10. Pharmacology in Neonatal Care*
Mary Miller-Bell, C. Michael Cotton and Deanne Buschbach
Optimal pharmacotherapy delivers the maximum intended beneficial effect with the minimum toxicity. Determining the optimal pharmacotherapy for neonates is problematic in that much of the data have been extrapolated from research in adults, children, and laboratory animals. Neonates show dramatic differences in the way they respond to drugs compared with older children and adults and within the neonatal population. 18 Gestational age, chronologic age, and disease state alter a neonate’s ability to metabolize medications and affect the infant’s response to the drug.
This chapter discusses pharmacology as it relates to the neonate and illustrates how rational medication decisions can be made for neonatal intensive care unit (NICU) patients. It also discusses strategies to avoid medication errors, strategies for drug delivery, and the new frontier of a genetic variation–based approach to pharmacotherapy.
PHYSIOLOGY
Pharmacodynamics and Pharmacokinetics
The drug-receptor theory states that the amount and duration of a drug’s availability to a receptor determine its effectiveness. Pharmacodynamics describes what the drug does to the body, whereas pharmacokinetics describes what the body does to the drug, how much drug is available to the receptors, and for how long (Figure 10-1). 12 A drug’s disposition can be described by four processes: drug entry (absorption), distribution, biotransformation, and elimination.
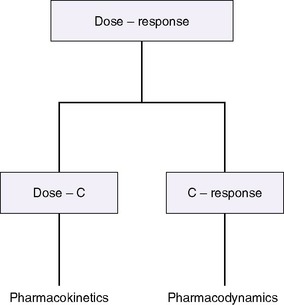
Pharmacodynamics relates the amount of available drug (or active metabolite) to the effect and depends on receptor availability, affinity of the drug for the receptor, and cellular function.
Antagonist drugs block a receptor’s cellular and physiologic activity (e.g., naloxone), whereas agonist drugs elicit the receptor’s action (e.g., cardiovascular agents such as dopamine and epinephrine). Some drugs act with receptors to increase or decrease gene expression (e.g., antenatal steroids), whereas others affect cell membrane permeability. Some drugs, such as methylxanthines, increase or decrease the amount or activity of “second messenger” molecules within cells. Antibiotics and antiviral agents act through some of these mechanisms to reduce the viability of pathogenic organisms by changing vital characteristics and functions. Readers should note that most drugs have more than one effect, so although the desired therapeutic effect may occur, the drug’s other effects can limit its usefulness. Side effects, which can vary from the minor to the prohibitive, occur within the therapeutic range of concentration. Toxic effects result from drug overdose or serum concentrations higher than the recommended therapeutic range.
Individual infants may have idiosyncratic responses to medications, as well as expected responses. Infants who are low sensitivity responders exhibit a drug response less than that expected for a usual dose, whereas infants who are extreme sensitivity responders exceed the expected response for a given dose and drug level. Unpredictable adverse reactions differ from expected responses. Patients may become tolerant to a given drug dosage, as is commonly seen with opiates. Tachyphylaxis, a rapid decrease in drug response without a dosage change, may be related to limited receptors or other intracellular mechanisms. 3,10
Developmental differences in number and function of receptors and intracellular mechanisms are critical to estimating drug actions. An example of developmental effect on pharmacodynamics is the diminished sensitivity of the cardiovascular system to digitalis in the youngest patients; the receptor number increases with age. Changes in alpha- and beta-adrenergic receptors also occur with gestational and chronologic age and must be considered in determining dosage with pressors and inotropes. 12
To elicit the desired therapeutic effect, the drug must be delivered to the receptor and remain available for an appropriate amount of time. 29
Pharmacokinetics describes the delivery and removal of the drug to and from the body. Doses and dose intervals are expressed mathematically by pharmacokinetic disposition parameters related to distribution, biotransformation, and elimination, such as clearance, volume of distribution, and half-life.
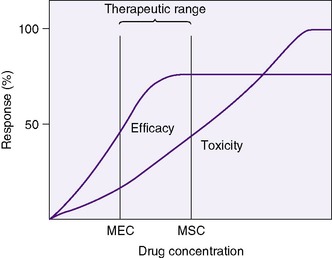
A clinician bases drug choice and dose regimen largely on the desired therapeutic response and toxic effect in “average” patients. Plasma concentration provides a surrogate for effect when the relationship between concentration (C) and effect has been demonstrated in similar patients. The minimum effective concentration (MEC) is that at which 50% of patients exhibit the desired response (Box 10-1). The maximum safe concentration (MSC) is that at which 50% of patients exhibit a toxic response (Figure 10-2). To continue the desired effect, the clinician aims to obtain a target plasma concentration at “steady state” (Css), somewhere between the MEC and the MSC, where most patients exhibit the desired effect and few suffer toxic effects. With ideal maintenance therapy, drug input equals drug elimination. The variability around the Css depends on dose, dose interval, and drug disposition.
BOX 10-1
| C | Drug concentration (plasma or serum) | mg/L |
| Css | Steady state concentration (average) | mg/L |
| MEC | Minimum effective concentration | mg/L |
| MSC | Maximum safe concentration | mg/L |
| F | Extent of drug availability (0-1): how much active drug gets to the systemic circulation | Unitless |
| Vd | Volume of distribution: relates to loading dose | L/kg |
| Cl | Clearance: relates to maintenance dose | L/kg/hr |
| t ½ | Drug elimination half-life: relates to the time course of changes in drug concentration | Hours |
L, Liter = 1000 milliliters; mg, milligram = 1000 micrograms.
The target Css is influenced by the amount of drug bound to plasma protein. In a newborn, free unconjugated bilirubin can displace numerous medicines of lower protein affinity, and numerous medicines can displace unconjugated bilirubin, increasing unconjugated bilirubin’s serum concentration and its potential for toxicity. Intravenous (IV) lipid infusions also can affect protein binding of both bilirubin and some medicines. The drug concentration measured in most available assays is usually the total, both protein-bound and free; therefore the available concentration at the receptor usually is somewhat less than the total serum concentration.
DOSE-CONCENTRATION CONSIDERATIONS RELATED TO AGE
The reported therapeutic range for theophylline in adults is 10 to 20 mcg/mL for bronchodilation. In neonates, the drug has been used to treat apnea of prematurity, and the effective range for this disorder has been 4 to 12 mcg/mL. 29 Theophylline is reported to be 36% bound to plasma protein at a total concentration of 8 mcg/mL in newborns, compared with 70% bound in adults. A total theophylline concentration of 10 mcg/mL in an adult represents 3 mcg/mL free theophylline available to receptors, whereas in a neonate, a total of 4.7 mcg/mL equals 3 mcg/mL of free theophylline (also, the metabolism of theophylline in neonates leads to measurable free caffeine). Decreased bound theophylline in neonates could explain why therapeutic effect is achieved with lower total serum concentration in neonates.
In the NICU, doses and intervals must be adjusted based on changes in the dose-concentration and the concentration-response relationships. This enables a more accurate and precise response to changes in dose-concentration effects, especially total concentrations, but as with the theophylline case, we must watch for clinical effects and estimate other factors’ influences to estimate the free concentration’s effectiveness and the receptor and cellular responsiveness. Potential causes of changes in dose-concentration relationships unique to newborns are described for the pharmacokinetic processes that follow.
Absorption
The process of absorption defines the rate and amount of drug that enters the bloodstream. The parameter F indicates the percentage of dispensed drug available in the systemic circulation, with F = 1 indicating the drug is 100% available. We lack systematic studies of absorption in sick newborns, and differences in absorptive processes are expected but remain unmeasured generally. Some differences in newborns that potentially affect bioavailability include developmental changes in surface area and permeability of gastrointestinal (GI) mucosa, age-dependent changes in acid secretion in the stomach (higher pH than in older children and adults), changes in gastric emptying time and total GI transit time, and the characteristics of GI flora. Drugs such as ranitidine and metoclopramide also affect absorption of other medications by means of the same mechanisms.
“First-pass” pharmacokinetics means the drug is absorbed through the GI mucosa and travels directly to the liver, where it is metabolized and excreted in significant amounts, limiting bioavailability. Different drugs are absorbed at different rates, and different formulations of the same medication may be protected from first-pass metabolism. Drugs also may be given by inhalation, intranasally, intrarectally, topically, intramuscularly, subcutaneously, and intravenously. 12
Distribution
Medications rely on blood flow and drug solubility for distribution to their sites of therapeutic effect. The volume of distribution for a drug is a parameter that relates total amount of drug distributed throughout the body to the serum or plasma concentration.3 It is an attempt to quantify the space in which the drug can go. Strictly defined, it is the hypothetical volume of body fluid necessary to dissolve the total amount of drug as found in the serum. Volume of distribution must be used to estimate the amount of a loading dose or a change in plasma concentration with any bolus dose:
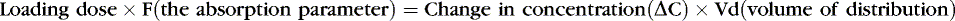
Or, put another way:
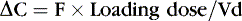
Volume of distribution usually is expressed as a function of body weight, with units of volume per kilogram. Major factors that affect distribution volume are plasma protein binding and body composition. 12,18,29 Changes in body composition happen throughout fetal and newborn life. Total body water decreases with increasing age: 85% in the smallest, most preterm infants; 70% in term infants; and 55% in most adults. Total body water may increase with such conditions as the syndrome of inappropriate antidiuretic hormone (SIADH) excretion, which increases total body water. Extracellular water composes about half this amount in a healthy term neonate. Large water-soluble molecules reach this compartment. Intravascular water composes about 10% of the body weight; protein-bound medications stay in this small compartment. Water-soluble drugs such as penicillins, aminoglycosides, and cephalosporins are distributed in a greater volume in smaller, more preterm infants, therefore requiring a higher loading dose per kilogram, if total body water were the only determinant of volume of distribution.
Plasma protein amounts and binding capacities also differ with gestational and chronologic age. Protein binding is decreased in newborns, because lower amounts of albumin are available than later in life, and fetal albumin has less capacity to bind some drugs. Acidic drugs such as ampicillin, phenytoin, and phenobarbital bind less well, thus increasing the free (available to receptor) fraction of the drug, with resultant increase in effect. Changes in pH also can affect a drug’s affinity for albumin. Fat content varies with gestational age and degree of illness; increased fat content increases the volume of distribution. Lipid-soluble molecules also are distributed in this space.
Of particular concern in newborns is the interaction of circulating unconjugated bilirubin and protein-bound drugs. Several anionic compounds bind to albumin and can displace bilirubin, increasing free bilirubin, thus increasing its potential for toxicity. Bilirubin has a higher affinity for albumin than some other medications; it may displace them from albumin, increasing the medication’s availability and potential to reach toxic levels.
Biotransformation
Biotransformation, or drug metabolism, occurs most commonly in the liver. Phase I metabolism describes the nonsynthetic metabolism of medications. Phase II, usually conjugation, or the addition of a substance to a medication, is synthetic metabolism. Oxidation, conjugation, glucuronidation, and hepatic blood flow change with gestational and chronologic age, diseased states, and use of certain medications. For example, oxidation and glucuronidation are decreased in newborns. Drugs such as acetaminophen, phenobarbital, and phenytoin, which require oxidation for elimination, remain available longer and may be transformed to other active metabolites (the neonatal liver metabolizes theophylline to caffeine), or the drug may remain at significant free concentrations for a prolonged period. The possibility of prolonged peak concentrations of available drug or active metabolites for many pharmaceuticals mandates careful monitoring of drug levels and clinical conditions to titrate dose intervals. To further the potential for confusion and trouble and further the argument for careful assessment of levels and clinical signs of effectiveness and toxicity, a decrease in plasma protein binding (or any other change in volume of distribution) may increase the hepatic clearance of a drug.
Clearance (Elimination)
Drug clearance or elimination occurs by excretion of unaltered drug or biotransformation to an inactive metabolite. Most drug elimination pathways can become saturated if the dose is high enough and dose intervals are too frequent. Most drugs in use in the NICU have therapeutic doses less than those necessary to saturate the elimination system. When clearance mechanisms are not saturated, the Css in plasma is proportional to the dose rate. Clearance equals the rate of drug elimination divided by the drug concentration. 19 Just as volume of distribution relates to loading dose and initial concentration, clearance relates to a maintenance dose that keeps a drug’s concentration at steady state. So for an ideal drug maintained at steady-state concentration:
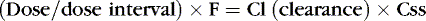
Stated another way:
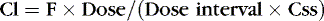
Or, to tailor the dose for a desired steady-state concentration:
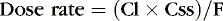
The appropriate dosing rate can be calculated if the clinician can specify the desired steady-state plasma concentration and knows the clearance and bioavailability of a drug (from peak and trough levels in a particular patient).
For example, clearance of theophylline in preterm infants is reported to be 0.017 L/kg/hr. If the desired Css = 8 mg/L, assuming F = 1, particularly if the dose is to be given intravenously:
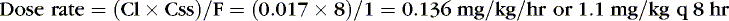
RENAL EXCRETION
The kidney is the primary route of excretion for many drugs commonly used in the NICU. The kidney clears drugs through glomerular filtration and tubular secretion. Examples of medications eliminated through the kidney are aminoglycosides, digoxin, diuretics, and penicillins. Doses and dose intervals of drugs that have renal excretion must change with age and disease state. The glomerular filtration rate (GFR) (i.e., the amount of blood filtered by the kidney in a unit of time) is low at birth and gradually increases over the first weeks. In preterm infants, the GFR starts even lower than in term infants, with a somewhat significant increase occurring at 34 weeks after conception. Tubular secretion also matures with increasing gestational age and depends on tubular function. In adults, aminoglycosides may be dispensed based on creatinine clearance, but in neonates less than 1 week old, serum creatinine may reflect maternal levels, as well as renal impairment. Acidosis and a history of hypoxia or ischemia also may modify an infant’s renal function, slowing excretion and altering pharmacokinetics. Again, measuring levels in cases of suspected renal impairment, whether from suspicious history or laboratory values, is important to determining an appropriate dosing strategy.
Half-Life
A drug’s “half-life” (t ½) is the time necessary for the drug level to decline by 50%. Half-life is related to both volume of distribution (Vd) and clearance (Cl), so that:
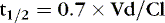
The t ½ is used to predict and interpret the time course of changes in plasma drug concentrations. For example, the time to steady state is 4 to 5 half-lives. The half-life is useful in selecting dose intervals. This concept is illustrated in Figure 10-3.
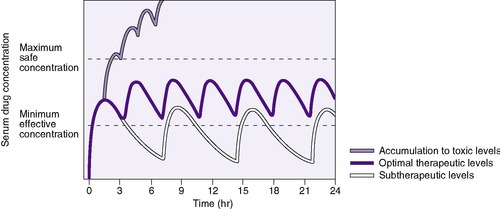 |
| FIGURE 10-3
(From Roberts RJ: Drug therapy in infants, Philadelphia, 1984, Saunders.)
|
Loading doses help expedite reaching desired therapeutic concentrations, especially for drugs with long half-lives, in which a desired effect is needed immediately. For drugs with one-compartment distribution that stay in the circulation and are not stored in cells or tissue, the loading dose may be given as a simple single dose. Drugs that are fat soluble or stored intracellularly are more difficult to assess, and therapeutic levels must be included in the loading dose assessment.
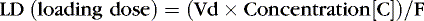
If the volume of distribution for theophylline in preterm infants is 0.7 L/kg, and 8 mg/L is the desired concentration, and F, for an intravenous dose, is assumed to be 1, a loading dose can be calculated.
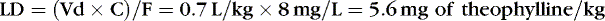
Pharmacogenetics and Pharmacogenomics
In the 30,000-plus known genes, there are over 4 million “common” variants (occurring in more than 1% of the population), many of which directly affect the function of the coded protein. In addition to the multiple factors like age, gender, disease, and concurrent medications, a patient’s genetic code can cause variation in the response to a particular drug. Once a drug is administered, it is absorbed and distributed to its site of action, then interacts with targets, and is finally metabolized and excreted. The enzymes and other compounds involved in each of these processes are subject to genetic variation that leads to variation in function. Pharmacogenetics is the study of the role of inheritance in the individual variation in drug response. Pharmacogenomics is the study of the influence of multiple genes and their interaction with each other and the environment on drug effects. 5,24,28
The first description, over 40 years ago, of a genetically caused variation in drug metabolism was for the enzyme responsible for hydrolysis of succinylcholine. One in 3500 people are homozygous for a gene encoding an atypical form of the enzyme butyrylcholinesterase, which is quite slow to hydrolyze the succinylcholine, leading to prolonged muscle paralysis. Concurrently, an enzyme responsible for N-acetylation, another form of drug metabolism, was found to have a common genetic variant, so some patients are fast metabolizers and others are slow metabolizers of drugs such as hydralazine.
The cytochrome P-450 enzymes are important in phase I drug metabolism. One in particular, CYP-450 2D6, responsible for metabolism of many drugs, has been extensively studied. About 5% to 10% of the adult Caucasian population have genetic variants of the enzyme, leading to decreased activity and higher and more prolonged active drug levels.
Recently, a genetic variant in mitochondrial DNA, the A1555G mutation gene, has been linked to risk for hearing loss associated with aminoglycoside toxicity. The risk-additive variant’s frequency in the general population is estimated between 1% and 3%, but among deaf subjects tested, concurrence of deafness with aminoglycoside treatment with this mutation is quite common. This could lead to future testing before use of aminoglycosides. It is unknown whether tight control of aminoglycoside levels would reduce risk for hearing loss. No recommendations can be made until more extensive, population-based studies are done. Such studies must include accounting for drug levels and duration and genotypes in assessment of risk for hearing loss. 21
In addition to genetic variation in drug metabolism, the genetic polymorphisms of drug targets—including adrenergic and dopamine receptors and enzymes such as acetylcholinesterase—are likely to have effects on the response to drugs targeting these proteins. This may occur with the use of medications such as bronchodilators, pressors, and inotropes, and ACE-inhibitors such as enalapril and captopril.
The study of pharmacogenetics and pharmacogenomics is new, especially to neonatology, in which little is known about pharmacokinetics and pharmacodynamics of commonly used medications. The genetic revolution that has come with completion of the sequencing of the entire human genome is on its way to reaching neonatology with the availability of rapid analysis of large population-based samples for thousands of genes and their variants and how they relate to drug response. In the future, these studies will lead to improved understanding of individual variation in drug response, which should allow development of strategies to individualize care and help avoid complications resulting from heretofore unexplained genetic variations. The challenge will be to understand how the multiple genes and environmental factors interact in individual infants to alter risk for disease and adverse drug responses in fragile infants.
DATA COLLECTION
Clinicians should be aware of a medication’s desired effects, side effects, and toxicities; know when they are expected to occur; and monitor for these effects. Whether a dose effect occurs or not should be noted. Dose–plasma concentration results should be recorded when therapeutic drug monitoring is done. If the drug’s serum concentration relates to clinical response, the blood concentration should be followed in addition to clinical signs. To optimally use drug serum levels, the expected blood concentration is calculated from the dosing history, and patient variables that may affect pharmacokinetics with the timing of blood samples are considered. A comparison of expected values with measured values allows rational adjustment of future dosing. 4 Potential explanations for differences between measured and expected concentrations are listed in Box 10-2.
BOX 10-2
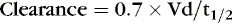
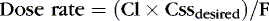
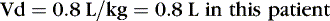
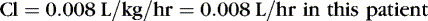
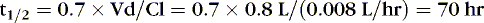
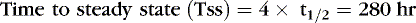
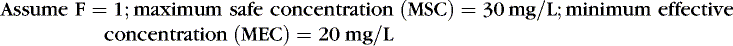
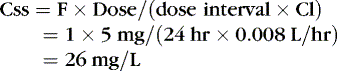
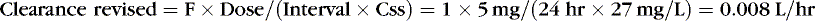
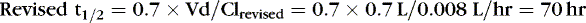
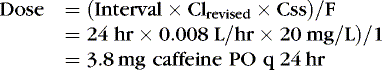
• Inadequate compliance
• Inadequate medication delivery
• Inappropriate timing of samples
• Laboratory error
• Revision in initial estimates of necessary pK required
pK, Pharmacokinetics.
Even if predictable pharmacokinetic and pharmacodynamic changes are considered, other factors may influence a drug’s effect. Clinical end-points must be followed and recorded and dose regimens adjusted accordingly. One example is the monitoring of renal function with indomethacin dosing: If clinical signs of renal dysfunction are noted, the drug is not administered. A pharmacist should be included in the caregiving team to clarify dose and disposition parameters for individual patients with their various conditions.
If a suboptimal clinical response is noted in conjunction with a subtherapeutic plasma concentration, revised estimates of clearance should be adjusted with one or two available plasma concentrations. If a single level is drawn after absorption and distribution is complete or near steady state, then the maintenance dose formula can be rearranged to calculate the revised clearance. The common-sense approach suggests that if a patient has half the expected concentration of a drug, then perhaps the clearance is twice the initial estimate. If the patient has twice the expected concentration, the clearance likely is half the initial estimate. However, this technique is misleading if steady state has not been reached. If a drug’s level is higher than expected and higher than what is considered safe or if toxicity is noted, the drug should be discontinued until the concentration decays to the appropriate target. If two concentrations are available after absorption and distribution, half-life is determined by plotting the concentrations on semilog paper. The revised clearance is calculated by rearranging the half-life formula:
Once the clearance and desired steady state are known, a new dose rate can be calculated:
Examples
The following examples illustrate the need to pay close attention to issues of drug delivery and clinical effects. The following examples are for caffeine citrate in neonates. The half-life of caffeine citrate in neonates is 3 to 4 days, the Vd is 0.8 to 0.9 L/kg, and clearance is 0.008 L/kg/hr.
EXAMPLE 1: A 15-day-old 1-kg preterm infant receives oral caffeine for apnea of prematurity. After the loading dose of 20 mg/kg, the infant has received 5 mg every 24 hours for 5 days. At 8 am on the fifth day, 4 hours after the last dose, the baby’s heart rate is more than 180 beats/min but apnea has not been a problem. Clinical and laboratory evaluation of tachycardia includes consideration of caffeine toxicity. A blood sample for caffeine is sent to the laboratory. Estimate the concentration.
Necessary data:
Therefore:
The Css was estimated using average Vd and Cl values reported in similar infants, adjusted for this infant’s weight. If this infant has diminished clearance relative to “average,” toxicity may result from the standard dose. Toxicity may not have been noted until day 5 because of the estimated time to steady state (Tss) of 115 hours.
EXAMPLE 2: At 4 am, 24 hours after the last dose, when the next dose is due, caffeine concentration of 27 mg/L is reported. This is higher than the therapeutic range for caffeine in neonates and is most likely the result of decreased clearance. Using this concentration, estimate the time when the concentration will decline to 20 mg/L and determine a 24-hour dose schedule to maintain that concentration. The 4 am dose is held, and the tachycardia resolves 24 hours later.
Therefore the concentration 70 hours later should be one half of the measured 27 mg/L, or about 13.5 mg/L. To maintain a concentration of 20 mg/L:
EXAMPLE 3: Before the new oral regimen is initiated, another blood specimen is drawn 70 hours after the first and is 14 mg/L. Because tachycardia has resolved, an oral regimen based on the last two levels is begun to maintain a caffeine concentration of 20 mg/L. Estimate the necessary maintenance dose: The concentration fell 50%, from 27 to 13.5 mg/L in 70 hours, which confirmed our original estimate of half-life.
DRUG CATEGORIES
Antimicrobial Agents
Antimicrobial agents inhibit growth or kill microorganisms; they include antibacterial, antiviral, and antifungal agents. Bacteriostatic agents limit growth, allowing host defenses to control spread; this will not reliably eliminate a pathogen. Bactericidal agents kill the pathogen. Bactericidal agents at low concentrations may be bacteriostatic. Minimum inhibitory concentration (MIC) is the lowest concentration of an antimicrobial that stops the spread of an organism in laboratory culture media. This cannot be directly measured in an infected neonate and depends on tissue concentration and numbers of bacteria present. Minimum bactericidal concentration (MBC) is the lowest concentration of antimicrobial that reduces microbial number in laboratory media by 99.9%. Pathogens can develop resistance to antimicrobials by changing their cellular structures or producing enzymes that reduce antimicrobial activity.
For effective antimicrobial action, the drug must reach an adequate concentration in the infected tissue. The ideal concentration elicits maximum effect on the pathogen with minimum effects on the patient. Selection criteria for antimicrobials include the microorganism’s sensitivity, the availability of the drug to the target tissue (some antibiotics do not cross the blood-brain barrier), bioactivity of the antimicrobial in the target tissue, the known MIC and MBC relative to side and toxic effect levels for the medication, and the individual infant’s biologic state—that is, whether the systems of absorbance and elimination are working adequately for effective and safe drug delivery and removal. When the use of antimicrobial agents is planned in a seriously ill infant, as with other drugs, greater consideration must be given to clinical status than to the gestational or chronologic age.
Diuretics
Diuretics are used in the NICU to remove excessive extracellular fluid. Diuretics commonly cause a loss of electrolytes along with water. Response to any diuretic depends on renal function and the drug’s ability to reach its target in adequate amounts. Most diuretics work within the tubule, but any drug that increases GFR can increase water loss. Drugs that increase cardiac output without decreasing renal perfusion and others that specifically increase renal blood flow also cause diuresis.
In infants, renal tubular function improves with increasing chronologic and gestational age. Because of poor absorption and response to aldosterone (especially in extremely preterm infants), electrolyte losses can be clinically significant with the addition of a loop diuretic such as furosemide or bumetanide. The ongoing losses may lead to hypochloremic metabolic alkalosis and less response to the diuretic.
Delivery of diuretics to the kidney loop increases with increasing chronologic and gestational age. Most diuretics rely on secretion from the proximal tubule and filtration through the glomerulus to reach their site of action. Both these functions improve with age. Enteral absorption of some diuretics is limited, so clinical effectiveness and electrolyte stability must be monitored closely to help determine safe and effective dosage regimens. The kidney also is responsible for diuretic excretion, again through tubular secretion and glomerular filtration. Because these functions are age dependent, the clinician must ensure that clearance time is adequate to avoid toxic levels.
Cardiovascular Drugs
Medicines used to improve cardiovascular function include digitalis and the sympathomimetic amines, which include drugs such as dopamine, dobutamine, and epinephrine. Antiarrhythmics, including digoxin, act to control the electrical conduction within the myocardium.
The sympathomimetic amines bind to β and G receptors; the number and availability of receptors determine response. β 1 receptor response leads to constriction of vascular smooth muscle. β 2 receptors cause decrease in GI motility. G1 receptor stimulation stimulates cardiac contractility, and G2 response includes vascular and bronchial smooth muscle relaxation. The response in any individual, and in any individual’s specific organ system, depends on the relative amount of these receptors. Receptor numbers and their linked response elements within cells vary with gestation and clinical condition, and response must be monitored to aid in dosing decisions. Prolonged administration of sympathomimetic amines can lead to decreased response—an example of tachyphylaxis.
Antihypertensive agents occasionally are used in neonates for essential hypertension and occasionally to decrease afterload in neonatal patients with heart failure. These include volume reducers such as diuretics, inhibitors of physiologic regulators of blood pressure like enalapril, and drugs that decrease vascular resistance through β and G receptors.
The pathophysiology of neonatal disease should direct choice of cardiovascular agent. Extremely close monitoring of physiologic effects helps determine safety and efficacy of therapy. Monitoring must include very frequent, if not continuous, monitoring of blood pressure, heart rate, perfusion, and oxygen saturation (preductal and postductal in some cases). Because other drugs are often given as a neonate receives cardiovascular medicines, thorough knowledge of possible drug interactions is mandatory. Absorption of cardiovascular drugs is unpredictable. The sympathomimetic amines must be given by the intravenous route unless used in an emergency situation when endotracheal (ET) administration of epinephrine is indicated. Once dosed, the drug must be delivered to the target organ system. Infants in shock may not have the circulatory wherewithal to deliver the medication to elicit the desired therapeutic response. Because of the variability in β and G receptor development and distribution, undesired side effects in various organ systems may accompany desired responses. Rapid metabolism of the sympathomimetic amines demands continuous IV infusion, and infiltration of IV fluids may lead to significant tissue damage. Along with the physiologic effects, these IV lines must be carefully monitored. 19,29
Central and Peripheral Nervous System Drugs
Nervous system drugs include analgesics, which decrease pain sensations; anesthetics, which control pain peripherally or in the central nervous system (CNS); sedatives/hypnotics including barbiturates (phenobarbital) and non-barbiturates (chloral hydrate, lorazepam), which do not control pain and can control some seizures; and antiepileptic agents, which are designed to control seizures (phenytoin, fosphenytoin). These drugs are associated with problems of addiction, tolerance, dependence, and withdrawal.
Addiction is a complex lifestyle change that involves drug-seeking behavior, which is not applicable to neonates. Tolerance occurs with many drug types. Tolerance exists when increasing doses and serum concentrations of a medicine are necessary to achieve a desired effect. A patient is dependent on a medication when regular drug administration is necessary for physical well-being. Withdrawal is a collection of physiologic and behavioral signs attributed to the absence of a medication in a dependent individual. Withdrawal has been identified for many medications, but it has been classified and described, along with weaning protocols, for opiate analgesics (see Chapter 11). 8
The mechanism of action of most CNS medications is not clearly known. Again, careful monitoring of therapeutic effects relative to dose, duration, and serum concentrations is extremely important. Significant respiratory depression can occur with most CNS medications, so appropriate resuscitation equipment must be available. Variations in hepatic metabolism and volume of distribution are important in the ongoing assessment of dose-response. Some medications are highly fat bound and are slowly released into the circulatory system, causing prolonged effects, both therapeutic and undesired (e.g., respiratory depression, poor gastric motility, and abnormal neurologic function, such as feeding difficulties).
If hypothermia is used for infants with hypoxic-ischemic encephalopathy (HIE), evidence suggests that opiates accumulate in the circulation in excess of accumulation in similar infants with HIE who are not cooled. Therefore, when using opiates in cooled infants with HIE, opiate levels are likely to be higher than expected for a given dose and expectations for neurologic examination must be modified given accumulation of high levels and delayed expectations.25
PREVENTION OF THERAPEUTIC MISHAPS
More individuals die each year in the United States from medical error than from traffic accidents. Many medical errors are medication errors. 16 Even after making a correct choice of medication, one must pay attention to the appropriate dose and interval based on factors that affect a drug’s pharmacokinetics and pharmacodynamics. Drug delivery must be ensured: This includes appropriate dose calculations; appropriately written and read orders; appropriate mixing with diluents; attention to drug interactions, incompatibilities, and contraindications; and drug delivery systems. In addition, effects of therapy at the chosen dose and systematic monitoring for therapeutic and toxic effects must be included in NICU care when medications are used.
Human error may occur, and it is in hospital areas of highest acuity, such as intensive care units and emergency departments, that the majority of medication errors have been described. In a review of medication errors in a large general hospital, pediatric medication errors occurred at a higher rate (5.89 errors per 1000 patients) than in the emergency department and medicine, surgery, and obstetric and gynecology (OB-GYN) units, with dosage calculation errors being the most common problem. 16 The authors suggested initiatives designed to prevent, detect, and avert problems associated with major factors associated with errors. In addition to calculation errors, problems included availability and information on drug therapy such as pharmacokinetic and drug interaction information, appreciation of patient characteristics that alter drug therapy, and confusing drug nomenclature. 1,2,6,15 In another review of hospital errors involving dosage equations, 17 antibiotics were the principal drug class involved. Errors in the equations used to calculate doses for all drug classes accounted for 29.5% of the errors.
Completing the “six rights” of medication administration (Box 10-3) in the NICU is complicated by the small doses and dosage adjustments based on infant weight or surface area. Investigators estimate that 8% of drug doses calculated and administered by competent NICU nurses are at least 10 times greater or less than the ordered dose.26 Another error risk arises from the fact that many drugs must be diluted because they are ordered in amounts that are not commercially available. The rate of drug entry, or absorption, also varies, depending on route of administration. Calculations can be difficult and at least must be double-checked. Examples should be readily available to those responsible for calculating doses. Other suggestions include the use of standardized drug preparations and dosing and standardized nomenclature or computer/digital order entry, with alerts for unusual doses. To avoid errors with emergency “code” medications, the doses of emergency medications should be calculated on admission, along with appropriate infusion rates (Figure 10-4). The calculated doses for the most commonly administered medications should be posted at the bedside; these should be updated routinely with the passage of days and weight changes (as the pharmacokinetics and pharmacodynamics change).
BOX 10-3
| Right drug | Right dose |
| Right patient | Right time |
| Right route | Right response |
 |
| FIGURE 10-4
Calculations for neonatal resuscitation medications. Other drugs and dosages could be added (see Table 4-2). ET, Endotracheal; IV, intravenous.
|
The Rule of Six was developed originally for use with vasopressor agents in code situations (its use has extended beyond that). The Rule, which allows nurses to estimate a pediatric dose by using a factor of 6, is prone to error. Standardized drug concentrations are less error prone and safer for patients than the Rule of Six. 22Because of medication errors, The Joint Commission (TJC) has required that standardized intravenous drug concentrations be used for pediatric patients receiving medications for which the Rule of Six was routinely used. Another error-avoidance strategy, not mandated by TJC but strongly recommended, is to integrate a clinical pharmacist into patient care rounds with physicians and nurses, particularly in intensive care and oncology, to provide more direct patient care and consultation rather than the traditional role of drug preparation and dispensing. Pharmacist interventions can reduce medication errors and adverse drug events.14,27 In addition, designing the ordering system to reduce complexity and provide rule-based order screening and double-checking of calculations and developing effective information delivery may be more effective than traditional education or process-improvement efforts that target interventions after an error occurs. 9,16
The American Academy of Pediatrics has published further recommendations for reducing medication errors for pediatric patients.2 These include some hospital-wide actions, including the establishment of a clearly defined system for drug ordering, dispensing, and administration, with review of the original drug order before dispensing and administration. Confirmation of patient weight and drug dosage and strength is also recommended. 17 Avoiding the use of the terminal zero to the right of the decimal point (e.g., writing 5 instead of 5.0), and using a zero to the left of a dose less than 1 (e.g., using 0.1 rather than .1) will help reduce medication errors. Avoid abbreviations of drug names (e.g., MS may mean either morphine sulfate or magnesium sulfate), spell out dosage units rather than using abbreviations (e.g., units rather than U, or mcg for microgram rather than μg), and use generic medication names rather than trade names. Avoid verbal orders whenever possible. 22 See Box 10-4 for other interventions to reduce medication errors. For pediatric nurses, recommendations include the following:
BOX 10-4
| Strategy | Examples |
|---|---|
| Develop a neonatal and pediatric formulary | Neonatal/pediatric dilutions of pediatric formulary for gentamicin, hydrocortisone, magnesium sulfate |
| Develop age-specific dosing guidelines | All medications commonly used to treat neonatal and pediatric patients |
| Use technology | Computerized physician order entry |
| Develop protocols and procedures | Fluid management, skin care, insulin |
| Support nonpunitive error reporting | Medication incident reports |
| Provide up-to-date references | Neonatal and pediatric drug dosage handbooks |
• Familiarizing oneself with the medication ordering and use system
• Verifying drug orders before administration
• Confirming patient identity before each dose
• Verifying calculations with a second individual
• Verifying any unusually large volumes or dosage units for a single patient dose
• Verifying verbal orders by “reading back” the complete order to the prescriber
• Listening to the patient, parent, or other caregiver
• Asking questions as to whether a drug should be administered
• Maintaining familiarity with the operation of administration devices and the potential for errors with such devices
METHODS OF ADMINISTRATION
Once a clinician orders a medication and the drug and dose are found to be appropriate for that particular infant, the nurse’s challenge is to administer the medication correctly. The following sections address some of the means of delivery that help improve accuracy of drug delivery.
Oral Administration
Variations in oral bioavailability and unanticipated and unmeasurable loss of drug complicate administering oral medications to newborns. Loss of medication occurs when infants regurgitate or require gastric suctioning and lose residual fluid that may include medication. If an infant is receiving orogastric (OG) or nasogastric (NG) feedings, medication should be placed into the center of the barrel of a syringe containing a small portion of the feeding. Medication may adhere to the plastic and decrease the amount of medicine delivered. The nurse must document drug administration attempts and any possible loss of drug, with an estimate of the amount lost. For infants receiving oral medications, documenting the color of the emesis or residual material helps determine presence of medications that have distinctive color.
If an infant is bottle fed, the nurse may put the medication in the full bottle. However, if the infant fails to take the whole volume, he or she has not received the full dose. One option is to finish the volume with gavage feeding. Another option is to gently introduce very small portions of a dose into the cheek pouch and wait for the infant to swallow. Another method is to put 5 to 10 mL of a feeding, with the medication, in a small bottle and let the infant take that amount; then continue with the remainder of the feeding. Medication also may be placed into a nipple with a small volume of formula and offered to an infant. For breast-feeding infants, medication may be administered into the mouth as just described, with or without a small volume of expressed breast milk. As with all dosing of medicines, it is imperative to record doses and volume and characteristics of any residual material or emesis.
Intramuscular Injection
A newborn infant has relatively little muscle mass to receive injections. When intramuscular (IM) injections are necessary, as with vitamin K, the anterior thigh is the site of choice. Comfort measures should be given before and after injection (seeChapters 5 and 12). Clean the site with alcohol, insert a 22- to 25-gauge needle into the muscle, and for most medications, draw back on the syringe to ensure safe needle placement (unless specifically contraindicated); then inject the medication. After injection, the area is massaged. For an infant weighing less than 1500 g, the volume injected into one leg should not exceed 0.5 mL. Document the administration.
Intravenous Administration
IV medication can be given by push or antegrade injection, pump infusion, and retrograde injection. Retrograde injection is no longer recommended to administer IV medications to neonates. 3 Although drugs directly enter the bloodstream, the time necessary to complete drug delivery to receptors is a function of dosage volume, IV flow rate, and injection site (depending on particular IV methods). 23,29 Failure to recognize these potential time lags could result in inappropriate expectations of the timing of physiologic responses and peak and trough concentrations. The use of microbore IV tubing will facilitate rapid drug delivery because the volume of the fluid in the tubing is reduced. An example of a pediatric syringe infusion preparation and delivery chart for common neonatal drugs may be useful to the reader. 29Some drugs should never be administered into the umbilical vein or artery, and drug incompatibilities should be recognized before setting up multiple drug dosing through the same IV line. Careful monitoring for infiltrates and knowledge of drug-specific treatment for this complication are essential to safe IV drug administration. Continuous IV infusion of pressors is common in the NICU, and because of their rapid clearance and physiologic importance, these infusions should never be interrupted without orders. Because of the sudden influx of potent medication, flushes to clear lines with continuous infusions of sympathomimetic amines should be avoided.
PUSH INJECTION
IV push medications must be mixed in appropriate volumes, delivered through appropriate-size syringes, and followed with an appropriate flush solution: heparin with normal saline solution (NS), 10% or 5% dextrose and water (D 10W or D 5W). If numerous flushes are given, care must be taken with the osmolality of the flush solution.
To administer an IV push injection, prepare the IV port closest to the patient. Administer a small volume of appropriate flush solution, and then administer the medication over 1 to 2 minutes. Slow pushes are ordered sometimes, but the rate should be specified by the ordering medical care provider. A post-medication flush is given at the same rate as the medication to clear the line of remaining medication. IV push administration of many medications used in the NICU is contraindicated because of the possibility of immediate adverse reactions associated with rapid bolus injections. Opiates and sedatives should be given with great care and with constant attention to respiratory and cardiovascular parameters. Check a pharmacology reference if there is any uncertainty.
ANTEGRADE INJECTION
Antegrade injection is the introduction of medication into an entry port along the course of the IV tubing. The flow of maintenance fluid carries the medication to the patient at its rate. Because infusion rates in neonates are characteristically low, significant delays in drug delivery result. If rapid infusion is necessary, as with emergency resuscitation medications, more rapid infusion rates are necessary. This can lead to a significant medication error if, after drug delivery, the IV rate is not returned to baseline.
PUMP INFUSION
To avoid delay of drug delivery, two methods of pump infusion using a mechanical infusion device allow control of drug amount and delivery rate. These devices consist of a pump that can be set to deliver a specific volume over a specific time, a syringe or other container that holds the medication or fluid to be delivered, and connecting tubing to connect the pump to a port for drug delivery. Because pumps vary by manufacturer and some may be used in a variety of ways, each NICU should have a policy to ensure that each staff member carries out pump infusions in the same manner. If different care providers start and end an infusion, the method used must be communicated.
Method 1
An exact ordered amount of medication is drawn into a syringe and diluted if necessary to provide the volume necessary for pump operation. This drug plus diluent fluid is flushed through the tubing, and the syringe is placed in the pump. After the pump finishes the infusion, some medication remains in the tubing and syringe hub. This medication needs to be flushed into the IV line with a flush solution to deliver the entire ordered dose.
Method 2
Medication is drawn into the syringe through the connecting tubing until the desired volume is in the syringe. The syringe then is placed on the pump, and a volume carrying the ordered amount of drug is infused. The tubing and syringe hub need not be flushed, because the infant has already received the entire ordered dose.
OTHER CONSIDERATIONS
Health care providers must remain attuned to additional concerns when administering IV medications. Medications may require filters or protection from light sources or have significant specific gravity osmolarity. A 0.22-mcg filter may provide “cold sterilization” (i.e., remove particulate matter and bacterial contamination). Some medications cannot be administered through a filter, because the filter removes the active ingredient. Medications with a specific gravity less than that of the IV fluid have a tendency to accumulate at high points in the IV tubing, whereas those with a higher specific gravity settle into low tubing loops, in both cases resulting in delayed and inaccurate drug delivery.
HOW TO GET INTRAVENOUS ACCESS: INSERTING PERIPHERAL INTRAVENOUS LINES
Common sites for IV placement in neonates include the hands, feet, arms, legs, or scalp veins. A transilluminator may help outline vessels in extremities. (When using a transilluminator, be mindful of potential burns from the high-intensity light source.)
Equipment
• Catheters with needles of appropriate size for the vessel
• Tape
• Alcohol
• Gauze
• Syringe with flush solution
• Tourniquet
• Arm board or leg board
• Restraints (as necessary)
• Gloves
• Comfort measures (see Chapter 12)
Procedure
Always consider comfort measures with any potentially painful procedure. Use of local anesthetic or other analgesic also should be considered (seeChapter 12). Assemble equipment at the bedside. Provide adequate temperature support. Tear two pieces of tape that are about 2 inches long and ½ inch wide; also tear three pieces that are 6 inches long. Select a vessel after confirming it is not an artery. Determine the direction of flow; veins fill toward the heart, arteries away from the heart.
Restrain the infant enough to avoid movement that prevents line placement. Some care providers place a leg board or arm board before the catheter is placed; others secure the limb after the catheter is in position. First flush the needle/catheter, and then remove the syringe. Place a tourniquet around the extremity, taking care to not pinch the skin. (Some caregivers prefer to not use a tourniquet and with practice may achieve success equal to that of caregivers using one.) Clean the site with alcohol, and allow it to dry. After gloving, insert the needle catheter into the vessel using your hand and fingers to anchor the skin surrounding the vessel. Insert the needle catheter at an acute angle and in the direction of blood flow. Observe for blood return or flashback into the tubing or cannula of the catheter. Some vessels do not provide blood return; babies in hemodynamic shock also may not have blood return. If the needle is thought to be in the vessel but no blood return is seen, then a small amount of flushing solution may be injected. If the needle is not in the vessel, the tissue will swell. If it blanches, the vessel is probably an artery. If blood return is seen, inject flushing solution to clear the needle, remove the needle, and gently advance the catheter.
Place a short piece of tape or small piece of transparent dressing across the catheter to secure it. Cross a longer piece of tape around the back of the catheter, and cross the ends across the front of the catheter. Check for proper position by disconnecting the IV catheter from the syringe to note blood return or by infusing a small amount of flushing solution. If necessary, use gauze under the IV catheter for support. Secure the IV catheter in place by using another long piece of tape. Cover the IV site to protect it. Leave adequate access to skin close to the IV site to allow monitoring for infiltrates.
If the medication is to be administered intermittently and the line is not otherwise used, it may be “heparin-locked” and flushed every shift with heparin solution (0.2 unit of heparin per milliliter of solution). Heparin solutions are available in several concentrations. It is good unit practice to standardize the volume and container type for each concentration and to individualize how each concentration-specific container looks and where it is kept. Controversy exists over the use of heparin versus NS for flushing lines. Two articles may be of interest: in one a rabbit model was used to determine the length of time for patency of catheters “locked” with heparin compared with those with NS; the second article examined the same issue in newborns and included a useful table comparing and contrasting the literature on the topic. Neither study found significant difference in length of time for catheter patency based solely on the infusate. 11,13
Teaching Model
Models for teaching IV insertion with various needles and catheters vary from the highly sophisticated (and expensive) computerized human patient simulator to the “low-tech” and inexpensive human placenta. The computerized human patient simulator is available in three models: adult, pediatric, and neonatal. The simulator allows lines to be inserted in vessels; then bar-coded syringes of “drugs” can be administered through the lines, and the simulator will respond with the appropriate physiologic response, which may include changes in blood pressure, heart rhythms, respiratory effort, and pupil dilation. Less advanced techniques include various models of neonate-size manikins with visible “vessels” in the scalp, arms, legs, and feet.
Critical Assessment
Criteria for Intravenous Extravasation
• Check all indwelling lines hourly for signs of extravasation.
• Look for phlebitis, edema, burns, adequacy of perfusion to site, hardness of tissue, or inflammation at needle site.
• For scalp veins, check dependent side of head for edema.
The fetal side of a human placenta is an inexpensive and easy-to-use model. The needed supplies and procedure follow.
Supplies
• Placenta
• Assorted needles and catheters
• Syringes with flush solution
• Gloves
• Tape
Procedure
After gloving, place the placenta fetal side up on drapes. Remove the fetal membranes, exposing the rich network of vessels. After re-gloving, insert IV needles with catheters into the larger vessels first and then into smaller vessels with improving technique. Tortuous or branching vessels can be used for various methods. Once catheters are in position, practice securing with various taping methods.
COMPLICATIONS OF INTRAVENOUS THERAPY
Complications of intravenous therapy include phlebitis, infiltration, hematomas, chemical burns, compartment syndrome, and emboli.19Long-term complications include disfigurement, contractions, and the need for surgical repair or amputation. Frequent (at least hourly) assessment of IV sites helps reduce, but does not absolutely prevent, IV complications (see the Critical Assessment box above). Swelling or discoloration of the extremity or skin at the needle tip is a sign of trouble, and the line should be removed. In the scalp, infiltration may be difficult to assess because swelling occurs not only at the IV site but also on the dependent side of the head. Scalp edema on the dependent side or a swollen eye is an indicator of scalp vein infiltration.
Footdrop7 and compartment syndrome, in which nerves and vessels are damaged by swelling of tissue within a limited space, have been associated with positioning a footboard along the lateral aspect of the fibula, with or without an IV infiltration. The use of rolled washcloths as footboards or extensive padding of IV boards with cotton or gauze may prevent excessive pressure. Unnoticed infiltrations may result in significant tissue loss. Warm soaks are contraindicated, because when extravasated fluid is warmed, it may exacerbate the burn, maceration, and necrosis. In addition, heat increases oxygen demand in already compromised tissues.
Elevating the infiltrated area increases venous and lymphatic drainage, which helps decrease the edema. Hyaluronidase destroys extracellular barriers, allowing rapid diffusion and absorption of the extravasated fluid. For vasoconstrictive substances that extravasate, local use of vasodilators like phentolamine can aid in reperfusion.
Table 10-1 lists treatment approaches for extravasation.
| Drug Supplied | Dosage/Administration | Comments |
|---|---|---|
|
Hyaluronidase (Amphadase)
150 units/mL
|
1 mL (150 units) given as 4 or 5 intradermal 0.2 mL injections with a 25-gauge needle around the periphery of the IV extravasation site |
Use with extravasation of hyperosmolar or extreme pH drugs.
Administer within 1 hr of event.
Not for use with vasoactive drugs
. |
|
Phentolamine (Regitine)
5 mg/mL in 1-mL vial
|
0.5 mg/mL given as 4 or 5 intradermal 0.2 mL injections with a 25-gauge needle around the periphery of the IV extravasation site |
Prepare a dilution.
Use with vasoactive drugs.
May be given up to 12 hr after an event.
|
PARENT TEACHING
IV lines in newborns may frighten parents, especially scalp vein lines (see the Parent Teaching box above). Without information, parents may mistakenly believe the fluid or a needle is going directly into their baby’s brain. It is helpful to reassure the parents that a needle, the fluid, and possibly medications are going into large veins. Also, reminding parents that although their infant has an IV line in place, they may still touch, hold, and feed him or her may help parents cope with interventions.
Parent Teaching
Indwelling Lines for Parents
Talk with parent about indwelling lines. Discuss:
• Type of line, purpose, and any limitations on holding, handling, or feeding the infant
• Pain control measures for the placement of lines
• That lines often need to be restarted
At discharge:
• The name of the medications, the dosages, purpose, routes, and any potential side effects
• Medication administration and what to do if the infant does not receive the full dose of medication
Parents should be made aware that pain assessment and control are part of the caregiver’s ongoing efforts, and both are addressed during IV placement and maintenance. Encourage parents to assist in pain management strategies during IV placement (see Chapter 12). They should be told that a newborn’s venous fragility, combined with the types of solutions used, makes restarting IV lines and multiple sticks per line relatively commonplace. The potential for infiltration also should be addressed, and parents should be included in the effort to monitor the appearance of IV sites.
As for an infant’s medications, the parents should be made aware of treatment choices in the NICU. They need not know the details of medication dosing but should be made aware of significant medications in their infant’s treatment regimen. At discharge, parents must know the names of their infant’s medications, their actions and the dosage, frequency of administration, and side effects, as well as where to obtain refills for each drug. Some of the medications given for infants are not readily available at some of the smaller pharmacies. Caregivers must teach parents to administer prescribed medicines, and the parents must demonstrate their ability to safely and reliably give their infant the recommended doses. The parents should receive written drug information instructions, which may be developed by the unit for their families or may be commercially available from such companies as Micromedex Thomson Reuters Healthcare (www.micromedex.com). Instructions must include actions, dosing amounts, routes of administration, dosing schedule, and potential side effects.
REFERENCES
1. American Academy of Pediatrics, Red Book: Report of the Committee on Infectious Diseases. ed 27 ( 2006)The Academy, Elk Grove Village, Ill.
2. American Academy of Pediatrics, Committee on Drugs and Committee on Hospital Care: Prevention of medication errors in the pediatric inpatient setting, Pediatrics 112 (2003) 431.
3. Brodsky, D.; Martin, C., Neonatology review. ( 2003)Hanley & Belfus, Philadelphia.
4. Capparelli, E.V., Clinical pharmacokinetics in infants and children, In: (Editors: Yaffe, S.J.; Aranda, J.V.) Neonatal and pediatric pharmacology ( 2005)Lippincott Williams & Wilkins, Philadelphia.
5. Evans, W.E.; McLeod, H.L., Drug therapy: pharmacogenomics—drug disposition, drug targets, and side effects, New Engl J Med 348 (2003) 538.
6. Fernandez, C.V.; Gillis-Ring, J., Strategies for the prevention of medical errors in pediatrics, J Pediatr 143 (2003) 155.
7. Fischer, A.Q.; Strasburger, J., Footdrop in the neonate secondary to the use of footboards, J Pediatr 101 (1982) 1003.
8. Franck, L.; Vilardi, J., Assessment and management of opioid withdrawal in ill neonates, Neonatal Netw 14 (1998) 39.
9. Glauber, J.; Goldmann, D.A.; Homer, C.J.; et al., Reducing medical error through systems improvement: the management of febrile infants, Pediatrics 105 (2000) 1330.
10. Hamzaui, F.H.; Murakawa, G.J., Topical medications, In: (Editors: Yaffe, S.J.; Aranda, J.V.) Neonatal and pediatric pharmacology ( 2005)Lippincott Williams & Wilkins, Philadelphia.
11. Hanrahan, K.S.; Kleiber, C.; Berends, S., Saline for peripheral intravenous locks in neonates: evaluating a change in practice, Neonatal Netw 19 (2000) 19.
12. Kauffman, R.E., Drug action and therapy in the infant and child, In: (Editors: Yaffe, S.J.; Aranda, J.V.) Neonatal and pediatric pharmacology ( 2005)Lippincott Williams & Wilkins, Philadelphia.
13. Kyle, L.A.; Turner, B.S., Efficacy of saline vs. heparin in maintaining 24-gauge intermittent intravenous catheters in a rabbit model, Neonatal Netw 18 (1999) 49.
14. Leape, L.L.; Cullen, D.J.; Clapp, M.D.; et al., Pharmacist participation on physician rounds and adverse drug events in the intensive care unit, JAMA 282 (1999) 267.
15. Lesar, T.; Briceland, L.; Stein, D.S., Factors related to errors in medication prescribing, JAMA 277 (1997) 312.
16. Lesar, T.S., Errors in the use of medication and dosage equations, Arch Pediatr Adolesc Med 152 (1998) 340.
17. Lucas, A.J., Improving medication safety in a neonatal intensive care unit, Am J Health Syst Pharm 61 (2004) 33.
18. Lugo, R.; Ward, R.M., Basic pharmacokinetic principles, In: (Editors: Polin, R.; Fox, W.; Abman, S.) Fetal and neonatal physiology ( 2004)Saunders, Philadelphia.
19. MacCara, M.E., Extravasation: a hazard of intravenous therapy, Drug Intell Clin Pharm 17 (1983) 713.
20. McCurdy, D.E.; Arnold, M.T., Development and implementation of a pediatric/neonatal IV syringe pump delivery system, J Neonatal Nurs 16 (1995) 9.
21. Nance, W.E., The genetics of deafness, Ment Retard Dev Dis Res Rev 9 (2003) 109.
22. National Patient Safety Goals for 2005 and 2004, n.d. Accessed August 12, 2004, fromwww.jcaho.org.
23. Nicholas, P.; Agius, C., Toward safer IV medication administration: the normal safety margins of many IV medications make this route particularly dangerous, J Infusion Nurs 28 (2005) 25.
24. Rioux, P.P., Clinical trials in pharmacogenetics and pharmacogenomics: methods and applications, Am J Health Syst Pharm 57 (2000) 887.
25. Roka, A.; Melinda, K.T.; Vasarhelyi, B.; et al., Elevated morphine concentrations in neonates treated with morphine and prolonged hypothermia for hypoxic ischemic encephalopathy, Pediatrics e844 (2008) 121.
26. Sakowski, J.; Newman, J.M.; Dozier, K., Severity of medication administration errors detected by bar-code medication administration system, Am J Health Syst Pharm 65 (2008) 1661.
27. Scarsi, K.K.; Fotis, M.A.; Noskin, G.A., Pharmacist participation in medical rounds reduces medication errors, Am J Health Syst Pharm 59 (2002) 2089.
28. Weinshilboum, R., Genomic medicine: inheritance and drug response, New Engl J Med 348 (2003) 529.
29. In: (Editors: Yaffe, S.J.; Aranda, J.V.) Neonatal and pediatric pharmacology ( 2005)Lippincott Williams & Wilkins, Philadelphia.