of vitamin B12. Children with 5,10-methylenetetrahydrofolate reductase (MTHFR) enzyme mutations may be at increased risk of nitrous oxide toxicity. Neurologic deterioration leading to death in a child following two exposures to nitrous oxide anesthesia has been reported [Selzer et al., 2003]. Neurologic toxicity, which occurs after months of daily use, presents as subacute combined degeneration of the spinal cord [Seppelt, 1995].
Hallucinogens
Phencyclidine, also known as angel dust or PCP, is a readily available drug of abuse that is commonly inhaled or ingested. The clinical syndrome of phencyclidine palmitate toxicity includes euphoria and a “high” appearance, at times with violent behavior associated with extreme muscular rigidity and Herculean displays of muscle strength. Vertical and horizontal nystagmus is common, along with marked pupillary dilatation. Instead of excitation, the intoxicated patient may manifest a syndrome of catatonic rigidity, opisthotonus, dystonic posturing, stupor or profound fluctuations in the level of consciousness, seizures, and myoclonus. Extreme elevations of blood pressure are common. Intracranial hemorrhage has been reported [Bessen, 1982; Boyko et al., 1987]. The phencyclidine palmitate-intoxicated patient should be sedated, especially if agitated or seizing, cooled for extreme degrees of hyperthermia, and treated for systolic hypertension with hydralazine or diazoxide [Kulberg, 1986]. Gastric lavage is helpful in removing ingested but unabsorbed drug. Urinary acidification promotes drug excretion.
Finally, new synthetic and naturally occuring hallucinogens continue to emerge. Bromo-dragonfly, or BDF (5-HT2A agonist and α1-agonist), K2/spice (cannabinoid receptor agonist), and salvia (kappa opioid receptor agonist) are but three of a myriad of newer agents. BDF and others have life-threatening and fatal side effects, including peripheral and visceral vasoconstriction and sympathetic toxidromic symptoms [Gussow, 2010]. Spice and salvia remain legal in many states. Recreational drug culture websites may be useful tools to help identify potential hallucinogens in the emergency department and clinic settings.
Amphetamines
Amphetamines, such as dextroamphetamine, benzedrine, and methamphetamine, cause clinically similar effects, including a sense of well-being and decreased fatigue. With increasing doses, acute paranoid psychosis may develop, with mania, hyperactivity, and severe sympathomimetic effects, including mydriasis, flushing, diaphoresis, and tachycardia. Convulsions are rare. Cerebral vasculitis, cerebral infarction, and intracranial hemorrhage have been reported with amphetamine use [Delaney and Estes, 1980; Matick et al., 1983; Rothrock et al., 1988]. Gastric lavage is useful in elimination of the toxin. Severe agitation or seizures may be treated with intravenous benzodiazepines; hallucinations may also be treated with haloperidol. Hyperthermia should be treated aggressively; diazoxide or nitroprusside may be indicated for severe hypertension. Adverse effects of medicinal amphetamine use are discussed later in this chapter.
“Ecstasy”
3,4-Methylenedioxymethamphetamine (commonly known as ecstasy) continues to be a popular drug of abuse. Its primary mode of action is to both stimulate and block reuptake of serotonin (5-HT) [Parrott, 2002]. It also has a lesser effect on other monoaminergic neurotransmitters. Taken at dance parties called raves, it promotes a feeling of closeness and heightens sensory experience. It also causes euphoria. Acutely, it commonly causes serotonergic symptoms of confusion, hyperkinesis, and increased body temperature. These symptoms may be exacerbated by conditions that may prevail at raves: overcrowding, high ambient temperature, and coincident use of other stimulants. Rarely, these symptoms can develop into a full-blown serotonergic syndrome and a medical emergency. Once the acute effects wear off, depressive symptoms resulting from monoaminergic depletion occur. There are both laboratory animal and human data suggesting subsequent serotonergic axonal damage [Parrott, 2002].
Ethanol
Ethanol is abused increasingly often by children; acute alcohol intoxication and even chronic alcoholism have become serious problems in children as young as 10 years of age. The signs of acute alcohol intoxication are familiar, but a pediatrician confronted with a child who exhibits incoordination, ataxia, wide mood swings, impaired awareness, and gastrointestinal distress may not consider the diagnosis. Profound CNS depression follows ingestion of large amounts of alcohol. Alcohol withdrawal syndromes are rare in children. Alcohol intoxication may occur in mixed substance abuse. Accidental alcohol intoxication also has been reported in young children drinking attractively packaged and flavored mouthwashes. These mouthwashes have a significant ethanol content (14.2–26.9 percent) [Selbst et al., 1985], as do a variety of medicinal preparations, including cough medicines, decongestants, and teething preparations [Pruitt, 1984]. Chronic ethanol exposure in pregnant women causes fetal alcohol syndrome, as well as lesser fetal alcohol effects (see later discussion); some studies have suggested radiographically demonstrable brain injury in infants born to pregnant women consuming large amounts of alcohol [Holzman et al., 1995; Mattson et al., 1994]. Alcohol abuse may have a relationship to teenage suicide [Esposito-Smythers and Spirito, 2004]. Alcohol intoxication may increase the risk for suicidal attempts acutely. Its long-term disruptive influence on the young person’s life also may lead to further depressive symptoms and subsequent suicidal behavior.
Other Medications
Benzodiazepines
Intoxication with benzodiazepines is common because of their ready availability. These compounds produce depression and sedation at low doses, and confusion, somnolence, and coma at higher doses. Respiratory depression and hypotension may occur. The combination of benzodiazepines and other CNS depressants is potentially fatal. Treatment consists of supportive care and administration of flumazenil, a specific benzodiazepine antagonist. An optimal dose is not defined, but an initial dose of 0.01–0.02 mg/kg (maximum dose, 0.125 mg) is suggested; repeated doses or continuous infusion may be necessary. Flumazenil-induced convulsions may occur in individuals with chronic benzodiazepine use [Leikin and Paloucek, 1995; Perry and Shannon, 1996].
Other Sedatives
Chloral hydrate, ethchlorvynol (Placidyl), methaqualone, and glutethimide (Doriden) are sedatives that can cause severe intoxication with CNS depression. With mild intoxication, the child may be lethargic, confused, and ataxic; with severe intoxication, profound hypotonia, coma, and ophthalmoplegia may occur. Glutethimide, in particular, is associated with prolonged periods of unconsciousness. Careful attention must be given to respiratory support, which may be prolonged, and premature weaning must be avoided [Nicholson, 1983]. Glutethimide and its metabolites persist in the blood because they recirculate from fat stores and undergo enterohepatic circulation, resulting in reabsorption from the gastrointestinal tract; however, there is a poor correlation between plasma levels of glutethimide and clinical effects. Activated charcoal and resin hemoperfusion have been used in patients who have not responded to standard supportive measures. There are no specific antidotes for these sedative agents, although methylene blue is used to treat glutethimide-induced methemoglobinemia.
Despite its notoriety as a teratogen, the hypnotic agent thalidomide has been used in many infectious and inflammatory conditions, including human immunodeficiency virus (HIV)-related complications, leprosy, graft-versus-host disease, and lupus erythematosus. Although it typically causes limb and craniofacial birth defects, fetal thalidomide exposure also can cause congenital cranial neuropathies. Its effects suggest a toxic neural cristopathy. Postnatally, thalidomide acts as a potent neurotoxin, causing a potentially severe and often prolonged, if not permanent, sensory neuropathy [Alexander and Wilcox, 1997; Forsyth et al., 1996; Haslett et al., 1997; LeQuesne, 1993; Rodier et al., 1997].
Antipsychotic Agents (Neuroleptics)
Major tranquilizers include phenothiazines and related compounds, as well as butyrophenones, which are associated with a variety of serious neurologic complications, including sedation, acute dystonic reactions, akathisia, pseudoparkinsonism, withdrawal, emergent dyskinesia, tardive dyskinesia, and neuroleptic malignant syndrome. Newer antipsychotic agents include clozapine, sertindole, olanzapine, and risperidone, and reportedly have fewer neurologic side effects [Caroff et al., 2002]. Acute intoxication in a young child usually results in a confusional state or depressed level of consciousness. Acute dystonic reactions, often observed in children after a single dose of phenothiazine or haloperidol, may manifest as an oculogyric crisis or sudden dystonic posturing of the head and neck, including opisthotonus, retrocollis, torticollis, facial grimacing, and tongue thrusting. These acute reactions are not necessarily dose-related, and are usually readily reversed with the intravenous administration of 25–50 mg of diphenhydramine. Miosis, coma, hypothermia or hyperthermia, and hypotension may occur after ingestion of a large dose of either phenothiazines or butyrophenones. Respiratory depression suggests the ingestion of additional agents, such as narcotics [Knight and Roberts, 1986].
Akathisia, an internal feeling of restlessness, is a dose-related neurologic complication of neuroleptic treatment and is thought to be rare in children [Sachdev, 1995]. It is one of the most distressing adverse effects of neuroleptic use but usually responds to treatment with anticholinergic drugs or blockers.
Neuroleptic malignant syndrome is a rare complication of neuroleptic drug therapy and is characterized by fever, muscular rigidity, autonomic dysfunction, and altered sensorium, with waxing and waning of consciousness. The patient may be in a catatonic-like stupor and may be mute. In children, neuroleptic malignant syndrome occurs during therapy with neuroleptics, as well as after accidental phenothiazine ingestion [Klein et al., 1985b]. Myoglobinuria and acute renal failure may supervene. Treatment consists of intensive supportive care, with monitoring of vital signs, aggressive fluid management, and, in selected cases, dantrolene administration [Caroff and Mann, 1993; Tueth, 1994]. Although reported more often in association with older antipsychotic medications, neuroleptic malignant syndrome also occurs with newer agents such as clozapine, albeit possibly with less dramatic clinical and laboratory findings [Sachdev et al., 1995]. A clinical presentation of confusion, myoclonus, hyperreflexia, diaphoresis, and diarrhea, progressing to fever, coma, and rigidity that improved over 10 days with bromocriptine and aggressive intensive care unit support, was seen in a young woman taking risperidone and fluvoxamine [Reeves et al., 2002]. It was thought that this may not have been neuroleptic malignant syndrome but rather serotonin syndrome. Aripiprazole has been reported to cause neuroleptic malignant syndrome as well, although the one pediatric report to date leaves open a broader differential diagnosis [Palakurthi et al., 2007].
Tardive dyskinesia after chronic phenothiazine or haloperidol use is extremely rare in children [Singer, 1986]. The propensity to develop tardive dyskinesia may be related to the patient’s underlying neural substrate, as found in a recent study. Juckel and colleagues [1995], using auditory event-related potentials, demonstrated an association between a small P300 evoked potential and subsequent tardive dyskinesia.
The newer “novel” antipsychotics, such as clozapine, risperidone, olanzapine, and sertindole, have clinically significant pharmacologic activity at multiple receptors, and fewer typical antidopaminergic side effects such as acute parkinsonism, akathisia, and tardive movement disorders [Baldessarini, 1996; Casey, 1996a; Krebs, 1995; Lindstrom et al., 1995; Schooler, 1996]. Clozapine, in particular, appears to be associated with less bradykinesia and akathisia than haloperidol, although not necessarily less tremor [Kurz et al., 1995]. Acute extrapyramidal effects have been seen in children after an overdose of 100 mg of clozapine [Goetz et al., 1993; POISINDEX, 2010]. Confusion, lethargy, and ataxia also have been reported in children. Although clozapine-associated seizures have been reported at doses of 250 mg/day, the prevalence of seizures is higher (5 percent) in adults taking 600–900 mg/day [Haller and Binder, 1990; Tueth, 1994]. Myoclonus also has been reported at higher doses [Bak et al., 1995]. Extrapyramidal side effects (acutely, dystonia) and tardive dyskinesia are reported infrequently with risperidone [Buzan, 1996; Daniel et al., 1996; Faulk et al., 1996]. Olanzapine appears to have an extrapyramidal side-effect profile intermediate between that of the older antipsychotics and its newer cousins [Casey, 1996b].
Antidepressants
Tricyclic antidepressants, such as amitriptyline, imipramine, and desipramine, have been associated with acute toxicity and significant morbidity and mortality. Acute encephalopathy occurs within 4 hours of ingestion and includes ataxia, hallucinations, and nystagmus. Somnolence occurs subsequently, and tremor or myoclonus may occur. Cardiac conduction abnormalities, including tachycardia, heart block, atrial fibrillation, and ventricular flutter, are the most common and serious non-neurologic results of tricyclic intoxication [Pimentel and Trommer, 1994]. Dry mouth, palpitations, and tachycardia occur as typical anticholinergic effects. Death may occur as a result of progressive coma, seizures, and cardiac conduction defects. In acute poisoning, activated charcoal with a cathartic may be helpful; lavage may be attempted if fewer than 6 hours have passed since ingestion. Up to 2 mg of physostigmine administered intravenously may awaken a comatose patient but should be avoided in patients with bradycardia. Keeping the blood pH slightly alkaline (e.g., 7.5) helps in the treatment of tachyarrhythmias. A prolonged QRS interval is a good predictor of seizures and cardiac dysrhythmias in acute intoxication with tricyclics, but serious CNS depression may occur with relatively normal QRS intervals [Boehnert and Lovejoy, 1985]. Amoxapine, a newer tricyclic antidepressant, also frequently induces seizures when taken in toxic amounts, but unlike the other drugs of this class, it does not seriously alter the electrocardiogram [Rogol et al., 1984].
SSRIs are some of the most frequently used medications in the United States at the present time. Such popularity stems from their broad range of efficacy and relatively favorable side-effect profile. Although cases of SSRI toxicity have increased as a result of this wide availability, serious neurotoxicity is limited generally to accidental ingestions or intentional overdoses, especially in combination with other medications or drugs of abuse [Klein-Schwartz and Anderson, 1996; Lavin et al., 1993; Bronstein et al., 2007]. Paradoxic behavioral reactions and SSRI-induced hyperkinetic movement disorders are perhaps the most likely adverse neurologic effects. In a review of 469 SSRI intoxication cases in adults, a more severe serotonergic syndrome, with coma or convulsions, occurred in 2.4 and 1.9 percent, respectively [Isbister et al., 2004]. Of note, of the five different SSRIs involved, citalopram had relative QTc prolongation, although its clinical significance was unclear. In a study of isolated accidental fluoxetine ingestion of 20 mg or more in 120 children younger than 6 years, 96 percent were asymptomatic. Sedation and vomiting were the only side effects reported in symptomatic children [Baker and Morgan, 2004]. A recent report of 26 cases of citalopram intoxication revealed generally mild cardiac toxicity (albeit with one death by cardiac arrest), but more severe encephalopathy and seizures [Jimmink et al., 2008]. Intensive care admission is recommended for all cases of citalopram intoxication. A neonatal withdrawal syndrome, including neonatal convulsions, has been described in newborns whose mothers were taking SSRIs during the pregnancy [Sanz et al, 2005].
Lithium
Lithium carbonate, used for the treatment of bipolar depression in adults, is effective in the treatment of certain behavioral and affective disorders in children [Goetting, 1985]. Most cases of lithium intoxication occur during the course of prolonged therapy. The typical patient has apathy, drowsiness, nystagmus, tinnitus, dysarthria, ataxia, coarse muscle tremors, vomiting, and diarrhea. Choreiform movements, dystonic posturing, and cogwheel rigidity may occur. Severity of symptoms correlates well with the serum lithium concentration. Seizures and coma indicate severe toxicity (i.e., serum lithium level >3.0 mmol/L). Treatment of acute poisoning (<30 minutes) should include induction of emesis or gastric lavage. However, because of the slow excretion of lithium, symptoms may progress, despite cessation of the medication. There is no specific treatment, other than attention to fluid balance and support. Sodium chloride administration enhances urinary excretion, particularly if the patient is hyponatremic. Peritoneal dialysis or hemodialysis is used in cases of clinically severe intoxication with serum lithium levels greater than 3 mmol/L [Tueth, 1994]. Prolonged hemodialysis may be necessary to prevent a rebound in serum lithium concentration [Sansone and Ziegler, 1985].
Salicylates and Acetaminophen
Salicylates are present in a myriad of medicinal preparations, ranging from aspirin tablets to oil of wintergreen and long-used Chinese medicinal oils [Chan et al., 1995]. Poisoning has been reported with all forms through both oral and topical routes [Abdel-Magid and el-Awad Ahmed, 1994]. Absorption rate depends on the preparation. Acute aspirin dosages of >200 mg/kg are toxic; doses of 500 mg/kg may be lethal [Cataletto, 2010]. Neurologic, hepatic, and pulmonary toxicity occurs in concert with other metabolic derangements [Bari, 1995]. Symptom onset in acute intoxications is within a few hours. Salicylates directly affect the CNS and cause central hyperventilation with respiratory alkalosis. Poisoning of the Krebs cycle and increased skeletal muscle metabolism result in increased oxygen consumption, increased carbon dioxide production, and metabolic acidosis. Salicylates also contribute directly to this acidosis. Mortality is related to brain salicylate concentration; respiratory decompensation and subsequent worsening acidosis facilitate salicylic acid passage into the CNS [Yip et al., 1994]. Severe intoxication is more likely in children younger than 4 years.
Chronic toxicity occurs after several days of use and sometimes may be more difficult to discern because of symptoms of the patient’s underlying illness. CNS disturbances occur in up to 60 percent of patients and include lethargy, coma, and convulsions; however, the predominant toxicity with chronic salicylism is pulmonary edema [English et al., 1996; Yip et al., 1994].
Poisoning also may result from chronic excessive use of bismuth subsalicylate (Pepto-Bismol). The combination of typical signs of chronic salicylate toxicity and radiopaque bismuth precipitate seen on radiographs of the abdomen suggests the diagnosis [Hearney et al., 1996; Vernace et al., 1994].
Failure to recognize salicylate poisoning and treat it aggressively contributes to morbidity and mortality. As in most intoxications, early recognition optimizes outcome. The classic triad of acute poisoning is hyperventilation, tinnitus, and gastrointestinal irritation. Supportive therapy and aggressive measures to speed elimination of salicylates from the body are the mainstays of treatment. Fluid diuresis, coupled with urinary alkalinization, is used in milder cases; more severe poisonings may require blood alkalinization. This alkalinization and early hemodialysis may be life-saving in severe intoxications [Yip et al., 1994]. Activated charcoal is beneficial, but the advantage of multiple-dose over single-dose regimens is not proved [Cienki et al., 1995]. Hypoglycemia may complicate salicylate poisoning. Intravenous fluids should contain at least 5 percent glucose; 10 percent glucose solutions should be used in young children with documented hypoglycemia or mental status changes. Aspirin tablets may form bezoars within the stomach; these provide a reservoir for prolonged salicylate absorption and may require dilutional catharsis. Gross overuse of bismuth-containing antacids may also cause a bismuth encephalopathy, characterized by delirium, myoclonus, tremor, and ataxia [Kumar, 2008].
Unlike salicylates, acetaminophen (and paracetamol) appears not to have a recognizable primary toxic effect on the brain. CNS effects result from acetaminophen-induced liver failure. Hepatic coma and life-threatening cerebral edema may be present in severe acute overdoses, although peak symptoms may not occur until 1–3 days after ingestion [Leikin and Paloucek, 1995]. Early lack of symptoms does not exclude a potentially fatal ingestion, and need for therapy is directly related to dose and measured acetaminophen blood level. Conservative estimates of the acute pediatric toxic dose are 140–150 mg/kg, but many authors use doses of 200 mg/kg, up to an adult toxic dose of about 7.5 g [Anker and Smilkstein, 1994]. Home ipecac administration for this and other suspected poisonings once was considered standard of care; however, the current American Academy of Pediatrics recommendation is to not have ipecac in the home [American Academy of Pediatrics Committee on Injury, Violence, and Poison Prevention, 2003; Bond, 1995; Shannon, 2003]. Gastric lavage is effective if performed within 1 hour after ingestion. Activated charcoal should be given up to 4 hours after ingestion. If dosage at or above toxic range is suspected, N-acetylcysteine should be started; efficacy of oral versus intravenous therapy is debated. A 4-hour postingestion blood acetaminophen level is compared with a standard nomogram to determine the need to continue N-acetylcysteine therapy [Brandwene et al., 1996]. N-acetylcysteine coupled with a molecular absorbant recirculating system, if started early, may allow hepatic regeneration and obviates the need for liver transplantation [Koivusalo et al., 2003]. After ingestion of extended-release acetaminophen preparations, a second blood acetaminophen level should be obtained 4–6 hours after the first (but at least 8–12 hours after ingestion) [Cetaruk et al., 1997; Leikin and Paloucek, 1995].
Stimulants
Stimulant medications, such as dextroamphetamine, pemoline, and methylphenidate, used for the treatment of attention-deficit hyperactivity disorder, may produce increased activity and varying dyskinesias, including motor tics and chorea [Jimenez-Jimenez et al., 1997; Nakamura et al., 2002; Stork and Cantor, 1997]. Some children receiving methylphenidate may develop Tourette’s syndrome, but a recent study of males with attention-deficit hyperactivity disorder and Tourette’s syndrome demonstrated that tics increased reversibly with stimulant use [Castellanos et al., 1997].
Atomoxetine is a nonstimulant agent approved for the treatment of attention-deficit hyperactivity disorder. Generally, side effects are mild, but limiting tachycardia can occur. Kashani and Ruha reported a single case of atomoxetine overdose (2840 mg with typical dose 1.2–1.8 mg/kg/day) in an adolescent who then presented with a seizure [Kashani and Ruha, 2007].
Theophylline
Theophylline and aminophylline, commonly used for treating apnea of prematurity and asthma, may cause neurologic symptoms as a result of either accidental ingestion or therapeutic error. Although theophylline concentrations higher than 20 mg/L are believed to be toxic, and clinical toxicity with nausea, vomiting, tachycardia, and agitation, followed by seizures, is more common when theophylline levels are greater than 30–40 mg/L, some children suffer lesser side effects with levels above 12–15 mg/L [Barr et al., 1994]. Gastrointestinal symptoms do not necessarily precede the onset of seizures, and seizures may occur with serum theophylline concentrations as low as 25 mg/L, although they are more common when concentrations exceed 50 mg/L [Gal et al., 1980; Jira et al., 1996]. Treatment of theophylline toxicity is supportive. Although activated charcoal absorbs the drug well within the gastrointestinal tract, hemoperfusion is considered the definitive treatment. Peritoneal dialysis has been used successfully in neonates and infants [Colonna et al., 1996; Jira et al., 1996]. Ipecac should be avoided. Seizures may be difficult to control but should be treated initially with benzodiazepines [Leikin and Paloucek, 1995]. Status epilepticus associated with increased theophylline levels carries a risk for increased morbidity over status epilepticus in general [Dunn and Parekh, 1991]. This risk may be secondary to theophylline-induced decreased cerebral blood flow or to increased risk for hypoxia due to underlying pulmonary disease.
Diphenhydramine
The antihistamine diphenhydramine is contained commonly in over-the-counter cold remedies and in topical preparations. Although neurologic side effects usually are mild (sedation or, paradoxically, hyperactivity), more overt symptoms can occur. Hallucinations and seizures have been reported. Rarely, diphenhydramine overdose results in death. Even excessive topical administration may lead to toxic blood levels. Risk factors may include use in a very young child, and anything that increases transdermal absorption, including excoriation due to pruritus and vasodilatation after a warm bath [Turner, 2009].
Atropine and Related Alkaloids
Many drugs with anticholinergic properties, such as atropine, may produce an acute anticholinergic syndrome if taken in excess (see previous toxidrome discussion). Examples of drugs with significant anticholinergic properties include atropine, scopolamine, and local mydriatics, such as cyclopentolate (Cyclogyl) and tropicamide (Mydriacyl); a few antihistaminics, such as diphenhydramine and tripelennamine; gastrointestinal antispasmodics, such as dicyclomine (Bentyl) and propantheline (Pro-Banthine); and older tricyclic antidepressants. The scopolamine-containing transdermal patches used for the prevention of motion sickness are a novel way to administer excessive amounts of scopolamine to children. The transdermal patch was developed primarily for adults and contains 1.5 mg of scopolamine, which can produce toxic psychosis in children and should be used with caution [Klein et al., 1985a; Lewis et al., 1994]. These patches also are used in some developmentally disabled children (e.g., with cerebral palsy) to decrease drooling. Poisoning also may occur after ingestion of plants containing belladonna alkaloids (Atropa belladonna, also known as deadly nightshade). Recreational abuse of such plants (e.g., tea made with Brugmansia [angel’s trumpet] and Datura spp.) in teenagers has been reported, although seizures or fatalities resulting from drinking the prepared tea appear unlikely [Isbister et al., 2003; Patel et al., 2003].
Supportive measures are the mainstay of therapy. Physostigmine may be used but may precipitate cholinergic toxicity, including seizures and bradyarrhythmias, and its use requires availability of adequate supportive therapy for these potential complications [Kulig and Rumack, 1983]. Physostigmine (0.5 mg intravenously) is administered slowly in children; it may be repeated every 10 minutes for a maximum total dose of 2 mg.
Drugs Used in Organ Transplantation
Organ transplantation may result in severe neurologic complications. Severe neurologic sequelae may occur in 10–15 percent of children who undergo hematopoietic stem-cell transplantation [Faraci et al., 2002]. In a large Italian study, risk factors were allogeneic donor, presence of graft-versus-host disease, and use of total brain irradiation.
Cyclosporine
Cyclosporine, a potent immunosuppressant, is being used increasingly in solid organ transplantation [de Groen et al., 1987; Lee and Vacanti, 1996]. It probably is implicated most in neurologic sequelae of transplantation drugs [Faraci et al., 2002]. Neurotoxicity, nephrotoxicity, and hypertension are complications of cyclosporine therapy. Cyclosporine is lipophilic, a property that allows it relatively easy access to the CNS. The most common side effects of cyclosporine on the nervous system are headache, confusion, and seizures, but cortical blindness, hemiparesis, ataxia, and tremor also are reported [Adams et al., 1987; Boon et al., 1988; Garg et al., 1993; Ghalie et al., 1990; Miller, 1996; Reece, 1991; Soy et al., 1995; Trzepacz et al., 1993]. A syndrome of encephalopathy, seizures, and cerebral white-matter changes also has been described. The white-matter changes are more prominent in the parieto-occipital areas and resemble posterior reversible encephalopathy syndrome [Miller, 1996; Truwit et al., 1991; Nishie et al., 2003]. Marchiori and associates reported a case of cyclosporine-associated posterior reversible encephalopathy syndrome and reversible opsoclonus in a teenager after orthotopic liver transplantation [Marchiori et al., 2004]. These complications seem to be reversible but may require a decrease in the cyclosporine dose. Side effects usually are associated with high levels of cyclosporine. Other factors that exacerbate cyclosporine toxicity include hypertension, fever, hypomagnesemia, hypocholesterolemia, aluminum overload (especially in patients undergoing renal transplantation), and concurrent methylprednisolone treatment [Bhatt et al., 1988; de Groen et al., 1987; Thompson et al., 1984; Trzepacz et al., 1993].
OKT3
OKT3 is a murine monoclonal anti-pan-T-cell antibody of the immunoglobulin G2a isotype. OKT3 has been used extensively as an immunosuppressant agent in acute cellular allograft rejection or graft-versus-host disease [Norman, 1989]. The target of OKT3 is CD3, a 17- to 20-kDa molecule found on mature T cells that is selectively removed during treatment with this monoclonal antibody. OKT3 blocks both the generation and function of cytotoxic T cells. The half-life of injected OKT3 is about 18 hours [Fung, 1991; Norman, 1989]. A picture of aseptic meningitis with fever, photophobia, cephalgia, and cerebrospinal fluid pleocytosis has been described with OKT3 use [Adair et al., 1991; Emmons et al., 1986; Martin et al., 1988]. Other complications have included cerebral edema, seizures, cerebritis, and tremor [Capone and Cohen, 1991; Garg et al., 1993; Thomas et al., 1987; Trzepacz et al., 1993].
Tacrolimus (FK-506)
Tacrolimus, or FK-506, is used primarily to prevent organ rejection in liver transplant recipients [Diasio and LoBuglio, 1996; Lee and Vacanti, 1996]. Like cyclosporine, it inhibits interleukin-2 and is thought to act by inhibiting T-cell activation. Serum levels of tacrolimus are altered significantly by multiple drugs, including cyclosporine, methylprednisolone, metoclopramide, some antiepileptic drugs, and various antifungal agents.
Tacrolimus has clinical toxicity similar to that of cyclosporine. At serum levels greater than 3 ng/mL, delirium has been reported [Trzepacz et al., 1993]. Other neurologic side effects include dysphasia, seizures, and tremor. The dose relationship of these side effects is unclear, although increased toxicity has been noted with concomitant use of agents that increase tacrolimus delivery to the small intestine (e.g., metoclopramide) [Backman et al., 1994; Trzepacz et al., 1993; Prescott et al., 2004]. MRI may indicate white-matter change, but reversibility may be predicted by apparent diffusion coefficient mapping [Shimono et al., 2003; Furukawa et al., 2001].
Antibiotics
Chloramphenicol
Chloramphenicol has been associated with the development of a reversible optic neuritis, demonstrable clinically and by visual-evoked potentials, in patients with cystic fibrosis who undergo long-term treatment with chloramphenicol [Leikin and Paloucek, 1995; Leitman et al., 1964; Spaide et al., 1987].
Nitrofurantoin
Polyneuropathy is a known complication in patients treated over prolonged periods with nitrofurantoin, particularly in the presence of impaired renal function. Patients have ascending sensory and motor polyneuropathy associated with increased spinal fluid protein [Spring et al., 2001].
Aminoglycosides
Aminoglycosides are potentially ototoxic. Recent data suggest that susceptibility to aminoglycoside-induced ototoxicity is associated with a mitochondrial mutation (nucleotide 1555) [Usami et al., 1997]. Symptoms may arise not only from oral or parenteral administration of the drugs, but also after wound irrigation. A myasthenic-like syndrome accompanied by generalized muscle weakness, which is partially antagonized by neostigmine, may result from the administration of aminoglycosides such as streptomycin, neomycin, kanamycin, bacitracin, colistin, and gentamicin. This reaction usually occurs in association with the simultaneous administration of a neuromuscular blocking agent (e.g., succinylcholine, d-tubocurarine, decamethonium bromide). This neuromuscular blocking effect is especially pronounced when these antibiotics are placed in the pleural or abdominal cavities, with resultant rapid absorption and high plasma levels. These antibiotics may also cause a transient clinical deterioration in patients with myasthenia gravis [Kaeser, 1984].
Beta-Lactam Antibiotics
The β-lactam antibiotics are a diverse group of medications with potential for myriad systemic and neurologic side effects. Seizures caused by the penicillins, cephalosporins, and carbapenems result from the inhibition of gamma-aminobutyric acid (GABA) receptors [Asensi et al., 1993; Sunagawa and Nouda, 1996]. Movement disorders and change in sensorium also may occur. Effects may be magnified in a child with an altered blood–brain barrier.
Antiviral Agents
Anti-HIV drugs have prolonged the lives of many patients with acquired immune deficiency syndrome. They also have been associated with significant neurotoxicity. Use of two older agents, dideoxycytidine and dideoxyinosine, as well as a newer reverse transcriptase inhibitor, stavudine, has been associated with peripheral neuropathy [Brew and Currie, 1993; Mueller, 1997]. So far, therapy with protease inhibitors has not demonstrated significant neurotoxicity, but only preliminary data are available [Mueller, 1997].
Antineoplastic Drugs
Use of chemotherapeutic agents has produced striking improvement in the outcome of childhood malignant disease but is associated with a number of neurologic complications in children and adults [Armstrong and Gilbert, 2004].
Vinca Alkaloids
Vincristine and vinblastine are potent antineoplastic agents with variable degrees of neurotoxicity, some of which is reversible. Impaired nutrition appears to increase susceptibility to vincristine toxicity. Greater neurotoxicity is also related to higher concentrations of drug per dose and increased frequency and duration of therapy. Some degree of symmetric peripheral neuropathy, with areflexia involving early and consistent loss of Achilles tendon reflexes, slapping gait, muscle atrophy, and leg weakness, develops frequently, even with customary doses of vincristine. Numbness and tingling paresthesias in hands and feet are common with preserved cutaneous sensation, although the child may complain of hand clumsiness before the onset of footdrop [Tuxen and Hansen, 1994]. Muscle cramps are also a frequent symptom of vincristine-associated motor neuropathies, occurring in both proximal and distal distributions [Haim et al., 1991]. Cranial nerve palsies also may occur, with involvement of cranial nerves II to VIII and X. The most common ocular findings are variable degrees of ophthalmoparesis and ptosis. Autonomic dysfunction is another aspect of vincristine neurotoxicity, with colicky abdominal pain, paralytic ileus, constipation (in 33–46 percent of patients), and bladder atony [Christensen et al., 1994; Tuxen and Hansen, 1994]. Generalized seizures and inappropriate secretion of antidiuretic hormone are additional but rare complications of vincristine treatment [Sandler et al., 1969; Shurin et al., 1982]. Vincristine’s peripheral neurotoxic effects may be potentiated by itraconazole [Ariffin et al., 2003; Kamaluddiin et al., 2001]. Vinorelbine has the potential to cause similar neurotoxic effects but, possibly because of its greater selectivity for mitotic rather than axonal microtubules, does so less often [Tuxen and Hansen, 1994].
Vincristine also causes a necrotizing myopathy [Le Quintrec and Le Quintrec, 1991]. Especially with concomitant steroid therapy, a combined myoneuropathy picture may develop [Bradley et al., 1970; DeAngelis et al., 1991]. Improvement usually ensues with completion of therapy.
Methotrexate
Myelopathy, headache, vomiting, nuchal rigidity with aseptic meningitis, truncal ataxia, tremor, paraplegia, delirium, and somnolence have been described as complications of both intrathecal and systemic therapy with methotrexate. Meningoencephalopathy can occur as late as several months after completion of methotrexate therapy. Scattered subcortical intracerebral calcifications may result from large cumulative doses of intravenous methotrexate, and concurrent administration of cytosine arabinoside may increase the risk for this complication [McIntosh et al., 1977]. The most devastating effect of methotrexate therapy, originally thought to be related to a combination of a high dose of intrathecal methotrexate and CNS irradiation, is a subacute and usually progressive leukoencephalopathy. A characteristic pattern of decreased density of periventricular white matter is evident on CT or as increased periventricular signal on T2-weighted MRI. Clinical manifestations include mental deterioration with dementia, seizures, and focal neurologic deficits. This complication may occur between 3 and 9 months after high-dose methotrexate therapy. An immediate and fatal necrotizing leukoencephalopathy has been reported in association with an inadvertent, more than 50-fold intrathecal overdose of methotrexate [Ettinger, 1982]. Leukoencephalopathy can occur with intravenous methotrexate therapy plus cranial irradiation without intrathecal administration of the methotrexate [McIntosh et al., 1977; Price and Jamieson, 1975; Weiss et al., 1974; Shuper et al., 2002]. Animal studies suggest that concomitant administration of steroids may play a role in this neurotoxicity [Mullenix et al., 1994]. In addition, underlying disease may potentiate methotrexate toxicity. Miller and Wilkinson describe 79–93 percent reduction in local cerebrospinal fluid clearance of intraventricularly administered methotrexate in three individuals with carcinomatous meningitis [Miller and Wilkinson, 1989]. Improvement also has been described in a single report using a combination of high-dose folinic acid and aminophylline [Jaksic et al., 2004].
l-Asparaginase
l-Asparaginase therapy may produce changes in mentation, somnolence, and EEG slowing [Moure et al., 1970; Tuxen and Hansen, 1994]. Intracranial thrombosis or hemorrhage and peripheral arterial thrombosis with headache, obtundation, hemiparesis, and seizures have also been recognized as complications in 1–2 percent of children receiving l-asparaginase [Priest et al., 1982]. The associated coagulation disorder is thought to be secondary to l-asparaginase-induced decrease in antithrombin III level [Kieslich et al., 2003]. Despite this, it is not yet clear that l-asparaginase-related cerebrovascular thrombosis responds to replacement of factors, or that it is resistant to low-molecular-weight heparin therapy.
Platinum Agents
Ototoxicity is a common complication of cisplatin therapy; peripheral sensory neuropathy may also occur, with dysesthesias and paresthesias, diminished reflexes, decreased vibratory and light touch sensation, and sensory ataxia [Cavaletti et al., 1996; Thompson et al., 1982; Steeghs et al., 2003; Markman, 2003]. Although retinal toxicity is reported in adults, it is rare in children and may be associated with abnormal renal function [Hilliard et al., 1997]. Autonomic neuropathy is rare. Oxaliplatin has similar neurotoxic potential; carboplatin appears somewhat less toxic to the nervous system [Tuxen and Hansen, 1994]. Possible neuroprotectant agents that may minimize this neurotoxicity include calcium channel antagonists, thiol compounds, adrenocorticotropic hormone analogs, and neurotropic factors [Cavaletti et al., 1996].
Cytosine Arabinoside
Cytosine arabinoside has been associated with transient paraplegia after intrathecal administration. In childhood acute myelogenous leukemia, patients given intrathecal and intravenous cytosine arabinoside have exhibited progressive ascending paralytic disease, with onset delayed up to 6 months after treatment, accompanied by spinal cord demyelination [Dunton et al., 1986]. Cytosine arabinoside may also cause cerebellar ataxia, change in sensorium (the latter at high doses), and, rarely, peripheral neuropathy or brachial plexopathy [Tuxen and Hansen, 1994].
Cyclophosphamide and Ifosfamide
Cyclophosphamide and ifosfamide both may cause CNS toxicity (10–30 percent incidence in the case of the latter) [DiMaggio et al., 1994; Tuxen and Hansen, 1994]. This toxicity is manifest as mental status changes, seizures, and ataxia. Cranial neuropathies also are possible. In the case of ifosfamide, toxicity may be evident within 2 hours after infusion is begun, and may continue for 1–3 days after a 5-day course of therapy [Pratt et al., 1989; Nicolao and Giometto, 2003; Rieger et al., 2004]. It is more likely to occur in the face of common concomitants of malignancy, including hepatic or renal dysfunction, hypoalbuminemia, acidosis, or prior CNS abnormality. Improvement has been reported with methylene blue administration [Raj et al., 2004.
Other Agents Used in Cancer Chemotherapy
Fludarabine, cladribine, and pentostatin are newer purine analogs that currently are used more often in adults than in children. They carry potential for fatal neurotoxicity at higher than recommended doses; at the recommended dosage, each carries a 15 percent risk for (usually reversible) neurotoxicity [Cheson et al., 1994].
Finally, as cytokines find increasing roles in cancer chemotherapy, their neurotoxicity is becoming more evident. Evidence is strongest for interferon and interleukin-2. Interferon therapy has been associated with frontal lobe dysfunction (decreased executive functions, transient aphasia), migraine, parkinsonism and akathisia, and peripheral neuropathy. Conversely, interleukin-2 use has been linked to neurobehavioral changes ranging from lethargy to delirium. The mechanism of neurotoxicity is unclear, although it is postulated that they circumvent the blood–brain barrier through a poorly understood mechanism. Quantification of the degree of toxicity awaits prospective studies [Forman, 1994; Margolin, 1994].
Steroids
Neurologic complications of steroid therapy include delirium, cortical atrophy, pseudotumor cerebri, and steroid myopathy. More subtle neurobehavioral effects of steroids, especially anabolic and androgenic steroids used (and abused) by athletes, are catalogued elsewhere [Lukas, 1996; Pope and Katz, 1994]. Pseudotumor cerebri related to steroids occurs primarily in children receiving prolonged corticosteroid treatment, at times after drug withdrawal. There does not appear to be any relationship between pseudotumor cerebri and the disorder for which steroids were being administered. Autonomic neuropathy also has been reported [Rosener et al., 1996]. Proximal myopathy may be a common complication of steroid therapy. Onset may be insidious, and it may occur after only a short duration (<14 days) of steroid therapy. Although often mild, myopathy may occur in up to 60 percent of patients treated with steroids [Batchelor et al., 1997].
Radiographic Contrast Agents
There is evidence that metrizamide reaches the intracranial subarachnoid space in these patients [Junck and Marshall, 1983]. Headache is common after metrizamide myelography. The incidence of seizures is 0.1 percent; many of the reported seizures have occurred in patients with a history of seizures [Torvik and Walday, 1995]. Rare transient adverse effects of metrizamide include paraparesis, loss of reflexes, blindness, speech disturbances, and diplopia. Cerebrospinal fluid pleocytosis and clinical evidence of aseptic meningitis may occur after metrizamide myelography [Junck and Marshall, 1983; DiMario, 1985]. Seizures also have been reported with diatrizoate meglumine (Hypaque) [Karl et al., 1994]. Newer nonionic contrast agents, including iohexol and iodixanol, appear to carry less risk, although similar side effects have been reported anecdotally [Torvik and Walday, 1995]. Symptoms occur 2–12 hours after injection and may last 24–72 hours.