Platelet Production, Structure, and Function
After completion of this chapter, the reader will be able to:
1. Diagram megakaryocytopoiesis, including megakaryocyte localization, growth factor control, endomitosis, and platelet shedding.
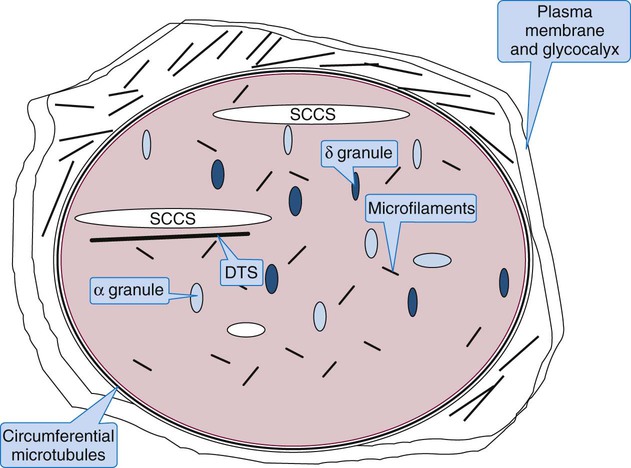
2. Describe the ultrastructure of resting platelets in the circulation, including the plasma membrane, tubules, microfibrils, and granules.
3. List the important platelet receptors and their ligands.
4. Recount platelet function, including adhesion, aggregation, and secretion.
5. Reproduce the biochemical pathways of platelet activation, including integrins, G proteins, the eicosanoid, and the diacylglycerol-inositol triphosphate pathway.
Megakaryocytopoiesis
Platelets are anucleate blood cells that circulate in amounts of 150 to 400 × 109/L, with mean counts slightly higher in women than in men.1 Platelets trigger primary hemostasis on exposure to endothelial, subendothelial, and plasma procoagulants in blood vessel injury. On a Wright-stained wedge-preparation blood film, platelets are distributed throughout the red blood cell monolayer at 7 to 21 per 100× field. They have an average diameter of 2.5 µm, corresponding to a mean platelet volume (MPV) of 8 to 10 fL in an isotonic suspension, as determined using laboratory profiling instruments.2 Their internal structure, although complex, is granular but scarcely visible using light microscopy.
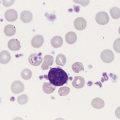
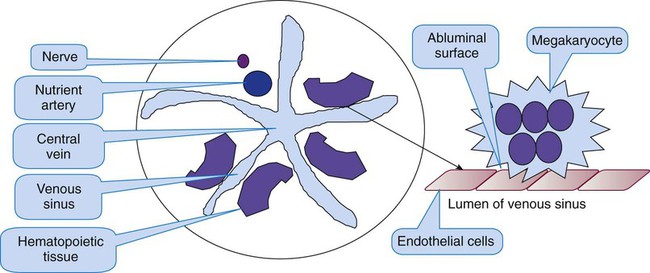
Platelets arise from unique bone marrow cells called megakaryocytes. Megakaryocytes are among the largest cells in the body and are polyploid, which means that they possess multiple chromosome copies within a single cell. On a Wright-stained bone marrow aspirate film, each megakaryocyte is 30 to 50 µm in diameter with a multilobulated nucleus and abundant granular cytoplasm. In healthy intact bone marrow tissue, megakaryocytes cluster in the extravascular compartment adjacent to the abluminal membrane (the surface opposite the lumen) of venous sinusoid endothelial cells (Figure 13-1).3 Myelocytic and erythrocytic precursor cells, which locate further from the endothelial cells, may cross the megakaryocyte cytoplasm to reach the sinusoid lumen, a faux phagocytosis known as emperopolesis.4 Megakaryocytes are also harvested from the lungs.5 In a normal Wright-stained bone marrow aspirate film, the microscopist may identify two to four megakaryocytes per 10× low-power field.
Endomitosis
Megakaryocyte maturation is marked by a mysterious form of mitosis that lacks telophase and cytokinesis (separation into daughter cells) and is called endomitosis. In endomitosis, DNA replication proceeds to the production of 8N, 16N, or 32N ploidy with duplicated sets of chromosomes, but no cell division.6 Some megakaryocyte nuclei replicate five times, reaching 128N; this level of ploidy is unusual, however, and may signal hematologic disease. Megakaryocytes employ their copious DNA to synthesize abundant cytoplasm, which differentiates into platelets. A single megakaryocyte may shed 2000 to 4000 platelets. In an average-size healthy human there are 108 megakaryocytes producing 1011 platelets per day. The key to endomitosis is the loss of spindle fiber orientation at the point of telophase, so that chromosomes do not proceed from equatorial plates to polar bodies, but rather duplicate in place. Cytokinesis is arrested, a cell-cycle adaptation found in no other normal human cell.
Megakaryocyte Progenitors
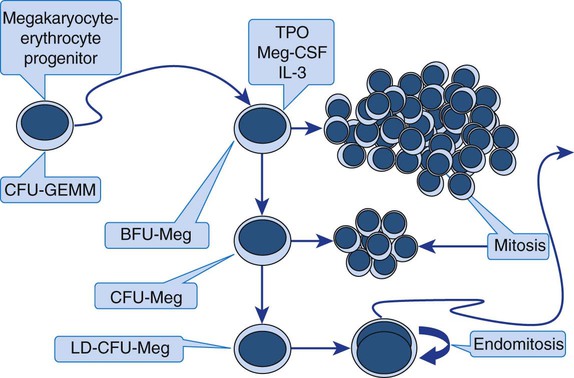
The megakaryocyte-erythrocyte progenitor (see Figure 7-13) differentiates into the megakaryocyte lineage under the influence of the hormone thrombopoietin (TPO) and a series of cytokines (see Table 13-3). There are three megakaryocyte lineage-committed progenitor stages, defined by their culture colony characteristics. In order of differentiation, these are the least mature burst-forming unit (BFU-Meg), the colony-forming unit (CFU-Meg), and the most mature light-density CFU (LD-CFU-Meg).7 All three progenitor stages resemble small lymphocytes and cannot be distinguished by Wright-stained light microscopy. The BFU-Meg and CFU-Meg are diploid and participate in normal mitosis, maintaining a pool of megakaryocyte progenitors. Their proliferative (mitotic) properties are reflected in their ability to form colonies of hundreds (BFUs) or scores (CFUs) of progeny in culture (Figure 13-2). In contrast to the BFU-Meg and CFU-Meg, the LD-CFU-Meg has little proliferative capacity and produces few cells, but begins the progress through endomitosis to reach increased nuclear ploidy.
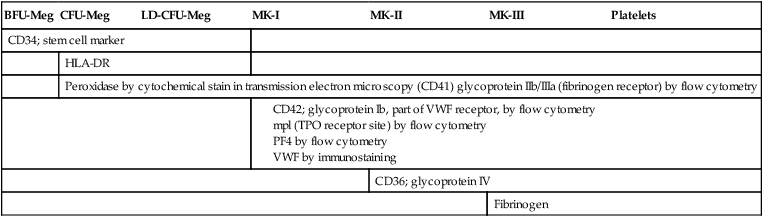
In specialty laboratories, immunologic probes and flow cytometry are employed to identify megakaryocyte progenitors. Useful flow cytometric progenitor markers are the general stem cell marker CD34, HLA-DR, and platelet glycoprotein IIIa (GP IIb/IIIa, CD41).8 Platelet peroxidase, localized in the endoplasmic reticulum of progenitors and megakaryoblasts, also may be identified by cytochemical stain in transmission electron microscopy. Identical peroxidase activity is localized to the dense tubular system of mature platelets.9
Terminal Megakaryocyte Differentiation
Megakaryocyte progenitors leave the proliferative phase and enter terminal differentiation, a series of stages in which microscopists begin to recognize their unique Wright-stained morphology in bone marrow aspirate films (Figure 13-3) or hematoxylin and eosin–stained bone marrow biopsy sections (Table 13-1).
TABLE 13-1
Features of the Three Terminal Megakaryocyte Differentiation Stages
| MK-I | MK-II | MK-III | |
| % Precursors | 20 | 25 | 55 |
| Diameter | 14-18 µm | 15-40 µm | 30-50 µm |
| Nucleus | Round | Indented | Multilobed |
| Nucleoli | 2-6 | Variable | Not visible |
| Chromatin | Homogeneous | Condensed | Deeply but variably condensed |
| Nucleus-to-cytoplasm ratio | 3 : 1 | 1 : 2 | 1 : 4 |
| Mitosis | Absent | Absent | Absent |
| Endomitosis | Present | Ends | Absent |
| Cytoplasm | Basophilic | Basophilic and granular | Eosinophilic and granular |
| α-granules | Present | Present | Present |
| δ-granules | Present | Present | Present |
| Demarcation system | Present | Present | Present |
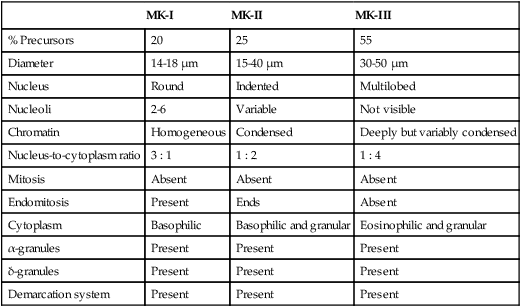
MK-I, Megakaryoblast; MK-II, promegakaryocyte; MK-III, megakaryocyte.
Morphologists call the least differentiated megakaryocyte precursor the MK-I stage or megakaryoblast. Although they no longer look like small lymphocytes, megakaryoblasts cannot be reliably distinguished from myeloblasts or pronormoblasts using light microscopy (Figure 13-4). The morphologist may see plasma membrane blebs, blunt projections from the margin that resemble platelets. At this stage the megakaryocyte begins to develop most of its cytoplasmic ultrastructure, including procoagulant-laden α-granules, δ-granules (dense bodies), and the demarcation system (DMS).10
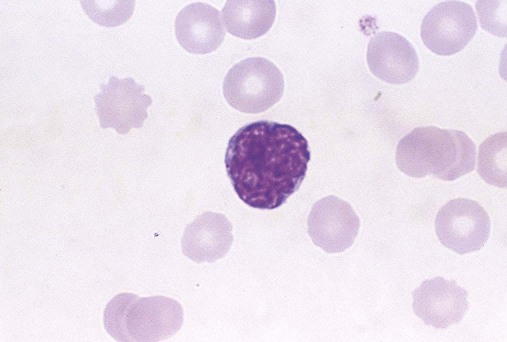
If there is a clinical need, immunologic probes or cytochemical markers are employed for definitive identification by flow cytometry (Table 13-2). In addition to the progenitor markers listed previously, the megakaryoblast may be identified using immunologic probes for the more differentiated membrane structures GP Ib, which is part of the GP Ib/IX/V von Willebrand factor (VWF) adhesion receptor (CD42); mpl, which is the TPO receptor site; or cytoplasmic VWF, detected by histochemical immunostaining rather than flow cytometry. CD36, which is GP IV, becomes visible in the developing MK-II stage, and fibrinogen may be detected by immunostaining in the fully developed megakaryocyte.
TABLE 13-2
Immunologic Probe or Cytochemical Markers at Each Stage of Megakaryocyte and Platelet Maturation
| BFU-Meg | CFU-Meg | LD-CFU-Meg | MK-I | MK-II | MK-III | Platelets |
| CD34; stem cell marker | ||||||
| HLA-DR | ||||||
| Peroxidase by cytochemical stain in transmission electron microscopy (CD41) glycoprotein IIb/IIIa (fibrinogen receptor) by flow cytometry | ||||||
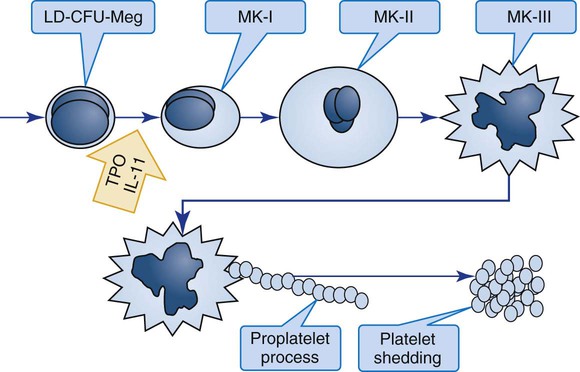
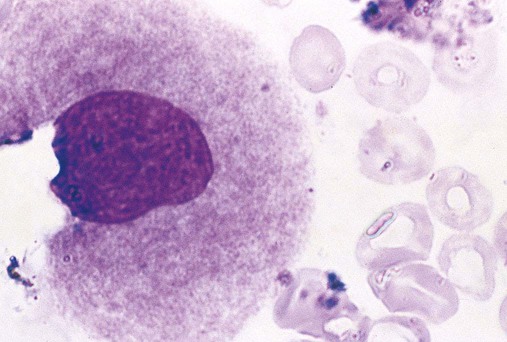
The megakaryocyte reaches its full ploidy level by the end of the MK-II stage. At the MK-III stage, the megakaryocyte is easily recognized at 10× magnification on the basis of its 30- to 50-µm diameter (Figures 13-5 and 13-6). The nucleus is intensely indented or lobulated, and the degree of lobulation is imprecisely proportional to ploidy. (When necessary, ploidy levels are measured using mepacrine, a nucleic acid dye, in megakaryocyte flow cytometry.11) The chromatin is variably condensed with light and dark patches. The cytoplasm is azurophilic (lavender), granular, and platelet-like, because of the spread of the DMS and α-granules. At full maturation, platelet shedding, or thrombocytopoiesis, proceeds.
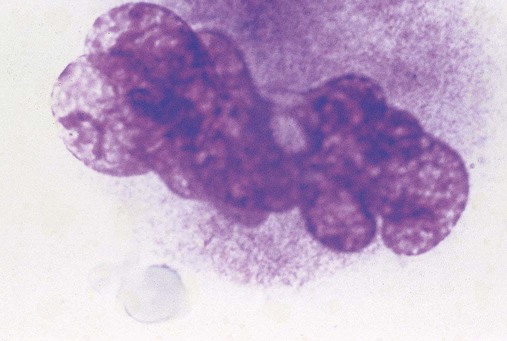
Thrombocytopoiesis (Platelet Shedding)
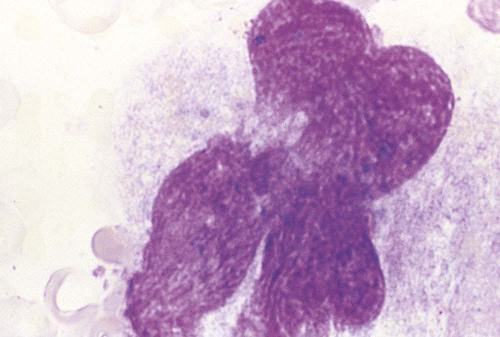
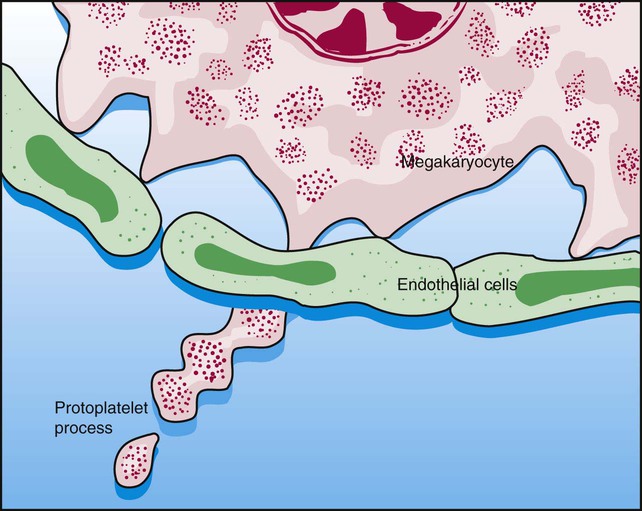
Figure 13-7 appears to show platelet shedding. One cannot find reliable evidence for platelet budding or shedding simply by examining megakaryocytes in situ, even in well-structured bone marrow biopsy preparations. In megakaryocyte cultures examined by transmission electron microscopy, the DMS dilates, longitudinal bundles of tubules form, cytoplasmic extensions called proplatelet processes develop, and transverse constrictions appear throughout the processes.12 The proplatelet processes pierce through or between sinusoid-lining endothelial cells, extend into the venous blood, and release platelets (Figure 13-8). Sometimes whole megakaryocytes escape the marrow in this fashion to lodge in other organs, such as the lungs. Morphologists presume that thrombopoiesis leaves behind naked megakaryocyte nuclei to be consumed by marrow macrophages, although these are rarely seen in bone marrow aspirate films. Their absence leads a few morphologists to speculate that the lung is the primary site of thrombopoiesis.13
Hormones and Cytokines of Megakaryocytopoiesis
TPO is a 70-kD molecule with 23% homology with erythropoietin.14 Messenger ribonucleic acid (RNA) for TPO has been found in the kidney, liver, stromal cells, and smooth muscle cells, though liver has the most copies. TPO circulates in plasma and is the ligand that binds a megakaryocyte and platelet membrane receptor protein, mpl, named for v-mpl, a viral oncogene associated with murine myeloproliferative leukemia. The concentration of TPO is inversely proportional to platelet and megakaryocyte mass, which implies that membrane binding and consequent disposal of TPO by platelets is the primary control mechanism.15 Investigators have used in vitro and in vivo experiments to show that TPO induces stem cells to differentiate into megakaryocyte progenitors in synergy with cytokines and that it further induces differentiation of megakaryocyte progenitors into megakaryocytes, induces the proliferation and maturation of megakaryocytes, and induces platelet release (Table 13-3).16,17 Recombinant TPO in several forms elevates the platelet count in healthy donors and in patients treated for a variety of neoplasms, including acute leukemia, and the commercial form NPlate (romiplostim, Amgen) is effective in raising the platelet count in immune thrombocytopenic purpura.18
TABLE 13-3
Hormones and Cytokines That Control Megakaryocytopoiesis
| Cytokine/Hormone | Differentiation to Progenitors | Differentiation to Megakaryocytes | Late Maturation | Thrombopoiesis | Clinical Use |
| TPO | + | + | + | 0 | Available |
| IL-3 | + | + | 0 | — | — |
| IL-6 | 0 | 0 | + | + | — |
| IL-11 | 0 | + | + | + | Available |

Cell-derived stimulators of megakaryocytopoiesis include interleukin-3 (IL-3), IL-6, and IL-11. IL-3 seems to act in synergy with TPO to induce early differentiation of stem cells, whereas IL-6 and IL-11 act in the presence of TPO to enhance the later phenomena of endomitosis, megakaryocyte maturation, and platelet release. IL-11 has been synthesized and marketed by Genetics Institute as Neumega (oprelvekin) and used to stimulate platelet production in patients with chemotherapy-induced thrombocytopenia.19 Other cytokines and hormones that participate synergistically with TPO and the interleukins are stem cell factor, also called kit ligand or mast cell growth factor; granulocyte-macrophage colony-stimulating factor (GM-CSF); granulocyte colony-stimulating factor (G-CSF); and erythropoietin (EPO). The list continues to grow.
Platelet factor 4 (PF4), β-thromboglobulin, neutrophil-activating peptide 2, IL-8, and other factors inhibit in vitro megakaryocyte growth, which indicates that they may have a role in the control of megakaryocytopoiesis in vivo. Internally, reduction in the transcription factors FOG, GATA-1, and NF-E2 diminish megakaryocytopoiesis at the progenitor, endomitosis, and terminal maturation phases.20
Platelets
The proplatelet process sheds platelets, cells consisting of granular cytoplasm with a membrane but no nuclear material, into the venous sinus. Their diameter in the monolayer of a Wright-stained peripheral blood wedge film averages 2.5 µm. MPV, as measured in an isotonic suspension flowing through the detector cell of a clinical profiling instrument, ranges from 8 to 10 fL. A frequency distribution of platelet volume is log-normal, however, which indicates a subpopulation of large platelets (see Figure 39-14). Volume heterogeneity in normal healthy humans reflects variation in platelet release volume and is not a function of platelet age or vitality, as many authors have previously assumed.5
Reticulated platelets, sometimes known as stress platelets, appear in compensation for thrombocytopenia.21 Reticulated platelets are markedly larger than ordinary mature circulating platelets; their diameter in blood films exceeds 6 µm and their MPV reaches 12 to 14 fL.22 Like ordinary platelets, they round up in EDTA, but in citrated whole blood, reticulated platelets are cylindrical and beaded, resembling megakaryocyte proplatelet processes. Reticulated platelets carry free ribosomes and fragments of rough endoplasmic reticulum, analogous to red blood cell reticulocytes, which has triggered speculation that they arise from early and rapid proplatelet extension and release. Nucleic acid dyes such as thiazole orange bind the RNA of the endoplasmic reticulum. This property is exploited by profiling instruments to provide a quantitative evaluation of reticulated platelet production under stress, a measurement that may be more useful than the MPV.23 This measurement is confounded by platelet dense granules, which falsely raise the reticulated platelet count by taking up nucleic acid dyes.
Platelet Ultrastructure
Resting Platelet Plasma Membrane
The platelet plasma membrane resembles any biologic membrane: a bilayer composed of proteins and lipids, as diagrammed in Figure 13-9. The predominant lipids are phospholipids, which form the basic structure, and cholesterol, which distributes asymmetrically throughout the phospholipids. The phospholipids form a bilayer with their polar heads oriented toward aqueous environments—toward the plasma externally and the cytoplasm internally. Their fatty acid chains, esterified to carbons 1 and 2 of the phospholipid triglyceride backbone, orient toward each other, perpendicular to the plane of the membrane, to form a hydrophobic barrier sandwiched within the hydrophilic layers.
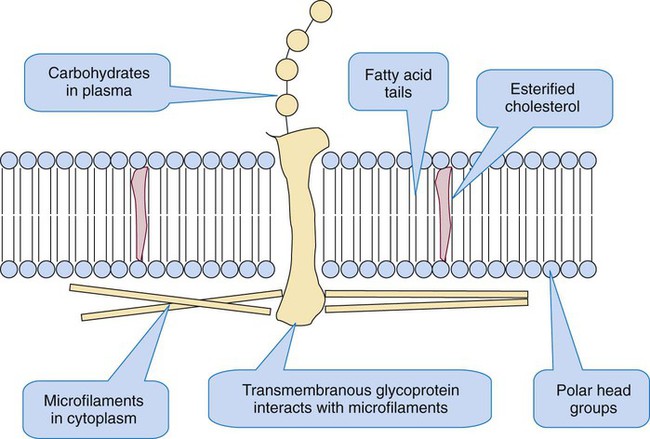
The neutral phospholipids phosphatidylcholine and sphingomyelin predominate in the plasma layer; the anionic or polar phospholipids phosphatidylinositol, phosphatidylethanolamine, and phosphatidylserine predominate in the inner, cytoplasmic layer. These phospholipids, especially phosphatidylinositol, support platelet activation by supplying arachidonic acid, an unsaturated fatty acid that is converted to the eicosanoids prostaglandin and thromboxane during platelet activation. Phosphatidylserine flips to the outer surface on activation and is the phospholipid surface on which coagulation enzymes assemble.24
Platelet Plasma Membrane Receptors That Provide for Adhesion
The platelet membrane supports more than 50 categories of receptors, including members of the cell adhesion molecule (CAM) integrin family, the CAM leucine-rich repeat family, the CAM immunoglobulin gene family, the CAM selectin family, the seven-transmembrane receptor (STR) family, and some miscellaneous receptors.25 Table 13-4 lists the receptors that support the initial phases of platelet adhesion and aggregation.
TABLE 13-4
| Electrophoresis Nomenclature | Current Nomenclature | Ligand | Cluster Designation | Comments |
| GP Ia/IIa | Integrin: α2β1 | Collagen | CD29, CD49b | |
| GP Ic/IIa | Integrin: α5β1 | Laminin | CD29, CD49e | |
| GP Ic/IIa | Integrin: α6β1 | Fibronectin | CD29, CD49f | |
| GP IV | Miscellaneous platelet receptor | Collagen II and thrombospondin | CD36 | GP IV provides signal transduction to trigger aggregation. It is distributed on the plasma membrane, SCCS, and 20% on α-granule membranes. |
| GP VI | CAM of the immunoglobulin gene family | Collagen | ||
| GP Ib/IX/V | CAM of the leucine-rich repeat family | VWF and thrombin bind GP Ibα; thrombin cleaves a site on GP V | CD42a, CD42b, CD42c, CD42d | GP Ib/IX/V is a 2 : 2 : 2 : 1 complex of GP Ibα and Ibβ, GP IX, and GP V. There are 25,000 copies on the resting platelet membrane surface, 5-10% on the α-granule membrane, but few on the SCCS membrane. GP Ibα is the VWF-specific site. Fifty percent of GP Ibα/Ibβ is cleared from the membrane on activation. Bernard-Soulier syndrome mutations are identified for all but GP V. Bound to subsurface actin-binding protein. |
| GP IIb/IIIa | Integrin: αIIbβ1 | Fibrinogen, VWF | CD41, CD61 | GP IIb and IIIa are distributed on the surface membrane, SCCS, and α-granule membranes (30%). Heterodimer forms on activation. |
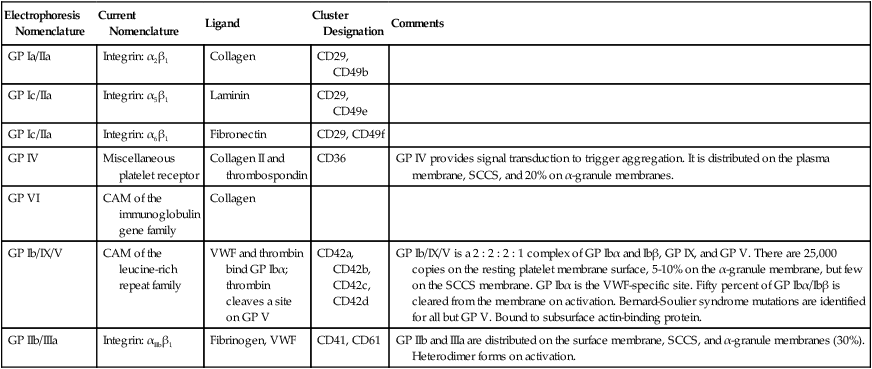
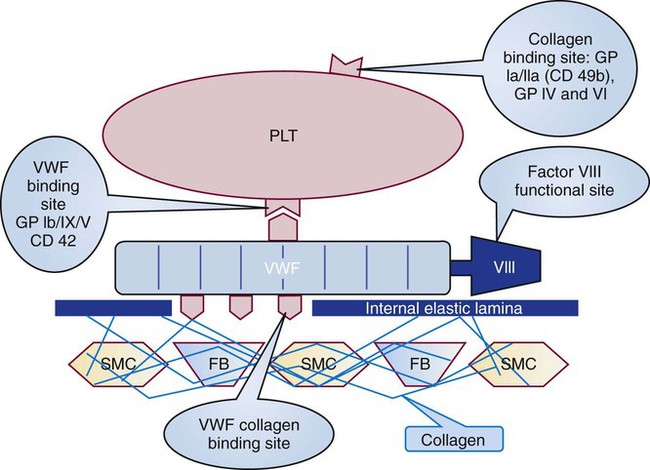
Several integrins bind collagen, enabling the platelet to adhere to the injured blood vessel lining. Integrins are heterodimeric (composed of two dissimilar proteins) CAMs that integrate their ligands, which they bind on the outside of the cell, with the internal cytoskeleton, triggering activation. GP Ia/IIa, or α2β1, is an integrin that binds subendothelial collagen in the damaged blood vessel wall to promote adhesion of the platelet to the vessel wall in regions of low blood flow, veins whose laminar flow shear rates are less than 1000 s–1. Likewise, α5β1 and α6β1 bind the adhesive endothelial cell proteins laminin and fibronectin, which further promotes platelet adhesion. Another collagen-binding receptor is GP VI, a member of the immunoglobulin gene family, so named because the genes of its members have multiple immunoglobulin-like domains. The unclassified platelet receptor GP IV also binds collagen and the adhesive protein thrombospondin.26
Perhaps the most important adhesion receptor is GP Ib/IX/V, a leucine-rich-repeat family CAM, named for its members’ multiple leucine-rich domains. GP Ib/IX/V arises from the genes GP1BA, GP1BB, GP9, and GP5. It is composed of two molecules each of GP Ibα, GP Ibβ, and GP IX, and one molecule of GP V. This totals seven noncovalently-bound subunits. The two copies of subunit GP Ibα account for VWF binding, necessary in regions with blood flow shear rates higher than 1000 s–1, as are found in capillaries and arterioles. Additional sites on the GP Ibα molecule bind thrombin. The accompanying GP Ibβ molecules cross the platelet membrane and interact with actin-binding protein to provide “outside-in” signaling. Two molecules of GP IX and one of GP V hold the four GP Ib molecules together. Mutations in GP Ibα, GP Ibβ, or GP IX (but not GP V) are associated with a moderate-to-severe mucocutaneous bleeding disorder, Bernard-Soulier syndrome. VWF deficiency is the basis for the most common inherited bleeding disorder, von Willebrand disease. Von Willebrand disease also is associated with mucocutaneous bleeding, although it is technically a plasma protein deficiency, not a platelet abnormality.27
The two subunits of GP IIb/IIIa, αIIb and β3, are initially inactive as they are distributed across the plasma membrane, the SCCS, and the internal layer of α-granule membranes. These form an active heterodimer, αIIbβ3, only when they encounter an “inside-out” signaling mechanism triggered by collagen binding to GP VI or VWF binding to GP Ib/IX/V. Although various agonists may activate the platelet, αIIbβ3 is a physiologic requisite because it binds fibrinogen, generating interplatelet aggregation. Mutations in αIIb or β3 cause a severe inherited mucocutaneous bleeding disorder, Glanzmann thrombasthenia. The αIIbβ3 integrin also binds VWF, vitronectin, and fibronectin, all adhesive proteins that share the target arginine-glycine-aspartate (RGD) amino acid sequence with fibrinogen.28 GP IIb/IIIa (αIIbβ3) is the target receptor for the intravenous “glycoprotein inhibitor” (GPI) drugs ReoPro (abciximab, Eli Lilly), Integrilin (eptifibatide, Millennium Pharmaceuticals) and Aggrastat (tirofiban, Medicure Pharma), any one of which may be employed during cardiac catherization. Use of the glycoprotein inhibitors requires platelet function assays for efficacy and safety (see Chapter 46).
Platelet Plasma Membrane Receptors That Provide for Activation: The Seven-Transmembrane Receptors
Thrombin, thrombin receptor activation peptide (TRAP), adenosine diphosphate (ADP), epinephrine, and the prostaglandin (eicosanoid, cyclooxygenase) pathway product thromboxane A2 (TXA2) all activate platelets. These platelet “agonists” are ligands for STRs, so named for their unique membrane-anchoring structure. The STRs have seven hydrophobic anchoring domains supporting an external binding site and an internal terminus that interacts with G proteins for outside-in platelet signaling. The STRs are listed in Table 13-5.29
TABLE 13-5
STRs* for Thrombin, Adenosine Diphosphate, Epinephrine, and the Prostaglandins
| STR Receptor-Ligand Interaction Coupled to Signaling | ||
| Receptor | Ligand | G proteins |
| PAR1 | Thrombin | Coupled to G1 protein that reduces cAMP; coupled to Gq and G12 proteins that increase IP3 and DAG |
| PAR4 | Thrombin | Coupled to Gq and G12 proteins that increase IP3 and DAG |
| P2Y1 | ADP | Coupled to Gq protein that increases IP3 and DAG |
| P2Y12 | ADP | Coupled to G1 protein that reduces cAMP |
| TPα and TPβ | TXA2 | Coupled to Gq protein that increases IP3 and DAG |
| α2-adrenergic | Epinephrine | Coupled to G1 protein that reduces cAMP; potentiates effects of ADP, thrombin, and TXA2 |
| IP | PGI2 | Coupled to GS protein that increases cAMP to inhibit activation |
*Named for their peculiar sevenfold membrane anchorage, these receptors mediate “outside-in” platelet activation by signaling G proteins.
There are about 600 copies of the high-affinity ADP receptors P2Y1 and P2Y12. These STRs activate the platelet through the G-protein signaling pathways. Platelets are partially inactivated by prasugrel (Effient), clopidogrel (Plavix) and ticlopidine (Ticlid), drugs that occupy P2Y12 (see Chapter 46). These drugs are standard oral antithrombotic treatments prescribed after acute myocardial infarctions, ischemic cerebrovascular accidents, and venous thromboembolic events. Platelet function assays are regularly employed to monitor prasugrel and clopidogrel efficacy and establish dosages.30
Additional Platelet Membrane Receptors
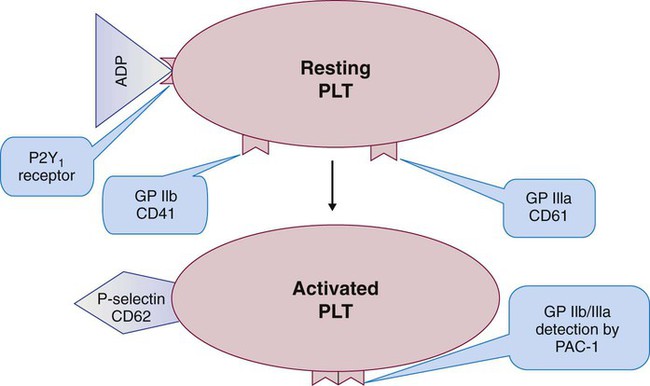
About 15 clinically relevant receptors were discussed in the preceding paragraphs. The platelet supports many additional receptors. The CAM immunoglobulin family includes the ICAMs (CD50, CD54, CD102), which play a role in inflammation and the immune reaction; PECAM (CD31), which mediates platelet–to–white blood cell and platelet–to–endothelial cell adhesion; and FcγRIIA (CD32), a low-affinity receptor for the immunoglobulin Fc portion that plays a role in a dangerous condition called heparin-induced thrombocytopenia (see Chapter 42).31 P-selectin (CD62) is an integrin that encourages platelets to bind endothelial cells, leukocytes, and each other.32 P-selectin is found on the α-granule membranes of the resting platelet, but migrates via the SCCS to the surface of activated platelets. P-selectin or CD62 quantification by flow cytometry is a successful clinical means for measuring in vivo platelet activation.
Platelet Cytoskeleton: Microfilaments and Microtubules
A thick circumferential bundle of microtubules maintains the platelet’s shape. The circumferential microtubules parallel the plane of the disc and reside just within, although not touching, the plasma membrane. There are 8 to 20 tubules composed of multiple subunits of tubulin that disassemble at refrigerator temperature or when treated with colchicine. When microtubules disassemble, platelets become round, but upon warming to 37° C, they recover their original disc shape. On cross section, microtubules are cylindrical, with a diameter of 25 nm, and hollow. The circumferential microtubules could be a single spiral tubule.33 Besides maintaining platelet shape, microtubules contract on activation to encourage expression of α-granule contents. They also reassemble in long parallel bundles to provide rigidity to pseudopods.
Platelet Granules: α-Granules, δ-Granules, and Lysosomes
There are 50 to 80 α-granules in each platelet. In contrast to the nearly opaque δ-granules, α-granules stain medium gray in osmium-dye transmission electron microscopy preparations (Figure 13-10). The α-granules are filled with proteins, some endocytosed, some synthesized within the megakaryocyte and stored in platelets (Table 13-6). Several α-granule proteins are membrane bound. As the platelet becomes activated, α-granule membranes fuse with the SCCS. Their contents flow to the nearby microenvironment, where they participate in adhesion and aggregation and support plasma coagulation.
TABLE 13-6
Representative α-Granule Proteins
| Coagulation Proteins | Noncoagulation Proteins | |
| Proteins Present in Plasma and α-Granules | ||
| Endocytosed | Fibronectin | Albumin |
| Fibrinogen | Immunoglobulins | |
| Megakaryocyte synthesized | Factor V | — |
| Thrombospondin | — | |
| VWF | — | |
| Present in α-Granules, But Not Plasma (Megakaryocyte Synthesized) | ||
| β-Thromboglobulin | EGF | |
| HMWK | Multimerin | |
| PAI-1 | PDC1 | |
| Plasminogen | PDGF | |
| PF4 | TGF-β | |
| Protein C inhibitor | VEGF/VPF | |
| Membrane-Bound Proteins | ||
| Restricted to α-granule membrane | P-selectin | GMP33 |
| — | Osteonectin | |
| In α-granule and plasma membrane | GP IIb/IIIa | cap1 |
| GP IV | CD9 | |
| GP Ib/IX/V | PECAM-1 | |
There are two to seven δ-granules per platelet. Also called dense bodies, these granules appear later than α-granules in megakaryocyte differentiation and stain black (opaque) when treated with osmium in transmission electron microscopy. Small molecules are probably endocytosed and are stored in the δ-granules; these are listed in Table 13-7. In contrast to the α-granules, which employ the SCCS, δ-granules migrate to the plasma membrane and release their contents directly into the plasma upon platelet activation. Membranes of δ-granules support the same integral proteins as the α-granules—P-selectin, GP IIb/IIIa, and GP Ib/IX/V, for instance—which implies a common source for the membranes of both types of granules.
TABLE 13-7
δ-Granule (Dense Body) Contents
| Small Molecule | Comment |
| ADP | Nonmetabolic, supports neighboring platelet aggregation by binding to ADP receptors P2Y1, P2Y12 |
| ATP | Function unknown, but ATP release is detected using firefly luciferase luminescence as an in vitro measure of platelet activation—a method called lumiaggregometry |
| Serotonin | Vasoconstrictor that binds endothelial cells and platelet membranes |
| Ca2+ and Mg2+ | Divalent cations support platelet activation and coagulation |
Platelet Activation
Although the following discussion seems to imply a linear process, adhesion, aggregation, and secretion are often simultaneous.34,35
Adhesion: Platelets Reversibly Bind Elements of the Vascular Matrix
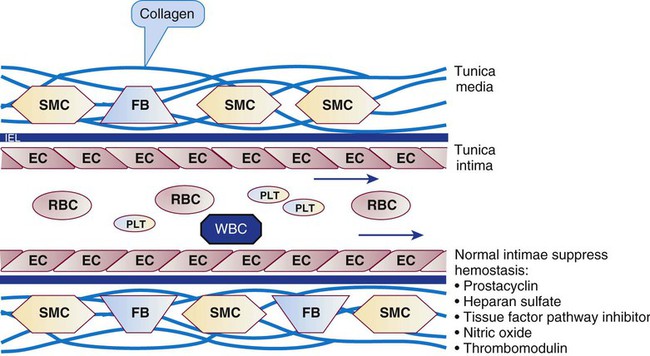
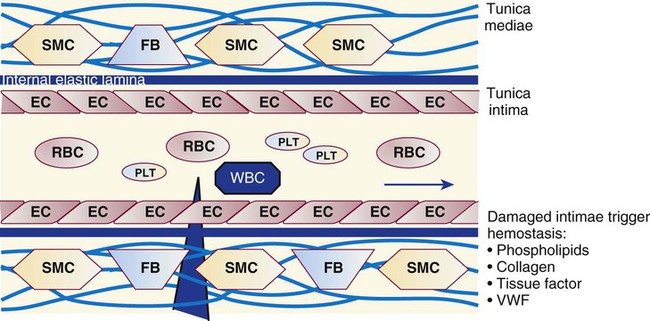
Platelets repair minor injuries to blood vessel linings, such as regular sloughing of senescent endothelial cells, through adhesion (Figures 13-11 and 13-12).36 Platelets may adhere directly to collagen of the vascular matrix in veins or venules through the CAMs described previously: GP Ia/IIa, GP IV, and GP VI.
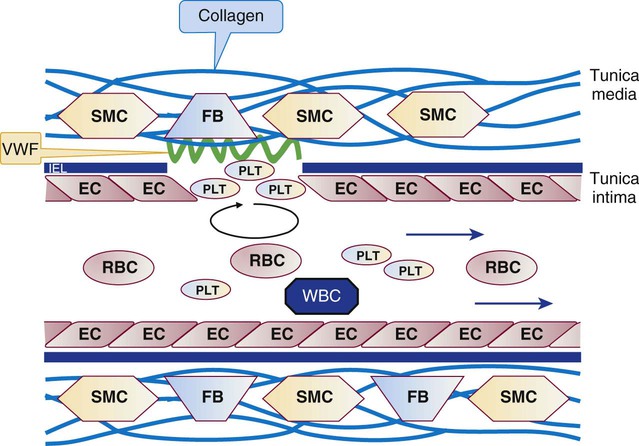
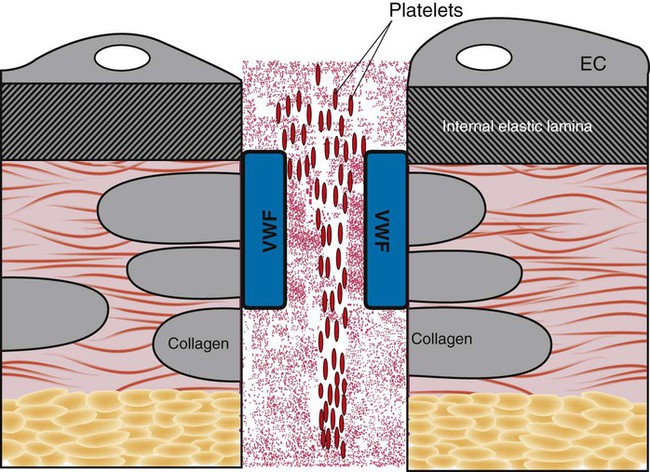
In capillaries and arterioles, where the blood flows rapidly and the shear rate exceeds 1000 s−1, the site of injury is first “carpeted” by VWF. VWF circulates as a multimeric globulin with a molecular weight of between 800,000 D and 20 million D, one of the largest proteins in the plasma. The combination of injury and shear stress “unrolls” the globular molecule to form fibrillar strands that coat injury sites and bind subendothelial collagen. A liver-secreted plasma enzyme, VWF-cleaving protease, controls thrombogenicity by rapidly digesting fibrillar VWF to inactive fragments.37 Platelets adhere to fibrillar VWF through their GP Ib/IX/V receptor, and the VWF-platelet layer fills in the space made by the injury to await replacement by normal vascular cells (Figure 13-13). Although seldom an isolated process, adhesion alone may exclude secretion of granule contents or the energy-dependent redistribution of CAMs such as GP IIb/IIIa (αIIbβ3) or P-selectin. Adhesion does require contraction of actin molecules, however, and the formation of pseudopods.
Aggregation: Platelets Irreversibly Bind Each Other
When vessel damage is extensive, platelets adhere and aggregate (Figures 13-14 and 13-15). Aggregation requires the active conformation of the GP IIb/IIIa (αIIbβ3) integrin and includes pseudopod formation and redistribution of P-selectin to the surface membrane (Figure 13-16). Membrane phospholipids also redeploy, with the more polar molecules, such as phosphatidylserine, flipping to the outer layer. GP IIb/IIIa binds the RGD sequence of any plasma protein; the protein most readily available is fibrinogen, which is the major player in aggregation. Membrane integrity is lost, and a syncytium of platelet cytoplasm forms as the platelet exhausts internal energy sources. Aggregation is a part of primary hemostasis, and its irreversible end point is the “white clot” or platelet-VWF plug. Although a normal part of vessel repair, white clots often imply inappropriate platelet activation in uninjured arterioles and arteries and are the pathologic basis for arterial thrombotic events, such as acute myocardial infarction, peripheral vascular disease, and strokes. Except for VWF, coagulation proteins are excluded from white clots.
The combination of polar phospholipid exposure, platelet microparticle dispersion, and secretion of the platelet’s α- and δ-granule contents triggers secondary hemostasis, called coagulation (see Chapter 40). Fibrin and red blood cells deposit around and within the platelet syncytium, forming a bulky “red clot.” The red clot is essential to wound repair, but is characteristic of inappropriate coagulation in venules and veins, resulting in deep vein thrombosis and pulmonary emboli.
Specialty and tertiary care coagulation laboratories provide in vitro whole blood or plasma platelet aggregometry and lumiaggregometry as a means for detecting and identifying aggregation abnormalities. The bleeding time test, although still used, has limited predictive value. Several systems for the physician’s office or near-patient laboratory, such as the Siemens PFA-100 (Deerfield, Ill.), and the Chronolog WBA (Havertown, Pa.), also are available to test for platelet function.38
Secretion: Activated Platelets Release Granular Contents
Outside-in platelet activation through ligand (agonist) binding to integrins and STRs triggers actin microfilament contraction. Intermediate filaments and microtubules contract, compressing granules. Contents of α-granules and lysosomes flow through the SCCS; meanwhile δ-granule contents are secreted through the plasma membrane (Figure 13-17). The δ-granule contents are vasoconstrictors and platelet agonists that amplify primary hemostasis; several of the α-granule contents are coagulation proteins (Table 13-8).
TABLE 13-8
Selected α-Granule Proteins and Their Properties
| α-Granule Protein | Properties |
| Platelet-derived growth factor | Supports mitosis of vascular fibroblasts and smooth muscle cells |
| Endothelial growth factor | Supports mitosis of vascular fibroblasts and smooth muscle cells |
| Transforming growth factor-β | Supports mitosis of vascular fibroblasts and smooth muscle cells |
| Fibronectin | Adhesion molecule |
| Thrombospondin | Adhesion molecule |
| Platelet factor 4 | Heparin neutralization |
| β-thromboglobulin | Found nowhere but platelet α-granules |
| Plasminogen | Fibrinolysis promotion and control |
| Plasminogen activation inhibitor-1 | Fibrinolysis promotion and control |
| α2-Antiplasmin | Fibrinolysis promotion and control |
| Protein C inhibitor | Coagulation control |
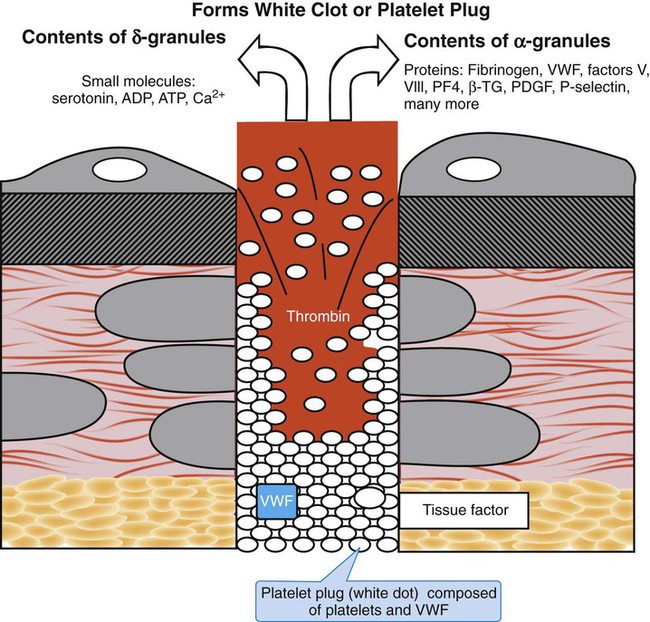
By presenting polar phospholipids on their membrane surfaces, platelets provide a localized cellular milieu that supports coagulation. Phosphatidylserine is the phospholipid on which two coagulation pathway complexes form: factor IX/VIII (tenase) and factor X/V (prothrombinase), supported by ionic calcium secreted by the δ-granules. The α-granule contents fibrinogen, factors V and VIII, and VWF (which binds and stabilizes factor VIII) are secreted and increase the localized concentrations of these essential coagulation proteins, which further supports the action of tenase and prothrombinase. Platelet secretions serve cell-based, controlled, localized coagulation. Table 13-8 lists some additional α-granule secretions that, although not proteins of the coagulation pathway, support hemostasis. Some proteins listed in Table 13-8 do not appear in Table 13-5. Neither list is exhaustive, because more α-granule contents are being identified.
Platelet Activation Pathways
G Proteins
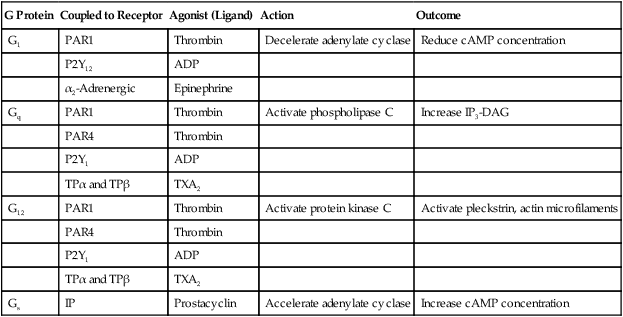
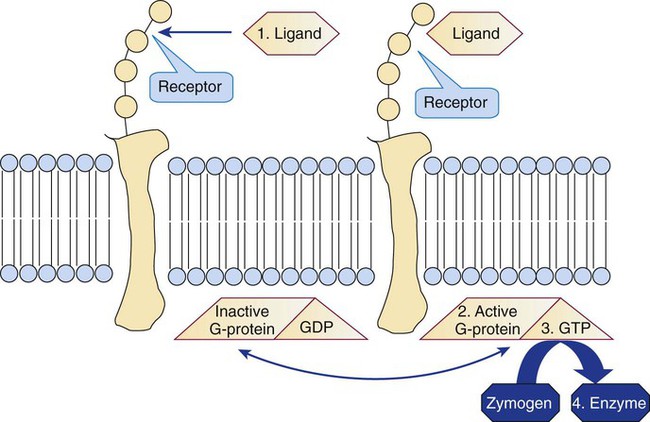
G proteins control cellular activation for all cells (not just platelets) at the inner membrane surface (Figure 13-18). G proteins are αβγ heterotrimers that bind guanosine diphosphate (GDP) when inactive. Membrane receptor-ligand (agonist) binding promotes GDP release and its replacement with guanosine triphosphate (GTP). The Gα monomer briefly disassociates, exerts guanosine triphosphatase activity, and hydrolyzes the bound GTP to GDP, releasing a phosphate radical. The G protein resumes the resting state, but the hydrolysis step provides the necessary phosphorylation trigger to energize the eicosanoid synthesis or the IP3-DAG pathway (Table 13-9).
TABLE 13-9
G Proteins in Platelets and Their Functions
| G Protein | Coupled to Receptor | Agonist (Ligand) | Action | Outcome |
| G1 | PAR1 | Thrombin | Decelerate adenylate cyclase | Reduce cAMP concentration |
| P2Y12 | ADP | |||
| α2-Adrenergic | Epinephrine | |||
| Gq | PAR1 | Thrombin | Activate phospholipase C | Increase IP3-DAG |
| PAR4 | Thrombin | |||
| P2Y1 | ADP | |||
| TPα and TPβ | TXA2 | |||
| G12 | PAR1 | Thrombin | Activate protein kinase C | Activate pleckstrin, actin microfilaments |
| PAR4 | Thrombin | |||
| P2Y1 | ADP | |||
| TPα and TPβ | TXA2 | |||
| Gs | IP | Prostacyclin | Accelerate adenylate cyclase | Increase cAMP concentration |
Eicosanoid Synthesis
The eicosanoid synthesis pathway, alternatively called the prostaglandin, cyclooxygenase, or thromboxane pathway, is one of two essential platelet activation pathways triggered by G protein (Figure 13-19). The platelet membrane’s inner leaflet is rich in phosphatidylinositol, a phospholipid whose number 2 carbon binds numerous unsaturated fatty acids, including 5,8,11,14-eicosatetraenoic acid, commonly called arachidonic acid. Membrane receptor-ligand binding and the consequent G-protein activation triggers phospholipase A2, a membrane enzyme that cleaves the ester bond connecting the number 2 carbon of the triglyceride backbone with arachidonic acid. Cleavage releases arachidonic acid to the cytoplasm, where it becomes the substrate for cyclooxygenase, anchored in the DTS. Cyclooxygenase converts arachidonic acid to prostaglandin G2 and prostaglandin H2, then thromboxane synthetase acts on prostaglandin H2 to produce TXA2. TXA2 binds membrane receptors TPα or TPβ, decelerating adenylate cyclase activity and reducing cAMP concentrations, which mobilizes ionic calcium from the DTS (Figure 13-20). The rising cytoplasmic calcium level causes contraction of actin microfilaments and platelet activation.
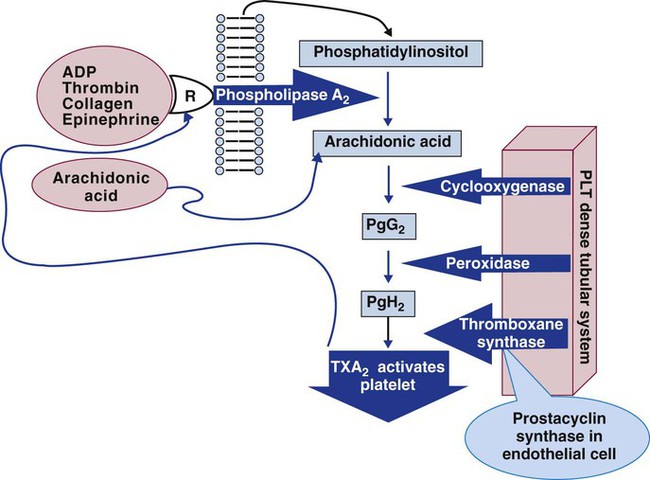
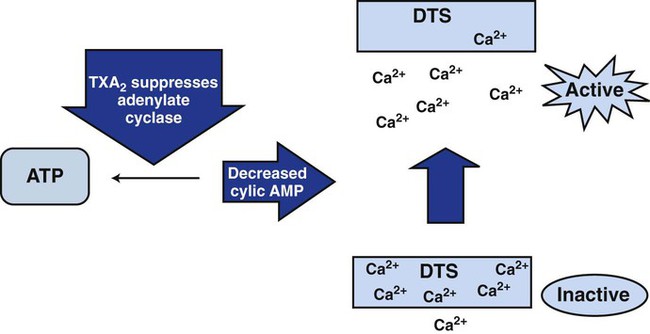
TXA2 has a half-life of 30 seconds, diffuses from the platelet, and spontaneously reduces to thromboxane B2, a stable, measurable plasma metabolite. Efforts to produce a clinical assay for plasma thromboxane B2 have been unsuccessful, because special specimen management is required to prevent ex vivo platelet activation with factitious unregulated release of thromboxane B2. Thromboxane B2 is acted on by a variety of liver enzymes to produce an array of soluble urine metabolites, including 11-dehydrothromboxane B2, which is stable and measurable.39 Immunoassays of urine 11-dehydrothromboxane B2 are used to characterize in vivo platelet activation and the suppressing effect of aspirin.40
Inositol Triphosphate–Diacylglycerol Activation Pathway
The IP3-DAG pathway is the second G protein–dependent platelet activation pathway (Figure 13-21). G-protein activation triggers the enzyme phospholipase C. Phospholipase C cleaves membrane phosphatidylinositol 4,5-bisphosphate to form IP3 and DAG, both second messengers for intracellular activation. IP3 promotes release of ionic calcium from the DTS, which triggers actin microfilament contraction. IP3 also may activate phospholipase A2. DAG triggers a multistep process: activation of phosphokinase C, which triggers phosphorylation of the protein pleckstrin, which regulates actin microfilament contraction.
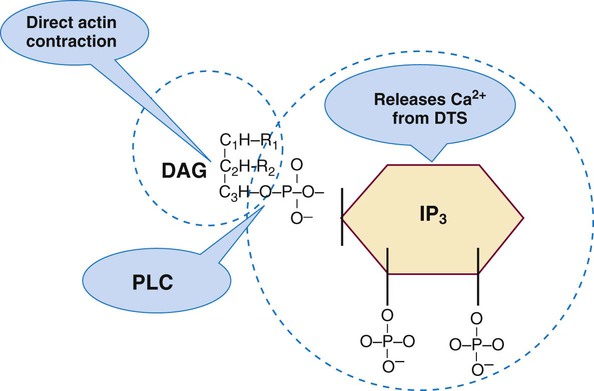
Summary
• Platelets arise from bone marrow megakaryocytes, which reside near the venous sinusoid. Megakaryocyte progenitors are recruited by IL-3, IL-6, IL-11, and TPO and mature via endomitosis. Platelets are released into the bone marrow through shedding from proplatelet processes, a process called thrombopoiesis.
• Circulating platelets are complex anucleate cells with a thick glycocalyx bearing an assortment of coagulation factors, extended by the SCCS. Inside is a system of microfibrils and microtubules that accomplish platelet contraction and pseudopod extension and the granules that provide coagulation factors and vasoactive chemicals.
• The platelet membrane supports many receptor sites that control platelet activation on binding their respective ligands.
• Platelets adhere to foreign surfaces, aggregate with each other, and secrete the substances stored within their granules.
• Platelet activation is internally managed through G proteins, the eicosanoid synthesis pathway, and the IP3-DAG pathway.
Review Questions
1. The megakaryocyte progenitor that undergoes endomitosis is:
2. The growth factor that is produced in the kidney and induces growth and differentiation of committed megakaryocyte progenitors is:
3. What platelet organelle sequesters ionic calcium and binds a series of enzymes of the eicosanoid pathway?
4. What platelet membrane receptor binds fibrinogen and supports platelet aggregation?
5. What platelet membrane phospholipid flips from the inner surface to the plasma surface on activation and serves as the assembly point for coagulation factors?
6. What is the name of the eicosanoid metabolite produced from endothelial cells that suppresses platelet activity?
7. Which of the following molecules is stored in platelet δ-granules (dense bodies)?
8. What plasma protein is essential to platelet adhesion?
9. Reticulated platelets can be enumerated in peripheral blood to detect: