21. Jaundice*
Beena D. Kamath, Elizabeth H. Thilo and Jacinto A. Hernandez
Jaundice, or hyperbilirubinemia, is an almost universal occurrence in neonates. All infants have a rise in their bilirubin levels after birth because of excessive bilirubin formation and an immature liver that cannot clear the bilirubin from the blood. Sixty percent of normal newborns become noticeably jaundiced sometime during the first week of life. 19
Severe hyperbilirubinemia, defined as total serum bilirubin above the 95th percentile for age in hours, occurs in 8% to 9% of infants during the first week of life.5 Experiences have shown the dangers of excessive levels of unconjugated bilirubin, such as the development of bilirubin encephalopathy and the devastating and irreversible effects of kernicterus. Although the severe sequelae remain rare, a resurgence of kernicterus was seen in the 1990s. 8,14 The increasing incidence of kernicterus was attributed to several causes: the advent of early postnatal discharge before establishment of effective breast feeding, a lack of recognition for the signs of bilirubin encephalopathy, a lack of appropriate follow-up for discharged infants, and a delay in the measurement of bilirubin levels. Therefore an understanding of the pathophysiology and clinical significance of hyperbilirubinemia is critical in the care of newborn infants.
This chapter provides the reader with a basic overview of the multiple causes and contributing factors in the development of hyperbilirubinemia; describes the diagnosis, clinical significance, and complications of hyperbilirubinemia; and discusses current treatment modalities and their complications.
PHYSIOLOGY
To understand the pathophysiology and clinical significance of hyperbilirubinemia, normal bilirubin metabolism in the newborn must be reviewed (Figure 21-1). A newborn has a rate of bilirubin production of 8 to 10 mg/kg/24 hr, which is 2 to 2½ times the production rate in adults. Red blood cells in newborns have a shortened life span of 70 to 90 days, compared with 120 days in adults. As the catabolism of 1 g of hemoglobin yields 35 mg of bilirubin, this accelerated red blood cell breakdown produces most of the bilirubin (75% to 85%) in newborns. The remaining 15% to 25% of bilirubin is derived from non-erythroid heme proteins found principally in the liver and heme precursors in the marrow and extramedullary hematopoietic areas that do not go on to form red blood cells (early peak or shunt bilirubin).
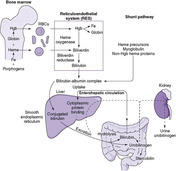 |
| FIGURE 21-1
(From Gartner LM, Hollander M: Disorders of bilirubin metabolism. In Assali NS, editor: Pathophysiology of gestation, vol 3, New York, 1972, Academic Press.)
|
Bilirubin metabolism is initiated in the reticuloendothelial system, principally in the liver and spleen, as old or abnormal red blood cells are removed from the circulation. The enzyme microsomal heme oxygenase will act on heme to produce biliverdin, and biliverdin reductase will convert this biliverdin into bilirubin. This bilirubin, in its unconjugated or indirect-reacting form, is released into the plasma. Exhaled carbon monoxide is an end product of these pathways.
At a normal plasma pH, bilirubin is very poorly soluble and binds tightly to circulating albumin, which serves as a carrier protein. Albumin contains one high-affinity site for bilirubin and one or more sites of lower affinity. Bilirubin binds to albumin in a molar ratio of between 0.5 and 1 mole of bilirubin per mole of albumin. A bilirubin/albumin molar ratio of 1 corresponds to approximately 8.5 mg bilirubin/g of albumin. This ratio may be somewhat lower in a sick very-low-birth-weight (VLBW) infant.7
Bilirubin bound to albumin is carried to the liver and transported into the hepatocyte by carrier-mediated diffusion. Intracellularly, bilirubin is bound to ligandin (Y protein) and, to a lesser extent, the Z protein. Conjugation occurs within the smooth endoplasmic reticulum of the cell. This reaction, catalyzed by the enzyme bilirubin uridine diphosphate glucuronosyl transferase (UDPGT), leads to the formation of water-soluble compounds called bilirubin glucuronides. In addition to UDPGT, conjugation requires glucuronic acid synthesized from glucose. Conjugated bilirubin is then actively secreted into bile and passes into the small intestine.
Conjugated bilirubin is not reabsorbed from the intestine, but the bowel lumen of the newborn contains the enzyme beta-glucuronidase, which can convert conjugated bilirubin back into glucuronic acid and unconjugated bilirubin, which may be absorbed. This pathway constitutes the enterohepatic circulation of bilirubin and contributes significantly to an infant’s bilirubin load.22
Factors That Affect Bilirubin Levels
The ability of albumin to bind bilirubin is affected by a number of different factors, including plasma pH, free fatty acid levels, and certain drugs (Table 21-1). Albumin binding of unconjugated bilirubin may be important in the prevention of toxicity (bilirubin encephalopathy or kernicterus). Once the high-affinity site is saturated, there is a rapid increase in potentially toxic free (nonbound) unconjugated bilirubin. Kernicterus has been clinically associated with administration of sulfisoxazole to newborns as a result of displacement of bilirubin from the primary binding site on albumin. Other drugs, such as ceftriaxone, also appear to displace bilirubin from this binding site. The effect on bilirubin-albumin binding of some but not all drugs used in newborn medicine has been studied in vitro. 23
| *From Hargreaves T: Effects of fatty acids on bilirubin conjugation, Arch Dis Child 48:446, 1973. |
|
| Factor | Mechanism |
|---|---|
| pH (acidosis) | Decreases binding by decreasing affinity at the binding site and increasing tissue affinity |
| Hematin | Competitively inhibits binding at primary site |
| Free fatty acid (intralipid) * | Competitively inhibits binding at primary site |
| Infection | Mechanism not established |
| Drugs such as sulfa compounds, sodium salicylate, phenylbutazone, and ceftriaxone | Primarily competitive binding; principally at secondary site; and best established for sulfisoxazole |
| Stabilizers for albumin preparations | Competitively inhibit binding at primary site |
| X-ray contrast media for cholangiography | Competitively inhibit binding at primary site |
Newborn monkeys have been shown to be deficient in the intracellular Y and Z proteins for the first few days of life, and this also may occur in the human newborn. The hormonal (estrogen) environment of the infant may inhibit liver function and bilirubin secretion. A rise in bilirubin levels shortly after birth is also partially attributable to a relative deficiency of UDPGT activity (0.1% of adult levels at 30 weeks’ gestation). Enzyme activity increases rapidly after birth independent of the infant’s gestational age.
Certain ethnic groups, including Eskimo, Asian, and Native American, have an increased incidence and severity of hyperbilirubinemia for reasons that are not clearly understood. In a hypoglycemic infant, glucuronide production may be limited and thus conjugation is impaired. The presence of beta-glucuronidase in the bowel lumen during fetal life enables bilirubin to be reabsorbed and transported across the placenta for excretion by the maternal liver.
ETIOLOGY
Bilirubin levels rise in newborn infants by three main mechanisms: increased production (accelerated red blood cell breakdown), decreased removal (transient liver enzyme insufficiency), and increased reabsorption (enterohepatic circulation) (Box 21-1). The normal pathways of bilirubin metabolism described earlier account for much of the increase in bilirubin levels in newborn infants; however, the following circumstances deserve special attention for infants who have prolonged or marked increases in bilirubin levels than would otherwise be expected.
BOX 21-1
Overproduction
• Hemolytic disease of the newborn
• Hereditary hemolytic anemias
• Membrane defects
• Hemoglobinopathies
• Enzyme defects
• Polycythemia
• Extravascular blood
• Swallowed
• Bruising or enclosed hemorrhage (e.g., cephalohematoma)
• Increased enterohepatic circulation
Slow Excretion
• Decreased hepatic uptake
• Decreased sinusoidal perfusion
• Ligandin deficiency
• Decreased conjugation
• Enzyme deficiency
• Enzyme inhibition, such as the Lucey-Driscoll syndrome
• Inadequate transport out of hepatocyte
• Biliary obstruction
Combined (Overproduction and Slow Excretion)
• Bacterial infection
• Congenital intrauterine infection
Breast Feeding
• Breast feeding jaundice
• Breast milk jaundice
Physiologic
Miscellaneous
• Hypothyroidism
• Galactosemia
• Infant of diabetic mother
From a management perspective, it is helpful to describe severe hyperbilirubinemia according to its time of onset, early or late, to determine its specific etiology. In general, early-onset severe hyperbilirubinemia is associated with increased bilirubin production, whereas late-onset hyperbilirubinemia is often associated with delayed bilirubin elimination with or without increased bilirubin production (Figure 21-2). 25
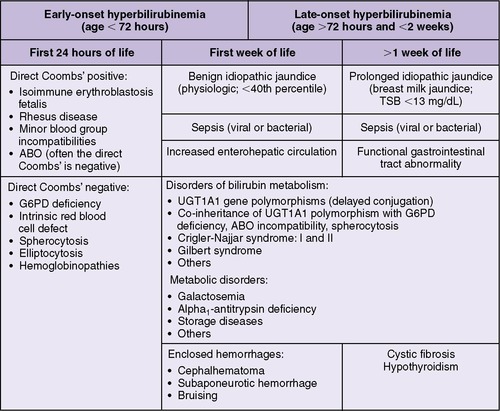 |
| FIGURE 21-2
(Modified from Smitherman H, Stark AR, Bhutani VK: Early recognition of neonatal hyperbilirubinemia and its emergent management, Semin Fetal Neonatal Med 11:214, 2006.)
|
Overproduction of Bilirubin
HEMOLYTIC DISEASE OF THE NEWBORN
Hemolytic disease of the newborn may occur when blood group incompatibilities such as Rh, ABO, or minor blood groups exist between a mother and her fetus. The classic example of hemolytic disease of the newborn has been erythroblastosis fetalis occurring as a result of Rh incompatibility. Fifteen percent of the white population is Rh negative. When an Rh-negative mother is sensitized to the Rh antigen by an improperly matched blood transfusion or the occurrence of fetal-maternal blood transfusion during pregnancy, delivery, abortion, or amniocentesis, the presence of the Rh antigen induces maternal antibody production. Because prior sensitization with the Rh antigen is necessary for antibody production, the first Rh-positive infant usually is not affected. Once a mother is sensitized, maternal immunoglobulin G (IgG) crosses the placenta into the fetal circulation where it reacts with the Rh antigen on fetal erythrocytes. These antibody-coated cells are recognized as abnormal and destroyed by the spleen. This results in increased amounts of hemoglobin, requiring metabolic degradation. As the destruction of erythrocytes and production of bilirubin progress, the ability of the fetus to compensate may be surpassed. Fortunately, the use of anti-D gamma globulin (RhoGAM), particularly antenatal administration at 26 to 28 weeks’ gestation to non-sensitized pregnant women, has markedly decreased the incidence of Rh isoimmunization and the resulting hyperbilirubinemia in newborn infants.
With the widespread use of RhoGAM, the most frequent cause of hemolytic disease of the newborn is now ABO blood group incompatibility. ABO incompatibility is limited to mothers of blood group O and affects infants of blood group A or B. All group O individuals have naturally occurring anti-A and anti-B (IgG) antibodies, so specific sensitization is not necessary. The resulting hyperbilirubinemia in the newborn is very variable and generally milder than that seen with Rh incompatibility.
HEREDITARY HEMOLYTIC ANEMIAS
Erythrocytes with abnormal membranes or containing abnormal hemoglobin variants have increased rates of red blood cell destruction. Individuals with enzyme defects, such as spherocytosis and elliptocytosis, cannot maintain the integrity of red blood cells because of abnormal osmotic fragility (generally increased) and an increased rate of splenic destruction. Glucose-6-phosphate dehydrogenase (G6PD) deficiency is the most common enzyme defect and is more commonly found in certain racial and ethnic groups, including Chinese, Greeks, and blacks. Pyruvate kinase deficiency is less common.
Individuals with hemoglobinopathies, which can be diagnosed by hemoglobin electrophoresis, also have increased splenic destruction. Often, the precipitating factor for the hemolysis cannot be found in infants. A family history is important, however, since it may be positive in as many as 80% of cases.
POLYCYTHEMIA
Polycythemia (with a central venous hematocrit value >65) is the condition in which an increased red blood cell mass, coupled with the shortened life span of these cells found in all newborns, results in an increased bilirubin load. Polycythemia may be idiopathic or may occur as a result of a maternal-fetal transfusion, twin-to-twin transfusion, chronic in utero hypoxia, or delayed clamping of the umbilical cord at the time of delivery.
EXTRAVASCULAR BLOOD
Enclosed hemorrhage includes cephalohematoma, subgaleal hemorrhage, cerebral hemorrhage, intraabdominal bleeding, and any occult internal bleeding, as well as extensive bruising. As these enclosed hemorrhages resolve, red blood cells trapped within are broken down and add to bilirubin production. Swallowed maternal blood is another possible source of increased bilirubin load.
INCREASED ENTEROHEPATIC CIRCULATION
As mentioned, the intestinal brush border contains the enzyme beta-glucuronidase, which can convert conjugated bilirubin back into its unconjugated (absorbable) form and glucuronic acid. Meconium contains a substantial amount of bilirubin, estimated at 1 mg of bilirubin per 1 g of meconium, or a total load of 100 to 200 mg. Any delay in the passage of meconium, as can occur with prematurity or bowel obstruction, increases the bilirubin load that must be metabolized. Hyperbilirubinemia requiring treatment due to these causes is rarely evident in the first 24 to 48 hours of life.
Slow Excretion of Bilirubin
Infants with normal bilirubin production rates may be unable to remove this load for a variety of reasons, as described in the following conditions.
DECREASED HEPATIC UPTAKE OF BILIRUBIN
Diminished hepatic uptake of bilirubin may be a result of inadequate perfusion of hepatic sinusoids or deficient carrier proteins (Y and Z). Certain drugs and compounds (e.g., steroid hormones, free fatty acids, chloramphenicol) may competitively bind to these proteins, creating a functional deficiency.
Inadequate perfusion of hepatic sinusoids occurs when there is a shunt through a persistent ductus venosus or extrahepatic portal vein thrombosis or with hyperviscosity and hypovolemia, as seen in infants with severe congestive heart failure. Although Y and Z proteins are decreased in some newborn primates, no actual deficiency has yet been demonstrated in the human newborn.
DECREASED BILIRUBIN CONJUGATION
Decreased bilirubin conjugation may be a result of UDPGT deficiency, as in the Crigler-Najjar syndromes or Gilbert syndrome. These disorders are caused by defects in the UDPGT1 gene complex recently identified on chromosome 2. Crigler-Najjar syndrome is rare and exists in two forms with either complete (type I) or partial (type II) absence of enzymatic activity. Type I is an autosomal recessive disorder. Phototherapy becomes ineffective in preventing excessive bilirubin levels, and liver transplantation is the only possible cure. Type II is inherited as an autosomal dominant disorder and responds to enzyme induction with phenobarbital. Gilbert syndrome is a milder and very common autosomal dominant disorder with partial enzyme activity generally becoming apparent beyond the newborn period with mild bilirubin elevation during times of stress or intercurrent illness. It may well be an important contributing factor, however, in cases of late-onset severe hyperbilirubinemia.
INADEQUATE TRANSPORT OUT OF THE HEPATOCYTE
Dubin-Johnson and Rotor syndromes are genetically inherited conditions (autosomal recessive and dominant, respectively) in which individuals can conjugate bilirubin normally but cannot excrete it, resulting in direct (conjugated) hyperbilirubinemia. These conditions, in addition to causing generalized hepatocellular damage, require specialized evaluation, including liver biopsy.
BILIARY OBSTRUCTION
Biliary obstruction often is seen as a diagnostic dilemma requiring differentiation between generalized hepatocellular damage and mechanical obstruction.
A variety of disorders can cause hepatocellular damage, including infections such as hepatitis and metabolic disorders such as galactosemia. In the neonatal intensive care unit (NICU), the most common cause of hepatocellular damage is the use of parenteral nutrition. The mechanism is not well established, but the damage takes at least 2 weeks to develop and is especially prominent in VLBW infants. Biliary atresia or, much less frequently, a choledochal cyst can cause mechanical obstruction to bile flow, resulting in a conjugated hyperbilirubinemia with light-colored stools.
Combined Overproduction and Slow Excretion
INFECTIONS
Bacterial infections (sepsis neonatorum, especially necrotizing enterocolitis [NEC] caused by toxin-producing organisms such as certain strains of Escherichia coli [E. coli]) or intrauterine viral infections can result in increased bilirubin production and decreased hepatic clearance.
Intrauterine infections, including syphilis, toxoplasmosis, rubella, cytomegalovirus, herpes simplex, Coxsackie B virus, and hepatitis virus, cause clinical jaundice with evidence for hepatocellular damage. Infants with these infections often have additional clinical stigmata of their infection such as thrombocytopenia and rash.
INFANT OF A DIABETIC MOTHER
The cause of hyperbilirubinemia in an infant of a diabetic mother (IDM) appears to be multifactorial. In addition to prematurity and a tendency to feed poorly, an IDM may have an increased bilirubin load as a result of an expanded red blood cell mass. Erythrocyte membrane composition may be altered, and macrosomic infants often are bruised during labor and delivery.
Jaundice Associated with Breast Feeding
Ideally, a trained observer should evaluate all breast-fed infants within 48 to 72 hours of discharge in either a home or office setting. 6 Early discharge of breast-fed infants with inadequate follow-up may result in excessive levels of bilirubin and the possibility of kernicterus. Thus promotion and support of successful breast feeding constitutes a key element of the American Academy of Pediatrics (AAP) clinical practice guideline on the management of hyperbilirubinemia. 1
BREAST-FEEDING JAUNDICE
In general, breast-fed infants have higher bilirubin levels than bottle-fed infants, especially in the first days of life. It has been postulated that this early jaundice is related to decreased caloric and fluid intake from colostrum and increased enterohepatic circulation resulting from low stool output and breast milk beta-glucuronidase. 1 Many studies show a relationship between the degree of hyperbilirubinemia and the amount of weight lost by the infant after birth.
Because of concern for breast-fed infants being underfed, it once was common practice in some institutions to supplement with glucose water or electrolyte solutions after nursing. Such supplementation should be avoided because it reduces breast-feeding frequency and maternal milk production, without improving the infant’s intestinal motility, leading to higher peak bilirubin levels. Optimal management of a breast-feeding mother and infant includes early and frequent nursing: 8 to 12 times each day. If the infant is unable to feed this frequently, the mother should be instructed in the use of a mechanical breast pump, and the infant supplemented with expressed breast milk or formula to improve both the milk supply and the infant’s nutritional status and intestinal motility.
BREAST MILK JAUNDICE
A small percentage (1% to 2%) of breast-fed infants exhibit prolonged and exaggerated jaundice possibly related to an inhibitor or inhibitory substance found in their mother’s breast milk that prolongs and increases enterohepatic circulation. The rate of recurrence in families approaches 70%.
Such infants have an unconjugated hyperbilirubinemia (>12 mg/dL) that becomes exaggerated and persistent toward the end of the first week of life. 10 Other causes of excessive hyperbilirubinemia should be ruled out. Elevated bilirubin levels may persist for 4 to 14 days, followed by a very gradual decline. For the vast majority of infants, it is not necessary to interrupt breast feeding, even if the bilirubin increases to a level that may require phototherapy.
Miscellaneous Causes
The following causes of hyperbilirubinemia are uncommon but important to consider in infants who have no other clear etiology to explain their elevated bilirubin levels. These conditions include hypothyroidism and galactosemia. States now require routine screening for these conditions, since early detection allows intervention before permanent adverse neurologic outcomes. Hyperbilirubinemia, unconjugated or mixed, may be the initial sign of these conditions.
HYPOTHYROIDISM
A prolonged period of unconjugated hyperbilirubinemia can be seen in infants with hypothyroidism. The mechanism of hyperbilirubinemia in hypothyroidism is not well understood, but in some animal studies, thyroxine was needed for the hepatic clearance of bilirubin.
GALACTOSEMIA
Galactosemia is an autosomal recessive disorder characterized by increased jaundice in infants fed breast milk or lactose-containing formulas. The mechanism of hyperbilirubinemia in galactosemia may be related to a lack of substrate for glucuronidation and the accumulation of abnormal hepatotoxic by-products. The presence of non–glucose-reducing substances in the urine suggests galactosemia.
INTERPRETATION OF HIGH BILIRUBIN LEVELS
Identification of those infants at risk for hyperbilirubinemia enables clinicians to provide timely treatment to prevent neuronal injury. The AAP has described risk factors for hyperbilirubinemia, which can be seen in Box 21-2.
BOX 21-2
Major Risk Factors
• Pre-discharge TSB or TcB level in the high-risk zone
• Jaundice observed in the first 24 hours of life
• Blood group incompatibility with positive direct antiglobulin test, other known hemolytic disease (e.g., G6PD deficiency), elevated ET coc
• Gestational age 35 to 36 weeks
• Previous sibling received phototherapy
• Cephalohematoma or significant bruising
• Exclusive breast feeding, especially if nursing is not going well and weight loss is excessive
• East Asian race*
Minor Risk Factors
• Pre-discharge TSB or TcB level in the high intermediate-risk zone
• Gestational age 37 to 38 weeks
• Jaundice observed before discharge
• Previous sibling with jaundice
• Macrosomic infant of a diabetic mother
• Maternal age ≥25 years
• Male gender
Decreased Risk (these factors are associated with decreased risk for significant jaundice, listed in order of decreasing importance)
• TSB or TcB level in the low-risk zone
• Gestational age ≥41 weeks
• Exclusive bottle feeding
• Black race*
• Discharge from the hospital after 72 hours
ET coc, End-tidal carbon monoxide corrected; G6PD, glucose-6-phosphate dehydrogenase; TcB, transcutaneous bilirubin; TSB, Total serum bilirubin.
From American Academy of Pediatrics, Subcommittee on Hyperbilirubinemia: Clinical Practice Guideline: Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation, Pediatrics 114:297, 2004.
The most important determinants of brain injury caused by hyperbilirubinemia are the concentrations of unconjugated bilirubin and free bilirubin, the concentration of serum albumin and its ability to bind unconjugated bilirubin, the concentration of hydrogen ion (pH), and neuronal susceptibility.24 The blood-brain barrier allows free bilirubin to pass; however, the blood-brain interface, consisting of capillary endothelium and astrocytic foot processes, and the choroid plexus have specific transporters that can pump free bilirubin out of the central nervous system, thereby protecting the brain from exposure to high bilirubin. Timing of exposure to excess bilirubin during neurodevelopment is important in determining the pattern of the neurologic damage; for example, because auditory pathways mature earlier than motor pathways, patterns of damage in premature infants may differ from those in mature infants. 24 Unconjugated bilirubin is fat soluble and can cross cell membranes; however, because most unconjugated bilirubin is bound to albumin, toxicity is avoided. 25 Therefore development of toxicity may also depend on albumin-bilirubin binding. Factors that interfere with albumin-bilirubin binding appear to predispose to the development of kernicterus (see Table 21-1). Once toxicity has occurred, it appears to be irreversible.
Although elevated levels of bilirubin occur in virtually all newborns, precise identification of what constitutes a “safe” level for an individual newborn, especially if sick or premature, remains elusive and is the subject of much ongoing investigation. A “pathologic” level for one infant may be a “physiologic” level for another infant; therefore it has been suggested that these terms be done away with altogether.
All bilirubin levels should be interpreted according to the infant’s age in hours. The AAP recommends a nomogram that designates risk for newborn infants at 35 weeks or greater, according to bilirubin level obtained at varying postnatal ages in hours . The nomogram (Figure 21-3), based on earlier work by Bhutani et al, designates whether an infant is at high, intermediate, or low risk for requiring further intervention for hyperbilirubinemia.1,5 This hour-specific bilirubin nomogram has been shown to be more accurate than a clinical risk factor scoring system for assessing risk for significant hyperbilirubinemia, 16 although the addition of gestational age to the risk assessment strategy can increase accuracy further. 17
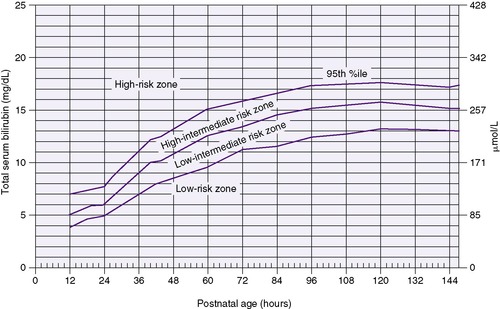 |
| FIGURE 21-3
(From Bhutani VK, Johnson L, Sivieri EM: Predictive ability of a predischarge hour-specific serum bilirubin for subsequent significant hyperbilirubinemia in healthy term and near-term newborns, Pediatrics 103:6, 1999.)
|
PREVENTION
Early Feeding
The physiologic mechanism is not entirely known but may be caused by a decrease in intestinal transit time and decreased enterohepatic circulation. When compared with infants not fed during the first 24 to 48 hours of life, infants fed earlier have lower peak bilirubin levels.
RhoGAM
Widespread use of RhoGAM has proven effective in preventing the sensitization of Rh-negative mothers after delivery or abortion of Rh-positive infants. RhoGAM, or anti-D gamma globulin, provides passive protection by preventing maternal production of anti-Rh antibodies that might affect subsequent Rh-positive pregnancies, causing destruction of fetal red blood cells.
Failures may occur if the amount of RhoGAM administered is insufficient compared with the load of fetal red blood cells received or if a significant fetal-maternal hemorrhage occurred before prophylaxis. Routine management of the Rh-negative mother now includes the administration of antenatal RhoGAM in the second trimester (26 to 28 weeks), at the time of amniocentesis, and after delivery.
Phenobarbital
Phenobarbital acts as an inducer of microsomal enzymes, increasing the levels of UDPGT. It also has a direct effect to stimulate bile secretion in infants with nonobstructive cholestasis and increases the concentration of ligandin. When used in conjunction with phototherapy, however, phenobarbital does not increase the rate of decline in bilirubin levels. Phenobarbital is effective when given to the mother before delivery. In infants with significant hemolytic disease of the newborn, it appears to slow the rate of rise of bilirubin and decrease the incidence of exchange transfusion. It is indicated in infants with Crigler-Najjar syndrome type II. Its use is not indicated on a routine prophylactic basis, because such use would overtreat many infants and other effects may be detrimental.
Tin Protoporphyrin and Tin Mesoporphyrin
Clinical trials have shown that administration of a single dose of tin protoporphyrin (SnPP) or tin mesoporphyrin (SnMP) to infants with ABO hemolytic disease or G6PD deficiency at birth effectively decreases bilirubin production. These compounds are potent competitive inhibitors of the enzyme heme oxygenase.15 Heme is excreted directly into bile when bilirubin production is suppressed. Infants receiving a single dose of SnMP (6 mcmol/kg of body weight intramuscularly [IM]) have lower peak serum bilirubin levels and a decreased need for phototherapy. Side effects have been minimal and include a transient erythema in those infants requiring phototherapy after receiving SnMP. SnMP also has been shown to be effective in controlling severe hyperbilirubinemia in breast-fed infants with high bilirubin levels between 48 and 96 hours of age. 20 Additional studies to determine what role these compounds will have in the management and prevention of neonatal jaundice are ongoing.
DATA COLLECTION
The history, physical examination, and laboratory data play an important role in the evaluation of the infant with hyperbilirubinemia (Box 21-3).
BOX 21-3
History
• Family
• Perinatal and obstetric
• Neonatal
Physical Examination
• Pallor
• Hepatosplenomegaly
• Enclosed hemorrhage
• Petechiae
• Congenital anomalies
Laboratory Data
All Jaundiced Infants
• Maternal and infant blood type
• Coombs’ test on cord blood
• Total/direct bilirubin (serial measurements)
• Complete blood count, including hematocrit, reticulocyte and platelet counts, white blood cell differential, and peripheral smear for red blood cell morphology
• Urinalysis, test for reducing substances
Selected Cases
• Protein, total/albumin
Sepsis Evaluation
• IgM
• Urine cytology for cytomegalovirus
• Viral cultures
New Techniques
• Transcutaneous bilirubinometry
• Bilirubin-binding tests
History
The evaluation of a jaundiced infant begins with a complete family, perinatal, and neonatal history. The family history should include the occurrence of disorders associated with hyperbilirubinemia in other family members, particularly siblings. The perinatal and obstetric history may provide clues or enable the clinician to anticipate possible hyperbilirubinemia. For example, hydrops fetalis is associated with Rh isoimmunization but is rarely seen with ABO incompatibility. The infant’s course during labor, delivery, and thereafter may be important. Items of interest include possible infection during the pregnancy, the use of oxytocin induction for delivery, or the occurrence of an asphyxial episode during labor or delivery. A history of medications used and the infant’s feeding and stooling patterns should also be obtained. The time of onset or detection of jaundice may be important, because jaundice in the first 24 hours of life always must be considered abnormal.
Signs and Symptoms and Clinical Approach
A wide spectrum of signs and symptoms may occur in a jaundiced infant, often depending on the cause of the jaundice. Jaundice in a newborn usually can be detected visually at a level of 6 to 7 mg/dL. Visible icterus appears first on the head and face and progresses in a cephalocaudal manner. The skin of the extremities, particularly the palmar and plantar surfaces, are the last skin surfaces to be affected. However, multiple studies show the inaccuracy of visual estimation of jaundice, even by experienced health care workers; thus all newborns should be assessed for hyperbilirubinemia with a serum or transcutaneous measurement if concern exists. Any measurement of bilirubin needs to be interpreted based on the infant’s age in hours at the time of measurement (seeFigure 21-3), which allows classification into high-risk, high intermediate-risk, low intermediate-risk, and low-risk zones. In addition, premature infants have a slightly later peak and are at risk for adverse neurologic outcomes at lower levels of bilirubin than older infants.
An infant with hemolytic disease of the newborn may show signs of jaundice and pallor in association with severe anemia and hydrops fetalis or may appear entirely normal at birth. Hepatosplenomegaly resulting from congestion and extramedullary hemopoiesis may be present. Infants affected by hemolytic disease of the newborn may also have pancreatic islet cell hyperplasia and are at increased risk for hypoglycemia. Careful physical examination may reveal the presence of a cephalohematoma or other lesion resulting from enclosed hemorrhage. The occurrence of petechiae or purpura raises the possibility of intrauterine infection or sepsis. Congenital anomalies or syndromic appearance should be noted, because an increased incidence of jaundice is noted in trisomic syndromes. Jaundice and umbilical hernia are also associated with congenital hypothyroidism.
SIGNS OF BILIRUBIN TOXICITY
Hyperbilirubinemia is of clinical concern because of the potential for brain injury. The spectrum of bilirubin-induced neurologic dysfunction (BIND) ranges from acute bilirubin encephalopathy to the devastating and irreversible syndrome of kernicterus.8,10,17 Acute bilirubin encephalopathy (ABE) describes the effects of hyperbilirubinemia seen during the hyperbilirubinemia and immediately thereafter. Clinical signs of ABE include lethargy, poor feeding, poor tone, a poor Moro reflex with incomplete flexion of the extremities, and a high-pitched cry. Opisthotonos posturing and retrocollis also may occur in the later stages. 24 As the symptoms of acute bilirubin encephalopathy worsen, the infant progresses to apnea, seizures, coma, and death.
Kernicterus, or chronic bilirubin encephalopathy, is an irreversible and devastating brain injury evidenced pathologically by yellowish staining in the deep nuclei of the central nervous system (CNS), particularly in the basal ganglia, cerebellum, and hippocampus. As opposed to other forms of perinatal brain injury, in the instance of kernicterus, a clear correlation exists between etiology, pathogenesis, and symptomatology. 24 Based on multiple studies, kernicterus has a mortality of 10% and at least 70% long-term morbidity. 11 Its clinical signs include extrapyramidal movement disorder, including dystonia and choreoathetoid movements (rapid, highly complex, involuntary, spasmodic movements); gaze abnormalities (especially affecting upward gaze); auditory disturbances (deafness); dysplasia of the enamel of deciduous teeth; and mild cognitive defects. The neuromotor abnormalities may be subtle, with the auditory abnormalities most apparent because the auditory pathways are the neural system most sensitive to bilirubin injury. 24
In later life, severely affected survivors with kernicterus may exhibit choreoathetosis, spastic cerebral palsy, mental retardation, sensory and perceptual deafness, and visual-motor incoordination. It is not likely that significant mental retardation alone, without the other features, is caused by bilirubin encephalopathy.
Whether more subtle long-term sequelae may occur in less severely affected infants and may not be apparent during the newborn period remains very controversial. There is speculation that some learning disabilities may be related to hyperbilirubinemia even at what had been previously considered “safe” levels. High bilirubin levels have been consistently associated with hearing impairment and abnormal brainstem auditory evoked potentials.11 Regarding other neurodevelopmental outcomes, extensive review of the literature dealing with full-term infants without hemolytic disease has found inconsistent evidence of adverse effects of bilirubin on intelligence quotient and neurologic examination; however, many of these studies have yielded mixed results and had limitations. 11,21 A recent study compared 140 term infants without kernicterus who had total serum bilirubin levels of at least 25 mg/dL with randomly selected controls. The study found no difference in adverse neurodevelopmental outcomes in infants born at or near term when the infants with hyperbilirubinemia were treated aggressively with phototherapy or exchange transfusion. 21
Infants with hemolytic disease and premature (especially VLBW) infants should receive phototherapy and exchange transfusion at lower bilirubin levels. Unfortunately, the “critical level” at which bilirubin toxicity occurs in either preterm or term infants has not been established.
Laboratory Data
A serum bilirubin level is the most reliable method upon which to make clinical decisions. Transcutaneous bilimeters have been used in newborn nurseries to screen for hyperbilirubinemia and work by emitting a beam of light onto the skin and measuring the light reflected, which is not absorbed by bilirubin in the skin. Transcutaneous bilimeters have been shown to be valid3; however, new studies are showing that bilimeter levels may significantly underestimate the severity of hyperbilirubinemia. Because of the uncertain accuracy of the transcutaneous bilirubin measurement, it is not recommended to consider this device reliable at levels greater than 14 mg/dL.18
Because the breakdown of bilirubin is the only chemical reaction in the body that results in formation of carbon monoxide (CO), this marker has been used to measure bilirubin production. 9 Measurement of end-tidal CO corrected for inhaled CO (ET coc) can identify infants with unusually high rates of bilirubin production and is the only clinical test providing direct measurement of the rate of heme catabolism and bilirubin production. It is not yet clear what role measurement of ETcoc will play in clinical management. The device is not commercially available in the United States.
In addition to a bilirubin level, mother’s and infant’s blood types and Rh status, as well as Coombs’ testing (antibody testing), both direct and indirect, are needed to evaluate for hemolytic disease. The direct Coombs’ test on cord blood is positive because of the presence of IgG on the surface of the infant’s red blood cells, whereas an indirect Coombs’ test is positive with the presence of IgG in the infant’s serum. An indirect positive only is generally less severe than a direct positive antibody screen.
In addition to measurements of hematocrit and reticulocytes, the peripheral blood smear should be carefully examined, seeking evidence of hemolysis (increased numbers of nucleated red blood cells or the presence of fragmented cells, poikilocytosis, and anisocytosis). Microspherocytosis is characteristic of ABO incompatibility and at times may be confused with hereditary spherocytosis. A knowledge of mother’s and baby’s blood types and the clinical course help differentiate the two.
Obtaining fractionated (total/direct) bilirubin levels and serial levels helps establish causes and enables the clinician to follow the rate of bilirubin rise, although the total bilirubin should be used for making clinical treatment decisions. Serum albumin levels should be determined and the bilirubin:albumin (B:A) ratio considered as an additional factor in deciding when to start phototherapy or perform an exchange transfusion. Clinical laboratory measurement of unbound (“free”) bilirubin is not available but may be a useful test in the future.
Evaluation for other potential causes of hyperbilirubinemia is essential when the etiology is not immediately clear. An elevated direct fraction of bilirubin, abnormal white blood cell count, left shifted differential, or thrombocytopenia may suggest infection. Urinalysis, including evaluation for reducing substances, may be helpful. Infants suspected of having congenital infection should have additional tests, including immunoglobulin M (IgM) levels. Blood, cerebrospinal fluid, and/or exudate from skin vesicles should be sent for viral cultures, and urine may be tested for cytomegalovirus (CMV). Newborns should be screened for hypothyroidism and galactosemia.
Minimum laboratory evaluation of the jaundiced newborn should include the mother’s and infant’s blood types, Rh status, and Coombs’ test (direct and indirect) on cord blood. A complete blood count (CBC) to include reticulocyte and platelet counts, white blood cell count and differential, peripheral smear for red blood cell morphology, and hematocrit should be performed. Infants suspected of having bacterial sepsis should receive antibiotic treatment and a complete sepsis evaluation, including cultures of blood, urine, and cerebrospinal fluid. Bilirubin levels (total and direct) should be measured serially and interpreted based on the infant’s age in hours at the time of measurement (seeFigure 21-3). Serum albumin levels may be helpful at higher bilirubin levels.
In addition to jaundice and anemia in the first few days of life, infants with hemolytic disease are at risk for late anemia after discharge from the nursery, potentially driving the infant’s physiologic nadir of red blood cell count even lower than usual, requiring treatment with erythropoietin or transfusion. These infants require close follow-up for anemia from their primary care provider.
TREATMENT
Treatment is aimed at lowering the concentration of circulating bilirubin or keeping it from increasing, thereby preventing the complications of acute bilirubin encephalopathy and the irreversible damage of kernicterus. Phototherapy, in particular, and exchange transfusions, now less so, are widely used in the treatment of hyperbilirubinemia; however, decisions to use these therapies are complicated by an incomplete understanding of bilirubin toxicity, especially as applied to an individual infant (Table 21-2). The AAP Subcommittee on Hyperbilirubinemia published clinical management guidelines that lend direction regarding the use of these therapies, and the recommendations are outlined in the algorithm detailed in Figure 21-4 and in Figure 21-5 (phototherapy) and Figure 21-6 (exchange transfusion).
| Weight (g) | Initiate Phototherapy (mg/dL) | Consider Exchange Transfusion (mg/dL) |
|---|---|---|
| 500-750 | 5-8 | 12-15 |
| 751-1000 | 6-10 | >15 |
| 1001-1250 | 8-10 | 15-18 |
| 1251-1500 | 10-12 | 17-20 |
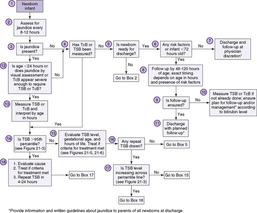 |
| FIGURE 21-4
(Modified from American Academy of Pediatrics, Subcommittee on Hyperbilirubinemia: Clinical Practice Guideline: Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation, Pediatrics 114:297, 2004.)
|
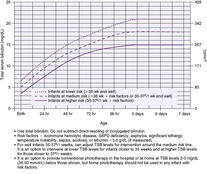 |
| FIGURE 21-5
(From American Academy of Pediatrics, Subcommittee on Hyperbilirubinemia: Clinical Practice Guideline: Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation, Pediatrics 114:297, 2004.)
|
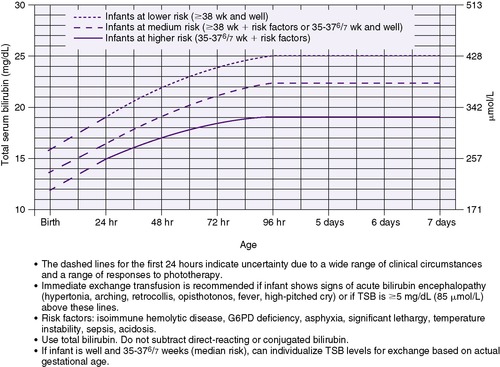 |
| FIGURE 21-6
(From American Academy of Pediatrics, Subcommittee on Hyperbilirubinemia: Clinical Practice Guideline: Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation, Pediatrics 114:297, 2004.)
|
Phototherapy
Phototherapy is the most commonly used treatment for hyperbilirubinemia. With the widespread use of phototherapy, the need for exchange transfusion in infants with non-hemolytic hyperbilirubinemia is almost obsolete. Hospital-based studies in the United States have shown that 5 to 40 infants per 1000 term and late preterm infants receive phototherapy before discharge from the nursery and an equal number are readmitted for phototherapy after discharge. 19 The decision to initiate phototherapy must be individualized for each newborn and should be based on the recent AAP guidelines (see discussion in legend for Figure 21-5).
With effective phototherapy, the infant’s bilirubin level should drop at a rate of 0.5 to 1 mg percent per hour, and by 30% to 40% after 24 hours of treatment, when applied at several days of age. The rate of decline of bilirubin in the first days (early hyperbilirubinemia, likely the result of increased bilirubin production) will not be as brisk, but the rate of rise will be significantly slowed. Bilirubin best absorbs light in the blue-green spectrum, particularly in the blue region of the spectrum near 460 nm 19; the spectrum of light at 425 to 475 nm is therefore most effective. Phototherapy uses this light energy to change the shape and structure of bilirubin, converting it to molecules that can be excreted, even when normal conjugation is deficient. 19 The most important of these molecules is lumirubin, a stable structural photoisomer. Lumirubin does not require conjugation and is rapidly excreted in both bile and urine. The production of lumirubin is an irreversible reaction that appears to be dose-related.
The efficacy of phototherapy depends on the energy output (irradiance) of the light source (measured with a radiometer in units of watts per square centimeter or microwatts per square centimeter per nanometer over a given wavelength band), the distance of the light source from the infant, and the surface area of the infant exposed to the light. Intensive phototherapy consists of 30 μW/cm 2/nm or more. 1 Fiberoptic blankets delivering phototherapy from a high-intensity light source are available for use by themselves or in conjunction with other sources of phototherapy but are unlikely to expose an adequate surface area on a term infant to provide intensive phototherapy. 19
The AAP discusses in their 2004 guidelines the most commonly used phototherapy units. These include daylight, cool white, blue, or “special blue” fluorescent tubes or tungsten-halogen lamps in different configurations, either free-standing or as part of a radiant warming device. A system using high-intensity gallium nitride light-emitting diodes has been introduced; in this model, six fiberoptic systems deliver light from a high-intensity lamp to a fiberoptic blanket. Most of these devices deliver enough output in the blue-green region of the visible spectrum to be effective for standard phototherapy use. The most effective light sources commercially available for phototherapy are those that use special blue fluorescent tubes or a specially designed light-emitting diode light (Natus Inc, San Carlos, Calif.). The special blue fluorescent tubes are labeled F20T12/BB (General Electric, Westinghouse, Sylvania) or TL52/20W (Phillips, Eindhoven, The Netherlands). It is important to note that special blue tubes provide much greater irradiance than regular blue tubes (labeled F20T12/B). Special blue tubes are most effective because they provide light predominantly in the blue-green spectrum. At these wavelengths, light penetrates skin well and is absorbed maximally by bilirubin. 1 Fiberoptic phototherapy blankets are available. These systems (Wallaby Phototherapy System, Fiberoptic Medical Products, Inc., Allentown, Pa.; and Biliblanket, Ohmeda, Columbia, Md.) use a high-intensity halogen light source for transmission of light by fiberoptic bundles. Irradiance and efficacy appear comparable with those for standard phototherapy. Purported advantages of these systems are elimination of the need for eye patches, exposure of greater surface area, and provision of phototherapy outside of the nursery with less interference in mother-infant bonding. These blankets are more convenient to use when phototherapy is necessary in an outpatient setting.
Physical and laboratory evaluation should be performed before initiating phototherapy in any infant. Once phototherapy has been initiated, serum levels of bilirubin must be monitored frequently (every 4 to 12 hours) because visual assessment of icterus is no longer valid. Hematocrit also must be monitored, especially in infants with hemolytic disease.
There are conflicting data in the literature on whether continuous or intermittent administration of phototherapy is most effective. Phototherapy may be interrupted during brief periods for feeding and parental contact. Table 21-3 outlines some of the nursing assessments and management to be performed in infants undergoing phototherapy.
| Nursing Assessment | |
|---|---|
| Area | Parameter |
| Physical status |
Intake and output
Color
Location of jaundice
Skin integrity
Stools (character, consistency)
Vital signs
Infant/environmental temperature
Hydration status
Signs of phototherapy side effects
Eye discharge and tearing
Position
Activity
|
| Neurobehavioral status |
Sleep-wake states
Sensory threshold
Behavioral responsiveness
Feeding behaviors
Consoling abilities
Stress responses
Interactive capabilities
|
| Nursing Management | |
| Nursing Diagnosis | Intervention |
| Deficient Fluid Volume (actual or potential) |
Monitor intake and output.
Monitor hydration status (weight, specific gravity, urine output).
Monitor stooling pattern, character.
Maintain adequate fluid intake (oral or parenteral).
|
| Imbalanced Nutrition: Less Than Body Requirements |
Assess feeding behavior and activity.
Monitor fluid and caloric intake, weight, abdominal girth.
Remove eye shields during feeding.
Hold during oral feedings as health and thermal status permit.
Bring to alert state before feeding.
Feed on demand if possible.
|
| Impaired Skin Integrity |
Observe color, rashes, excoriation.
Clean skin with warm water.
Clean perineal area after stooling.
Turn frequently (also increases skin exposure to phototherapy).
Ensure Plexiglas shield is in place between light source and infant to reduce exposure to UV light.
|
| Risk for Injury |
Observe for side effects associated with phototherapy.
Observe for signs of sepsis.
Provide care to minimize side effects of phototherapy.
Shield eyes from lights with opaque patches.
Ensure eyelids are closed when shield is applied to prevent corneal injury.
Remove eye shield and observe eyes regularly.
Monitor position of eye shield to prevent occlusion of nose.
Avoid tight head band on eye shield to reduce risk of increased intracranial pressure especially in preterm infants.
Observe for eye discharge, tearing.
Shield testes and possibly ovaries (data unclear about need to do this) with diaper.
|
| Ineffective Thermoregulation |
Place in warm, thermoneutral environment.
Monitor environmental and infant temperature.
Observe for hypothermia and hyperthermia.
Reduce heat losses from environmental sources.
Use servocontrol for infants in incubator or under radiant warmer.
Shield servocontrol thermistor from direct exposure to phototherapy lights.
|
After phototherapy ceases, bilirubin levels should be followed for at least 24 hours to rule out the occurrence of significant rebound. A rebound in the total serum bilirubin level of 1 to 2 mg/dL, and occasionally more, can occur after phototherapy is discontinued. Infants at increased risk for rebound are those less than 37 weeks’ gestation, those with hemolytic disease, and those treated with phototherapy during the birth hospitalization, since the bilirubin is still expected to rise at the time phototherapy is discontinued. 19
Despite its widespread use since 1958, questions about the safety and side effects of phototherapy remain. However, reports of clinically significant toxicity are rare. 19 Animal studies have demonstrated a potential retinal toxicity of light. Although it is not established that this occurs in the human newborn, the possibility remains a concern and the infant’s eyes should be covered while phototherapy is in use. Patches should completely cover the eyes without placing excessive pressure on the eyes and should be carefully positioned to avoid occluding the nares. Eye patches should be removed every 4 hours to permit evaluation of the infant’s eyes. The patches should be left off during feedings and parental visits.
Infants exposed to phototherapy, particularly low-birth-weight infants and infants under a radiant warmer, have significant increases in their insensible water losses (IWLs). Infants in incubators or servocontrolled care centers may become overheated. The servocontrol probe should be shielded by an opaque covering. Infants treated in open cribs may become cold stressed. Fluid balance must be monitored carefully in an infant receiving phototherapy. Infants under phototherapy also have increased stool water losses and may develop temporary lactose intolerance. The infant’s temperature, weight, and intake and output should be monitored frequently. The presence of reducing substances in the stool can be treated with a non–lactose-containing formula.
Infants who have an associated cholestatic jaundice and are exposed to phototherapy may develop the bronze baby syndrome, presumably caused by retention of a bilirubin breakdown product produced by phototherapy, although the mechanism is unclear. An infant with bronze baby syndrome develops a dark gray-brown discoloration of the skin, urine, and serum. There are generally no clinical symptoms with this syndrome, but at least one death has been reported. After phototherapy ceases, the bronzing gradually resolves.
Transient skin rashes and tanning resulting from increased melanin production have been reported, as have bullous skin eruptions in infants treated with tin mesoporphyrin who are subsequently exposed to sunlight or daylight fluorescent bulbs. 19 A recent study has suggested that intensive phototherapy might increase the number of melanocytic nevi identified at school age. 19 Other potential problems include interference with biologic (circadian) rhythms and maternal-infant bonding. Although there may be some transient, short-term growth effects, long-term growth effects and development appear unaffected by phototherapy.
Intravenous Immunoglobulin
When Coombs’-positive hemolysis is present and the total serum bilirubin is rising despite intensive phototherapy or is approaching the exchange level, intravenous immunoglobulin (IVIG) should be administered to the infant to decrease the severity of hemolysis. The dose is 500 mg/kg to 1 g/kg IV over 2 to 4 hours and may be repeated one time after 12 hours. This intervention has been shown in multiple trials to decrease the need for exchange transfusion by approximately 70% and is recommended for either Rh or ABO isoimmunization. 1,2
Exchange Transfusion
An exchange transfusion is indicated for correction of severe anemia and removal of antibody-coated red blood cells in hemolytic disease or removal of excessive unconjugated bilirubin regardless of its cause. Phototherapy cannot be used in place of an exchange transfusion in those infants with severe hemolytic disease. A packed red blood cell exchange transfusion using type O Rh-negative blood will correct anemia and hypoxemia, as well as remove sensitized cells and bilirubin, leading to a more complete therapy for the problem.
It must be stressed that the decision to perform an exchange transfusion must be individualized for each patient. Particularly in VLBW infants, the indications to perform an exchange transfusion vary from nursery to nursery. The recent AAP guidelines for performing exchange transfusion in infants of 35 or more weeks of gestation are shown in Figure 21-6 (see discussion in legend).
Usually a double-volume exchange transfusion is performed using 160 mL/kg of appropriate whole blood product. ABO type-specific Rh-negative blood should be used in cases with Rh incompatibility. Type O Rh-specific cells are indicated when ABO incompatibility exists.
The blood bank can prepare this blood for the infant with a predetermined hematocrit, usually 50% to 55%. An exchange transfusion will reduce bilirubin levels by approximately 45% to 85%, according to various sources. Administration of 1 g/kg of 25% albumin 1 hour before the exchange transfusion has been shown in some studies to increase the efficiency of exchange by about 40%. As plasma and tissue levels equilibrate post-transfusion, the bilirubin rises to about 60% of the pre-exchange level.
Exchange transfusion trays are commercially available and include a four-way stopcock, necessary tubing and syringes, 10% calcium gluconate, and a plastic bag for discarded blood.
The infant should be in a NICU for close observation during and immediately after the procedure. The procedure is performed by removing small aliquots of the infant’s blood and replacing similar small aliquots of transfused blood product while blood pressure, heart rate, and general condition are monitored. Generally, 5-mL to 20-mL aliquots of blood are used, depending on the size and condition of the infant. The initial aliquot should be withdrawn and sent to the laboratory for bilirubin, hematocrit, calcium, and cultures. The rate of exchange is usually 5 to 8 mL/min. Blood used in the exchange should be warmed and mixed in the bag after every 50 to 100 mL.
The final aliquot from an exchange should be sent for complete blood count (CBC), fractionated bilirubin, calcium ion, electrolytes, culture, and repeat type and crossmatch studies for potential additional exchange transfusion. In addition to the individuals performing the exchange, one person must keep an accurate record of time, volumes withdrawn and infused, vital signs, and medications administered.
Exchange transfusion is a procedure with many potential complications and carries a mortality risk of about 0.5%. For this reason and because so few exchange transfusions are performed today, this procedure should be done only by personnel familiar with it and its complications, preferably in a tertiary care unit. Vascular complications are related to the use of umbilical catheters (discussed in Chapter 7). Necrotizing enterocolitis has been reported as a post-exchange complication, probably as a result of bowel ischemia during the procedure.
Electrolyte and glucose disturbances are related to the blood preparation used for the exchange. Citrate used as part of the anticoagulant solution binds divalent ions such as calcium and magnesium; thus laboratory evaluation of calcium and magnesium during the procedure is essential. The infant should be evaluated for hypocalcemia after each 100 mL of the exchange has been completed. Clinical signs and symptoms of hypocalcemia include irritability, tachycardia, or prolongation of the Q-oTc interval. If hypocalcemia is detected, 1 mL of a 10% calcium gluconate solution is infused slowly.
Acid-citrate-dextrose and citrate-phosphate-dextrose blood have high levels of sodium and glucose and sometimes potassium. Initial hyperglycemia may be followed by reactive hypoglycemia as a result of an insulin response. Although acidic at the time of infusion, a post-exchange alkalosis may occur as citrate is metabolized to bicarbonate in the liver.
Many of the electrolyte and acid-base disturbances may be avoided by the use of fresh, heparinized blood. Bleeding may occur in an overheparinized infant but is reversible with protamine sulfate. Thrombocytopenia may occur, especially in the infant needing repeated exchange transfusions. Bacterial infection is rare, and routine antibiotic prophylaxis is not indicated. Most complications are avoidable if careful attention to technique is observed.
PARENT TEACHING
Providing parents with written information about jaundice and its therapy may be a beneficial adjunct to verbal explanations and is a key element of recent AAP guidelines on management of hyperbilirubinemia (Box 21-4). Because early discharge policies (<48 hours) have increased the need for outpatient evaluation or management of neonatal hyperbilirubinemia, it is important that parents feel empowered to ask questions about hyperbilirubinemia and its symptoms so that they can bring any concerns to the attention of health care providers. Indeed, early discharge of infants has now led to hyperbilirubinemia being the most common cause for hospital readmission in term infants.
BOX 21-4
Important Points for the Management of Jaundice
• Promote and support successful breastfeeding.
• Establish nursery protocols for the identification and evaluation of hyperbilirubinemia.
• Measure the total serum bilirubin (TSB) or transcutaneous bilirubin (TcB) level on infants jaundiced in the first 24 hours.
• Recognize that visual estimation of the degree of jaundice can lead to errors, particularly in darkly pigmented infants.
• Interpret all bilirubin levels according to the infant’s age in hours.
• Recognize that infants at less than 38 weeks’ gestation, particularly those who are breastfed, are at higher risk of developing hyperbilirubinemia.
• Perform a systematic assessment on all infants before discharge for the risk of severe hyperbilirubinemia.
• Provide parents with written and verbal information about newborn jaundice.
• Provide appropriate follow-up based on the time of discharge and risk assessment.
• Treat newborns, when indicated, with phototherapy or exchange transfusion.
From American Academy of Pediatrics, Subcommittee on Hyperbilirubinemia: Clinical Practice Guideline: Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation, Pediatrics 114:297, 2004.
Hyperbilirubinemia and its treatment can be disturbing to parents. Parents often feel guilty that something they did or failed to do may have resulted in their infant’s jaundice. Providing parents and families with consistent information, reassurance, and support is essential. This is especially true for the nursing mother, who may be questioning her ability to provide adequate nourishment for her infant.
The use of phototherapy can be distressing for parents and should be explained to them before they see the infant under phototherapy lights for the first time. Parents often worry that the bright lights will cause permanent damage to their infant’s eyes despite reassurances to the contrary.
In addition, incubators, bili-masks, and phototherapy lights can all contribute to a sense of separation between parents and their infant by creating a physical and emotional barrier. Parents may avoid coming to the nursery to be with their infant. If they do come to their infant’s bedside, they may be reluctant to touch or participate in care for fear of interfering with phototherapy and potentially hindering their infant’s progress. Phototherapy lights should be turned off and eye patches removed for brief periods during feedings and social times to facilitate face-to-face interaction with parents.
As with many disorders in newborn infants, time and energy spent providing parents with information and support can alleviate much fear, guilt, and anger. It also can help facilitate the development of a healthy family relationship in a time of crisis. Signs and symptoms of jaundice should be explained in a manner that is understandable and meaningful for parents, emphasizing that neonatal hyperbilirubinemia is usually a transient condition and one to which all infants must adapt after birth.
HEALTH SYSTEMS APPROACH TO BILIRUBIN
In the 1970s and 1980s, few health care providers had the opportunity to see a patient with kernicterus. 24 In recent times, however, rates of kernicterus have been rising and is seen as a systems failure in neonatal services. 4 For that reason, the Joint Commission on Accreditation of Healthcare Organizations issued Sentinel Event Alerts on kernicterus in 2001 and again in 2004. 12,13 However, neither hyperbilirubinemia nor kernicterus has been a reportable disease, and no reliable information source exists to produce national annual estimates. 11
The root cause analysis for the reappearance of kernicterus revealed several factors. First, health services are provided by multiple providers at multiple sites, many of whom do not have a sufficient understanding of bilirubin and its potential for toxicity. Early discharge of newborn infants younger than 72 hours has the consequence that infants will be discharged before the natural peak of bilirubin rise in term infants and before the establishment of adequate breast feeding. A lack of knowledge by parents regarding hyperbilirubinemia and its symptoms and limitations within health care systems to provide appropriate pre-discharge screening of at-risk infants only serves to complicate issues. 4
The AAP published guidelines on the management of hyperbilirubinemia in infants 35 weeks or more in 2004. 1 The overall aim was to promote an approach that would (1) reduce the frequency of severe hyperbilirubinemia and bilirubin encephalopathy, (2) minimize the risk for unintended harm (e.g., increased anxiety, decreased breast feeding, unnecessary treatment for the general population), and (3) avoid excessive cost and waste. These guidelines emphasize the importance of universal systematic assessment for the risk for severe hyperbilirubinemia, close follow-up, and prompt intervention when indicated. The ten key elements of the AAP practice guidelines are listed in Box 21-4. Bhutani and Johnson went further to recommend a five-step nationwide strategy to prevent severe neonatal hyperbilirubinemia as follows4:
• An institutional curriculum for the systems approach, including universal prenatal, pre-discharge, and post-discharge risk assessment of severe neonatal hyperbilirubinemia
• Advocacy for on-site services that promote breast feeding in the context of supervised and seamless health care delivery during the first month of life
• Effective parent-provider partnerships for safer management of neonatal jaundice
• Statewide (or regional) reporting of birthing institution outcome assessment for severe neonatal hyperbilirubinemia along with outcomes for neonatal screening for other inherited disorders
• Nationwide surveillance in which all severe cases of severe neonatal hyperbilirubinemia are reported
REFERENCES
1. American Academy of Pediatrics, Subcommittee on Hyperbilirubinemia: Clinical Practice Guideline: Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation, Pediatrics 114 (2004) 297.
2. Anderson, D.; Ali, K.; Blanchette, V.; et al., Guidelines on the use of intravenous immunoglobulin for hematologic conditions, Transfus Med Rev 21 (2 Suppl 1) ( 2007) S9.
3. Bhutani, V.K.; Gourley, G.M.; Adler, S.; et al., Noninvasive measurement of total serum bilirubin in a multiracial predischarge newborn population to assess the risk of severe hyperbilirubinemia, Pediatrics 106 (2000) e17.
4. Bhutani, V.K.; Johnson, L., Prevention of severe neonatal hyperbilirubinemia in healthy infants of 35 or more weeks gestation: implementation of a systems based approach, J Pediatr (Rio J) 83 (2007) 289.
5. Bhutani, V.K.; Johnson, L.; Sivieri, E.M., Predictive ability of a predischarge hour-specific serum bilirubin for subsequent significant hyperbilirubinemia in healthy term and near-term newborns, Pediatrics 103 (1999) 6.
6. Blackburn, S., Hyperbilirubinemia and neonatal jaundice, Neonatal Netw 14 (1995) 15.
7. Cashore, W.J., Bilirubin and jaundice in the micropremie, Clin Perinatol 27 (2000) 178.
8. Centers for Disease Control and Prevention, Kernicterus in full term infants: United States, 1994–1998, MMWR Morb Mortal Wkly Rep 50 (2001) 494.
9. Dennery, P.A.; Seidman, D.S.; Stevenson, D.K., Neonatal hyperbilirubinemia, N Engl J Med 344 (2001) 581.
10. Gartner, L.M., Jaundice and breastfeeding, Pediatr Clin North Am 48 (2001) 389.
11. Ip, S.; Chung, M.; Kulig, J.; et al., An evidence-based review of important issues concerning neonatal hyperbilirubinemia, Pediatrics e130 (2004) 114.
12. Joint Commission on Accreditation of Healthcare Organizations (JCAHO), Kernicterus threatens healthy newborns, Sentinel Event Alert ( Issue 18) ( 2001); April 1.
13. Joint Commission on Accreditation of Healthcare Organizations (JCAHO), Revised guidelines to help prevent kernicterus, Sentinel Event Alert ( Issue 31) ( 2004); August 31.
14. Juretschke, L.J., Kernicterus: still a concern, Neonatal Netw 24 (2005) 7.
15. Kappas, A.; Drummond, G.S.; Henschke, C.; et al., Direct comparison of Sn-mesoporphyrin, an inhibitor of bilirubin production, and phototherapy in controlling hyperbilirubinemia in term and near-term newborns, Pediatrics 95 (1995) 468.
16. Keren, R.; Bhutani, V.K.; Luan, X.; et al., Identifying newborns at risk of significant hyperbilirubinemia: a comparison of two recommended approaches, Arch Dis Child 90 (2005) 415.
17. Keren, R.; Luan, X.; Friedman, S.; et al., A comparison of alternative risk-assessment strategies for predicting significant neonatal hyperbilirubinemia in term and near-term infants, Pediatrics 121 (2008) e170.
18. Leite, M.J.; Granato Vde, A.; Facchini, F.P.; et al., Comparison of transcutaneous and plasma bilirubin measurement, J Pediatr (Rio J) 83 (2007) 283.
19. Maisels, M.J.; McDonagh, A.F., Phototherapy for neonatal jaundice, N Engl J Med 358 (2008) 920.
20. Martinez, J.C.; Garcia, H.O.; Otheguy, L.E.; et al., Control of severe hyperbilirubinemia in full-term newborns with the inhibitor of bilirubin production Sn-mesoporphyrin, Pediatrics 103 (1999) 1.
21. Newman, T.B., Outcomes among newborns with total serum bilirubin levels of 25 mg per deciliter or more, N Engl J Med 354 (2006) 1889.
22. Poland, R.L.; Odell, G.B., Physiologic jaundice: the enterohepatic circulation of bilirubin, N, Engl J Med 284 (1971) 1.
23. Robertson, A.; Karp, W.; Brodersen, R., Bilirubin displacing effect of drugs used in neonatology, Acta Paediatr Scand 80 (1991) 1119.
24. Shapiro, S.M.; Bhutani, V.K.; Johnston, L., Hyperbilirubinemia and kernicterus, Clin Perinatol 33 (2006) 387.
25. Smitherman, H.; Stark, A.R.; Bhutani, V.K., Early recognition of neonatal hyperbilirubinemia and its emergent management, Semin Fetal Neonatal Med 11 (2006) 214.
SELECTED READINGS
Auerbach, K.; Gartner, L., Breastfeeding and human milk: their association with jaundice in the neonate, Clin Perinatol 14 (1987) 89.
Catz, C.; Hanson, J.W.; Simpson, L.; et al., Summary of workshop: early discharge and neonatal hyperbilirubinemia, Pediatrics 96 (1995) 743.
Kopelman, A.E.; Brown, R.S.; Odell, G.B., The “bronze” baby syndrome: a complication of phototherapy, J Pediatr 8 (1972) 466.
Maisels, M.J., Phototherapy: 25 years later, In: (Editors: Fanaroff, A.A.; Klaus, M.H.) The yearbook of neonatal and perinatal medicine ( 1996)Mosby, St Louis.
Maisels, M.J., Neonatal jaundice, In: (Editor: Avery, G.B.) Neonatology: pathophysiology and management of the newborned 5 ( 1999)Lippincott, Philadelphia.
Martin, G.I., Proceedings from the International Congress on Neonatal Jaundice March 16-18, 2001, J Perinatol 21 (Suppl 1) ( 2001) S1–S27.
Scheidt, P.C.; Bryla, D.A.; Nelson, K.B.; et al., Phototherapy for neonatal hyperbilirubinemia: six-year follow-up of the National Institute of Child Health and Human Development Clinical Trial, Pediatrics 85 (1990) 455.
Stevenson, D.K.; Vreman, H.J., Carbon monoxide and bilirubin production in neonates, Pediatrics 100 (1997) 252.
Volpe, J., Bilirubin and brain injury, In: (Editor: Volpe, J.) Neurology of the newborned 5 ( 2008)Saunders, Philadelphia.
Yao, T.C.; Stevenson, D.K., Advances in the diagnosis and treatment of neonatal hyperbilirubinemia, Clin Perinatol 22 (1995) 741.



