CHAPTER 24 Plasma Membrane Receptors
Cells use about 20 different families of receptor proteins (Fig. 24-1) to detect and respond to the myriad of incoming chemical and physical stimuli (Appendix 24-1). Most receptors are plasma membrane proteins that interact with chemical ligands or are stimulated by physical events such as light absorption. A few chemical stimuli, including steroid hormones and the gas nitric oxide, cross the plasma membrane and bind receptors inside the cell.
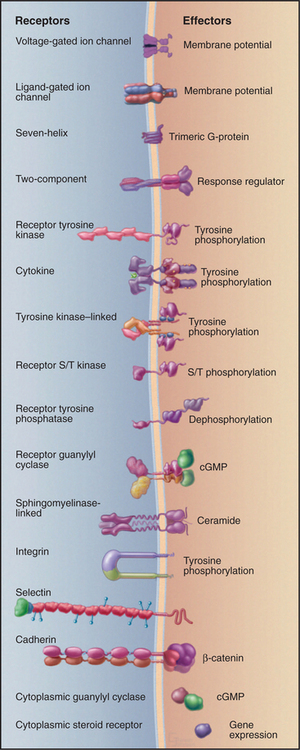
Figure 24-1 sixteen classes of receptors with signal transduction mechanisms. S/T, serine/threonine.
Gene duplication and divergent evolution within each family have produced genes for multiple receptor isoforms that interact with different ligands. In multicellular organisms, selective expression of certain receptors and their associated cytoplasmic transduction machinery allows differentiated cells to respond specifically to particu-lar ligands but not others (see Fig. 27-1). Fortunately, the mechanisms of the best-characterized receptors usually apply to the rest of their family. Thus, learning about a few examples provides a working knowledge of many related receptors.
One cannot predict the type of receptor, signal transduction mechanism, or nature of the response from the chemical nature of a stimulus (Appendix 24-1). Although proteins and peptides are the only known ligands for receptor kinases and kinase-linked receptors, proteins and peptides also stimulate some seven-helix receptors and guanylyl cyclase receptors. A particularly wide range of stimuli activate seven-helix receptors, including photons, amino acids, nucleotides, biogenic amines, lipids, peptides, proteins, and hundreds of different organic molecules. Some ligands bind distinct receptors on different cells. For example, acetylcholine activates muscle contraction by opening a ligand-gated ion channel (see Fig. 10-12). It also binds seven-helix receptors on other cells, activating signaling pathways mediated by guanosine triphosphate (GTP)–binding proteins. Some ligands with similar names bind to different types of receptors. For example, several interleukins (IL-2 through IL-6) bind to cytokine receptors, but IL-1 activates a sphingomyelinase-linked receptor, and IL-8 binds a seven-helix, G-protein-coupled receptor.
This chapter covers nine families of well-characterized receptors that transfer signals across the plasma membrane. Other chapters describe additional recep-tor families: Chapter 10, ligand-gated and voltage-gated ion channels; Chapter 15, nuclear receptors for steroids and other ligands; Chapter 25, receptors with protein-phosphatase activity; Chapter 26, cytoplas-mic nitric oxide receptors with guanylyl cyclase activity; Chapter 27, two-component receptors and tyro-sine kinase–linked receptors; and Chapter 30, cell adhesion receptors, including integrins, cadherins, and selectins.
Seven-Helix Receptors
Members of the largest family of plasma membrane receptors are built from a serpentine arrangement of seven transmembrane α-helices. These diverse receptors use trimeric GTP-binding proteins (see Fig. 25-9) to relay signals to effector proteins inside cells. Seven-helix receptors are found in slime molds, so the genes for these proteins originated in early eukaryotes more than 1 billion years ago. Four percent of the genes of the nematode Caenorhabditis elegans (790) encode seven-helix receptors, the largest family of proteins in the organism. In mammals, olfactory cells alone use 500 to 1000 different seven-helix receptors to discriminate odorant molecules (see Fig. 27-1). Other cells are estimated to express another 375 seven-helix receptors to respond to light, amino acids, peptide and protein hormones, catecholamines, and lipids. The chemical ligand remains to be determined for about 40% of these 375 receptors, which are termed orphan receptors. A majority of medically useful drugs bind seven-helix receptors.
Seven hydrophobic sequences traverse the plasma membrane as α-helices (Fig. 24-2). The topology is the same as that of bacteriorhodopsin (see Fig. 7-8), but this might be an example of convergent evolution. Comparative analysis of amino acid sequences suggests that all seven-helix receptors have the same arrangement of helices. For example, the minimum length of sequences connecting the helices is compatible only with the helices being arranged sequentially from I to VII in a serpentine fashion as they cross the lipid bilayer. The N-terminus is outside the cell and varies from 7 to 6000 residues. Some of the larger N-terminal domains participate in ligand binding. The C-terminal segment of the polypeptide is in the cytoplasm and varies in length from 12 to more than 350 residues. Seven-helix receptors are shown throughout this book as individual proteins, but multiple lines of evidence show that many seven-helix receptors function as dimers or larger oligomers, allowing for cross talk between the subunits.
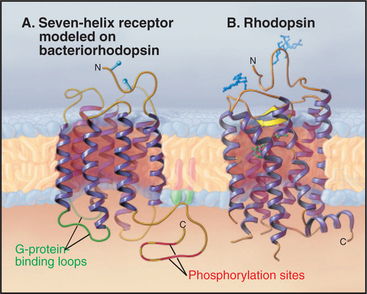
Most seven-helix receptors are activated by binding a soluble chemical ligand, but some interesting variations exist. Biochemical and mutagenesis experiments indicate that most small ligands bind in a central pocket among the extracellular ends of the helices. Residues lining this pocket are highly variable between receptors, providing specificity for each receptor to bind a particular ligand. The light-absorbing pigment 11- cis retinal, of the photoreceptor protein rhodopsin, is the best-characterized “ligand.” 11-cis retinal is unusual in that it is bound covalently to the receptor (Fig. 24-2B) and is activated by absorbing a photon (see Fig. 27-2). In other respects, it is a good model for other ligands (Fig. 24-3). Neurotransmitters, such as norepinephrine, and drugs also bind between the helices about one third of the way across the membrane. Peptide hormones bind deep in the helical pocket but probably also interact with residues that are more exposed on the cell surface. Receptors for some large ligands (pituitary glycoprotein hormones, such as luteinizing hormone, follicle-stimulating hormone, and thyroid-stimulating hormone) and some small ligands (glutamate, g-amino butyric acid, calcium) bind with high affinity to extracellular N-terminal domains of their seven-helix receptor. The N-terminal domain with bound ligand then stimulates the transmembrane domain of the receptor. The blood-clotting enzyme thrombin activates its receptor on platelets by proteolysis of the receptor rather than by direct binding (see Fig. 30-14). The N-terminal peptide cleaved from the receptor disso-ciates and activates other receptors; what is left of the newly truncated N-terminus folds back and activates its own receptor.
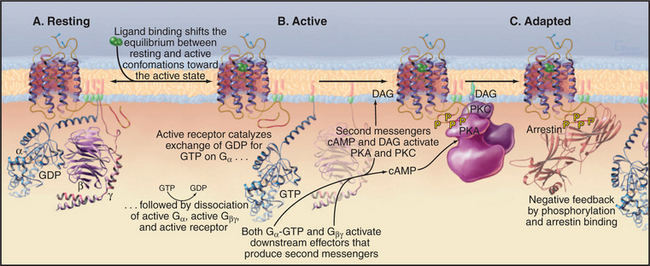
Seven-helix receptors exist in an equilibrium between two conformations: a resting state and an activated state with the ability to catalyze the exchange of nucleotide bound to trimeric G-proteins (Fig. 24-3). Without bound ligand, the resting state is strongly favored. Ligand binding to the receptor (or the isomerization of retinal after absorbing light) initiates signal transduction by shifting the equilibrium to the active state. Activation involves movement of at least two transmembrane helices, but the structural details of this conformational change are not yet well defined. In any event, activation must rearrange the cytoplasmic ends of the helices and the loops connecting them to create a binding site for a target G-protein.
Active receptors transfer the signal to the cyto-plasm by activating trimeric G-proteins. Cytoplasmic loops of active receptors catalyze the dissociation of guanosine diphosphate bound (EDP) to an inactive Ga subunit. GTP then binds and activates Ga (see Fig. 25-9). A single active seven-helix receptor can amplify the signal by activating up to 100 G-proteins. After dissociating from the receptor and each other, both Ga-GTP and Gbg stimulate downstream effector proteins, further amplifying the signal (see Fig. 27-3 for an example of amplification).
Most seven-helix receptors adapt to sustained stimulation. In the short term, phosphorylation of the C-terminal tail of the receptor provides negative feedback that inactivates receptors with ligands still bound (Fig. 24-3C). Along one pathway, second messengers—produced in response to receptor activation—stimulate protein kinases, including cyclic adenosine monophosphate (cAMP)–activated protein kinase A and protein kinase C (see Fig. 25-4). These kinases phosphorylate the C-terminal tails of active receptors, inhibiting interactions with G-proteins. This mechanism allows for cross talk between receptors, as activation of one class of receptors can inactivate other receptors. A second pathway uses Gbg subunits released in response to receptor stimulation to activate protein kinases specific for the receptors themselves, called G-protein-coupled receptor kinases. These kinases phosphorylate serines or threonines on the C-terminal tails of active (but not inactive) receptors.
Phosphorylation of receptor tails creates a binding site for arrestin, a protein with multiple functions. First, arrestin blocks interactions of the receptor with G-proteins, terminating signaling through the main pathway downstream of most seven-helix receptors. In some cases, arrestin initiates a new signal through the MAP kinase pathway (see Figs. 27-6 and 27-7 for two other pathways). Arrestin also promotes the removal of seven-helix receptors from the plasma membrane by endocytosis in clathrin-coated vesicles, a longer-term mechanism that turns down the response of a cell to continuous stimulation. Some internalized receptors recycle to the plasma membrane, but others modified by ubiquitin are directed to lysosomes for destruction. Chapter 27 covers three dramatic examples of seven-helix receptor adaptation.
Mutations of more than 30 seven-helix receptors have been linked to human diseases (Table 24-1). More than 600 mutations are known to inactivate seven-helix receptors in humans by every conceivable means from failure to synthesize the full length protein to reduced affinity for ligands to failure to activate G-proteins. These inherited loss of function mutations are recessive. For example, loss of function mutations in rhodopsin cause retinitis pigmentosa, a degeneration of photoreceptor cells. Another example is severe obesity associated with loss of function mutations of a seven-helix receptor that participates in the neural circuits controlling eating. More than a hundred different mutations produce receptors that are constitutively active without ligand. Particular mutations of rhodopsin cause night blindness, and mutations in a calcium receptor cause dysfunction of the parathyroid gland. The physiology of these activating mutations is complicated, because cells use feedback mechanisms to compensate for the continually active receptors.
| Defective Receptor | Disease Phenotype |
|---|---|
| Activating Mutations | |
| Parathyroid Ca2+ sensor | Hypoparathyroidism |
| Rhodopsin | Night blindness |
| Thyroid hormone receptor | Hyperthyroidism, thyroid cancer |
| Loss of Function Mutations | |
| Cone cell opsin | Color blindness; no response to certain wavelengths |
| Parathyroid Ca2+ sensor | Hyperparathyroidism, failure to respond to high levels of serum Ca2+ |
| Rhodopsin | Retinitis pigmentosa, retinal degeneration |
| Thyroid hormone receptor | Hypothyroidism |
| Vasopressin receptor | Nephrogenic diabetes insipidus; failure of kidneys to resorb water |
Receptor Tyrosine Kinases
Many polypeptide growth factors activate cells by binding plasma membrane receptors with cytoplasmic protein tyrosine kinase activity (Fig. 24-4). Ligand binding to extracellular domains allows the cytoplasmic kinase domains of pairs receptors to activate each other and to phosphorylate each other and downstream proteins that control cellular proliferation and differentiation. Mammals have 20 families of receptor tyrosine kinases with distinct structural features.
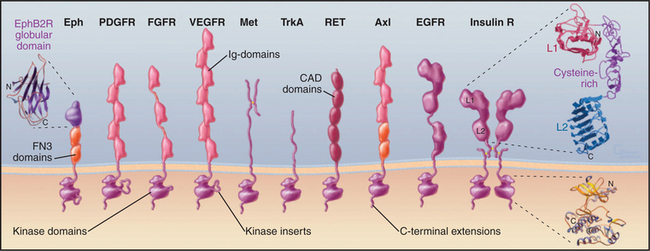
The growth factors that activate receptor tyrosine kinases regulate development and differentiation. For example, epidermal growth factor (EGF) stimulates proliferation and differentiation of epithelial cells. Platelet-derived growth factor stimulates growth of smooth muscle cells, glial cells, and fibroblasts (see Fig. 32-11). Some growth factors were discovered by biochemical purification of proteins that stimulate cellular growth or differentiation. EGF was discovered with a bioassay, as it causes the eyelids of newborn mice to open prematurely. A homolog of the EGF receptor, HER2/ErbB2, was discovered as the normal version of a cancer-causing viral oncogene. Other ligands and receptors were discovered as genes in flies or worms required for development. The Drosophila sevenless gene encodes a receptor tyrosine kinase that is related to insulin receptor. Mutations in the sevenless gene result in failure to develop photoreceptor cell number 7 in the fly’s eye.
Receptor tyrosine kinases consist of an extracellular ligand-binding domain connected to a cytoplasmic tyrosine kinase domain by a single transmembrane helix (Fig. 24-4). The ligand binding is mediated by immunoglobulin domains, fibronectin III domains (see Fig. 3-13), cadherin domains (see Fig. 30-5), and less familiar domains such as β-helical and cysteine-rich domains. This domain architecture illustrates that genes for receptor tyrosine kinases were assembled from sequences for familiar domains followed by divergence to allow for interactions with diverse ligands.
Ligand binding activates all well-characterized receptor tyrosine kinases by bringing together a pair of kinase domains on the cytoplasmic face of the membrane. Dimeric ligands such as platelet-derived growth factor recruit a pair of receptors from the pool of subunits diffusing in the plane of the membrane and connect them physically. This induced dimerization juxtaposes two kinase domains in the cytoplasm. An EGF monomer binds a receptor and induces a conformational change that favors the formation of a receptor dimer but is not a physical part of the connection between the subunits (Fig. 24-5). Insulin binding induces a conformational change in a preformed receptor dimer held together by a disulfide bond. The conformational change brings together the kinase domains (see Fig. 27-7).
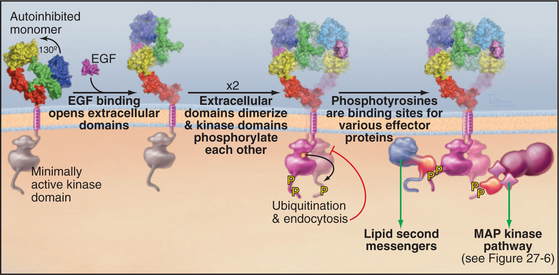
The juxtaposition of kinase domains allows the partners to activate each other by direct interaction and by phosphorylating each other on tyrosine residues. In most cases, phosphorylation of tyrosines on the activation loop of the catalytic domain refolds the loop into an active conformation (see Fig. 25-3D), although this is not required for the EGF receptor. In all cases, the paired kinases phosphorylate tyrosines on inserts and C-terminal extensions of the kinase domain, creating phosphotyrosine-binding sites for downstream effector and adapter proteins with SH2 and PTB domains (Fig. 24-5; also see Figs. 27-6 and 27-7). Each SH2 and PTB domain binds preferentially to a certain phosphotyrosine site by virtue of a pocket that recognizes both phosphotyrosine adjacent residues (see Fig. 25-11). For example, each of five phosphotyrosines of the platelet-derived growth factor receptor favors binding of a different effector or adapter protein.
Receptor tyrosine kinases activate effector proteins in two different ways. First, binding of an effector protein to a receptor phosphotyrosine favors its phosphorylation by the receptor kinase. In the case of phospholipase C g, tyrosine phosphorylation both activates its catalytic activity and dissociates the enzyme from its phosphotyrosine-binding site, allowing it to move to its site of action on the membrane. Second, binding to the receptor may promote activity by bringing an effector protein near its substrate. This applies to both phosphoinositide-3-kinase, which acts on lipid substrates in the membrane bilayer (see Fig. 27-7), and the nucleotide exchange protein that activates the Ras guanosine triphosphatase (GTPase), which is anchored to the membrane bilayer (see Fig. 27-6).
Multiple mechanisms turn off receptor tyrosine kinases. In the short term, lipid second messengers produced by phospholipase Cg activate protein kinase C, which inhibits the kinase by phosphorylation. Active kinases are also targets for the addition of a single ubiquitin to one or more lysine side chains. These single ubiquitins are the signal for the receptor to be removed from the cell surface by endocytosis in clathrin-coated vesicles (see Fig. 23-2).
Mutations in receptor tyrosine kinases cause human disease. Many cancers have activating mutations or overexpression of EGF receptors. Activating mutations in a fibroblast growth factor receptor lead to a variety of congenital abnormalities of the skeleton, including a form of dwarfism and premature fusion of the sutures between the bones of the skull. Some of these mutations activate by promoting receptor dimerization through disulfide bonds or association of transmembrane helices. Others change ligand specificity.
Cytokine Receptors
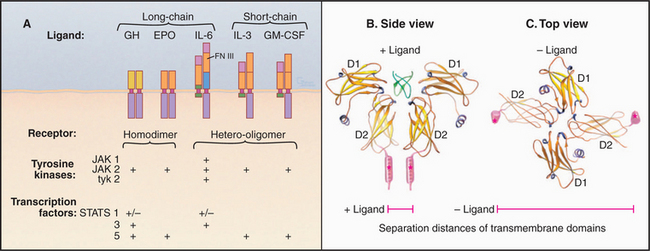
Cytokines are a diverse family of polypeptide hormones and growth factors that regulate many cellular processes. Although they differ in detail, all cytokines are four-helix bundles. Pituitary growth hormone controls body growth and development of mammals, so loss of function receptor mutations cause one type of dwarfism. Erythropoietin regulates the proliferation and differentiation of red blood cell precursors (see Fig. 28-7). Interleukins modulate cells of the immune system, so loss of function receptor mutations result in deficient immune cells.
Cytokine receptors are homodimers or heterodimers, all with two extracellular fibronectin III domains that bind the ligand (Fig. 24-6). A single polypeptide segment, likely an α-helix, crosses the membrane. Cytoplasmic domains lack enzyme activity but bind one of several protein tyrosine kinases called JAKs (“just another kinase”). It has not been settled whether inactive, ligand-free receptors diffuse separately in the plane of the membrane or form dimers with widely separated transmembrane domains, as observed in crystals of erythropoietin receptor without ligand (Fig. 24-6C). This separation of cytoplasmic domains could explain the lack of activity of ligand-free dimers.
Ligand binding activates cytokine receptors by bringing together two JAK kinases bound to the cytoplasmic domains—either by changing the conformation of preformed dimers or by inducing the dimerization of separate receptors (Fig. 24-7). A conformational change of a preformed dimer would explain how erythropoietin can activate cells with low concentrations of receptors on the cell surface. Close proximity allows JAKs to activate each other by transphosphorylation. The signal is then propagated to the nucleus when JAKs phosphorylate selected members of a family of transcription factors called STATs, which migrate to the nucleus to regulate gene expression (see Fig. 27-9).
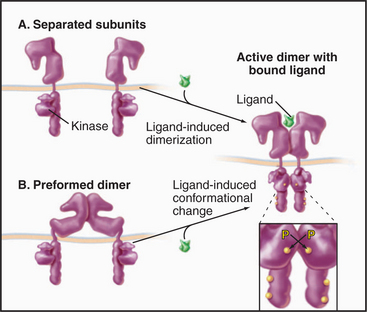
Figure 24-7 cytokine receptor activation models. A, Ligand-induced dimerization of separate subunits. B, Ligand-induced conformational change in a preformed dimer. In either case, proximity of the cytoplasmic domains allows associated JAK tyrosine kinases to activate each other by transphosphorylation. See Fig. 27-9 for details.
(Reference: Remy I, Wilson IA, Michnick SW, et al: Erythropoietin receptor activation by a ligand-induced conformational change. Science 283:990–993, 1999.)
Receptor Serine/Threonine Kinases
A third class of growth factor receptors uses cytoplasmic serine/threonine kinase domains to transduce signals (Figs. 24-8 and 24-9). Dimeric protein ligands bring together two different types of receptor subunits to turn on kinase activity. Humans have genes for seven type I receptors and five type II receptors. Active receptors phosphorylate transcription factors called Smads, stimulating their movement from cytoplasm into the nucleus, where they regulate genes controlling cellular proliferation and differentiation (see Fig. 27-10).
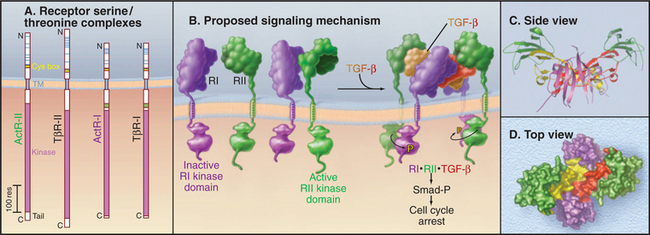
Figure 24-8 receptor serine/threonine kinases. A, Drawing of domain architecture of activin and transforming growth factor-b (TGF-b) receptors. (TM, transmembrane domain.) B, Mechanism of activation of receptor serine/threonine kinases. Ligand binds two type II receptors (RII) and two type I receptors (RI). Within this hexameric complex, the type II receptor phosphorylates and activates the type I receptors, which in turn phosphorylate cytoplasmic transcription factors called Smads. Phosphorylated Smads move to the nucleus to activate particular genes. See Fig. 27-10 for details. C–D, Ribbon and space-filling models of BMP7 bound to type I and type II receptors. This model is based on crystal structures of dimeric BMP7 bound to two extracellular domains of the type II activin receptor and of BMP2 bound to two extracellular domains of BMP type I receptor.
(References: Greenwald J, Groppe J, Gray P, et al: The BMP7/ActRII extracellular domain complex provides new insights into the cooperative nature of receptor assembly. Mol Cell 11:605–617, 2003; and Shi Y, Massague J: Mechanisms of TGFb signaling from cell membranes to the nucleus. Cell 113:685–700, 2003. PDB files: 1LX5 and 1S4Y.)
Humans have 40 proteins that bind receptor serine/threonine kinases, including transforming growth factor- b (TGF-b), activin, inhibins, and bone morphogenetic proteins. These dimeric growth factors are particularly important during embryonic development. Activin was discovered as a releasing factor for pituitary follicle-stimulating hormone but also has a strong influence on the differentiation of early embryonic cells into primitive germ layers. Bone morphogenetic proteins influence the differentiation of osteoblasts, which lay down bone matrix, among many other cells. TGF-b has two distinct effects. First, it inhibits proliferation of most adult cells. Mice with null mutations of one of their three TGF-b genes die with inflammation in multiple organs caused by excessive proliferation of lymphocytes. Second, TGF-b stimulates production of extracellular matrix, including collagen, proteoglycans, and adhesive glycoproteins (see Chapter 29). These proteins are essential for the development of organs. Overproduction of extracellular matrix is a common feature of many chronic inflammatory diseases, so inappropriate expression of TGF-b might provide a link between inflammation and the pathological fibrosis that scars diseased organs. On the other hand, loss of TGF-b receptors during progression of some tumors makes them unresponsive to growth inhibition by TGF-b and contributes, in part, to their ability to replicate autonomously.
Four receptor subunits bind independently to a dimeric ligand, beginning with high-affinity binding of two type II receptors followed by lower-affinity interactions with two type I receptors (Fig. 24-8B). Within this complex, the constitutively active type II receptor kinases phosphorylate the cytoplasmic domain of the type I receptors on serine and threonine residues. This activates the type I receptor kinase, which phosphorylates cytoplasmic transcription factors called Smads. Phosphorylated Smads move to the nucleus, where they cooperate with other transcription factors to regulate gene expression (see Fig. 27-10).
Guanylyl Cyclase Receptors
Animals have a family of cell surface receptors (Fig. 24-9) with intracellular domains that catalyze the formation of 3′–5′-cGMP from GTP. Vertebrates have at least seven guanylyl cyclase receptor isoforms; nematode worms have more than 25. The gases nitric oxide and carbon monoxide activate related cytoplasmic guanylyl cyclases that participate in additional signal transduction pathways (see Fig. 26-1). Regardless of its enzymatic origin, cGMP regulates the same targets: cGMP-gated ion channels (see Fig. 10-10), cGMP-stimulated protein kinases (see Fig. 25-4), and cyclic nucleotide phosphodiesterases (see Fig. 26-1).
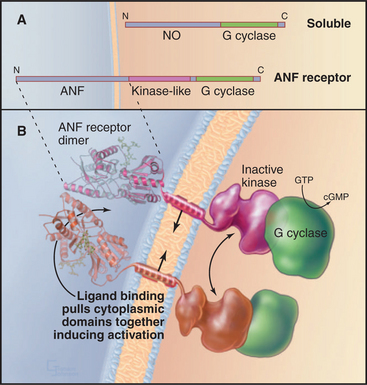
Guanylyl cyclase receptors are homodimers with novel, ligand-binding extracellular domains and two cytoplasmic domains—an enzymatically inactive kinase domain and a guanylyl cyclase domain (Fig. 24-9). The cyclase domain is closely related to adenylyl cyclases (see Fig. 26-2). In the absence of ligand, the extracellular domains hold the cytoplasmic domains apart. Ligand binding closes the cleft between the extracellular domains, pulling together the transmembrane and cytoplasmic domains in a way that stimulates guanylyl cyclase activity.
Guanylyl cyclase receptor A (GC-A) binds atrial natriuretic factor, a polypeptide hormone that is secreted mainly by the heart to control blood pressure. It stimulates excretion of salt and water by the kidney and dilates blood vessels. Mice with null mutations for GC-A have high blood pressure and enlarged hearts and fail to respond when overloaded with fluid and salt administered intravenously. Intestinal guanylyl cyclase receptor C (GC-C) binds bacterial enterotoxin, the mediator of fluid secretion in bacterial dysentery. The bacterial toxin mimics endogenous peptides of un-known function, which are secreted by various tissues but principally by the intestine. Mice with null mutations for GC-C are completely resistant to enterotoxin but have no apparent physiological defects. Guanylyl cyclase receptors E and F (GC-E, GC-F) are restricted to the eye; a null mutation for GC-E results in loss of cone visual receptor cells. Guanylyl cyclase receptor D is restricted to olfactory neuroepithelium. Sea urchin spermatozoa use a guanylyl cyclase receptor to respond to peptides secreted by eggs.
Tumor Necrosis Factor Receptor Family
TNF and its receptor are the prototypes for a diverse group of cell-signaling partners (Fig. 24-10). Lymphocytes produce three isoforms of TNF (also called lymphotoxin and cachectin), a trimeric lymphokine with many functions, including roles in shock and inflammation, protection from bacterial infections, killing tumor cells, and wasting in chronic disease. Mice with a genetic deletion of the lymphotoxin-a gene have no lymph nodes, so TNF participates in the development of the immune system. Other ligands for the TNF class of receptors are also trimers of subunits composed of β-strands, such as nerve growth factor and Fas-ligand. These ligands and receptors regulate many processes, including cell proliferation and death (see Fig. 46-17).
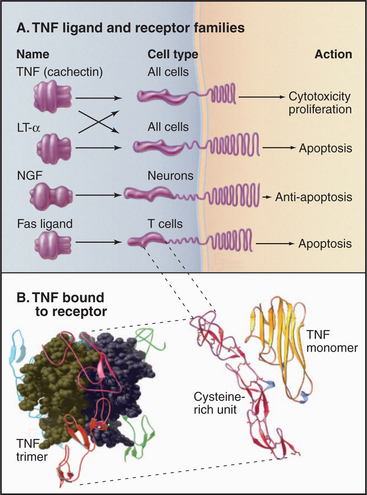
Figure 24-10 tumor necrosis factor receptor family. A, Domain architecture of a sample of members from the TNF receptor family. (LT-a, lymphotoxin-a; NGF, nerve growth factor.) Fas and Fas ligand are presented in Chapter 46. B, Atomic model of TNF bound to its receptor. TNF is a trimer of three identical b sandwich subunits arranged in a pear-like structure. The four extracellular cysteine-rich domains of the receptor grasp TNF-like prongs.
(A, Adapted with permission from Beutler B, vanHuffel C: Unraveling function in the TNF ligand and receptor family. Science 264:667–668, 1994. Copyright 1994 AAAS. B, Reference: Banner DW, D’Arcy A, Janes W, et al: Crystal structure of the soluble human 55 kD TNF receptor-human TNF beta complex: Implications for TNF receptor activation. Cell 73:431–445, 1993. PDB file: 1TNR.)
Human cells express two types of TNF receptors that bind the same ligands but generate different responses. The two receptors have similar ligand-binding domains coupled by single transmembrane segments to different cytoplasmic domains. The extracellular part of these receptors consists of four similar repeats of about 40 amino acids, each with six conserved cysteines (Fig. 24-10B). The cysteines form three disulfide bridges, arranged like the rungs of a ladder, to stabilize these small domains. In the absence of ligand, individual receptor subunits are presumed to diffuse independently in the plane of the membrane.
The structure of TNF bound to the extracellular domains of its receptor showed how the receptor works. Three finger-like receptors grasp one trimeric TNF molecule by binding along the interfaces between TNF subunits. The tapered shape of TNF brings together the transmembrane segments and cytoplasmic domains of three receptors. Something about this arrangement of TNF receptors activates a plasma membrane phospholipase that hydrolyzes sphingomyelin, producing the second messenger ceramide (see Fig. 26-11). Adapter proteins associated with active TNF receptors recruit protein kinases that alter gene expression by activating the transcription factor NF-kB (see Fig. 15-22). Other receptors with linear arrays of cysteine-rich subdomains, similar to the TNF receptor, also bind to multimeric ligands, so receptor activation by clustering their cytoplasmic domains might be a general theme. For example, Fas ligand triggers cell death by clustering Fas and activating a cascade of intracellular proteolysis (see Fig. 46-17).
Toll-Like Receptors
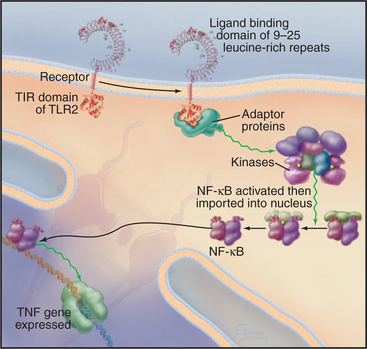
Metazoan organisms use a small family of receptors named Toll-like receptors (TLRs) to sense and respond to infection by a wide variety of microorganisms including viruses, bacteria, fungi, and protozoa. The Toll gene was discovered in Drosophila encoding a receptor that was first linked to dorsal-ventral polarity in early development and was later found to be required for resistance to fungal infections. Mammals have about a dozen TLRs (Fig. 24-11) that bind certain macromolecules associated with microorganisms: double-stranded RNA from viruses, flagellin from bacteria, lipopolysaccharide from the outer membrane of gram-negative bacteria, and zymosan from the cell walls of fungi. Dimeric TLRs on the plasma membrane of white blood cells (including lymphocytes) and antigen-processing cells called dendritic cells sense these foreign macromolecules and stimulate the cell to respond by secreting inflammatory mediators such as TNF and interleukin-1 and -6. TLR3 is located in endosomes, where it can bind double-stranded RNA released from viruses. The signaling pathway from TLRs to TNF involves cytoplasmic adapter proteins and kinases that activate transcription factors including NF-kB (see Fig. 15-22C). TNF and interleukins then alert distant cells to respond to the infection.
Notch Receptors
Genetic analysis established that Delta/Notch signaling is vital for animal development, but less is known about the mechanisms than those of the other receptors presented in this chapter. Some Delta is active as a cell surface protein that interacts locally with adjacent cells, but a matrix metalloproteinase (see Fig. 29-20) cleaves some Delta from the membrane, allowing it to act at a distance from its cell of origin. The receptors, called Notch (flies, vertebrates) or Lin-12 (worms), consist of several extracellular EGF-like domains and leucine-rich repeats, a single transmembrane span, and an intracellular region of ankyrin repeats that lacks any known enzyme activity. Notch is synthesized as a single polypeptide chain and is cleaved once before transport to the plasma membrane. The two polypeptides remain covalently associated, presumably by a disulfide bond. Cells carrying Delta activate Notch receptors on adjacent cells. This leads to proteolytic cleavage that frees the intracellular domains from the membrane. These cytoplasmic domains move into the nucleus and join a complex of proteins, including CSL, that activate transcription of certain genes.
Hedgehog Receptors
Genetic studies of Drosophila revealed a novel class of protein ligands, called Hedgehog, and two membrane proteins, called Patched and Smoothened, that are required for signal transduction during development. The Hedgehog receptor Patched consists of 12 transmembrane segments related to proton-driven bacterial antiporters, but the transported substrate, if any, is not known. Smoothened is an unusual seven-helix receptor, because it is constitutively active. Substoichiometric quantities of Patched inhibit the activity of Smoothened, perhaps by transporting out of the cell a ligand that binds and inhibits Smoothened. In flies, the Hedgehog pathway cooperates with the Wnt system (see Fig. 30-8) to establish boundaries between segments of the embryo and maintain a pool of stem cells.
Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257-263.
Bray D. Signaling complexes: Biophysical constraints on intracellu-lar communication. Annu Rev Biophys Biomol Struct. 1998;27:59-75.
Burgess AW, Cho H-S, Eigenbrot C, et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541-552.
Carpenter G. Nuclear localization and possible functions of receptor tyrosine kinases. Curr Opin Cell Biol. 2003;15:143-148.
Haglund K, Di Fiore PP, Dikic I. Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem Sci. 2003;28:598-604.
Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373-398.
Kadesch T. Notch signaling: The demise of elegant simplicity. Curr Opin Genet Dev. 2004;14:506-512.
Krzysztof P. G protein-coupled receptor rhodopsin. Annu Rev Biochem. 2006;75:743-767.
Lefkowitz RJ, Whalen EJ. Beta-arrestins: Traffic cops of cell signaling. Curr Opin Cell Biol. 2004;16:162-168.
Lum L, Beachy PA. The Hedgehog response network: Sensors, switches and routers. Science. 2004;304:1755-1759.
Marchese A, Chen C, Kim Y-M, Benovic JL. The ins and outs of G protein-coupled receptor trafficking. Trends Biochem Sci. 2003;28:369-376.
Park PS, Filipek S, Wells JW, Palczewski K. Oligomerization of G protein-coupled receptors: Past, present, and future. Biochemistry. 2004;43:15643-15656.
Penn RB, Pronin AN, Benovic JL. Regulation of G protein-coupled receptor kinases. Trends Cardiovasc Med. 2000;10:81-89.
Sakmar TP. Structure of rhodopsin and the superfamily of seven-helical receptors: The same and not the same. Curr Opin Cell Biol. 2002;14:189-195.
Schoneberg T, Schulz A, Biebermann H, et al. Mutant G-protein-coupled receptors as a cause of human diseases. Pharmacol Ther. 2004;104:173-206.
Sebald W, Mueller TD. The interaction of BMP-7 and ActRII implicates a new mode of receptor assembly. Trends Biochem Sci. 2003;28:518-521.
Shi Y, Massague J. Mechanisms of TGFb signaling from cell membranes to the nucleus. Cell. 2003;113:685-700.
Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335-376.
Wells JA, de Vos AM. Hematopoietic receptor complexes. Annu Rev Biochem. 1996;65:609-634.
Zhang G. Tumor necrosis factor family ligand-receptor binding. Curr Opin Struct Biol. 2004;14:154-160.
Zhang X, Gureasko J, Shen K, et al. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137-1149.
APPENDIX 24-1 Receptors and Ligands
| Classes of Receptors Activators | Nature of Activation | Examples of Biological Function |
|---|---|---|
| Voltage-Gated Ion Channels→Membrane Depolarization or Repolarization | ||
| Voltage-gated potassium channel | Electrical | Membrane repolarization |
| Voltage gated sodium channel | Electrical | Action potential |
| Ligand-Gated Ion Channels→Changes in Membrane Permeability | ||
| Acetylcholine (nicotinic) | Biogenic amine | Action potential |
| Adenosine triphosphate | Nucleotide | Change in membrane potential |
| Glutamate (N-methyl-d-aspartate) | Amino acid | Change in membrane potential |
| Glutamate (non–N-methyl-d-aspartate) | Amino acid | Change in membrane potential |
| Glycine | Amino acid | Change in membrane potential |
| Serotonin | Biogenic amine | Change in membrane potential |
| Seven-Helix Receptors→Trimeric G Proteins→Diverse Responses | ||
| Acetylcholine (muscarinic) | Biogenic amine | Slows heart; stimulates intestinal secretion |
| Adrencorticotropic hormone | Peptide | Stimulates adrenal cortisol production |
| Adenosine | Nucleoside | Dilates blood vessels |
| Angiotensin II | Peptide | Stimulates aldosterone secretion; contracts smooth muscle |
| Bradykinin | Protein | Stimulates intestinal secretion |
| Calcitonin | Protein | Inhibits calcium resorption from bone |
| Cholecystokinin | Peptide | Stimulates intestinal secretion |
| Complement (C5a, C3a) | Protein | Leukocyte chemoattractant |
| Dopamine | Biogenic amine | Neurotransmitter; inhibits prolactin secretion |
| Eicosanoids (prostaglandins) | Lipid | Promote or inhibit platelet aggregation, many other actions |
| Endothelins | Protein | Vasoconstriction |
| Epinephrine | Biogenic amine | Glycogenolysis; increases cardiac contractility |
| F-met-leu-phe | Peptide | Leukocyte chemotaxis |
| Follicle-stimulating hormone | Protein | Growth of ovarian follicle |
| γ-aminobutyric acid | Amino acid | Inhibitory neurotransmitter; stimulates intestinal secretion |
| Glucagon | Peptide | Glycogenolysis; stimulates intestinal secretion |
| Glutamate | Amino acid | Modulates synaptic transmission |
| Growth hormone–releasing factor | Peptide | Stimulates secretion of growth hormone |
| Histamine | Amino acid | Allergic responses; vasodilation; stimulates secretion |
| Interleukin, IL-8 | Protein | Chemotaxis of leukocytes |
| Leutinizing hormone | Protein | Steroid production by ovarian granulosa cells |
| Light absorption by rhodopsin | Photon | Vision |
| Lysophosphatidic acid | Lipid | Fibroblast proliferation, neurite retraction |
| Melanocyte-stimulating hormone | Protein | Melanin synthesis |
| Neurokinins (substance P) | Peptide | Stimulates gastrointestinal and pancreatic secretion; neurotransmitter |
| Norepinephrine | Biogenic amine | Smooth muscle relaxation |
| Odorants | Organics | Olfaction |
| Opioids | Alkaloids | Alters mood |
| Oxytocin | Peptide | Contraction of uterus |
| Parathyroid hormone | Protein | Bone calcium resorption |
| Peptide-releasing factors | Protein | Secretion of pituitary hormones |
| Platelet-activating factor | Lipid | Platelet activation |
| Serotonin | Biogenic amine | Stimulates intestinal secretion |
| Somatostatin | Peptide | Inhibits secretion of growth hormone, insulin, and glucagon |
| Thrombin | Protein | Activates platelets |
| Thyroid-stimulating hormone | Protein | Thyroid hormone secretion |
| Vasoactive intestinal peptide | Peptide | Stimulates intestinal secretion |
| Melanocyte-stimulating hormone | Biogenic amine | Stimulates gastrointestinal and pancreatic secretion; neurotransm |
| Vasopressin | Peptide | Regulates the permeability of the renal tubule to water |
| Wingless (Wnt) | Protein | Modulates gene expression |
| Two-Component Systems: Receptor/Histidine Kinase→Response Regulator→Diverse Responses | ||
| Aspartate | Amino acid | Controls flagellar motor and chemotaxis |
| Osmotic pressure | Physical | Regulates gene expression |
| Receptor Tyrosine Kinase→Ras, MAP Kinase, PLC, PI3 Kinase | ||
| Epidermal growth factor | Protein | Epithelial cell proliferation and differentiation |
| Fibroblast growth factor-α | Protein | Mesoderm differentiation; fibroblast mitogen |
| Fibroblast growth factor-β | Protein | Fibroblast mitogen |
| Hepatocyte growth factor (scatter factor) | Protein | Epithelial cell mitogenesis, motility |
| Insulin | Protein | Glucose uptake; cell growth |
| Insulin-like growth factor I | Protein | General body growth |
| Macrophage colony-stimulating factor | Protein | Growth and differentiation of monocytes |
| Neurotrophins (nerve growth) | Protein | Neural growth; neuron survival |
| Platelet-derived growth factor | Protein | Smooth muscle, fibroblast, glial growth and differentiation |
| Steel ligand | Protein | Development of melanocytes, germ cells |
| Transforming growth factor-α | Protein | Differentiation of connective tissue |
| Vascular endothelial cell growth factor | Protein | Endothelial cell growth |
| Cytokine Receptors→JAK Kinase→STAT Transcription Factors→Gene Expression | ||
| Ciliary neurotrophic factor | Protein | Survival/differentiation of neurons and glial cells |
| Erythropoietin | Protein | Growth and differentiation of red cell precursors |
| Granulocyte (colony-stimulating factor) | Protein | Growth and differentiation of granulocyte precursors |
| Granulocyte-monocyte (colony-stimulating factor) | Protein | Growth and differentiation of leukocyte precursors |
| Growth hormone | Protein | Cell growth and differentiation of somatic cells |
| Interleukin, IL-2 | Protein | Growth factor for lymphocytes |
| Interleukin, IL-3 | Protein | Growth factor for hematopoietic stem cells |
| Interleukin, IL-4 | Protein | Regulates gene expression |
| Interleukin, IL-5 | Protein | Regulates gene expression |
| Interleukin, IL-6 | Protein | Regulates gene expression |
| Interferon α/β | Protein | Regulates gene expression |
| Interferon γ | Protein | Macrophage and lymphocyte gene expression |
| Prolactin | Protein | Stimulates milk synthesis |
| Tyrosine Kinase–Linked Receptors→Cytoplasmic Tyrosine Kinase→Gene Expression | ||
| MHC-peptide complex → T-cell receptor | Protein | Growth and differentiation of T lymphocytes |
| Antigens → B-cell receptor | Various | Growth and differentiation of B lymphocytes |
| Receptor Serine/Threonine Kinase→Smad Transcription Factors→Control of Gene Expression | ||
| Activin | Peptide | Mesoderm development |
| Bone morphogenetic protein | Protein | Mesoderm development |
| Inhibins | Protein | Inhibition of gonadal stromal mitogenesis |
| Transforming growth factor-β | Protein | Growth arrest, mesoderm development |
| Membrane Guanylyl Cyclase Receptors→cGMP→Regulation of Kinases and Channels | ||
| Atrial natriuretic peptide | Peptide | Vasodilation; sodium excretion; intestinal secretion |
| Heat-stable endotoxin, guanylin | Unknown | |
| Sea urchin egg peptides | Peptide | Fertilization |
| Sphingomyelinase-Linked Receptors →Ceramide-Activated Kinases→Gene Expression | ||
| Interleukin, IL-1 | Protein | Inflammation, wound healing |
| Tumor necrosis factor | Protein | Inflammation, tumor cell death |
| Integrins→Nonreceptor Tyrosine Kinases→Diverse Responses | ||
| Fibronectin, other matrix proteins | Protein | Cell motility, gene expression |
| Selectins | ||
| Mucins | Glycoproteins | Cell adhesion |
| Cadherins | ||
| Like cadherins on another cell | Protein | Contact inhibition |
| Notch | ||
| Delta | Cell surface protein | Cell fate determination |
| Cytoplasmic Guanylyl Cyclase Receptors→cGMP→Kinases, cGMP-Gated Channels | ||
| Nitric oxide | Gas | Smooth muscle relaxation |
| Cytoplasmic Steroid Receptors→Active Transcription Factor→Gene Expression | ||
| Retinoic acid | Organic | Cell growth and differentiation |
| Steroid hormones | Steroids | Cell growth and differentiation |
| Thyroid hormone | Amino acid | Cell growth and differentiation |