Chapter 1 Pharmacodynamics: Receptors and Concentration-Response Relationships
| Abbreviations | |
|---|---|
| β-ARK | β adrenergic receptor kinase |
| cAMP | Cyclic adenosine monophosphate |
| GABA | γ-aminobutyric acid |
| GABAA | γ-aminobutyric acid type A receptor |
| GPCR | G-protein–coupled receptor |
| LGIC | Ligand-gated ion channel |
| Epi | Epinephrine |
| NE | Norepinephrine |
| RTK | Receptor tyrosine kinase |
For most drugs, the site of action is a specific macromolecule, generally termed a receptor or a drug target, which may be a membrane protein, a cytoplasmic or extracellular enzyme, or a nucleic acid. A drug may show organ or tissue selectivity as a consequence of selective tissue expression of the drug target. For example, the action of the proton pump inhibitor esomeprazole occurs specifically in the parietal cells that line the gastric pits of the stomach because that is where its target, the potassium/hydrogen adenosine triphosphatase (K+/H+-ATPase), is expressed. Although the actions of a few drug types, such as osmotic diuretics (see Chapter 21), may not involve receptors as they are usually defined, the concept of receptors as sites of drug action is critical to understanding pharmacology.
Receptors (i.e., drug targets) fall into many classes, but two types predominate:
This lock and key hypothesis is still relevant to how we understand receptors today. It emphasizes the idea that the drug and receptor must be structurally complementary to recognize each other and initiate an effect.
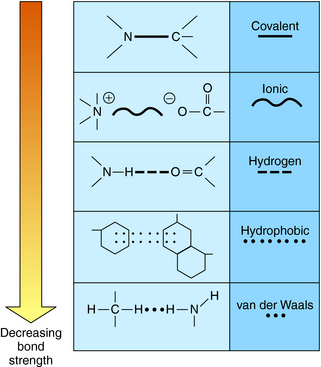
The vast majority of drugs are small molecules with molecular weights below 500 to 800. These molecules interact with their protein targets via a number of different chemical bonds. The principal types of chemical bonds are depicted in Figure 1-1. These bonds apply to the interactions between drugs and classical receptors. Covalent bonds require considerable energy to break and are classified as irreversible when formed in drug-receptor complexes. Ionic bonds are also strong but may be reversed by a change in pH. Most drug-receptor interactions involve multiple weak bonds.
Molecules that bind to receptors may have two major effects on the conformation of the receptor molecule. Agonists will bind to the receptor and activate it, like a key will fit into a lock and turn it. Activation of the receptor by agonist binding initiates a conformational change in the receptor and activation of one or more downstream signaling pathways. An example of the action of an agonist is provided by the effect of acetylcholine on the nicotinic cholinergic receptor at the neuromuscular junction. When acetylcholine binds to its binding sites on the external surface of this receptor, the channel opens and allows Na+ to flow down its electrochemical gradient and depolarize the muscle cell.
Antagonists are drugs that bind to the receptor but do not have the unique structural features necessary to activate it. In the lock and key analogy, antagonists can fit in the lock but cannot open it. Like agonists, antagonists fit into a specific binding site within the receptor but lack the proper structural features to initiate a conformational change leading to receptor activation. However, because they occupy the binding site of the receptor, antagonists inhibit activation by agonists. An example of antagonists is the class of neuromuscular blocking drugs used in the operating room to relax skeletal muscles during surgery. These drugs are analogs of curare, the active molecule in plant extracts used as arrow poisons by Native South Americans and studied by early European explorers. Curare is an antagonist at nicotinic cholinergic receptors at the neuromuscular junction and blocks the ability of acetylcholine or similar agonists to activate this receptor. This blockade inhibits muscle depolarization and causes paralysis of skeletal muscle, including the diaphragm and intercostal muscles needed for respiration. Several modern analogs of curare are available and used routinely during general anesthesia for relaxing muscle tone in patients undergoing surgery (see Chapter 12).
A third class of drugs that interact with receptors are allosteric modulators. These compounds bind to a site on the receptor distinct from that which normally binds agonist, called an allosteric site. Occupation of this site can either increase or decrease the response to the natural agonist, depending on whether it is a positive or negative modulator. Because allosteric modulators bind to sites different from where agonists bind, interactions between agonists and allosteric modulators are not competitive. The binding sites for agonists, antagonists, and allosteric modulators are depicted in Figure 1-2.
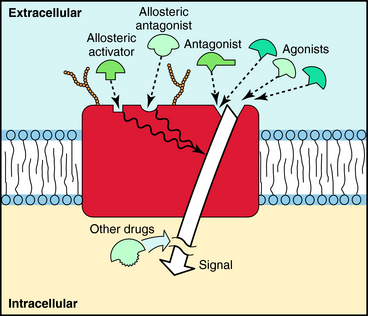
Receptors are a primary focus for investigating the mechanisms by which drugs act. With the sequencing of the human genome, the structures and varieties of most receptors have now been identified. This advance has revealed many new receptors that could be potential drug targets for further pharmaceutical development. The major features of receptors are listed in Box 1-1. Three major concepts are illuminated by the concept of drug receptors.
The first is the quantitative relationship between drug concentration and the subsequent physiological response. This response is determined primarily by the affinity of the drug for the receptor, which is a measure of the binding constant of the drug for the receptor protein. A high affinity means that a low concentration of drug is needed to occupy receptor sites, whereas a low affinity means that much higher concentrations of drug are needed. The concentration-response curve is also influenced by the number of receptors available for binding. In general, more receptors can produce a greater response, although this is not always the case.
The second key concept is that receptors and their distribution in the tissues of the body are responsible for the specificity of drug action. The size, shape, and charge of a receptor determine its affinity for binding any of the vast array of chemically different hormones, neurotransmitters, or drug molecules it may encounter. If the structure of the drug changes even slightly, the type of receptor the drug binds to will also often change. Drug binding to receptors often exhibits stereoselectivity, in which stereoisomers of a drug that are chemically identical, but have different orientations around a single bond, can have very different affinities. For example, the L-isomer of narcotic analgesics is approximately 1000 times more potent than the D-isomer, which is essentially inactive for pain relief (see Chapter 36). The presence or absence of a single hydroxyl group, methyl group, or other apparently minor structural change can also dramatically alter the affinity of a drug for a receptor.
Receptors also explain the key concept of pharmacological antagonists, which prevent agonist activation by binding to a receptor. Administration of an antagonist will block tonic or stimulated activity of endogenous neurotransmitters and hormones, thus interfering with their normal physiological functions. An example is propranolol, which, by antagonizing β1 adrenergic receptors, prevents the normal increase in heart rate associated with activation of the sympathetic nervous system (see Chapter 11).
Specialized receptors are usually involved in the normal regulation of cell function by hormones, neurotransmitters, growth factors, steroids, and autocoids. Although they are often found on the cell surface, where they are easily accessible to hydrophilic messengers, many hormone receptors are located inside the cell, and ligands for these molecules easily cross the cell membrane (see Part V). An example of an intracellular receptor is the glucocorticoid receptor (see Chapter 39). For most receptor types, multiple distinct subtypes can cause similar or distinct responses. This diversity of receptors and responses provides new targets for drug development.
In most cases, a drug (D) binds to a receptor (R) in a reversible bimolecular reaction described as:
Four major superfamilies of receptors are involved in signal transduction, representing the targets of clinically useful drugs. These include ligand-gated ion channels (LGICs), G-protein–coupled receptors (GPCRs), receptor tyrosine kinase (RTKs), and nuclear hormone receptors. Table 1-1 contains a list of receptors in these classes that are important in the actions of several therapeutically useful drugs.
| Type | Subtype | Endogenous Ligand |
|---|---|---|
| LGICs | ||
| Acetylcholine | Nicotinic | Acetylcholine |
| GABA | A, C | GABA |
| Glutamate | NMDA, kainate, AMPA | Glutamate or aspartate |
| Serotonin | 5-HT3 | Serotonin |
| GPCRs | ||
| ACTH | – | ACTH |
| Acetylcholine | Muscarinic | Acetylcholine |
| Adrenergic | α1-2, β1-3 | Epi and NE |
| GABA | B | GABA |
| Glucagon | – | Glucagon |
| Glutamate | Metabotropic | Glutamate |
| Opioid | μ, κ, δ | Enkephalins |
| Serotonin | 5-HT1-2,4,5-7 | 5-HT |
| Dopamine | D1-5 | Dopamine |
| Adenosine | A1, A2a, A2b, A3 | Adenosine |
| Histamine | H1-4 | Histamine |
| RTKs | ||
| Insulin | – | Insulin |
| NGF | – | NGF |
| EGF | – | EGF |
| Nuclear Hormone Receptors | ||
| Estrogen | α, β | Estrogen |
| Glucocorticoid | – | Cortisol |
| Androgens | – | Testosterone |
ACTH, Adrenocorticotrophic hormone; EGF, epidermal growth factor; Epi, epinephrine; NGF, nerve growth factor; NE, norepinephrine.
LGICs are most important in the central and peripheral nervous systems, excitable tissues such as the heart, and the neuromuscular junction. They include nicotinic cholinergic receptors (Fig. 1-3) at the neuromuscular junction, many of the γ-aminobutyric acid (GABA) and glutamate receptors in the brain, and one type of serotonin receptor. These receptors are responsible for fast synaptic transmission, where release of a transmitter causes an electrical effect on the postsynaptic neuron by opening a specific ion channel and leading to a change in membrane potential. LGICs are complex proteins composed of four or five subunits, and the specific subunit combinations differ at different sites in the body, allowing for selectivity of effects.

For example, the subunits of the nicotinic receptor at the neuromuscular junction differ from those of the nicotinic receptor at autonomic ganglia. As a consequence, although the responses to acetylcholine and ion gating properties of these two channels are similar, these receptors are activated and antagonized by different drugs. This property allows selective blockade of the neuromuscular junction by drugs that do not block the channel at autonomic ganglia (see Chapter 12). Typically, LGICs have two binding sites for agonist and binding sites for allosteric modulators, which increase or decrease the ability of the transmitter to open the channel. One important class of allosteric modulators is the benzodiazepines, which are used extensively for the treatment of anxiety and sleep disorders (see Chapter 31). These drugs bind to γ-aminobutyric acid type A (GABAA) receptors, which are ligand-gated chloride ion channels that are activated by the inhibitory neurotransmitter GABA. Benzodiazepines have no effect on channel opening by themselves, but their binding dramatically increases the ability of GABA to open the channel.
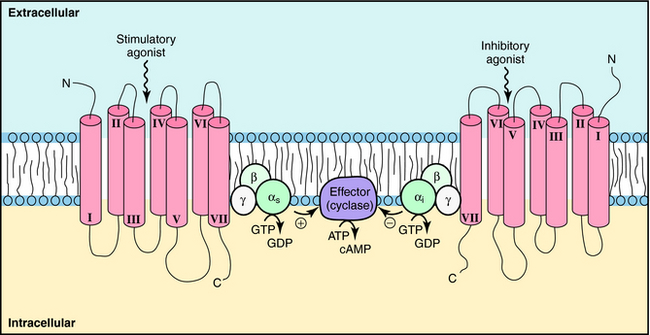
GPCRs are probably the most important class of receptors in pharmacology, because most currently marketed drugs target this receptor superfamily. GPCRs are much simpler than ligand-gated ion channels, being usually composed of a single subunit that contains seven transmembrane spanning domains. They are thought to have a single binding site, and as yet, there are only a few allosteric modulators for this class of receptors. GPCRs activate signals by inducing a conformational change that activates a large family of G-proteins to regulate signaling pathways (Fig. 1-4). These events regulate a host of important cellular functions (Box 1-2). GPCRs represent the largest protein family in the human genome, accounting for approximately 2% of all human genes. Approximately half of these receptors are olfactory receptors for detecting odorants; most of the remaining GPCRs respond to neurotransmitters, hormones, autocoids, and cytokines.
BOX 1–2 GPCR Signaling
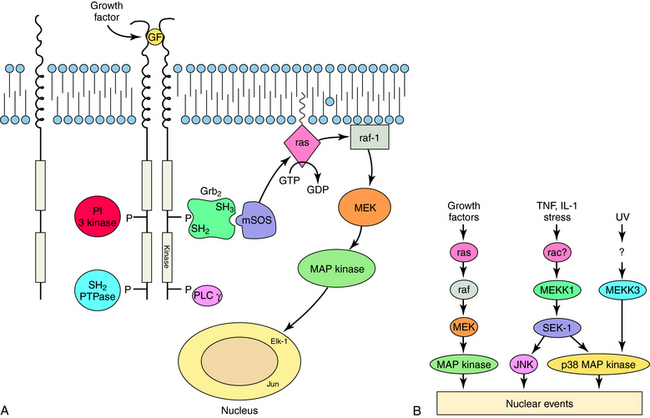
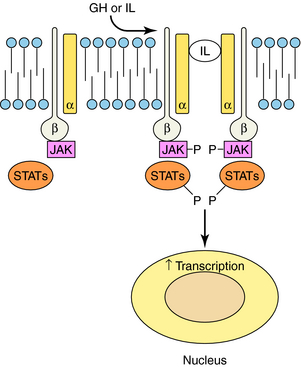
RTKs contain an extracellular ligand binding domain, one transmembrane spanning segment, and an intracellular tyrosine kinase domain. Binding of a ligand to the extracellular domain causes dimerization of the receptor and stimulates a tyrosine kinase activity within the intracellular domain. The best examples of these receptors are the growth factor receptors such as epidermal growth factor or nerve growth factor receptors (Fig. 1-5). When growth factors bind to these receptors, they cause tyrosine phosphorylation of the receptor, other proteins, or both, which leads to activation of a large number of cellular pathways. Cytokine receptors are part of this subfamily because they are structurally very similar. However, instead of intrinsic enzymatic activity within the receptor molecule, they have docking sites where tyrosine kinase enzymes bind (Fig. 1-6). These receptors are activated by a variety of molecules such as erythropoietin, interleukins, and growth hormone. RTKs are an increasingly important target for drugs to treat neoplastic diseases, where cell growth is uncontrolled (see Chapters 53 and 54).
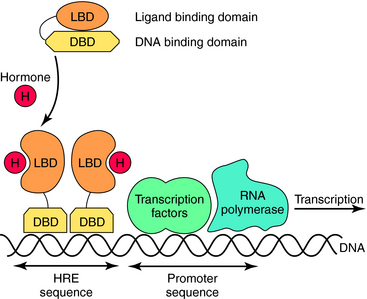
Nuclear hormone receptors are located in the cytosol, and unlike the other receptor superfamilies that are activated on the cell surface, these receptors bind their ligand in the cytoplasm and translocate to the nucleus. Intracellular receptors respond to highly hydrophobic compounds that easily cross cell membranes, including various classes of steroids, but there are also nuclear receptors for other compounds, such as retinoic acid. These receptors are usually ligand-activated transcription factors that, when bound by ligand, dimerize, enter the nucleus, and bind directly to specific DNA recognition sequences and increase or decrease transcription of particular genes (Fig. 1-7).
In contrast to the four classes of specialized receptors that have been honed by evolution to provide the selectivity and specificity required for intercellular communication, there are also intracellular enzymes that provide drug targets with excellent specificity. Two notable examples include esomeprazole, which inhibits the parietal cell H+/K+-ATPase and is very useful for treatment of gastric hyperacidity (see Chapter 18), and imatinib mesylate, which inhibits the abl tryrosine kinase and is useful in the treatment of leukemias (see Chapter 55). Neither the H+/K+ -ATPase nor the abl tyrosine kinase are specialized receptors for transmembrane signaling, but both are central to control of a primary function in the cells in which they are expressed. Thus both of these drugs act with excellent specificity.
Receptors are commonly named after the natural agonist that activates them. For example, acetylcholine acts through cholinergic receptors; Epi (adrenaline) and NE (noradrenaline) act through adrenergic receptors; and serotonin acts through serotonergic receptors. Receptor activation is very specific, and there is little cross-reactivity between natural compounds and other receptors. For instance, acetylcholine binds only to cholinergic receptors and does not bind to adrenergic receptors or members of other receptor families. This is true for essentially all transmitters and hormones. However, a transmitter like dopamine (the immediate precursor of NE), which has its own family of dopaminergic receptors, also binds with low affinity to adrenergic receptors as a consequence of its structural similarity to NE. This is advantageous, because dopamine is used clinically to stimulate β1 adrenergic receptors in cardiac failure (see Chapter 11).
As mentioned, each receptor family typically contains multiple subtypes that may be characterized pharmacologically by the use of selective agonists, antagonists, or both. For example, there are two major subfamilies of cholinergic receptors, nicotinic and muscarinic. Nicotinic cholinergic receptors are selectively activated by the agonist nicotine and are selectively blocked by drugs like curare. In contrast, muscarinic cholinergic receptors are selectively activated by the agonist muscarine and are selectively blocked by the antagonist atropine (see Chapter 10). Nicotine and curare have essentially no effect on muscarinic cholinergic receptors, and muscarine and atropine have essentially no effect on nicotinic cholinergic receptors. Both nicotinic and muscarinic cholinergic receptor subfamilies, like that for most other neurotransmitters, consist of multiple subtypes. These are discussed in subsequent chapters.
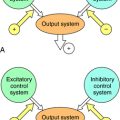
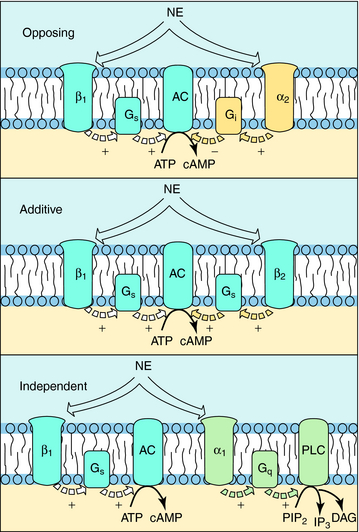
The situation is complicated because multiple receptor subtypes for one transmitter can coexist on a single cell, raising the possibility that one transmitter can deliver multiple messages to the same cell. These messages may be opposing, complementary, or independent. For example, various combinations of adrenergic receptors can be present on the same cell. The β1 adrenergic receptor activates adenylyl cyclase through a G-protein (Gs). Because the α2 adrenergic receptor inhibits adenylyl cyclase through a different G-protein (Gi), mutually antagonistic signals will be generated by the presence of both subtypes in response to the same neurotransmitter, NE. In a like manner, additive signals can be generated by the presence of the β2 adrenergic receptor, which also activates adenylyl cyclase through Gs, or independent signals can be generated by the presence of the α1 adrenergic receptor, which activates phospholipase C (Fig. 1-8). Overall, the response of a cell to a single transmitter (or a drug that mimics a neurotransmitter) depends on the types and relative proportions of receptor subtypes present in the cell.
CONCENTRATION-RESPONSE RELATIONSHIPS
Binding of a drug to a receptor is a reversible bimolecular interaction, as described in Equation 1-1. This equation follows the law of mass action, which states that at equilibrium, the product of the active masses on one side of the equation divided by the product of active masses on the other side of the equation is a constant. Therefore concentrations of both drug and receptor are important in determining the extent of receptor occupation and subsequent tissue response.
Quantification of the amount of drug necessary to produce a given response is referred to as a concentration–response relationship. Practically, one rarely knows the concentration of drug at the active site, so it is usually necessary to work with dose-response relationships. The dose of a drug is simply the amount administered (e.g., 10 mg), whereas the concentration is the amount per unit volume (e.g., mg/ml). To achieve similar concentrations in patients, it is often necessary to adjust the dose based on patient size, weight, and other factors (see Chapter 3).
Dose-response curves are usually assumed to be at equilibrium, or steady-state, when the rate of drug influx equals the rate of drug efflux, although this is an ideal that is not often achieved in practice. Once the drug reaches its receptors, many responses are graded; that is, they vary from minimum to maximum response. Most concentration- and dose-response curves are plotted on log scales rather than linear scales to make it easier to compare drug potencies; the log scale yields an S-shaped curve, as shown (Fig. 1-9).
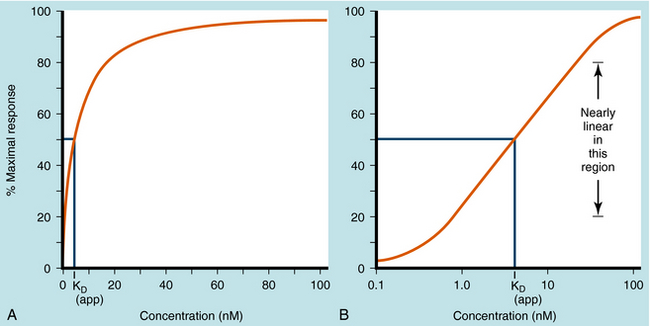
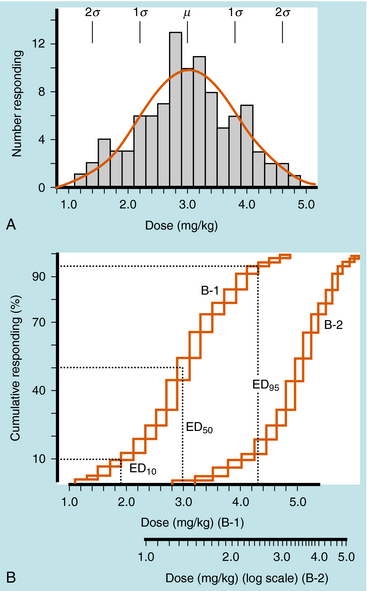
Quantal responses are all-or-none responses to a drug. For example, after the administration of a hypnotic drug, a patient is either asleep or not. Construction of dose-response curves for quantal responses requires the use of populations of subjects who are characterized by interindividual variability. A few subjects demonstrate an initial response at a low dose, most subjects demonstrate an initial response at an intermediate dose, and a few subjects demonstrate an initial response at a high dose (Fig. 1-10, A), resulting in a bell-shaped “Gaussian” distribution of sensitivity. Quantal dose-response curves are often plotted in a cumulative manner, comparing the dose of drug on the x-axis with the cumulative percentage of subjects responding to that dose on the y-axis (Fig. 1-10, B).
Occupation of a receptor by a drug is derived from the mass action law (see Equation 1-1) and is:
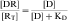
The KD value is a fixed parameter describing the affinity of a drug for the receptor binding site. From Equation 1-2, it is clear that when KD equals [D], half of the receptors are occupied; thus KD is the concentration of drug that achieves half maximal saturation of the receptor population. This means that drugs with a high KD (low affinity) will require a high concentration for occupancy, whereas drugs with a low KD (high affinity) require lower concentrations. KD is also equal to the ratio of the rate of dissociation of the [DR] complex to its rate of formation. Thus the KD of a drug is a reflection of the structural affinity of the drug and its receptor, that is, how quickly the drug binds to the receptor and how long it stays bound. Every drug/receptor combination will have a characteristic KD, although these values can differ by many orders of magnitude. For example, glutamate has approximately millimolar affinity for its receptors, whereas some β adrenergic antagonists have nanomolar affinities for their receptors.
There are two major classes of antagonists, competitive and noncompetitive. Most antagonists are competitive antagonists. These drugs compete with agonists for the same binding site on a given receptor. If the receptor is occupied by a competitive antagonist, then agonist binding to the receptor is reduced. Likewise, if the receptor is bound by an agonist, antagonist binding will be diminished. When present alone, each drug will occupy the receptors in a concentration-dependent manner as described in Equation 1-2. However, when both drugs are present and competing for the same binding site, the equation describing agonist occupancy is:
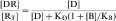
Equation 1-3 also demonstrates the very important point that the presence of a competitive antagonist causes a shift to the right in the agonist dose-response curve (decreased apparent KD) but no change in the shape of the curve. This parallel rightward shift is diagnostic of competitive antagonists and means that every portion of the dose-response curve is shifted by exactly the same amount (Fig. 1-11). The magnitude of rightward shift is dependent on the concentration of antagonist, which is variable, divided by its affinity constant, which is fixed. Therefore, if the concentration of drug is known, measuring the magnitude of the rightward shift allows direct calculation of the KB for the antagonist. This is very important because the affinity constant is essentially a molecular description of how well a drug binds to a particular receptor and is constant for any drug/receptor pair. Thus comparison of affinity constants for antagonists at receptors in different tissues is the best way to determine whether they are the same or different, making antagonists extremely useful in subclassifying receptors.
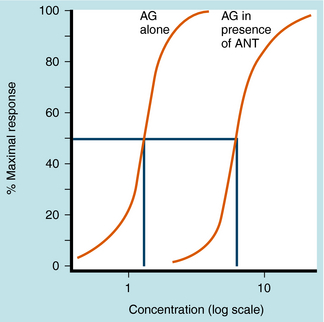
The second, less common type of antagonist is the noncompetitive antagonist. There are a number of different noncompetitive antagonists, but most drugs in this class are irreversible alkylating agents. An example of this class of drug is phenoxybenzamine, a drug that acts predominantly on α1 adrenergic receptors and is used to mitigate the effects of catecholamines secreted by adrenal tumors (see Chapter 11). Drugs of this class contain highly reactive groups, and when they bind to receptors, they form covalent bonds, occupying the binding site in an essentially irreversible, nonsurmountable manner. Because they decrease the number of available receptors, noncompetitive antagonists will usually decrease the maximum response to an agonist without affecting its EC50 (concentration causing half-maximal effect). An advantage of a noncompetitive antagonist is its long-lasting effect. Because the drug binds a receptor irreversibly, the drug effect lasts until new receptors are synthesized.
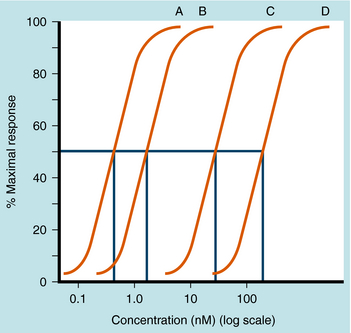
As discussed, agonists can participate in both equilibria shown in Equation 1-1, binding to and activating receptors to cause a conformational change. Partial agonists have a dual activity, that is, they act partially as agonists and partially as antagonists. When bound to their receptors, partial agonists are only partly able to shift the receptor to its activated conformational state. Efficacy is the proportion of receptors that are forced into their active conformation when occupied by a particular drug. It is used to describe the maximal effect of partial agonists in causing a receptor conformational change and can range from 0 to 1. Drugs with full efficacy are called “full agonists,” drugs with some efficacy are “partial agonists,” whereas drugs with zero efficacy are “antagonists.”
Partial agonists can also partially inhibit the response to full agonists acting at the same receptor type (Fig. 1-12). If both full and partial agonists are present, as the concentration of partial agonist is increased, more receptors will be occupied by the partial agonist. This will cause a decrease in response, because some of the receptors will no longer be activated. At very high concentrations of partial agonist relative to its affinity constant, all of the receptors will be occupied by partial agonist, and the full agonist becomes essentially irrelevant. Therefore a diagnostic feature of a partial agonist is that it inhibits the action of a full agonist to the level of its own maximal effect.
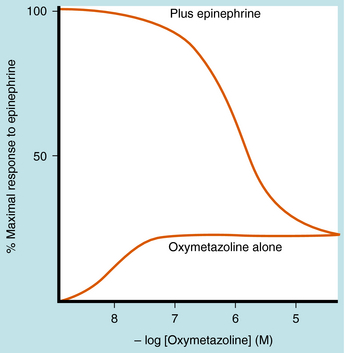
SIGNAL AMPLIFICATION—SPARE RECEPTORS
Spare receptors are important in all-or-none responses, where it is especially important that activation does not fail (e.g., the neuromuscular junction or the heart). The presence of spare receptors shifts the agonist dose-response curve to the left of the KD for binding of agonist to receptor, and the degree of shift is proportional to the proportion of spare receptors (Fig. 1-13). Thus spare receptors make a tissue more sensitive to an agonist without changing its affinity for the receptor. Because the existence of spare receptors is fairly common, the EC50 for an agonist is usually not equal to its KD. For example, the β1 adrenergic receptor has the same chemical and physical properties in every tissue in which it is expressed. However, the EC50 for a particular drug in activating responses mediated by this receptor can vary by orders of magnitude in different tissues, depending on the degree of receptor reserve.
RECEPTOR DESENSITIZATION AND SUPERSENSITIVITY
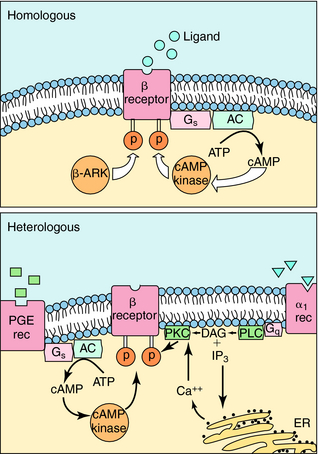
The response of any cell to hormones or neurotransmitters is tightly regulated and can vary depending on other stimuli impinging on the cell. Very often, the number of receptors in the membrane of a cell or responsiveness of the receptors themselves is regulated. One hormone can sensitize a cell to the effects of another hormone, and more commonly, when a cell is continuously exposed to stimulation by a transmitter or hormone, it may become desensitized. An example of this phenomenon is the loss of the ability of inhaled β2 adrenergic agonists to dilate the bronchi of asthmatic patients after repeated use of the drug (see Chapter 16). A hormone or agonist can affect the way a cell responds to itself (homologous effects) or how it responds to other hormones (heterologous effects). As an example of the latter phenomenon, exposure of a cell to estrogen sensitizes many cells to the effects of progesterone.
The mechanisms involved in homologous and heterologous desensitization of β2 adrenergic receptors are well understood (Fig. 1-14). Receptor phosphorylation on serine-threonine residues by three different protein kinases plays a role in the loss of responsiveness. These include β adrenergic receptor kinase (β-ARK), cAMP-dependent protein kinase, and protein kinase C. Phosphorylation of the β adrenergic receptor inhibits its ability to interact with G-proteins and subsequently leads to its sequestration or internalization in a compartment where it cannot interact with extracellular hormone. β-ARK is particularly important in homologous desensitization. It is capable only of phosphorylating the active, agonist-bound form of the receptor.
In the absence of hormone, most receptors are not localized to particular regions of the cell membrane. When a hormone binds, receptors rapidly migrate to coated pits. These are specialized invaginations of the membrane surrounded by an electron-dense cage formed by the protein clathrin; this is where receptor-mediated endocytosis occurs. Segments of membranes within coated pits rapidly pinch off to form intracellular vesicles rich in receptor-ligand complexes (Fig. 1-15). Vesicles then fuse with tubular-reticular structures. In most cases dissociated hormone is incorporated into vesicles that fuse with lysosomes, with the hormone then degraded by lysosomal enzymes. Dissociated receptor recirculates to the cell surface. However, a fraction of internalized hormone may also be recirculated to the cell surface along with receptor, and then released. This process is termed retroendocytosis. Free receptors may recirculate to the cell surface or may be sequestered temporarily in an intracellular membrane compartment. Alternatively, receptor may be transported to lysosomes, where it is also degraded. The latter two cases result in a net decrease in cell receptor number.
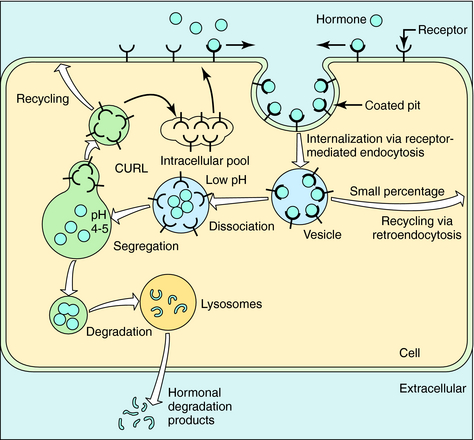
FIGURE 1–15 Pathways of receptor internalization and recycling. CURL, Compartment of uncoupling of receptor and ligand.
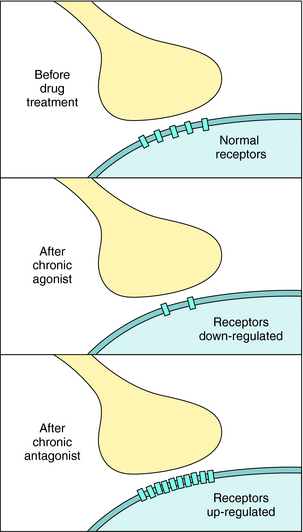
Finally, it is important to realize that the number of receptors in the plasma membrane of cells is not static (Fig. 1-16). Receptor number may be increased or decreased under the influence of hormonal mechanisms. Altered receptor number attributable to internalization or degradation has an intermediate time course, whereas an altered rate of receptor synthesis occurs more slowly. Receptors may also be up regulated, and this phenomenon can result in receptor supersensitivity. Up regulation can occur after exposure of the receptor to an antagonist, or inhibition of transmitter synthesis or release. In addition, other hormones can increase receptor number. For example, excessive production of thyroid hormone can increase the synthesis of β adrenergic receptors in cardiac tissue, leading to some of the signs and symptoms of Graves’ disease (see Chapter 42). Thus the number of cell-surface receptors, and thereby hormone sensitivity, can be continuously regulated. This property of receptor biology can be exploited therapeutically. For example, during the third trimester of pregnancy, under the influence of nuclear hormones, the number of β2 adrenergic receptors on uterine smooth muscle is dramatically increased, allowing the use of selective β2 adrenergic agonists, like terbutaline, to delay premature labor (see Chapter 11).
Kenakin T. A pharmacology primer: Theory, applications, and methods, 2 ed. San Diego: Academic Press, 2006.
For the latest information on receptor nomenclature, readers are referred to the database maintained by the International Union of Basic and Clinical Pharmacology at: http://www.iuphar-db.org/index.jsp.