Brain Resuscitation
Perspective
Recognition of the dominant role of the brain in determining the quality of human life dates back to the dawn of recorded medical history. Until recently, however, medical efforts after cardiac arrest have focused exclusively on cardiac resuscitation. Recent advances in the understanding of the pathophysiologic mechanisms of brain ischemia have encouraged attention to cerebral resuscitation as a critical component of what is now recognized as a complex multisystem pathophysiologic state called post–cardiac arrest syndrome.1 This chapter reviews the pathophysiology of postischemic encephalopathy and discusses therapies for improving neurologic recovery after cardiac arrest.
Pathophysiology
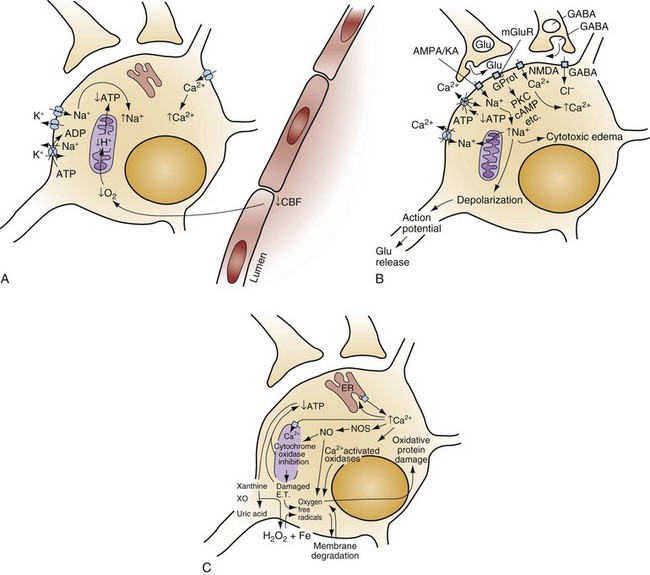
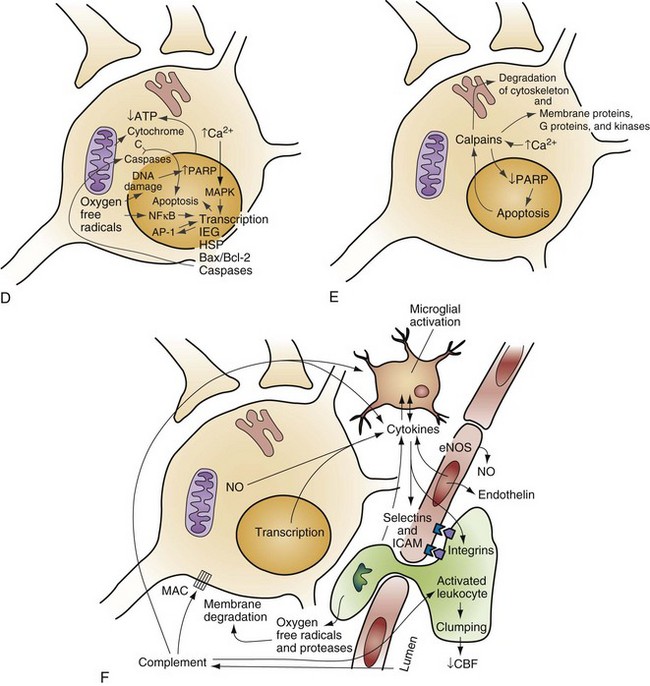
When the brain is deprived of adequate blood flow, the resulting ischemia is characterized by a bewildering array of inter-related physiologic and cellular responses that ultimately result in neuronal cell death (Fig. 8-1).2,3 Although this complex cascade of events can be triggered by periods of ischemia lasting only a few minutes, the resulting neuronal death is usually delayed by hours or days. Furthermore, the biology of cerebral cell death after global cerebral ischemia follows (with slight variations) the pattern of delayed cerebral cell death that follows stroke, traumatic brain injury, and other forms of hypoxic or toxic brain injury. Increased understanding of the brain’s response to injury during the period between insult and neuronal cell death will eventually allow more specific brain resuscitation therapies.
Management
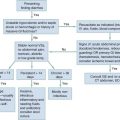
Standard management of cardiac arrest and subsequent ischemic brain damage involves restoring cerebral blood flow (CBF) and preventing secondary insult. These treatments have generally not been studied in prospective, randomized controlled trials, but they are supported by clinical experience and limited experimental data. Although proposed and experimental therapies are generally aimed at specific molecular interventions in the pathophysiology of ischemic brain injuries, none of these as yet have proven effective in clinical trials. The most comprehensive review and consensus guideline statement on care of patients with post–cardiac arrest syndrome was produced by the International Liaison Committee on Resuscitation and its constituent bodies, with the endorsement of the American College of Emergency Physicians, Society for Academic Emergency Medicine, Society of Critical Care Medicine, and Neurocritical Care Society.1 Improvements in post–cardiac arrest care, through an inclusive multisystem approach, can increase the likelihood of meaningful recovery in these patients. Implementation of standardized protocols for postresuscitation care that include many or all of the following components have demonstrated increases in survival with a favorable neurologic outcome of up to 27 to 30% in repeated (although poorly controlled) before-and-after studies.4,5
Standard Strategies
Return of Spontaneous Circulation
The efficacy of closed-chest CPR in generating adequate cerebral perfusion is somewhat controversial. Cardiac output during optimal standard closed-chest CPR has previously been estimated to be only 20 to 30% of normal, but more recent data suggest that higher cardiac outputs are possible in clinical practice, and, unquestionably, effective CPR is essential to neurologic recovery after cardiac arrest. Considerable effort has been directed toward the investigation of improved CPR techniques that will prove to be even more effective and for longer periods (see Chapter 9).
Treatment of Hypotension, Hypoperfusion, and Hypoxia
Hypotension in the postarrest period can dangerously lower cerebral perfusion pressure. Although CBF is normally independent of perfusion pressure over a wide range of arterial blood pressure, such autoregulation is often lost in the injured brain. As a result, perfusion of ischemic tissue becomes passively dependent on arterial pressure, and hypotension can compromise CBF and result in significant additional brain damage.6 Therefore, after return of spontaneous circulation (ROSC), low arterial pressures should be rapidly normalized, with intravascular volume administration and vasopressors used as needed. Because elevated arterial pressures may be needed to provide sufficient CBF, hypertension usually should not be treated in the postresuscitation period. Very high blood pressures may require treatment, but specific cutoffs are controversial. In general, diastolic pressures may be allowed to run as high as 120 mm Hg without requiring treatment. In fact, hypertension is sometimes induced clinically or experimentally with vasopressors in an attempt to raise cerebral perfusion pressure and improve neurologic recovery.6 Because it is unproved, and because risks of this therapy include blood-brain barrier disruption and worsening of vasogenic edema, induced hypertension is not currently a standard therapy.
CVR after resuscitation from cardiac arrest is another determinant of CBF and may be affected by hyperventilation and microvascular patency. Although the cerebral circulation may lose its ability to adjust to blood pressure changes after ischemia, attenuated responsiveness to carbon dioxide and oxygen levels in arterial blood can still be present.7 Carbon dioxide is a potent vasoactive agent, and lowering of the arterial carbon dioxide partial pressure (PaCO2) by hyperventilation results in rapid reduction of CBF. Because reductions in CBF reduce total cerebral blood volume, hyperventilation may transiently abort brainstem herniation in the presence of critically elevated intracranial pressure (ICP) until osmotherapy or ventriculostomy can be initiated. When ICP is not elevated, however, the vasoconstriction and increased CVR caused by hyperventilation can cause potentially dangerous reductions in CBF.7 Although controversy surrounds whether increases in ICP are clinically significant after global ischemia, the measurement of ICP is generally not recommended in the management of adult cardiac arrest survivors.8 In general, ventilation to maintain a PaCO2 of 35 to 40 mm Hg is safe and appropriate, and inadvertent hyperventilation should be avoided. CVR may also be elevated after cardiac arrest by endothelin-induced vasospasm or microvascular occlusion by leukocyte clumping or coagulation. Hemodilution, anticoagulation, and antiplatelet agents have been studied in animals for effectiveness in mitigating microvascular occlusion with mostly negative results.9 Acute use of these agents to improve microcirculatory flow has not been studied in human cerebral ischemia.
Normal arterial oxygen saturation should be maintained after resuscitation from cardiac arrest. Because the injured brain may not be able to compensate for hypoxia by augmenting CBF, cerebral oxygen delivery may diminish rapidly as the oxygen content of blood decreases. Hyperoxia secondary to the use of 100% oxygen in the immediate postarrest period, however, has also been shown to increase oxidative brain injury in animal models of cardiac arrest and resuscitation.10 Normoxia or mild hyperoxia (arterial partial pressure of oxygen [PaO2] of 80-120 mm Hg with oxyhemoglobin saturation percentage maintained in the high nineties) should be maintained through use of the lowest fraction of inspired oxygen (FIO2) possible. The use of 100% oxygen is appropriate during cardiac arrest, but FIO2 should be titrated downward shortly after the ROSC. A large multicenter cohort study found hyperoxia (defined as PaO2 300 mm Hg or above on first intensive care unit [ICU]–obtained arterial blood gas [ABG] measurement) to be associated with an 18% (95% confidence interval [CI] 14-22%) higher absolute mortality.11 An important clinical trial to compare earlier use of normoxia after ROSC with the more typical prolonged period of postresuscitative hyperoxia has been proposed. Because hypoxia, hypocapnia, and hypercapnia must be avoided, controlled ventilation is appropriate in the period after resuscitation, with muscle relaxation and sedation if needed.
Maintenance of Body Temperature
Hyperthermia (or fever) exacerbates brain injury and worsens neurologic outcome.12,13 Elevated body temperature increases cerebral metabolic demand by 8 to 13% per degree Celsius, escalates glutamate release, increases oxygen free radical production, and increases cytoskeletal and blood-brain barrier breakdown with increased vasogenic edema.14 Core body temperature (usually rectal, bladder, or esophageal) should be accurately measured in patients resuscitated from cerebral ischemia.15 Hyperthermia may be treated with antipyretics, circulating air or water cooling systems, or evaporative cooling via water mist and fans.15 Aggressive treatment should, at a minimum, be used to prevent temperature increases in the postischemic period in all patients, and the practice of inducing therapeutic hypothermia has emerged as a therapy for comatose survivors of cardiac arrest.
Resuscitative Mild Hypothermia
Hypothermia was first reported more than 50 years ago to have a protective effect in global and focal brain ischemia and ranges from mild (32-34° C) to profound (5° C).16 The mechanism by which hypothermia conveys protection is uncertain, but several possibilities have been suggested. Hypothermia reduces glutamate release, metabolic demand, free radical formation, and production of inflammatory cytokines.14 Cell-signaling and genetic responses to cellular injury are also affected by hypothermia, and hypothermia may protect the brain from programmed neuronal cell death.17,18
Mild hypothermia is easier to achieve and has fewer adverse effects than lower temperatures and has consistently been found to be neuroprotective in experimental cerebral ischemia.17 Two multicenter prospective, randomized, controlled trials of mild hypothermia have now shown marked improvements in neurologic outcome in comatose survivors of out-of-hospital cardiac arrest.19,20 The improvements in neurologic outcome (recovery with minimal or no neurologic deficits at hospital discharge) and in mortality in patients treated with hypothermia in these trials are shown in Table 8-1. In these trials, the number needed to treat to have one additional patient with a good neurologic outcome was only about seven. The 2005 and 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care and those of the International Liaison Committee on Resuscitation recommend cooling unconscious adult patients after cardiac arrest to 33° C for 12 to 24 hours.21,22 Implementation of these guidelines has been slow and represents a failure of clinical translation. Barriers to adoption and the public health impact of widespread use have been reported.23
Table 8-1
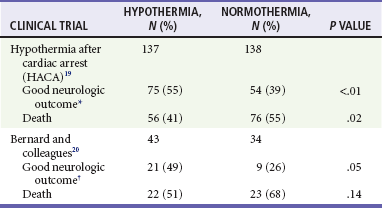
*Good neurologic outcome defined as recovery with no neurologic deficits or with moderate disability but living independently and working at least part time at 6 months.
†Good neurologic outcome defined as recovery with moderate, minimal, or no neurologic deficits at hospital discharge and discharge to home or acute rehabilitation facility.
Techniques used to induce hypothermia in comatose survivors of cardiac arrest, including traditional and advanced surface cooling, administration of ice-cold saline, and endovascular cooling, are discussed in Chapter 9. The optimal method of cooling patients resuscitated from cardiac arrest has not been established.
The rate of cooling needed for effective neuroprotection, the optimal duration of cooling, and the best process for rewarming after hypothermia are all unknown.24,25 Animal experimentation and the consensus recommendations suggest that cooling should be initiated as early and as rapidly as possible.26 Cooling may begin during the out-of-hospital phase of resuscitation and may even be initiated before ROSC. In the positive clinical trials, hypothermia at 33 ± 1° C was achieved by 2 hours or 8 hours after ROSC and was maintained for either 12 hours or 24 hours.19,20 Patients were then allowed to rewarm passively or with a combination of passive and active rewarming.19,20 Rebound hyperthermia is common with passive rewarming and should be avoided.27 Although the clinical trials enrolled only patients resuscitated from cardiac arrest caused by ventricular fibrillation, there is clinical experience with resuscitative hypothermia in patients with cardiac arrest from other causes,28 and consensus recommendations support its use in such cases.26
The clinical importance of initiating hypothermia in the prehospital setting remains unclear. Implementation of prehospital cooling in patients with cardiac arrest appeared effective as part of a comprehensive postarrest program in an observational before-and-after study at Wake Forest,5 but a randomized Australian trial of prehospital cooling versus rapid cooling shortly after emergency department (ED) arrival in Australia did not demonstrate any added benefit in those cooled in the field.29
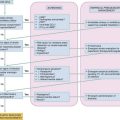
Cooling of comatose survivors of out-of-hospital cardiac arrest requires a multidisciplinary hospital policy and should be initiated as early as possible (Box 8-1). In hospitals capable of hypothermic resuscitation, cooling is optimally started in the field or the ED and maintained in the ICU.8,25 Regardless of cooling technique, patients cooled after cardiac arrest require pharmacologic therapy to prevent shivering because shivering effectively warms the patient. In the two clinical trials, shivering was prevented with a nondepolarizing paralytic, and sedation was maintained with midazolam with or without fentanyl.19,20 When paralysis is not clinically desirable, it may be possible to sufficiently lower the shivering threshold in awake patients with meperidine, buspirone, dexmedetomidine, or a combination of these.30,31 Shivering can sometimes also be reduced in a patient cooled by an endovascular catheter even when the core temperature is 33° C by applying a warming blanket, because surface temperature receptors control thermoregulation to a much greater extent than core temperature receptors. Other pharmacologic treatments that may be used during induced hypothermia include antipyretics, which lower the core body temperature set point, even in normothermic patients, although to such a small degree that the effect is unlikely to be clinically relevant.32 More effective pharmacologic lowering of core body temperature is, however, being investigated. Neurotensin is an endogenous neuropeptide involved in thermoregulation that can induce hypothermia and neuroprotection in experimental models of cerebral ischemia.33
Therapeutic hypothermia has also been proven effective in neonatal hypoxic ischemic encephalopathy, a condition similar in many ways to the global cerebral ischemic injury resulting from cardiac arrest. Cooling after resuscitation from cardiac arrest is also currently being studied in other infants and children. Clinical trials of mild hypothermia in adults and children with traumatic brain injury have repeatedly shown no benefit, but another trial focusing on even earlier and more rapid cooling after injury is underway in Australia and New Zealand. Cooling during the resuscitation of patients with acute ischemic stroke is also being investigated.34 Ongoing efforts to develop feasible methods of selective cooling of the brain after cardiac arrest35,36 and studies of profound systemic hypothermia are still experimental.
Treatment of Hyperglycemia
Postischemic hyperglycemia has detrimental effects on CBF, metabolism, edema formation, and neurologic outcome. In experimental focal cerebral ischemia, profound hyperglycemia (>500 mg/dL) causes a more pronounced decrease in intracellular pH, increases brain lactate levels, and increases neuronal loss. Increased neuronal damage from hyperglycemia in global cerebral ischemia may also be glutamate mediated. Observational studies in patients with stroke and survivors of cardiac arrest have shown that hyperglycemia after brain ischemia is strongly associated with worse outcomes in both diabetics and nondiabetics.37,38 In experimental studies, normoglycemia and mild insulin-induced hypoglycemia have been shown to improve neurologic function after focal and global ischemia. It is interesting to note that insulin itself may have a neuronal growth factor–like effect that may theoretically also be neuroprotective. Thus the best available evidence supports active treatment of hyperglycemia after global brain ischemia, and the administration of glucose should be avoided except in verified hypoglycemia.22
Seizure Management
Seizures may result from global cerebral ischemia and may exacerbate the underlying brain injury. Seizure activity can increase brain metabolism by 300 to 400%, worsening the mismatch between oxygen delivery and demand in the postarrest period, with greater metabolic failure and neuronal loss and worsened neurologic outcome. Although prevention of seizures has not been demonstrated to improve neurologic recovery, seizures are clearly not desirable in the postischemic period.8 The prophylactic use of anticonvulsant drugs in patients resuscitated from cardiac arrest is controversial and is not standard care, but it is generally agreed that seizures should be quickly and effectively treated. Common therapeutic agents include benzodiazepines, phenytoin, and barbiturates. Each of these anticonvulsant drugs has also been considered as specific therapy for cerebral ischemia because of the antagonism of excitatory amino acids, sodium channel blockade, or effects on cerebral metabolism. Although these drugs are of proven value as anticonvulsants, other uses in cerebral ischemia are experimental and unproved.
Clinical Outcomes
The published experience in Olmsted County, Minnesota, between 1990 and 2000 may represent the best possible outcomes with currently available therapy.39 First responders (including police and firefighters) with automated external defibrillators in that county responded to 330 patients with cardiac arrest, 200 (61%) of whom had ventricular fibrillation at presentation. The majority of patients with ventricular fibrillation (145 patients, 44% of all arrests) survived to hospital admission, and 84 patients (25%) were discharged alive. Remarkably, among these 84 survivors, 79 (24% of all arrests) left the hospital neurologically intact.39
More typical outcomes were identified by a recent very large cohort study of outcomes in 8091 patients with cardiac arrest in Ontario, Canada.40 Survival was 5.2% at hospital discharge, and was 4.0% at 1 year. The vast majority of 1-year survivors (3% of all those with cardiac arrest), however, had no or minimal neurologic deficits, and the average quality of life indices of all survivors were as good as those for patients without a history of cardiac arrest.40 The Ontario experience echoes that of a Portuguese study,41 but other recent data on 6240 patients from studies in the Netherlands,42 Norway,43 and Switzerland all confirm a rate of survival to discharge home with minimal or no deficit of 8% in all out-of-hospital cardiac arrests.44 Quality of life among long-term survivors of cardiac arrest is consistently high in all of these studies, with low rates of patients in persistent vegetative states or requiring skilled nursing care. Overall, among those surviving to hospital admission, 14 to 55% of patients will have good long-term neurologic outcomes.39,41 A large, prospective observational study from the Resuscitation Outcomes Consortium found significant regional variation in survival to hospital discharge among out-of-hospital cardiac arrest patients.45 Across the 10 regions from North America, survival ranged from 3 to 16.3%, with a median of 8.4%.
Despite these data, nihilism is common among physicians treating patients with cardiac arrest and ischemic brain injury. This may arise in part from the fact that most survivors of cardiac arrest are comatose at the time of admission and are without early prognostic findings suggesting which patients will have a favorable outcome. Guidelines for neurologists regarding prognostication are largely driven by data from the pre–therapeutic hypothermia era, but absent cortical somatosensory evoked potentials and an unreactive electroencephalographic background pattern at 72 hours appear to be incompatible with long-term recovery; on the other hand, hypothermia-treated patients with absent motor responses to pain and incomplete recovery of brainstem reflexes were found to survive, in contrast to findings of prior meta-analyses.46–48 The prognostic usefulness of the examination in the immediate postresuscitation period is likely quite limited, with little consensus on the minimum observation period required to establish brain death.49 Despite continued work to identify an imaging biomarker for outcome after cardiac arrest, there is no established role for either early magnetic resonance imaging or computed tomography in prognostication in survivors of cardiac arrest.50,51 Nihilism is of particular concern, however, and should be avoided because of the potential for poor prognoses to be self-fulfilling. In the near future, serum biomarkers of brain injury may identify the potential for neurologic recovery early in a patient’s course and help guide therapy. Until early predictions of outcome can be accurately made, the emergency physician should consider every survivor of cardiac arrest as having a significant chance of full recovery (14-55%) and should know that bad neurologic outcomes are usually fatal rather than chronically debilitating.
Summary
Key Concepts




References
1. Neumar, RW, et al. Post-cardiac arrest syndrome. Circulation. 2008;118:2452–2483.
2. Lipton, P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568.
3. Harukuni, I, Bhardwaj, A. Mechanisms of brain injury after global cerebral ischemia. Neurol Clin. 2006;24:1–21.
4. Sunde, K, et al. Implementation of a standardised treatment protocol for post resuscitation care after out-of-hospital cardiac arrest. Resuscitation. 2007;73:29–39.
5. Hinchey, PR, et al. Improved out-of-hospital cardiac arrest survival after the sequential implementation of 2005 AHA guidelines for compressions, ventilations, and induced hypothermia: The Wake County experience. Ann Emerg Med. 2010;56:348–357.
6. Marzan, AS, Hungerbühler, HJ, Studer, A, Baumgartner, RW, Georgiadis, D. Feasibility and safety of norepinephrine-induced arterial hypertension in acute ischemic stroke. Neurology. 2004;62:1193–1195.
7. O’Neill, JF, Deakin, CD. Do we hyperventilate cardiac arrest patients? Resuscitation. 2007;73:82–85.
8. Geocadin, RG, Koenig, MA, Jia, X, Stevens, RD, Peberdy, MA. Management of brain injury after resuscitation from cardiac arrest. Neurol Clin. 2008;26:487–506.
9. Chaves, C, Caplan, L. Heparin and oral anticoagulants in the treatment of brain ischemia. J Neurol Sci. 2000;173:3–9.
10. Shi, H, Liu, KJ. Cerebral tissue oxygenation and oxidative brain injury during ischemia and reperfusion. Front Biosci. 2007;12:1318–1328.
11. Kilgannon, JH, et al. Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA. 2010;303:2165–2171.
12. Hajat, C, Hajat, S, Sharma, P. Effects of poststroke pyrexia on stroke outcome: A meta-analysis of studies in patients. Stroke. 2000;31:410–414.
13. Hickey, RW, et al. Induced hyperthermia exacerbates neurologic neuronal histologic damage after asphyxial cardiac arrest in rats. Crit Care Med. 2003;31:531–535.
14. Corbett, D, Thornhill, J. Temperature modulation (hypothermic and hyperthermic conditions) and its influence on histologic and behavioral outcomes following cerebral ischemia. Brain Pathol. 2000;10:145–152.
15. Marion, DW. Controlled normothermia in neurologic intensive care. Crit Care Med. 2004;32(2 Suppl):S43–S45.
16. Marshall, SB, Owens, JC, Swan, H. Temporary circulatory occlusion to the brain of the hypothermic dog. AMA Arch Surg. 1956;72:98.
17. Hicks, S, DeFranco, D, Callaway, C. Hypothermia during reperfusion after asphyxial cardiac arrest improves functional recovery and selectively alters stress protein expression. J Cereb Blood Flow Metab. 2000;20:520–530.
18. Yenari, M, et al. Gene therapy and hypothermia for stroke treatment. Ann N Y Acad Sci. 2003;993:54–68.
19. The Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549–556.
20. Bernard, SA, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557–563.
21. Peberdy, MA, et al. Part 9: Post-cardiac arrest care. Circulation. 2010;122(18 Suppl 3):S768–S786.
22. Morrison, LJ, et al. Part 8: Advanced life support. Circulation. 2010;122(16 Suppl 2):S345–S421.
23. Majersik, JJ, et al. Public health impact of full implementation of therapeutic hypothermia after cardiac arrest. Resuscitation. 2008;77:189–194.
24. Bernard, S. Hypothermia after cardiac arrest: How to cool and for how long? Crit Care Med. 2004;32:897–899.
25. Dine, CJ, Abella, BS. Therapeutic hypothermia for neuroprotection. Emerg Med Clin North Am. 2009;27:137–149.
26. Nolan, JP, et al. Therapeutic hypothermia after cardiac arrest: An advisory statement by the Advanced Life Support Task Force of the International Liaison Committee on Resuscitation. Circulation. 2003;108:118–121.
27. Felberg, RA, et al. Hypothermia after cardiac arrest: Feasibility and safety of an external cooling protocol. Circulation. 2001;104:1799–1804.
28. Silfvast, T, Tiainen, M, Poutiainen, E, Roine, RO. Therapeutic hypothermia after prolonged cardiac arrest due to non-coronary causes. Resuscitation. 2003;57:109–112.
29. Bernard, SA, et al. Induction of therapeutic hypothermia by paramedics after resuscitation from out-of-hospital ventricular fibrillation cardiac arrest. Circulation. 2010;122:737–742.
30. Mokhtarani, M, et al. Buspirone and meperidine synergistically reduce the shivering threshold. Anesth Analg. 2001;93:1233–1239.
31. Doufas, AG, et al. Dexmedetomidine and meperidine additively reduce the shivering threshold in humans. Stroke. 2003;34:1218–1223.
32. Kasner, SE, et al. Acetaminophen for altering body temperature in acute stroke: A randomized clinical trial. Stroke. 2002;33:130–135.
33. Katz, LM, Young, A, Frank, JE, Wang, Y, Park, K. Neurotensin-induced hypothermia improves neurologic outcome after hypoxic-ischemia. Crit Care Med. 2004;32:806–810.
34. Guluma, KZ, et al. Effect of endovascular hypothermia on acute ischemic edema: Morphometric analysis of the ICTuS trial. Neurocrit Care. 2008;8:42–47.
35. Wagner, KR, Zuccarello, M. Local brain hypothermia for neuroprotection in stroke treatment and aneurysm repair. Neurol Res. 2005;27:238–245.
36. Wang, H, et al. Rapid and selective cerebral hypothermia achieved using a cooling helmet. J Neurosurg. 2004;100:272–277.
37. Capes, SE, Hunt, D, Malmberg, K, Pathak, P, Gerstein, HC. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview. Stroke. 2001;32:2426–2432.
38. Skrifvars, MB, Pettila, V, Rosenberg, PH, Castren, M. A multiple logistic regression analysis of in-hospital factors related to survival at six months in patients resuscitated from out-of-hospital ventricular fibrillation. Resuscitation. 2003;59:319–328.
39. Bunch, TJ, et al. Long-term outcomes of out-of-hospital cardiac arrest after successful early defibrillation. N Engl J Med. 2003;348:2626–2633.
40. Stiell, I, et al. Health-related quality of life is better for cardiac arrest survivors who received citizen cardiopulmonary resuscitation. Circulation. 2003;108:1939–1944.
41. Granja, C, Cabral, G, Pinto, AT, Costa-Pereira, A. Quality of life 6-months after cardiac arrest. Resuscitation. 2002;55:37–44.
42. van Alem, AP, Waalewijn, RA, Koster, RW, de Vos, R. Assessment of quality of life and cognitive function after out-of-hospital cardiac arrest with successful resuscitation. Am J Cardiol. 2004;93:131–135.
43. Naess, AC, Steen, PA. Long term survival and costs per life year gained after out-of-hospital cardiac arrest. Resuscitation. 2004;60:57–64.
44. Saner, H, Borner Rodriguez, E, Kummer-Bangerter, A, Schuppel, R, von Planta, M. Quality of life in long-term survivors of out-of-hospital cardiac arrest. Resuscitation. 2002;53:7–13.
45. Nichol, G, et al. Regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA. 2008;300:1423–1431.
46. Rossetti, AO, Oddo, M, Logroscino, G, Kaplan, PW. Prognostication after cardiac arrest and hypothermia: A prospective study. Ann Neurol. 2010;67:301–307.
47. Booth, CM, Boone, RH, Tomlinson, G, Detsky, AS. Is this patient dead, vegetative, or severely neurologically impaired? Assessing outcome for comatose survivors of cardiac arrest. JAMA. 2004;291:870–879.
48. Wijdicks, EFM, Hijdra, A, Young, GB, Bassetti, CL, Wiebe, S. Practice parameter: Prediction of outcome in comatose survivors after cardiopulmonary resuscitation (an evidence-based review). Neurology. 2006;67:203–210.
49. Wijdicks, EFM, Varelas, PN, Gronseth, GS, Greer, DM. Evidence-based guideline update: Determining brain death in adults. Neurology. 2010;74:1911–1918.
50. Weiss, N, Galanaud, D, Carpentier, A, Naccache, L, Puybasset, L. Clinical review: Prognostic value of magnetic resonance imaging in acute brain injury and coma. Crit Care. 2007;11:230.
51. Metter, RB, Rittenberger, JC, Guyette, FX, Callaway, CW. Association between a quantitative CT scan measure of brain edema and outcome after cardiac arrest. Resuscitation. 2011;82:1180–1185.