Chapter 132 Phakomatoses
Introduction
The phakomatoses are a group of syndromes characterized by systemic hamartomas of the eye, brain, skin, and sometimes the viscera and bones.1–4 As a result of the multisystem involvement, the phakomatoses are also known as oculoneurocutaneous syndromes. Variable clinical manifestations of these syndromes are recognized and comprehensive care of the patient often involves coordination of many specialists.
The term “phakoma” was first used by Van der Hoeve in 1932 to indicate a mother spot, or birthmark, a characteristic finding in many of these entities.4 At that time, retinal and cerebellar hemangiomatosis (von Hippel–Lindau syndrome), neurofibromatosis (von Recklinghausen syndrome), and tuberous sclerosis complex (Bourneville syndrome) were all classified as phakomatoses. Later, encephalofacial hemangiomatosis (Sturge–Weber syndrome), racemose angiomatosis (Wyburn-Mason syndrome), and cavernous hemangioma of the retina with cutaneous and central nervous system involvement were included with these conditions. More recently, other entities such as organoid nevus syndrome and oculocutaneous melanocytosis have been categorized with these classic oculoneurocutaneous syndromes.
Definition of hamartia, hamartoma, chorista, choristoma
To better understand the phakomatoses, certain terms such as hamartia, hamartoma, chorista, and choristoma should be defined.1 Hamartia and hamartoma are terms that refer to a malformation that is composed of tissues normally present at the location where it develops. A hamartia is a nontumorous anomaly and a hamartoma is a tumorous malformation. The term systemic hamartomatosis is used to designate multiple organ involvement. Examples of hamartomas include the retinal capillary hemangioma (hemangioblastoma) that occurs from already present vascular tissue in the retina in von Hippel Lindau syndrome, or the cutaneous peripheral nerve tumors that occur from the already present neural tissue within the skin in patients with neurofibromatosis.
Most of the tumors that develop with the phakomatoses are benign. They are usually stationary or slowly progressive lesions that generally lack the capacity for limitless proliferation found with cancers.1 Some phakomatoses can be associated with malignant neoplasms. For example, there is an increased incidence of malignant schwannomas of the peripheral nerves in patients with neurofibromatosis. Renal cell carcinoma occurs with greater frequency in patients with von Hippel Lindau disease.
Neurofibromatosis (von recklinghausen syndrome)
Neurofibromatosis is an oculoneurocutaneous syndrome characterized by multisystem involvement.5,6 It is transmitted by an autosomal dominant mode of inheritance with about 80% penetrance. About one-half of the cases occur initially as spontaneous mutations with a negative family history.
Neurofibromatosis type 1
General considerations
Neurofibromatosis type 1 occurs at a rate of 1 in 3000 persons, but it is estimated that the frequency could be higher as some individuals manifest only mild features. Approximately one-half of affected patients represent a new mutation. This condition is caused by an autosomal dominant mutation in the NF1 gene that leads to decreased production of the protein neurofibromin, which has a tumor suppressor function. Only one deleted is necessary to manifest this condition. The NF1 gene is on long arm of chromosome 17. There have been more that 250 mutations identified. Complete gene deletion leads to severe phenotype. This highly penetrant phenotype has a wide variety of manifestations and can vary within families. Another locus, the SPRED1 gene, has been found in patients with “mild neurofibromatosis” and this represents Legius syndrome.7
The criteria for diagnosis of neurofibromatosis type 1 are listed in Table 132.1.8,9 This condition more often affects patients of the Caucasian race and equally in boys and girls. Scoliosis can be a prominent feature.
Table 132.1 Diagnostic criteria for neurofibromatosis type 1 (NF1) – at least two of the following seven criteria should be present for diagnosis*
| Feature | |
|---|---|
| 1.Café au lait | ≥6 café au lait spots larger than 5 mm diameter in prepubertal children (<10 years) |
| or | |
| ≥6 café au lait spots larger than 15 mm in diameter in postpubertal individuals (adults) | |
| 2.Freckles in axilla or inguinal region | Crowe sign |
| 3.Skin neurofibroma | ≥2 typical neurofibroma |
| or | |
| ≥1 plexiform neurofibroma | |
| 4.Optic nerve glioma | |
| 5.Iris Lisch nodules | ≥2 lesions |
| 6.Osseous lesion | Sphenoid dysplasia |
| or | |
| Long bone abnormalities (cortex thinning or pseudoarthrosis) | |
| 7.Relative (1st degree) with NF1 by above criteria | Parent, sibling or offspring |
* Some manifestations do not appear until later life, delaying diagnosis. (Adapted from Stumpf DA, Alksne JF, Annegers JF. Neurofibromatosis. Conference statement. National Institute of Health Consensus Development Conference. Arch Neurol 1988; 45:575–88 and Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;278:51–7.9)
Ophthalmologic features
Neurofibromatosis has the most diversified ocular findings among the phakomatoses.10–17 This condition can involve the eyelid, conjunctiva, aqueous outflow channels, uveal tract, retina, orbit, and optic nerve (Fig. 132.1).
Eyelid involvement is characterized by nodular or plexiform neurofibroma. Nodular neurofibroma appears as a solitary or multifocal painless, smooth-surfaced and well-defined mass, often the size of a pea, and without color change. Plexiform neurofibroma presents as a diffuse thickening of the eyelid that can produce the typical S-shaped curvature to the eyelid, a finding highly characteristic of neurofibromatosis. The conjunctiva can be involved by diffuse or localized neurofibromas. Patients with neurofibromatosis have an increased incidence of congenital glaucoma, which can be secondary to several mechanisms.10 There is a high association of neurofibromatosis type 1 with optic nerve glioma.12
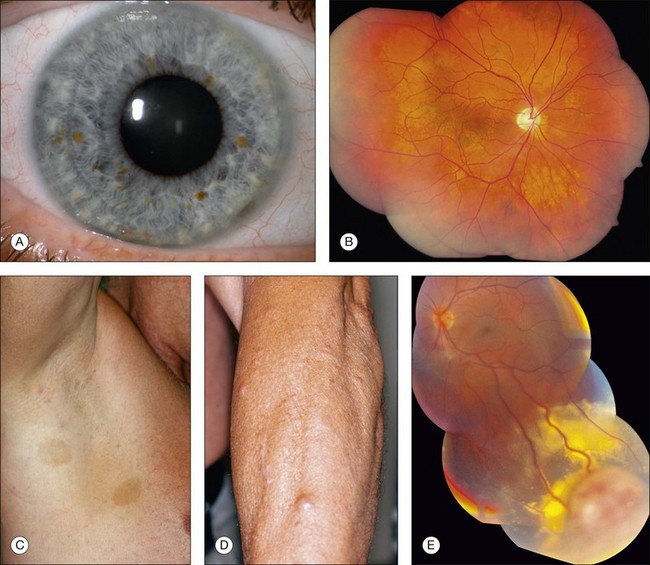
A variety of ocular fundus lesions can occur in patients with neurofibromatosis. Multiple iris hamartomas, known as Lisch nodules, are the most common uveal abnormality.11 Iris Lisch nodules are the most common ophthalmic abnormality of neurofibromatosis type 1. These characteristically orange-tan nodules appear in early childhood (usually by age 6 years) as discrete, multiple, bilateral tumors of the anterior border layer of the iris, classically measuring less than 1 mm diameter and best detected by slit lamp biomicroscopy. Histopathologically, iris Lisch nodules are hamartomas composed of aggregates of melanocytes on the anterior border layer of the iris.
The choroidal findings in patients with neurofibromatosis type 1 include unifocal or multifocal choroidal nevus, diffuse plexiform neurofibroma, neurilemoma, and melanoma. Multiple bilateral, choroidal nevi are highly suggestive of neurofibromatosis type 1. They are usually small, ill-defined, and randomly distributed. Choroidal neurofibroma usually appears as a diffuse thickening of the uveal tract from an increased number of neurofibromatous and melanocytic elements. Choroidal neurilemoma (Schwannoma) is a rare finding and manifests as a circumscribed, amelanotic elevated tumor.14 Most solitary choroidal neurilemomas, however, are not associated with neurofibromatosis. There appears to be a higher incidence of uveal melanoma in patients with neurofibromatosis.15,16
Several retinal and optic disc lesions can occur with neurofibromatosis. Retinal astrocytic hamartoma is a manifestation of neurofibromatosis, but is more common with tuberous sclerosis complex. Retinal vasoproliferative tumor can occur with neurofibromatosis, leading to exudative retinopathy and risk for blindness.17 Fundus changes can occur secondary to optic nerve glioma including optic disc edema, optic atrophy, opticociliary shunt vessels and, rarely, central retinal vein obstruction.
Neurofibromatosis type 2
General considerations
Neurofibromatosis type 2 is a multisystem disorder with prominent features of central nervous system tumors including bilateral vestibular schwannomas (acoustic neuromas), spinal cord schwannomas, meningiomas, gliomas, and juvenile posterior subcapsular cataract. This condition is also referred to as MISME syndrome, a mnemonic referring to related tumors of MIS (multiple inherited schwannomas), M-meningiomas, and E-ependymomas.18 Cutaneous features are less often seen with this form of neurofibromatosis. The criteria for diagnosis of neurofibromatosis type 2 are listed in Table 132.2.
Table 132.2 Diagnostic criteria for neurofibromatosis type 2 (NF2). Diagnosis is established with at least one of the three items listed below
|
Feature 1 Bilateral 8th cranial nerve tumors confirmed on magnetic resonance imaging or computed tomography |
(Adapted from Stumpf DA, Alksne JF, Annegers JF. Neurofibromatosis. Conference statement. National Institute of Health Consensus Development Conference. Arch Neurol 1988;45:575–88 and Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;278:51–7.9)
Neurofibromatosis type 2 can be associated with reduced life span secondary to central nervous system tumors, particularly if they are present at a young age and are multiple. The average age at symptom onset is approximately 20 years but can be delayed. Early age at symptoms and the presence of intracranial meningioma at diagnosis are two signs of higher risk for disease severity and mortality. In an analysis of 150 affected patients in 1992, more than 40% were expected to die by 50 years.19 Recent advances in treatment have extended life prognosis.
The incidence of neurofibromatosis type 2 is 1 in 25 000 live births and has nearly 100% penetrance by 60 years of age.20 It is estimated that this condition has a diagnostic prevalence of 1 in 100 000 people in 2005. Neurofibromatosis type 2 is related to a mutation in the NF2 gene at chromosome 22q12.2. This gene produces merlin (also called neurofibromin-2), a tumor suppressor. When mutated, decreased function of merlin leads to the uncontrolled development of tumors, particularly in the central nervous system. One-half of affected patients have a de novo mutation.
Ophthalmologic features
Neurofibromatosis type 2 displays three important ophthalmologic findings, notably posterior subcapsular cataract in childhood, combined hamartoma of the retina and retinal pigment epithelium, and epiretinal membranes (Table 132.3). The juvenile posterior subcapsular cataract (<50 years) is a criterion for diagnosis of this condition. Other lens opacity in the capsular or cortical region of young patients are believed related to neurofibromatosis type 2. Lisch nodules are not a feature of neurofibromatosis type 2.
Table 132.3 Frequency of clinical features in patients with neurofibromatosis type 2
| Feature | Frequency associated with neurofibromatosis type 2 (%) |
|---|---|
| Ophthalmologic features | |
| Juvenile posterior subcapsular cataracts | 60–81 |
| Epiretinal membranes | 12–40 |
| Retinal hamartomas | 6–22 |
| Cutaneous features | |
| Cutaneous tumors | 59–68 |
| Cutaneous plaques | 41–48 |
| Subcutaneous tumors | 43–48 |
| Intradermal tumors | Rare |
| Central nervous system features | |
| Bilateral vestibular (cranial nerve 8) schwannomas | 90–95 |
| Other cranial nerve schwannomas | 24–51 |
| Intracranial meningiomas | 45–58 |
| Spinal tumors | 63–90 |
| Extramedullary | 55–90 |
| Intramedullary | 18–53 |
| Peripheral neuropathy | 66 |
(Adapted from Asthagiri AR, Butman JA, Kim HJ, et al. Neurofibromatosis type 2. Lancet 2009;373:1974–86.20)
Epiretinal membranes and combined hamartoma of the retina and retinal pigment epithelium can have overlapping clinical phenotype and can be multifocal. Of those with severe clinical features of neurofibromatosis type 2, 80% display epiretinal membranes.21 The hamartomas are along the inner retina but lead to prominent retinal dragging, corkscrew retinal vessels, gray-green appearance and tumor formation.22,23
Management
Patients with neurofibromatosis type 2 should have annual ophthalmic, neurologic, dermatologic, and auditory examinations. This requires a multidisciplinary team. Surgical resection of symptomatic neurologic tumors is performed, but radiotherapy or chemotherapy can be used, particularly for ependymomas. Erlotinib has been used for unresectable progressive vestibular schwannomas and this medication is under trial. Additionally, bevacizumab and Gleevec have been investigated for treatment of schwannomas. Regarding ophthalmic care, cataract surgery can be beneficial. Additionally, monitoring of epiretinal membrane with clinical examination and optical coherence tomography, with surgical removal if progressive, can be considered.22
Encephalofacial hemangiomatosis (sturge–weber syndrome)
General considerations
The Sturge–Weber (SW) syndrome is characterized by congenital hamartomas of the eye, skin, and brain that manifest at different times during life, mostly in childhood. These include choroidal hemangioma, facial nevus flammeus (port wine stain), and brain hemangioma with intracranial calcification.24–28 Some patients display a forme fruste, or partial expression, rather than the entire syndrome. In contrast to the other systemic hamartomatosis, there is no recognizable hereditary pattern associated with SW syndrome. There is no predisposition for sex or race.
This condition affects 1 per 50 000 persons. Roach has designed a classification for Sturge–Weber syndrome (Table 132.4).29
Table 132.4 Roach diagnostic scale for classification of encephalotrigeminal angiomatosis (Sturge–Weber syndrome)
| Type | Title | Involvement of skin, eye, and brain |
|---|---|---|
| Type I | Classic Sturge–Weber syndrome | Leptomeningeal angioma present |
| Cutaneous facial angioma present | ||
| Glaucoma likely present | ||
| Type II | Sturge–Weber syndrome | Leptomeningeal angioma absent |
| Cutaneous facial angioma present | ||
| Glaucoma possibly present | ||
| Type III | Sturge–Weber syndrome forme fruste | Leptomeningeal angioma present |
| Cutaneous facial angioma absent | ||
| Glaucoma absent |
(Adapted from Roach ES. Neurocutaneous syndromes. Pedatr Clin North Am 1992;39:591–620.29)
In 1987, Happle proposed that Sturge–Weber syndrome was an example of somatic mosaicism of a genetic trait.30 In 2003, Comi and associates supported this theory but demonstrating abnormality in the gene expression of fibronectin in fibroblasts from the region of the port wine stain compared with normal skin.31
Ophthalmologic features
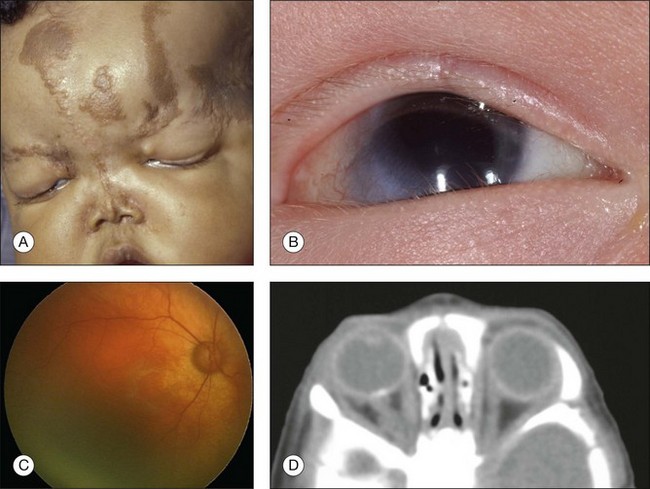
The ocular findings associated with Sturge–Weber syndrome include eyelid involvement with the nevus flammeus, prominent epibulbar blood vessels, glaucoma, retinal vascular tortuosity and diffuse choroidal hemangioma (Fig. 132.2). Glaucoma is more common in patients with Sturge–Weber syndrome than it is in the other systemic hamartomatoses. In a study of 50 patients with nevus flammeus, there was an 8% overall incidence of glaucoma. If the facial hemangioma involved both the first and second division of the trigeminal nerve, the incidence was 15%.26 The glaucoma occurs unilaterally on the side of the facial hemangioma. It may gradually lead to extensive cupping of the optic disc and blindness. We have been referred patients with facial nevus flammeus who developed complete blindness from unrecognized glaucoma. The associated glaucoma can be either congenital or juvenile.
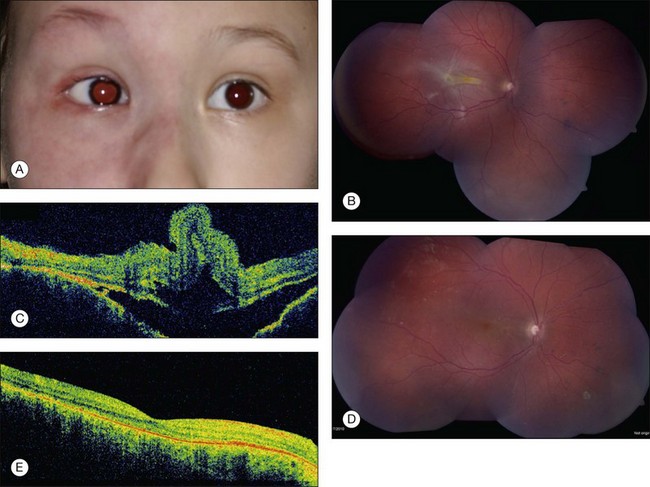
Sullivan and associates reviewed eye findings in 51 patients with Sturge–Weber syndrome and found 71% with glaucoma (onset before 2 years of age in most cases), 69% with conjunctival or episcleral prominent vessels, and 55% with diffuse choroidal hemangioma.32
The main abnormality of the uveal tract in patients in Sturge–Weber syndrome is diffuse choroidal hemangioma.33 Patients with this tumors display bright red pupillary reflex in the involved eye compared with the contralateral eye. This phenomenon, which is due to the light reflex from the highly vascularized tumor in the posterior pole, has been called the “tomato catsup” fundus. B-scan ultrasonography typically demonstrates a diffuse, highly echogenic thickening of the choroid. The tumor is usually unilateral but bilateral cases associated with bilateral facial nevus flammeus have been recognized. Diffuse tumor is usually diagnosed when the affected patient is young (median age 8 years), either because the associated facial hemangioma prompts a fundus examination or because visual impairment occurs from hyperopic amblyopia or from a secondary retinal detachment. Diffuse choroidal hemangioma can lead to a total retinal detachment and secondary neovascular glaucoma.
Central nervous system features
The typical CNS feature associated with Sturge–Weber syndrome is a diffuse leptomeningeal hemangioma that is ipsilateral to the facial hemangioma.1 This lesion can show secondary calcification that appears radiographically as a radio-opaque double line which has been called the “railroad track” sign. The calcification often increases during the first 20 years of life and usually becomes stable in adulthood. Seizures, stroke, headache, learning disorders, and developmental delay can occur as a result of the leptomeningeal hemangioma (Table 132.5).
| Feature | Percentage |
|---|---|
| Cutaneous facial nevus flammeus | 87 |
| Bilateral cerebral involvement | 15 |
| Seizures | 72–93 |
| Hemiparesis | 25–93 |
| Hemianopia | 44 |
| Headache | 44–62 |
| Developmental delay or mental retardation | 50–75 |
| Glaucoma | 30–71 |
| Choroidal hemangioma | 40 |
Of all patients with facial port wine stain, only 8% show complete features of Sturge–Weber syndrome.
Management
The management of the diffuse choroidal hemangioma varies with the extent of the tumor and secondary retinal detachment. If the tumor is minimally elevated and without subretinal fluid, observation is advised and correction of the induced hyperopic change may be considered. Monitoring for amblyopia is important. Thicker tumors with secondary retinal detachment are treated with photodynamic therapy or external beam radiotherapy.34 Newer information on the use of oral propranolol for treatment of exudative retinal detachment from diffuse choroidal hemangioma may offer another alternative. External beam radiotherapy or plaque radiotherapy with a total dose of 20–40 Gy has been successful in achieving resolution of fluid and return of some visual acuity, depending on the chronicity. The treatment of related glaucoma should be undertaken by an ophthalmologist who is experienced in the management of complicated glaucoma problems.
Racemose hemangiomatosis (wyburn-mason syndrome)
General considerations
Racemose hemangioma of the midbrain and ipsilateral retina is called the Wyburn-Mason syndrome.35 In contrast to the other oculoneurocutaneous syndromes, it has few skin changes except for occasional small hemangiomas. The ocular and central nervous system changes, however, can be quite striking. Like Sturge–Weber syndrome, this congenital condition does not appear to be familial and does not exhibit a hereditary pattern. The characteristic arteriovenous communications can range from subtle asymptomatic lesions to more extensive lesions that form tumor-like vascular masses, often referred to as racemose or cirsoid hemangiomas.
The exact association of the retinal and central nervous system hemangiomas is not clear but it is estimated that 30% of patients with the retinal findings have brain findings.36 On the other hand, it is estimated that 8% of patients with brain findings have retinal findings.36
Ophthalmologic features
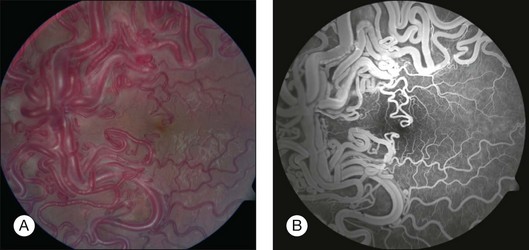
The classic ocular finding is the racemose hemangioma of the retina (Fig. 132.3).37–39 Similar vascular malformations can occur in the orbit and adjacent structures. These retinal arteriovenous communications have been divided into three groups according to the Archer classification (Table 132.6).39 Group I is characterized by interposition of an abnormal capillary plexus between the major vessels. It is not a true tumor and the affected patient is often asymptomatic. Group II is typified by a direct arteriovenous communication without interposition of capillary or arteriolar elements. The dilated blood vessels in this group can superficially resemble a retinal capillary hemangioma, but no tumor, exudation, or retinal detachment is present. In general, these patients have few visual symptoms, but they can have associated cerebral arteriovenous malformations. Group III is characterized by a more extensive complex arteriovenous communication, which is often associated with visual loss. In this group, the most striking feature is one or more dilated arteries that emerge from the optic disc, pass for a variable distance into the retina, form distinct arteriovenous communications, and then pass back to the optic disc as large veins. There is characteristically no exudation or retinal detachment and the vessels do not leak fluorescein. Although these lesions are believed to remain stationary indefinitely, we have observed changes in the distribution of the blood vessels over several years. Branch retinal vein obstruction can sometimes occur.40,41 Iris racemose hemangioma has not been associated with retinal racemose hemangioma or the Wyburn-Mason syndrome.42
| Group | Feature | Comments |
|---|---|---|
| I | Abnormal capillary plexus between the major vessels of the arteriovenous malformations | Such lesions tend to be small, patients asymptomatic, and intracranial involvement uncommon |
| II | Arteriovenous malformations lack any intervening capillary bed between the artery and vein | Risk of retinal decompensation resulting in retina edema, hemorrhage, and vision loss. Low risk for intracranial arteriovenous malformations |
| III | Extensive arteriovenous malformations with dilated and tortuous vessels and no distinction between artery and vein | High risk for visual loss due to retinal decompensation or retinal compression of nerve fiber layer, optic nerve, or other vessels. High risk for intracranial arteriovenous malformations |
(Adapted from Archer DM, Deutman A, Ernest JT, Krill AE. Arteriovenous communications of the retina. Am J Ophthalmol 1973;75:224–41.39)
Retinal cavernous hemangiomatosis
Ophthalmologic features
The only ocular manifestation of this syndrome is the retinal cavernous hemangioma (Fig. 132.4). Ophthalmoscopically, it appears as a dark grape-like cluster of venous intraretinal aneurysms either at the optic disc, macula, or in the peripheral retina.43–45 There is no obvious feeding artery and the lesion is typically centered along the course of a vein. This lesion produces no exudation or subretinal fluid, likely due to the fact that the thin-walled channels are lined by nonfenestrated endothelium. Repetitive vitreous hemorrhages (often mild and subclinical) can lead to white fibroglial tissue on the surface. This non-progressive thin-walled, aneurysmal retinal tumor can remain hidden in the fundus for decades until dilation is performed or the patient develops vitreous hemorrhage.
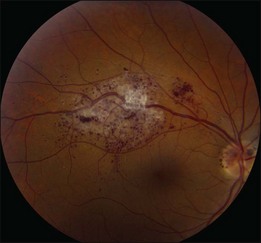
Other rare ophthalmic findings include iris cavernous hemangiomatosis with repetitive hemorrhages.46
Central nervous system features
Cavernous hemangioma can involve any region of the central nervous system but is more common in the supratentorial rather than infratentorial region. Spinal cord involvement can occur. Symptoms include seizures, hemorrhage, and focal neurologic defect most commonly. About 25% of patients display no neurologic symptom despite having a cavernous hemangioma within the brain.47 Intracranial hemorrhage occurs in 12% to 48%. Magnetic resonance imaging can provide information to the location and size of the mass.
Organoid nevus syndrome
General considerations
The organoid nevus syndrome (ONS) is an oculoneurocutaneous condition without clear genetic abnormality. The features include the sebaceous nevus of Jadassohn, cerebral atrophy, arachnoidal cyst, epibulbar complex choristoma, eyelid coloboma, posterior calcified scleral cartilage, and occasionally other features.48,49 The sebaceous nevus of Jadassohn is a well-recognized dermatologic entity, but the complete syndrome of organoid nevus syndrome is relatively uncommon. This sporadic condition displays more choristomas than hamartomas, unlike other phakomatoses. Despite the lack of genetic understanding, there have been exceptionally rare cases of multiple generations affected with organoid nevus syndrome.
Ophthalmologic features
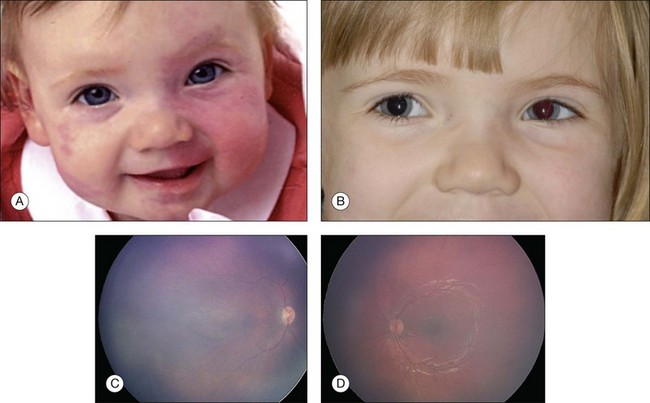
Although there are several ophthalmic features of the organoid nevus syndrome, two most important ones are the epibulbar complex choristoma and posterior scleral cartilage (Fig. 132.5). The epibulbar complex choristoma is a fleshy lesion of the conjunctiva that often extends onto the cornea. It is composed histopathologically of a dermolipoma that contains variable combinations of ectopic lacrimal gland and hyaline cartilage. The posterior scleral cartilage produces a peculiar yellow-white discoloration of the fundus in the area of involvement that has been reported erroneously in the literature as optic nerve coloboma or choroidal osteoma. With ultrasonography and CT, there is a bone density plaque at the level of the choroid and sclera that corresponds to cartilage in the posterior sclera and not bone as seen with an osteoma.
Dermatologic features
The main dermatologic feature of the organoid nevus syndrome is the sebaceous nevus of Jadassohn. It appears as a geographic yellow-brown lesion that often involves the preauricular or brow region and extends onto the scalp where it is associated with alopecia and often down along the neck and nose. This lesion becomes hypertrophied with time. There are three stages of this cutaneous lesion dependent on patient age.48 The first stage occurs in infancy with underdevelopment of hair, sebaceous glands and adnexal structures. The second stage occurs at puberty with massive overdevelopment of adnexal structures. The third stage occurs in adulthood with benign and malignant tumors including basal cell carcinoma, syringocystadenoma papilliferum, sebaceous adenoma, keratoacanthoma, and others. It is estimated that 20% of patients with sebaceous nevus of Jadassohn will develop basal cell carcinoma arising from the nevus.
Phacomatosis pigmentovascularis
General considerations
Phacomatosis pigmentovascularis is a rare condition representing the coexistence of cutaneous vascular malformation (usually nevus flammeus) with melanocytic nevus (usually melanocytosis). Since the first report on this condition by Ota in 1947, there have been few reports, mostly in the dermatologic literature.50 In 2005, Tran and Zografos reported three cases, all with uveal melanoma.51 In 2005, Happle proposed reclassification (Table 132.7).52 According to Happle, phacomatosis pigmentovascularis is divided into three types: phacomatosis cesioflammea, phacomatosis spilorosea, and phacomatosis cesiomarmorata (Table 132.7). Phacomatosis cesioflammea is characterized by coexistence of a dermal melanocytosis (blue spot) and nevus flammeus (port wine stain). “Caesius” is Latin for “bluish gray” and “flammea” for “flame” or “fire.” The other two types have less relationship to the eye.
Table 132.7 Classification of phacomatosis pigmentovascularis (PPV)
| New classification | Findings | Old classification |
|---|---|---|
| Phacomatosis cesioflammea | Nevus caesius (blue spot) or melanocytosis with nevus flammeus | PPV II a/b |
| Phacomatosis spilorosea | Nevus spilus (speckled lentiginous nevus) with pale pink telangiectatic nevus | PPV II a/b |
| Phacomatosis cesiomarmorata | Nevus caesius (blue spot) with cutis marmorata | PPV V a/b |
| Phacomatosis pigmentovascularis unclassifiable | Various pigmentary and vascular nevi | PPV IV a/b and no name |
(Adapted from Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol 2005;141:385–8.52)
The association of dermal melanocytosis with cutaneous nevus flammeus is believed to be due to the “twin spotting” phenomenon, which is the association of two genetically different clones of cells within a region of normal cells. This involves sporadic somatic recombination and a mosaic distribution of lesions. Moutray and associates describe monozygotic twins discordant for phacomatosis cesioflammea, supporting post-zygotic twin spotting theory.53 In that report, twin #1 was normal and twin #2 had cutaneous nevus flammeus of the arm, maxilla, and periocular region in addition to Mongolian spot and bilateral ocular melanocytosis.
In 2011, Shields and associates reported seven patients with this condition: three with related uveal melanoma; one with primary acquired melanosis of the conjunctiva (melanoma in situ); and one with optic disc melanocytoma.54 The serious relationship of this phakomatosis to melanoma was emphasized.
Ophthalmologic features
The ophthalmologic features of phacomatosis pigmentovascularis includes a spectrum of findings with unilateral or bilateral periocular facial nevus flammeus (port wine stain), ocular or oculodermal melanocytosis, secondary glaucoma, and risk for uveal or conjunctival melanoma (Fig. 132.6). There are only few reports in the ophthalmic literature.
Dermatologic features
Fernandez-Guarino reviewed the published dermatologic literature on 216 cases of phacomatosis pigmentovascularis and classified 77% as cesioflammea, 13% as spilorosea, 1% as cesiomarmorata, and 8% unclassifiable.55 The cutaneous manifestations included mostly pigmented lesions of nevus of Ota (melanocytosis), Mongolian spot, or café-au-lait spot, nonpigmentation of vitiligo, as well as vascular lesions of nevus flammeus (port wine stain) or nevus anemicus. These cutaneous findings tended to be patchy without midline separation and scattered over the surface of the body.
Central nervous system features
Neurologic features of phacomatosis pigmentovascularis include seizures, cortical atrophy, Arnold Chiari type 1, bilateral deafness, idiopathic facial paralysis, hydrocephalus, diabetes insipidus, plexiform neurofibroma, psychomotor developmental delay, and electroencephalogram alterations.55
Other features
Miscellaneous features include scoliosis, extremity length asymmetry, syndactyly, macrocephaly, renal agenesis, renal angiomatosis, hepatosplenomegaly, cavernous hemangioma, umbilical hernia, hypoplasia of leg veins, IgA deficit, hyper IgE syndrome, eczema, and premature eruption of teeth.55
Oculodermal melanocytosis
General considerations
Ocular or oculodermal melanocytosis is a congenital pigmentary condition that can affect the periocular skin, sclera, uvea, orbit, palate, ear drum, and meninges.1–3,56–61 This condition predisposes the patient to melanoma, particularly uveal melanoma and occasionally orbital or meningeal melanoma.61–66 Ocular or oculodermal melanocytosis can occur as a diffuse condition, affecting all quadrants of the eye in a homogeneous fashion, or as a sectoral condition, affecting only a portion of the eye.67 This is a sporadic condition with no genetic mutation identified.
Ophthalmologic features
The ophthalmologic features of ocular or oculodermal melanocytosis include dermal periocular pigmentation (flat blue nevus) with occasional pigment in the temporal fossa along the hair line, scleral pigmentation, and uveal pigmentation involving the iris, ciliary body, and choroid (Fig. 132.7). Orbital pigmentation with blue nevus cells is occasionally found. The iris can manifest mammilation, or clumping of tissue into a “polka dot” appearance, related to the dense pigment.68 Early onset equatorial drusen is often found. The main concern of this excessive pigmentation is the risk for the development of uveal melanoma (1/400 risk for Caucasians), orbital melanoma, and meningeal melanoma. Occasionally, optic disc melanocytoma and ipsilateral headache are a part of this syndrome.
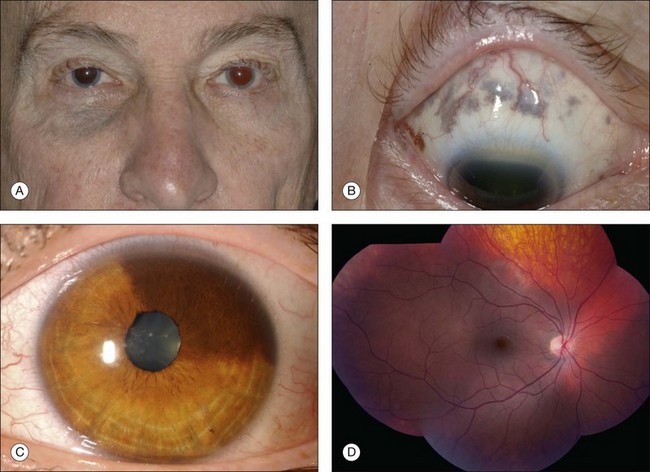
Genetic evaluation following enucleation of an eye with melanoma arising from melanocytosis revealed normal disomy chromosome 3 in the melanocytosis and monosomy chromosome 3 in the melanoma.68
Central nervous system features
The blue nevus can extend to involve the meninges and carries a small risk for the development of meningeal melanoma.69
Other features
Similar blue nevus pigmentation can involve the ipsilateral palate and tympanic membrane.
1 Shields JA, Shields CL. Systemic hamartomatoses (“Phakomatoses”). In: Intraocular tumors. A text and atlas. Philadelphia: WB Saunders; 1992:513–539.
2 Shields JA, Shields CL. Intraocular tumors. An atlas and textbook, 2nd ed, Philadelphia: Lippincott Williams and Wilkins, 2008.
3 Shields JA, Shields CL. The systemic hamartomatoses (“Phakomatoses”). In: Mannis MJ, Macsai MS, Huntley AC. Eye and skin disease. New York: Lippincott-Raven; 1996:367–380.
4 Van der Hoeve J. The Doyne memorial lecture. Eye symptoms in phakomatoses. Trans Ophthalmol Soc UK. 1932;52:380–401.
5 von Recklinghausen FD. Uber die multiplen fibrome der haut und ihre beziehungen zu den neurrommen. Festschr Feier fundfund-zwanzigjahrigen. Berlin Institute of Pathology. Berlin: Hirschwald; 1882.
6 Brasfield RD, Das Gupta TK. von Recklinghausen’s disease: A clinicopathological study. Ann Surg. 1972;175:86–104.
7 Spurlock G, Bennett E, Chuzhanova N, et al. SPRED1 mutations (Legius syndrome): another clinically useful genotype for dissecting the neurofibromatosis type 1 phenotype. J Med Genet. 2009;46:431–437.
8 Stumpf DA, Alksne JF, Annegers JF. Neurofibromatosis. Conference statement. National Institute of Health Consensus Development Conference. Arch Neurol. 1988;45:575–578.
9 Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997;278:51–57.
10 Grant WM, Walton DS. Distinctive gonioscopic findings in glaucoma due to neurofibromatosis. Arch Ophthalmol. 1968;79:127–134.
11 Lewis RA, Riccardi VM. von Recklinghausen neurofibromatosis. Incidence of iris hamartoma. Ophthalmology. 1981;88:348–354.
12 Lewis RA, Gerson LP, Axelson KA, et al. Von Recklinghausen neurofibromatosis II. Incidence of optic gliomata. Ophthalmology. 1984;91:929–935.
13 Kaiser-Kupfer MI, Freidlin V, Dariles MB, et al. The association of posterior capsular lens opacities with bilateral acoustic neuromas in patients with neurofibromatosis type 2. Arch Ophthalmol. 1989;107:541–544.
14 Shields JA, Sanborn GE, Kurz GH, et al. Benign peripheral nerve tumor of the choroid. Ophthalmology. 1981;88:1322–1329.
15 Wiznia RA, Freedman JE, Mancini AD, et al. Malignant melanoma of the choroid in neurofibromatosis. Am J Ophthalmol. 1978;86:684–687.
16 Honavar S, Singh AD, et al. Iris melanoma in a patient with neurofibromatosis. Surv Ophthalmol. 2000;45:231–236.
17 Shields CL, Shields JA, Barrett J, et al. Vasoproliferative tumors of the ocular fundus. Classification and clinical manifestations in 103 patients. Arch Ophthalmol. 1995;113:615–623.
18 Evans DG. Neurofibromatosis 2 (Bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II). Genet Med. 2009;11:599–610.
19 Evans DGR, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84:603–618.
20 Asthagiri AR, Butman JA, Kim HJ, et al. Neurofibromatosis type 2. Lancet. 2009;373:1974–1986.
21 Meyers SM, Gutman FA, Kaye LD, et al. Retinal changes associated with neurofibromatosis 2. Trans Am Ophthalmol Soc. 1995;93:245–257.
22 Shields CL, Mashayekhi A, Dai VV, et al. Optical coherence tomography findings of combined hamartoma of the retina and retinal pigment epithelium in 11 patients. Arch Ophthalmol. 2005;123:1746–1750.
23 Shields CL, Thangappan A, Hartzell K, et al. Combined hamartoma of the retina and retinal pigment epithelium in 77 consecutive patients. Visual outcome based on macular versus extramacular tumor location. Ophthalmology. 2008;115:2246–2252.
24 Sturge WA. A case of partial epilepsy apparently due to a lesion of one of the vasomotor centers of the brain. Trans Clin Soc Lond. 1879;12:162–167.
25 Weber FP. Right-sided hemihypertrophy resulting from right-sided congenital spastic hemiplegia with a morbid condition of the left side of the brain revealed by radiogram. J Neurol Psycho-Pathol (London). 1922;37:301–311.
26 Stevenson RF, Morin JD. Ocular findings in nevus flammeus. Can J Ophthalmol. 1975;10:136–139.
27 Susac JO, Smith JL, Scelfo RJ. The “tomato catsup” fundus in Sturge–Weber syndrome. Arch Ophthalmol. 1974;92:69–70.
28 Lindsey P, Shields JA, Goldberg RE, et al. Bilateral nevus flammeus and bilateral choroidal hemangiomas. Retina. 1981;1:88–95.
29 Roach ES. Neurocutaneous syndromes. Pedatr Clin North Am. 1992;39:591–620.
30 Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Serm. 1987;16:899–906.
31 Comi AM, Hunt P, Vawter MP, et al. Increased fibronectin expression in Sturge–Weber syndrome fibroblasts and brain tissue. Pediatr Res. 2003;53:762–769.
32 Sullivan TJ, Clarke MP, Morin JD. The ocular manifestations of the Sturge–Weber syndrome. J Pediatr Ophthalmol Strabismus. 1992;29:349–356.
33 Witschel H, Font RL. Hemangioma of the choroid. A clinicopathologic study of 71 cases and a review of the literature. Surv Ophthalmol. 1976;20:415–431.
34 Ramasubramanian A, Shields CL. The current management of choroidal hemangioma. Retina Today. 2010 December:52–55.
35 Wyburn-Mason R. Arteriovenous aneurysm of midbrain and retina, facial naevi and mental changes. Brain. 1943;66:163–203.
36 Theron J, Newton TH, Hoyt WF. Unilateral retinocephalic vascular malformations. Neuroradiology. 1974;7:185–196.
37 Cameron ME. Congenital arterio-venous aneurysm of the retina. Br J Ophthalmol. 1958;42:655–666.
38 Cameron ME, Greer CH. Congenital arterio-venous aneurysm of the retina. A post-mortem report. Br J Ophthalmol. 1968;52:768–772.
39 Archer DM, Deutman A, Ernest JT, et al. Arteriovenous communications of the retina. Am J Ophthalmol. 1973;75:224–241.
40 Shah GK, Shields JA, Lanning R. Branch retinal vein obstruction secondary to retinal arteriovenous communication. Am J Ophthalmol. 1998;126:446–448.
41 Materin MA, Shields CL, Marr BP, et al. Retinal racemose hemangioma. Retina. 2005;25:936–937.
42 Shields JA, Streicher TFE, Spirkova JHJ, et al. Arteriovenous malformation of the iris in 14 cases. The 2004 Alvaro Rodriquez MD Gold Medal Award Lecture. Arch Ophthalmol. 2006;124:370–375.
43 Gass JDM. Cavernous hemangioma of the retina. A neuro-oculocutaneous syndrome. Am J Ophthalmol. 1971;71:799–814.
44 Lewis RA, Cohen MH, Wise GN. Cavernous hemangioma of the retina. A report of three cases and a review of the literature. Br J Ophthalmol. 1975;59:422–434.
45 Goldberg RE, Pheasant TR, Shields JA. Cavernous hemangioma of the retina. A four generation pedigree with neurocutaneous involvement. Arch Ophthalmol. 1979;97:2321–2324.
46 Thangappan A, Shields CL, Gerontis CC, et al. Iris cavernous hemangioma associated with multiple cavernous hemangiomas in the brain, kidney, and skin. Cornea. 2007;26:481–483.
47 Seigel AM. Familial cavernous angioma: an unknown disease. Acta Neurol Scand. 1998;98:369–371.
48 Shields JA, Shields CL, Eagle RC, Jr., et al. Ocular manifestations of the organoid nevus syndrome. Ophthalmology. 1997;104:549–557.
49 Kraus NJ, Ramasubramanian A, Shields CL, et al. Ocular features of the organoid nevus syndrome. Retinal Cases & Brief Reports. 2010;4:385–386.
50 Ota M, Kawamura T, Ito N. [Phakomatosis pigmentovascularis.]. Jpn J Dermatol. 1947;52:1–3.
51 Tran HV, Zografos L. Primary choroidal melanoma in phacomatosis pigmentovascularis IIa. Ophthalmology. 2005;112:1232–1235.
52 Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385–388.
53 Moutray T, Napier M, Shafiq A, et al. Monozygotic twins discordant for phacomatosis pigmentovascularis: evidence for the concept of twin spotting. Am J Med Genet. 2010;152:718–720.
54 Shields CL, Kligman BE, Suriano M, et al. Phacomatosis Pigmentovascularis of Cesioflammea Type in 7 Cases. Combination of ocular pigmentation (melanocytosis, melanosis) and nevus flammeus with risk for melanoma. Ophthalmology. 2011;129:746–750.
55 Fernandez-Guarino M, Boixeda P, de las Heras B, et al. Phacomatosis pigmentovascularis: Clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88–93.
56 Shields CL, Shields JA. Tumors of the conjunctiva and cornea. SurvOphthalmol. 2004;49:3–24.
57 Gonder JR, Nichol J, Augsburger JJ, Shields JA. Ocular and oculodermal melanocytosis. Can J Ophthalmol. 1985;20:176–178.
58 Gonder JR, Ezell PC, Shields JA, et al. Ocular melanocytosis: a study to determine the prevalence rate of ocular melanocytosis. Ophthalmology. 1982;89:950–952.
59 Velazquez N, Jones IS. Ocular and oculodermal melanocytosis associated with uveal melanoma. Ophthalmology. 1983;90:1472–1476.
60 Gonder JR, Shields JA, Albert DM, et al. Uveal malignant melanoma associated with ocular and oculodermal melanocytosis. Ophthalmology. 1982;89:953–960.
61 Singh AD, De Potter P, Fijal BA, et al. Lifetime prevalence of uveal melanoma in Caucasian patients with ocular (dermal) melanocytosis. Ophthalmology. 1998;105:195–198.
62 Gunduz K, Shields JA, Shields CL, et al. Choroidal melanoma in a 14 year-old patient with ocular melanocytosis. Arch Ophthalmol. 1998;116:1112–1114.
63 Shields JA, Shields CL, Eagle RC, Jr., et al. Malignant melanoma arising from a large uveal melanocytoma in a patient with oculodermal melanocytosis. Arch Ophthalmol. 2000;118:990–993.
64 Honavar SG, Shields CL, Singh AD, et al. Two discrete choroidal melanomas in an eye with ocular melanocytosis. Ophthalmology. 2002;47:36–41.
65 Shields JA, Shields CL, Naseripor M, et al. Choroidal melanoma in a black patient with oculodermal melanocytosis. Retina. 2002;22:126–128.
66 Shields CL, Eagle RC, Ip MS, et al. Two discrete uveal melanomas in a child with ocular melanocytosis. Retina. 2006;26:684–687.
67 Shields CL, Qureshi A, Mashayekhi A, et al. Sector (partial) oculo (dermal) melanocytosis in 89 eyes. Ophthalmology. 2011;118:2474–2479.
68 Horgan N, Shields CL, Swanson L, et al. Altered chromosome 3 expression of uveal melanoma in the setting of melanocytosis. Acta Ophthalmol Scand. 2009;87:578–580.
69 Kiratli H, Bilgic S, Satilmis M. Ocular melanocytosis associated with intracranial melanoma. Br J Ophthalmol. 1996;80:1025.