[level-membership-for-neurosurgery-category]
CHAPTER 215 Lipomyelomeningocele
Johnson first described a lipomyelomeningocele (LMM) in 1857,1 although it was not until 1971 that Rogers and colleagues first introduced the term lipomyelomeningocele in their series of spinal lipomas.2 Since its original description, several authors have subsequently reported on their experience with this very common form of occult spinal dysraphism.3,4 LMM occurs in approximately 1 in every 4000 births in the United States, with a female-to-male prevalence ratio of 1.5 : 1.5 The classification, embryology, diagnosis, and surgical management of LMM are outlined in this chapter.
Definition and Classification
The literature regarding LMM is very confusing. The term lipomyelomeningocele has mistakenly been grouped into the spectrum of all lipomas of the lumbosacral spine and is often misleading because it implies herniation of neural elements through a spina bifida defect into a meningeal sac. Unlike myelomeningoceles or meningoceles, however, there is neither neural nor meningeal tissue found outside the spinal canal. The neural elements remain within the spinal canal, and there is typically an associated subcutaneous lipomatous mass that protrudes through a midline bony defect, displaces the dura, and infiltrates and tethers the spinal cord.5,6 In 1982, Chapman classified three anatomic variants of LMM according to the relationship of the lipoma–spinal cord interface: dorsal, caudal, and transitional.7
Lipomas that insert onto the dorsal surface of the conus medullaris are classified as dorsal-type LMMs (Fig. 215-1). In this variant there is typically a substantial subcutaneous fat component attached to the underlying spinal cord via a fibrolipomatous stalk of varying thickness. The posterior half of the lower spinal cord at the site of fusion of the neural fold is unfused (i.e., partial dorsal myeloschisis is present), and the stalk attaches directly to the exposed alar and basal cord regions, just posterior and medial to the posterior columns and dorsal to the central canal.8 The lipoma can be located either in the midline (68%) or eccentrically (32%) and can extend into the central canal and expand it for variable distances.9 Along the lateral interface of the attachment of the lipoma to the spinal cord, the dura and pia are fused. Sensory nerve roots emerge just anterior to this lateral line of fusion, which can be traced circumferentially. There is often no evidence of leptomeninges posteriorly, and no sensory or motor roots are found within the actual substance of the lipoma.
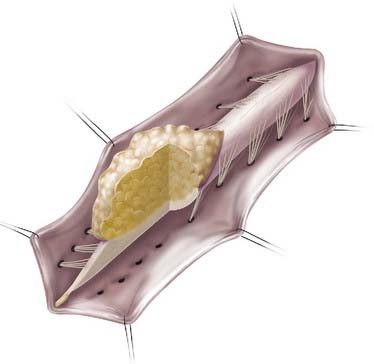
In caudal- or terminal-type LMM, the lipoma–spinal cord interface is located at the caudal end of the conus, almost like a continuation of the spinal cord itself (Fig. 215-2). The remaining lipomatous mass may then lie entirely within the spinal canal or extend dorsally through a defect in the dura and bone into the subcutaneous space. The fatty tumor may either replace or surround the filum terminale, or a separate filum may lie anteriorly. The nerve roots may lie ventral to the lipoma or pass through the mass itself. A caudal-type LMM is often asymmetrical and involves more of the cord and nerve roots on one side, and unlike the discrete lipoma-cord interface of a dorsal-type LMM, the lipoma-cord interface in a caudal-type LMM is diffuse.8
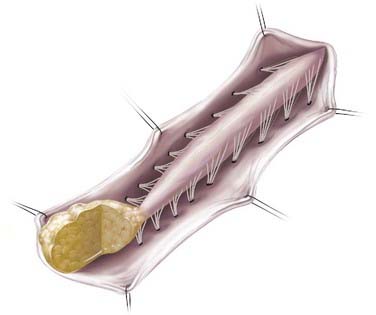
In addition to the dorsal and caudal LMM forms originally described by Chapman, transitional forms between the two variants may exist (Fig. 215-3). Frequently in these cases, the more cephalad portion is of the dorsal type with posterior rootlets emerging from the cord just ventral to the line of fusion of the lipoma to the cord and leptomeninges. Caudally, however, as the line of fusion is displaced ventrally, nerve roots emerge from the anterolateral portion of the lipoma mass. Magnetic resonance imaging (MRI) can often be helpful in defining the type and may be useful in determining the operative approach. The transitional form is considered by some to simply represent a continuum between the other two forms.7,8,10,11
Embryology
Congenital lumbosacral lipomas develop as a result of disturbances in the precise sequence of events encompassing the embryologic stages of primary and secondary neurulation, which occur between postovulatory day (POD) 25 and 48.8 Closure of the posterior neuropore marks the completion of the first phase (primary neurulation) of neural tube formation, at which point the fetal spine is covered by ectoderm but the lumbar, sacral, and coccygeal segments have not yet developed.12 The caudal end of the neural tube and the notochord combine into a large aggregate of undifferentiated cells in a process called condensation. This caudal cell mass extends to the level of the tail fold and represents a solid neural cord containing neurons, neural crest, glial cells, and ependymal cells. In a process called canalization, a series of small vacuoles within the mass begin to coalesce and enlarge to create an ependymal-lined tube in continuity with the rostral central canal of the previously formed neural tube.13 This process takes place between POD 27 and 29 and leads to formation of the ventriculus terminalis.14,15
The third phase in formation of the caudal neural tube involves regression of the structures previously derived during canalization. During retrogressive differentiation, the embryologic tail disappears, whereas the filum terminale, coccygeal ligament, and ventriculus terminalis of the conus remain. This process is thought to complete by POD 48 to 52.12,13,15–17 Because cell rests within the caudal cell mass have totipotent characteristics, a small clone of lipomatous cells developing in the region would not necessarily inhibit the growth of surrounding structures. The bony vertebrae develop subsequently, and the sacrococcygeal segments also undergo regressive changes to decrease the number of segments originally present.15 As a result, vertebral maldevelopments are found in conjunction with the neural defects arising during this period.
McClone and Naidich proposed that the process of dysjunction, in which the closed neural tube detaches from the cutaneous ectoderm, occurs prematurely in patients with LMM. Consequently, a dorsal cleft is maintained and paraxial mesenchyme tissue has access to the prospective lumen of the neural tube, thereby further preventing its closure. The luminal surface of the ectoderm induces mesenchymal cells to differentiate along a path that results in adipocyte formation, whereas the outer surface of the incompletely closed neural tube causes the mesenchyme to form the piaarachnoid and dura.18,19 Other tissues have been found in the substance of an LMM, including striated muscle, cartilage, nerve cells, ependyma, and even cerebellum.6 Similar to other spinal dysraphisms, a lumbosacral lipoma that tethers the spinal cord at the upper lumbar level probably occurs at a slightly earlier embryologic period than does a lesion that tethers the cord lower down in the spine.8 Several authors have found that patients with LMM can have other associated dysraphic conditions of the spinal cord, including split cord malformation, terminal syringomyelia, neurenteric and intraspinal dermoid/epidermoid cysts, a tight filum terminale, and tethering tracts.20,21
Clinical Findings
Cutaneous Signs
Although patients with LMM can have a spectrum of cutaneous anomalies, infants are seen most often with a cutaneous sign that suggests a potential underlying abnormality. The most common finding (>90% of patients) is a subcutaneous fatty mass covered with skin that is located in or near the midline of the lumbosacral spine (Fig. 215-4). This mass is not tender and appears to be in continuity with the normal subcutaneous tissues. Although the overlying skin is typically intact, LMMs can also be associated with a hairy patch (hypertrichosis), hemangioma, dermal sinus, skin dimple, skin tag, atretic meningocele, or caudal appendage (Table 215-1).20,22 The most superficial portion of the mass typically consists of lobulated and unencapsulated subcutaneous fat, which becomes more fibrous as the deeper structures are approached. The stalk of the fibrous fatty tissue can then be found to penetrate the dura and is associated with the spinal cord, conus, cauda equina, or filum terminale, as described earlier. Additional malformations may also be present, including cloacal exstrophy, bifid uterus, duplicated vagina, anterior sacral meningocele, anomalies of the lower limbs, and imperforate anus.15 It is rare for patients with LMM to have intellectual impairment or associated hydrocephalus, Chiari malformations, or other brain anomalies.15,23,24
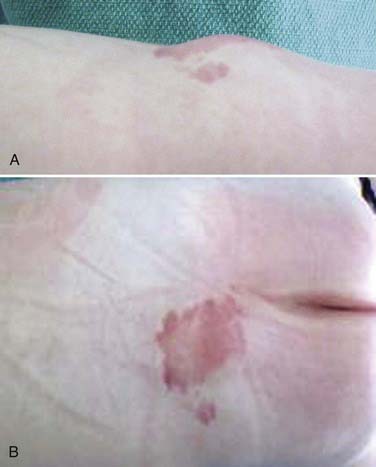
TABLE 215-1 Lumbosacral Cutaneous Manifestations Found in Patients with Lipomyelomeningocele
| SERIES | HOFFMAN ET AL.,20 1985 (N = 97) | KANEV ET AL.,22 1990 (N = 80) |
|---|---|---|
| Soft tissue mass | 97 | 80 |
| Skin dimple | 26 | 14 |
| Hemangioma | 24 | 9 |
| Hypertrichosis | 1 | 11 |
| Skin tag/tail-like appendage | 5 | 6 |
| Atretic or denuded skin patch | 1 | |
| Dermal sinus hypopigmentation | 3 | 3 |
Symptoms
LMMs typically cause tethering of the spinal cord via the associated fibrous subcutaneous lipoma. As infants age, symptoms reminiscent of tethered cord syndrome typically develop, along with any combination of neurologic, orthopedic, and urologic issues. Slightly more than one third of patients will demonstrate an asymmetrical subcutaneous mass with regard to the midline; these patients have a higher incidence of neurological deficits in the lower limb ipsilateral to the side of the mass.25 Symptoms depend on the level of the cord-lipoma interface, as well as patient age at the time of diagnosis, and may be related to several possible causes working independently or in combination.15 Deficits present at birth may become manifested as a result of abnormal development of the spinal cord and nerve roots. The presence of a mass within the confines of the bony spinal canal may also contribute to patient symptoms in more ways than one. Namely, progressive mechanical distortion or ischemia of the neural elements may result from tethering of the neural elements or, alternatively, from a local mass effect with compression of the cord, conus, or cauda equina. Similar to other fatty growths, these tumors can enlarge with age in proportion to normal body growth.26
Although an unknown number of patients may initially be seen with asymptomatic LMMs, it is crucial for clinicians to understand that progressive neurological impairment can develop in patients who at first seem normal. In infants and toddlers, accurate assessment of clinical involvement is very difficult. Yamada demonstrated that spinal cord tethering interferes with normal energy metabolism of the spinal cord, which can lead to ischemia and progressive neural damage.27 In addition, although most patients demonstrate slow loss of function over a period of years, sudden and acute loss of function has also been described. For example, this has been reported in previously asymptomatic women placed in the lithotomy position during pregnancy or in patients with hyperflexion injury from a motor vehicle accident.
Muscle weakness and gait disturbance are often present by the time that the patient starts ambulating. Clinical examination may reveal muscular atrophy, leg length discrepancy, varus or valgus foot deformity of one or both legs, hammertoes or claw toes, scoliosis, mixed deep tendon reflexes (absent, normal, increased), mixed/patchy sensory abnormalities, back pain, or any combination thereof. Abnormal voiding patterns with urinary or fecal incontinence or the presence of urinary tract infections may also be an initial symptom. Although patients can exhibit any of the aforementioned signs and symptoms, infants typically have cutaneous anomalies; older children have orthopedic, neurologic, or urologic symptoms; and adults often have pain.15
Evaluation
Prenatal Diagnosis
LMMs are increasingly being diagnosed in utero as a result of the frequent use of screening antenatal ultrasonography (US).28 Accurate prenatal diagnosis allows appropriate planning for delivery and neonatal care.29 However, because these defects are closed with no communication between fetal spinal fluid and maternal amniotic fluid, amniotic fluid analysis for α-fetoprotein levels is not a reliable diagnostic measure.30–32
Ultrasonography
US has the ability to differentiate among fat, spinal fluid, and spinal cord as a result of their different echogenic properties. Because infants younger than 6 months are skeletally immature with poorly calcified bone, US can image through the spine in these individuals, which can be a very useful tool for defining the attachment of lipomas to the spinal cord.33,34 Several benefits of US include its relative inexpensiveness, ease of use, and lack of need for patient sedation. It is also beneficial in determining spinal cord motion for possible evidence of spinal cord tethering. US is unlikely ever to replace other modalities such as MRI, however, because it is limited in its ability to provide the appropriate preoperative information. In addition, US can be operator dependent and possibly miss subtle lesions unless the user is well experienced.
Plain Radiographs
Plain radiographs can play a beneficial role in the initial assessment of patients suspected of having closed spinal dysraphism; however, as an initial evaluation in most pediatric neurosurgery centers, they are largely being replaced by MRI.35 Anteroposterior and lateral radiographs at the appropriate spinal levels almost always reveal abnormalities such as a dorsal fusion defect in the lamina (bony spina bifida) or widening of the spinal canal.8,36 Varying degrees of agenesis or deformity of the sacrum have also been reported in the literature. Some surgeons prefer to have plain radiographs available for surgical planning. Absence or incomplete calcification of bony elements limits the utility of plain radiographs in children younger than 18 months.37
Magnetic Resonance Imaging
MRI is the study of choice for diagnosing and monitoring patients with LMMs.35,38 MRI is especially beneficial in children with congenital disorders because of its capability of three-dimensional imaging and ability to visualize and differentiate neural tissue. T1-weighted imaging provides clear anatomic detail of the spinal cord and filum terminale, which allows visualization of the vertebral level of the conus; the absence, presence, and location of fat within the cord, spinal canal, or filum; and the size of the filum (Fig. 215-5). T2-weighted images allow identification of spinal cord tumors, such as dermoids and epidermoids, as well as the presence of fluid-containing structures such as syringomyelia.36 Performance of MRI to determine spinal cord motion with the patient supine and prone may help one decide whether ventral movement of the cord is adequate and rule out dorsal tethering. The use of gadolinium is typically not required.

Computed Tomography
Computed tomography (CT) is rarely used at our institution for children, either for diagnostic or for planning purposes. There is no information that can be obtained from a CT scan that cannot be obtained from high-quality MRI and plain films except for bony details in a skeletally immature infant. The risk-to-benefit ratio in obtaining a CT scan is, in our opinion, not high enough to warrant the additional cost and, most importantly, radiation exposure to developing infants and children. We consider CT an appropriate adjunct for patients in whom the information cannot be obtained from other imaging modalities, including plain radiographs, or when the index of suspicion for additional bony abnormalities is high (e.g., septum in a split cord malformation). Some authors use CT myelography; however, this technique is invasive and requires a lumbar puncture, which adds another element of risk because the conus is low-lying and the bone may be incompletely ossified.39
Urodynamics
The difficulty in determining the extent of urologic involvement in young patients with LMM who are not yet toilet-trained warrants objective urodynamic functional assessment. The use of urodynamic testing, predominantly cystometrography, at our institution is routine for both preoperative evaluation and postoperative long-term follow-up. Other studies designed to evaluate structural abnormalities or non-neurogenic functional problems within the urologic system, such as voiding cystourethrography and renal US, are performed at the discretion of the urologist. Although objective monitoring of bladder function can be difficult to interpret in children, mainly because of their inability to cooperate, studies have shown that comparison of postoperative studies with their preoperative baselines is useful. In fact, there is evidence that release of tethered cord in patients with occult spina bifida and urodynamic derangements often results in postoperative improvement.40–43 Abnormalities that may be detected include a spastic (older children and adults), flaccid (younger children), or dyssynergetic bladder.15
Surgical Treatment
Indications
The true natural history of LMMs is unknown. Despite considerable clinical experience suggesting that surgery is superior to observation, ethical considerations have precluded randomized prospective study of the efficacy of repair of LMM.3 Long-term follow-up in the presurgery era and retrospective analyses of these data, however, support the notion that patients with LMM undergo progressive neurological, orthopedic, and urologic decline. Keating and coworkers reported urinary incontinence in 26% of infants and neonates with LMM as opposed to 92% of older children.15,44 Hoffman and colleagues demonstrated an 85% rate of deterioration in children with untreated LMM by 5 years of age.20 Kanev and Bierbrauer found that approximately two thirds of children younger than 6 months were intact whereas only a limited number of patients older than 4 years retained normal neurological and bladder function.3
Even though some surgeons still question the utility of prophylactic surgery, several studies have advocated early surgery and aggressive therapy because of its relative safety and effectiveness.3,4,7,9,45,46 Surgical intervention, thus, is primarily intended to prevent the progression of fixed deficits or the development of new ones. Kulkarni and colleagues demonstrated that there is little difference in the deterioration-free interval between patients who are treated surgically and those managed conservatively.47 There have also been reports of spontaneous reduction in the size of LMMs during follow-up without surgical intervention.47,48 Regardless, if surgery is undertaken, there is no justification for superficial cosmetic excision of the fatty tissue alone because this will not address the underlying cause of future neurological injury and may make future surgical exploration more difficult.49
Most pediatric neurosurgeons choose to operate on patients before 6 months of age if possible.9,10,20,49–51 The principal goals of surgery are to (1) detach the spinal cord from all tethering structures, (2) decompress the intramedullary mass, and (3) reconstruct the spinal cord and dural sac.15 These three goals should be the principal aims of the surgeon, all while minimizing the risk for neurological deficits. At the point where resection of the lipoma may cause new or increased neurological problems, the procedure should be terminated.
Operative Technique
 We start with a straight incision in the vertical axis overlying the subcutaneous mass (an elliptical incision is occasionally needed to resect redundant skin). The skin is initially taken down with a scalpel blade and the dissection continued by undermining the subcutaneous tissues with monopolar cautery to carefully identify the entry of the fibrolipomatous stalk through the medial fascial defect (Fig. 215-6, Video 215-1). An adequate amount of fat is left under the skin to minimize devascularization and necrosis. A self-retaining retractor is placed and the lumbodorsal fascia then opened on both sides of the midline while leaving the cephalad interspinous ligaments intact. An elliptical incision is made around the stalk. Large lipomatous masses are often amputated at the level of the stalk to help in visualization of the interface between the spinal cord and lipoma. Care is taken during all stages of the surgical resection to identify the most intact caudal spinous process and all laminar defects or bifid processes because one can accidentally dissect into the dura. The paraspinous muscles are then dissected off the spinous process and laminae bilaterally; the dissection is maintained medial to the articular facet joints to avoid disrupting the facet capsule. There is often a fibrous band of tissue corresponding to the periosteum of an incompletely formed bony element immediately caudal to the last intact lamina.52 To prevent postlaminectomy scoliosis or kyphosis, we prefer performing an osteoplastic laminectomy in young children. Although there are several ways to do this, we cut the lamina bilaterally, and the whole segment of posterior elements is reflected cranially and held out of the field with a stay suture (the interspinous ligament between the most cephalad segments are kept intact). Careful inspection of the epidural space before lifting the bone will allow identification of other tethering bands or structures that could be attached to the underside of the lamina.
We start with a straight incision in the vertical axis overlying the subcutaneous mass (an elliptical incision is occasionally needed to resect redundant skin). The skin is initially taken down with a scalpel blade and the dissection continued by undermining the subcutaneous tissues with monopolar cautery to carefully identify the entry of the fibrolipomatous stalk through the medial fascial defect (Fig. 215-6, Video 215-1). An adequate amount of fat is left under the skin to minimize devascularization and necrosis. A self-retaining retractor is placed and the lumbodorsal fascia then opened on both sides of the midline while leaving the cephalad interspinous ligaments intact. An elliptical incision is made around the stalk. Large lipomatous masses are often amputated at the level of the stalk to help in visualization of the interface between the spinal cord and lipoma. Care is taken during all stages of the surgical resection to identify the most intact caudal spinous process and all laminar defects or bifid processes because one can accidentally dissect into the dura. The paraspinous muscles are then dissected off the spinous process and laminae bilaterally; the dissection is maintained medial to the articular facet joints to avoid disrupting the facet capsule. There is often a fibrous band of tissue corresponding to the periosteum of an incompletely formed bony element immediately caudal to the last intact lamina.52 To prevent postlaminectomy scoliosis or kyphosis, we prefer performing an osteoplastic laminectomy in young children. Although there are several ways to do this, we cut the lamina bilaterally, and the whole segment of posterior elements is reflected cranially and held out of the field with a stay suture (the interspinous ligament between the most cephalad segments are kept intact). Careful inspection of the epidural space before lifting the bone will allow identification of other tethering bands or structures that could be attached to the underside of the lamina.

The operating microscope is typically brought in at this point to provide better magnification and illumination. The epidural fat is melted with bipolar cautery and the abnormal tract penetrating the dura identified. The key to preventing neurological deficits is to locate and open the dura in an area that appears the most normal or that has never previously been violated (i.e., in reoperations). Sometimes this means extending the laminectomy one or two levels higher. The normal dura is then opened with a scalpel until the spinal cord is identified below and the opening carried caudally toward the penetrating tract (Fig. 215-7). Once the incision approaches the point where the lipoma penetrates the dura matter, the dura is opened on either side of the stalk circumferentially. Care is taken to identify the underlying neural structures throughout the dissection. This is crucial because nerve roots may enter the cord at the same point where the dura, lipoma, and spinal cord all join together; an incision too close to the point of dural penetration into the lipoma could result in transection of the dorsal nerve roots.53 Dural tack-ups are placed.

Circumferential dissection around the lipoma stalk is continued until the dura is completely separated from the cord. The dural opening is then continued caudally below the dural-stalk-cord junction until the filum is identified. Traditionally, the CO2 laser has been used to debulk the fatty mass. More authors, however, are using an yttrium-aluminum-garnet laser. The latter is less effective in pulverizing fat than the CO2 laser, but it is more practical and less hazardous. At our institution, the Cavitron ultrasonic surgical aspirator is used routinely because it is safe and effective, particularly when resecting along the fat-cord interface. An yttrium-aluminum-garnet laser is also occasionally beneficial.54,55
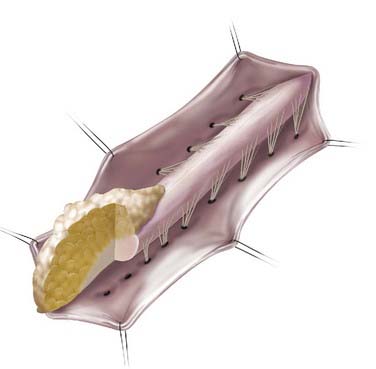
The fatty mass is debulked through a midline dorsal myelotomy until all visible fatty tissue is resected and the plane of gliotic tissue between the lipoma and glial fibrous tissue is encountered. Once all resectable intramedullary lipoma has been debulked, the filum terminale is identified and divided. The midline myeloschisis defect is then closed dorsally with 6-0 or 7-0 nonabsorbable suture to reconstruct the tubular configuration and thus minimize exposed raw edges (Fig. 215-8). This may contribute to reduced future tethering secondary to scar tissue.

Intraoperative Electrophysiology
The use of intraoperative electrophysiologic monitoring techniques is controversial among surgeons because many untether the spinal cord on the basis of anatomic rather than electrophysiologic criteria. No prospective controlled trials have been conducted to determine the efficacy of this modality in avoiding injury during treatment.55 Anecdotal data have provided some support for its use; we find it to be a useful adjunct to surgery when distinguishing among functioning nerve roots, spinal cord tissue, and nonfunctional tethering structures.56,57 Some authors argue, however, that neuromonitoring techniques are cumbersome and of questionable utility because stimulation of putative nervous tissue may result in the spread of current and thus the potential to mistake scar tissue or filum terminale for neural structures.15 Other authors use neuromonitoring in a limited fashion (e.g., electromyography) when sectioning the filum or identifying nerve roots in a complex lipoma.55
Neuromonitoring techniques include (1) posterior tibial and peroneal somatosensory evoked potentials, which theoretically detect excessive traction or lateral pressure on the conus medullaris; (2) pudendal sensory evoked potentials, which detect injury to the S2-4 segments, an area that is especially vulnerable to injury during untethering procedures; and (3) bladder and external anal sphincter manometry, as well as lower limb electromyography, which help differentiate between nerve roots and other structures such as tethering bands and scar tissue (Fig. 215-9).11,35,58

Complications
Surgery for LMM is relatively safe. Although no mortalities have been reported in recent studies, potential complications from surgery include CSF leakage, development of a pseudomeningocele, and new neurological deficits (more commonly seen with transitional lipomas).5,22,59 When a CSF leak occurs, the wound is often oversewn as a first step. If the CSF leak persists, however, the wound is re-explored. Some authors advocate placement of an external spinal drainage catheter during the healing period.15 Other reported complications include aseptic meningitis from host-graft inflammation (which can be treated with a short course of steroids), meningitis, intradural abscess, wound infection, and wound breakdown.3,5,22 Again, significant debulking of the subcutaneous portion of the lipoma is contraindicated because of the risk for necrosis of the overlying skin.
Outcomes
A goal of neurological improvement as a result of the surgical intervention should not be considered unreasonable. Anderson reported that improvement in stance and gait occurred in approximately 20% of symptomatic patients after surgery.60 Previous studies have also found that patients younger than 18 months demonstrate a higher rate of improvement in urodynamic and urinary function than do older children and adults.61,62 Similarly, the likelihood of reversing neurological deficits with surgical treatment decreases with age. Symptoms that commonly improve after surgery include low back pain, buttock pain, and radiculopathy.15
Foot deformities may stabilize or even worsen over time after surgery secondary to persistent muscular weakness and imbalance.15 Long-term neurosurgical and radiologic observation are important even after successful repair of LMM because of the possibility of cord retethering from scar tissue. This phenomenon has been reported in up to approximately 15% of patients, although the true incidence is not known.3,5,9,22,45,59,63 Patients who complain of increasing or new back or leg pain or the onset of new neurological problems (e.g., extremity weakness or urinary incontinence) should be reimaged with MRI of the spine. The main reason for MRI is to rule out syringomyelia. Imaging the potentially tethered cord itself may not be of much value for two main reasons: first, the termination of the spinal cord rarely changes after the first surgery; second, the incidence of imaging-proven retethering secondary to scar tissue is high, but only a small percentage of patients have clinical symptoms and signs that require reoperation. In fact, although retethering can occur any time from several months to several years after surgery, it is treated surgically only if it causes clinical dysfunction.46,64 Repeated surgical interventions for retethering becomes increasingly difficult with each subsequent operation because of the development of additional postoperative scarring and arachnoiditis.64 There is some evidence that tight closure of the dural sac and incomplete release of tethering structures are risk factors for recurrent tethered cord symptoms.64 In addition, surgeons have attempted to minimize retethering by minimizing bleeding onto the spinal canal during surgery, the use of various dural substitutes, and other nuances in their techniques. However, none of these methods have proved effective thus far, and the problem of retethering after LMM repair remains a major concern in the treatment and follow-up of these patients.
Atala A, Bauer S, Dyro F, et al. Bladder functional changes resulting from lipomyelomeningocele repair. J Urol. 1992;148:592-594.
Blount JP, Elton S. Spinal lipomas. Neurosurg Focus. 2001;10(1):e3.
Cecchini A, Locatelli D, Bonfanti N, et al. Lipomyelomeningoceles: a neuroradiological approach. J Neuroradiol. 1988;15:49-61.
Chapman PH, Beyerl B. The tethered spinal cord, with particular reference to spinal lipoma and diastematomyelia. In: Hoffman HJ, Epstein F, editors. Disorders of the Developing Nervous System: Diagnosis and Treatment. Boston: Blackwell Scientific; 1986:109-131.
Colak A, Pollack I, Albright A. Recurrent tethering: a common long-term problem after lipomyelomeningocele repair. Pediatr Neurosurg. 1998;29:184-190.
Finn MA, Walker ML. Spinal lipomas: clinical spectrum, embryology, and treatment. Neurosurg Focus. 2007;23(2):1-12.
Foster L, Kogan B, Cogen P, et al. Bladder function in patients with lipomyelomeningocele. J Urol. 1990;143:984-986.
French BN. The embryology of spinal dysraphism. Clin Neurosurg. 1983;30:295-365.
Hoffman H, Taecholarn C, Hendrick E, et al. Lipomyelomeningoceles and their management. Concept Pediatr Neurosurg. 1985;5:107-117.
Hoffman H, Taecholarn C, Hendrick E, et al. Management of lipomyelomeningoceles. Experience at the Hospital for Sick Children, Toronto. J Neurosurg. 1985;61:1-8.
Iskandar BJ, Fulmer BB, Hadley MN, et al. Congenital tethered spinal cord syndrome in adults. Neurosurg Focus. 2001;10(1):e7.
Iskandar BJ, Oakes WJ. Anomalies of the spine and spinal cord. In: McClone DG, editor. Pediatric Neurosurgery. Philadelphia: WB Saunders; 2001:307-324.
Kanev P, Bierbrauer K. Reflections on the natural history of lipomyelomeningocele. Pediatr Neurosurg. 1995;22:137-140.
Kanev PM, Lemire RJ, Loeser JD, et al. Management and long-term follow-up review of children with lipomyelomeningocele, 1952-1987. J Neurosurg. 1990;73:48-52.
Kothbauer K, Schmid UD, Seiler RW, et al. Intraoperative motor and sensory monitoring of the cauda equine. Neurosurgery. 1994;34:702-707. discussion 707
Lemire R, Loeser J, Leech R. Normal and Abnormal Development of the Human Nervous System. Hagerstown, MD: Harper & Row; 1975.
McClone DG, Mutluer S, Naidlich T. Lipomyelomeningoceles of the conus medullaris. In: Concepts in Pediatric Neurosurgery. Basel: S Karger; 1983:170-177.
Oakes WJ. Tethered spinal cord, intramedullary spinal lipoma, and lipomyelomeningocele. In: Rengachary SS, Wilkins RH, editors. Neurosurgical Operative Atlas. Chicago: American Association of Neurological Surgeons; 1992:133-141.
Oakes WJ. Management of spinal cord lipomas and lipomyelomeningoceles. In: Wilkins RH, Rengachary SS, editors. Neurosurgery Update II, vol 3. New York: McGraw-Hill; 1991:3497-3504.
Pang D. Intraoperative neurophysiological monitoring of the lower sacral nerve roots and spinal cord. In: Yamada S, editor. Tethered Cord Syndrome. Park Ridge, IL: American Association of Neurological Surgeons; 1993:135-147.
Sutton LN. Spinal dysraphism. In: Rengachary SS, Ellenbogen RG, editors. Principles of Neurosurgery. 2nd ed. Edinburgh: CV Mosby; 2005:105-115.
Sutton LN. LMM. Neurosurg Clin North Am. 1995;6:325-338.
Yamada S. Tethered spinal cord: pathophysiology and management. In: Park T, editor. Spinal Dysraphism. Boston: Blackwell Scientific; 1992:74-92.
1 Johnson A. Fatty tumor from the sacrum of a child, connected with the spinal membranes. Trans Pathol Soc London. 1857;8:16-18.
2 Rogers HM, Long DM, Chou SN, et al. Lipomas of the spinal cord and cauda equine. J Neurosurg. 1971;34:349-354.
3 Kanev P, Bierbrauer K. Reflections on the natural history of lipomyelomeningocele. Pediatr Neurosurg. 1995;22:137-140.
4 Schut L, Bruce DA, Sutton LN. The management of the child with a lipomyelomeningocele. Clin Neurosurg. 1983;30:446-476.
5 Hoffman H, Taecholarn C, Hendrick E, et al. Lipomyelomeningoceles and their management. Concept Pediatr Neurosurg. 1985;5:107-117.
6 Blount JP, Elton S. Spinal lipomas. Neurosurg Focus. 2001;10(1):e3.
7 Chapman PH. Congenital intraspinal lipomas. Anatomic considerations and surgical treatment. Childs Brain. 1982;9:37-47.
8 Walsh JW, Osterdock RJ. Lipomyelomeningocele. In: Winn HR, editor. Youmans Neurological Surgery. 5th ed. Philadelphia: Elsevier; 2004:3229-3244.
9 McClone DG, Mutluer S, Naidlich T. Lipomyelomeningoceles of the conus medullaris. In: Concepts in Pediatric Neurosurgery. Basel: S Karger; 1983:170-177.
10 Chapman PH, Beyerl B. The tethered spinal cord, with particular reference to spinal lipoma and diastematomyelia. In: Hoffman HJ, Epstein F, editors. Disorders of the Developing Nervous System: Diagnosis and Treatment. Boston: Blackwell Scientific; 1986:109-131.
11 Pang D. Tethered cord syndrome. In: Wilkins RH, Rengachary SS, editors. Neurosurgery. New York: McGraw-Hill; 1996:3465-3496.
12 Lemire R, Loeser J, Leech R. Normal and Abnormal Development of the Human Nervous System. Hagerstown, MD: Harper & Row; 1975.
13 Muller F, O’Rahilly R. The development of the human brain, the closure of the caudal neuropore, and the beginning of secondary neurulation at stage 12. Anat Embryol. 1987;176:413-430.
14 Ciappetta P, D’Urso PI, Luzzi S, et al. Cystic dilation of the ventriculus terminalis in adults. J Neurosurg. 2008;8:92-99.
15 Sutton LN. LMM. Neurosurg Clin North Am. 1995;6:325-338.
16 Schoenwolf GC. Histological and ultrastructural studies of secondary neurulation in mouse embryos. Am J Anat. 1984;169:361-376.
17 French BN. The embryology of spinal dysraphism. Clin Neurosurg. 1983;30:295-365.
18 McClone DG, Naidich TP. Terminal myelocystocele. Neurosurgery. 1985;16:36-43.
19 Naidich TP, McClone DG. Spinal dysraphism. In: Newton TH, Potts DG, editors. Modern Neuroradiology, Computed Tomography of the Spine and Spinal Cord, vol 1. San Francisco: Clavadel Press; 1983:299-315.
20 Hoffman H, Taecholarn C, Hendrick E, et al. Management of lipomyelomeningoceles. Experience at the Hospital for Sick Children, Toronto. J Neurosurg. 1985;61:1-8.
21 Iskandar BJ, Fulmer BB, Hadley MN, et al. Congenital tethered spinal cord syndrome in adults. Neurosurg Focus. 2001;10(1):e7.
22 Kanev PM, Lemire RJ, Loeser JD, et al. Management and long-term follow-up review of children with lipomyelomeningocele, 1952-1987. J Neurosurg. 1990;73:48-52.
23 Naidich TP, McClone DG, Mutluer S. A new understanding of dorsal dysraphism with lipoma (lipomyeloschisis): radiologic evaluation and surgical correction. AJR Am J Roentgenol. 1984;140:1065-1078.
24 Friedrich WN, Shurtleff DB, Shaffer J. Cognitive abilities and lipomyelomeningocele. Psychol Rep. 1993;73:467-470.
25 Oakes WJ. Management of spinal cord lipomas and lipomyelomeningoceles. In: Wilkins RH, Rengachary SS, editors. Neurosurgery Update II, vol 3. New York: McGraw-Hill; 1991:3497-3504.
26 James CCM, Lassman LP. Spina Bifida Occulta: Orthopedic, Radiological and Neurosurgical Aspects. London: Academic Press; 1981.
27 Yamada S. Tethered spinal cord: pathophysiology and management. In: Park T, editor. Spinal Dysraphism. Boston: Blackwell Scientific; 1992:74-92.
28 Seeds J, Powers S. Early prenatal diagnosis of familial lipomyelomeningocele. Obstet Gynecol. 1988;72:469-471.
29 Seeds JW, Jones FD. Lipomyelomeningocele: prenatal diagnosis and management. Obstet Gynecol. 1986;67:34S-37S.
30 Bennett MJ, Johnson RD, Blau K, et al. Some problems of alpha-fetoprotein screening. Lancet. 1978;2:1296-1297.
31 Boulot P, Ferran JL, Charlier C, et al. Prenatal diagnosis of diastematomyelia. Pediatr Radiol. 1993;23:67-68.
32 Burton BK. Alpha-fetoprotein screening. Adv Pediatr. 1986;33:181-183.
33 Naidich TP, Fernbach SK, McClone DG, et al. (John Caffey Award) Sonography of the caudal spine and back: congenital anomalies in children. AJNR Am J Neuroradiol. 1984;5:221-234.
34 Naidich TP, McCline DG. Ultrasonography versus computer tomography. In: Holtzman R, Stein B, editors. The Tethered Spine Cord. New York: Thieme-Stratton; 1985:47-58.
35 Iskandar BJ, Oakes WJ. Anomalies of the spine and spinal cord. In: McClone DG, editor. Pediatric Neurosurgery. Philadelphia: WB Saunders; 2001:307-324.
36 Hedlund GL. The dysraphic pediatric spine. Radiologist. 1996;3:265-277.
37 Gupta RK, Sharma A, Jena A, et al. Magnetic resonance evaluation of spinal dysraphism in children. Childs Nerv Syst. 1990;6:161-165.
38 Brophy J, Sutton L, Zimmerman R, et al. Magnetic resonance imaging of lipomyelomeningocele surgery. Neurosurgery. 1989;25:336-340.
39 Cecchini A, Locatelli D, Bonfanti N, et al. Lipomyelomeningoceles: a neuroradiological approach. J Neuroradiol. 1988;15:49-61.
40 Dias MS, Pang D. Split cord malformations. Neurosurg Clin North Am. 1996;6:339-358.
41 Fidas A, MacDonald HL, Elton RA, et al. Neurophysiologic measurements in patients with genuine stress incontinence of urine and the relation of neurogenic defects to the presence of spina bifida occulta. Br J Urol. 1988;62:46-50.
42 Khoury E, Hendrick EB, McLorie GA, et al. Occult spinal dysraphism: clinical and urodynamic outcome after division of the filum terminale. J Urol. 1990;144:426-428.
43 Lassman LP, James CCM. Meningocele manqué. Childs Brain. 1977;3:1-11.
44 Keating MA, Rink RC, Bauer SB, et al. Neurourological implications of the changing approach in management of occult spinal lesions. J Urol. 1988;140:1299-1301.
45 Pierre-Kahn A, Lacombe J, Pichon J. Intraspinal lipomas with spina bifida: prognosis and treatment in 73 cases. J Neurosurg. 1986;65:756-761.
46 Sutton LN. Spinal dysraphism. In: Rengachary SS, Ellenbogen RG, editors. Principles of Neurosurgery. 2nd ed. Edinburgh: CV Mosby; 2005:105-115.
47 Kulkarni AV, Pierre-Kahn A, Zerah M. Conservative management of asymptomatic spinal lipomas of the conus. Neurosurgery. 2004;54:868-873.
48 Klein O, Thompson D. Spontaneous regression of lipomyelomeningocele associated with terminal syringomyelia in a child. J Neurosurg. 2007;107:244-247.
49 Lassman LP, James CCM. Lumbosacral lipomas: critical surgery of 26 cases submitted to laminectomy. J Neurol Neurosurg Psychiatry. 1967;30:174-181.
50 Bruce D, Schut L. Spinal lipomas in infancy and childhood. Childs Brain. 1979;5:192-203.
51 Dubowitz V, Lorber J, Zachary RB. lipoma of the cauda equina. Arch Dis Child. 1969;40:207-213.
52 Oakes WJ. Tethered spinal cord, intramedullary spinal lipoma, and lipomyelomeningocele. In: Rengachary SS, Wilkins RH, editors. Neurosurgical Operative Atlas. Chicago: American Association of Neurological Surgeons; 1992:133-141.
53 McClone DG. LMM repair. In: McClone DG, editor. Pediatric Neurosurgery. Philadelphia: WB Saunders; 2001:302-306.
54 McClone DG. Laser resection of fifty spinal lipomas. Neurosurgery. 1986;18:611-615.
55 Finn MA, Walker ML. Spinal lipomas: clinical spectrum, embryology, and treatment. Neurosurg Focus. 2007;23(2):1-12.
56 Kothbauer K, Schmid UD, Seiler RW, et al. Intraoperative motor and sensory monitoring of the cauda equine. Neurosurgery. 1994;34:702-707. discussion 707
57 Paradiso G, Lee GY, Sargeant R, et al. Multimodality intraoperative neurophysiologic monitoring findings during surgery for adult tethered cord syndrome: analysis of a series of 44 patients with long term follow-up. Spine. 2006;31:2095-2102.
58 Pang D. Intraoperative neurophysiological monitoring of the lower sacral nerve roots and spinal cord. In: Yamada S, editor. Tethered Cord Syndrome. Park Ridge, IL: American Association of Neurological Surgeons; 1993:135-147.
59 Sutton L, Duhaime AC, Schut L. LMM. In: Park T, editor. Spinal Dysraphism. Boston: Blackwell Scientific; 1992:59-73.
60 Anderson F. Occult spinal dysraphism. A series of 73 cases. Pediatrics. 1975;55:826-834.
61 Atala A, Bauer S, Dyro F, et al. Bladder functional changes resulting from lipomyelomeningocele repair. J Urol. 1992;148:592-594.
62 Foster L, Kogan B, Cogen P, et al. Bladder function in patients with lipomyelomeningocele. J Urol. 1990;143:984-986.
63 Sakamoto H, Hakuba A, Ujitani K, et al. Surgical treatment of the retethered spinal cord after repair of LMM. J Neurosurg. 1991;74:709-714.
64 Colak A, Pollack I, Albright A. Recurrent tethering: a common long-term problem after lipomyelomeningocele repair. Pediatr Neurosurg. 1998;29:184-190.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 215 Lipomyelomeningocele
Johnson first described a lipomyelomeningocele (LMM) in 1857,1 although it was not until 1971 that Rogers and colleagues first introduced the term lipomyelomeningocele in their series of spinal lipomas.2 Since its original description, several authors have subsequently reported on their experience with this very common form of occult spinal dysraphism.3,4 LMM occurs in approximately 1 in every 4000 births in the United States, with a female-to-male prevalence ratio of 1.5 : 1.5 The classification, embryology, diagnosis, and surgical management of LMM are outlined in this chapter.
Definition and Classification
The literature regarding LMM is very confusing. The term lipomyelomeningocele has mistakenly been grouped into the spectrum of all lipomas of the lumbosacral spine and is often misleading because it implies herniation of neural elements through a spina bifida defect into a meningeal sac. Unlike myelomeningoceles or meningoceles, however, there is neither neural nor meningeal tissue found outside the spinal canal. The neural elements remain within the spinal canal, and there is typically an associated subcutaneous lipomatous mass that protrudes through a midline bony defect, displaces the dura, and infiltrates and tethers the spinal cord.5,6 In 1982, Chapman classified three anatomic variants of LMM according to the relationship of the lipoma–spinal cord interface: dorsal, caudal, and transitional.7
Lipomas that insert onto the dorsal surface of the conus medullaris are classified as dorsal-type LMMs (Fig. 215-1). In this variant there is typically a substantial subcutaneous fat component attached to the underlying spinal cord via a fibrolipomatous stalk of varying thickness. The posterior half of the lower spinal cord at the site of fusion of the neural fold is unfused (i.e., partial dorsal myeloschisis is present), and the stalk attaches directly to the exposed alar and basal cord regions, just posterior and medial to the posterior columns and dorsal to the central canal.8 The lipoma can be located either in the midline (68%) or eccentrically (32%) and can extend into the central canal and expand it for variable distances.9 Along the lateral interface of the attachment of the lipoma to the spinal cord, the dura and pia are fused. Sensory nerve roots emerge just anterior to this lateral line of fusion, which can be traced circumferentially. There is often no evidence of leptomeninges posteriorly, and no sensory or motor roots are found within the actual substance of the lipoma.
In caudal- or terminal-type LMM, the lipoma–spinal cord interface is located at the caudal end of the conus, almost like a continuation of the spinal cord itself (Fig. 215-2). The remaining lipomatous mass may then lie entirely within the spinal canal or extend dorsally through a defect in the dura and bone into the subcutaneous space. The fatty tumor may either replace or surround the filum terminale, or a separate filum may lie anteriorly. The nerve roots may lie ventral to the lipoma or pass through the mass itself. A caudal-type LMM is often asymmetrical and involves more of the cord and nerve roots on one side, and unlike the discrete lipoma-cord interface of a dorsal-type LMM, the lipoma-cord interface in a caudal-type LMM is diffuse.8
In addition to the dorsal and caudal LMM forms originally described by Chapman, transitional forms between the two variants may exist (Fig. 215-3). Frequently in these cases, the more cephalad portion is of the dorsal type with posterior rootlets emerging from the cord just ventral to the line of fusion of the lipoma to the cord and leptomeninges. Caudally, however, as the line of fusion is displaced ventrally, nerve roots emerge from the anterolateral portion of the lipoma mass. Magnetic resonance imaging (MRI) can often be helpful in defining the type and may be useful in determining the operative approach. The transitional form is considered by some to simply represent a continuum between the other two forms.7,8,10,11
Embryology
Congenital lumbosacral lipomas develop as a result of disturbances in the precise sequence of events encompassing the embryologic stages of primary and secondary neurulation, which occur between postovulatory day (POD) 25 and 48.8 Closure of the posterior neuropore marks the completion of the first phase (primary neurulation) of neural tube formation, at which point the fetal spine is covered by ectoderm but the lumbar, sacral, and coccygeal segments have not yet developed.12 The caudal end of the neural tube and the notochord combine into a large aggregate of undifferentiated cells in a process called condensation. This caudal cell mass extends to the level of the tail fold and represents a solid neural cord containing neurons, neural crest, glial cells, and ependymal cells. In a process called canalization, a series of small vacuoles within the mass begin to coalesce and enlarge to create an ependymal-lined tube in continuity with the rostral central canal of the previously formed neural tube.13 This process takes place between POD 27 and 29 and leads to formation of the ventriculus terminalis.14,15
The third phase in formation of the caudal neural tube involves regression of the structures previously derived during canalization. During retrogressive differentiation, the embryologic tail disappears, whereas the filum terminale, coccygeal ligament, and ventriculus terminalis of the conus remain. This process is thought to complete by POD 48 to 52.12,13,15–17 Because cell rests within the caudal cell mass have totipotent characteristics, a small clone of lipomatous cells developing in the region would not necessarily inhibit the growth of surrounding structures. The bony vertebrae develop subsequently, and the sacrococcygeal segments also undergo regressive changes to decrease the number of segments originally present.15 As a result, vertebral maldevelopments are found in conjunction with the neural defects arising during this period.
McClone and Naidich proposed that the process of dysjunction, in which the closed neural tube detaches from the cutaneous ectoderm, occurs prematurely in patients with LMM. Consequently, a dorsal cleft is maintained and paraxial mesenchyme tissue has access to the prospective lumen of the neural tube, thereby further preventing its closure. The luminal surface of the ectoderm induces mesenchymal cells to differentiate along a path that results in adipocyte formation, whereas the outer surface of the incompletely closed neural tube causes the mesenchyme to form the piaarachnoid and dura.18,19 Other tissues have been found in the substance of an LMM, including striated muscle, cartilage, nerve cells, ependyma, and even cerebellum.6 Similar to other spinal dysraphisms, a lumbosacral lipoma that tethers the spinal cord at the upper lumbar level probably occurs at a slightly earlier embryologic period than does a lesion that tethers the cord lower down in the spine.8 Several authors have found that patients with LMM can have other associated dysraphic conditions of the spinal cord, including split cord malformation, terminal syringomyelia, neurenteric and intraspinal dermoid/epidermoid cysts, a tight filum terminale, and tethering tracts.20,21
Clinical Findings
Cutaneous Signs
Although patients with LMM can have a spectrum of cutaneous anomalies, infants are seen most often with a cutaneous sign that suggests a potential underlying abnormality. The most common finding (>90% of patients) is a subcutaneous fatty mass covered with skin that is located in or near the midline of the lumbosacral spine (Fig. 215-4). This mass is not tender and appears to be in continuity with the normal subcutaneous tissues. Although the overlying skin is typically intact, LMMs can also be associated with a hairy patch (hypertrichosis), hemangioma, dermal sinus, skin dimple, skin tag, atretic meningocele, or caudal appendage (Table 215-1).20,22 The most superficial portion of the mass typically consists of lobulated and unencapsulated subcutaneous fat, which becomes more fibrous as the deeper structures are approached. The stalk of the fibrous fatty tissue can then be found to penetrate the dura and is associated with the spinal cord, conus, cauda equina, or filum terminale, as described earlier. Additional malformations may also be present, including cloacal exstrophy, bifid uterus, duplicated vagina, anterior sacral meningocele, anomalies of the lower limbs, and imperforate anus.15 It is rare for patients with LMM to have intellectual impairment or associated hydrocephalus, Chiari malformations, or other brain anomalies.15,23,24
TABLE 215-1 Lumbosacral Cutaneous Manifestations Found in Patients with Lipomyelomeningocele
| SERIES | HOFFMAN ET AL.,20 1985 (N = 97) | KANEV ET AL.,22 1990 (N = 80) |
|---|---|---|
| Soft tissue mass | 97 | 80 |
| Skin dimple | 26 | 14 |
| Hemangioma | 24 | 9 |
| Hypertrichosis | 1 | 11 |
| Skin tag/tail-like appendage | 5 | 6 |
| Atretic or denuded skin patch | 1 | |
| Dermal sinus hypopigmentation | 3 | 3 |
Symptoms
LMMs typically cause tethering of the spinal cord via the associated fibrous subcutaneous lipoma. As infants age, symptoms reminiscent of tethered cord syndrome typically develop, along with any combination of neurologic, orthopedic, and urologic issues. Slightly more than one third of patients will demonstrate an asymmetrical subcutaneous mass with regard to the midline; these patients have a higher incidence of neurological deficits in the lower limb ipsilateral to the side of the mass.25 Symptoms depend on the level of the cord-lipoma interface, as well as patient age at the time of diagnosis, and may be related to several possible causes working independently or in combination.15 Deficits present at birth may become manifested as a result of abnormal development of the spinal cord and nerve roots. The presence of a mass within the confines of the bony spinal canal may also contribute to patient symptoms in more ways than one. Namely, progressive mechanical distortion or ischemia of the neural elements may result from tethering of the neural elements or, alternatively, from a local mass effect with compression of the cord, conus, or cauda equina. Similar to other fatty growths, these tumors can enlarge with age in proportion to normal body growth.26
Although an unknown number of patients may initially be seen with asymptomatic LMMs, it is crucial for clinicians to understand that progressive neurological impairment can develop in patients who at first seem normal. In infants and toddlers, accurate assessment of clinical involvement is very difficult. Yamada demonstrated that spinal cord tethering interferes with normal energy metabolism of the spinal cord, which can lead to ischemia and progressive neural damage.27 In addition, although most patients demonstrate slow loss of function over a period of years, sudden and acute loss of function has also been described. For example, this has been reported in previously asymptomatic women placed in the lithotomy position during pregnancy or in patients with hyperflexion injury from a motor vehicle accident.
Muscle weakness and gait disturbance are often present by the time that the patient starts ambulating. Clinical examination may reveal muscular atrophy, leg length discrepancy, varus or valgus foot deformity of one or both legs, hammertoes or claw toes, scoliosis, mixed deep tendon reflexes (absent, normal, increased), mixed/patchy sensory abnormalities, back pain, or any combination thereof. Abnormal voiding patterns with urinary or fecal incontinence or the presence of urinary tract infections may also be an initial symptom. Although patients can exhibit any of the aforementioned signs and symptoms, infants typically have cutaneous anomalies; older children have orthopedic, neurologic, or urologic symptoms; and adults often have pain.15
Evaluation
Prenatal Diagnosis
LMMs are increasingly being diagnosed in utero as a result of the frequent use of screening antenatal ultrasonography (US).28 Accurate prenatal diagnosis allows appropriate planning for delivery and neonatal care.29 However, because these defects are closed with no communication between fetal spinal fluid and maternal amniotic fluid, amniotic fluid analysis for α-fetoprotein levels is not a reliable diagnostic measure.30–32
Ultrasonography
US has the ability to differentiate among fat, spinal fluid, and spinal cord as a result of their different echogenic properties. Because infants younger than 6 months are skeletally immature with poorly calcified bone, US can image through the spine in these individuals, which can be a very useful tool for defining the attachment of lipomas to the spinal cord.33,34 Several benefits of US include its relative inexpensiveness, ease of use, and lack of need for patient sedation. It is also beneficial in determining spinal cord motion for possible evidence of spinal cord tethering. US is unlikely ever to replace other modalities such as MRI, however, because it is limited in its ability to provide the appropriate preoperative information. In addition, US can be operator dependent and possibly miss subtle lesions unless the user is well experienced.
Plain Radiographs
Plain radiographs can play a beneficial role in the initial assessment of patients suspected of having closed spinal dysraphism; however, as an initial evaluation in most pediatric neurosurgery centers, they are largely being replaced by MRI.35 Anteroposterior and lateral radiographs at the appropriate spinal levels almost always reveal abnormalities such as a dorsal fusion defect in the lamina (bony spina bifida) or widening of the spinal canal.8,36 Varying degrees of agenesis or deformity of the sacrum have also been reported in the literature. Some surgeons prefer to have plain radiographs available for surgical planning. Absence or incomplete calcification of bony elements limits the utility of plain radiographs in children younger than 18 months.37
[/not-level-membership-for-neurosurgery-category]
