Chapter 2 Pharmacokinetics: Absorption, Distribution, Metabolism, and Elimination
| Abbreviations | |
|---|---|
| CLh | Hepatic clearance |
| CLr | Renal clearance |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| GI | Gastrointestinal |
| Km | Michaelis constant |
| NAD(P) | Nicotinamide adenine dinucleotide (phosphate) |
| pH | Logarithm of the reciprocal of the hydrogen ion concentration |
| pKa | Logarithm of the reciprocal of the dissociation constant |
| UDP | Uridine diphosphate |
| Vmax | Maximum rate of reaction |
WHAT HAPPENS TO DRUGS IN THE BODY?
In nearly all cases drugs must traverse membranes to reach their site of action. The ease by which a compound crosses membranes is key to assessing the rates and extent of absorption and distribution throughout multiple compartments of the body. This chapter considers factors for assessing how specific drugs cross membranes and what variables are most important.
Numerous factors influence the rate of delivery, distribution, and disappearance of drug to and from its site of action. All these processes and variables, depicted in Figure 2-1, are described in this chapter.
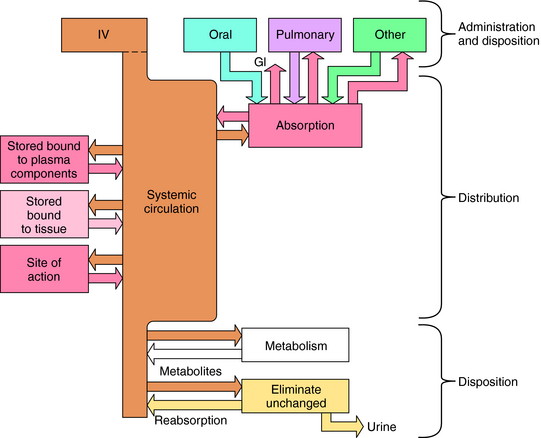
Transport of Drugs Across Membranes
some affinity for H2O (i.e., hydrophilicity), or they cannot dissolve and be transported to their sites of action by the blood and other body fluids.
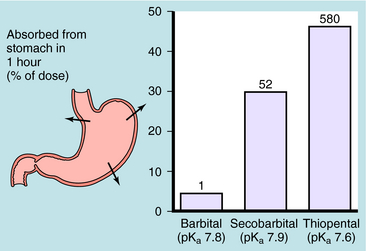
Generally, drugs that have high lipid solubility cross membranes better than those with low lipid solubility. This is exemplified in Figure 2-2 for three different barbiturates. The oil/water equilibrium partition coefficient is a measure of lipid solubility, or hydrophobicity. Drug is added to a mixture of equal volumes of oil and H2O, and the mixture is agitated to promote solubilization of the compound in each phase. When equilibrium is attained, the phases are separated and undergo assay for drug. The ratio of the concentration in the two phases is the partition coefficient. Therefore the larger the partition coefficient is, the greater the lipid solubility is. Figure 2-2 shows that absorption across the stomach wall is greater for the barbiturate with the largest lipid solubility.
Influence of pH on Drug Absorption and Distribution
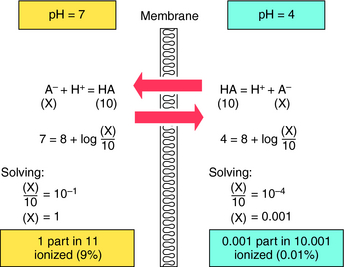
Passive diffusion of a drug that is a weak electrolyte is generally a function of the pKa of the drug and the pH of the two compartments, because only the uncharged form of the drug can diffuse across membranes. The pH values of the major body fluids, which range from 1 to 8, are shown in Table 2-1. To predict how a drug will be distributed between gastric juice (pH 1.0) on one side of the membrane and blood (pH 7.4) on the other side, the degree of dissociation of the drug at each pH value is determined.
| Fluids | pH |
|---|---|
| Gastric juice | 1.0-3.0 |
| Small intestine: duodenum | 5.0-6.0 |
| Small intestine: ileum | 8 |
| Large intestine | 8 |
| Plasma | 7.4 |
| Cerebrospinal fluid | 7.3 |
| Urine | 4.0-8.0 |
An acid is defined as a compound that can dissociate and release a hydrogen ion, whereas a base can take up a hydrogen ion. By this definition, RCOOH and RNH3+ are acids and RCOO–and RNH2 are bases. The equilibrium dissociation expression and the equilibrium dissociation constant (Ka) can be described for an acid HA or BH+ and a base A– or B, as shown below. The convention for Ka requires that the acid appear on the left and the base appear on the right of the dissociation equation as:
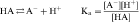
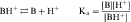
Taking the negative log of both sides yields:
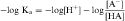
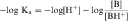
By definition the negative log of [H+] is pH and the negative log of Ka is pKa. Therefore, equations 2-3 and 2-4 can be simplified and rearranged to give:
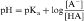
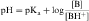
Equations 2-5 and 2-6 are the acid and base forms, respectively, of the Henderson-Hasselbach equation, and they can be used to calculate the pH of the solution when the pKa and the ratios of [A–]/[HA] or [B]/[BH+] are known. In pharmacology it is often of interest to calculate the ratios of [A–]/[HA] or [B]/[BH+] when the pH and the pKa are known. For this calculation, equations 2-5 and 2-6 are rearranged to equations 2-7 and 2-8 as follows:
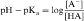
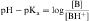
The results are plotted in Figure 2-3 to show the fraction of the nonionized (HA or B) forms. The pKa is the pH when the drug is 50% dissociated. Applying equations 2-7 and 2-8 to an acidic drug with a pKa of 6.0 enables one to calculate the degree of ionization for this drug in the stomach or blood (assuming the blood pH is 7.0 for ease of calculation), as follows:
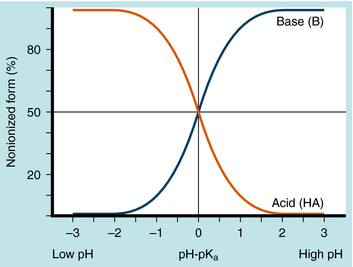
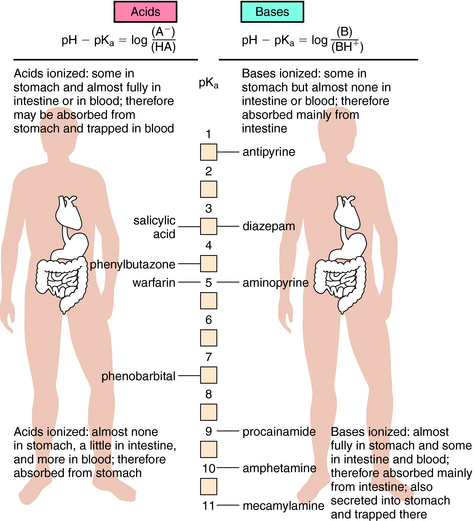
Another example is shown in Figure 2-4 for a basic drug. This approach is particularly useful for predicting whether drugs can be absorbed in the stomach, the upper intestine, or not at all. Figure 2-5 provides a summary of the effects of pH on drug absorption in the GI tract for several acidic and basic drugs. It also assists in predicting which drugs will undergo tubular reabsorption, which is discussed later.
Most drugs are transported across membranes by simple passive diffusion. The concentration gradient across the membrane is the driving force that establishes the rate of diffusion from high to low concentrations. Other mechanisms, including active transport, facilitated diffusion, or pinocytosis, also exist. Active transport involves specific carrier molecules in the membrane that bind to and carry the drug across the lipid bilayer. Because there are a finite number of carrier molecules, they exhibit classical saturation kinetics. Drugs may also compete with a specific carrier molecule for transport, which can lead to drug-drug interactions that modify the time and intensity of action of a given drug. An active transport system may concentrate a drug on one side of a membrane, because cellular energy is used to drive transport, with no dependence on a concentration gradient. The primary active drug transport systems are present in renal tubule cells, biliary tract, blood-brain barrier, and the GI tract.
Distribution to Special Organs and Tissues
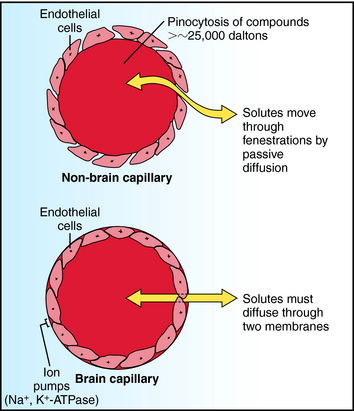
Two compartments of special importance are the brain and the fetus. Many drugs do not readily enter brain. Capillaries in brain differ structurally from those in other tissues, with the result that a barrier exists between blood within brain capillaries and the extracellular fluid in brain tissue. This blood-brain barrier hinders transport of drugs and other materials from blood into brain tissue. The blood-brain barrier is found throughout brain and spinal cord at all regions central to the arachnoid membrane, except for the floor of the hypothalamus and the area postrema. Structural differences between brain and non-brain capillaries, and how these differences influence blood-brain transport of solutes, are shown schematically in Figure 2-6. Non-brain capillaries have fenestrations (openings) between the endothelial cells through which solutes move readily by passive diffusion, with compounds having molecular weights greater than approximately 25,000 daltons (Da) undergoing transport by pinocytosis. In brain capillaries, tight junctions are present because there are no fenestrations, and pinocytosis is greatly reduced. Special transport systems are available at brain capillaries for glucose, amino acids, amines, purines, nucleosides, and organic acids; all other materials must cross two endothelial membranes plus the endothelial cytoplasm to move from capillary blood to tissue extracellular fluid. Thus the main route of drug entry into central nervous system (CNS) tissue is by passive diffusion across membranes, restricting the available compounds used to treat brain disorders. At the same time, the potential deleterious effects of many compounds on the CNS are not realized, because the blood-brain barrier acts as a safety buffer. Generally, only highly lipid-soluble drugs cross the blood-brain barrier, and thus for these drugs no blood-brain barrier exists. In infants and the elderly, the blood-brain barrier may be compromised, and drugs may diffuse into brain.
METABOLISM AND ELIMINATION OF DRUGS
Oxidation and conjugation are the two most important and are discussed further. Simpler examples are given for reduction and hydrolysis.
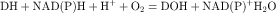
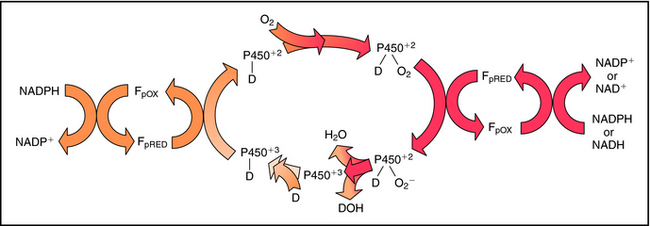
In most cells the cytochrome P450s are associated with the endoplasmic reticulum. More than 50 isoforms of human P450 exist, with various substrate specificities and different mechanisms regulating their expression. This plethora of enzyme systems provides the body with the ability to metabolize large numbers of different drugs. The common feature of P450 substrates is their lipid solubility. Most lipophilic drugs and environmental chemicals are substrates for one or more forms of P450. During the catalytic reaction, when the drug binds to cytochrome P450, the heme iron in the enzyme undergoes a cycle that begins in the ferric oxidation state. The heme iron undergoes reduction to the ferrous state and binds oxygen, and the molecular oxygen bound to the active site is reduced to a reactive form that inserts one oxygen atom into the drug substrate with the other oxygen being reduced to H2O, with the eventual regeneration of the ferric state of the heme iron. Free radical or iron-radical groups are formed at one or more parts of the cycle. The reaction cycle is summarized in Figure 2-7.
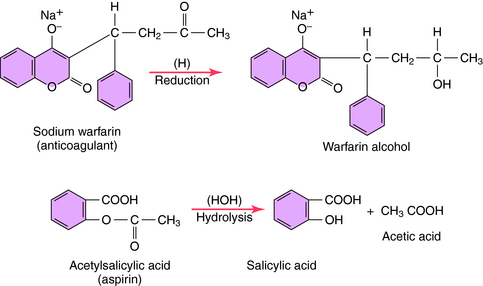
Typical metabolic reactions involving reduction and hydrolysis of drugs are shown in Figure 2-8. Oxidations, reductions, and hydrolytic reactions are commonly referred to as phase I reactions.
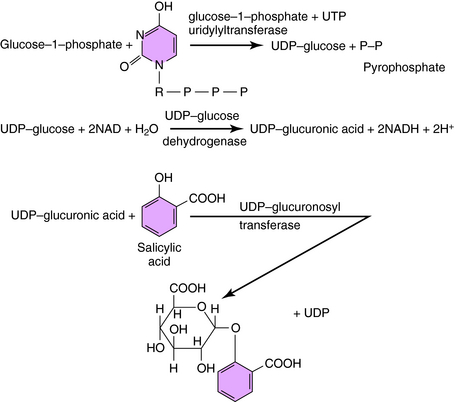
Conjugation, the second class of reactions to drug metabolism, involves coupling the drug molecule to an endogenous substituent group so that the resulting product will have greater H2O solubility, leading to enhanced renal or biliary elimination. Conjugation reactions, like other metabolic processes, are catalyzed by phase II drug-metabolizing enzymes. In addition, the groups that are being coupled need to be “activated” by the transfer of energy from high-energy phosphate compounds. For example, glucuronic acid can be conjugated by the enzyme UDP-glucuronosyl transferase to compounds of the general types ROH, RCOOH, RNH2, or RSH, where R represents the remainder of the drug molecule. However, glucuronic acid must first be activated by the reaction of glucose-1-phosphate with uridine triphosphate to form UDP-glucose followed by oxidation to activated UDP glucuronic acid. The reaction sequence is shown in Figure 2-9 for the formation of the ROH glucuronide of salicylic acid. Another glucuronide could be formed through conjugation with the RCOOH group. Many drugs, as well as endogenous materials, including bilirubin, thyroxine, and steroids, also undergo conjugation with activated glucuronic acid in the presence of UDP-glucuronosyl transferase. In addition to glucuronate, conjugation may also occur with activated glycine, acetate, sulfate, and other groups besides glucuronate, leading to drug conjugates that will be readily excreted.
Factors Regulating Rates of Drug Metabolism
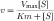
Vmax is directly proportional to the concentration of the enzyme. If a change occurs in the concentration of enzyme, there should be a similar change in the rate of metabolism. Because different drugs may be substrates for the same metabolizing enzyme, they can competitively inhibit each other’s metabolism. However, this is usually not a significant problem, because the capacity of the metabolizing system is large, and drugs are usually present in concentrations less than their Km.
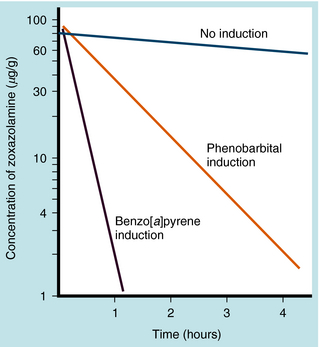
Many drugs, environmental chemicals, air pollutants, and components of cigarette smoke stimulate the synthesis of drug-metabolizing enzymes. This process, termed enzyme induction, may elevate the level of hepatic drug-metabolizing enzymes. In most cases the inducers are also substrates for the enzymes they induce. However, the induction is generally nonspecific and may result in increases in the metabolism of a variety of substrates. For example, phenobarbital and the highly reactive air pollutant 3,4-benzo[a]pyrene can increase the rate of oxidation of the CNS muscle relaxant zoxazolamine in animals (Fig. 2-10). Because cigarette smoke contains compounds that can promote induction, chronic smokers have considerably higher levels of some hepatic and lung drug-metabolizing enzymes. Induction of P450 by polycyclic aromatic hydrocarbons in smoke causes female smokers to have lower circulating estrogen than nonsmokers.
For nearly all drugs, the normal therapeutic range of concentrations is much smaller than the Km. Thus hepatic or other drug-metabolizing enzymes are operating at concentration levels far below saturation, where equation 2-9 reduces to a first-order reaction. Thus drug metabolism typically follows first-order kinetics. An exception is the metabolism of salicylic acid, in which enzyme saturation can occur at elevated drug concentrations. Aspirin (acetylsalicylic acid) is used extensively for the treatment of inflammatory diseases, with the optimum therapeutic concentration only slightly below the concentration where signs of toxicity appear. Aspirin is hydrolyzed to salicylic acid, which in turn has several routes of metabolism before elimination (Fig. 2-11). Two pathways are subject to saturation in humans:
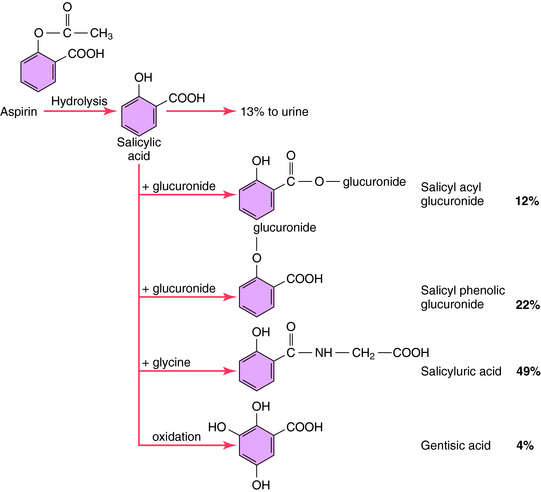
For enzyme saturation the kinetics become zero order, and the rate of reaction becomes constant at Vmax. This is consistent with equation 2-9, when [S] is much larger than Km. Saturation of drug-metabolizing enzymes has a pronounced influence on drug-plateau concentrations. With zero-order kinetics, elimination rates no longer depend on dose or blood concentration.
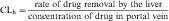
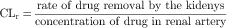
The mechanisms by which the renal clearance of drugs takes place (glomerular filtration, tubular secretion, and tubular reabsorption) are the same as those responsible for the renal elimination of endogenous substances (Fig. 2-12).
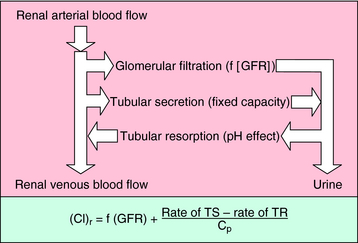
The third mechanism affecting renal clearance is reabsorption of filtered or secreted drug from the tubules back into the venous blood of the nephrons. Although this process may be either active or passive, for most drugs it occurs by passive diffusion. Drugs that are readily reabsorbed are characterized by high lipid solubility or by a significant fraction in a nonionized form at urine pH and in the ionized form at plasma pH. For example, salicylic acid (pKa = 3.0) is approximately 99.99% ionized at pH 7.4 (see equation 2-7) but only approximately 90% ionized at pH 4.0. Thus some reabsorption of salicylic acid could be expected from acidic urine. In drug overdose, the manipulation of urine pH is sometimes used to prevent reabsorption. Ammonium chloride administration leads to acidification of the urine; sodium bicarbonate administration leads to alkalinization of the urine. Some additional examples are given in Table 2-2.
TABLE 2–2 Effect of Urine pH on Renal Clearance for Drugs that Undergo Tubular Resorption
| Bases Cleared Rapidly by Making Urine More Acidic | Acids Cleared Rapidly by Making Urine More Alkaline |
|---|---|
| Amphetamine | Acetazolamide |
| Chloroquine | Nitrofurantoin |
| Imipramine | Phenobarbital |
| Levorphanol | Probenecid |
| Mecamylamine | Salicylates |
| Quinine | Sulfathiazole |
Modified Renal Function and Drug Elimination
Renal clearance of drugs may be decreased in neonates, geriatric patients, and individuals with improperly functioning kidneys. The effect of patient age on renal clearance of drugs is discussed in Chapter 3. In the case in which it is desirable to administer a drug that is disposed of primarily by renal elimination, and the individual has impaired renal function, the extent of renal function must be determined. Creatinine clearance is the standard clinical determination used to obtain an approximate measure of renal function. To determine the rate of urinary excretion of creatinine, urine is collected over a known period (often 24 hours) and pooled, its volume is measured, and it undergoes assay for creatinine. At the midpoint of the urine collection period, a serum sample is obtained and undergoes assay for creatinine. Creatinine clearance is calculated by dividing the rate of urinary excretion of creatinine (mg/min) by the serum concentration of creatinine (mg/mL), resulting in units of mL/min.
Determination of creatinine clearance provides a measure of glomerular filtration. In addition, the relationship between the rate constant for renal elimination of unchanged drug and creatinine clearance must be demonstrated. For the usual case of first-order renal elimination, that relationship is linear; thus a creatinine clearance of 50% of normal means that renal elimination of this drug would be expected to operate at 50%, and the rate of drug input should be reduced accordingly. For example, a drug administered 100 mg every 6 hours (400 mg in 24 hours) to a patient with normal creatinine clearance could be given 40 mg every 12 hours (80 mg in 24 hours), if the creatinine clearance decreased to only 20% of normal. It is assumed that other pathways for disappearance of this drug retain normal function. Clearance is considered further in Chapter 3.
A plot of the plasma drug concentration curve in a population of patients receiving the same drug dosage results in a normal bell-shaped curve. However, if a genetic factor or factors are involved, the population distribution curve is bimodal (or sometimes multimodal)—an indication of separate populations, one drug sensitive and one less drug sensitive (Fig. 2-13).
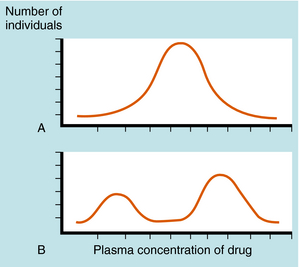
Genetic differences in enzyme activity associated with biotransformation of specific drugs is often responsible for differences in pharmacogenetics. An example is acetylation polymorphism. N-Acetylation of aromatic amines and hydrazines is one of several reactions for drug and chemical detoxification. Primary sites for acetylation are the liver and GI mucosa. Differences in N-acetylation were originally recognized in patients with tuberculosis who were treated with isoniazid, a drug metabolized principally by this mechanism. By determining the plasma concentration at a specific time after a fixed dose of isoniazid, patients could be classified as slow or rapid acetylators, indicating that N-acetylating activity is distributed bimodally. Acetylation polymorphisms are now known to influence the metabolism of many drugs and chemicals in addition to isoniazid (Box 2-1). This phenomenon varies widely with race and geographical distribution; 45% of whites and blacks in the United States are slow acetylators, whereas 10% of Asians in the United States are slow acetylators.
Drugs can induce hemolytic anemia in patients genetically deficient in red blood cell glucose-6-phosphate dehydrogenase (Box 2-2). This enzyme, part of the red blood cell hexose monophosphate shunt, is a primary source of reduced NADPH, a cofactor for glutathione reductase. Hemolysis of red blood cells results from the cells’ inability to maintain sufficient reduced glutathione critical for maintaining reduced protein sulfhydryl groups. The oxidized state of glutathione promotes enzyme denaturation and erythrocyte membrane instability. Many glucose-6-phosphate dehydrogenase variants have been identified, and it is estimated that more than 200 million people worldwide have a variant enzyme. These specific examples emphasize the importance of considering genetic variation in evaluating abnormal responses to drugs in patients.
Guengerich FP. Mechanisms of cytochrome P450 substrate oxidation: MiniReview. J Biochem Mol Toxicol. 2007;21:163-168.
Tompkins LM, Wallace AD. Mechanisms of cytochrome P450 induction. J Biochem Mol Toxicol. 2007;21:176-181.
Wilke RA, Lin DW, Roden DM, et al. Identifying genetic risk factors for serious adverse drug reactions: Current progress and challenges. Nat Rev Drug Discov. 2007;6:904-916.