CHAPTER 134 Pituitary Tumors
Functioning and Nonfunctioning
Pituitary Surgery: Historical Considerations
Most accounts of the history of pituitary surgery begin with an operation performed more than a century ago (1892) when Paul,1 a British surgeon, undertook the surgical management of an acromegalic patient. The patient, a 34-year-old woman, suffered from headache, facial pain, and vision loss. At the suggestion of Horsely, Paul performed a subtemporal decompression, but the tumor proved inaccessible. The patient’s symptoms, presumably caused by elevated intracranial pressure, were alleviated. The patient died about 3 months later, and the autopsy revealed a tangerine-sized pituitary tumor. The histologic diagnosis was “round cell sarcoma,” the prevailing nosology for pituitary adenomas at the time. Horsely2 subsequently used temporal and subfrontal approaches to treat 10 patients with pituitary tumors and reported this series in 1906.
The next major technical and conceptual advance came from Vienna, where a number of surgeons devised and successfully applied extracranial approaches to the sella. Schloffer3 was the first in 1907, employing an extensive lateral rhinotomy type of incision, with resection of the septum and turbinates en route to the sella. A variation of this approach was advocated by von Eisenberg4 and subsequently modified by Hochenegg,5 who accessed the sella through the frontal sinus.5 In 1909 Cushing6 used a variation of Schloffer’s method in an acromegalic patient, subsequently refining the technique and adopting the sublabial incision that was later described by Halstead7,8 and Kanavel.9 Contemporaneously, major contributions to the evolution of the transsphenoidal approach were provided by Hirsch,10 who described the endonasal approach in 1910 and later described the submucosal transseptal transsphenoidal approach.
As cranial neurosurgery continued to evolve, so did transcranial approaches to pituitary tumors. The second decade of the 20th century brought an escalating enthusiasm for transcranial approaches, and transsphenoidal approaches were all but abandoned, particularly in North America. Owing to technical contributions by Heuer, Frazier, Krause, Elsberg, Cushing, and others, transcranial approaches to the pituitary became routine. Fortunately, transsphenoidal methods were sustained by a few. Most notable among these was Dott of Edinburgh. A former pupil of Cushing, Dott continued to practice, refine, and teach transsphenoidal techniques. One of his students, Guiot,11 popularized the procedure in France and became one of its major advocates. Guiot passed on transsphenoidal methods to Hardy12 of Montreal, who reintroduced this approach to the neurosurgical mainstream in North America. In doing so, Hardy merged the procedure with the operating microscope, illuminating the microsurgical aspects of the technique, promulgating the concept of microadenoma, and showing the feasibility of selective tumor removal while preserving normal pituitary tissue and function. Together, Guiot and Hardy popularized their work in the 1960s and established the technical and conceptual elements of the procedure that form the basis of transsphenoidal microsurgery as it is practiced today.
The procedure has continued to evolve.13,14 Technical adjuncts include the application of transsphenoidal endoscopy, frameless stereotaxy for transsphenoidal surgery, and intrasellar ultrasonography.15–18 Transsphenoidal microsurgery is the preferred approach for more than 95% of pituitary tumors and for a large proportion of other sellar abnormalities. Further evolution of the transsphenoidal approach occurred with the introduction of the endoscope. Although used intermittently as an adjunct to microscopic transsphenoidal surgery,19,20 the concept of a pure endoscopic transsphenoidal technique (without the use of the operating microscope) was introduced in the 1990s and has recently become popular.21–30 The multidisciplinary neurosurgical and otolaryngologic team at the University of Pittsburgh Medical Center has broadened the scope of pathology that can be approached endonasally, including complex skull base pathology extending from the crista galli to the upper cervical spine.28,31 Whether performed microscopically or endoscopically, the transsphenoidal approach is preferred for more than 95% of pituitary tumors and for an expanding proportion of parasellar pathologies as well.
Pituitary Tumors: General Considerations
Epidemiology
Pituitary tumors are common lesions believed to account for 10% to 15% of all primary brain tumors.32 More precise estimates on their incidence and prevalence vary to some degree, depending on the means of survey, the population studied, and the period of the study. Data from academic medical centers suggest that pituitary tumors represent as many as 20% of surgically resected primary brain tumors.33 Epidemiologic estimates indicate an annual incidence of 8.2 to 14.7 cases per 100,000 people.34 A recent Belgian study reported a prevalence of 94 cases per 100,000.35 By these measures, pituitary tumors are the third most common primary intracranial tumor, preceded in frequency only by gliomas and meningiomas. These data underestimate the true frequency of pituitary tumors, however. Unselected autopsy studies have repeatedly shown that 20% to 25% of the general population harbor small pituitary microadenomas. These lesions are clinically silent; occur in patients without apparent endocrine symptoms; have the morphology of null cell, gonadotroph, or lactotroph adenomas; and are identified only after careful microscopic postmortem examination of a serially sectioned pituitary gland.32,36 The high subclinical prevalence of these so-called incidentalomas has also been validated with magnetic resonance imaging (MRI). When carefully sought, subtle signal changes in the pituitary gland indicative of a clinically occult microadenoma can be identified in 10% or more of routine MRI scans.37–39 It therefore follows that neoplastic transformation is a frequent occurrence in the pituitary, although it reaches clinical attention and requires intervention in relatively few cases.
Although pituitary adenomas occur in all age groups, the highest incidence is between the third and sixth decades of life. As a general rule, functioning pituitary tumors tend to be more common among younger adults, whereas nonfunctioning adenomas become more prominent with increasing age.40–42 Pituitary adenomas are uncommon in the pediatric population, representing only 2% of all primary pediatric brain tumors.43 The relative rarity of these lesions in the pediatric population is also reflected in several large institutional series of pituitary adenomas. At the Centre Foch, the Mayo Clinic, and the University of San Francisco, the respective proportions of pediatric pituitary tumors were 2.1% (66 of 3200 patients), 2.03% (36 of 1776), and 5.34% (119 of 2330).41,44,45 In most surgical series, pituitary tumors are distinctly more common among women, particularly premenopausal women. The basis of the higher prevalence in women is unclear, especially because incidental pituitary tumors are equally distributed between the sexes in autopsy series.36 One plausible explanation is that the clinical manifestations of pituitary adenomas, most notably menstrual dysfunction, are more conspicuous in women and are more readily diagnosed in premenopausal women than in men. The fact that Cushing’s disease is much more prevalent in women than men may contribute to this phenomenon.
Genetic predisposition to pituitary tumors is restricted to a single uncommon condition: multiple endocrine neoplasia type 1 (MEN-1) syndrome. Only 3% of all pituitary tumors occur in the context of this disorder. An autosomal dominant condition, MEN-1 syndrome is characterized by tumors of the parathyroid glands, pancreatic islet cells, and pituitary. Because the condition is variably penetrant, only 25% of affected patients develop pituitary adenomas, and most of these are macroadenomas associated with excess levels of growth hormone (GH) or prolactin (PRL), or both.46,47
Pathology
Because pituitary adenoma is clinically and numerically the most significant neoplastic process affecting the pituitary gland, only this lesion is discussed in this chapter. However, a diverse collection of other abnormalities can occur in the sellar region. Often masquerading as nonfunctioning pituitary adenomas, these lesions include a wide variety of neoplasms, developmental lesions, cysts, inflammatory processes, and aneurysms (Table 134-1).
|
Tumors of Adenohypophysial Origin Tumors of Neurohypophysial Origin Rare Tumors of Nonpituitary Origin |
Normal Adenohypophysial Morphology
Collectively, the adenohypophysis includes the pars distalis (anterior lobe), pars intermedia (intermediate lobe), and pars tuberalis (funnel-shaped upward extension of anterior lobe cells on the anterior face of the pituitary stalk). It is the site of meticulously regulated hormone synthesis and release. Representing approximately 80% of the entire pituitary, the adenohypophysis is the primary intrasellar site for most pathologic processes, including neoplastic disease. The anterior lobe is composed of five principal secretory cell types, each functionally and ultrastructurally distinct, and each distributed in a fairly consistent topologic arrangement within the gland. These five cell types are somatotrophs, lactotrophs, corticotrophs, thyrotrophs, and gonadotrophs, and they are distinguished functionally by their secretion of GH, PRL, adrenocorticotropic hormone (ACTH), thyroid-stimulating hormone (TSH), and gonadotropins (luteinizing hormone [LH] and follicle-stimulating hormone [FSH]), respectively. In a remarkably integrated fashion, the secretory and proliferative capabilities of these cells are governed by a precise and continuously regulated balance between stimulatory and suppressive hypothalamic influences and the negative feedback effects imposed by target organ hormones.48,49 Although susceptibilities vary, any of these cell types may be subject to neoplastic transformation. The resulting adenoma generally retains the secretory capabilities, some of the morphologic characteristics, and the nomenclature of the cell of origin.48,49
Microscopically, the anterior pituitary exhibits a delicate acinar architecture, and each acinus is composed of an admixture of various secretory cell types. Regionally within the gland, a definite and consistent topologic arrangement exists, in which different cell types are distributed at preferential intraglandular locations.32 Given these regional differences in the density of various adenohypophysial cells, different pituitary adenoma types vary correspondingly in their preferential site of intraglandular origin. An awareness of this topologic organization is important to neurosurgeons, who must occasionally dissect through a seemingly normal pituitary gland in search of a microadenoma. When visualized in horizontal cross section, the anterior lobe is composed of two lateral wings and a trapezoidal, central mucoid wedge. GH-producing cells populate the lateral wings and are especially abundant along its anterior face. Somatotroph adenomas usually arise at this site. PRL-producing cells can be found anywhere within the anterior lobe, although the densest accumulation occurs along the posterior aspect of the lateral wings, just anterior to the neural lobe. Most lactotroph adenomas also originate here. Corticotrophs, representing 10% to 15% of all adenohypophysial cells, usually reside within the central wedge, just anterior to the posterior lobe. This is a common but not invariable site for corticotroph adenomas. Thyrotrophs, accounting for less than 5% of all adenohypophysial cells, occupy a small zone in the anteromedial aspect of the central wedge. Although thyrotroph adenomas are seldom discovered while still microadenomas, most can be presumed to have originated at this site. Gonadotrophs are widely distributed throughout the pars distalis and have no favored site of accumulation. Gonadotroph adenomas do not have a stereotypical site of origin. They may be related to pituitary stem cells, “null cells,” and folliculostellate cells.
Histopathology of Pituitary Adenomas
Grossly, pituitary tumors are yellow-gray to purple; they often have a soft, fluid to creamy texture, in contrast to the firmness of the normal gland. The histologic growth pattern of pituitary adenomas varies, ranging from diffuse to sinusoidal to papillary. Beyond their descriptive merit, such designations are without prognostic significance. The most important histologic characteristics of pituitary adenomas are cellular monomorphism and a lack of acinar organization. In contrast, the normal pituitary exhibits an intimate admixture of different cell types arranged in a well-organized acinar pattern. Disruption of this acinar structure in adenomas is particularly well seen with silver stains for reticulin fibers. When studied in this manner, the presence of the interface between adenoma and nontumorous pituitary gland can be especially eye-catching, because the compressed rim of the latter forms a pseudocapsule around the former. This pseudocapsule of compressed normal gland, although thin, is often robust and in many cases fully contains the tumor.50
For the basic diagnosis of pituitary adenoma, light microscopy and hormone immunohistochemistry generally suffice. More precise classification requires ultrastructural examination. The ultrastructural profile of any given tumor provides information concerning tumor cytogenesis, degree of differentiation, and cellular constitution.32 The principal ultrastructural features used to distinguish and classify adenomas include cell size and shape, nuclear morphology, and the distribution and morphology of secretory granules, rough endoplasmic reticulum, Golgi apparatus, and microfilament accumulation. Morphologic details of this ultrastructural classification are presented elsewhere51; pertinent clinicopathologic aspects are discussed in the following sections.
Classification of Pituitary Adenomas
Pathologic Classification
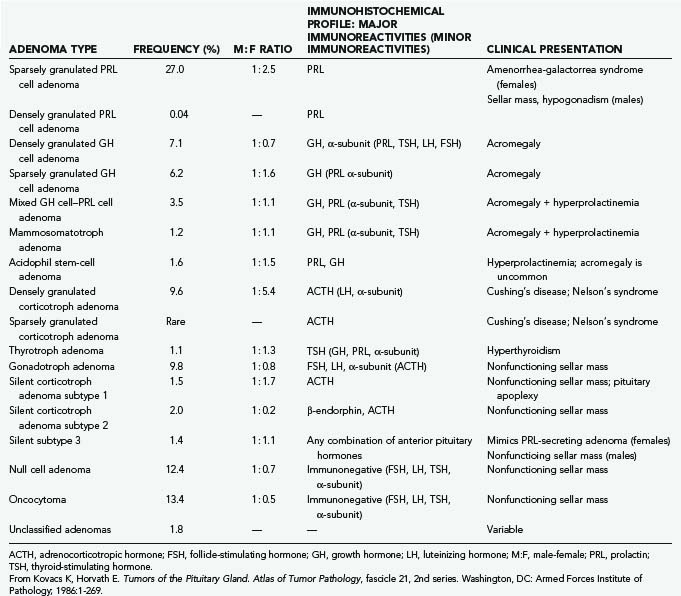
Immunohistochemistry and electron microscopy represent the “gold standard” methods of classifying pituitary adenomas. Delineating tumors on the basis of hormonal content, ultrastructural morphology, and cellular derivation, these methods have led to a new classification of pituitary adenomas that reliably correlates structure with function and cytogenesis with biology.32 As outlined in Table 134-2, this classification recognizes 14 principal pituitary adenoma subtypes, each having its own immunohistochemical and ultrastructural profile. Adenomas are stratified first on the basis of cellular origin and hormonal content and secondarily on the basis of ultrastructure.32,51–54 Subtypes that appear to have a higher recurrence rate include sparsely granulated GH adenomas, mammosomatotroph adenomas, acidophil stem-cell adenomas, and silent adenomas (especially silent ACTH adenomas and silent subtype 3 tumors).32,53,54
The morphologic appraisal of pituitary adenomas has its limitations. Whether considered from the standpoints of routine histology, hormonal immunophenotype, or ultrastructural morphology, none of these techniques permits reliable inferences about tumor behavior or prognosis.51 Accordingly, pituitary adenomas are not amenable to any form of histopathologic grading, such as that available for astrocytic tumors, that could reliably distinguish aggressive variants from indolent ones. The usual morphologic markers of tumor aggressiveness—pleomorphism, nuclear atypia, increased cellularity, and mitotic activity—correlate poorly with an adenoma’s invasive tendency, proliferative capacity, potential for recurrence, and overall biologic behavior. In general, mitotic figures and other morphologic markers of aggressive behavior are neither sufficiently common among invasive tumors nor sufficiently rare among noninvasive tumors to be of routine prognostic use.55 Nevertheless, pituitary adenomas are graded as “typical adenomas,” “atypical adenomas,” and “pituitary carcinomas.”53 Atypical adenomas are characterized by atypical morphology, an elevated mitotic index, a Ki-67 (MIB-1) labeling index greater than 3%, and extensive p53 nuclear staining. Pituitary carcinoma is characterized by the presence of noncontiguous metastatic spread of an adenoma.53,54
Imaging Classification
From a surgical standpoint, pituitary tumors can be classified on the basis of their size and growth characteristics, as determined by imaging studies. The simplest system, based purely on size, classifies tumors as microadenomas (<1 cm in diameter) or macroadenomas (>1 cm in diameter). This system fails to account for the tremendous variability in the size and growth characteristics of macroadenomas. The most enduring classification is that devised by Hardy12,56 and modified by Wilson.57 This five-tiered radiologic classification first differentiates tumors as microadenomas or macroadenomas. Microadenomas are designated grade 0 or grade I tumors, depending on whether the sellar appearance is normal or minor sellar changes are present, respectively. Macroadenomas causing diffuse enlargement, focal destruction, and extensive destruction of the sella are referred to as grade II, grade III, and grade IV tumors, respectively. In this system, macroadenomas are also staged according to the degree and direction of extrasellar extension—extending to the suprasellar cistern only (stage A), to third ventricle floor (stage B), or into the third ventricle (stage C). Tumors that have extensive lateral intradural or extradural extension are referred to as stage D and stage E, respectively.
With the advent of high-quality MRI, Knosp and colleagues58 proposed a grading system that predicts cavernous sinus invasion based on the position of the tumor in relation to tangents drawn through the supra- and intracavernous carotid arteries in the coronal view. Tumors that encase the carotid artery are universally found to invade the cavernous sinus at surgery. Tumors that do not pass beyond a tangent along the medial border of the supra- and intracavernous sinus do not invade the cavernous sinus. However, no other radiographic findings can definitively predict cavernous sinus invasion or the lack thereof.59
World Health Organization Classification
A variety of approaches can be used to classify pituitary tumors, ranging from simple to sophisticated, with each having its own merits and limitations. The management of pituitary tumors is a multidisciplinary endeavor, and nosologic preferences vary among specialties; the pathologist may favor a morphologic classification, the endocrinologist may favor one based on endocrine activity, and the neurosurgeon may favor one based on tumor size and invasiveness. In a bid to create a comprehensive classification scheme universally acceptable to clinicians, pathologists, and surgeons alike, a seven-tiered classification of adenohypophysial tumors has been proposed by the World Health Organization.53 In this scheme, seven descriptive levels are applied to each adenoma:
Clinical Manifestation of Pituitary Tumors
The clinical manifestation of pituitary adenomas usually centers on one or more of three clinical scenarios.49,60,61 The first involves pituitary hyperfunction in the form of several characteristic hypersecretory states. Hypersecretion of PRL, GH, ACTH, and TSH (rarely) produces corresponding clinical syndromes: amenorrhea-galactorrhea syndrome, acromegaly or gigantism, Cushing’s disease, and secondary hyperthyroidism. Because as many as 70% of pituitary adenomas are endocrinologically active, the presence of a hypersecretory endocrine state is the most common mode of presentation.
The second type of manifestation involves pituitary insufficiency and is typically associated with larger tumors that compress the nontumorous pituitary gland or its stalk or, in the case of giant pituitary adenomas, compress hypophysiotropic areas of the hypothalamus. In general, the pituitary gland displays remarkable functional resilience to even chronic compression and distortion. Eventually, however, anterior pituitary failure supervenes. Each pituitary endocrine axis appears to have a different tolerance of chronic compression. Gonadotrophs are most vulnerable and are affected first. Thereafter, thyrotroph, somatotroph, and eventually corticotroph functions are sequentially compromised.49 Curiously, regardless of how large the tumor is or how extreme the glandular or stalk compression is, posterior pituitary failure (i.e., diabetes insipidus) is rarely a presenting feature of pituitary adenomas; its preoperative presence virtually excludes a diagnosis of pituitary adenoma. Hypopituitarism that accompanies pituitary adenomas is usually a chronic process, but in the setting of pituitary apoplexy, it can be an acute, unexpected, and immediately life-threatening event.
Pituitary tumors are occasionally discovered incidentally, typically in a patient suffering from headache or other nonspecific symptoms in whom routine brain imaging reveals an abnormality in the size, shape, or contents of the sella. The situation is especially common today because of the superior resolution of MRI. When carefully sought, subtle signal changes in the pituitary gland can be identified in 10% to 12% or more of routine MRI scans, but the mere presence of an incidental abnormality on MRI does not imply the presence of an adenoma or the need for immediate intervention.37,39 Instead, careful clinical and endocrinologic correlations are required for such incidentalomas. Incidentally discovered adenomas may not always be asymptomatic. Up to 15% of incidentally discovered tumors are associated with endocrinopathy.62 Although few incidental microadenomas show interval growth, more than one third of macroadenomas increase in size over time.39,62–66 The decision whether to treat these tumors depends on tumor size, the presence or absence of neurological or endocrinologic symptoms, and the patient’s age, sex, and preference.
Diagnostic and Therapeutic Principles
Diagnostic Approach
Establish an Endocrine Diagnosis
Ordinarily, the history and physical examination provide some indication of the patient’s endocrine status. Suspicions of hormone excess or deficiency must then be validated by careful endocrine testing. An endocrine diagnosis is reached by measuring pituitary and target gland hormones in basal and dynamic states. These measurements are sensitive indicators of disturbed pathophysiologic activity and generally indicate whether a pituitary tumor is present and, if so, its secretory type. As an initial endocrine screen, basal measurements of PRL, GH, ACTH, LH, FSH, TSH, α-subunit, thyroxine, cortisol, insulin-like growth factor type 1 (IGF-1), testosterone, and estradiol should be obtained. By identifying states of relative excess or deficiency, this survey can provide a preliminary estimate of the integrity of various pituitary–target gland axes. Thereafter, additional provocative, dynamic, and special hormonal assays are performed to precisely define a specific endocrinopathy.49
Establish an Anatomic Diagnosis
Patients with Cushing’s disease may not have identifiable tumors on standard pituitary protocol MRI. In these patients, spoiled gradient recalled acquisition in the steady state (SPGR)-sequences with thinner slices can identify a larger percentage of small tumors and provide useful surgical information.67,68 In Cushing’s disease patients without identifiable tumors, inferior petrosal sinus sampling may be required to establish the diagnosis of pituitary-dependent Cushing’s disease. Although the initial hope was that inferior petrosal sinus sampling would reliably lateralize the tumor, it does so accurately in only 60% to 70% of cases.69,70 In certain circumstances, such as cases involving Cushing’s disease and acromegaly, extracranial imaging may also be required to secure the correct anatomic diagnosis. Exclusion of an ectopic hormone-secreting tumor in the chest, abdomen, or retroperitoneum may be necessary to confirm a pituitary-dependent source of hormonal excess.
Surgery for Pituitary Adenomas
Surgical Indications
The most urgent indication for surgical intervention is related to pituitary apoplexy.71–74 Patients may present with hemorrhage into an existing pituitary tumor or with acute necrosis of the tumor and subsequent swelling. In either case, the presentation includes sudden headache, precipitous loss of vision, ophthalmoplegia, altered level of consciousness, and collapse from acute adrenal insufficiency. In such situations, urgent glucocorticoid replacement and surgical decompression constitute the most reliable and effective form of therapy. Nevertheless, clinical apoplexy should be distinguished from subclinical apoplexy. Many tumors undergo microhemorrhage or infarction without clinically devastating results. Other patients may present with headache and pituitary insufficiency but without visual decline. Supportive nonsurgical treatment with analgesia and appropriate hormone replacement can be considered for these patients. Indeed, some patients managed in this manner experience tumor involution with serial imaging.
Among hyperfunctioning pituitary adenomas, surgery is the treatment of choice for Cushing’s disease, acromegaly, and secondary hyperthyroidism. In the case of Cushing’s disease, medical management is invariably suboptimal, and surgery provides the best means of obtaining prompt and lasting remission. In the case of somatotroph and thyrotroph adenomas, some latitude exists for the use of somatostatin analogues as the initial intervention, but surgery remains the preferred, primary, and definitive treatment for these conditions.75–78 For most prolactinomas, medical therapy is the preferred option unless the tumor is unresponsive or the patient is intolerant of medical therapy.
Failure of prior therapy often represents an indication for surgical intervention and usually occurs in one of several situations. A minority of patients are intolerant of the side effects of medical therapy. Other patients treated with radiotherapy have a favorable initial response followed by recurrence of symptoms in the form of mass effect or recurrent hormonal hypersecretion. For others treated with pharmacologic therapy, the response might have been suboptimal. For example, in some patients with a presumed prolactinoma, medical therapy can normalize PRL levels, but tumor growth continues. At surgery, these patients are usually found to have lesions other than prolactinoma. In other patients with genuine prolactinomas, a suboptimal response to medical therapy may take the form of persistently elevated PRL levels. Surgery may reduce the tumor burden and lead to a more effective pharmacologic response.79 The same also applies to acromegalic patients treated with somatostatin analogues.80
A somewhat generic surgical indication is the need for tissue diagnosis. Although seldom required in the case of functioning pituitary adenomas, this indication may be important when the surgeon is confronted with a nonfunctioning sellar mass whose pathologic identity cannot be confirmed without histologic examination.51
Choice of Surgical Approach
Surgical approaches to the sellar region can be broadly divided into three groups: transsphenoidal approaches, conventional craniotomy, and alternative skull base approaches (Table 134-3). For historical completeness, stereotactic approaches constitute a fourth category, but this approach is seldom indicated. Within each of the three groups, there is at least one standard procedure, as well as a menu of technical variations and options that allow the operation to be precisely tailored to the situation at hand. The overwhelming majority of all pituitary adenomas can be accessed through a transsphenoidal approach. The remainder require transcranial approaches consisting of standard pterional or subfrontal craniotomy or various skull base approaches that may be transcranial, extracranial, or a combination of the two.
|
Standard Transsphenoidal Approaches Standard Transcranial Approaches Alternative Skull Base Approaches |
If any of these features exist, a transcranial procedure should at least be considered.
Occasionally, the configuration of the tumor is such that a single approach, transsphenoidal or transcranial, is insufficient to achieve complete tumor removal (Fig. 134-1). This situation is uncommon and is typically associated with giant tumors having a significant intracranial component that may be inaccessible from below. In such situations, a combined transsphenoidal-transcranial approach can be used. Barrow and colleagues81 described the simultaneous performance of both approaches by two surgical teams. Alternatively, the two can be performed in a staged fashion. When considering such combined approaches, it is important to ascertain that the risks of aggressive surgery are justified by the benefits of radical tumor removal. In some instances, particularly those involving elderly patients with large, slow-growing, nonfunctioning tumors, a less radical and safer subtotal decompression may be preferable. However, in carefully selected patients, the combined approach represents an effective alternative.
Transsphenoidal Approaches
Details of anesthetic management have been described elsewhere.82 Special consideration must be given to the intubation of acromegalic patients, who may require awake intubation to safely secure an airway. Perioperative prophylactic antibiotics are routinely employed. Traditionally, perioperative “stress dose” exogenous corticosteroids were administered to all patients except those with Cushing’s disease whose serum cortisol was to be monitored in the postoperative period. However, routine administration appears to be required only in those patients who are adrenally insufficient preoperatively; in others, we no longer administer perioperative exogenous steroids. Instead, patients are monitored for clinical symptoms of adrenal insufficiency, and morning serum cortisol levels are drawn on each postoperative day to determine hypothalamic-pituitary-adrenal axis reserve. Levels less than 8 µg/dL are considered low and are replaced accordingly.
For most pituitary tumors, some variation of the transsphenoidal approach is the most appropriate route.25,83–85 All the variations (e.g., endonasal versus sublabial, endoscopic versus microscopic, submucosal versus direct sphenoidotomy) are minimally invasive, and the true standard of care is simply some route involving the transsphenoidal corridor. The virtues of the transsphenoidal approach are many. Most important, it represents the most physiologic and minimally traumatic corridor of surgical access to the sella, providing direct and superior visualization of the pituitary gland and adjacent pathology. This is true regardless of which variation is used. The techniques described here do not represent a compendium of all variations; they include the approaches we use in the treatment of pituitary adenomas and the relative indications for each. Virtually all tumors can be resected using these techniques, although each technique (and its variations) has particular advantages and disadvantages.
Adjuncts
Image guidance is an important surgical consideration. Videofluoroscopic guidance in the lateral view confirms the appropriate trajectory to the sphenoid sinus and sella and is used in microscopic transsphenoidal approaches. In endoscopic cases, the panoramic view of the sphenoid sinus often provides adequate information regarding the location of the sella. These cues are less apparent in presellar sphenoid sinuses and are inapparent in conchal-type sinuses. Therefore, endoscopic surgeons should at least consider using image guidance (either videofluoroscopy or frameless stereotactic methods) in presellar or conchal-type sphenoid sinuses. In addition, large tumors that fill the sphenoid sinus disrupt the normal anatomy and are relative indications for image guidance. Frameless stereotactic image guidance is particularly useful in repeat transsphenoidal surgery, in which the intraoperative cues regarding the anatomic midline are often disrupted.16,17,86 Intraoperative MRI can also be a valuable adjunct.
Positioning
The patient’s head is supported by a Mayfield headrest with a horseshoe (Fig. 134-2). Because the head is not fixed, gentle lateral movements of the head can be used to optimize intraoperative visualization, especially of the cavernous sinus area. This is particularly useful in microscopic cases because it allows the surgeon to move the patient’s head instead of stopping the operation to move the microscope. Although this is not a factor when the endoscope is used (because it provides a panoramic view), we do not fix the patient’s head in endoscopic cases either, to avoid the postoperative scalp discomfort associated with the cranial clamp. Fixation of the head may be necessary when certain forms of semirecumbent guidance are used.
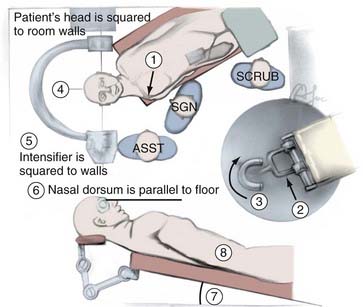
A semirecumbent position is used, with the patient’s back at a 20-degree angle from the horizontal and the head above the heart (see Fig. 134-2). This facilitates venous drainage and decreases venous pressure within the cavernous sinus. The right shoulder is placed at the upper right-hand corner of the table; the patient’s left ear is pointed toward the left shoulder, and the table is turned so that the patient’s head remains parallel to the wall of the room. This moves the foot of the table away from the surgeon and allows him or her to stand or sit in close proximity to the surgical field without bending over the patient. It also provides a spatial cue regarding the midline during a microscopic approach. If both the patient’s head and the microscope are perpendicular to the wall, the operative line of sight is in the midline. The head may be tilted to the right in some endoscopic approaches.
Nasal Preparation
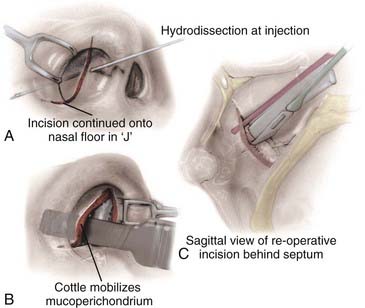
All surgeries can benefit from some degree of nasal decongestion, with the possible exception of patients with a history of hypertension and coronary artery disease. Before and immediately after induction of anesthesia, patients are given oxymetazoline intranasally. During positioning, oxymetazoline-soaked patties are placed between the middle turbinates and septum using a nasal speculum and bayonet forceps. The pledgets are allowed to remain in contact with the nasal mucosa for 5 to 10 minutes, during which draping of the patient is completed. The patties are removed after prepping and draping, and the nasal mucosa can be injected with 0.2% ropivacaine with 1 : 200,000 epinephrine. In microscopic cases, 10 to 20 mL of the solution is infiltrated, first along the upper gingiva, then along the inferior portion of the nasal septum, and finally along the lateral aspects of the nasal septum. A conscious effort is made to dissect the nasal mucosa away from the cartilaginous septum with the injection solution (Fig. 134-3A and B). This step is performed under direct vision with the aid of a nasal speculum, headlight, and loupes. In endoscopic cases, the middle turbinate, posterior septum, and sphenoid rostrum are injected (Fig. 134-4).
Sphenoid Sinus Access and Exposure
Microscopic Approaches
The next major consideration in the transsphenoidal procedure is the precise route of entry into the sphenoid sinus. For microscopic approaches, the two basic options are the endonasal approach and the sublabial approach. Selection of one over the other depends on the size of the nostril, the size of the lesion, and the preference of the surgeon. We tend to favor endonasal approaches in most instances, reserving the sublabial incision for pediatric patients or adults with small nostrils in whom the broader corridor afforded by the piriform aperture improves the visualization of the surgical field and the maneuverability of the surgical instruments. The endonasal microscopic approaches include the transseptal submucosal, septal pushover, and direct sphenoidotomy (see Fig. 134-4). These differ based on the location of the initial incision. With the transeptal submucosal technique, the incision is made just within the nostril, posterior to the columella; with the septal pushover, it is fashioned at the junction of the bony and cartilaginous septum; and with direct sphenoidotomy, the incision is made at the junction of the septum and the rostrum of the sphenoid. As the incision is carried farther back, the amount of septal dissection, and therefore the number of nasal complications, necessarily decreases. This decrease in septal dissection comes at the cost of a progressively narrower and potentially off-midline trajectory to the sella.
Endonasal Microscopic Approaches
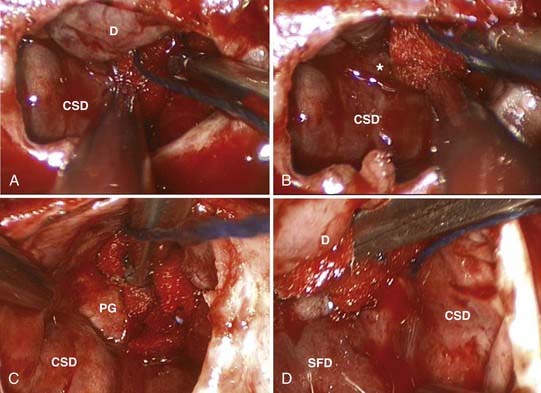
The endonasal submucosal transseptal approach begins with a right-sided hemitransfixion incision in the nostril, with the columella retracted to the patient’s left and the ala retracted toward the right (see Fig. 134-3A). The inferior border of the cartilaginous septum is exposed with sharp dissection, and one side of the septum is exposed submucosally with a combination of sharp and blunt dissection, thereby creating a unilateral anterior tunnel (Fig. 134-5). The dissection continues posteriorly, elevating the nasal mucosa away from the cartilaginous septum back to its junction with the bony septum. A vertical incision is made at this junction, and bilateral posterior submucosal tunnels are created on either side of the perpendicular plate of the ethmoid. The articulation of the cartilaginous septum with the maxilla is then dissected free, and an attempt is made to raise the inferior mucosal tunnel on the opposite side so that the cartilaginous septum can be displaced laterally without creating inferior mucosal tears. A self-retaining nasal speculum can be introduced to straddle the perpendicular plate of the ethmoid, exposing the face of the sphenoid sinus.
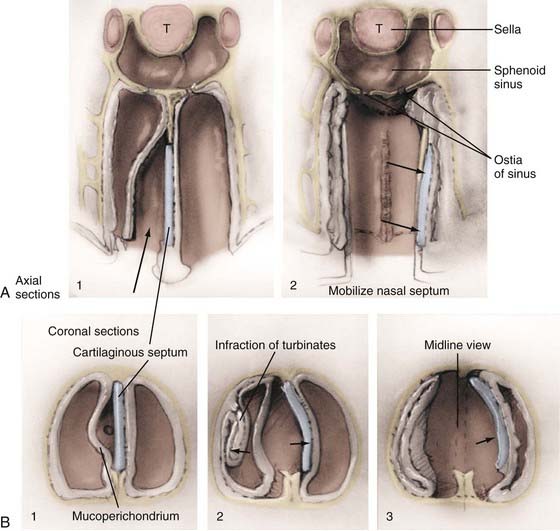
In some patients, particularly those who have had previous nasal, septal, or transsphenoidal surgery, we have used an alternative endonasal approach called the endonasal septal pushover technique (Fig. 134-3C).83,87,88 Instead of a submucosal incision for the creation of an anterior nasal tunnel, the nostril is entered, and an incision is made through the lateral mucous membrane of the nasal septum at the base of the septal insertion onto the maxillary ridge. The incision is carried back to the junction of the cartilaginous and bony septa or back to the face of the sphenoid if this bone has previously been removed. The nasal septum is carefully disarticulated, an opposite-side inferior tunnel is developed, and the septum together with the two layers of attached mucous membrane is reflected laterally to expose the perpendicular plate of the ethmoid and the sphenoid face. This rapid method may lack the elegance of the standard submucosal approach, but it can be very effective when the conventional approach proves difficult. If necessary, it can also be converted to a sublabial approach.
The most rapid of all endonasal approaches is the direct sphenoidotomy.89,90 In this approach, the speculum is inserted under fluoroscopic guidance directly anterior to the sphenoid rostrum. A sharp incision is made at the attachment of the septum to the sphenoid rostrum, and the septum is reflected laterally, exposing the rostrum of the sphenoid. Because there is no submucosal septal dissection, there is rarely any need for nasal packing with its resultant postoperative discomfort. The primary advantages of the septal pushover and direct sphenoidotmy techniques are the rapidity of the approaches and the avoidance of anterior septal dissection and its potential complications. However, these more direct approaches provide a narrower exposure and an off-midline trajectory—factors that should be considered when choosing the endonasal technique.
Endoscopic Approaches
Although the transsphenoidal approach has always been considered minimally invasive, particularly when compared with conventional transcranial approaches, the concept has been redefined in the context of endoscopic approaches to the sella.21,25,28,31,91 These approaches use straight and angled endoscopes as the sole visualization tools (a pure endoscopic approach) or as a supplement to the operating microscope (an endoscopic-assisted microscopic approach).
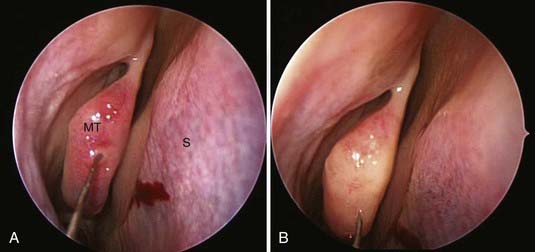
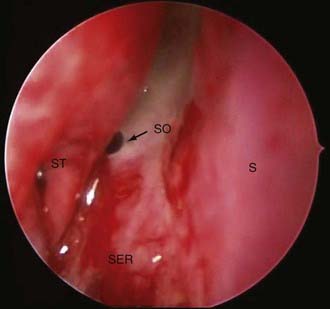
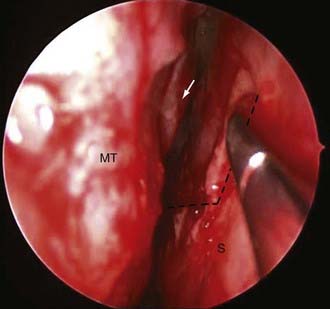
The 0-degree endoscope is used for the majority of exposure and tumor resection. The endoscope is brought within the nostril, and the sinonasal anatomy is identified, including the nasal floor and both the inferior and middle turbinates. The middle turbinate is lateralized, and the choana and sphenoethmoid recess are identified. The sphenoid ostium is then identified posterior to the inferior third of the superior turbinate (Fig. 134-6). Once identified, the posterior septum can be incised and reflected contralaterally to identify the contralateral sphenoid ostium (Figs. 134-7 and 134-8). The bone between the two ostia is then removed, providing the initial sphenoidotomy. A posterior septectomy is then completed, with care taken not to remove the septum more anterior than the anterior limit of the middle turbinate.
Sphenoidotomy
Endoscopic
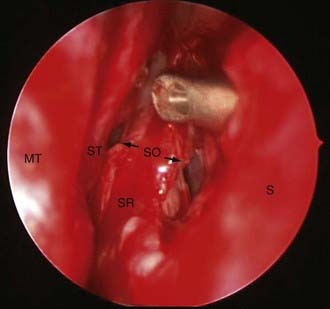
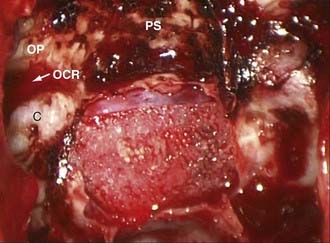
The endoscopic anterior sphenoidotmy is generally larger than that required for microscopic approaches. This relates to the fact that endoscopic exposures must account for the added instrument in the operative field (the endoscope). Once the initial sphenoidotomy has been performed, the intersphenoid sinus septae should be removed so that the sella can be identified. The extent of the sphenoidotomy can then be tailored to the location of the sella. The inferior extent of the sphenoidotomy should allow a sucker to be placed on the clivus below the level of the tumor. The proximal vomer should be protected as a reference point for the anatomic midline. Care must also be taken not to injure the posterior nasal branches of the sphenopalatine artery at the inferolateral margins of the sphenoidotomy. The superior extent of the sphenoidotomy provides room for the endoscope during tumor resection. The sphenoidotomy should continue superiorly until the tuberculum sellae, lateral opticocarotid recesses, and planum sphenoidale are readily observed (Fig. 134-9).
Sellar Entry
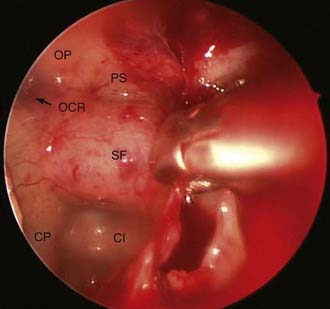
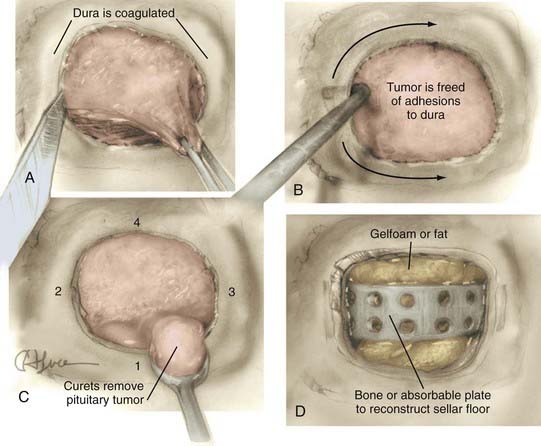
After the sellar floor has been penetrated, the opening is widened with a Kerrison-type punch. An adequate bony exposure is crucial to the success of the transsphenoidal approach, particularly when dealing with large tumors. For recurrent tumors in particular, a wide bony opening can allow virgin dura to be uncovered. Identification of the latter greatly assists in establishing a plane between dura and scar tissue. We favor a wide removal of the sellar floor in virtually every case, extending from one cavernous sinus to the other. A small, bony margin of the sellar floor should be left, because this facilitates sellar reconstruction at the end of the procedure (Fig. 134-10).
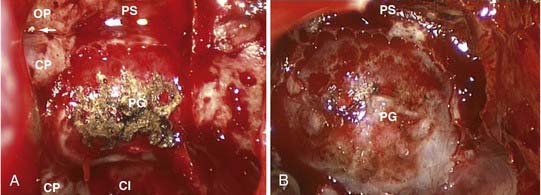
Tumor Removal
For the typical macroadenoma, the tumor is entered with a ring curet; tissue is loosened and then removed with a relatively blunt curet and forceps. Regardless of whether a micrscopic or endoscopic surgery is performed, the surgeon should attempt tumor removal in an orderly fashion (Fig. 134-11). Our practice is to first remove tumor in the inferior aspect and then proceed laterally, from inferior to superior aspects on both sides, removing tumor along the medial side of the cavernous sinus. The main distinction during endoscopic removal is the ability to directly visualize tumor removal from the cavernous sinus walls and suprasellar space (Fig. 134-12). The surgeon must resist coring out the central and most accessible portion of the tumor first, because this may cause premature descent of the diaphragma and entrapment of more laterally situated tumor. It is also important to delay the superior dissection until the lesion is relatively free elsewhere, because this minimizes trauma to the pituitary stalk and secondarily transmitted trauma to the hypothalamus. The surgeon may occasionally be required to follow a tumor into a cavernous sinus or deal with a tumor directly involving the diaphragma. In either case, any maneuver more forceful than gentle cureting may be dangerous; pulling of adherent fragments must be avoided. Decompression of the intrasellar portion of the tumor frequently permits a suprasellar extension to prolapse into view within the sella. After this has been resected, the diaphragma prolapses and generally signifies that the resection is complete. When spontaneous prolapse of the tumor capsule or diaphragma does not occur, instillation of 15 to 20 mL of air or lactated Ringer’s solution into a lumbar subarachnoid catheter may facilitate descent. Alternatively, bilateral jugular vein compression or application of positive end-expiratory pressure can help in delivering a suprasellar tumor. If the tumor still fails to descend, a ring curet can be used cautiously in the intracranial space. These maneuvers are well suited for the endoscopic technique, which allows both cavernous sinus and suprasellar portions of tumors to be removed under direct visualization. Bleeding from the tumor bed can usually be controlled by precise tamponade with cotton patties or Gelfoam.
In all cases, a concerted effort is made to preserve normal pituitary tissue. In a large, diffuse adenoma, normal glandular tissue usually appears as a thin membrane situated superolaterally against the sellar wall. The orange-yellow gland, together with its firm consistency, distinguishes it from the grayish color and finely granular texture typical of the tumor (see Fig. 134-12). A biopsy of the suspected glandular remnant may be taken for confirmation, but the appearance is often so typical that this tissue can be left behind with confidence.
Microadenomas necessitate a different operative strategy because many are not immediately visualized on opening the dura. A systematic search through a seemingly normal-appearing gland is often required. We begin with a transverse glandular incision, followed by subdural dissection and mobilization of the lateral wings. If the incision in the gland is deep enough, lateral pressure with a Hardy dissector usually causes the microadenoma to herniate into the operative field. Its location can therefore be delineated, its cavity entered, and its removal completed by use of a small ring curet and cup forceps. All suspicious tissue is removed, and a biopsy specimen is occasionally obtained from the residual and presumably normal pituitary gland. It is also possible to search for and find the pseudocapsule in many cases. If preserved, an extracapsular resection ensures complete removal of the tumor.50,92
Reconstruction and Closure
After the tumor has been removed and hemostasis has been achieved, the sella must be carefully evaluated with regard to reconstruction and closure. We prefer not to leave dead space in the sella. If no CSF leak has occurred, the sella is loosely packed with Gelfoam. If a CSF leak has occurred, some form of tissue graft becomes mandatory. Reconstruction of the sellar roof can be attempted with a piece of homologous dural graft material or fascia lata; however, this step probably adds little to the security of the seal. Current practice favors simple packing of the sella with fat taken from the right lower quadrant or subumbilical region of the abdomen. The fat is soaked in a chloramphenicol solution, may be rolled in Avitene, is trimmed to appropriate size, and is snugly placed within the sella. In either case, the sellar floor is then carefully reconstructed using a suitably trimmed piece of cartilage, bone, or tailored artificial grafts (Fig. 134-13; see Fig. 134-11). Some surgeons use fibrin glue as part of the closure in patients who have had CSF leaks, but we have found that an impressive mound of fibrin glue is a poor substitute for an inadequate structural repair; the integrity of the latter and not the adhesive properties of the former is the essential deterrent to postoperative CSF leaks. Likewise, we seldom use continuous lumbar drainage in patients who have had postoperative CSF leaks. Instead, we have adopted a policy of immediate reexploration, repacking of the sella, and reconstruction of the floor.
Complications of Transsphenoidal Surgery
Of the many virtues of the transsphenoidal approach, its safety and low complication rate are among the most important. With its lack of visible scars, lower mortality and morbidity compared with conventional transcranial approaches, and greater patient tolerance, transsphenoidal surgery is appealing to both patient and physician. Transsphenoidal surgery is among the safest procedures in neurosurgical practice. As determined by several retrospective cumulative series, operative mortality and major morbidity rates are 0.5% and 2.2%, respectively.93 In a series of transsphenoidal operations performed for Cushing’s disease during the past decade, mortality and permanent morbidity rates were 0.9% and 1.8%, respectively.94
Operative deaths are usually the result of intracranial hemorrhage, hypothalamic damage, or meningitis related to CSF fistulas. Although uncommon, a wide variety of other complications can occur with this approach (Table 134-4).95
| COMPLICATION | PATIENTS* |
|---|---|
| OPERATIVE MORTALITY (30 DAY) | |
| Hypothalamic injury or hemorrhage | 5 |
| Meningitis | 2 |
| Vascular injury or occlusion | 4 |
| CSF leak or pneumocephalus, SAH or spasm, MI | 1 |
| Postoperative MI, postoperative seizure | 2 |
| Total | 14 (1.0%) |
| MAJOR MORBIDITY | |
| Vascular occlusion, stroke, SAH or spasm | 5 |
| Vision loss (new) | 11 |
| Vascular injury (repaired) | 8 |
| Meningitis (nonfatal) | 8 |
| Sellar abscess | 1 |
| Sellar pneumatocele | 1 |
| Sixth cranial nerve palsy | 2 |
| Third cranial nerve palsy | 1 |
| CSF rhinorrhea | 49 |
| Total | 86 (3.4%) |
| LESSER MORBIDITY | |
| Hemorrhage (intra- or postoperative) | 9 |
| Postoperative psychosis | 5 |
| Nasal septal perforation | 16 |
| Sinusitis, wound infection | 5 |
| Transient cranial nerve (III or IV) palsy | 5 |
| Diabetes insipidus (usually transient) | 35 |
| Cribriform plate fracture | 2 |
| Maxillary fracture | 2 |
| Hepatitis | 1 |
| Symptomatic SIADH | 37 |
| Total | 117 (4.6%) |
CSF, cerebrospinal fluid; MI, myocardial infarction; SAH, subarachnoid hemorrhage; SIADH, syndrome of inappropriate antidiuretic hormone.
* From the authors’ series of 2562 pituitary adenomas.
Hypothalamic Injury
Damage to the hypothalamus may result from direct surgical injury and from hemorrhage or ischemia provoked by the procedure. Clinical manifestations of hypothalamic damage include death, coma, diabetes insipidus, memory loss, and disturbances of vegetative functions (e.g., morbid obesity, uncontrollable hunger or thirst, disturbances in temperature regulation). Such complications are more common in patients with prior craniotomy or radiation therapy.96 Gentle surgical technique and avoidance of traction on the tumor capsule and pituitary stalk minimize the occurrence of such injuries.
Visual Damage
Damage to the optic nerves and chiasm can also occur from direct surgical trauma, hemorrhage, or ischemia. Fractures of bony structures at the base of the skull can damage optic nerves and can occur from misdirected placement and aggressive opening of transsphenoidal retractors. Many patients have preoperative compromise of visual function, making them more vulnerable to further damage. Such complications are more likely to occur in patients with adhesions from prior cranial surgery or radiation.96 Assessment of the bony anatomy, careful and gentle technique, confirmation of surgical landmarks, and effective use of fluoroscopic guidance to direct the approach are the major methods of avoiding these complications. At the time of sellar reconstruction, overpacking the sella can cause chiasmal compression, whereas underpacking can lead to a secondary empty sella with the late onset of vision loss due to chiasmatic prolapse.
Vascular Complications
Although rare, arterial injury is a well-known complication of transsphenoidal surgery and is one of the main sources of operative mortality accompanying the procedure.97,98 Virtually every transsphenoidal series includes at least one example of arterial injury, which is often fatal. The intracavernous portion of the carotid tends to be most vulnerable, followed by other components of the circle of Willis. Because the tumor may be quite adherent to arterial structures, arteries may be lacerated, perforated, avulsed, or damaged, with the subsequent development of spasm or intraluminal thrombosis. Intracranial hemorrhage, thrombotic stroke, embolic stroke, and the development of false aneurysms or carotid-cavernous fistulas are the usual sequelae of such injuries. When vascular injury is suspected, tamponade should be used to control hemorrhage, and an immediate postoperative angiogram should be obtained. Gentle technique devoid of aggressive traction on the tumor capsule, not deviating from the midline, and repeated assessment of bony landmarks are the most effective means of avoiding these frequently devastating complications.
Iatrogenic Hypopituitarism
In most instances, existing pituitary function can be preserved. For microadenomas, our experience indicates that loss of one or more anterior pituitary functional axes occurs in approximately 3% of cases.99,100 For macroadenomas, we have found that anterior pituitary function can be preserved in more than 95% of cases, provided pituitary function was normal preoperatively. Partial or complete restoration of endocrine deficits is achieved in only 16% to 35% of patients with established preoperative endocrine deficits. Although diabetes insipidus occurs temporarily in as many as one third of all patients, posterior pituitary failure is permanent in no more than 3% of patients. As a rule, postoperative endocrine deficits tend to be more common and more severe in patients who have undergone reoperation or in those treated by craniotomy.
Complications Associated with Reoperation
It is generally true in neurosurgery and certainly true for pituitary tumors that reoperations are associated with somwhat higher complication rates and less favorable outcomes than initial operations.101 This issue was carefully studied in a series of 158 patients, most of whom had pituitary tumors and all of whom required secondary transsphenoidal surgery for recurrence, persistent disease, or treatment of complications related to unsuccessful prior therapy.96 Although the scope of that study extended beyond the single issue of repeat surgery for recurrent pituitary tumors, many of the conclusions are applicable in the current context. Of the 158 patients treated with repeat transsphenoidal surgery, operative mortality was 2.5%, and the new complication rate was 29%; this contrasts with the 0.5% mortality and 2.2% morbidity rates that are frequently quoted for initial transsphenoidal operations.96
Operative complications are notably more frequent and more serious in patients whose initial approach was transcranial. In such situations, the diaphragma may be partially or totally absent, leading to scarring and dense arachnoid adhesions between the tumor and the optic apparatus, hypothalamus, and basal vasculature, all of which add to the hazards of a transsphenoidal procedure.95–97 For patients whose original approach was transsphenoidal, scarring, adhesions, and distorted anatomy also predispose to complications, although to a lesser degree. Prior radiotherapy or pharmacologic therapy can be expected to induce fibrotic changes in the tumor, although we have not found this to have a consistently adverse effect or be an overly important predisposing factor for surgical complications. Most cases of operative mortality have been the result of intracranial hemorrhage, hypothalamic damage, or meningitis related to CSF fistulas.
Transcranial Approaches
Indications
There are three basic transcranial approaches to pituitary tumors: pterional, subfrontal, and subtemporal. Selection of one approach over the others depends on the precise geometry and growth trajectory of the tumor, as well as the preference and experience of the surgeon. Probably the most versatile approach, and the one that we prefer, is the pterional approach. Occasionally other skull base craniotomy approaches are useful (see Table 134-3).
Pterional Approach
In most instances, obvious tumor is encountered behind the tuberculum and between the optic nerves. The pituitary stalk, with its portal vessels producing a characteristic vertically striated appearance, can usually be identified behind the tumor in the triangle between the lateral border of the right optic nerve and the carotid triangle. Every effort is made not to disturb this structure. The tumor capsule is then carefully dissected away from surrounding structures. Great care should be exercised in dealing with portions of the tumor attached to the optic apparatus, the dissection of which may damage these structures or their microvasculature. The tumor can be entered through several operative corridors. Usually, the capsule is incised between the optic nerves. The tumor is entered and its contents removed by curets, suction, or, for unusually fibrous tumors, sharp dissection. Manipulation of the capsule and additional internal decompression can be performed through the opticocarotid triangle as well. A trans–lamina terminalis approach can also be performed if a third ventricular component fails to descend. If the intrasellar portion of the tumor is difficult to remove because of its consistency, a prefixed chiasm, or other factors, a transtuberculum, transsphenoidal corridor can be used.102 This requires drilling just behind the jugum sphenoidale to expose the sphenoid sinus, followed by opening the anterior wall of the sella. When such a maneuver is anticipated, a subfrontal approach is preferred over a pterional one.