Pesticides
Organophosphate and Carbamate Insecticides
The first organophosphate insecticide, triethyl pyrophosphate, was synthesized in 1859 but did not replace nicotine as a pesticide until World War II. After World War II, these compounds were used as chemical warfare agents, as organophosphorus and carbamate insecticides, and as medicinal agents. With concern about the long half-life of the organochlorine dichlorodiphenyltrichloroethane (DDT), causing it to accumulate in the environment, the organophosphate insecticides became the most common pesticides for home and industrial use. Since the increased awareness of terrorism in the 1990s, nerve agents have gained prominence as weapons of mass destruction.1
Principles of Disease
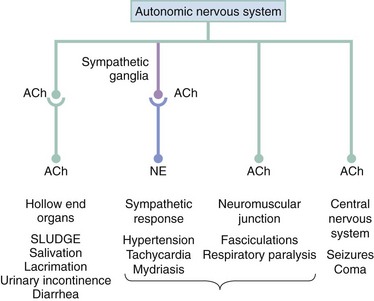
Organophosphorus pesticides work by persistently inhibiting the enzyme acetylcholinesterase, the enzymatic deactivator of the ubiquitous neurotransmitter acetylcholine. Because of the global penetration of organophosphorus compounds, inhibition occurs at tissue sites (true acetylcholinesterase and represented by erythrocyte or red blood cell [RBC] cholinesterase) and in plasma (circulating pseudocholinesterase).2 Inhibition of cholinesterase results in the accumulation and subsequent prolonged effect of acetylcholine at a variety of neurotransmitter receptors, including sympathetic and parasympathetic ganglionic nicotinic sites, postganglionic cholinergic sympathetic and parasympathetic muscarinic sites, skeletal muscle nicotinic sites, and central nervous system sites (Fig. 163-1).
Clinical Features
The accumulation of acetylcholine results in the classic SLUDGE cholinergic syndrome, manifested by hyperactivity of cholinergic responses at the receptor sites indicated previously. The clinical syndrome of muscarinic acetylcholinesterase inhibition (Table 163-1) causes postganglionic acetylcholine-induced hollow end-organ general hypersecretion, resulting in clinical findings that include miotic pupils, lacrimation, rhinorrhea, sialorrhea, bronchorrhea, vomiting, diarrhea, and urinary incontinence. Bradycardia is a classic sign of the cholinergic syndrome, but the increased release of norepinephrine from postganglionic sympathetic neurons precipitated by excess cholinergic activity at sympathetic ganglia may result in normal or even tachycardic heart rates (nicotinic effect). Sympathetic hyperactivity can cause diffuse diaphoresis, although this response is mediated by cholinergic receptors at preganglionic (nicotinic) and postganglionic (muscarinic) sites. The most lethal components of acetylcholinesterase inhibition occur in the brain and neuromuscular junction. A combination of sympathetic stimulation, involvement of the N-methyl-D-aspartate (NMDA) receptor, and enhanced acetylcholine concentrations can induce seizures.3 At the neuromuscular junction, excess acetylcholine causes hyperstimulation of the muscles with secondary paralysis, and when the diaphragm is affected, cholinesterase poisoning leads to respiratory arrest.4
Table 163-1
| Salivation | Diarrhea/Diaphoresis |
| Lacrimation | Urination |
| Urinary incontinence | Miosis |
| Defecation | Bradycardia/Bronchorrhea/Bronchospasm |
| Gastrointestinal cramps | Emesis |
| Emesis | Lacrimation |
| Salivation |
Complications
Nicotinic hyperstimulation of skeletal muscle leads to significant morbidity and mortality associated with acetylcholinesterase inhibitor toxicity. Signs of skeletal muscle hyperactivity include involuntary twitches, fasciculations, and hyperactive reflexes. Muscle hyperactivity eventually progresses to muscle fatigue and paralysis, including the respiratory musculature and particularly the diaphragm.4 Respiratory insufficiency may be delayed and result in death if it is not anticipated and corrected by mechanical or pharmacologic means.
Acetylcholinesterase inhibitors produce direct toxic effects on the central nervous system, leading to neurologic signs of confusion, combativeness, seizures, and coma. Status epilepticus may occur in severely poisoned patients. Structural central nervous system damage may occur if seizures are not terminated rapidly.5
Diagnostic Strategies
After acute exposures, the plasma cholinesterase levels decrease first, followed by decreases in RBC cholinesterase levels. The RBC cholinesterase level correlates best with activity at the nerve terminal.2 Patients with chronic exposures may have a normal plasma cholinesterase level but have reduced RBC cholinesterase activity, which will confirm the poisoning. The true reflection of depressed cholinesterase activity is found in the RBC activity, and even a mild acute exposure may result in severe clinical poisoning. The RBC cholinesterase level recovers at a rate of 1% per day in untreated patients and takes approximately 6 to 12 weeks to normalize, whereas plasma cholinesterase levels may recover in 4 to 6 weeks. Other laboratory studies should focus on the evaluation of pulmonary, cardiovascular, and renal function and fluid and electrolyte balance. Patients presenting with no acidosis or only a metabolic acidosis on the arterial blood gas analysis have mortality lower than that of patients with a respiratory or mixed acidosis.6
Management
Decontamination should start in the out-of-hospital phase to prevent further absorption and subsequent toxicity and to protect care providers. Because dermal exposure is most likely, removal and destruction of clothing and thorough flushing of exposed skin may limit absorption and subsequent toxicity. Alternatively, dermal decontamination can be done with dry agents, such as military resins, flour, sand, or bentonite. Caregivers are at risk for contamination from splashes or handling of contaminated clothing. Treating personnel may be rotated to limit their exposure to the organophosphates.4 Caregivers should use universal precautions, including eye shields, protective clothing, and nitrile or butyl rubber gloves. In the case of ingestion, gastrointestinal decontamination procedures are of questionable benefit because of the rapid absorption of these compounds. Profuse vomiting and diarrhea are seen early in ingestion and may limit7 or negate any beneficial effect of additional gastrointestinal decontamination.8,9 The administration of activated charcoal after ingestion has no proven benefit in these poisonings.10
Because death is due to airway and respiratory failure, supportive care should be directed primarily toward airway management and include suctioning of secretions and vomitus, oxygenation, and, when necessary, ventilatory support. Succinylcholine can be used for intubation but may have an extremely prolonged duration (up to 3 to 7 hours) as it is metabolized by acetylcholinesterase, which is inhibited in this setting.2 It is preferable to use a competitive neuromuscular blocking agent, such as rocuronium, for rapid sequence intubation in these patients, but increased dosing may be necessary. Although some authors have advocated the use of beta-blockers to control tachycardia, this may increase cardiovascular instability and worsen bronchospasm.8,11 Most cardiovascular effects from organophosphates rarely require specific therapy.
The definitive treatment of acetylcholinesterase inhibition starts with atropine. A competitive inhibitor of acetylcholine at muscarinic receptor sites, atropine reverses the clinical effects of cholinergic excess at parasympathetic end-organs and sweat glands. Large doses of atropine may be required.12 Data suggest that the more rapid the atropinization, the faster control is obtained.8,9 Suggested dosing is 1 or 2 mg of atropine (0.02-0.05 mg/kg) intravenously, with doubling of each subsequent dose every 5 minutes until there is control of mucous membrane hypersecretion and the airway clears.4,8,9,13 If intravenous access is not immediately available, atropine may be administered intramuscularly. Patients may require 200 to 500 mg of atropine intravenously during the first hour, followed by prolonged continuous infusions of 5 to 100 mg/hr to maintain adequate secretion control.13 Tachycardia and mydriasis may occur at these doses, but they are not indications to stop atropine administration. The endpoint of atropinization is drying of respiratory secretions, easing of respiration, and mean arterial pressure greater than 60 mm Hg.13 Animal evidence suggests that early rapid atropinization may limit seizure propagation and, in conjunction with diazepam, prevent status epilepticus.5 Atropine is not active at nicotinic sites and does not reverse the skeletal muscle effects (e.g., muscle fatigue and respiratory failure).2,9 Other anticholinergic medications, such as diphenhydramine or ophthalmic agents, may have benefit if atropine is scarce or unavailable; however, optimal intravenous dosing is not known.14
The second part of acetylcholinesterase inhibition treatment is the use of an oxime, such as pralidoxime (2-PAM, Protopam) or obidoxime (Toxogonin), to regenerate the organophosphate-acetylcholinesterase complex and to restore cholinesterase activity at muscarinic and nicotinic sites.2,4,9,15 There are several dosing regimens; the most common dose of pralidoxime is 1 or 2 g intravenously (pediatric dose, 25-50 mg/kg); additional doses may be given on the basis of clinical response. The medication may be given in a bolus of 1 or 2 g intravenously during 30 to 60 minutes every 4 to 8 hours or 500 mg/hr (pediatric dose, 10-25 mg/kg/hr).15,16 The World Health Organization recommends an initial dose of 30 mg/kg, followed by 8 mg/kg/hr continued for at least 24 hours or, if an infusion cannot be used, 30 mg/kg every 4 hours.17 The infusion may be continued for several days with no adverse effects attributable to the pralidoxime; however, rapid administration can lead to hypertension, vomiting, and a transient reversible neuromuscular blockade.18 The ideal dose of pralidoxime should be determined by monitoring of the clinical condition of the patient and serial cholinesterase levels; the patient may require higher doses of oxime than are recommended here. The World Health Organization–recommended infusion dose of obidoxime is 4 mg/kg, followed by 0.5 mg/kg/hr; alternatively, intermittent intravenous doses of 4 mg/kg, then 2 mg/kg, every 4 hours are given.17 Pralidoxime and obidoxime can be administered by intramuscular injection. Indications for oxime therapy include respiratory depression or apnea, fasciculations, seizures, arrhythmias, cardiovascular instability, and use of large amounts of atropine. Oxime therapy can be used whenever the patient requires more than a limited amount of atropine (2-4 mg) to completely reverse the signs and symptoms of intoxication or in any patient who requires repeated doses of atropine. Oxime therapy and atropine are synergistic.
In the past, pralidoxime was used only within the first 24 hours because of aging of the organophosphate-acetylcholinesterase complex, but not all organophosphates behave in a similar manner. Dimethyl and diethyl phosphoryl insecticides react differently at variable rates with acetylcholinesterase and oxime therapy. Many organophosphates are highly lipid soluble and slowly leach out of fat stores for up to 6 weeks, resulting in newly formed complexes with excellent clinical reversal of the cholinesterase inhibition by pralidoxime and by measurements of cholinesterase activity. Pralidoxime can also combine with unbound organophosphates and prevent their subsequent binding to nerve terminals. Even with optimal treatment, seriously intoxicated patients may require long-term supportive care, including ventilator support.4,16
Several studies have looked at the efficacy of pralidoxime.19,20 The results have been mixed and may be due to the variability among different organophosphates. Until this variability is further elucidated, pralidoxime administration in patients with organophosphate toxicity is still recommended.
In conjunction with atropine and the oxime pralidoxime, patients with agitation, seizures, and coma should be treated with adequate doses of a benzodiazepine after the airway has been secured.4,5,21 Although diazepam is most studied, any parenteral benzodiazepine may be used. The military has classically used diazepam autoinjectors for intramuscular injection, but midazolam is the best intramuscular agent, with lorazepam as an alternative.
Sarin, soman, tabun, and VX are nerve agents that might be used in a terrorist attack. These agents have important differences from the common household or commercial organophosphorus insecticides. These agents tend to age very quickly; tabun (GA) ages in 14 hours, sarin (GB) in 5 hours, soman (GD) in 5 or 6 minutes, and VX in 48 hours. Because of this rapid aging, reversal of nerve agent poisoning with pralidoxime is time sensitive. VX is an oily but highly toxic agent with low volatility. It does not readily vaporize, and because it has a low risk of inhalation, exposure is predominantly transcutaneous. The other agents can be mostly dispersed into the air by explosion or vaporization, resulting in inhalation exposure. These agents do not require the extremely large doses of atropine but do require pralidoxime.22–24
New therapies for treatment of organophosphorus poisoning, including the use of N-acetylcysteine and exogenous acetylcholinesterase, show promise in research studies.25,26 When they are added to anticholinergics, NMDA receptor antagonists may decrease organophosphorus compound–induced seizures.27
Disposition
Because of the prolonged effects of acetylcholinesterase inhibition, most patients with significant exposures require hospital admission. On occasion, a person with chronic exposure, depressed cholinesterase levels, and mild visual or gastrointestinal symptoms may be observed on an outpatient basis; however, some patients, particularly those exposed to fenthion, initially present with signs and symptoms of mild exposure and progress to severe, life-threatening toxicity over time.28 If plasma cholinesterase levels are available, they may be useful for treatment and disposition decisions. Asymptomatic or minimally symptomatic patients with normal or minimally depressed levels may be discharged after 4 to 6 hours with close outpatient follow-up to ensure that progressive toxicity does not occur. Patients who arrive with known severely depressed levels (usually associated with significant symptoms) require admission and close monitoring, usually in a high-intensity care unit. Patients may have rebound toxicity several days after apparently satisfactory response to initial treatment. Rebound toxicity may occur for many reasons, including persistent release of organophosphates from lipid stores.
A secondary syndrome, the intermediate syndrome (IMS), occurs 24 to 96 hours after exposure and consists of proximal muscle weakness specifically of the respiratory muscles. It is believed to be an abnormality at the neuromuscular junction. Patients with IMS present with respiratory failure several days after the acute cholinergic symptoms have resolved and may require several weeks of ventilatory support. It is theorized that this may be a result of inadequate initial oxime treatment or premature discontinuation of oxime therapy.4,29 Oximes may be beneficial for IMS; however, this is controversial.30
Finally, organophosphorus delayed neuropathy has been reported as a different entity. It affects an axonal enzyme, neurotoxic esterase, with a peripheral sensorimotor neuropathy 7 to 21 days after exposure.4
Carbamate Insecticides
Carbamate insecticides are another class of acetylcholinesterase inhibitors and are differentiated from the organophosphorus compounds by their relatively short duration of toxic effects. Carbamates inhibit acetylcholinesterase for minutes to 48 hours, and the carbamate-cholinesterase binding is reversible. Although the clinical picture of acute carbamate poisoning may be identical to that of organophosphate poisoning, the toxic effects are limited in duration, and patients may require only decontamination, supportive care, and treatment with adequate doses of atropine. Although the duration is limited, patients may become just as ill and require assisted ventilation and seizure therapy. The use of pralidoxime is controversial in carbamate poisoning; an animal study suggests that pralidoxime administration may produce greater toxicity in cases of carbaryl (Sevin) poisoning.31 Nevertheless, if doubt exists as to whether a severe poisoning is due to a carbamate or organophosphate, pralidoxime should be administered.
Chlorinated Hydrocarbon Insecticides
DDT, the prototype of chlorinated hydrocarbon insecticides (sometimes referred to as organochlorine insecticides), was first used extensively during World War II for control of typhus and malaria and widely used in the United States as a general insecticide after the war. Because of the effectiveness of DDT, many other chlorinated hydrocarbon insecticides were developed and used extensively in agricultural, commercial, and residential pest control. Although these insecticides were effective, their widespread use, long half-life, and persistence had negative ecologic repercussions. Many of these insecticides have been targeted as persistent organic pollutants by international agencies, leading to their restricted use.32
Although chlorinated hydrocarbon insecticides are no longer used in the United States for agricultural use, γ-hexachlorobenzene, better known as lindane (Kwell), is still used as a topical medicinal agent for the treatment of head lice and scabies. As a result, lindane is probably the most common cause of toxicity from an organochlorine compound in the United States. Given its toxicity, lindane is no longer a first-line agent for the treatment of scabies.33 In 2001, California issued a ban on the use and sale of lindane, and other states are considering a ban on lindane.32
Clinical Features
The primary clinical picture of acute or cumulative toxicity from chlorinated hydrocarbon pesticides is related to their neurotoxicity. Premonitory peripheral signs and symptoms, such as tremor or paresthesias, may be absent, and the first sign of toxicity may be seizure activity.34 Additional signs include confusion, combativeness, and muscle twitching. Untreated, continued muscle activity can lead to hyperthermia, metabolic acidosis, and rhabdomyolysis with secondary acute tubular necrosis. Because many of these agents are halogenated, ventricular dysrhythmias may occur from catecholamine sensitization and direct myocardial toxic effects. Immediate hepatotoxicity is unlikely without secondary hyperthermia or other metabolic complications. Long-term exposure may result in neuropsychiatric symptoms.35 Diagnosis may be difficult in chlorinated hydrocarbon pesticide exposure because the patient may be unable to provide a history. Nonhospital personnel are often in the best position to obtain information on pesticide availability and the situation surrounding the exposure. Another clue is the solvent odor and oily feel of the hydrocarbon solvent containing the highly lipid-soluble chlorinated hydrocarbon pesticides.
Management and Disposition
Skin decontamination with soap and water may reduce toxicity in acute dermal exposure. High lipid solubility results in rapid absorption, and gastrointestinal decontamination is not of benefit. Elimination of some chlorinated hydrocarbon insecticides can be increased, and repeated doses of cholestyramine (4 g orally every 8 hours) given during a mass exposure of chlordecone (a chlorinated hydrocarbon insecticide) enhanced the fecal elimination of this compound.35–37
The primary objective is seizure control, best accomplished with short-acting benzodiazepines or barbiturates. Recurrent seizures or status epilepticus may require high-dose barbiturates and paralyzing agents (e.g., pancuronium or vecuronium) to prevent secondary morbidity from continuous motor activity in prolonged seizures. The seizure activity is usually self-limited, lasting only 1 or 2 days even in severe cases.34,36,38
Substituted Phenols
The substituted phenols include dinitrophenol (DNP), pentachlorophenol, and dinitrocresol. These compounds have been used since the 1930s as insecticides, termiticides, herbicides, and wood preservatives. They are currently used in agricultural, commercial, and residential applications, including over-the-counter preparations for home gardeners. Substituted phenols such as DNP are abused as weight reduction agents and occasionally in illegitimate weight reduction operations.39
Principles of Disease
Substituted phenols produce their toxicity by uncoupling cellular oxidative phosphorylation; this leads to inefficient production of high-energy phosphate substrates and increased cellular use of oxygen, glucose, and water, with subsequent excess heat production. These compounds are commonly used during the summer when the external heat predisposes users to increased toxicity.40 In addition, nitro-substituted phenols may produce methemoglobinemia.
Chlorophenoxy Compounds
The chlorophenoxy pesticides were developed in the early 1940s and hailed as a selective herbicide particularly effective against broadleaf weeds. This class of herbicide developed a special notoriety during the Vietnam War as Agent Orange, a defoliant used in aerial spraying, consisting of a mixture of 2,4-dichlorophenoxyacetic acid (2,4-D) and 2,4,5-trichlorophenoxyacetic acid (2,4,5-T). Unfortunately, 2,4,5-T is almost always contaminated with isomers of tetrachlorodibenzodioxin. This concern for dioxin exposure has led to the extensive medical investigations of Vietnam veterans and severe restrictions on the production and use of 2,4,5-T.41 Because of the relative safety and broadleaf selectivity of 2,4-D, however, most home gardeners have at least one chlorophenoxy compound on a shelf in their garage, and some old cans may contain 2,4,5-T or a mixture of both compounds.
Principles of Disease
Chlorophenoxy compounds may be absorbed through the skin, gastrointestinal tract, and respiratory tract, but almost all significant poisonings are a result of accidental or intentional ingestion. The lipid solubility of these compounds is low, and excretion is fairly rapid, so cumulative toxicity from repeated exposures does not occur.42
Clinical Features
Similar to most organic pesticides in an organic solvent, the chlorophenoxy herbicides may produce mild, nonspecific dermal and gastrointestinal irritation with nausea, vomiting, and gastrointestinal distress. Large exposures are likely to cause systemic symptoms ranging from diffuse myotonia and muscle fasciculations progressing to rhabdomyolysis, hyperthermia, and a hypermetabolic state with metabolic acidosis.43
Bipyridyl Compounds
The bipyridyl (also called dipyridyl) compounds paraquat and diquat were first investigated in the late 1950s and early 1960s. They are extremely effective contact herbicides that are rapidly inactivated by the surrounding soil in the event of overspray. Paraquat is activated when it is exposed to sunlight, which led to its use as the herbicide of choice during aerial spraying of marijuana by the U.S. and Mexican governments. After spraying, however, growers simply would harvest the crops before the plants were exposed to enough sunlight to damage them, resulting in an apparently healthy harvest but one contaminated with paraquat. The burning of marijuana pyrolyzes paraquat into a nontoxic form, a fact that was lost in the warning messages dispensed by the government at that time.44
Principles of Disease
Of the two bipyridyl compounds in use, paraquat is the most significant in terms of number of cases and toxic effects. Paraquat use is tightly regulated in the United States but is widespread throughout the world. Diquat is less regulated in the United States and is included in some formulations of herbicides sold for residential use. Paraquat is absorbed through the skin, gastrointestinal tract, and respiratory tract. Almost all fatal exposures have resulted from the ingestion of paraquat, although a few case reports have involved extensive skin contamination. Toxicity has occurred, but no fatal cases have been reported from inhalation of paraquat vapor or aerosols. Diquat is poorly absorbed through intact skin, and most cases of toxicity result from ingestion.45
Paraquat’s toxic effect is from the production of superoxides created during cyclic oxidation-reduction reactions of the compound in tissues. Lipid peroxidation of cellular membranes seems to be one significant pathway of cellular injury.46,47
Paraquat selectively concentrates in the lungs because of an amine uptake mechanism in alveolar cells. In addition, high concentrations of oxygen significantly increase the extent of paraquat-induced injury so that the lungs are the major target organs. The pathophysiologic lesions include direct injury to the alveolar-capillary membrane followed by surfactant loss, adult respiratory distress syndrome, progressive pulmonary fibrosis, and respiratory failure. Paraquat damages other major organ systems by the same cellular membrane effects, including the liver, kidneys, heart, and central nervous system. Diquat has similar effects, with most of its toxicity concentrated in the kidneys rather than in lung tissue.45
Clinical Features
The paraquat-induced pulmonary injury usually progresses during 1 to 3 weeks, although the clinical course varies considerably with severity of poisoning, involvement of other organ systems, and underlying medical problems. This is not a factor in the emergency department, and the delayed pulmonary injury is not discussed here. Diquat usually spares the lungs but produces similar toxicity in all other organ systems.45
Diagnostic Strategies
Paraquat is measurable in the blood. As long as the time of acute exposure is known, the level serves as an accurate prognostic marker. The assay is not readily available in the United States, and by the time the results are obtained, nothing can be done to change the eventual outcome. There is a qualitative bedside test that uses the reduction of paraquat or diquat in alkalinized urine by sodium dithionite, but the reagent frequently is not available.43 Studies should evaluate caustic gastrointestinal injury and pulmonary and renal damage.
Management and Disposition
Cyclophosphamides in combination with corticosteroids have also been investigated as a means to attenuate paraquat toxicity.48,49 Whereas the results have been mixed, this therapy has been recommended by some experts in the setting of significant paraquat toxicity.
There are other suggested treatment adjuncts, such as N-acetylcysteine, nitric oxide, deferoxamine, and cytoprotective agents such as amifostine, but no single therapy has proved consistently successful.50
Pyrethrins and Pyrethroids
Principles of Disease
Pyrethrins and pyrethroids have a variety of effects in humans and other mammals.51,52 Clinically, the naturally occurring pyrethrins can cause sensitization and allergic phenomena, but this does not occur with the synthetic pyrethroids. Both classes are associated with sodium channel blockade, slowing the rate of activation of the sodium channel and extending the time during which the channel is open. In addition, both classes affect GABA receptors, inhibiting chloride channel function. Less significant effects include potentiation of nicotinic cholinergic neurotransmission, enhancement of norepinephrine release, and inhibition of calcium adenosine triphosphatase interference with sodium-calcium exchange across membranes.51,52
Clinical Features
Sodium channel–mediated and GABA-mediated chloride channel effects mediate neurologic signs and symptoms. Facial paresthesias have been reported, and seizures occur with massive ingestions.51,52 Nonspecific symptoms, such as headache, fatigue, dizziness, and weakness, are also reported.
Diagnostic Strategies
No laboratory tests are available to measure pyrethrins or pyrethroids in a clinical setting.
Glyphosate
Principles of Disease
Glyphosate is toxic to plants by inhibition of the enzyme 5-enolpyruvylshikimate-3-phosphate synthase in the shikimic acid metabolic pathway. After application of glyphosate on the leaves, it is transported to the roots, where the enzyme is active. Humans lack this enzyme and do not have enzyme-related toxicity. Reported toxicity is believed to result largely from the surfactant POEA and may reflect the direct corrosive effect from the amine salt, or it may uncouple oxidative phosphorylation.53
Clinical Features
Most ingestions of the dilute solution cause only minimal symptoms, including gastrointestinal distress. Patients ingesting large volumes of dilute solutions or moderate volumes of concentrated solutions complain of sore throat, nausea, abdominal pain, and fever. They may have vomiting, diarrhea, respiratory distress, noncardiogenic pulmonary edema, dysrhythmias, shock, coma, and renal failure. Acidosis reflects poor tissue perfusion and cardiovascular compromise.53 Negative prognostic indicators include shock, acidosis, and persistent hyperkalemia.53
Diagnostic Strategies
The critical element in diagnosis is history of ingestion. Laboratory analysis may demonstrate an anion gap metabolic acidosis, hypoxemia, and hyperkalemia. Elevated transaminases may occur in 30% of ill patients, and signs of renal failure may develop in persistent shock states. The electrocardiogram may show ventricular dysrhythmias and secondary signs of hypoxemia.53
DEET
N,N-Diethyl-m-toluamide or N,N-diethyl-3-methylbenzamide (DEET) is not a pesticide but an insect repellent. It is the most widely used chemical insect repellent in the United States. DEET was developed by scientists at the U.S. Department of Agriculture in 1946, patented by the U.S. Army soon thereafter, and released to the general public in 1957.54 With the prevalence of Lyme disease and other concerning arthropod-borne diseases, the use of DEET has greatly increased. Formulations containing DEET range from 5 to 100%. The U.S. Army routinely used 75% solutions until 1987 but now uses a 35% time-release, polymer-based formulation. The American Academy of Pediatrics recommends 30% as the maximum concentration for use in infants and children and does not recommend use of DEET in infants younger than 2 months.55
Principles of Disease
DEET is lipophilic and can be absorbed through the skin. Skin absorption and toxicity increase with repeated applications, increased ambient temperatures, sweating, and abraded, thin skin. Ingestion may lead to toxicity.56 DEET primarily affects the central nervous system. Its mechanism of action is unknown. It may sensitize the skin and cause allergic reactions.
Clinical Features
Prolonged skin contact may lead to contact dermatitis, and prolonged contact with high concentrations has led to skin blisters. Patients who have ingested DEET or have repeated skin applications in a hot enclosed environment that enhances absorption have developed liver function test abnormalities and neurologic findings, including encephalopathy, seizures, movement disorders, and coma.56 Most exposures to DEET result in no or minimal toxicity and should not preclude its use in susceptible populations in which significant arthropod-borne diseases are prevalent.57
Management and Disposition
Key Concepts
 All patients exposed to cholinesterase inhibitors should have skin decontamination. Health care personnel need to be protected during this process.
All patients exposed to cholinesterase inhibitors should have skin decontamination. Health care personnel need to be protected during this process.


 The clinical endpoint for atropine administration is drying of airway secretions.
The clinical endpoint for atropine administration is drying of airway secretions.



 Rapid cooling and glucose are the two most important therapies in substituted phenol toxicity.
Rapid cooling and glucose are the two most important therapies in substituted phenol toxicity.


 The predominant form of pyrethrin and pyrethroid toxicity is allergic.
The predominant form of pyrethrin and pyrethroid toxicity is allergic.



Acknowledgment
I would like to thank Cynthia K. Aaron for her contributions as a previous author to this chapter.
References
1. The Persian Gulf experience and health. NIH Technology Assessment Workshop Panel. JAMA. 1994;272:391.
2. Sener, EB, Ustun, E, Kocamanoglu, S, Tur, A. Prolonged apnea following succinylcholine administration in undiagnosed acute organophosphate poisoning. Acta Anaesthesiol Scand. 2002;46:1046–1048.
3. Johnson, PS, Michaelis, EK. Characterization of organophosphate interactions at N-methyl-D-aspartate receptors in brain synaptic membranes. Mol Pharmacol. 1992;41:750.
4. Johnson, MK, et al. Evaluation of antidotes for poisoning by organophosphorus pesticides. Emerg Med. 2000;12:22.
5. Shih, TM, Duniho, SM, McDonough, JH. Control of nerve agent–induced seizures is critical for neuroprotection and survival. Toxicol Appl Pharmacol. 2003;188:69.
6. Liu, JH, et al. Acid-base interpretation can be the predictor of outcome among patients with acute organophosphate poisoning before hospitalization. Am J Emerg Med. 2008;26:24.
7. Petroianu, G, Ruefer, R. Poisoning with organophosphorus compounds. Emerg Med (Fremantle). 2001;13:258.
8. Eddleston, M, Roberts, D, Buckley, N. Management of severe organophosphorus pesticide poisoning. Crit Care. 2002;6:259.
9. Eddleston, M, Singh, S, Buckley, N. Acute organophosphorus poisoning. Clin Evid. 2003;9:1542.
10. Eddleston, M, et al. Multiple-dose activated charcoal in acute self poisoning: A randomised controlled trial. Lancet. 2008;371:579.
11. Sungur, M, Guven, M. Intensive care management of organophosphate insecticide poisoning. Crit Care. 2001;5:211.
12. Golsousidis, H, Kokkas, V. Use of 19 590 mg of atropine during 24 days of treatment, after a case of unusually severe parathion poisoning. Hum Toxicol. 1985;4:339.
13. Eddleston, M, et al. Speed of initial atropinisation in significant organophosphorus pesticide poisoning: A systematic comparison of recommended regimens. J Toxicol Clin Toxicol. 2004;42:865.
14. Bryant, SM, et al. Pretreating rats with parenteral ophthalmic antimuscarinic agents decreases mortality from lethal organophosphate poisoning. Acad Emerg Med. 2007;14:370.
15. Eddleston, M, et al. Oximes in acute organophosphorus pesticide poisoning: A systematic review of clinical trials. QJM. 2002;95:275.
16. Worek, F, et al. Reappraisal of indications and limitations of oxime therapy in organophosphate poisoning. Hum Exp Toxicol. 1997;16:466.
17. Feldman, RJ, Szajewski, J. Cholingeric Syndrome. http://www.intox.org/databank/documents/treat/treate/trt15_e.htm.
18. Medicis, JJ, et al. Pharmacokinetics following a loading plus a continuous infusion of pralidoxime compared with the traditional short infusion regimen in human volunteers. J Toxicol Clin Toxicol. 1996;34:289.
19. Eddleston, M, et al. Differences between organophosphorus insecticides in human self poisoning: A prospective cohort study. Lancet. 2005;366:1422.
20. Eddleston, M, et al. Pralidoxime in acute organophosphorus insecticide poisoning—a randomised controlled trial. PLoS Med. 2009;6:e1000104.
21. Shih, T, McDonough, JH, Jr., Koplovitz, I. Anticonvulsants for soman-induced seizure activity. J Biomed Sci. 1999;6:86.
22. Sidell, FR, Borak, J. Chemical warfare agents: II. Nerve agents. Ann Emerg Med. 1992;21:865.
23. Markenson, D, Redlener, I. Pediatric terrorism preparedness national guidelines and recommendations: Findings of an evidenced-based consensus process. Biosecur Bioterror. 2004;2:301.
24. Rotenberg, JS, Newmark, J. Nerve agent attacks on children: Diagnosis and management. Pediatrics. 2003;112:648.
25. Evron, T, et al. Plant-derived human acetylcholinesterase-R provides protection from lethal organophosphate poisoning and its chronic aftermath. FASEB J. 2007;21:2961.
26. Yurumez, Y, et al. Beneficial effect of N-acetylcysteine against organophosphate toxicity in mice. Biol Pharm Bull. 2007;30:490.
27. Dekundy, A, et al. NMDA antagonists exert distinct effects in experimental organophosphate or carbamate poisoning in mice. Toxicol Appl Pharmacol. 2007;219:114.
28. Davies, JO, Eddleston, M, Buckley, NA. Predicting outcome in acute organophosphorus poisoning with a poison severity score or the Glasgow coma scale. QJM. 2008;101:371.
29. Senanayake, N, Sanmuganathan, PS. Extrapyramidal manifestations complicating organophosphorus insecticide poisoning. Hum Exp Toxicol. 1995;14:600.
30. Karalliedde, L, Baker, D, Marrs, TC. Organophosphate-induced intermediate syndrome: Aetiology and relationships with myopathy. Toxicol Rev. 2006;25:1.
31. Kurtz, PH. Pralidoxime in the treatment of carbamate intoxication. Am J Emerg Med. 1990;8:68.
32. Humphreys, EH, et al. Outcomes of the California ban on pharmaceutical lindane: Clinical and ecologic impacts. Environ Health Perspect. 2008;116:297.
33. Karr, CJ, Solomon, GM, Brock-Utne, AC. Health effects of common home, lawn, and garden pesticides. Pediatr Clin North Am. 2007;54:63.
34. Jaeger, U, et al. Acute oral poisoning with lindane-solvent mixtures. Vet Hum Toxicol. 1984;26:11.
35. Kilburn, KH. Chlordane as a neurotoxin in humans. South Med J. 1997;90:299.
36. Aks, SE, et al. Acute accidental lindane ingestion in toddlers. Ann Emerg Med. 1995;26:647.
37. Cohn, WJ, et al. Treatment of chlordecone (Kepone) toxicity with cholestyramine: Results of a controlled clinical trial. N Engl J Med. 1978;298:243.
38. Cable, GG, Doherty, S. Acute carbamate and organochlorine toxicity causing convulsions in an agricultural pilot: A case report. Aviat Space Environ Med. 1999;70:68.
39. Miranda, EJ, et al. Two deaths attributed to the use of 2,4-dinitrophenol. J Anal Toxicol. 2006;30:219.
40. Jorens, PG, Schepens, PJ. Human pentachlorophenol poisoning. Hum Exp Toxicol. 1993;12:479.
41. Kahn, PC, et al. Dioxins and dibenzofurans in blood and adipose tissue of Agent Orange–exposed Vietnam veterans and matched controls. JAMA. 1988;259:1661.
42. Reifenrath, WG, Hawkins, GS, Kurtz, MS. Percutaneous penetration and skin retention of topically applied compounds: An in vitro–in vivo study. J Pharm Sci. 1991;80:526.
43. Bradberry, SM, et al. Mechanisms of toxicity, clinical features, and management of acute chlorophenoxy herbicide poisoning: A review. J Toxicol Clin Toxicol. 2000;38:111.
44. Landrigan, PJ, et al. Paraquat and marijuana: Epidemiologic risk assessment. Am J Public Health. 1983;73:784.
45. Jones, GM, Vale, JA. Mechanisms of toxicity, clinical features, and management of diquat poisoning: A review. J Toxicol Clin Toxicol. 2000;38:123.
46. Wilks, MF, et al. Improvement in survival after paraquat ingestion following introduction of a new formulation in Sri Lanka. PLoS Med. 2008;5:e49.
47. Fukushima, T, et al. Mechanism of cytotoxicity of paraquat: I. NADH oxidation and paraquat radical formation via complex I. Exp Toxicol Pathol. 1993;45:345.
48. Lin, JL, et al. A prospective clinical trial of pulse therapy with glucocorticoid and cyclophosphamide in moderate to severe paraquat-poisoned patients. Am J Respir Crit Care Med. 1999;159:357.
49. Perriens, JH, et al. High-dose cyclophosphamide and dexamethasone in paraquat poisoning: A prospective study. Hum Exp Toxicol. 1992;11:129.
50. Wills, BK, et al. The effect of amifostine, a cytoprotective agent, on paraquat toxicity in mice. J Med Toxicol. 2007;3:1.
51. Ray, DE, Forshaw, PJ. Pyrethroid insecticides: Poisoning syndromes, synergies, and therapy. J Toxicol Clin Toxicol. 2000;38:95.
52. Soderlund, DM, et al. Mechanisms of pyrethroid neurotoxicity: Implications for cumulative risk assessment. Toxicology. 2002;171:3.
53. Lee, HL, et al. Clinical presentations and prognostic factors of a glyphosate-surfactant herbicide intoxication: A review of 131 cases. Acad Emerg Med. 2000;7:906.
54. Fradin, MS. Mosquitoes and mosquito repellents: A clinician’s guide. Ann Intern Med. 1998;128:931.
55. American Academy of Pediatrics Committee on Environmental Health. Follow safety precautions when using DEET on children. AAP News. 2003;22:99.
56. Tenenbein, M. Severe toxic reactions and death following the ingestion of diethyltoluamide-containing insect repellents. JAMA. 1987;258:1509.
57. Koren, G, Matsui, D, Bailey, B. DEET-based insect repellents: Safety implications for children and pregnant and lactating women. CMAJ. 2003;169:209.