CHAPTER 135 Craniopharyngioma
“Craniopharyngioma” was the name introduced by Cushing for tumors derived “from epithelial rests ascribable to an imperfect closure of the hypophysial or craniopharyngeal duct.”1 It remains a rare but challenging tumor given that its location, difficulties associated with treatment, and a high recurrence rate can lead to significant morbidity in patients whom it affects. The treatment and general therapeutic approach to craniopharyngioma have undergone several transformations, even within the modern neurosurgical era, with proponents of both radicalism and conservatism. Significant advances in preoperative imaging, surgical techniques, and adjuvant therapies have enabled neurosurgeons and neuro-oncologists to improve the quality of care that these patients receive.
Historical Review
One of the earliest descriptions of craniopharyngioma is credited to Zenker, who recognized a suprasellar lesion containing cholesterol crystals in an 1857 autopsy study.2 In 1892, Onanoff coined the term pituitary adamantinoma after appreciating the similarities between tumors of the jaw and tumors of the pituitary region.3 In 1899, the pathologists Mott and Barrett began to investigate a group of epithelial-type tumors that occupied the sellar region. They postulated that these tumors arose from either Rathke’s pouch or the hypophysial duct.4 In the next few years, these tumors were reported by both Babinski and Frohlich as suprasellar lesions without acromegaly.5,6 In 1909, Halsteadt was credited with the first transsphenoidal removal of a craniopharyngioma from a patient with symptoms of a sellar mass.7
These tumors were initially named craniopharyngeal pouch tumors in 1921 by McKenzie and Sosman.8 Other terms such as ameloblastoma, epidermoid tumor, and craniopharyngeal fat tumor were also applied before Frazier, Alpers, and Cushing began to use the term craniopharyngioma.1,9 This term is now well entrenched in the neurosurgical literature, although embryologically these tumors are remnants of the primitive stomodeum and not the pharynx.10
The surgical philosophy regarding the treatment of craniopharyngioma has also vacillated significantly over the past 5 decades. Early operative series demonstrated an extremely high mortality rate of 40%, with only 15% of patients undergoing total removal.11 By the early 1960s, many believed that aggressive surgery should be abandoned in favor of cytoreduction combined with radiotherapy.12 In the mid-1970s, with improvements in both postoperative endocrinologic care and overall surgical technique, there was renewed support for an aggressive surgical approach.13–15 Even today, controversy exists between those who advocate aggressive surgical resection and those who support a more conservative approach.
Craniopharyngioma is thought to arise from ectodermally derived epithelial cell remnants of Rathke’s pouch and the craniopharyngeal duct. Neoplastic transformation of cells derived from tooth primordia gives rise to adamantinomatous craniopharyngioma, whereas such transformation in cells derived from buccal mucosa primordia gives rise to the papillary type.16 By the fourth week of gestation, invagination of the stomodeum, lined by epithelial cells, takes place. This upward migration is met by a downward movement of neuroepithelium from the hypothalamus. This upward invagination, termed Rathke’s pouch, is responsible for development of the adenohypophysis, whereas the downward growth of neuroepithelium is the precursor of the future neurohypophysis.
As Rathke’s pouch meets the infundibulum, it separates from the stomodeum and rotates around the anterolateral surface of the infundibulum. This rotation, which occurs during formation of the adenohypophysis, is responsible for delivering embryonic rests to suprasellar or parasellar locations. This migration pathway from the primitive oral cavity is termed the craniopharyngeal duct. In 1904, Erdheim reported that the origin of craniopharyngiomas was based on incomplete involution of this pathway.17
Recent evidence supports the hypothesis that embryonic rests of cells from the craniopharyngeal duct produce the pituitary gland, Rathke’s pouch, and craniopharyngiomas.18 Both human chorionic gonadotropin and P-glycoprotein have been demonstrated to be produced by all these structures.18,19
Challenges to the craniopharyngeal duct hypothesis have centered on evidence that children younger than 10 years rarely have any squamous cell rests around the pituitary. It has been demonstrated that only 3% of neonates harbor these cell rests; however, they are found more often in older individuals.20,21
The second major hypothesis proposed for the pathogenesis of craniopharyngioma centers on the premise that existing cell rests in the adenohypophysis undergo metaplasia. This concept partially accounts for the observation that squamous papillary tumors occur predominantly in adults and lack any resemblance to tooth-forming epithelium. Arguments against this hypothesis are based on evidence that mixed tumors demonstrating both adamantinomatous and papillary squamous characteristics exist.22,23
Incidence
Craniopharyngiomas are relatively uncommon tumors, with the annual incidence rate being between 0.5 and 2.5 new cases per million population per year.24 Craniopharyngioma has a higher annual incidence of 5.25 cases per million in the pediatric population,25 in whom it accounts for 6% to 13% of intracranial tumors. In 60% of patients, craniopharyngioma is diagnosed after the age of 16.26–29
Age
Craniopharyngioma has two histopathologic subtypes, with significant implications regarding age at diagnosis. Adamantinomatous craniopharyngioma is observed to have a bimodal distribution, with one peak in children between 5 and 15 years old and a second peak in adults 45 to 60 years old. The papillary subtype occurs almost exclusively in adults at a mean age of 40 to 55.24,30,31
Sex
Most large series do not show any consistent sex predilection for craniopharyngiomas. Some studies have shown a greater preponderance in males in childhood and in females in adulthood,24,32 and a large series in England suggested that males are affected 30% more often than females.33
Location
Craniopharyngiomas may arise anywhere along the craniopharyngeal duct, but they most commonly arise in the sellar/parasellar region. Four percent are purely intrasellar, 21% are sellar and suprasellar, and 75% are suprasellar alone, often with extension up into the third ventricle.34–36 In very rare cases, craniopharyngiomas may develop in an ectopic fashion, with reports of sites such as the sphenoid sinus,37 nasopharynx,38 clivus,39 and extradural temporal region.40
Geography
Geographic variation may also exist, with various reports citing craniopharyngiomas as accounting for 1.5% to 6.5% of all primary brain tumors. The low incidence of 1.5% in Australia contrasts with the 6.5% rate in China and a possibly higher incidence in Africa.41–43 An American study examining three different databases calculated similar incidence rates of 0.13 per 100,000 person-years.24 Other groups have published comparable incidence rates of 0.5 to 2.0 new cases per year per million population.32,44
Given the predominant location of these tumors in the sellar/parasellar region, craniopharyngioma is characteristically manifested as three clinical syndromes: visual dysfunction, disturbance of the hypothalamic-pituitary axis, and raised intracranial pressure as a result of obstruction of flow of cerebrospinal fluid (CSF) and hydrocephalus.26,45–47 The median duration of symptoms in a series of 34 adult patients was 10 months. The pediatric cohort of patients in this mixed series had a median symptom duration of just 3 months.48 Adults are more likely than children to have symptoms of visual or endocrine abnormalities, whereas children more often have symptoms of increased intracranial pressure. In adults, headache, vomiting, and visual disturbance (hemianopia, uniocular visual loss, and diplopia) are the most common initial complaints.49 More than 80% of adult patients complain of some visual loss at initial evaluation and have evidence of a visual deficit on formal testing. In children, the most common initial symptom is headache, which occurs in 50% to 80%,26,50 followed by vomiting (21% to 68%) and visual deterioration (47% to 80%).
The most common neurological signs relate to visual disturbance: visual field defects (35% to 79%), papilledema (10% to 50%), optic atrophy, and eye movement disorders. In adult patients, 29% have evidence of papilledema at initial encounter, compared with more than 50% in the pediatric population.48 In very young children, increased head circumference or a bulging fontanelle may be seen. Ataxia is described in about 20% of instances.
Approximately 30% of adults will initially have symptoms of endocrine disturbance.51 Gonadal insufficiency is the most common endocrine abnormality at diagnosis and consists of loss of libido and reduced masculine hair growth pattern in men. Women may complain of irregular menstrual periods or even amenorrhea. Other endocrinologic problems at initial evaluation include diabetes insipidus, hyperprolactinemia, adrenal insufficiency, and thyroid insufficiency.
Less than 15% of children with craniopharyngioma have complaints attributable to an endocrinologic deficit,52 even though almost 90% have some endocrine abnormality. Delayed puberty (4% to 24% of patients), obesity (8% to 15%), anorexia, short stature, and precocious puberty are all potential manifestations of craniopharyngioma.23,26,53 Growth hormone deficiency is the most frequently observed deficit, with more than 75% of patients being affected. Specific hormonal deficiencies are identified, with luteinizing hormone/follicle-stimulating hormone (40% of patients) more commonly affected than adrenocorticotropic hormone (25%) and thyroid-stimulating hormone (25%).54 Diabetes insipidus is reported in up to 17% of children and 30% of adults.
Hydrocephalus is an important concomitant factor, particularly in pediatric craniopharyngioma. Hydrocephalus is identified at diagnosis in approximately a third of craniopharyngioma patients overall and in almost one third of children with craniopharyngioma36,54–56 and may require definitive treatment if primary tumor surgery fails to resolve the issue. In one series, 43% of children went on to require long-term treatment of hydrocephalus.56 In another series, only 29% of adult patients had any evidence of hydrocephalus at initial evaluation, although there was a greater than 50% incidence of hydrocephalus in the pediatric population.48 Adequate treatment of hydrocephalus is imperative to minimize long-term cognitive deficits in these children, especially in those undergoing radiation therapy. There would seem to be a correlation between shunt requirement and worse outcome; however, it remains to be clarified whether this is directly related to shunting or is due to the presence of generally larger tumors with hypothalamic involvement in this group.54
Neurobehavioral abnormalities appear to be more common in adults than in children. It has been estimated that more than 30% of adult patients with craniopharyngioma older than 45 years have dementia or suffer from intermittent confusion, hypersomnia, apathy, or depression.57,58 Children may also suffer from neurocognitive decline and exhibit common clinical features such as abulia, psychomotor retardation, and flattening of affect.
Very rarely, craniopharyngioma may develop acutely after intratumoral hemorrhage59 and rarely may rupture and result in aseptic meningitis or spontaneous drainage through the nasopharynx.60–62
There is considerable difficulty in diagnosing craniopharyngioma in young children because some may have relatively nonspecific symptoms such as vomiting and irritability, and visual field loss is often overlooked. A high degree of clinical suspicion is needed in such cases. It has been recognized that the diagnosis of brain tumors in children is frequently delayed in comparison to other childhood tumors.63–65
In 2007, the World Health Organization (WHO) grading system for craniopharyngioma defined two major subtypes of the tumor, adamantinomatous and papillary, with both corresponding to WHO grade I.66 Transitional or mixed forms have also been described but are rare.30,67 The classic adamantinomatous type appears to be present in more than 95% of pediatric cases.55 It is distinctly uncommon for children to display the papillary squamous subtype in isolation or mixed with adamantinomatous histology. The papillary squamous variant is almost exclusively seen in adults and represents close to 30% of all craniopharyngiomas seen in this population.30,55
Histologically, adamantinomatous craniopharyngioma is composed of squamous epithelium in cords, lobules, and trabeculae surrounded by palisaded columnar epithelium.68 “Wet keratin” nodules may be found within the solid portion of the tumor, and a gliotic reaction with abundant Rosenthal fibers is often seen in adjacent brain tissue (Fig. 135-1).
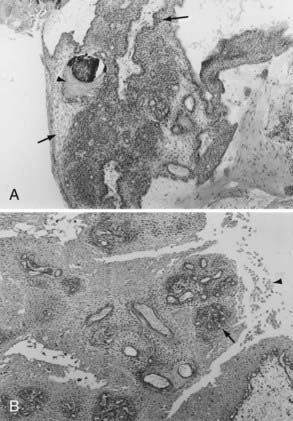
The typical papillary subtype has a well-circumscribed solid appearance with a lower likelihood of cysts and absence of calcification and cholesterol deposits. Histologically, the papillary subtype is a bland mass of well-differentiated squamous epithelium forming pseudopapillae with an anastomosing fibrovascular stroma.30,31,55
Electron microscopy is rarely needed for the diagnosis of craniopharyngioma but typically reveals glycogen and tonofilaments in epithelial cells, which are joined by desmosomes.69 Mineral precipitates are seen in membrane-bound vesicles. Immunohistochemistry is positive for cytokeratins70 and epithelial membrane antigen.68
Malignant transformation of craniopharyngioma has been discussed in the literature but is thought to be very rare and may require additional oncogenic insults, such as exposure to radiation.71–73 The molecular genetic abnormalities associated with craniopharyngioma remain poorly characterized, although there are several reports of clustering of craniopharyngioma in families suggestive of a putative genetic basis.74–76 Some craniopharyngiomas are monoclonal in origin,77,78 and cytogenetic abnormalities have been reported in chromosomes 2 and 12,77,79,80 but additional studies have not shown significant chromosomal imbalances in either type of craniopharyngioma.81,82
Evidence is emerging that implicates specific genes and cell signaling pathways in the development of craniopharyngioma. Mutations of the β-catenin gene have been identified in 70% of adamantinomatous craniopharyngiomas,83,84 whereas none were seen in papillary craniopharyngiomas. β-Catenin is a downstream component of the Wnt intracellular signaling pathway involved in proliferation, morphogenesis, and differentiation,83 and it has been postulated that disruption of this pathway may be important in the pathogenesis of adamantinomatous craniopharyngioma.
Information regarding the natural history of craniopharyngioma is sparse because most patients eventually undergo treatment. In an Oxford (U.K.) series, 13 patients did not initially receive treatment, and 92% of them ultimately required treatment for evidence of disease progression during the study period.36
Plain radiography of the skull is now rarely performed in patients suspected of having a craniopharyngioma; however, radiographs may provide information regarding the size and shape of the sella and may demonstrate lesional calcification. Approximately 65% of adults and 90% of children with craniopharyngiomas will have abnormal findings on skull radiographs. Calcification of the tumor is seen in approximately 40% of adults and 85% of children.85 Calcification is not a unique feature of adamantinomatous lesions inasmuch as it has been demonstrated in histologically confirmed papillary tumors.30,55
Today, MRI is more commonly used to fully characterize a craniopharyngioma. Classically, the solid tumor is isointense to hypointense on T1-weighted images, and T2-weighted images show mixed hypointensity or hyperintensity; reticular enhancement is seen after the injection of gadolinium.86 Edema may be identified in adjacent brain tissue or along the optic tract and is useful in differentiating craniopharyngioma from other tumors in this region.87
In a series of 86 adult patients with craniopharyngiomas at the University of Erlangen, 80% of the tumors were heterogeneous in appearance.51 Most of the tumors were either monocystic with a solid component or predominantly solid with a cystic component. A minority were entirely cystic or entirely solid. All these tumors were suprasellar, with 57% of them harboring an intrasellar component. In addition, 60% of the suprasellar tumors had a component that was retrosellar, and 40% had a component that was in either the posterior fossa or the parasellar space.
There are no radiologic features that can absolutely discriminate among the subtypes of craniopharyngioma. Yet lobulated shape, vessel encasement, and calcification have all been postulated to be indicative of the adamantinomatous subtype.30,67,88
Classification and Grading Schemes
A number of classification schemes have been proposed to assist in decision making regarding surgery, particularly concerning the approach and its concomitant risks.54,89–91 Hoffman and colleagues proposed intrasellar, prechiasmatic, and retrochiasmatic as three basic subtypes,15 whereas Samii and Bini devised a five-tier grading system based on vertical height of the tumor.92 Yasargil and associates recommended grading with respect to the diaphragma and the third ventricle (Fig. 135-2).54 The most recently proposed grading system, from Puget and coworkers, relates to the degree of involvement of the hypothalamus to further define resectability (Fig. 135-3).90 No one system has come into widespread use, but all have useful elements for approaching these tumors surgically, considering their position in relation to the chiasm, or taking into account their vertical height and hypothalamic involvement.
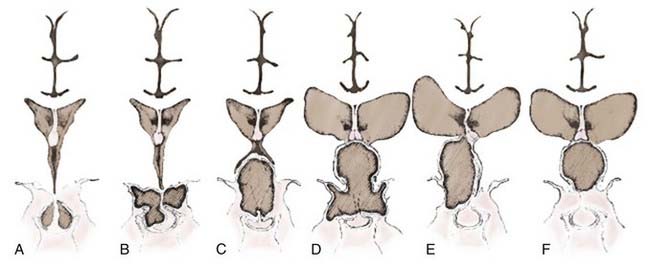
Giant craniopharyngiomas have been defined as tumors that have a maximal diameter greater than 5 cm.93 They are frequently cystic in nature and may extend into the anterior, middle, and posterior fossa.
Differential Diagnosis
Treatment
Full endocrinologic assessment is required, and appropriate replacement therapy should be provided, with particular emphasis on identifying diabetes insipidus and hypocortisolemia. An estimate of bone age and, in young women, ovarian ultrasonography are advocated.94 Likewise, accurate neuro-ophthalmologic assessment, including visual field testing, is critical to have a thorough understanding of the tumor and its relationship to the visual apparatus and as a baseline for analysis after treatment.
Traditionally, surgical resection has been the preferred first option for treatment of these lesions. Most would recommend that if complete surgical resection can be performed without injury to the anterior hypothalamus and without effect on personality and quality of life, it should be performed. The difficulty lies in achieving this goal in a high percentage of cases. An increasing number of experts advocate subtotal resection because of its lower perioperative complication rate, supplemented by adjuvant radiotherapy.56,90,95 Such management has been shown to be associated with equivalent cognitive outcomes and less neurological impairment and deterioration of quality of life.
Occasionally, one may encounter an adult patient who is asymptomatic and found to have a craniopharyngioma on imaging studies. The overall age and medical status of the patient, along with the imaging characteristics of the tumor, are important factors when considering whether treatment is necessary. If one is quite certain about the pathology and the tumor is not threatening the visual apparatus, a period of observation may be warranted. Careful endocrinologic, ophthalmologic, and imaging follow-up can then be used to assess the need for treatment. In a series of 85 predominantly adult patients seen between 1938 and 1970, Bartlett used observation as an approach in 11 patients.57 Five of these patients were monitored for more than 15 years and required no treatment.
Complete surgical resection appears to be an important prognostic factor for recurrence and is the best management method for craniopharyngioma if it can be done safely. It requires great surgical skill and judgment, but considerable effort should be exerted to perform as complete a resection as safely possible. Unfortunately, complete resection may be very difficult to achieve.96
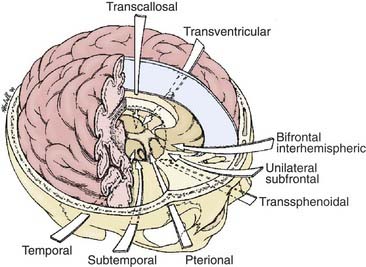
The most appropriate surgical corridor for resecting a craniopharyngioma is determined by numerous factors, including the shape and size of tumor, the presence of cysts, its location, the degree of calcification, and the surgeon’s preference and experience (Fig. 135-4). The standard approaches for attempted GTR include pterional, orbitozygomatic, subfrontal/trans–lamina terminalis, interhemispheric-transcallosal, and transcortical-transventricular; reports of endoscopic endonasal transsphenoidal removal of craniopharyngiomas are emerging as well.
Gross-Total Resection
Subfrontal/Trans–Lamina Terminalis Approach
Suitability
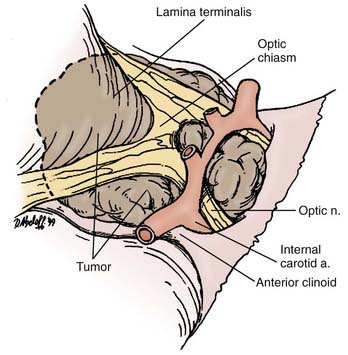
This approach is most favored in the presence of a midline prechiasmatic tumor that extends into the anterior cranial fossa floor or superiorly into the suprasellar cistern and the third ventricle.97 Its advantages include the midline orientation of the approach with access to both the optic nerves and internal carotid arteries, as well as the third ventricle via the lamina terminalis.
Operative Details
The patient is placed in a supine position with the head slightly extended such that the anterior skull base is perpendicular to the floor. Head fixation is preferred, and the use of neuronavigation equipment may be helpful, particularly for solid tumors. A lumbar drain may be considered. Mannitol and dexamethasone are administered. A bicoronal incision is made, with preservation of a vascularized pericranial graft for use in reconstruction if necessary. Once adequate bony exposure has been achieved, a low unilateral or bilateral frontal craniotomy is performed, and if present (in older children and adults), the frontal sinus is opened. In general, we prefer a unilateral craniotomy to avoid bihemispheric injury. Cranialization may need to be performed on the frontal sinus. Additional removal of the orbital bar may provide greater exposure.98 The dural opening is created behind the orbital bar, and the right olfactory nerve will usually be dissected from the undersurface of the frontal lobe or divided. Elevation of the frontal lobes from the anterior cranial fossa floor may be added by dividing the superior sagittal sinus at the floor. Exposure of the optic nerves, optic chiasm, and carotid arteries bilaterally is then afforded. Opening of the lamina terminalis immediately posterior to the chiasm then allows access to tumors that extend into the third ventricle.
Difficulties
Both frontal lobes are elevated during the bifrontal approach, which may be associated with increased postoperative morbidity. This approach is also compromised by the potential for injury or the necessity for sacrifice of one of the olfactory nerves. The frontal sinus will be opened when present (in older children and adults) and will require cranialization; there is a risk for infection or CSF leakage with this procedure. Removal of an intrasellar tumor can be difficult when using a subfrontal approach, but it is possible and may be facilitated by the use of an angled endoscope (Fig. 135-5).
Bifrontal Basal Interhemispheric Approach
Suitability
Although the subfrontal approach is favored by many neurosurgeons, the basal bifrontal interhemispheric approach has been proposed by others.99 This approach is well suited for large, midline, retrochiasmatic tumors that may have retrosellar extension. Although it may be more technically challenging, this approach allows wider visualization of the optic pathway and anterior circle of Willis. Proponents of the bifrontal basal interhemispheric approach cite its ability to avoid the potential blind spots encountered with unilateral approaches. This technique has also been combined with division of the anterior communicating artery in an effort to maximize removal of the retrochiasmatic portion of these tumors.99
Transsphenoidal and Extended Transsphenoidal Approaches
Suitability
Smaller midline tumors within the sella or with an infradiaphragmatic suprasellar component may be removed by the transsphenoidal route. This route is most favorable for patients with enlargement of the sella. Significant suprasellar extension of craniopharyngiomas can be difficult to access from a transsphenoidal approach; however, the presence of cystic rather than solid disease makes its use more favorable.100 Advantages of this approach include the lack of need for brain retraction and potentially better visual outcomes than observed when using cranial approaches.54,101,102
Operative Details
The patient is placed supine on the operating table with the head elevated and slightly angled toward the surgeon. Use of a Mayfield headrest and neuronavigation devices can obviate the need for fluoroscopy, although either or both can be used. The procedure can be performed with an endonasal endoscope103 or by using the operating microscope with a nasal speculum. Sublabial, transseptal, and direct approaches to the sphenoid face have all been described. The mucosa is reflected, and the sphenoid sinus is opened with a drill or Kerrison punch. The mucosa of the sinus is excised, and the anterior wall of the sella is removed to expose the sella dura. Anterior compression of the anterior pituitary is a common finding, and the gland may need to be divided to provide access to the tumor.104
For the extended transsphenoidal procedure, the tuberculum sellae can be removed, and additionally, excision of the planum sphenoidale will provide improved access to the suprasellar region. The dura may then be opened over the gland and the circular sinus and anterior to the sella to provide access to the suprasellar cistern. The dural defect is closed with a graft and supported by bone or synthetic plates, with fat placed in the sphenoid and a vascularized nasal flap added for coverage of the site of surgical access from the nasopharynx.105 Some authors advocate the use of postoperative lumbar drainage to reduce the risk for delayed CSF rhinorrhea.106
Difficulties
Lateral extension of the tumor can be difficult to access from this approach, as can large tumors. Tumor involvement in the region of the anterior cerebral complex may expose the perforating vessels to risk for injury during surgery. Direct control of intracranial neural and vascular structures is inferior when using the transsphenoidal or extended transsphenoidal approach (Fig. 135-6). Suprasellar calcifications are thought to be a contraindication to the use of this approach.104 This approach can be difficult or impossible to use in children with a poorly pneumatized sinus and in patients with nasal and sinus pathology. Reconstruction after tumor removal can be complicated, and high rates of CSF leakage have been reported in the literature, a problem that has not been completely resolved after extended transsphenoidal surgery. Use of a vascularized nasal mucosal flap is showing promise in this regard.105
Pterional Approach
Suitability
The pterional approach has traditionally been the most favored one for resection of craniopharyngiomas,54,90,96 and it would seem to best be suited for smaller tumors confined to the suprasellar space.98 It does allow access to both prechiasmatic and retrochiasmatic lesions, along with those above and below the diaphragma, and may be used in combination with interhemispheric-transcallosal or transcortical-transventricular approaches to remove larger lesions with significant suprasellar extension.
Interhemispheric-Transcallosal Approach
Suitability
Large midline tumors with suprasellar extension into the third and, potentially, the lateral ventricle can be removed if the interhemispheric-transcallosal approach is combined with a basal procedure. It may be satisfactorily used alone in the rare setting (3% to 10%) of a purely third ventricular craniopharygioma.54
Transcortical-Transventricular Approach
Difficulties
The transcortical-transventricular approach involves cortical injury with an increased postoperative risk for seizures. The size of the ventricle is important, and hence in the absence of significant hydrocephalus, this approach is unfavorable. Ipsilateral dissection of the wall of the third ventricle is also difficult with this approach.54
Results of Attempted Gross-Total Resection
Rates of radiologically confirmed GTR vary significantly in the microsurgical neurosurgery era. Yasargil and coauthors reported performing GTR in 90% of 144 surgical patients,54 whereas Fahlbusch and others have reported rates closer to 50%,53,90,96 and Karavitaki and associates documented 18% of 103 tumors as being completely resected.36 Some authors have identified greater GTR rates in children (77%) than in adults (27%).55 Large tumor size, supradiaphragmatic location, hydrocephalus, and recurrence are adverse prognostic factors for radical resection.13,53,55,96,107
In addition to variation in achievement of gross-total control among series, there is significant variation in reported recurrence rates after GTR. The lowest rates document 0% recurrence after GTR at 10 years,36 15% at 10 years,98 19% at 10 years,96 17% at a mean follow-up of 49 months,55 and 36% at a median follow-up of 7 years.90 Additionally, other authors have reported higher rates, including a 43% recurrence rate after GTR at a median follow-up of 78 months.108 In the Iowa series, local control rates after complete resection were reported to be 50%,109 and in a Pittsburgh study, the local control rate at 10 years after surgery alone was 42%.110
Postoperative Mortality and Morbidity
Postoperative morbidity and mortality are central issues in the management of craniopharyngioma. Postoperative mortality in some early series was reported to be as high as 41%.32,111–113 More recent series have reported mortality of 1.7% to 5.4%,15,53,96 although a mortality rate of about 15% in the first postoperative year was reported in a group of adult patients undergoing aggressive microsurgical resection.54
In general, radical excision has been associated with higher morbidity and mortality, but there are exceptions.36,96 Mortality in adults is most often the result of hypothalamic damage or coexisting medical illness. Patients who undergo transsphenoidal resection consistently have lower overall mortality rates than do those who undergo craniotomy.96,114
Visual decline after an aggressive surgical attempt occurs in 4% to 30% of patients.15,51,54,55 Important variables predicting visual outcome appear to include the extent and duration of preoperative visual deficits, the relationship of the tumor to the optic apparatus, and performance of transsphenoidal versus transcranial surgery. In Yasargil and colleagues’ series of adult patients, all who had normal vision preoperatively retained normal visual acuity postoperatively.54 In contrast, vision in 50% of patients with preoperative visual deficits either stabilized or declined postoperatively. Rates of visual decline in patients treated by subtotal resection followed by radiation therapy varied from 2% in most series to as high as 30% in some series.115,116
One of the most common initial symptoms in adults with craniopharyngioma is endocrinopathy, but relatively few studies have methodically looked at preoperative and postoperative hormonal status in the adult population. In general, most studies state that surgical intervention leads to a significant degree of endocrine dysfunction.15,54,117 The endocrine requirements of patients have been found to be greater after undergoing GTR than after subtotal resection or after subtotal resection plus radiation therapy,108 in a manner similar to results that document visual impairment and hypothalamic obesity to be greater after undergoing GTR.
Deficiencies of four or more hormones develop in more than 75% of patients who undergo aggressive tumor resection.52,118 Diabetes insipidus is seen postoperatively in about 80% of patients who undergo aggressive tumor removal, as opposed to 38% of those who undergo partial resection and 22% of those treated by radiotherapy alone.116 Other studies have claimed improvement in the patient’s endocrine status after transsphenoidal resection, as long as the pituitary stalk is left intact.119
One study examined the preoperative and postoperative endocrine status of 140 predominantly adult patients with craniopharyngiomas.120 Preoperatively, approximately 75% of patients had evidence of growth hormone deficiency and secondary hypogonadism. Hyperprolactinemia, secondary adrenal failure, and hypothyroidism were found in 41%, 32%, and 25% of patients, respectively. Preoperatively, diabetes insipidus was relatively uncommon, being clinically evident in approximately 15% of patients. Postoperatively, diabetes insipidus was common, with an incidence of 59% at 3 months. This finding is consistent with the series of Yasargil and colleagues,54 wherein permanent postoperative diabetes insipidus developed in 68% of patients. In general, it is rare to witness any improvement in endocrine status, and those with a partially deficient hypothalamic-pituitary axis frequently deteriorate.120
Many proponents of limited surgery point to evidence that radical surgery has the potential to inflict significant neuropsychological damage. The majority of these data have been retrospective in nature and are derived from craniopharyngioma series made up predominantly of children. Up to a third of children exhibit impaired frontal lobe function postoperatively.15,121 Studies including both adults and children that have controlled for age have suggested an association between the extent of tumor resection and Karnofsky Performance Scale (KPS) score. In one study it was found during follow-up that those who underwent GTR were more likely to have a KPS score in the 80 to 100 range.55 Only one prospective study has been conducted that exclusively looked at neuropsychological outcome in adults.122 Within this small series of 13 patients, 10 underwent transcranial surgery in an attempt to achieve GTR. A battery of tests were used in this study, both preoperatively and postoperatively. No significant impairment in neuropsychological performance or quality of life could be demonstrated postoperatively.
Because of the potential adherence of craniopharyngiomas to blood vessels, postoperative vascular complications have also been reported. Fusiform dilation of the carotid artery is believed to result from possible adventitial damage that occurs when craniopharyngiomas are peeled off this vessel.123,124 Although most of these cases involve pediatric patients, it can also occur in the adult population.54 In addition to fusiform dilation, significant postoperative vasospasm resulting in permanent cerebral ischemia has been reported.15
Subtotal Resection
Subtotal resection of craniopharyngioma involves limited surgery to decompress cystic or solid components of the tumor to alleviate symptoms, restore vision, reestablish CSF flow, and reduce the volume of the radiation field. Subtotal resection can be considered to incorporate a range of procedures, from stereotactic or endoscopic cyst aspiration and biopsy, to open biopsy and decompression, to more aggressive resection of tumor without the aim of GTR, and to preservation of the infundibulum, hypothalamus, and brainstem. It has been proposed as an approach to reduce the morbidity of surgery for craniopharyngioma and, with the addition of adjuvant conformal irradiation, to maintain effective disease control.56,95
Stereotactic Cyst Aspiration
In many instances, one encounters a craniopharyngioma that is mainly cystic in nature. These large cysts can be decompressed as either a primary or a definitive procedure. Frequently, this maneuver rapidly alleviates many of the symptoms of visual compromise, hypothalamic dysfunction, and increased intracranial pressure. It is often possible to pass a catheter through the ventricular system and into the cyst. The catheter is then connected to a reservoir system. Once the cyst is decompressed, the solid portion of the tumor is removed. Because most cysts reaccumulate within 2 months, definitive surgical resection is often done within 4 weeks of cyst decompression.96 Another option is to repeatedly tap the reservoir when there is clinical or radiographic evidence of recurrence or to use this portal of entry for intracavitary radiotherapy or chemotherapy.
Radiation therapy has long been used as an adjuvant treatment of craniopharyngioma after subtotal resection and as primary treatment of recurrence.125 Evidence in support of this therapy is derived from several retrospective series,109,116,126–128 in which 10- and 20-year progression-free survival rates for limited resection plus radiotherapy have been superior to those for surgery alone. Early work has shown that conventional fractionated radiotherapy appears to be beneficial when administered in doses between 45 and 55 Gy delivered to the primary tumor in fractions of 1.8 to 2 Gy.33
The risks of delivering conventionally fractionated radiotherapy to the sella and parasellar region are not insignificant. Pituitary dysfunction, hypothalamic dysfunction,129,130 and visual and cognitive deficits are well-recognized complications, in addition to radiation necrosis, vasculopathy, and second malignancies. As a result, newer, more accurate modalities of radiation delivery have been preferred for craniopharyngioma.
Conformal radiotherapy, intensity-modulated radiotherapy, stereotactic radiotherapy, stereotactic radiosurgery (SRS), and multisession SRS131 have all been used to maximize treatment effect and minimize damage to surrounding tissue.
Conformal radiotherapy at a total dose of 54 to 55.8 Gy with a 1-cm tumor margin has been shown to be as effective in preventing recurrence as external beam radiation using conventional methods.56
Stereotactic radiotherapy has been documented to be feasible, efficacious, and associated with low morbidity in adults and children with craniopharyngioma.132,133 Doses of between 9 and 21.8 Gy have been applied.134–139 A single dose of greater than 8 to 10 Gy delivered to a segment of the optic apparatus140 and 14 to 24 Gy to the brainstem is thought to be likely to result in injury; consequently, SRS is not typically used for tumors located less than 3 to 5 mm from the optic apparatus or brainstem.131,137,139,141,142 Generally, tumors less than 3 cm in maximal diameter are considered suitable for SRS. In a retrospective series of 98 patients treated by surgery and Gamma Knife SRS, overall 5- and 10-year survival rates were 94% and 91%, respectively, and the respective progression-free survival rates were 60.8% and 53.8%.139 Deterioration in both vision and endocrinologic function was documented as side effects in 6.1% of patients. These results are inferior to those seen with fractionated radiotherapy.
The use of proton radiotherapy alone or in combination with photons has been reported to achieve favorable results in patients with craniopharyngioma. The physical characteristics of the proton beam provide the benefit of reducing the dose to normal tissues surrounding a lesion, which is of central importance in treating children and those with benign lesions in particular.143–146
Cysts are a common component of craniopharyngioma and have traditionally been thought to not respond well to radiotherapy. Indeed, there are a significant number of reports of cyst enlargement during and after radiotherapy that require treatment, in some cases in up to 50% of patients.95,132,147 The importance of repeating imaging studies during treatment has been emphasized to ensure that the radiation treatment plan achieves adequate coverage.56,144
Intracavitary radiotherapy using radioactive colloidal chromic phosphate134,148–150 and yttrium 90 colloid151–154 has been evaluated in the modern treatment of craniopharyngioma. Julow and colleagues analyzed 60 patients treated over a 30-year period with 73 stereotactically administered injections of yttrium 90 colloid at average dose of 300 Gy to the cyst wall after postoperative tumor recurrence.151 Cyst size decreased in all cases, and the reduction was greater than 80% in 47 cases. Three patients (5%) had deterioration of vision attributed to radiation, and transient oculomotor palsy developed in 6. One patient had injury to the internal carotid artery wall secondary to radiation.153 Hasegawa and coauthors reported the treatment of 49 patients with intracavitary 32P (with an average of 224 Gy delivered to the cyst wall), in just over half of whom this represented the primary treatment of craniopharyngioma.149 The tumor control rate was 87%, with 80% of patients showing regression of cyst size and no perioperative or treatment-related morbidity. They concluded that this therapy was safe and effective but failed to treat the solid component of the tumor. In general, it is thought that large, multicystic tumors adjacent to the optic nerves and chiasm are not amenable to intracavitary radiotherapy.56
Because of potentially devastating neurological sequelae, radiation therapy has generally been avoided entirely in children younger than 3 years, with some authors advocating reoperation at tumor recurrence to delay radiotherapy in very young children.98
Overall Complications of Radiation Therapy
There are reports of early acute deterioration in patients with craniopharyngioma who undergo radiotherapy. In 26 of 188 patients (14%), acute deterioration developed around the time of radiotherapy.147 Most of these patients suffered from visual deterioration related to cystic enlargement or hydrocephalus and required surgical intervention to deal with this decline.
The delayed effects of radiotherapy are also significant, although with conformal radiotherapy and newer modalities of treatment their incidence is thought to be very low.132 Optic neuropathy was observed in early series of patients undergoing radiation therapy for craniopharyngioma, but in modern series with a dosage of 54 Gy and fractions of 1.8 Gy, this is very rare.155 It is difficult to assess the cause of the endocrinopathy in patients with craniopharyngioma and whether the disease or the treatment is implicated. In one series, impairment of hormone function secondary to radiation treatment developed in 3 of 18 patients at a median follow-up of 43 months.132
Development of a second neoplasm is an important consideration in patients who are treated for a benign neoplasm at an early age, and rates are difficult to determine, but in at least one patient in the Boston series a high-grade glioma developed 8 years after irradiation.156 In the Marsden series33 with 12 years of follow-up, no cases were seen, but the latency of such events is significant. The information available from large series of patients undergoing radiotherapy administered to the sella region for pituitary adenoma, which would seem applicable to craniopharyngioma, suggests a 1.9% rate of second malignancy at 20 years.157
The recurrence rate after subtotal surgery and radiotherapy varies from 7%156 to 22%98 in children and may be lower in adult series. Combs and coworkers reported 100% local control rates at both 5 and 10 years in a predominantly adult series of 40 patients treated at recurrence after surgery at a median follow-up of 40 months by fractionated stereotactic radiotherapy with a median target dose of 52.2 Gy.155
Given concerns regarding morbidity from attempted complete resection, there has been a trend in the past decade to attempt minimally invasive options in the treatment of cystic craniopharyngioma. More than two thirds of craniopharyngiomas are predominantly cystic, and the cystic component of craniopharyngioma in children can account for up to 80% to 90% of the tumor volume.158,159 The cystic component is the most dynamic component of the tumor and is often responsible for the compression of adjacent critical structures and production of symptoms. Accordingly, various approaches have been explored to specifically combat the cystic component, particularly in children, so that aggressive surgical strategies or radiation therapy, or both, may be avoided until the child is older.
Insertion of Ommaya or other reservoirs into the cystic component of craniopharyngiomas has an established history.148,160,161 There are nuances in catheter and reservoir placement, and this procedure is not without complications.162,163 Aspiration of cyst contents can be performed repeatedly to reduce mass effect. Additionally, the Ommaya reservoir provides access to the cyst for administration of chemotherapeutic agents. Performance of a reservoir permeability test is an important step in assessing suitability for intracystic therapy with any given catheter. Generally, this involves a 1-mL injection of Omnipaque (GE Healthcare) into the reservoir, followed by CT evaluation.164
Bleomycin
Intracystic administration of bleomycin has been used as an alternative therapeutic approach to aggressive surgical resection at diagnosis and as an option at recurrence since the early 1980s.161,162,165–168 Bleomycin is primarily an inhibitor of DNA and protein synthesis. It has been proposed to be most suitable in children younger than 10 years, especially those with cystic retrochiasmatic tumors and those with hypothalamic involvement, in whom surgical risks are highest.162 Bleomycin is administered at a dose of less than 0.9 mg/kg per week or 1 mg/mL per dose, or both. A review of the literature in 2004 revealed that 50% of 70 patients who were treated with this modality had a complete response and that 50% of patients had been treated with bleomycin alone at 5 years’ follow-up, with some studies reporting patients being monitored for up to 16 years.161,162,166,169
There are well-described complications from the use of intracystic bleomycin. Acute side effects can include headache, nausea, and vomiting and occur in 10% to 70% of patients.162,167,170 Leakage of bleomycin beyond the cyst wall after injection has been described and may be associated with both transient and significant permanent complications and even death.164,171 Hypothalamic dysfunction, cerebral vasculopathy, hypopituitarism, blindness, and hemiparesis have all been reported as complications of this treatment.98,172 The appearance on imaging is one of bilateral symmetrical changes in the basal ganglia, thalami, and internal capsule, with a hyperintense fluid-attenuated inversion recovery (FLAIR) and T2 signal. The use of steroids has been proposed as an effective treatment of this condition.164 The question regarding the concomitant use of bleomycin and radiation therapy has yet to be resolved, but it has been suggested that it may be associated with greater toxicity.162
Interferon
In an attempt to avoid the potential complications of neurotoxic bleomycin, other agents have been investigated. Reports of the use of subcutaneous and intracystic interferon alfa for the treatment of craniopharyngioma have emerged, with encouraging results.173–175 Interferon alfa is thought to exert its biologic effect by inducing apoptosis in tumor cells and by inhibition of angiogenesis.176,177 Of 21 children treated with this modality by Ierardi and coworkers,174 complete reduction was achieved in 11, partial response in 7, and a minor response in 3 at an average follow-up of 2 years and 3 months. The side effects from interferon alfa would appear to be less significant than those seen with bleomycin, with fatigue, fever, and loss of appetite being the most common.173,174 This agent has been used in a small group of patients, and further analysis of its effectiveness is awaited.
Systemic chemotherapy has not been a traditional adjuvant treatment of craniopharyngioma, but it has been used.175,178–180 Subcutaneous administration of interferon alfa-2a was reported in 2000, and 3 of 15 patients exhibited a clinical response after 1 year of therapy, with significant but reversible toxicity.175 Initial results have not been encouraging, as reflected by the paucity of studies in the literature.
Recurrence and Patterns of Failure
Craniopharyngioma carries a significant risk for local recurrence and may rarely recur at a distant site, either as a result of direct implantation during surgery or by CSF dissemination.181–184 Tumor recurrence in the intrasellar space in patients who have undergone transcranial procedures highlights the importance of adequate intrasellar exposure during transcranial operations.185
The diagnosis of recurrence may be made by routine imaging, by reappearance of symptoms, or in more novel ways such as through screening assessment of urinary matrix metalloproteinase levels.186
As discussed previously, the rate of recurrence of craniopharyngioma at 10 years after GTR (confirmed on postoperative MRI) ranges from 0% to 69%.36,53,55,96,187–189 An unresolved issue is exactly what surveillance protocol is optimal for patients with craniopharyngioma to maximize detection of recurrence without unduly wasting resources. Most recurrences seem to occur within the first 5 years after surgery, but delayed recurrence has been reported, and lifelong follow-up of most of these patients is advocated.98 One suggested approach is to perform annual MRI after GTR, with studies at 6-month intervals if resection of the tumor was subtotal.106
Age
Most studies have suggested that age at diagnosis is not a significant factor for recurrence.33,36,55,66 However, this statement should be qualified by noting that being younger than 5 years was found to be a significant predictive factor for recurrence in children with craniopharyngioma in one study of 75 pediatric cases.189
Sex
There is no evidence to suggest a difference in recurrence rates based on sex.33,36
Tumor Size
In the Boston Children’s Hospital series, the presence of a tumor larger than 5 cm in maximal diameter was a significant prognostic factor for recurrence.116
Histologic Subtype
There are some reports that the recurrence rate is higher for the adamantinomatous type than for the papillary type of craniopharyngioma,31,190–192 but other studies have failed to find such differences.30,55
Brain Invasion
In patients who have undergone GTR for craniopharyngioma, evidence of brain invasion does not correlate with a higher recurrence rate.55
MIB-1
Conflicting information has been published regarding the significance of MIB-1 staining, with most studies finding no significant association of either high or low MIB-1 labeling indices with recurrence.193–198
Molecular Markers
Information is emerging regarding molecular markers that predict the biologic behavior of craniopharyngioma.192 Positive immunostaining (immunoreactivity) for p53 has been shown to be associated with recurrence.199 Confirmation of such preliminary data is required.
Tumor Resection
The extent of tumor resection appears to be an important factor. Most studies seem to suggest that the extent of resection, particularly GTR, is associated with reduced risk for recurrence.36,54,128 In the Oxford series,36 no patients who underwent GTR experienced recurrence. It is important to determine exactly how one ascertains the degree of surgical resection and, therefore, the amount of postoperative residual tumor. With modern operative techniques, GTR rates ranging from 18% to 90% have been reported.36,54,96
Radiotherapy
Studies comparing subtotal resection alone and resection combined with radiotherapy have demonstrated the ability of radiotherapy to increase the time to local tumor recurrence and to improve overall survival.33,36,85,156,200 The risk of recurrence for subtotally resected lesions treated with adjuvant radiation therapy is 15% to 30%.28,33 Rajan and associates,33 in a large retrospective study in which 55% of the patients were adults, examined the results of limited surgery combined with radiotherapy for the treatment of craniopharyngioma. Only 4 of the 173 patients in this series underwent complete resection, as reported by the surgeon. All patients in this series received radiotherapy. The progression-free survival rate was 83% at 10 years.
Miscellaneous Factors
Although age and extent of surgical resection are by far the most well investigated potentially prognostic factors, others have been suggested in the literature, including factors such as growth hormone administration. The location of the craniopharyngioma and its subsequent treatment result in a significant number of these patients being deficient in growth hormone (54% to 100%).36,201,202 There is concern that patients receiving recombinant growth hormone may be at greater risk for recurrence of tumors and, by inference, for recurrence of craniopharyngioma based on in vitro, animal, and epidemiologic data in humans.203–205 Most recent reports suggest that such is not likely to be the case.206–209 In the Oxford series,208 32 patients with a mean follow-up interval of longer than 10 years received growth hormone, whereas 53 patients with a mean follow-up of greater than 8 years did not receive it. During subsequent monitoring, tumor recurrence developed in 4 patients who received growth hormone and in 22 who did not. Multivariate analysis, however, did not show growth hormone administration to be a significant independent predictor of tumor recurrence.208
Treatment of Recurrence
If tumor recurrence has been identified after attempted GTR and radiotherapy has not been administered, the treatment options would consist of radiotherapy or reoperation followed by radiotherapy. Initial consideration can be given to reoperation in cases of local recurrence, although the likelihood of achieving GTR decreases to 0% to 25%,36,53,96 and the mortality and morbidity related to second surgery have variably been documented to be higher in most series.36,96 Mortality was nearly 10 times higher in the reoperation group than in the primary surgery group in Erlangen96 and was 24% in the Oxford series.36 Radiotherapy after re-resection has been documented to be beneficial, with 72% of patients remaining progression free at 10 years after radiotherapy, although this was not affected by whether the second surgery was performed.126
If the recurrence is primarily cystic, intracystic therapies may be used, be it intracavitary irradiation149,153 or chemotherapy with bleomycin or interferon, as described earlier.173 If radiotherapy has already been administered, the treatment options become relatively limited in both scope and efficacy. Further radiation therapy was thought to no longer be feasible, but reports of the use of radiosurgery after irradiation are emerging.210
Outcome
Mortality
Modern overall survival rates with craniopharyngioma are 80% to 91% at 5 years, 83% to 96% at 10 years,36,53,96,108,156 and 84% at 30 years.36 Karavitaki and associates found no difference in overall survival rates among patients undergoing GTR (100%), partial resection and radiotherapy (87%), and partial resection alone (86%).36
Prognostic Factors
Age as a prognostic factor is controversial. Neonatal occurrence of craniopharyngioma is associated with very high mortality,211 although for patients beyond that stage, the data are conflicting.26 There is no evidence suggesting a difference in survival based on sex.33,36
The tumor’s histologic type, location, and size and the presence of hydrocephalus do not appear to be independent predictors of survival.26
Long-Term Morbidity and Quality of Life
Although current therapeutic modifications must be aimed at improving survival, the issue of quality of life must also be given strong consideration.36 Craniopharyngioma patients must endure significant lifelong morbidity from visual loss, endocrinopathy, obesity, and neurocognitive deficits as a result of the disease or the therapy. In the Oxford series of both adults and children,36 at a follow-up of 10 years 48% of patients had major visual field deficits, 39% had hyperphagia and weight gain, 11% had permanent motor deficits, 12% had epilepsy, 15% had psychological disorders requiring treatment, and 9% were completely dependent for basal daily activities. These patients are at risk for cognitive decline with time212 and have been demonstrated to have problems with working memory and long-term memory that vary in severity.213,214
Both adults215 and children216 treated for craniopharyngioma have been documented to have a significant reduction in quality of life, as determined by a number of health-related questionnaires.
Conclusion
Bunin GR, Surawicz TS, Witman PA, et al. The descriptive epidemiology of craniopharyngioma. J Neurosurg. 1998;89:547-551.
Buslei R, Nolde M, Hofmann B, et al. Common mutations of beta-catenin in adamantinomatous craniopharyngiomas but not in other tumours originating from the sellar region. Acta Neuropathol. 2005;109:589-597.
Cavalheiro S, Dastoli PA, Silva NS, et al. Use of interferon alpha in intratumoral chemotherapy for cystic craniopharyngioma. Childs Nerv Syst. 2005;21:719-724.
Combs SE, Thilmann C, Huber PE, et al. Achievement of long-term local control in patients with craniopharyngiomas using high precision stereotactic radiotherapy. Cancer. 2007;109:2308-2314.
Defoort-Dhellemmes S, Moritz F, Bouacha I, et al. Craniopharyngioma: ophthalmological aspects at diagnosis. J Pediatr Endocrinol Metab. 2006;19(suppl 1):321-324.
Dekkers OM, Biermasz NR, Smit JW, et al. Quality of life in treated adult craniopharyngioma patients. Eur J Endocrinol. 2006;154:483-489.
Fahlbusch R, Honegger J, Paulus W, et al. Surgical treatment of craniopharyngiomas: experience with 168 patients. J Neurosurg. 1999;90:237-250.
Fitzek MM, Linggood RM, Adams J, et al. Combined proton and photon irradiation for craniopharyngioma: long-term results of the early cohort of patients treated at Harvard Cyclotron Laboratory and Massachusetts General Hospital. Int J Radiat Oncol Biol Phys. 2006;64:1348-1354.
Hargrave DR. Does chemotherapy have a role in the management of craniopharyngioma? J Pediatr Endocrinol Metab. 2006;19(suppl 1):407-412.
Harwood-Nash DC. Neuroimaging of childhood craniopharyngioma. Pediatr Neurosurg. 1994;21(suppl 1):2-10.
Hoffman HJ, De, Silva M, Humphreys RP, et al. Aggressive surgical management of craniopharyngiomas in children. J Neurosurg. 1992;76:47-52.
Honegger J, Barocka A, Sadri B, et al. Neuropsychological results of craniopharyngioma surgery in adults: a prospective study. Surg Neurol. 1998;50:19-29.
Honegger J, Buchfelder M, Fahlbusch R. Surgical treatment of craniopharyngiomas: endocrinological results. J Neurosurg. 1999;90:251-257.
Hopper N, Albanese A, Ghirardello S, et al. The pre-operative endocrine assessment of craniopharyngiomas. J Pediatr Endocrinol Metab. 2006;19(suppl 1):325-327.
Julow J, Backlund EO, Lanyi F, et al. Long-term results and late complications after intracavitary yttrium-90 colloid irradiation of recurrent cystic craniopharyngiomas. Neurosurgery. 2007;61:288-296.
Karavitaki N, Brufani C, Warner JT, et al. Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clin Endocrinol (Oxf). 2005;62:397-409.
Karavitaki N, Cudlip S, Adams CB, et al. Craniopharyngiomas. Endocr Rev. 2006;27:371-397.
Kassam AB, Gardner PA, Snyderman CH, et al. Expanded endonasal approach, a fully endoscopic transnasal approach for the resection of midline suprasellar craniopharyngiomas: a new classification based on the infundibulum. J Neurosurg. 2008;108:715-728.
Kobayashi T, Kida Y, Mori Y, et al. Long-term results of gamma knife surgery for the treatment of craniopharyngioma in 98 consecutive cases. J Neurosurg. 2005;103:482-488.
Merchant TE, Kiehna EN, Sanford RA, et al. Craniopharyngioma: the St. Jude Children’s Research Hospital experience 1984-2001. Int J Radiat Oncol Biol Phys. 2002;53:533-542.
Prabhu VC, Brown HG. The pathogenesis of craniopharyngiomas. Childs Nerv Syst. 2005;21:622-627.
Puget S, Garnett M, Wray A, et al. Pediatric craniopharyngiomas: classification and treatment according to the degree of hypothalamic involvement. J Neurosurg. 2007;106:3-12.
Rajan B, Ashley S, Gorman C, et al. Craniopharyngioma—a long-term results following limited surgery and radiotherapy. Radiother Oncol. 1993;26:1-10.
Van, Effenterre R, Boch AL. Craniopharyngioma in adults and children: a study of 122 surgical cases. J Neurosurg. 2002;97:3-11.
Yasargil MG, Curcic M, Kis M, et al. Total removal of craniopharyngiomas. Approaches and long-term results in 144 patients. J Neurosurg. 1990;73:3-11.
1 Cushing H. The craniopharyngiomas. In: Intracranial Tumors: Notes upon a Series of Two Thousand Verified Cases with Surgical Mortality Percentages Pertaining Thereto. Springfield, IL: Charles C Thomas; 1932:93-98.
2 Zenker F. Enormcystenbildung im gehirn, von hiranhang ausgehend. Arch Pathol Anat Physiol Klin Med. 1857;2:454-466.
3 Onanoff J. Sur un Cas d’Epithelioma (Etude Histologique). Paris, 1892.
4 Mott FW, Barrett JOW. Three cases of tumors of the third ventricle. Arch Neurol (London). 1899;1:417-440.
5 Frohlich A. Ein Fall von Tumor der Hypophysis Cerebri ohne Akromegalie. Wien Klin Rundschau. 1901;15:883-906.
6 Babinski J. Tumeur du corps pituitaire sans acromegalie et avec arret de development des organes genitaux. Rev Neurol (Paris). 1900;8:531-533.
7 Halsteadt A. Remarks on an operative treatment of tumors of the hypophysis. Surg Gynecol Obstet. 1910;10:494-502.
8 McKenzie K, Sosman MC. The roentgenological diagnosis of craniopharyngeal pouch tumors. AJR Am J Roentgenol. 1921;11:171-176.
9 Frazier CH, Alpers BJ. Adamantinoma of the craniopharyngeal duct. Arch Neurol Psychiatry. 1931;26:905-965.
10 Russell DS, Rubinstein LJ, editors. Pathology of Tumors of the Nervous System. 5th ed. Baltimore: Williams & Wilkins. 1989:695-702.
11 Grant F. Surgical experience with tumors of the pituitary gland. JAMA. 1948;136:668-672.
12 Kramer S, McKissock W, Concannon JP. Craniopharyngiomas. Treatment by combined surgery and radiation therapy. J Neurosurg. 1961;18:217-226.
13 Katz EL. Late results of radical excision of craniopharyngiomas in children. J Neurosurg. 1975;42:86-93.
14 Sweet W. Radical surgical treatment of craniopharyngiomas. Clin Neurosurg. 1975;23:52-79.
15 Hoffman HJ, De, Silva M, Humphreys RP, et al. Aggressive surgical management of craniopharyngiomas in children. J Neurosurg. 1992;76:47-52.
16 Prabhu VC, Brown HG. The pathogenesis of craniopharyngiomas. Childs Nerv Syst. 2005;21:622-627.
17 Erdheim J. Uber Hypophysenganggeschwulste und Hirncholesteatome. Sitzungsbericht der Kaiserlichen Akademie der Wissenchaften Mathematisch-Naturwissenschaftliche Classe (Wien). 1904:537-726.
18 Tachibana O, Yamashima T, Yamashita J, et al. Immunohistochemical expression of human chorionic gonadotropin and P-glycoprotein in human pituitary glands and craniopharyngiomas. J Neurosurg. 1994;80:79-84.
19 Zumkeller W, Saaf M, Rahn T, et al. Demonstration of insulin-like growth factors I, II and heterogeneous insulin-like growth factor binding proteins in the cyst fluid of patients with craniopharyngioma. Neuroendocrinology. 1991;54:196-201.
20 Luse SA, Kernohan JW. Squamous-cell nests of the pituitary gland. Cancer. 1955;8:623-628.
21 Goldberg GM, Eshbaugh DE. Squamous cell nests of the pituitary gland as related to the origin of craniopharyngiomas. A study of their presence in the newborn and infants up to age four. Arch Pathol. 1960;70:293-299.
22 Miller DC. Pathology of craniopharyngiomas: clinical import of pathological findings. Pediatr Neurosurg. 1994;21(suppl 1):11-17.
23 Petito CK, DeGirolami U, Earle KM. Craniopharyngiomas: a clinical and pathological review. Cancer. 1976;37:1944-1952.
24 Bunin GR, Surawicz TS, Witman PA, et al. The descriptive epidemiology of craniopharyngioma. J Neurosurg. 1998;89:547-551.
25 Kuratsu J, Ushio Y. Epidemiological study of primary intracranial tumors in childhood. A population-based survey in Kumamoto Prefecture, Japan. Pediatr Neurosurg. 1996;25:240-246.
26 Karavitaki N, Cudlip S, Adams CB, et al. Craniopharyngiomas. Endocr Rev. 2006;27:371-397.
27 Matson DD, Crigler. Management of craniopharyngioma in childhood. J Neurosurg. 1969;30:377-390.
28 Choux M, Lena G, Genitori L. Le craniopharyngiome de l’enfant. Neurochirurgie. 1991;37:12-165.
29 Garre ML, Cama A. Craniopharyngioma: modern concepts in pathogenesis and treatment. Curr Opin Pediatr. 2007;19:471-479.
30 Crotty TB, Scheithauer BW, Young, et al. Papillary craniopharyngioma: a clinicopathological study of 48 cases. J Neurosurg. 1995;83:206-214.
31 Adamson TE, Wiestler OD, Kleihues P, et al. Correlation of clinical and pathological features in surgically treated craniopharyngiomas. J Neurosurg. 1990;73:12-17.
32 Sorva R, Heiskanen O. Craniopharyngioma in Finland. A study of 123 cases. Acta Neurochir (Wien). 1986;81:85-89.
33 Rajan B, Ashley S, Gorman C, et al. Craniopharyngioma—a long-term results following limited surgery and radiotherapy. Radiother Oncol. 1993;26:1-10.
34 Jane, Laws ER. Craniopharyngioma. Pituitary. 2006;9:323-326.
35 Harwood-Nash DC. Neuroimaging of childhood craniopharyngioma. Pediatr Neurosurg. 1994;21(suppl 1):2-10.
36 Karavitaki N, Brufani C, Warner JT, et al. Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clin Endocrinol (Oxf). 2005;62:397-409.
37 Koral K, Weprin B, Rollins NK. Sphenoid sinus craniopharyngioma simulating mucocele. Acta Radiol. 2006;47:494-496.
38 Shuman AG, Heth JA, Marentette LJ, et al. Extracranial nasopharyngeal craniopharyngioma: case report. Neurosurgery. 2007;60:E780-781.
39 Kawamata T, Kubo O, Kamikawa S, et al. Ectopic clival craniopharyngioma. Acta Neurochir (Wien). 2002;144:1221-1224.
40 Banczerowski P, Balint K, Sipos L. Temporal extradural ectopic craniopharyngioma. Case report. J Neurosurg. 2007;107:178-180.
41 Cheng MK. Brain tumors in the People’s Republic of China: a statistical review. Neurosurgery. 1982;10:16-21.
42 Stiller CA, Nectoux J. International incidence of childhood brain and spinal tumours. Int J Epidemiol. 1994;23:458-464.
43 Ohaegbulam SC. Craniopharyngioma. J Neurosurg. 1999;90:984-985.
44 Banna M. Craniopharyngioma in adults. Surg Neurol. 1973;1:202-204.
45 Haupt R, Magnani C, Pavanello M, et al. Epidemiological aspects of craniopharyngioma. J Pediatr Endocrinol Metab. 2006;19(suppl 1):289-293.
46 Defoort-Dhellemmes S, Moritz F, Bouacha I, et al. Craniopharyngioma: ophthalmological aspects at diagnosis. J Pediatr Endocrinol Metab. 2006;19(suppl 1):321-324.
47 Hopper N, Albanese A, Ghirardello S, et al. The pre-operative endocrine assessment of craniopharyngiomas. J Pediatr Endocrinol Metab. 2006;19(suppl 1):325-327.
48 Bulow B, Attewell R, Hagmar L, et al. Postoperative prognosis in craniopharyngioma with respect to cardiovascular mortality, survival, and tumor recurrence. J Clin Endocrinol Metab. 1998;83:3897-3904.
49 Larijani B, Bastanhagh MH, Pajouhi M, et al. Presentation and outcome of 93 cases of craniopharyngioma. Eur J Cancer Care (Engl). 2004;13:11-15.
50 de, Vries L, Lazar L, Phillip M. Craniopharyngioma: presentation and endocrine sequelae in 36 children. J Pediatr Endocrinol Metab. 2003;16:703-710.
51 Fahlbusch R, Honegger J, Buchfelder M. Clinical features and management of craniopharyngiomas in adults. In: Tindall GT, Cooper PR, Barrow DL, editors. The Practice of Neurosurgery. Baltimore: Williams & Wilkins; 1996:1159-1173.
52 Sklar CA. Craniopharyngioma: endocrine abnormalities at presentation. Pediatr Neurosurg. 1994;21(suppl 1):18-20.
53 Van, Effenterre R, Boch AL. Craniopharyngioma in adults and children: a study of 122 surgical cases. J Neurosurg. 2002;97:3-11.
54 Yasargil MG, Curcic M, Kis M, et al. Total removal of craniopharyngiomas. Approaches and long-term results in 144 patients. J Neurosurg. 1990;73:3-11.
55 Weiner HL, Wisoff JH, Rosenberg ME, et al. Craniopharyngiomas: a clinicopathological analysis of factors predictive of recurrence and functional outcome. Neurosurgery. 1994;35:1001-1010.
56 Merchant TE, Kiehna EN, Sanford RA, et al. Craniopharyngioma: the St. Jude Children’s Research Hospital experience 1984-2001. Int J Radiat Oncol Biol Phys. 2002;53:533-542.
57 Bartlett JR. Craniopharyngiomas. An analysis of some aspects of symptomatology, radiology and histology. Brain. 1971;94:725-732.
58 Russell RW, Pennybacker JB. Craniopharyngioma in the elderly. J Neurol Neurosurg Psychiatry. 1961;24:1-13.
59 Ishii K, Isono M, Hori S, et al. [A case of craniopharyngioma with intratumoral hemorrhage.]. No Shinkei Geka. 1999;27:73-77.
60 Maier HC. Craniopharyngioma with erosion and drainage into the nasopharynx. An autobiographical case report. J Neurosurg. 1985;62:132-134.
61 Yamamoto T, Yoneda S, Funatsu N. Spontaneous haemorrhage in craniopharyngioma. J Neurol Neurosurg Psychiatry. 1989;52:803-804.
62 Okamoto H, Harada K, Uozumi T, et al. Spontaneous rupture of a craniopharyngioma cyst. Surg Neurol. 1985;24:507-510.
63 Flores LE, Williams DL, Bell BA, et al. Delay in the diagnosis of pediatric brain tumors. Am J Dis Child. 1986;140:684-686.
64 Thulesius H, Pola J, Hakansson A. Diagnostic delay in pediatric malignancies—a population-based study. Acta Oncol. 2000;39:873-876.
65 Haimi M, Peretz, Nahum M, Ben, Arush MW. Delay in diagnosis of children with cancer: a retrospective study of 315 children. Pediatr Hematol Oncol. 2004;21:37-48.
66 Louis D, Ohgaki H. WHO Classification of Tumours of the Central Nervous System, ed 3. Basel: World Heath Organization; 2007.
67 Eldevik OP, Blaivas M, Gabrielsen TO, et al. Craniopharyngioma: radiologic and histologic findings and recurrence. AJNR Am J Neuroradiol. 1996;17:1427-1439.
68 Burger PC, Scheithauer BW, Vogel SF. Surgical Pathology of the Central Nervous System and Its Coverings, ed 4. New York: Churchill Livingstone. 2002.
69 Sato K, Kubota T. Fine structure of ossification in craniopharyngiomas. Ultrastruct Pathol. 1999;23:395-399.
70 Kurosaki M, Saeger W, Ludecke DK. Immunohistochemical localisation of cytokeratins in craniopharyngioma. Acta Neurochir (Wien). 2001;143:147-151.
71 Nelson GA, Bastian FO, Schlitt M, et al. Malignant transformation in craniopharyngioma. Neurosurgery. 1988;22:427-429.
72 Suzuki F, Konuma I, Matsumoto M, et al. [Craniopharyngioma with malignant transformation—a report of two cases.]. Gan No Rinsho. 1989;35:723-728.
73 Virik K, Turner J, Garrick R, et al. Malignant transformation of craniopharyngioma. J Clin Neurosci. 1999;6:527-530.
74 Vargas JR, Pino JA, Murad TM. Craniopharyngioma in two siblings. JAMA. 1981;246:1807-1808.
75 Combelles G, Ythier H, Wemeau JL, et al. [Craniopharyngioma in the same family.]. Neurochirurgie. 1984;30:347-349.
76 Boch AL, van, Effenterre R, Kujas M. Craniopharyngiomas in two consanguineous siblings: case report. Neurosurgery. 1997;41:1185-1187.
77 Rienstein S, Adams EF, Pilzer D, et al. Comparative genomic hybridization analysis of craniopharyngiomas. J Neurosurg. 2003;98:162-164.
78 Sarubi JC, Bei H, Adams EF, et al. Clonal composition of human adamantinomatous craniopharyngiomas and somatic mutation analyses of the patched (PTCH), Gsalpha and Gi2alpha genes. Neurosci Lett. 2001;310:5-8.
79 Karnes PS, Tran TN, Cui MY, et al. Cytogenetic analysis of 39 pediatric central nervous system tumors. Cancer Genet Cytogenet. 1992;59:12-19.
80 Gorski GK, McMorrow LE, Donaldson MH, et al. Multiple chromosomal abnormalities in a case of craniopharyngioma. Cancer Genet Cytogenet. 1992;60:212-213.
81 Yoshimoto M, de, Toledo SR, da, Silva NS, et al. Comparative genomic hybridization analysis of pediatric adamantinomatous craniopharyngiomas and a review of the literature. J Neurosurg. 2004;101:85-90.
82 Rickert CH, Paulus W. Lack of chromosomal imbalances in adamantinomatous and papillary craniopharyngiomas. J Neurol Neurosurg Psychiatry. 2003;74:260-261.
83 Kato K, Nakatani Y, Kanno H, et al. Possible linkage between specific histological structures and aberrant reactivation of the Wnt pathway in adamantinomatous craniopharyngioma. J Pathol. 2004;203:814-821.
84 Buslei R, Nolde M, Hofmann B, et al. Common mutations of beta-catenin in adamantinomatous craniopharyngiomas but not in other tumours originating from the sellar region. Acta Neuropathol. 2005;109:589-597.
85 Sung K, Chang CH, Harisiadis L, et al. Treatment results of craniopharyngiomas. Cancer. 1981;47:847-852.
86 Choi SH, Kwon BJ, Na DG, et al. Pituitary adenoma, craniopharyngioma, and Rathke cleft cyst involving both intrasellar and suprasellar regions: differentiation using MRI. Clin Radiol. 2007;62:453-462.
87 Nagahata M, Hosoya T, Kayama T, et al. Edema along the optic tract: a useful MR finding for the diagnosis of craniopharyngiomas. AJNR Am J Neuroradiol. 1998;19:1753-1757.
88 Sartoretti-Schefer S, Wichmann W, Aguzzi A, et al. MR differentiation of adamantinous and squamous-papillary craniopharyngiomas. AJNR Am J Neuroradiol. 1997;18:77-87.
89 Ciric IS, Cozzens JW. Craniopharyngiomas: transsphenoidal method of approach—for the virtuoso only? Clin Neurosurg. 1980;27:169-187.
90 Puget S, Garnett M, Wray A, et al. Pediatric craniopharyngiomas: classification and treatment according to the degree of hypothalamic involvement. J Neurosurg. 2007;106:3-12.
91 Kassam AB, Gardner PA, Snyderman CH, et al. Expanded endonasal approach, a fully endoscopic transnasal approach for the resection of midline suprasellar craniopharyngiomas: a new classification based on the infundibulum. J Neurosurg. 2008;108:715-728.
92 Samii M, Bini W. Surgical treatment of craniopharyngiomas. Zentralbl Neurochir. 1991;52:17-23.
93 Konovalov AN. Technique and strategies of direct surgical management of craniopharyngiomas. In: Apuzzo MLJ, editor. Surgery of the Third Ventricle. Baltimore: Williams & Wilkins; 1998:1133-1142.
94 Garnett MR, Puget S, Grill J, et al. Craniopharyngioma. Orphanet J Rare Dis. 2007;2:18.
95 Merchant TE, Kiehna EN, Kun LE, et al. Phase II trial of conformal radiation therapy for pediatric patients with craniopharyngioma and correlation of surgical factors and radiation dosimetry with change in cognitive function. J Neurosurg. 2006;104:94-102.
96 Fahlbusch R, Honegger J, Paulus W, et al. Surgical treatment of craniopharyngiomas: experience with 168 patients. J Neurosurg. 1999;90:237-250.
97 Couldwell WT. Surgery of the anterior skull base. Otolaryngol Clin North Am. 1993;26:673-693.
98 Scott RM. Craniopharyngioma: a personal (Boston) experience. Childs Nerv Syst. 2005;21:773-777.
99 Shibuya M, Takayasu M, Suzuki Y, et al. Bifrontal basal interhemispheric approach to craniopharyngioma resection with or without division of the anterior communicating artery. J Neurosurg. 1996;84:951-956.
100 Chakrabarti I, Amar AP, Couldwell W, et al. Long-term neurological, visual, and endocrine outcomes following transnasal resection of craniopharyngioma. J Neurosurg. 2005;102:650-657.
101 Honegger J, Buchfelder M, Fahlbusch R, et al. Transsphenoidal microsurgery for craniopharyngioma. Surg Neurol. 1992;37:189-196.
102 Laws. Transsphenoidal microsurgery in the management of craniopharyngioma. J Neurosurg. 1980;52:661-666.
103 Jho HD, Carrau RL. Endoscopic endonasal transsphenoidal surgery: experience with 50 patients. J Neurosurg. 1997;87:44-51.
104 Samii M, Tatagiba M. Surgical management of craniopharyngiomas: a review. Neurol Med Chir (Tokyo). 1997;37:141-149.
105 Kassam AB, Thomas A, Carrau RL, et al. Endoscopic reconstruction of the cranial base using a pedicled nasoseptal flap. Neurosurgery. 2008;63:ONS44-53.
106 Honegger J, Tatagiba M. Craniopharyngioma surgery. Pituitary. 2008;11:361-373.
107 Di, Rocco C, Caldarelli M, Tamburrini G, et al. Surgical management of craniopharyngiomas—experience with a pediatric series. J Pediatr Endocrinol Metab. 2006;19(suppl 1):355-366.
108 Lin LL, El, Naqa I, Leonard JR, et al. Long-term outcome in children treated for craniopharyngioma with and without radiotherapy. J Neurosurg Pediatr. 2008;1:126-130.
109 Wen BC, Hussey DH, Staples J, et al. A comparison of the roles of surgery and radiation therapy in the management of craniopharyngiomas. Int J Radiat Oncol Biol Phys. 1989;16:17-24.
110 Stripp DC, Maity A, Janss AJ, et al. Surgery with or without radiation therapy in the management of craniopharyngiomas in children and young adults. Int J Radiat Oncol Biol Phys. 2004;58:714-720.
111 Svolos DG. Craniopharyngiomas. A study based on 108 verified cases. Acta Chir Scand Suppl. 1969;403:1-44.
112 Love JG, Marshall TM. Craniopharyngiomas. Surg Gynecol Obstet. 1950;90:591-601.
113 Hoff JT, Patterson RHJr. Craniopharyngiomas in children and adults. J Neurosurg. 1972;36:299-302.
114 Laws. Transsphenoidal removal of craniopharyngioma. Pediatr Neurosurg. 1994;21(suppl 1):57-63.
115 Scott RM, Pomeroy SL, Tarbell NJ. Craniopharyngioma. In: Black PM, Loeffler JS, editors. Cancer of the Nervous System. Cambridge: Blackwell Science; 1997:414-422.
116 Hetelekidis S, Barnes PD, Tao ML, et al. 20-year experience in childhood craniopharyngioma. Int J Radiat Oncol Biol Phys. 1993;27:189-195.
117 Sorva R, Heiskanen O, Perheentupa J. Craniopharyngioma surgery in children: endocrine and visual outcome. Childs Nerv Syst. 1988;4:97-99.
118 Sklar CA. Craniopharyngioma: endocrine sequelae of treatment. Pediatr Neurosurg. 1994;21(suppl 1):120-123.
119 Konig A, Ludecke DK, Herrmann HD. Transnasal surgery in the treatment of craniopharyngiomas. Acta Neurochir (Wien). 1986;83:1-7.
120 Honegger J, Buchfelder M, Fahlbusch R. Surgical treatment of craniopharyngiomas: endocrinological results. J Neurosurg. 1999;90:251-257.
121 Cavazzuti V, Fischer EG, Welch K, et al. Neurological and psychophysiological sequelae following different treatments of craniopharyngioma in children. J Neurosurg. 1983;59:409-417.
122 Honegger J, Barocka A, Sadri B, et al. Neuropsychological results of craniopharyngioma surgery in adults: a prospective study. Surg Neurol. 1998;50:19-29.
123 Sutton LN. Vascular complications of surgery for craniopharyngioma and hypothalamic glioma. Pediatr Neurosurg. 1994;21(suppl 1):124-128.
124 Sutton LN, Gusnard D, Bruce DA, et al. Fusiform dilatations of the carotid artery following radical surgery of childhood craniopharyngiomas. J Neurosurg. 1991;74:695-700.
125 Kramer S, Southard M, Mansfield CM. Radiotherapy in the management of craniopharyngiomas: further experiences and late results. Am J Roentgenol Radium Ther Nucl Med. 1968;103:44-52.
126 Jose CC, Rajan B, Ashley S, et al. Radiotherapy for the treatment of recurrent craniopharyngioma. Clin Oncol (R Coll Radiol). 1992;4:287-289.
127 Moon SH, Kim IH, Park SW, et al. Early adjuvant radiotherapy toward long-term survival and better quality of life for craniopharyngiomas—a study in single institute. Childs Nerv Syst. 2005;21:799-807.
128 Habrand JL, Ganry O, Couanet D, et al. The role of radiation therapy in the management of craniopharyngioma: a 25-year experience and review of the literature. Int J Radiat Oncol Biol Phys. 1999;44:255-263.
129 DeVile CJ, Grant DB, Hayward RD, et al. Growth and endocrine sequelae of craniopharyngioma. Arch Dis Child. 1996;75:108-114.
130 Muller HL, Emser A, Faldum A, et al. Longitudinal study on growth and body mass index before and after diagnosis of childhood craniopharyngioma. J Clin Endocrinol Metab. 2004;89:3298-3305.
131 Lee M, Kalani MY, Cheshier S, et al. Radiation therapy and CyberKnife radiosurgery in the management of craniopharyngiomas. Neurosurg Focus. 2008;24(5):E4.
132 Schulz-Ertner D, Frank C, Herfarth KK, et al. Fractionated stereotactic radiotherapy for craniopharyngiomas. Int J Radiat Oncol Biol Phys. 2002;54:1114-1120.
133 Tarbell NJ, Barnes P, Scott RM, et al. Advances in radiation therapy for craniopharyngiomas. Pediatr Neurosurg. 1994;21(suppl 1):101-107.
134 Lunsford LD, Pollock BE, Kondziolka DS, et al. Stereotactic options in the management of craniopharyngioma. Pediatr Neurosurg. 1994;21(suppl 1):90-97.
135 Prasad D, Steiner M, Steiner L. Gamma knife surgery for craniopharyngioma. Acta Neurochir (Wien). 1995;134:167-176.
136 Chung WY, Pan DH, Shiau CY, et al. Gamma knife radiosurgery for craniopharyngiomas. J Neurosurg. 2000;93(suppl 3):47-56.
137 Chiou SM, Lunsford LD, Niranjan A, et al. Stereotactic radiosurgery of residual or recurrent craniopharyngioma, after surgery, with or without radiation therapy. Neuro Oncol. 2001;3:159-166.
138 Amendola BE, Wolf A, Coy SR, et al. Role of radiosurgery in craniopharyngiomas: a preliminary report. Med Pediatr Oncol. 2003;41:123-127.
139 Kobayashi T, Kida Y, Mori Y, et al. Long-term results of gamma knife surgery for the treatment of craniopharyngioma in 98 consecutive cases. J Neurosurg. 2005;103:482-488.
140 Girkin CA, Comey CH, Lunsford LD, et al. Radiation optic neuropathy after stereotactic radiosurgery. Ophthalmology. 1997;104:1634-1643.
141 Selch MT, DeSalles AA, Wade M, et al. Initial clinical results of stereotactic radiotherapy for the treatment of craniopharyngiomas. Technol Cancer Res Treat. 2002;1:51-59.
142 Ulfarsson E, Lindquist C, Roberts M, et al. Gamma knife radiosurgery for craniopharyngiomas: long-term results in the first Swedish patients. J Neurosurg. 2002;97:613-622.
143 Fitzek MM, Linggood RM, Adams J, et al. Combined proton and photon irradiation for craniopharyngioma: long-term results of the early cohort of patients treated at Harvard Cyclotron Laboratory and Massachusetts General Hospital. Int J Radiat Oncol Biol Phys. 2006;64:1348-1354.
144 Winkfield KM, Linsenmeier C, Yock TI, et al. Surveillance of craniopharyngioma cyst growth in children treated with proton radiotherapy. Int J Radiat Oncol Biol Phys. 2009;73:716-721.
145 Baumert BG, Norton IA, Lomax AJ, et al. Dose conformation of intensity-modulated stereotactic photon beams, proton beams, and intensity-modulated proton beams for intracranial lesions. Int J Radiat Oncol Biol Phys. 2004;60:1314-1324.
146 Austin-Seymour M, Munzenrider J, Linggood R, et al. Fractionated proton radiation therapy of cranial and intracranial tumors. Am J Clin Oncol. 1990;13:327-330.
147 Rajan B, Ashley S, Thomas DG, et al. Craniopharyngioma: improving outcome by early recognition and treatment of acute complications. Int J Radiat Oncol Biol Phys. 1997;37:517-521.
148 Trippi AC, Garner JT, Kassabian JT, et al. A new approach to inoperable craniopharyngiomas. Am J Surg. 1969;118:307-310.
149 Hasegawa T, Kondziolka D, Hadjipanayis CG, et al. Management of cystic craniopharyngiomas with phosphorus-32 intracavitary irradiation. Neurosurgery. 2004;54:813-822.
150 Shahzadi S, Sharifi G, Andalibi R, et al. Management of cystic craniopharyngiomas with intracavitary irradiation with 32P. Arch Iran Med. 2008;11:30-34.
151 Julow J, Lanyi F, Hajda M, et al. Treatment of cystic craniopharyngiomas with yttrium-90 colloid solution. Neurosurg Focus. 1997;3(6):e6.
152 Van, den, Berge JH, Blaauw G, Breeman WA, et al. Intracavitary brachytherapy of cystic craniopharyngiomas. J Neurosurg. 1992;77:545-550.
153 Julow J, Backlund EO, Lanyi F, et al. Long-term results and late complications after intracavitary yttrium-90 colloid irradiation of recurrent cystic craniopharyngiomas. Neurosurgery. 2007;61:288-296.
154 Julow J, Lanyi F, Hajda M, et al. Stereotactic intracavitary irradiation of cystic craniopharyngiomas with yttrium-90 isotope. Prog Neurol Surg. 2007;20:289-296.
155 Combs SE, Thilmann C, Huber PE, et al. Achievement of long-term local control in patients with craniopharyngiomas using high precision stereotactic radiotherapy. Cancer. 2007;109:2308-2314.
156 Fischer EG, Welch K, Shillito JJr, et al. Craniopharyngiomas in children. Long-term effects of conservative surgical procedures combined with radiation therapy. J Neurosurg. 1990;73:534-540.
157 Brada M, Ford D, Ashley S, et al. Risk of second brain tumour after conservative surgery and radiotherapy for pituitary adenoma. BMJ. 1992;304:1343-1346.
158 Hukin J, Visser J, Sargent M, et al. Childhood craniopharyngioma: Vancouver experience. Childs Nerv Syst. 2005;21:758-765.
159 Backlund EO. Treatment of craniopharyngiomas: the multimodality approach. Pediatr Neurosurg. 1994;21(suppl 1):82-89.
160 Fox JL. Intermittent drainage of intracranial cyst via the subcutaneous Ommaya reservoir. Technical note. J Neurosurg. 1967;27:272-273.
161 Hader WJ, Steinbok P, Hukin J, et al. Intratumoral therapy with bleomycin for cystic craniopharyngiomas in children. Pediatr Neurosurg. 2000;33:211-218.
162 Hukin J, Steinbok P, Lafay-Cousin L, et al. Intracystic bleomycin therapy for craniopharyngioma in children: the Canadian experience. Cancer. 2007;109:2124-2131.
163 Zanon N, Cavalheiro S, da, Silva MC. Does the choice of surgical approach to insert an intratumoral catheter influence the results of intratumoral cystic treatment? Surg Neurol. 2008;70:66-69.
164 Lafay-Cousin L, Bartels U, Raybaud C, et al. Neuroradiological findings of bleomycin leakage in cystic craniopharyngioma. Report of three cases. J Neurosurg. 2007;107:318-323.
165 Umezawa H, Maeda K, Takeuchi T, et al. New antibiotics, bleomycin A and B. J Antibiot (Tokyo). 1966;19:200-209.
166 Takahashi H, Nakazawa S, Shimura T. Evaluation of postoperative intratumoral injection of bleomycin for craniopharyngioma in children. J Neurosurg. 1985;62:120-127.
167 Broggi G, Giorgi C, Franzini A, et al. Preliminary results of intracavitary treatment of craniopharyngioma with bleomycin. J Neurosurg Sci. 1989;33:145-148.
168 Cavalheiro S, Sparapani FV, Franco JO, et al. Use of bleomycin in intratumoral chemotherapy for cystic craniopharyngioma. Case report. J Neurosurg. 1996;84:124-126.
169 Haisa T, Ueki K, Yoshida S. Toxic effects of bleomycin on the hypothalamus following its administration into a cystic craniopharyngioma. Br J Neurosurg. 1994;8:747-750.
170 Broggi G, Franzini A. Bleomycin for cystic craniopharyngioma. J Neurosurg. 1996;84:1080-1081.
171 Savas A, Erdem A, Tun K, et al. Fatal toxic effect of bleomycin on brain tissue after intracystic chemotherapy for a craniopharyngioma: case report. Neurosurgery. 2000;46:213-217.
172 Belen D, Er U, Yigitkanli K, et al. Delayed neurotoxic complication of intracavitary bleomycin therapy for craniopharyngioma in a child who had previously undergone radiosurgery. Case report. J Neurosurg. 2007;106:391-393.
173 Cavalheiro S, Dastoli PA, Silva NS, et al. Use of interferon alpha in intratumoral chemotherapy for cystic craniopharyngioma. Childs Nerv Syst. 2005;21:719-724.
174 Ierardi DF, Fernandes MJ, Silva IR, et al. Apoptosis in alpha interferon (IFN-alpha) intratumoral chemotherapy for cystic craniopharyngiomas. Childs Nerv Syst. 2007;23:1041-1046.
175 Jakacki RI, Cohen BH, Jamison C, et al. Phase II evaluation of interferon-alpha-2a for progressive or recurrent craniopharyngiomas. J Neurosurg. 2000;92:255-260.
176 Chawla-Sarkar M, Lindner DJ, Liu YF, et al. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237-249.
177 Muhr C, Gudjonsson O, Lilja A, et al. Meningioma treated with interferon-alpha, evaluated with [(11)C]-L-methionine positron emission tomography. Clin Cancer Res. 2001;7:2269-2276.
178 Bremer AM, Nguyen TQ, Balsys R. Therapeutic benefits of combination chemotherapy with vincristine, BCNU, and procarbazine on recurrent cystic craniopharyngioma. A case report. J Neurooncol. 1984;2:47-51.
179 Lippens RJ, Rotteveel JJ, Otten BJ, et al. Chemotherapy with Adriamycin (doxorubicin) and CCNU (lomustine) in four children with recurrent craniopharyngioma. Eur J Paediatr Neurol. 1998;2:263-268.
180 Hargrave DR. Does chemotherapy have a role in the management of craniopharyngioma? J Pediatr Endocrinol Metab. 2006;19(suppl 1):407-412.
181 Barloon TJ, Yuh WT, Sato Y, et al. Frontal lobe implantation of craniopharyngioma by repeated needle aspirations. AJNR Am J Neuroradiol. 1988;9:406-407.
182 Malik JM, Cosgrove GR, VandenBerg SR. Remote recurrence of craniopharyngioma in the epidural space. Case report. J Neurosurg. 1992;77:804-807.
183 Ito M, Jamshidi J, Yamanaka K. Does craniopharyngioma metastasize? Case report and review of the literature. Neurosurgery. 2001;48:933-936.
184 Gupta DK, Mahapatra AK. Re: Ectopic recurrence of craniopharyngioma. J Clin Neurosci. 2006;13:405.
185 Yonekawa Y, Ogata N, Imhof HG, et al. Selective extradural anterior clinoidectomy for supra- and parasellar processes. Technical note. J Neurosurg. 1997;87:636-642.
186 Smith ER, Manfredi M, Scott RM, et al. A recurrent craniopharyngioma illustrates the potential usefulness of urinary matrix metalloproteinases as noninvasive biomarkers: case report. Neurosurgery. 2007:60-E1148.
187 Kim SK, Wang KC, Shin SH, et al. Radical excision of pediatric craniopharyngioma: recurrence pattern and prognostic factors. Childs Nerv Syst. 2001;17:531-537.
188 Maira G, Anile C, Rossi GF, et al. Surgical treatment of craniopharyngiomas: an evaluation of the transsphenoidal and pterional approaches. Neurosurgery. 1995;36:715-724.
189 De, Vile CJ, Grant DB, Kendall BE, et al. Management of childhood craniopharyngioma: can the morbidity of radical surgery be predicted? J Neurosurg. 1996;85:73-81.
190 Kahn EA, Gosch HH, Seeger JF, et al. Forty-five years experience with the craniopharyngiomas. Surg Neurol. 1973;1:5-12.
191 Tavangar SM, Larijani B, Mahta A, et al. Craniopharyngioma: a clinicopathological study of 141 cases. Endocr Pathol. 2004;15:339-344.
192 Xu J, Zhang S, You C, et al. Expression of human MCM6 and DNA Topo II alpha in craniopharyngiomas and its correlation with recurrence of the tumor. J Neurooncol. 2007;83:183-189.
193 Pan J, Qi ST, Deng YJ, et al. [Expression of proliferating cell nuclear antigen in craniopharyngioma and tumor recurrence.]. Di Yi Jun Yi Da Xue Xue Bao. 2002;22:363-365.
194 Duo D, Gasverde S, Benech F, et al. MIB-1 immunoreactivity in craniopharyngiomas: a clinico-pathological analysis. Clin Neuropathol. 2003;22:229-234.
195 Nishi T, Kuratsu J, Takeshima H, et al. Prognostic significance of the MIB-1 labeling index for patient with craniopharyngioma. Int J Mol Med. 1999;3:157-161.
196 Agozzino L, Ferraraccio F, Accardo M, et al. Morphological and ultrastructural findings of prognostic impact in craniopharyngiomas. Ultrastruct Pathol. 2006;30:143-150.
197 Losa M, Vimercati A, Acerno S, et al. Correlation between clinical characteristics and proliferative activity in patients with craniopharyngioma. J Neurol Neurosurg Psychiatry. 2004;75:889-892.
198 Raghavan R, Dickey WTJr, Margraf LR, et al. Proliferative activity in craniopharyngiomas: clinicopathological correlations in adults and children. Surg Neurol. 2000;54:241-248.
199 Tena-Suck ML, Salinas-Lara C, Arce-Arellano RI, et al. Clinico-pathological and immunohistochemical characteristics associated to recurrence/regrowth of craniopharyngiomas. Clin Neurol Neurosurg. 2006;108:661-669.
200 Regine WF, Mohiuddin M, Kramer S. Long-term results of pediatric and adult craniopharyngiomas treated with combined surgery and radiation. Radiother Oncol. 1993;27:13-21.
201 Curtis J, Daneman D, Hoffman HJ, et al. The endocrine outcome after surgical removal of craniopharyngiomas. Pediatr Neurosurg. 1994;21(suppl 1):24-27.
202 Paja M, Lucas T, Garcia-Uria J, et al. Hypothalamic-pituitary dysfunction in patients with craniopharyngioma. Clin Endocrinol (Oxf). 1995;42:467-473.
203 Khandwala HM, McCutcheon IE, Flyvbjerg A, et al. The effects of insulin-like growth factors on tumorigenesis and neoplastic growth. Endocr Rev. 2000;21:215-244.
204 Ogilvy-Stuart AL, Gleeson H. Cancer risk following growth hormone use in childhood: implications for current practice. Drug Saf. 2004;27:369-382.
205 Renehan AG, Zwahlen M, Minder C, et al. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. Lancet. 2004;363:1346-1353.
206 Clayton PE, Price DA, Shalet SM, et al. Craniopharyngioma recurrence and growth hormone therapy. Lancet. 1988;1:642.
207 Darendeliler F, Karagiannis G, Wilton P, et al. Recurrence of brain tumours in patients treated with growth hormone: analysis of KIGS (Pfizer International Growth Database). Acta Paediatr. 2006;95:1284-1290.
208 Karavitaki N, Warner JT, Marland A, et al. GH replacement does not increase the risk of recurrence in patients with craniopharyngioma. Clin Endocrinol (Oxf). 2006;64:556-560.
209 Price DA, Wilton P, Jonsson P, et al. Efficacy and safety of growth hormone treatment in children with prior craniopharyngioma: an analysis of the Pharmacia and Upjohn International Growth Database (KIGS) from 1988 to 1996. Horm Res. 1998;49:91-97.
210 Kondziolka D, Nathoo N, Flickinger JC, et al. Long-term results after radiosurgery for benign intracranial tumors. Neurosurgery. 2003;53:815-822.
211 Arai T, Ohno K, Takada Y, et al. Neonatal craniopharyngioma and inference of tumor inception time: case report and review of the literature. Surg Neurol. 2003;60:254-259.
212 Merchant TE. Craniopharyngioma radiotherapy: endocrine and cognitive effects. J Pediatr Endocrinol Metab. 2006;19(suppl 1):439-446.
213 Carpentieri SC, Waber DP, Scott RM, et al. Memory deficits among children with craniopharyngiomas. Neurosurgery. 2001;49:1053-1058.
214 Waber DP, Pomeroy SL, Chiverton AM, et al. Everyday cognitive function after craniopharyngioma in childhood. Pediatr Neurol. 2006;34:13-19.
215 Dekkers OM, Biermasz NR, Smit JW, et al. Quality of life in treated adult craniopharyngioma patients. Eur J Endocrinol. 2006;154:483-489.
216 Kendall-Taylor P, Jonsson PJ, Abs R, et al. The clinical, metabolic and endocrine features and the quality of life in adults with childhood-onset craniopharyngioma compared with adult-onset craniopharyngioma. Eur J Endocrinol. 2005;152:557-567.






