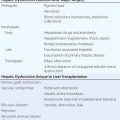
459 |
Peripheral Neuropathy |
Peripheral nerves are composed of sensory, motor, and autonomic elements. Diseases can affect the cell body of a neuron or its peripheral processes, namely the axons or the encasing myelin sheaths. Most peripheral nerves are mixed and contain sensory and motor as well as autonomic fibers. Nerves can be subdivided into three major classes: large myelinated, small myelinated, and small unmyelinated. Motor axons are usually large myelinated fibers that conduct rapidly (approximately 50 m/s). Sensory fibers may be any of the three types. Large-diameter sensory fibers conduct proprioception and vibratory sensation to the brain, while the smaller-diameter myelinated and unmyelinated fibers transmit pain and temperature sensation. Autonomic nerves are also small in diameter. Thus, peripheral neuropathies can impair sensory, motor, or autonomic function, either singly or in combination. Peripheral neuropathies are further classified into those that primarily affect the cell body (e.g., neuronopathy or ganglionopathy), myelin (myelinopathy), and the axon (axonopathy). These different classes of peripheral neuropathies have distinct clinical and electrophysiologic features. This chapter discusses the clinical approach to a patient suspected of having a peripheral neuropathy, as well as specific neuropathies, including hereditary and acquired neuropathies. The inflammatory neuropathies are discussed in Chap. 460.
GENERAL APPROACH
In approaching a patient with a neuropathy, the clinician has three main goals: (1) identify where the lesion is, (2) identify the cause, and (3) determine the proper treatment. The first goal is accomplished by obtaining a thorough history, neurologic examination, and electrodiagnostic and other laboratory studies (Fig. 459-1). While gathering this information, seven key questions are asked (Table 459-1), the answers to which can usually identify the category of pathology that is present (Table 459-2). Despite an extensive evaluation, in approximately half of patients, no etiology is ever found; these patients typically have a predominately sensory polyneuropathy and have been labeled as having idiopathic or cryptogenic sensory polyneuropathy (CSPN).
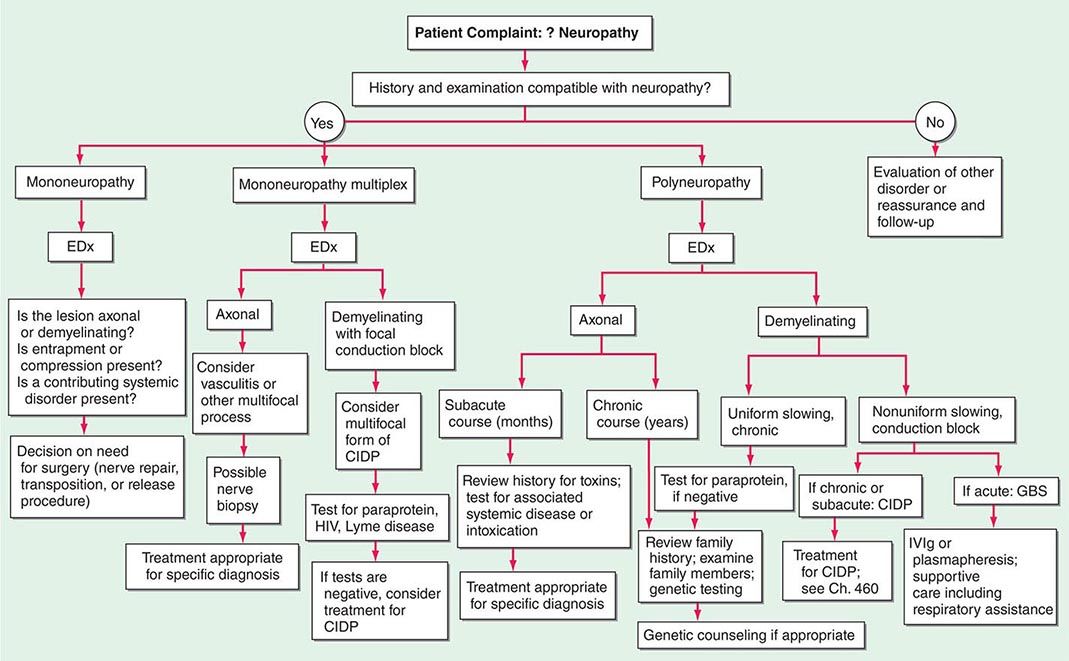
FIGURE 459-1 Approach to the evaluation of peripheral neuropathies. CIDP, chronic inflammatory demyelinating polyradiculoneuropathy; EDx, electrodiagnostic; GBS, Guillain-Barré syndrome; IVIg, intravenous immunoglobulin.
|
APPROACH TO NEUROPATHIC DISORDERS: SEVEN KEY QUESTIONS |
|
PATTERNS OF NEUROPATHIC DISORDERS |
Abbreviations: CIDP, chronic inflammatory demyelinating polyneuropathy; CMT, Charcot-Marie-Tooth disease; CMV, cytomegalovirus; GBS, Guillain-Barré syndrome; HIV, human immunodeficiency virus; HNA, hereditary neuralgic amyotrophy; HNPP, hereditary neuropathy with liability to pressure palsies; HSAN, hereditary sensory and autonomic neuropathy; SMA, spinal muscular atrophy.
INFORMATION FROM THE HISTORY AND PHYSICAL EXAMINATION: SEVEN KEY QUESTIONS (TABLE 459-1)
1. What Systems are Involved? It is important to determine if the patient’s symptoms and signs are motor, sensory, autonomic, or a combination of these. If the patient has only weakness without any evidence of sensory or autonomic dysfunction, a motor neuropathy, neuromuscular junction abnormality, or myopathy should be considered. Some peripheral neuropathies are associated with significant autonomic nervous system dysfunction. Symptoms of autonomic involvement include fainting spells or orthostatic lightheadedness; heat intolerance; or any bowel, bladder, or sexual dysfunction (Chap. 454). There will typically be an orthostatic fall in blood pressure without an appropriate increase in heart rate. Autonomic dysfunction in the absence of diabetes should alert the clinician to the possibility of amyloid polyneuropathy. Rarely, a pandysautonomic syndrome can be the only manifestation of a peripheral neuropathy without other motor or sensory findings. The majority of neuropathies are predominantly sensory in nature.
2. What is the Distribution of Weakness? Delineating the pattern of weakness, if present, is essential for diagnosis, and in this regard two additional questions should be answered: (1) Does the weakness only involve the distal extremity, or is it both proximal and distal? and (2) Is the weakness focal and asymmetric, or is it symmetric? Symmetric proximal and distal weakness is the hallmark of acquired immune demyelinating polyneuropathies, both the acute form (acute inflammatory demyelinating polyneuropathy [AIDP], also known as Guillain-Barré syndrome [GBS]) and the chronic form (chronic inflammatory demyelinating polyneuropathy [CIDP]). The importance of finding symmetric proximal and distal weakness in a patient who presents with both motor and sensory symptoms cannot be overemphasized because this identifies the important subset of patients who may have a treatable acquired demyelinating neuropathic disorder (i.e., AIDP or CIDP).
Findings of an asymmetric or multifocal pattern of weakness narrow the differential diagnosis. Some neuropathic disorders may present with unilateral extremity weakness. In the absence of sensory symptoms and signs, such weakness evolving over weeks or months would be worrisome for motor neuron disease (e.g., amyotrophic lateral sclerosis [ALS]), but it would be important to exclude multifocal motor neuropathy that may be treatable (Chap. 452). In a patient presenting with asymmetric subacute or acute sensory and motor symptoms and signs, radiculopathies, plexopathies, compressive mononeuropathies, or multiple mononeuropathies (e.g., mononeuropathy multiplex) must be considered.
3. What is the Nature of the Sensory Involvement? The patient may have loss of sensation (numbness), altered sensation to touch (hyperpathia or allodynia), or uncomfortable spontaneous sensations (tingling, burning, or aching) (Chap. 31). Neuropathic pain can be burning, dull, and poorly localized (protopathic pain), presumably transmitted by polymodal C nociceptor fibers, or sharp and lancinating (epicritic pain), relayed by A-delta fibers. If pain and temperature perception are lost, while vibratory and position sense are preserved along with muscle strength, deep tendon reflexes, and normal nerve conduction studies, a small-fiber neuropathy is likely. This is important, because the most likely cause of small-fiber neuropathies, when one is identified, is diabetes mellitus or glucose intolerance. Amyloid neuropathy should be considered as well in such cases, but most of these small-fiber neuropathies remain idiopathic in nature despite extensive evaluation.
Severe proprioceptive loss also narrows the differential diagnosis. Affected patients will note imbalance, especially in the dark. A neurologic examination revealing a dramatic loss of proprioception with vibration loss and normal strength should alert the clinician to consider a sensory neuronopathy/ganglionopathy (Table 459-2, Pattern 8). In particular, if this loss is asymmetric or affects the arms more than the legs, this pattern suggests a non-length-dependent process as seen in sensory neuronopathies.
4. Is There Evidence of Upper Motor Neuron Involvement? If the patient presents with symmetric distal sensory symptoms and signs suggestive of a distal sensory neuropathy, but there is additional evidence of symmetric upper motor neuron involvement (Chap. 30), the physician should consider a disorder such as combined system degeneration with neuropathy. The most common cause for this pattern is vitamin B12 deficiency, but other causes of combined system degeneration with neuropathy should be considered (e.g., copper deficiency, HIV infection, severe hepatic disease, adrenomyeloneuropathy).
5. What is the Temporal Evolution? It is important to determine the onset, duration, and evolution of symptoms and signs. Does the disease have an acute (days to 4 weeks), subacute (4–8 weeks), or chronic (>8 weeks) course? Is the course monophasic, progressive, or relapsing? Most neuropathies are insidious and slowly progressive in nature. Neuropathies with acute and subacute presentations include GBS, vasculitis, and radiculopathies related to diabetes or Lyme disease. A relapsing course can be present in CIDP and porphyria.
6. Is There Evidence for a Hereditary Neuropathy? In patients with slowly progressive distal weakness over many years with very little in the way of sensory symptoms yet with significant sensory deficits on clinical examination, the clinician should consider a hereditary neuropathy (e.g., Charcot-Marie-Tooth disease [CMT]). On examination, the feet may show arch and toe abnormalities (high or flat arches, hammertoes); scoliosis may be present. In suspected cases, it may be necessary to perform both neurologic and electrophysiologic studies on family members in addition to the patient.
7. Does the Patient Have Any Other Medical Conditions? It is important to inquire about associated medical conditions (e.g., diabetes mellitus, systemic lupus erythematosus); preceding or concurrent infections (e.g. diarrheal illness preceding GBS); surgeries (e.g., gastric bypass and nutritional neuropathies); medications (toxic neuropathy), including over-the-counter vitamin preparations (B6); alcohol; dietary habits; and use of dentures (e.g., fixatives contain zinc that can lead to copper deficiency).
PATTERN RECOGNITION APPROACH TO NEUROPATHIC DISORDERS
Based on the answers to the seven key questions, neuropathic disorders can be classified into several patterns based on the distribution or pattern of sensory, motor, and autonomic involvement (Table 459-2). Each pattern has a limited differential diagnosis. A final diagnosis is established by using other clues such as the temporal course, presence of other disease states, family history, and information from laboratory studies.
ELECTRODIAGNOSTIC STUDIES
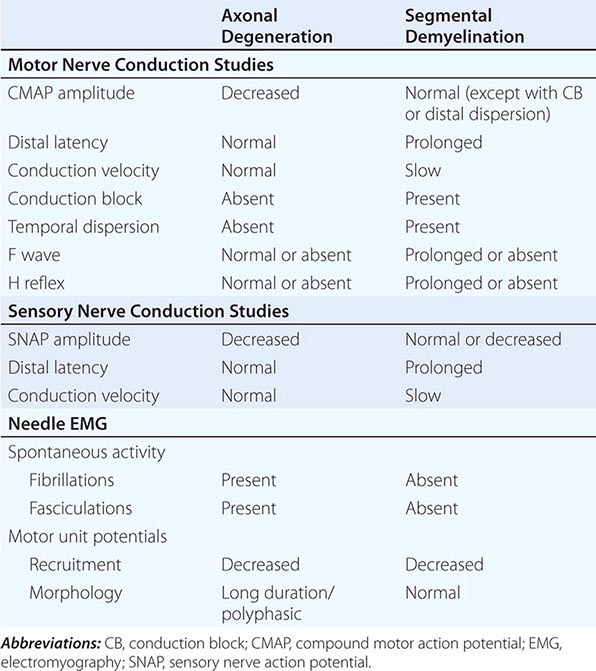
The electrodiagnostic (EDx) evaluation of patients with a suspected peripheral neuropathy consists of nerve conduction studies (NCS) and needle electromyography (EMG). In addition, studies of autonomic function can be valuable. The electrophysiologic data provide additional information about the distribution of the neuropathy that will support or refute the findings from the history and physical examination; they can confirm whether the neuropathic disorder is a mononeuropathy, multiple mononeuropathy (mononeuropathy multiplex), radiculopathy, plexopathy, or generalized polyneuropathy. Similarly, EDx evaluation can ascertain whether the process involves only sensory fibers, motor fibers, autonomic fibers, or a combination of these. Finally, the electrophysiologic data can help distinguish axonopathies from myelinopathies as well as axonal degeneration secondary to ganglionopathies from the more common length-dependent axonopathies.
NCS are most helpful in classifying a neuropathy as being due to axonal degeneration or segmental demyelination (Table 459-3). In general, low-amplitude potentials with relatively preserved distal latencies, conduction velocities, and late potentials, along with fibrillations on needle EMG, suggest an axonal neuropathy. On the other hand, slow conduction velocities, prolonged distal latencies and late potentials, relatively preserved amplitudes, and the absence of fibrillations on needle EMG imply a primary demyelinating neuropathy. The presence of nonuniform slowing of conduction velocity, conduction block, or temporal dispersion further suggests an acquired demyelinating neuropathy (e.g., GBS or CIDP) as opposed to a hereditary demyelinating neuropathy (e.g., CMT type 1).
|
ELECTROPHYSIOLOGIC FEATURES: AXONAL DEGENERATION VERSUS SEGMENTAL DEMYELINATION |
Autonomic studies are used to assess small myelinated (A-delta) or unmyelinated (C) nerve fiber involvement. Such testing includes heart rate response to deep breathing, heart rate, and blood pressure response to both the Valsalva maneuver and tilt-table testing and quantitative sudomotor axon reflex testing (Chap. 454). These studies are particularly useful in patients who have pure small-fiber neuropathy or autonomic neuropathy in which routine NCS are normal.
OTHER IMPORTANT LABORATORY INFORMATION
In patients with generalized symmetric peripheral neuropathy, a standard laboratory evaluation should include a complete blood count, basic chemistries including serum electrolytes and tests of renal and hepatic function, fasting blood glucose (FBS), HbA1c, urinalysis, thyroid function tests, B12, folate, erythrocyte sedimentation rate (ESR), rheumatoid factor, antinuclear antibodies (ANA), serum protein electrophoresis (SPEP) and immunoelectrophoresis or immunofixation, and urine for Bence Jones protein. Quantification of the concentration of serum free light chains and the kappa/lambda ratio is more sensitive than SPEP, immunoelectrophoresis, or immunofixation in looking for a monoclonal gammopathy and therefore should be done if one suspects amyloidosis. A skeletal survey should be performed in patients with acquired demyelinating neuropathies and M-spikes to look for osteosclerotic or lytic lesions. Patients with monoclonal gammopathy should also be referred to a hematologist for consideration of a bone marrow biopsy. An oral glucose tolerance test is indicated in patients with painful sensory neuropathies even if FBS and HbA1c are normal, as the test is abnormal in about one-third of such patients. In addition to the above tests, patients with a mononeuropathy multiplex pattern of involvement should have a vasculitis workup, including antineutrophil cytoplasmic antibodies (ANCA), cryoglobulins, hepatitis serology, Western blot for Lyme disease, HIV, and occasionally a cytomegalovirus (CMV) titer.
There are many autoantibody panels (various antiganglioside antibodies) marketed for screening routine neuropathy patients for a treatable condition. These autoantibodies have no proven clinical utility or added benefit beyond the information obtained from a complete clinical examination and detailed EDx. A heavy metal screen is also not necessary as a screening procedure, unless there is a history of possible exposure or suggestive features on examination (e.g., severe painful sensorimotor and autonomic neuropathy and alopecia—thallium; severe painful sensorimotor neuropathy with or without gastrointestinal [GI] disturbance and Mee’s lines—arsenic; wrist or finger extensor weakness and anemia with basophilic stippling of red blood cells—lead).
In patients with suspected GBS or CIDP, a lumbar puncture is indicated to look for an elevated cerebral spinal fluid (CSF) protein. In idiopathic cases of GBS and CIDP, there should not be pleocytosis in the CSF. If cells are present, one should consider HIV infection, Lyme disease, sarcoidosis, or lymphomatous or leukemic infiltration of nerve roots. Some patients with GBS and CIDP have abnormal liver function tests. In these cases, it is important to also check for hepatitis B and C, HIV, CMV, and Epstein-Barr virus (EBV) infection. In patients with an axonal GBS (by EMG/NCS) or those with a suspicious coinciding history (e.g., unexplained abdominal pain, psychiatric illness, significant autonomic dysfunction), it is reasonable to screen for porphyria.
In patients with a severe sensory ataxia, a sensory ganglionopathy or neuronopathy should be considered. The most common causes of sensory ganglionopathies are Sjögren’s syndrome and a paraneoplastic neuropathy. Neuropathy can be the initial manifestation of Sjögren’s syndrome. Thus, one should always inquire about dry eyes and mouth in patients with sensory signs and symptoms. Further, some patients can manifest sicca complex without full-blown Sjögren’s syndrome. Thus, patients with sensory ataxia should have a senile systemic amyloidosis (SSA) and single strand binding (SSB) in addition to the routine ANA. To work up a possible paraneoplastic sensory ganglionopathy, antineuronal nuclear antibodies (e.g., anti-Hu antibodies) should be obtained (Chap. 122). These antibodies are most commonly seen in patients with small-cell carcinoma of the lung but are seen also in breast, ovarian, lymphoma, and other cancers. Importantly, the paraneoplastic neuropathy can precede the detection of the cancer, and detection of these autoantibodies should lead to a search for malignancy.
NERVE BIOPSIES
Nerve biopsies are now rarely indicated for evaluation of neuropathies. The primary indication for nerve biopsy is suspicion for amyloid neuropathy or vasculitis. In most instances, the abnormalities present on biopsies do not help distinguish one form of peripheral neuropathy from another (beyond what is already apparent by clinical examination and the NCS). Nerve biopsies should only be done if the NCS are abnormal. The sural nerve is most commonly biopsied because it is a pure sensory nerve and biopsy will not result in loss of motor function. In suspected vasculitis, a combination biopsy of a superficial peroneal nerve (pure sensory) and the underlying peroneus brevis muscle obtained from a single small incision increases the diagnostic yield. Tissue can be analyzed by frozen section and paraffin section to assess the supporting structures for evidence of inflammation, vasculitis, or amyloid deposition. Semithin plastic sections, teased fiber preparations, and electron microscopy are used to assess the morphology of the nerve fibers and to distinguish axonopathies from myelinopathies.
SKIN BIOPSIES
Skin biopsies are sometimes used to diagnose a small-fiber neuropathy. Following a punch biopsy of the skin in the distal lower extremity, immunologic staining can be used to measure the density of small unmyelinated fibers. The density of these nerve fibers is reduced in patients with small-fiber neuropathies in whom NCS and routine nerve biopsies are often normal. This technique may allow for an objective measurement in patients with mainly subjective symptoms. However, it adds little to what one already knows from the clinical examination and EDx.
SPECIFIC DISORDERS
HEREDITARY NEUROPATHIES
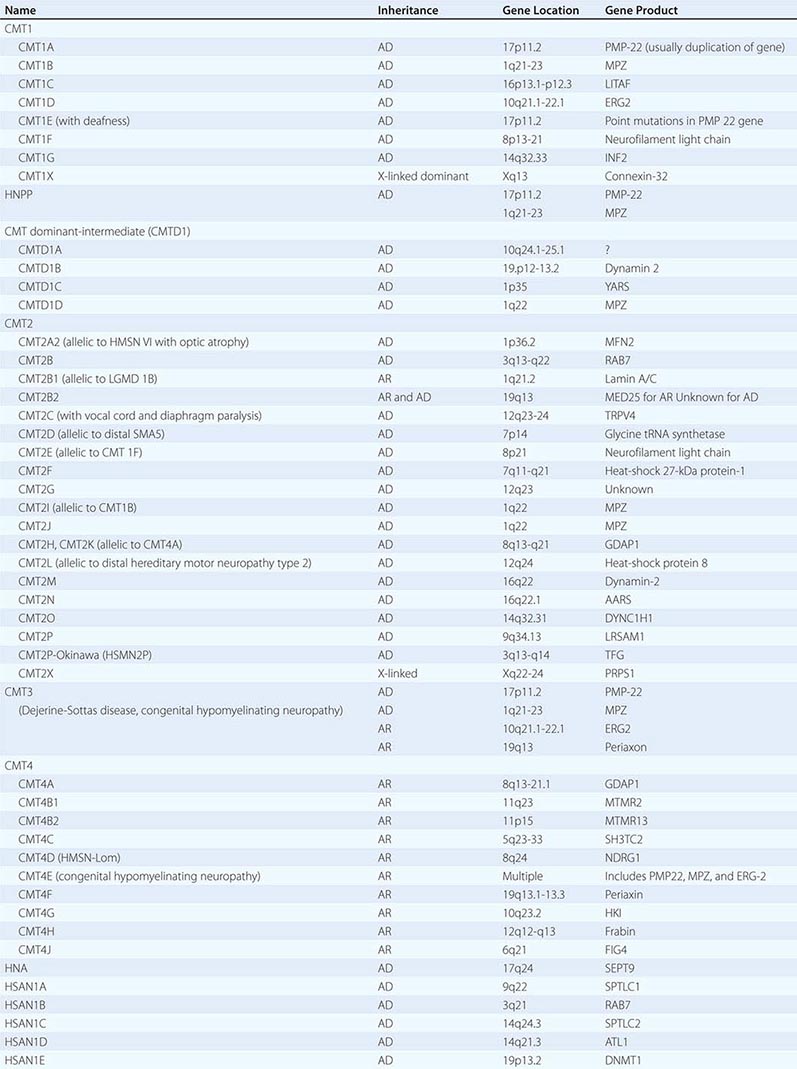
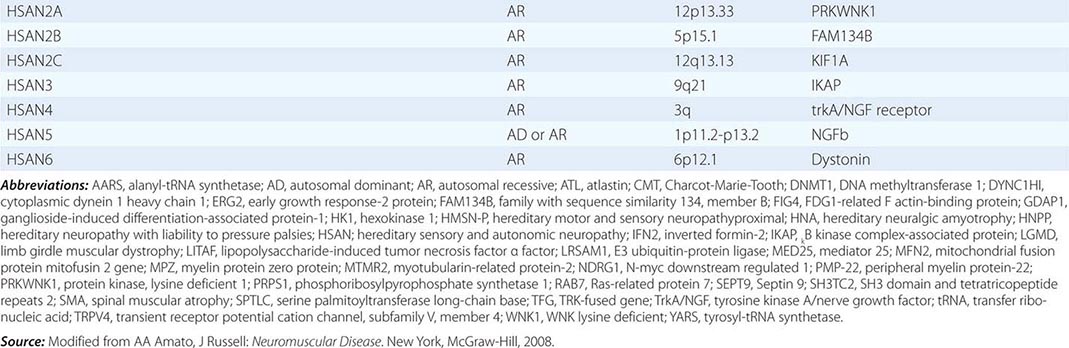
Charcot-Marie-Tooth (CMT) disease is the most common type of hereditary neuropathy. Rather than one disease, CMT is a syndrome of several genetically distinct disorders (Table 459-4). The various subtypes of CMT are classified according to the nerve conduction velocities and predominant pathology (e.g., demyelination or axonal degeneration), inheritance pattern (autosomal dominant, recessive, or X-linked), and the specific mutated genes. Type 1 CMT (or CMT1) refers to inherited demyelinating sensorimotor neuropathies, whereas the axonal sensory neuropathies are classified as CMT2. By definition, motor conduction velocities in the arms are slowed to less than 38 m/s in CMT1 and are greater than 38 m/s in CMT2. However, most cases of CMT1 actually have motor nerve conduction velocities (NCVs) between 20 and 25 m/s. CMT1 and CMT2 usually begin in childhood or early adult life; however, onset later in life can occur, particularly in CMT2. Both are associated with autosomal dominant inheritance, with a few exceptions. CMT3 is an autosomal dominant neuropathy that appears in infancy and is associated with severe demyelination or hypomyelination. CMT4 is an autosomal recessive neuropathy that typically begins in childhood or early adult life. There are no medical therapies for any of the CMTs, but physical and occupational therapy can be beneficial, as can bracing (e.g., ankle-foot orthotics for footdrop) and other orthotic devices.
|
CLASSIFICATION OF CHARCOT-MARIE-TOOTH DISEASE AND RELATED NEUROPATHIES |
CMT1 CMT1 is the most common form of hereditary neuropathy, with the ratio of CMT1:CMT2 being approximately 2:1. Affected individuals usually present in the first to third decade of life with distal leg weakness (e.g., footdrop), although patients may remain asymptomatic even late in life. People with CMT generally do not complain of numbness or tingling, which can be helpful in distinguishing CMT from acquired forms of neuropathy in which sensory symptoms usually predominate. Although usually asymptomatic in this regard, reduced sensation to all modalities is apparent on examination. Muscle stretch reflexes are unobtainable or reduced throughout. There is often atrophy of the muscles below the knee (particularly the anterior compartment), leading to so-called inverted champagne bottle legs.
Motor NCVs are usually in the 20–25 m/s range. Nerve biopsies usually are not performed on patients suspected of having CMT1, because the diagnosis usually can be made by less invasive testing (e.g., NCS and genetic studies). However, when done, the biopsies reveal reduction of myelinated nerve fibers with a predilection for the loss of the large-diameter fibers and Schwann cell proliferation around thinly or demyelinated fibers, forming so-called onion bulbs.
CMT1A is the most common subtype of CMT1, representing 70% of cases, and is caused by a 1.5-megabase (Mb) duplication within chromosome 17p11.2-12 wherein the gene for peripheral myelin protein-22 (PMP-22) lies. This results in patients having three copies of the PMP-22 gene rather than two. This protein accounts for 2–5% of myelin protein and is expressed in compact portions of the peripheral myelin sheath. Approximately 20% of patients with CMT1 have CMT1B, which is caused by mutations in the myelin protein zero (MPZ). CMT1B is for the most part clinically, electrophysiologically, and histologically indistinguishable from CMT1A. MPZ is an integral myelin protein and accounts for more than half of the myelin protein in peripheral nerves. Other forms of CMT1 are much less common and again indistinguishable from one another clinically and electrophysiologically.
CMT2 CMT2 tends to present later in life compared to CMT1. Affected individuals usually become symptomatic in the second decade of life; some cases present earlier in childhood, whereas others remain asymptomatic into late adult life. Clinically, CMT2 is for the most part indistinguishable from CMT1. NCS are helpful in this regard; in contrast to CMT1, the velocities are normal or only slightly slowed. The most common cause of CMT2 is a mutation in the gene for mitofusin 2 (MFN2), which accounts for one-third of CMT2 cases overall. MFN2 localizes to the outer mitochondrial membrane, where it regulates the mitochondrial network architecture by fusion of mitochondria. The other genes associated with CMT2 are much less common.
CMTDI In dominant-intermediate CMTs (CMTDIs), the NCVs are faster than usually seen in CMT1 (e.g., >38 m/s) but slower than in CMT2.
CMT3 CMT3 was originally described by Dejerine and Sottas as a hereditary demyelinating sensorimotor polyneuropathy presenting in infancy or early childhood. Affected children are severely weak. Motor NCVs are markedly slowed, typically 5–10 m/s or less. Most cases of CMT3 are caused by point mutations in the genes for PMP-22, MPZ, or ERG-2, which are also the genes responsible for CMT1.
CMT4 CMT4 is extremely rare and is characterized by a severe, childhood-onset sensorimotor polyneuropathy that is usually inherited in an autosomal recessive fashion. Electrophysiologic and histologic evaluations can show demyelinating or axonal features. CMT4 is genetically heterogenic (Table 459-4).
CMT1X CMT1X is an X-linked dominant disorder with clinical features similar to CMT1 and CMT2, except that the neuropathy is much more severe in men than in women. CMT1X accounts for approximately 10–15% of CMT overall. Men usually present in the first two decades of life with atrophy and weakness of the distal arms and legs, areflexia, pes cavus, and hammertoes. Obligate women carriers are frequently asymptomatic, but can develop signs and symptoms. Onset in women is usually after the second decade of life, and the neuropathy is milder in severity.
NCS reveal features of both demyelination and axonal degeneration that are more severe in men compared to women. In men, motor NCVs in the arms and legs are moderately slowed (in the low to mid 30-m/s range). About 50% of men with CMT1X have motor NCVs between 15 and 35 m/s with about 80% of these falling between 25 and 35 m/s (intermediate slowing). In contrast, about 80% of women with CMT1X have NCVs in the normal range and 20% have NCVs in the intermediate range. CMT1X is caused by mutations in the connexin 32 gene. Connexins are gap junction structural proteins that are important in cell-to-cell communication.
Hereditary Neuropathy with Liability to Pressure Palsies (HNPP) HNPP is an autosomal dominant disorder related to CMT1A. Although CMT1A is usually associated with a 1.5-Mb duplication in chromosome 17p11.2 that results in an extra copy of PMP-22 gene, HNPP is caused by inheritance of the chromosome with the corresponding 1.5-Mb deletion of this segment, and thus affected individuals have only one copy of the PMP-22 gene. Patients usually manifest in the second or third decade of life with painless numbness and weakness in the distribution of single peripheral nerves, although multiple mononeuropathies can occur. Symptomatic mononeuropathy or multiple mononeuropathies are often precipitated by trivial compression of nerve(s) as can occur with wearing a backpack, leaning on the elbows, or crossing one’s legs for even a short period of time. These pressure-related mononeuropathies may take weeks or months to resolve. In addition, some affected individuals manifest with a progressive or relapsing, generalized and symmetric, sensorimotor peripheral neuropathy that resembles CMT.
Hereditary Neuralgic Amyotrophy (HNA) HNA is an autosomal dominant disorder characterized by recurrent attacks of pain, weakness, and sensory loss in the distribution of the brachial plexus often beginning in childhood. These attacks are similar to those seen with idiopathic brachial plexitis (see below). Attacks may occur in the postpartum period, following surgery, or at other times of stress. Most patients recover over several weeks or months. Slightly dysmorphic features, including hypotelorism, epicanthal folds, cleft palate, syndactyly, micrognathia, and facial asymmetry, are evident in some individuals. EDx demonstrate an axonal process. HNA is caused by mutations in septin 9 (SEPT9). Septins may be important in formation of the neuronal cytoskeleton and have a role in cell division, but the mechanism of causing HNA is unclear.
Hereditary Sensory and Autonomic Neuropathy (HSAN) The HSANs are a very rare group of hereditary neuropathies in which sensory and autonomic dysfunction predominates over muscle weakness, unlike CMT, in which motor findings are most prominent (Table 459-4). Nevertheless, affected individuals can develop motor weakness and there can be overlap with CMT. There are no medical therapies available to treat these neuropathies, other than prevention and treatment of mutilating skin and bone lesions.
Of the HSANs, only HSAN1 typically presents in adults. HSAN1 is the most common of the HSANs and is inherited in an autosomal dominant fashion. Affected individuals with HSAN1 usually manifest in the second through fourth decades of life. HSAN1 is associated with the degeneration of small myelinated and unmyelinated nerve fibers leading to severe loss of pain and temperature sensation, deep dermal ulcerations, recurrent osteomyelitis, Charcot joints, bone loss, gross foot and hand deformities, and amputated digits. Although most people with HSAN1 do not complain of numbness, they often describe burning, aching, or lancinating pains. Autonomic neuropathy is not a prominent feature, but bladder dysfunction and reduced sweating in the feet may occur. HSAN1A, which is most common, is caused by mutations in the serine palmitoyltransferase long-chain base 1 (SPTLC1) gene.
OTHER HEREDITARY NEUROPATHIES (TABLE 459-5)
|
RARE HEREDITARY NEUROPATHIES |
FABRY’S DISEASE
Fabry’s disease (angiokeratoma corporis diffusum) is an X-linked dominant disorder. Although men are more commonly and severely affected, women can also show severe signs of the disease. Angiokeratomas are reddish-purple maculopapular lesions that are usually found around the umbilicus, scrotum, inguinal region, and perineum. Burning or lancinating pain in the hands and feet often develops in males in late childhood or early adult life. However, the neuropathy is usually overshadowed by complications arising from the associated premature atherosclerosis (e.g., hypertension, renal failure, cardiac disease, and stroke) that often lead to death by the fifth decade of life. Some patients also manifest primarily with a dilated cardiomyopathy.
Fabry’s disease is caused by mutations in the a-galactosidase gene that leads to the accumulation of ceramide trihexoside in nerves and blood vessels. A decrease in a-galactosidase activity is evident in leukocytes and cultured fibroblasts. Glycolipid granules may be appreciated in ganglion cells of the peripheral and sympathetic nervous systems and in perineurial cells. Enzyme replacement therapy with a-galactosidase B can improve the neuropathy if patients are treated early, before irreversible nerve fiber loss.
ADRENOLEUKODYSTROPHY/ADRENOMYELONEUROPATHY
Adrenoleukodystrophy (ALD) and adrenomyeloneuropathy (AMN) are allelic X-linked dominant disorders caused by mutations in the peroxisomal transmembrane adenosine triphosphate-binding cassette (ABC) transporter gene. Patients with ALD manifest with central nervous system (CNS) abnormalities. However, 30% with mutations in this gene present with the AMN phenotype that typically manifests in the third to fifth decade of life with mild to moderate peripheral neuropathy combined with progressive spastic paraplegia (Chap. 456). Rare patients present with an adult-onset spinocerebellar ataxia or only with adrenal insufficiency.
EDx is suggestive of a primary axonopathy with secondary demyelination. Nerve biopsies demonstrate a loss of myelinated and unmyelinated nerve fibers with lamellar inclusions in the cytoplasm of Schwann cells. Very long chain fatty acid (VLCFA) levels (C24, C25, and C26) are increased in the urine. Laboratory evidence of adrenal insufficiency is evident in approximately two-thirds of patients. The diagnosis can be confirmed by genetic testing.
Adrenal insufficiency is managed by replacement therapy; however, there is no proven effective therapy for the neurologic manifestations of ALD/AMN. Diets low in VLCFAs and supplemented with Lorenzo’s oil (erucic and oleic acids) reduce the levels of VLCFAs and increase the levels of C22 in serum, fibroblasts, and liver; however, several large, open-label trials of Lorenzo’s oil failed to demonstrate efficacy.
REFSUM’S DISEASE
Refsum’s disease can manifest in infancy to early adulthood with the classic tetrad of (1) peripheral neuropathy, (2) retinitis pigmentosa, (3) cerebellar ataxia, and (4) elevated CSF protein concentration. Most affected individuals develop progressive distal sensory loss and weakness in the legs leading to footdrop by their 20s. Subsequently, the proximal leg and arm muscles may become weak. Patients may also develop sensorineural hearing loss, cardiac conduction abnormalities, ichthyosis, and anosmia.
Serum phytanic acid levels are elevated. Sensory and motor NCS reveal reduced amplitudes, prolonged latencies, and slowed conduction velocities. Nerve biopsy demonstrates a loss of myelinated nerve fibers, with remaining axons often thinly myelinated and associated with onion bulb formation.
Refsum disease is genetically heterogeneous but autosomal recessive in nature. Classical Refsum disease with childhood or early adult onset is caused by mutations in the gene that encodes for phytanoyl-CoA α-hydroxylase (PAHX). Less commonly, mutations in the gene encoding peroxin 7 receptor protein (PRX7) are responsible. These mutations lead to the accumulation of phytanic acid in the central and peripheral nervous systems. Refsum’s disease is treated by removing phytanic precursors (phytols: fish oils, dairy products, and ruminant fats) from the diet.
TANGIER DISEASE
Tangier disease is a rare autosomal recessive disorder that can present as (1) asymmetric multiple mononeuropathies, (2) a slowly progressive symmetric polyneuropathy predominantly in the legs, or (3) a pseudo-syringomyelia pattern with dissociated sensory loss (i.e., abnormal pain/temperature perception but preserved position/vibration in the arms [Chap. 456]). The tonsils may appear swollen and yellowish-orange in color, and there may also be splenomegaly and lymphadenopathy.
Tangier disease is caused by mutations in the ATP-binding cassette transporter 1 (ABC1) gene, which leads to markedly reduced levels of high-density lipoprotein (HDL) cholesterol levels, whereas triacylglycerol levels are increased. Nerve biopsies reveal axonal degeneration with demyelination and remyelination. Electron microscopy demonstrates abnormal accumulation of lipid in Schwann cells, particularly those encompassing umyelinated and small myelinated nerves. There is no specific treatment.
PORPHYRIA
Porphyria is a group of inherited disorders caused by defects in heme biosynthesis (Chap. 430). Three forms of porphyria are associated with peripheral neuropathy: acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), and variegate porphyria (VP). The acute neurologic manifestations are similar in each, with the exception that a photosensitive rash is seen with HCP and VP but not in AIP. Attacks of porphyria can be precipitated by certain drugs (usually those metabolized by the P450 system), hormonal changes (e.g., pregnancy, menstrual cycle), and dietary restrictions.
An acute attack of porphyria may begin with sharp abdominal pain. Subsequently, patients may develop agitation, hallucinations, or seizures. Several days later, back and extremity pain followed by weakness ensues, mimicking GBS. Weakness can involve the arms or the legs and can be asymmetric, proximal, or distal in distribution, as well as affecting the face and bulbar musculature. Dysautonomia and signs of sympathetic overactivity are common (e.g., pupillary dilation, tachycardia, and hypertension). Constipation, urinary retention, and incontinence can also be seen.
The CSF protein is typically normal or mildly elevated. Liver function tests and hematologic parameters are usually normal. Some patients are hyponatremic due to inappropriate secretion of antidiuretic hormone (Chap. 401e). The urine may appear brownish in color secondary to the high concentration of porphyrin metabolites. Accumulation of intermediary precursors of heme (i.e., d-aminolevulinic acid, porphobilinogen, uroporphobilinogen, coproporphyrinogen, and protoporphyrinogen) is found in urine. Specific enzyme activities can also be measured in erythrocytes and leukocytes. The primary abnormalities on EDx are marked reductions in compound motor action potential (CMAP) amplitudes and signs of active axonal degeneration on needle EMG.
The porphyrias are inherited in an autosomal dominant fashion. AIP is associated with porphobilinogen deaminase deficiency, HCP is caused by defects in coproporphyrin oxidase, and VP is associated with protoporphyrinogen oxidase deficiency. The pathogenesis of the neuropathy is not completely understood. Treatment with glucose and hematin may reduce the accumulation of heme precursors. Intravenous glucose is started at a rate of 10–20 g/h. If there is no improvement within 24 h, intravenous hematin 2–5 mg/kg per day for 3–14 days should be given.
FAMILIAL AMYLOID POLYNEUROPATHY
Familial amyloid polyneuropathy (FAP) is phenotypically and genetically heterogeneous and is caused by mutations in the genes for transthyretin (TTR), apolipoprotein A1, or gelsolin (Chap. 137). The majority of patients with FAP have mutations in the TTR gene. Amyloid deposition may be evident in abdominal fat pad, rectal, or nerve biopsies. The clinical features, histopathology, and EDx reveal abnormalities consistent with a generalized or multifocal, predominantly axonal but occasionally demyelinating, sensorimotor polyneuropathy.
Patients with TTR-related FAP usually develop insidious onset of numbness and painful paresthesias in the distal lower limbs in the third to fourth decade of life, although some patients develop the disorder later in life. Carpal tunnel syndrome (CTS) is common. Autonomic involvement can be severe, leading to postural hypotension, constipation or persistent diarrhea, erectile dysfunction, and impaired sweating. Amyloid deposition also occurs in the heart, kidneys, liver, and corneas. Patients usually die 10–15 years after the onset of symptoms from cardiac failure or complications from malnutrition. Because the liver produces much of the body’s TTR, liver transplantation has been used to treat FAP related to TTR mutations. Serum TTR levels decrease after transplantation, and improvement in clinical and EDx features has been reported.
Patients with apolipoprotein A1-related FAP (Van Allen type) usually present in the fourth decade with numbness and painful dysesthesias in the distal limbs. Gradually, the symptoms progress, leading to proximal and distal weakness and atrophy. Although autonomic neuropathy is not severe, some patients develop diarrhea, constipation, or gastroparesis. Most patients die from systemic complications of amyloidosis (e.g., renal failure) 12–15 years after the onset of the neuropathy.
Gelsolin-related amyloidosis (Finnish type) is characterized by the combination of lattice corneal dystrophy and multiple cranial neuropathies that usually begin in the third decade of life. Over time, a mild generalized sensorimotor polyneuropathy develops. Autonomic dysfunction does not occur.
ACQUIRED NEUROPATHIES
PRIMARY OR AL AMYLOIDOSIS (SEE CHAP. 137)
Besides FAP, amyloidosis can also be acquired. In primary or AL amyloidosis, the abnormal protein deposition is composed of immunoglobulin light chains. AL amyloidosis occurs in the setting of multiple myeloma, Waldenström’s macroglobulinemia, lymphoma, other plasmacytomas, or lymphoproliferative disorders, or without any other identifiable disease.
Approximately 30% of patients with AL primary amyloidosis present with a polyneuropathy, most typically painful dysesthesias and burning sensations in the feet. However, the trunk can be involved, and some patients manifest with a mononeuropathy multiplex pattern. CTS occurs in 25% of patients and may be the initial manifestation. The neuropathy is slowly progressive, and eventually weakness develops along with large-fiber sensory loss. Most patients develop autonomic involvement with postural hypertension, syncope, bowel and bladder incontinence, constipation, impotence, and impaired sweating. Patients generally die from their systemic illness (renal failure, cardiac disease).
The monoclonal protein may be composed of IgG, IgA, IgM, or only free light chain. Lambda (λ) is more common than κ light chain (>2:1) in AL amyloidosis. The CSF protein is often increased (with normal cell count), and thus the neuropathy may be mistaken for CIDP (Chap. 460). Nerve biopsies reveal axonal degeneration and amyloid deposition in either a globular or diffuse pattern infiltrating the perineurial, epineurial, and endoneurial connected tissue and in blood vessel walls.
The median survival of patients with primary amyloidosis is less than 2 years, with death usually from progressive congestive heart failure or renal failure. Chemotherapy with melphalan, prednisone, and colchicine, to reduce the concentration of monoclonal proteins, and autologous stem cell transplantation may prolong survival, but whether the neuropathy improves is controversial.
DIABETIC NEUROPATHY
Diabetes mellitus (DM) is the most common cause of peripheral neuropathy in developed countries. DM is associated with several types of polyneuropathy: distal symmetric sensory or sensorimotor polyneuropathy, autonomic neuropathy, diabetic neuropathic cachexia, polyradiculoneuropathies, cranial neuropathies, and other mononeuropathies. Risk factors for the development of neuropathy include long-standing, poorly controlled DM and the presence of retinopathy and nephropathy.
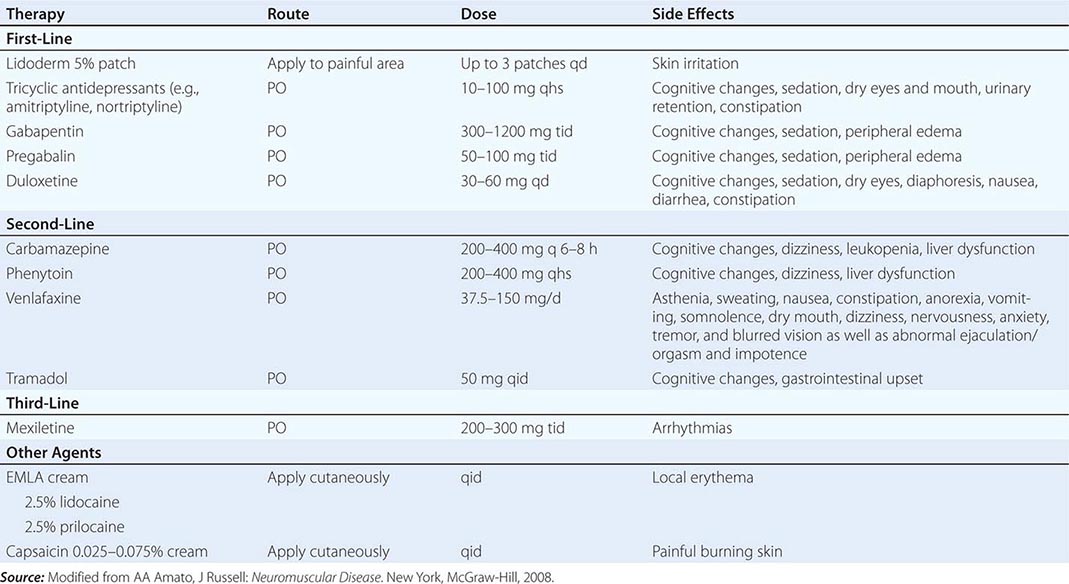
Diabetic Distal Symmetric Sensory and Sensorimotor Polyneuropathy (DSPN) DSPN is the most common form of diabetic neuropathy and manifests as sensory loss beginning in the toes that gradually progresses over time up the legs and into the fingers and arms. When severe, a patient may develop sensory loss in the trunk (chest and abdomen), initially in the midline anteriorly and later extending laterally. Tingling, burning, deep aching pains may also be apparent. NCS usually show reduced amplitudes and mild to moderate slowing of conduction velocities (CVs). Nerve biopsy reveals axonal degeneration, endothelial hyperplasia, and, occasionally, perivascular inflammation. Tight control of glucose can reduce the risk of developing neuropathy or improve the underlying neuropathy. A variety of medications have been used with variable success to treat painful symptoms associated with DSPN, including antiepileptic medications, antidepressants, sodium channel blockers, and other analgesics (Table 459-6).
|
TREATMENT OF PAINFUL SENSORY NEUROPATHIES |
Diabetic Autonomic Neuropathy Autonomic neuropathy is typically seen in combination with DSPN. The autonomic neuropathy can manifest as abnormal sweating, dysfunctional thermoregulation, dry eyes and mouth, pupillary abnormalities, cardiac arrhythmias, postural hypotension, GI abnormalities (e.g., gastroparesis, postprandial bloating, chronic diarrhea or constipation), and genitourinary dysfunction (e.g., impotence, retrograde ejaculation, incontinence). Tests of autonomic function are generally abnormal, including sympathetic skin responses and quantitative sudomotor axon reflex testing. Sensory and motor NCS generally demonstrate features described above with DSPN.
Diabetic Radiculoplexus Neuropathy (Diabetic Amyotrophy or Bruns-Garland Syndrome) Diabetic radiculoplexus neuropathy is the presenting manifestation of DM in approximately one-third of patients. Typically, patients present with severe pain in the low back, hip, and thigh in one leg. Rarely, the diabetic polyradiculoneuropathy begins in both legs at the same time. Atrophy and weakness of proximal and distal muscles in the affected leg become apparent within a few days or weeks. The neuropathy is often accompanied or heralded by severe weight loss. Weakness usually progresses over several weeks or months, but can continue to progress for 18 months or more. Subsequently, there is slow recovery but many are left with residual weakness, sensory loss, and pain. In contrast to the more typical lumbosacral radiculoplexus neuropathy, some patients develop thoracic radiculopathy or, even less commonly, a cervical polyradiculoneuropathy. CSF protein is usually elevated, while the cell count is normal. ESR is often increased. EDx reveals evidence of active denervation in affected proximal and distal muscles in the affected limbs and in paraspinal muscles. Nerve biopsies may demonstrate axonal degeneration along with perivascular inflammation. Patients with severe pain are sometimes treated in the acute period with glucocorticoids, although a randomized controlled trial has yet to be performed, and the natural history of this neuropathy is gradual improvement.
Diabetic Mononeuropathies or Multiple Mononeuropathies The most common mononeuropathies are median neuropathy at the wrist and ulnar neuropathy at the elbow, but peroneal neuropathy at the fibular head, and sciatic, lateral femoral, cutaneous, or cranial neuropathies also occur. In regard to cranial mononeuropathies, seventh nerve palsies are relatively common but may have other, nondiabetic etiologies. In diabetics, a third nerve palsy is most common, followed by sixth nerve, and, less frequently, fourth nerve palsies. Diabetic third nerve palsies are characteristically pupil-sparing (Chap. 39).
HYPOTHYROIDISM
Hypothyroidism is more commonly associated with a proximal myopathy, but some patients develop a neuropathy, most typically CTS. Rarely, a generalized sensory polyneuropathy characterized by painful paresthesias and numbness in both the legs and hands can occur. Treatment is correction of the hypothyroidism.
SJÖGREN’S SYNDROME
Sjögren’s syndrome, characterized by the sicca complex of xerophthalmia, xerostomia, and dryness of other mucous membranes, can be complicated by neuropathy (Chap. 383). Most common is a length-dependent axonal sensorimotor neuropathy characterized mainly by sensory loss in the distal extremities. A pure small-fiber neuropathy or a cranial neuropathy, particularly involving the trigeminal nerve, can also be seen. Sjögren’s syndrome is also associated with sensory neuronopathy/ganglionopathy. Patients with sensory ganglionopathies develop progressive numbness and tingling of the limbs, trunk, and face in a non-length-dependent manner such that symptoms can involve the face or arms more than the legs. The onset can be acute or insidious. Sensory examination demonstrates severe vibratory and proprioceptive loss leading to sensory ataxia.
Patients with neuropathy due to Sjögren’s syndrome may have ANAs, SS-A/Ro, and SS-B/La antibodies in the serum, but most do not. NCS demonstrate reduced amplitudes of sensory studies in the affected limbs. Nerve biopsy demonstrates axonal degeneration. Nonspecific perivascular inflammation may be present, but only rarely is there necrotizing vasculitis. There is no specific treatment for neuropathies related to Sjögren’s syndrome. When vasculitis is suspected, immunosuppressive agents may be beneficial. Occasionally, the sensory neuronopathy/ganglionopathy stabilizes or improves with immunotherapy, such as IVIg.
RHEUMATOID ARTHRITIS
Peripheral neuropathy occurs in at least 50% of patients with rheumatoid arthritis (RA) and may be vasculitic in nature (Chap. 380). Vasculitic neuropathy can present with a mononeuropathy multiplex, a generalized symmetric pattern of involvement, or a combination of these patterns (Chap. 385). Neuropathies may also be due to drugs used to treat the RA (e.g., tumor necrosis blockers, leflunomide). Nerve biopsy often reveals thickening of the epineurial and endoneurial blood vessels as well as perivascular inflammation or vasculitis, with transmural inflammatory cell infiltration and fibrinoid necrosis of vessel walls. The neuropathy often is responsive to immunomodulating therapies.
SYSTEMIC LUPUS ERYTHEMATOSUS (SLE)
Between 2 and 27% of individuals with SLE develop a peripheral neuropathy (Chap. 378). Affected patients typically present with a slowly progressive sensory loss beginning in the feet. Some patients develop burning pain and paresthesias with normal reflexes, and NCS suggest a pure small-fiber neuropathy. Less common are multiple mononeuropathies presumably secondary to necrotizing vasculitis. Rarely, a generalized sensorimotor polyneuropathy meeting clinical, laboratory, electrophysiologic, and histologic criteria for either GBS or CIDP may occur. Immunosuppressive therapy is beneficial in SLE patients with neuropathy due to vasculitis. Immunosuppressive agents are less likely to be effective in patients with a generalized sensory or sensorimotor polyneuropathy without evidence of vasculitis. Patients with a GBS or CIDP-like neuropathy should be treated accordingly (Chap. 385).
SYSTEMIC SCLEROSIS (SCLERODERMA)
A distal symmetric, mainly sensory, polyneuropathy complicates 5–67% of scleroderma cases (Chap. 382). Cranial mononeuropathies can also develop, most commonly of the trigeminal nerve, producing numbness and dysesthesias in the face. Multiple mononeuropathies also occur. The EDx and histologic features of nerve biopsy are those of an axonal sensory greater than motor polyneuropathy.
MIXED CONNECTIVE TISSUE DISEASE (MCTD)
A mild distal axonal sensorimotor polyneuropathy occurs in approximately 10% of patients with MCTD.
SARCOIDOSIS
The peripheral or central nervous system is involved in about 5% of patients with sarcoidosis (Chap. 390). The most common cranial nerve involved is the seventh nerve, which can be affected bilaterally. Some patients develop radiculopathy or polyradiculopathy. With a generalized root involvement, the clinical presentation can mimic GBS or CIDP. Patients can also present with multiple mononeuropathies or a generalized, slowly progressive, sensory greater than motor polyneuropathy. Some have features of a pure small-fiber neuropathy. EDx reveals an axonal neuropathy. Nerve biopsy can reveal noncaseating granulomas infiltrating the endoneurium, perineurium, and epineurium along with lymphocytic necrotizing angiitis. Neurosarcoidosis may respond to treatment with glucocorticoids or other immunosuppressive agents.
HYPEREOSINOPHILIC SYNDROME
Hypereosinophilic syndrome is characterized by eosinophilia associated with various skin, cardiac, hematologic, and neurologic abnormalities. A generalized peripheral neuropathy or a mononeuropathy multiplex occurs in 6–14% of patients.
CELIAC DISEASE (GLUTEN-INDUCED ENTEROPATHY OR NONTROPICAL SPRUE)
Neurologic complications, particularly ataxia and peripheral neuropathy, are estimated to occur in 10% of patients with celiac disease (Chap. 349). A generalized sensorimotor polyneuropathy, pure motor neuropathy, multiple mononeuropathies, autonomic neuropathy, small-fiber neuropathy, and neuromyotonia have all been reported in association with celiac disease or antigliadin/antiendomysial antibodies. Nerve biopsy may reveal a loss of large myelinated fibers. The neuropathy may be secondary to malabsorption of vitamins B12 and E. However, some patients have no appreciable vitamin deficiencies. The pathogenic basis for the neuropathy in these patients is unclear but may be autoimmune in etiology. The neuropathy does not appear to respond to a gluten-free diet. In patients with vitamin B12 or vitamin E deficiency, replacement therapy may improve or stabilize the neuropathy.
INFLAMMATORY BOWEL DISEASE
Ulcerative colitis and Crohn’s disease may be complicated by GBS, CIDP, generalized axonal sensory or sensorimotor polyneuropathy, small-fiber neuropathy, or mononeuropathy (Chap. 351). These neuropathies may be autoimmune, nutritional (e.g., vitamin B12 deficiency), treatment related (e.g., metronidazole), or idiopathic in nature. An acute neuropathy with demyelination resembling GBS, CIDP, or multifocal motor neuropathy may occur in patients treated with tumor necrosis factor α blockers.
UREMIC NEUROPATHY
Approximately 60% of patients with renal failure develop a polyneuropathy characterized by length-dependent numbness, tingling, allodynia, and mild distal weakness. Rarely, a rapidly progressive weakness and sensory loss very similar to GBS can occur that improves with an increase in the intensity of renal dialysis or with transplantation. Mononeuropathies can also occur, the most common of which is CTS. Ischemic monomelic neuropathy (see below) can complicate arteriovenous shunts created in the arm for dialysis. EDx in uremic patients reveals features of a length-dependent, primarily axonal, sensorimotor polyneuropathy. Sural nerve biopsies demonstrate a loss of nerve fibers (particularly large myelinated nerve fibers), active axonal degeneration, and segmental and paranodal demyelination. The sensorimotor polyneuropathy can be stabilized by hemodialysis and improved with successful renal transplantation.
CHRONIC LIVER DISEASE
A generalized sensorimotor neuropathy characterized by numbness, tingling, and minor weakness in the distal aspects of primarily the lower limbs commonly occurs in patients with chronic liver failure. EDx studies are consistent with a sensory greater than motor axonopathy. Sural nerve biopsy reveals both segmental demyelination and axonal loss. It is not known if hepatic failure in isolation can cause peripheral neuropathy, as the majority of patients have liver disease secondary to other disorders, such as alcoholism or viral hepatitis, which can also cause neuropathy.
CRITICAL ILLNESS POLYNEUROPATHY
The most common causes of acute generalized weakness leading to admission to a medical intensive care unit (ICU) are GBS and myasthenia gravis (Chap. 461). However, weakness developing in critically ill patients while in the ICU is usually caused by critical illness polyneuropathy (CIP) or critical illness myopathy (CIM) or, much less commonly, by prolonged neuromuscular blockade. From a clinical and EDx standpoint, it can be quite difficult to distinguish these disorders. Most specialists suggest that CIM is more common. Both CIM and CIP develop as a complication of sepsis and multiple organ failure. They usually present as an inability to wean a patient from a ventilator. A coexisting encephalopathy may limit the neurologic exam, in particular the sensory examination. Muscle stretch reflexes are absent or reduced.
Serum creatine kinase (CK) is usually normal; an elevated serum CK would point to CIM as opposed to CIP. NCS reveal absent or markedly reduced amplitudes of motor and sensory studies in CIP, whereas sensory studies are relatively preserved in CIM. Needle EMG usually reveals profuse positive sharp waves and fibrillation potentials, and it is not unusual in patients with severe weakness to be unable to recruit motor unit action potentials. The pathogenic basis of CIP is not known. Perhaps circulating toxins and metabolic abnormalities associated with sepsis and multiorgan failure impair axonal transport or mitochondrial function, leading to axonal degeneration.
LEPROSY (HANSEN’S DISEASE)
![]() Leprosy, caused by the acid-fast bacteria Mycobacterium leprae, is the most common cause of peripheral neuropathy in Southeast Asia, Africa, and South America (Chap. 203). Clinical manifestations range from tuberculoid leprosy at one end to lepromatous leprosy at the other end of the spectrum, with borderline leprosy in between. Neuropathies are most common in patients with borderline leprosy. Superficial cutaneous nerves of the ears and distal limbs are commonly affected. Mononeuropathies, multiple mononeuropathies, or a slowly progressive symmetric sensorimotor polyneuropathy may develop. Sensory NCS are usually absent in the lower limb and are reduced in amplitude in the arms. Motor NCS may demonstrate reduced amplitudes in affected nerves but occasionally can reveal demyelinating features. Leprosy is usually diagnosed by skin lesion biopsy. Nerve biopsy can also be diagnostic, particularly when there are no apparent skin lesions. The tuberculoid form is characterized by granulomas, and bacilli are not seen. In contrast, with lepromatous leprosy, large numbers of infiltrating bacilli, TH2 lymphocytes, and organism-laden, foamy macrophages with minimal granulomatous infiltration are evident. The bacilli are best appreciated using the Fite stain, where they can be seen as red-staining rods often in clusters free in the endoneurium, within macrophages, or within Schwann cells.
Leprosy, caused by the acid-fast bacteria Mycobacterium leprae, is the most common cause of peripheral neuropathy in Southeast Asia, Africa, and South America (Chap. 203). Clinical manifestations range from tuberculoid leprosy at one end to lepromatous leprosy at the other end of the spectrum, with borderline leprosy in between. Neuropathies are most common in patients with borderline leprosy. Superficial cutaneous nerves of the ears and distal limbs are commonly affected. Mononeuropathies, multiple mononeuropathies, or a slowly progressive symmetric sensorimotor polyneuropathy may develop. Sensory NCS are usually absent in the lower limb and are reduced in amplitude in the arms. Motor NCS may demonstrate reduced amplitudes in affected nerves but occasionally can reveal demyelinating features. Leprosy is usually diagnosed by skin lesion biopsy. Nerve biopsy can also be diagnostic, particularly when there are no apparent skin lesions. The tuberculoid form is characterized by granulomas, and bacilli are not seen. In contrast, with lepromatous leprosy, large numbers of infiltrating bacilli, TH2 lymphocytes, and organism-laden, foamy macrophages with minimal granulomatous infiltration are evident. The bacilli are best appreciated using the Fite stain, where they can be seen as red-staining rods often in clusters free in the endoneurium, within macrophages, or within Schwann cells.
Patients are generally treated with multiple drugs: dapsone, rifampin, and clofazimine. Other medications that are used include thalidomide, pefloxacin, ofloxacin, sparfloxacin, minocycline, and clarithromycin. Patients are generally treated for 2 years. Treatment is sometimes complicated by the so-called reversal reaction, particularly in borderline leprosy. The reversal reaction can occur at any time during treatment and develops because of a shift to the tuberculoid end of the spectrum, with an increase in cellular immunity during treatment. The cellular response is upregulated as evidenced by an increased release of tumor necrosis factor α, interferon γ, and interleukin 2, with new granuloma formation. This can result in an exacerbation of the rash and the neuropathy as well as in appearance of new lesions. High-dose glucocorticoids blunt this adverse reaction and may be used prophylactically at treatment onset in high-risk patients. Erythema nodosum leprosum (ENL) is also treated with glucocorticoids or thalidomide.
LYME DISEASE
Lyme disease is caused by infection with Borrelia burgdorferi, a spirochete usually transmitted by the deer tick Ixodes dammini (Chap. 210). Neurologic complications may develop during the second and third stages of infection. Facial neuropathy is most common and is bilateral in about half of cases, which is rare for idiopathic Bell’s palsy. Involvement of nerves is frequently asymmetric. Some patients present with a polyradiculoneuropathy or multiple mononeuropathies. EDx is suggestive of a primary axonopathy. Nerve biopsies can reveal axonal degeneration with perivascular inflammation. Treatment is with antibiotics (Chap. 210).
DIPHTHERITIC NEUROPATHY
Diphtheria is caused by the bacteria Corynebacterium diphtheriae (Chap. 175). Infected individuals present with flulike symptoms of generalized myalgias, headache, fatigue, low-grade fever, and irritability within a week to 10 days of the exposure. About 20–70% of patients develop a peripheral neuropathy caused by a toxin released by the bacteria. Three to 4 weeks after infection, patients may note decreased sensation in their throat and begin to develop dysphagia, dysarthria, hoarseness, and blurred vision due to impaired accommodation. A generalized polyneuropathy may manifest 2 or 3 months following the initial infection, characterized by numbness, paresthesias, and weakness of the arms and legs and occasionally ventilatory failure. CSF protein can be elevated with or without lymphocytic pleocytosis. EDx suggests a diffuse axonal sensorimotor polyneuropathy. Antitoxin and antibiotics should be given within 48 h of symptom onset. Although early treatment reduces the incidence and severity of some complications (i.e., cardiomyopathy), it does not appear to alter the natural history of the associated peripheral neuropathy. The neuropathy usually resolves after several months.
HUMAN IMMUNODEFICIENCY VIRUS (HIV)
HIV infection can result in a variety of neurologic complications, including peripheral neuropathies (Chap. 226). Approximately 20% of HIV-infected individuals develop a neuropathy either as a direct result of the virus itself, other associated viral infections (e.g., CMV), or neurotoxicity secondary to antiviral medications (see below). The major presentations of peripheral neuropathy associated with HIV infection include (1) distal symmetric polyneuropathy, (2) inflammatory demyelinating polyneuropathy (including both GBS and CIDP), (3) multiple mononeuropathies (e.g., vasculitis, CMV-related), (4) polyradiculopathy (usually CMV-related), (5) autonomic neuropathy, and (6) sensory ganglionitis.
HIV-Related Distal Symmetric Polyneuropathy (DSP) DSP is the most common form of peripheral neuropathy associated with HIV infection and usually is seen in patients with AIDS. It is characterized by numbness and painful paresthesias involving the distal extremities. The pathogenic basis for DSP is unknown but is not due to actual infection of the peripheral nerves. The neuropathy may be immune mediated, perhaps caused by the release of cytokines from surrounding inflammatory cells. Vitamin B12 deficiency may contribute in some instances but is not a major cause of most cases of DSP. Some antiretroviral agents (e.g., dideoxycytidine, dideoxyinosine, stavudine) are also neurotoxic and can cause a painful sensory neuropathy.
HIV-Related Inflammatory Demyelinating Polyradiculoneuropathy Both AIDP and CIDP can occur as a complication of HIV infection. AIDP usually develops at the time of seroconversion, whereas CIDP can occur any time in the course of the infection. Clinical and EDx features are indistinguishable from idiopathic AIDP or CIDP (discussed in Chap. 460). In addition to elevated protein levels, lymphocytic pleocytosis is evident in the CSF, a finding that helps distinguish this HIV-associated polyradiculoneuropathy from idiopathic AIDP/CIDP.
HIV-Related Progressive Polyradiculopathy An acute, progressive lumbosacral polyradiculoneuropathy usually secondary to CMV infection can develop in patients with AIDS. Patients present with severe radicular pain, numbness, and weakness in the legs, which is usually asymmetric. CSF is abnormal, demonstrating an increased protein along with reduced glucose concentration and notably a neutrophilic pleocytosis. EDx studies reveal features of active axonal degeneration. The polyradiculoneuropathy may improve with antiviral therapy.
HIV-Related Multiple Mononeuropathies Multiple mononeuropathies can also develop in patients with HIV infection, usually in the context of AIDS. Weakness, numbness, paresthesias, and pain occur in the distribution of affected nerves. Nerve biopsies can reveal axonal degeneration with necrotizing vasculitis or perivascular inflammation. Glucocorticoid treatment is indicated for vasculitis directly due to HIV infection.
HIV-Related Sensory Neuronopathy/Ganglionopathy Dorsal root ganglionitis is a very rare complication of HIV infection, and neuronopathy can be the presenting manifestation. Patients develop sensory ataxia similar to idiopathic sensory neuronopathy/ganglionopathy. NCS reveal reduced amplitudes or absence of sensory nerve action potentials (SNAPs).
HERPES VARICELLA-ZOSTER VIRUS
Peripheral neuropathy from herpes varicella-zoster (HVZ) infection results from reactivation of latent virus or from a primary infection (Chap. 217). Two-thirds of infections in adults are characterized by dermal zoster in which severe pain and paresthesias develop in a dermatomal region followed within a week or two by a vesicular rash in the same distribution. Weakness in muscles innervated by roots corresponding to the dermatomal distribution of skin lesions occurs in 5–30% of patients. Approximately 25% of affected patients have continued pain (postherpetic neuralgia [PHN]). A large clinical trial demonstrated that vaccination against zoster reduces the incidence of HVZ among vaccine recipients by 51% and reduces the incidence of PHN by 67%. Treatment of PHN is symptomatic (Table 459-6).
CYTOMEGALOVIRUS
CMV can cause an acute lumbosacral polyradiculopathy and multiple mononeuropathies in patients with HIV infection and in other immune deficiency conditions (Chap. 219).
EPSTEIN-BARR VIRUS
EBV infection has been associated with GBS, cranial neuropathies, mononeuropathy multiplex, brachial plexopathy, lumbosacral radiculoplexopathy, and sensory neuronopathies (Chap. 218).
HEPATITIS VIRUSES
Hepatitis B and C can cause multiple mononeuropathies related to vasculitis, AIDP, or CIDP (Chap. 362).
NEUROPATHIES ASSOCIATED WITH MALIGNANCY
Patients with malignancy can develop neuropathies due to (1) a direct effect of the cancer by invasion or compression of the nerves, (2) remote or paraneoplastic effect, (3) a toxic effect of treatment, or (4) as a consequence of immune compromise caused by immunosuppressive medications. The most common associated malignancy is lung cancer, but neuropathies also complicate carcinoma of the breast, ovaries, stomach, colon, rectum, and other organs, including the lymphoproliferative system.
PARANEOPLASTIC SENSORY NEURONOPATHY/GANGLIONOPATHY
Paraneoplastic encephalomyelitis/sensory neuronopathy (PEM/SN) usually complicates small-cell lung carcinoma (Chap. 122). Patients usually present with numbness and paresthesias in the distal extremities that are often asymmetric. The onset can be acute or insidiously progressive. Prominent loss of proprioception leads to sensory ataxia. Weakness can be present, usually secondary to an associated myelitis, motor neuronopathy, or concurrent Lambert-Eaton myasthenic syndrome (LEMS). Many patients also develop confusion, memory loss, depression, hallucinations or seizures, or cerebellar ataxia. Polyclonal antineuronal antibodies (IgG) directed against a 35- to 40-kDa protein or complex of proteins, the so-called Hu antigen, are found in the sera or CSF in the majority of patients with paraneoplastic PEM/SN. CSF may be normal or may demonstrate mild lymphocytic pleocytosis and elevated protein. PEM/SN is probably the result of antigenic similarity between proteins expressed in the tumor cells and neuronal cells, leading to an immune response directed against both cell types. Treatment of the underlying cancer generally does not affect the course of PEM/SN. However, occasional patients may improve following treatment of the tumor. Unfortunately, plasmapheresis, intravenous immunoglobulin, and immunosuppressive agents have not shown benefit.
NEUROPATHY SECONDARY TO TUMOR INFILTRATION
Malignant cells, in particular leukemia and lymphoma, can infiltrate cranial and peripheral nerves, leading to mononeuropathy, mononeuropathy multiplex, polyradiculopathy, plexopathy, or even a generalized symmetric distal or proximal and distal polyneuropathy. Neuropathy related to tumor infiltration is often painful; it can be the presenting manifestation of the cancer or the heralding symptom of a relapse. The neuropathy may improve with treatment of the underlying leukemia or lymphoma or with glucocorticoids.
NEUROPATHY AS A COMPLICATION OF BONE MARROW TRANSPLANTATION
Neuropathies may develop in patients who undergo bone marrow transplantation (BMT) because of the toxic effects of chemotherapy, radiation, infection, or an autoimmune response directed against the peripheral nerves. Peripheral neuropathy in BMT is often associated with graft-versus-host disease (GVHD). Chronic GVHD shares many features with a variety of autoimmune disorders, and it is possible that an immune-mediated response directed against peripheral nerves is responsible. Patients with chronic GVHD may develop cranial neuropathies, sensorimotor polyneuropathies, multiple mononeuropathies, and severe generalized peripheral neuropathies resembling AIDP or CIDP. The neuropathy may improve by increasing the intensity of immunosuppressive or immunomodulating therapy and resolution of the GVHD.
LYMPHOMA
Lymphomas may cause neuropathy by infiltration or direct compression of nerves or by a paraneoplastic process. The neuropathy can be purely sensory or motor, but most commonly is sensorimotor. The pattern of involvement may be symmetric, asymmetric, or multifocal, and the course may be acute, gradually progressive, or relapsing and remitting. EDx can be compatible with either an axonal or demyelinating process. CSF may reveal lymphocytic pleocytosis and an elevated protein. Nerve biopsy may demonstrate endoneurial inflammatory cells in both the infiltrative and the paraneoplastic etiologies. A monoclonal population of cells favors lymphomatous invasion. The neuropathy may respond to treatment of the underlying lymphoma or immunomodulating therapies.
MULTIPLE MYELOMA
Multiple myeloma (MM) usually presents in the fifth to seventh decade of life with fatigue, bone pain, anemia, and hypercalcemia (Chap. 136). Clinical and EDx features of neuropathy occur in as many as 40% of patients. The most common pattern is that of a distal, axonal, sensory, or sensorimotor polyneuropathy. Less frequently, a chronic demyelinating polyradiculoneuropathy may develop (see POEMS, Chap. 460). MM can be complicated by amyloid polyneuropathy and should be considered in patients with painful paresthesias, loss of pinprick and temperature discrimination, and autonomic dysfunction (suggestive of a small-fiber neuropathy) and CTS. Expanding plasmacytomas can compress cranial nerves and spinal roots as well. A monoclonal protein, usually composed of γ or μ heavy chains or κ light chains, may be identified in the serum or urine. EDx usually shows reduced amplitudes with normal or only mildly abnormal distal latencies and conduction velocities. A superimposed median neuropathy at the wrist is common. Abdominal fat pad, rectal, or sural nerve biopsy can be performed to look for amyloid deposition. Unfortunately, the treatment of the underlying MM does not usually affect the course of the neuropathy.
NEUROPATHIES ASSOCIATED WITH MONOCLONAL GAMMOPATHY OF UNCERTAIN SIGNIFICANCE (SEE CHAP. 460)
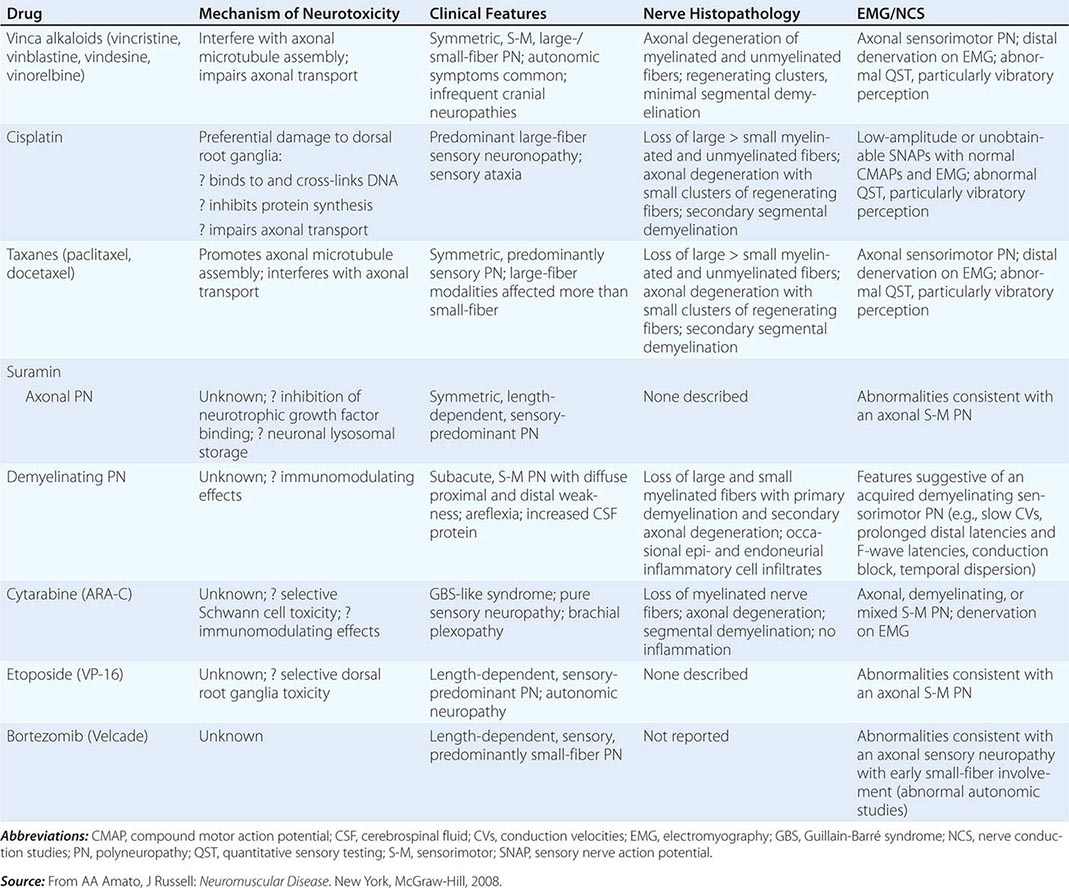
Toxic Neuropathies Secondary to Chemotherapy Many of the commonly used chemotherapy agents can cause a toxic neuropathy (Table 459-7). The mechanisms by which these agents cause toxic neuropathies vary, as does the specific type of neuropathy produced. The risk of developing a toxic neuropathy or more severe neuropathy appears to be greater in patients with a preexisting neuropathy (e.g., Charcot-Marie-Tooth disease, diabetic neuropathy) and those who also take other potentially neurotoxic drugs (e.g., nitrofurantoin, isoniazid, disulfiram, pyridoxine). Chemotherapeutic agents usually cause a sensory greater than motor length-dependent axonal neuropathy or neuronopathy/ganglionopathy.
|
TOXIC NEUROPATHIES SECONDARY TO CHEMOTHERAPY |
OTHER TOXIC NEUROPATHIES
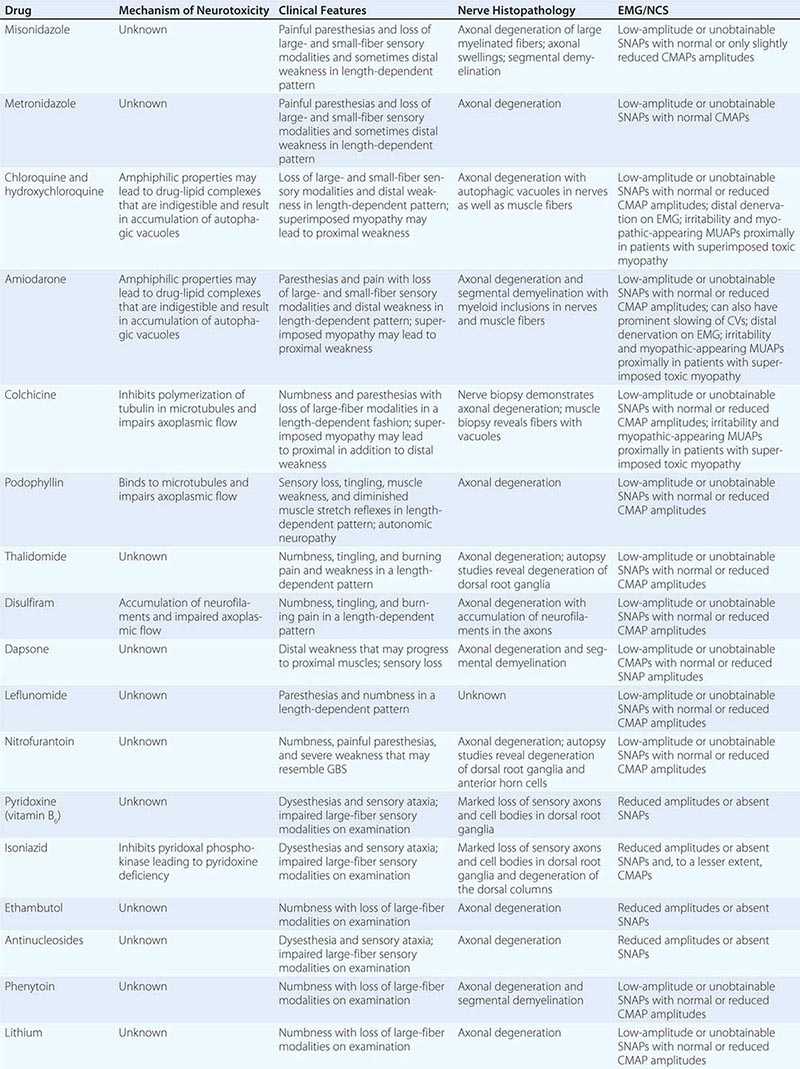
Neuropathies can develop as complications of toxic effects of various drugs and other environmental exposures (Table 459-8). The more common neuropathies associated with these agents are discussed here.
|
TOXIC NEUROPATHIES |
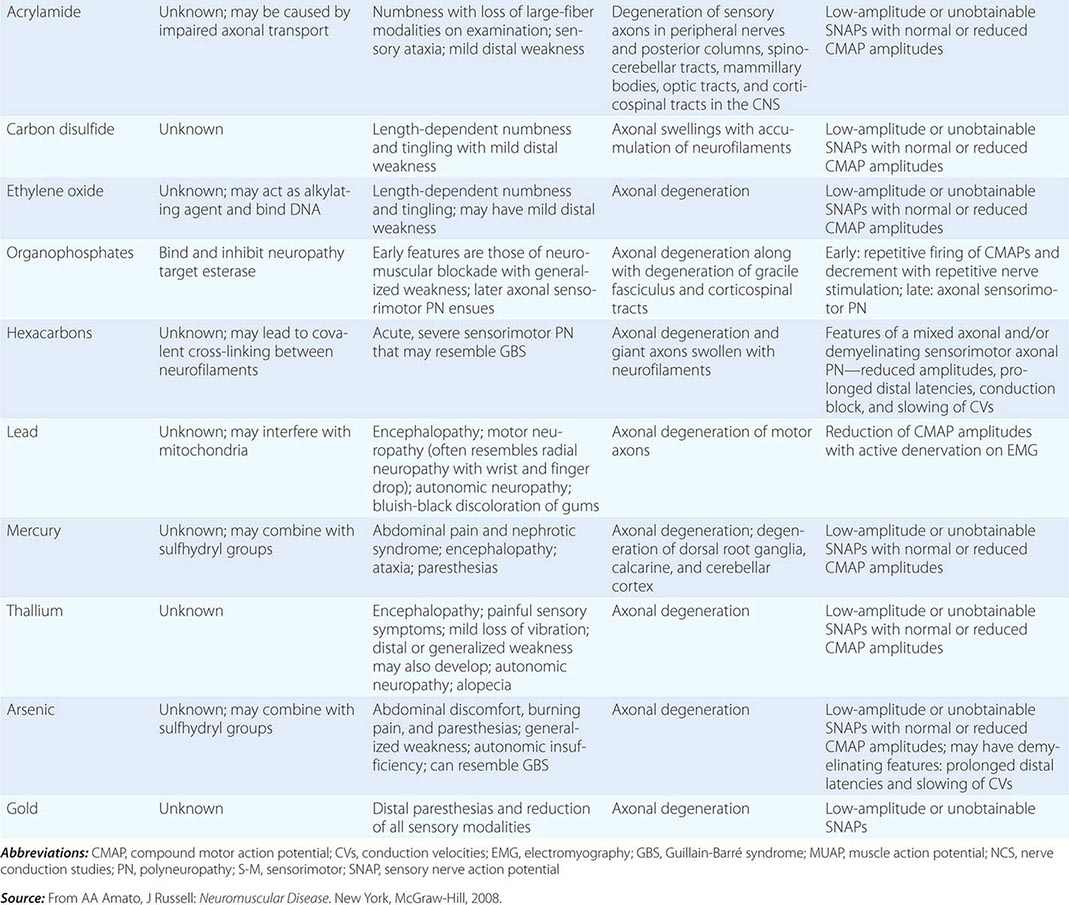
CHLOROQUINE AND HYDROXYCHLOROQUINE
Chloroquine and hydroxychloroquine can cause a toxic myopathy characterized by slowly progressive, painless, proximal weakness and atrophy, which is worse in the legs than the arms. In addition, neuropathy can also develop with or without the myopathy leading to sensory loss and distal weakness. The “neuromyopathy” usually appears in patients taking 500 mg daily for a year or more but has been reported with doses as low as 200 mg/d. Serum CK levels are usually elevated due to the superimposed myopathy. NCS reveal mild slowing of motor and sensory NCVs with a mild to moderate reduction in the amplitudes, although NCS may be normal in patients with only the myopathy. EMG demonstrates myopathic muscle action potentials (MUAPs), increased insertional activity in the form of positive sharp waves, fibrillation potentials, and occasionally myotonic potentials, particularly in the proximal muscles. Neurogenic MUAPs and reduced recruitment are found in more distal muscles. Nerve biopsy demonstrates autophagic vacuoles within Schwann cells. Vacuoles may also be evident in muscle biopsies. The pathogenic basis of the neuropathy is not known but may be related to the amphiphilic properties of the drug. These agents contain both hydrophobic and hydrophilic regions that allow them to interact with the anionic phospholipids of cell membranes and organelles. The drug-lipid complexes may be resistant to digestion by lysosomal enzymes, leading to the formation of autophagic vacuoles filled with myeloid debris that may in turn cause degeneration of nerves and muscle fibers. The signs and symptoms of the neuropathy and myopathy are usually reversible following discontinuation of medication.
AMIODARONE
Amiodarone can cause a neuromyopathy similar to chloroquine and hydroxychloroquine. The neuromyopathy typically appears after patients have taken the medication for 2–3 years. Nerve biopsy demonstrates a combination of segmental demyelination and axonal loss. Electron microscopy reveals lamellar or dense inclusions in Schwann cells, pericytes, and endothelial cells. The inclusions in muscle and nerve biopsies have persisted as long as 2 years following discontinuation of the medication.
COLCHICINE
Colchicine can also cause a neuromyopathy. Patients usually present with proximal weakness and numbness and tingling in the distal extremities. EDx reveals features of an axonal polyneuropathy. Muscle biopsy reveals a vacuolar myopathy, whereas sensory nerves demonstrate axonal degeneration. Colchicine inhibits the polymerization of tubulin into microtubules. The disruption of the microtubules probably leads to defective intracellular movement of important proteins, nutrients, and waste products in muscle and nerves.
THALIDOMIDE
Thalidomide is an immunomodulating agent used to treat multiple myeloma, GVHD, leprosy, and other autoimmune disorders. Thalidomide is associated with severe teratogenic effects as well as peripheral neuropathy that can be dose-limiting. Patients develop numbness, painful tingling, and burning discomfort in the feet and hands and less commonly muscle weakness and atrophy. Even after stopping the drug for 4–6 years, as many as 50% patients continue to have significant symptoms. NCS demonstrate reduced amplitudes or complete absence of SNAPs, with preserved conduction velocities when obtainable. Motor NCS are usually normal. Nerve biopsy reveals a loss of large-diameter myelinated fibers and axonal degeneration. Degeneration of dorsal root ganglion cells has been reported at autopsy.
PYRIDOXINE (VITAMIN B6) TOXICITY
Pyridoxine is an essential vitamin that serves as a coenzyme for transamination and decarboxylation. However, at high doses (116 mg/d), patients can develop a severe sensory neuropathy with dysesthesias and sensory ataxia. NCS reveal absent or markedly reduced SNAP amplitudes with relatively preserved CMAPs. Nerve biopsy reveals axonal loss of fiber at all diameters. Loss of dorsal root ganglion cells with subsequent degeneration of both the peripheral and central sensory tracts have been reported in animal models.
ISONIAZID
One of the most common side effects of isoniazid (INH) is peripheral neuropathy. Standard doses of INH (3–5 mg/kg per day) are associated with a 2% incidence of neuropathy, whereas neuropathy develops in at least 17% of patients taking in excess of 6 mg/kg per day. The elderly, malnourished, and “slow acetylators” are at increased risk for developing the neuropathy. INH inhibits pyridoxal phosphokinase, resulting in pyridoxine deficiency and the neuropathy. Prophylactic administration of pyridoxine 100 mg/d can prevent the neuropathy from developing.
ANTIRETROVIRAL AGENTS
The nucleoside analogues zalcitabine (dideoxycytidine or ddC), didanosine (dideoxyinosine or ddI), stavudine (d4T), lamivudine (3TC), and antiretroviral nucleoside reverse transcriptase inhibitor (NRTI) are used to treat HIV infection. One of the major dose-limiting side effects of these medications is a predominantly sensory, length-dependent, symmetrically painful neuropathy. Zalcitabine (ddC) is the most extensively studied of the nucleoside analogues, and at doses greater than 0.18 mg/kg per day, it is associated with a subacute onset of severe burning and lancinating pains in the feet and hands. NCS reveal decreased amplitudes of the SNAPs with normal motor studies. The nucleoside analogues inhibit mitochondrial DNA polymerase, which is the suspected pathogenic basis for the neuropathy. Because of a “coasting effect,” patients can continue to worsen even 2–3 weeks after stopping the medication. Following dose reduction, improvement in the neuropathy is seen in most patients after several months (mean time about 10 weeks).
HEXACARBONS (n-HEXANE, METHYL n-BUTYL KETONE)/GLUE SNIFFER’S NEUROPATHY
n-Hexane and methyl n-butyl ketone are water-insoluble industrial organic solvents that are also present in some glues. Exposure through inhalation, accidentally or intentionally (glue sniffing), or through skin absorption can lead to a profound subacute sensory and motor polyneuropathy. NCS demonstrate decreased amplitudes of the SNAPs and CMAPs with slightly slow CVs. Nerve biopsy reveals a loss of myelinated fibers and giant axons that are filled with 10-nm neurofilaments. Hexacarbon exposure leads to covalent cross-linking between axonal neurofilaments that result in their aggregation, impaired axonal transport, swelling of the axons, and eventual axonal degeneration.
LEAD
Lead neuropathy is uncommon, but it can be seen in children who accidentally ingest lead-based paints in older buildings and in industrial workers exposed to lead-containing products. The most common presentation of lead poisoning is an encephalopathy; however, symptoms and signs of a primarily motor neuropathy can also occur. The neuropathy is characterized by an insidious and progressive onset of weakness usually beginning in the arms, in particular involving the wrist and finger extensors, resembling a radial neuropathy. Sensation is generally preserved; however, the autonomic nervous system can be affected. Laboratory investigation can reveal a microcytic hypochromic anemia with basophilic stippling of erythrocytes, an elevated serum lead level, and an elevated serum coproporphyrin level. A 24-h urine collection demonstrates elevated levels of lead excretion. The NCS may reveal reduced CMAP amplitudes, while the SNAPs are typically normal. The pathogenic basis may be related to abnormal porphyrin metabolism. The most important principle of management is to remove the source of the exposure. Chelation therapy with calcium disodium ethylene-diaminetetraacetic acid (EDTA), British anti-Lewisite (BAL), and penicillamine also demonstrates variable efficacy.
MERCURY
Mercury toxicity may occur as a result of exposure to either organic or inorganic mercurials. Mercury poisoning presents with paresthesias in hands and feet that progress proximally and may involve the face and tongue. Motor weakness can also develop. CNS symptoms often overshadow the neuropathy. EDx shows features of a primarily axonal sensorimotor polyneuropathy. The primary site of neuromuscular pathology appears to be the dorsal root ganglia. The mainstay of treatment is removing the source of exposure.
THALLIUM
Thallium can exist in a monovalent or trivalent form and is primarily used as a rodenticide. The toxic neuropathy usually manifests as burning paresthesias of the feet, abdominal pain, and vomiting. Increased thirst, sleep disturbances, and psychotic behavior may be noted. Within the first week, patients develop pigmentation of the hair, an acne-like rash in the malar area of the face, and hyperreflexia. By the second and third week, autonomic instability with labile heart rate and blood pressure may be seen. Hyporeflexia and alopecia also occur but may not be evident until the third or fourth week following exposure. With severe intoxication, proximal weakness and involvement of the cranial nerves can occur. Some patients require mechanical ventilation due to respiratory muscle involvement. The lethal dose of thallium is variable, ranging from 8 to 15 mg/kg body weight. Death can result in less than 48 h following a particularly large dose. NCS demonstrate features of a primarily axonal sensorimotor polyneuropathy. With acute intoxication, potassium ferric ferrocyanide II may be effective in preventing absorption of thallium from the gut. However, there may be no benefit once thallium has been absorbed. Unfortunately, chelating agents are not very efficacious. Adequate diuresis is essential to help eliminate thallium from the body without increasing tissue availability from the serum.
ARSENIC
Arsenic is another heavy metal that can cause a toxic sensorimotor polyneuropathy. The neuropathy manifests 5–10 days after ingestion of arsenic and progresses for several weeks, sometimes mimicking GBS. The presenting symptoms are typically an abrupt onset of abdominal discomfort, nausea, vomiting, pain, and diarrhea followed within several days by burning pain in the feet and hands. Examination of the skin can be helpful in the diagnosis as the loss of the superficial epidermal layer results in patchy regions of increased or decreased pigmentation on the skin several weeks after an acute exposure or with chronic low levels of ingestion. Mee’s lines, which are transverse lines at the base of the fingernails and toenails, do not become evident until 1 or 2 months after the exposure. Multiple Mee’s lines may be seen in patients with long fingernails who have had chronic exposure to arsenic. Mee’s lines are not specific for arsenic toxicity as they can also be seen following thallium poisoning. Because arsenic is cleared from blood rapidly, the serum concentration of arsenic is not diagnostically helpful. However, arsenic levels are increased in the urine, hair, and fingernails of patients exposed to arsenic. Anemia with stippling of erythrocytes is common, and occasionally pancytopenia and aplastic anemia can develop. Increased CSF protein levels without pleocytosis can be seen; this can lead to misdiagnosis as GBS. NCS are usually suggestive of an axonal sensorimotor polyneuropathy; however, demyelinating features can be present. Chelation therapy with BAL has yielded inconsistent results; therefore, it is not generally recommended.
NUTRITIONAL NEUROPATHIES
COBALAMIN (VITAMIN B12)
Pernicious anemia is the most common cause of cobalamin deficiency. Other causes include dietary avoidance (vegetarians), gastrectomy, gastric bypass surgery, inflammatory bowel disease, pancreatic insufficiency, bacterial overgrowth, and possibly histamine-2 blockers and proton pump inhibitors. An underappreciated cause of cobalamin deficiency is food-cobalamin malabsorption. This typically occurs in older individuals and results from an inability to adequately absorb cobalamin in food protein. No apparent cause of deficiency is identified in a significant number of patients with cobalamin deficiency. The use of nitrous oxide as an anesthetic agent or as a recreational drug can produce acute cobalamin deficiency neuropathy and subacute combined degeneration.
Complaints of numb hands typically appear before lower extremity paresthesias are noted. A preferential large-fiber sensory loss affecting proprioception and vibration with sparing of small-fiber modalities is present; an unsteady gait reflects sensory ataxia. These features, coupled with diffuse hyperreflexia and absent Achilles reflexes, should always focus attention on the possibility of cobalamin deficiency. Optic atrophy and, in severe cases, behavioral changes ranging from mild irritability and forgetfulness to severe dementia and frank psychosis may appear. The full clinical picture of subacute combined degeneration is uncommon. CNS manifestations, especially pyramidal tract signs, may be missing, and in fact some patients may only exhibit symptoms of peripheral neuropathy.
EDx shows an axonal sensorimotor neuropathy. CNS involvement produces abnormal somatosensory and visual evoked potential latencies. The diagnosis is confirmed by finding reduced serum cobalamin levels. In up to 40% of patients, anemia and macrocytosis are lacking. Serum methylmalonic acid and homocysteine, the metabolites that accumulate when cobalamin-dependent reactions are blocked, are elevated. Antibodies to intrinsic factor are present in approximately 60%, and antiparietal cell antibodies in about 90%, of individuals with pernicious anemia.
Cobalamin deficiency can be treated with various regimens of cobalamin. One typical regimen consists of 1000 μg cyanocobalamin IM weekly for 1 month and monthly thereafter. Patients with food cobalamin malabsorption can absorb free cobalamin and therefore can be treated with oral cobalamin supplementation. An oral cobalamin dose of 1000 μg per day should be sufficient. Treatment for cobalamin deficiency usually does not completely reverse the clinical manifestations, and at least 50% of patients exhibit some permanent neurologic deficit.
THIAMINE DEFICIENCY
Thiamine (vitamin B1) deficiency is an uncommon cause of peripheral neuropathy in developed countries. It is now most often seen as a consequence of chronic alcohol abuse, recurrent vomiting, total parenteral nutrition, and bariatric surgery. Thiamine deficiency polyneuropathy can occur in normal, healthy young adults who do not abuse alcohol but who engage in inappropriately restrictive diets. Thiamine is water-soluble. It is present in most animal and plant tissues, but the greatest sources are unrefined cereal grains, wheat germ, yeast, soybean flour, and pork. Beriberi means “I can’t, I can’t” in Singhalese, the language of natives of what was once part of the Dutch East Indies (now Sri Lanka). Dry beriberi refers to neuropathic symptoms. The term wet beriberi is used when cardiac manifestations predominate (in reference to edema). Beriberi was relatively uncommon until the late 1800s when it became widespread among people for whom rice was a dietary mainstay. This epidemic was due to a new technique of processing rice that removed the germ from the rice shaft, rendering the so-called polished rice deficient in thiamine and other essential nutrients.
Symptoms of neuropathy follow prolonged deficiency. These begin with mild sensory loss and/or burning dysesthesias in the toes and feet and aching and cramping in the lower legs. Pain may be the predominant symptom. With progression, patients develop features of a nonspecific generalized polyneuropathy, with distal sensory loss in the feet and hands.
Blood and urine assays for thiamine are not reliable for diagnosis of deficiency. Erythrocyte transketolase activity and the percentage increase in activity (in vitro) following the addition of thiamine pyrophosphate (TPP) may be more accurate and reliable. EDx shows nonspecific findings of an axonal sensorimotor polyneuropathy. When a diagnosis of thiamine deficiency is made or suspected, thiamine replacement should be provided until proper nutrition is restored. Thiamine is usually given intravenously or intramuscularly at a dose of 100 mg/d. Although cardiac manifestations show a striking response to thiamine replacement, neurologic improvement is usually more variable and less dramatic.
VITAMIN E DEFICIENCY
The term vitamin E is usually used for a-tocopherol, the most active of the four main types of vitamin E. Because vitamin E is present in animal fat, vegetable oils, and various grains, deficiency is usually due to factors other than insufficient intake. Vitamin E deficiency usually occurs secondary to lipid malabsorption or in uncommon disorders of vitamin E transport. One hereditary disorder is abetalipoproteinemia, a rare autosomal dominant disorder characterized by steatorrhea, pigmentary retinopathy, acanthocytosis, and progressive ataxia. Patients with cystic fibrosis may also have vitamin E deficiency secondary to steatorrhea. There are genetic forms of isolated vitamin E deficiency not associated with lipid malabsorption. Vitamin E deficiency may also occur as a consequence of various cholestatic and hepatobiliary disorders as well as short-bowel syndromes resulting from the surgical treatment of intestinal disorders.
Clinical features may not appear until many years after the onset of deficiency. The onset of symptoms tends to be insidious, and progression is slow. The main clinical features are spinocerebellar ataxia and polyneuropathy, thus resembling Friedreich’s ataxia or other spinocerebellar ataxias. Patients manifest progressive ataxia and signs of posterior column dysfunction, such as impaired joint position and vibratory sensation. Because of the polyneuropathy, there is hyporeflexia, but plantar responses may be extensor as a result of the spinal cord involvement. Other neurologic manifestations may include ophthalmoplegia, pigmented retinopathy, night blindness, dysarthria, pseudoathetosis, dystonia, and tremor. Vitamin E deficiency may present as an isolated polyneuropathy, but this is very rare. The yield of checking serum vitamin E levels in patients with isolated polyneuropathy is extremely low, and this test should not be part of routine practice.
Diagnosis is made by measuring a-tocopherol levels in the serum. EDx shows features of an axonal neuropathy. Treatment is replacement with oral vitamin E, but high doses are not needed. For patients with isolated vitamin E deficiency, treatment consists of 1500–6000 IU/d in divided doses.
VITAMIN B6 DEFICIENCY
Vitamin B6, or pyridoxine, can produce neuropathic manifestations from both deficiency and toxicity. Vitamin B6 toxicity was discussed above. Vitamin B6 deficiency is most commonly seen in patients treated with isoniazid or hydralazine. The polyneuropathy of vitamin B6 is nonspecific, manifesting as a generalized axonal sensorimotor polyneuropathy. Vitamin B6 deficiency can be detected by direct assay. Vitamin B6 supplementation with 50–100 mg/d is suggested for patients being treated with isoniazid or hydralazine. This same dose is appropriate for replacement in cases of nutritional deficiency.
PELLAGRA (NIACIN DEFICIENCY)
Pellagra is produced by deficiency of niacin. Although pellagra may be seen in alcoholics, this disorder has essentially been eradicated in most Western countries by means of enriching bread with niacin. Nevertheless, pellagra continues to be a problem in a number of underdeveloped regions, particularly in Asia and Africa, where corn is the main source of carbohydrate. Neurologic manifestations are variable; abnormalities can develop in the brain and spinal cord as well as peripheral nerves. When peripheral nerves are involved, the neuropathy is usually mild and resembles beriberi. Treatment is with niacin 40–250 mg/d.
COPPER DEFICIENCY
A syndrome that has only recently been described is myeloneuropathy secondary to copper deficiency. Most patients present with lower limb paresthesias, weakness, spasticity, and gait difficulties. Large-fiber sensory function is impaired, reflexes are brisk, and plantar responses are extensor. In some cases, light touch and pinprick sensation are affected, and NCS indicate sensorimotor axonal polyneuropathy in addition to myelopathy.
Hematologic abnormalities are a known complication of copper deficiency; these can include microcytic anemia, neutropenia, and occasionally pancytopenia. Because copper is absorbed in the stomach and proximal jejunum, many cases of copper deficiency are in the setting of prior gastric surgery. Excess zinc is an established cause of copper deficiency. Zinc upregulates enterocyte production of metallothionine, which results in decreased absorption of copper. Excessive dietary zinc supplements or denture cream containing zinc can produce this clinical picture. Other potential causes of copper deficiency include malnutrition, prematurity, total parenteral nutrition, and ingestion of copper-chelating agents.
Following oral or IV copper replacement, some patients show neurologic improvement, but this may take many months or not occur at all. Replacement consists of oral copper sulfate or gluconate 2 mg one to three times a day. If oral copper replacement is not effective, elemental copper in the copper sulfate or copper chloride forms can be given as 2 mg IV daily for 3–5 days, then weekly for 1–2 months until copper levels normalize. Thereafter, oral daily copper therapy can be resumed. In contrast to the neurologic manifestations, most of the hematologic indices completely normalize in response to copper replacement therapy.
NEUROPATHY ASSOCIATED WITH GASTRIC SURGERY
Polyneuropathy may occur following gastric surgery for ulcer, cancer, or weight reduction. This usually occurs in the context of rapid, significant weight loss and recurrent, protracted vomiting. The clinical picture is one of acute or subacute sensory loss and weakness. Neuropathy following weight loss surgery usually occurs in the first several months after surgery. Weight reduction surgical procedures include gastrojejunostomy, gastric stapling, vertical banded gastroplasty, and gastrectomy with Roux-en-Y anastomosis. The initial manifestations are usually numbness and paresthesias in the feet. In many cases, no specific nutritional deficiency factor is identified.
Management consists of parenteral vitamin supplementation, especially including thiamine. Improvement has been observed following supplementation, parenteral nutritional support, and reversal of the surgical bypass. The duration and severity of deficits before identification and treatment of neuropathy are important predictors of final outcome.
CRYPTOGENIC (IDIOPATHIC) SENSORY AND SENSORIMOTOR POLYNEUROPATHY
CSPN is a diagnosis of exclusion, established after a careful medical, family, and social history; neurologic examination; and directed laboratory testing. Despite extensive evaluation, the cause of polyneuropathy in as many as 50% of all patients is idiopathic. CSPN should be considered a distinct diagnostic subset of peripheral neuropathy. The onset of CSPN is predominantly in the sixth and seventh decades. Patients complain of distal numbness, tingling, and often burning pain that invariably begins in the feet and may eventually involve the fingers and hands. Patients exhibit a distal sensory loss to pinprick, touch, and vibration in the toes and feet, and occasionally in the fingers. It is uncommon to see significant proprioception deficits, even though patients may complain of gait unsteadiness. However, tandem gait may be abnormal in a minority of cases. Neither subjective nor objective evidence of weakness is a prominent feature. Most patients have evidence of both large- and small-fiber loss on neurologic exam and EDx. Approximately 10% of patients have only evidence of small-fiber involvement. The ankle muscle stretch reflex is frequently absent, but in cases with predominantly small-fiber loss, this may be preserved. The EDx findings range from isolated sensory nerve action potential abnormalities (usually with loss of amplitude), to evidence for an axonal sensorimotor neuropathy, to a completely normal study (if primarily small fibers are involved). Therapy primarily involves the control of neuropathic pain (Table 459-6) if present. These drugs should not be used if the patient has only numbness and tingling but no pain.
Although no treatment is available that can reverse an idiopathic distal peripheral neuropathy, the prognosis is good. Progression often does not occur or is minimal, with sensory symptoms and signs progressing proximally up to the knees and elbows. The disorder does not lead to significant motor disability over time. The relatively benign course of this disorder should be explained to patients.
MONONEUROPATHIES/PLEXOPATHIES/RADICULOPATHIES
MEDIAN NEUROPATHY
CTS is a compression of the median nerve in the carpal tunnel at the wrist. The median nerve enters the hand through the carpal tunnel by coursing under the transverse carpal ligament. The symptoms of CTS consist of numbness and paresthesias variably in the thumb, index, middle, and half of the ring finger. At times, the paresthesias can include the entire hand and extend into the forearm or upper arm or can be isolated to one or two fingers. Pain is another common symptom and can be located in the hand and forearm and, at times, in the proximal arm. CTS is common and often misdiagnosed as thoracic outlet syndrome. The signs of CTS are decreased sensation in the median nerve distribution; reproduction of the sensation of tingling when a percussion hammer is tapped over the wrist (Tinel’s sign) or the wrist is flexed for 30–60 s (Phalen’s sign); and weakness of thumb opposition and abduction. EDx is extremely sensitive and shows slowing of sensory and, to a lesser extent, motor median potentials across the wrist. Treatment options consist of avoidance of precipitating activities; control of underlying systemic-associated conditions if present; nonsteroidal anti-inflammatory medications; neutral (volar) position wrist splints, especially for night use; glucocorticoid/anesthetic injection into the carpal tunnel; and surgical decompression by dividing the transverse carpal ligament. The surgical option should be considered if there is a poor response to nonsurgical treatments; if there is thenar muscle atrophy and/or weakness; and if there are significant denervation potentials on EMG.
Other proximal median neuropathies are very uncommon and include the pronator teres syndrome and anterior interosseous neuropathy. These often occur as a partial form of brachial plexitis.
ULNAR NEUROPATHY AT THE ELBOW—“CUBITAL TUNNEL SYNDROME”
The ulnar nerve passes through the condylar groove between the medial epicondyle and the olecranon. Symptoms consist of paresthesias, tingling, and numbness in the medial hand and half of the fourth and the entire fifth fingers, pain at the elbow or forearm, and weakness. Signs consist of decreased sensation in an ulnar distribution, Tinel’s sign at the elbow, and weakness and atrophy of ulnar-innervated hand muscles. The Froment sign indicates thumb adductor weakness and consists of flexion of the thumb at the interphalangeal joint when attempting to oppose the thumb against the lateral border of the second digit. EDx may show slowing of ulnar motor NCV across the elbow with prolonged ulnar sensory latencies. Treatment consists of avoiding aggravating factors, using elbow pads, and surgery to decompress the nerve in the cubital tunnel. Ulnar neuropathies can also rarely occur at the wrist in the ulnar (Guyon) canal or in the hand, usually after trauma.
RADIAL NEUROPATHY
The radial nerve winds around the proximal humerus in the spiral groove and proceeds down the lateral arm and enters the forearm, dividing into the posterior interosseous nerve and superficial nerve. The symptoms and signs consist of wristdrop; finger extension weakness; thumb abduction weakness; and sensory loss in the dorsal web between the thumb and index finger. Triceps and brachioradialis strength is often normal, and triceps reflex is often intact. Most cases of radial neuropathy are transient compressive (neuropraxic) injuries that recover spontaneously in 6–8 weeks. If there has been prolonged compression and severe axonal damage, it may take several months to recover. Treatment consists of cock-up wrist and finger splints, avoiding further compression, and physical therapy to avoid flexion contracture. If there is no improvement in 2–3 weeks, an EDx study is recommended to confirm the clinical diagnosis and determine the degree of severity.
LATERAL FEMORAL CUTANEOUS NEUROPATHY (MERALGIA PARESTHETICA)
The lateral femoral cutaneous nerve arises from the upper lumbar plexus (spinal levels L2/3), crosses through the inguinal ligament near its attachment to the iliac bone, and supplies sensation to the anterior lateral thigh. The neuropathy affecting this nerve is also known as meralgia paresthetica. Symptoms and signs consist of paresthesias, numbness, and occasionally pain in the lateral thigh. Symptoms are increased by standing or walking and are relieved by sitting. There is normal strength, and knee reflexes are intact. The diagnosis is clinical, and further tests usually are not performed. EDx is only needed to rule out lumbar plexopathy, radiculopathy, or femoral neuropathy. If the symptoms and signs are classic, EMG is not necessary. Symptoms often resolve spontaneously over weeks or months, but the patient may be left with permanent numbness. Treatment consists of weight loss and avoiding tight belts. Analgesics in the form of a lidocaine patch, nonsteroidal agents, and occasionally medications for neuropathic pain can be used (Table 459-6). Rarely, locally injecting the nerve with an anesthetic can be tried. There is no role for surgery.
FEMORAL NEUROPATHY
Femoral neuropathies can arise as complications of retroperitoneal hematoma, lithotomy positioning, hip arthroplasty or dislocation, iliac artery occlusion, femoral arterial procedures, infiltration by hematogenous malignancy, penetrating groin trauma, pelvic surgery including hysterectomy and renal transplantation, and diabetes (a partial form of lumbosacral diabetic plexopathy); some cases are idiopathic. Patients with femoral neuropathy have difficulty extending their knee and flexing the hip. Sensory symptoms occurring either on the anterior thigh and/or medial leg occur in only half of reported cases. A prominent painful component is the exception rather than the rule, may be delayed, and is often self-limited in nature. The quadriceps (patellar) reflex is diminished.
SCIATIC NEUROPATHY
Sciatic neuropathies commonly complicate hip arthroplasty, pelvic procedures in which patients are placed in a prolonged lithotomy position, trauma, hematomas, tumor infiltration, and vasculitis. In addition, many sciatic neuropathies are idiopathic. Weakness may involve all motions of the ankles and toes as well as flexion of the leg at the knee; abduction and extension of the thigh at the hip are spared. Sensory loss occurs in the entire foot and the distal lateral leg. The ankle jerk and on occasion the internal hamstring reflex are diminished or more typically absent on the affected side. The peroneal subdivision of the sciatic nerve is typically involved disproportionately to the tibial counterpart. Thus, patients may have only ankle dorsiflexion and eversion weakness with sparing of knee flexion, ankle inversion, and plantar flexion; these features can lead to misdiagnosis of a common peroneal neuropathy.
PERONEAL NEUROPATHY
The sciatic nerve divides at the distal femur into the tibial and peroneal nerve. The common peroneal nerve passes posterior and laterally around the fibular head, under the fibular tunnel. It then divides into the superficial peroneal nerve, which supplies the ankle evertor muscles and sensation over the anterolateral distal leg and dorsum of the foot, and the deep peroneal nerve, which supplies ankle dorsiflexors and toe extensor muscles and a small area of sensation dorsally in the area of the first and second toes.
Symptoms and signs consist of footdrop (ankle dorsiflexion, toe extension, and ankle eversion weakness) and variable sensory loss, which may involve the superficial and deep peroneal pattern. There is usually no pain. Onset may be on awakening in the morning. Peroneal neuropathy needs to be distinguished from L5 radiculopathy. In L5 radiculopathy, ankle invertors and evertors are weak and needle EMG reveals denervation. EDx can help localize the lesion. Peroneal motor conduction velocity shows slowing and amplitude drop across the fibular head. Management consists of rapid weight loss and avoiding leg crossing. Footdrop is treated with an ankle brace. A knee pad can be worn over the lateral knee to avoid further compression. Most cases spontaneously resolve over weeks or months.
RADICULOPATHIES
Radiculopathies are most often due to compression from degenerative joint disease and herniated disks, but there are a number of unusual etiologies (Table 459-9). Degenerative spine disease affects a number of different structures, which narrow the diameter of the neural foramen or canal of the spinal column and compromise nerve root integrity; these are discussed in detail in Chap. 22.
|
CAUSES OF RADICULOPATHY |
PLEXOPATHIES
BRACHIAL PLEXUS
The brachial plexus is composed of three trunks (upper, middle, and lower), with two divisions (anterior and posterior) per trunk (Fig. 459-2). Subsequently, the trunks divide into three cords (medial, lateral, and posterior), and from these arise the multiple terminal nerves innervating the arm. The anterior primary rami of C5 and C6 fuse to form the upper trunk; the anterior primary ramus of C7 continues as the middle trunk, while the anterior rami of C8 and T1 join to form the lower trunk. There are several disorders commonly associated with brachial plexopathy.
FIGURE 459-2 Brachial plexus anatomy. L, lateral; M, medial; P, posterior. (From J Goodgold: Anatomical Correlates of Clinical Electromyography. Baltimore, Williams and Wilkins, 1974, p. 126, with permission.)
Immune-Mediated Brachial Plexus Neuropathy Immune-mediated brachial plexus neuropathy (IBPN) goes by various terms, including acute brachial plexitis, neuralgic amyotrophy, and Parsonage-Turner syndrome. IBPN usually presents with an acute onset of severe pain in the shoulder region. The intense pain usually lasts several days to a few weeks, but a dull ache can persist. Individuals who are affected may not appreciate weakness of the arm early in the course because the pain limits movement. However, as the pain dissipates, weakness and often sensory loss are appreciated. Attacks can occasionally recur.
Clinical findings are dependent on the distribution of involvement (e.g., specific trunk, divisions, cords, or terminal nerves). The most common pattern of IBPN involves the upper trunk or a single or multiple mononeuropathies primarily involving the suprascapular, long thoracic, or axillary nerves. Additionally, the phrenic and anterior interosseous nerves may be concomitantly affected. Any of these nerves may also be affected in isolation. EDx is useful to confirm and localize the site(s) of involvement. Empirical treatment of severe pain with glucocorticoids is often used in the acute period.
Brachial Plexopathies Associated with Neoplasms Neoplasms involving the brachial plexus may be primary nerve tumors, local cancers expanding into the plexus (e.g., Pancoast lung tumor or lymphoma), and metastatic tumors. Primary brachial plexus tumors are less common than the secondary tumors and include schwannomas, neurinomas, and neurofibromas. Secondary tumors affecting the brachial plexus are more common and are always malignant. These may arise from local tumors, expanding into the plexus. For example, a Pancoast tumor of the upper lobe of the lung may invade or compress the lower trunk, whereas a primary lymphoma arising from the cervical or axillary lymph nodes may also infiltrate the plexus. Pancoast tumors typically present as an insidious onset of pain in the upper arm, sensory disturbance in the medial aspect of the forearm and hand, and weakness and atrophy of the intrinsic hand muscles along with an ipsilateral Horner’s syndrome. Chest computed tomography (CT) scans or magnetic resonance imaging (MRI) can demonstrate extension of the tumor into the plexus. Metastatic involvement of the brachial plexus may occur with spread of breast cancer into the axillary lymph nodes with local spread into the nearby nerves.
Perioperative Plexopathies (Median Sternotomy) The most common surgical procedures associated with brachial plexopathy as a complication are those that involve median sternotomies (e.g., open-heart surgeries and thoracotomies). Brachial plexopathies occur in as many as 5% of patients following a median sternotomy and typically affect the lower trunk. Thus, individuals manifest with sensory disturbance affecting the medial aspect of forearm and hand along with weakness of the intrinsic hand muscles. The mechanism is related to the stretch of the lower trunk, so most individuals who are affected recover within a few months.
Lumbosacral Plexus The lumbar plexus arises from the ventral primary rami of the first to the fourth lumbar spinal nerves (Fig. 459-3). These nerves pass downward and laterally from the vertebral column within the psoas major muscle. The femoral nerve derives from the dorsal branches of the second to the fourth lumbar ventral rami. The obturator nerve arises from the ventral branches of the same lumbar rami. The lumbar plexus communicates with the sacral plexus by the lumbosacral trunk, which contains some fibers from the fourth and all of the fibers from the fifth lumbar ventral rami (Fig. 459-4).
FIGURE 459-3 Lumbar plexus. Posterior divisions are in orange, and anterior divisions are in yellow. (From J Goodgold: Anatomical Correlates of Clinical Electromyography. Baltimore, Williams and Wilkins, 1974, p. 126, with permission.)
FIGURE 459-4 Lumbosacral plexus. Posterior divisions are in orange, and anterior divisions are in yellow. (From J Goodgold: Anatomical Correlates of Clinical Electromyography. Baltimore, Williams and Wilkins, 1974, p. 126, with permission.)
The sacral plexus is the part of the lumbosacral plexus that is formed by the union of the lumbosacral trunk with the ventral rami of the first to fourth sacral nerves. The plexus lies on the posterior and posterolateral wall of the pelvis with its components converging toward the sciatic notch. The lateral trunk of the sciatic nerve (which forms the common peroneal nerve) arises from the union of the dorsal branches of the lumbosacral trunk (L4, L5) and the dorsal branches of the S1 and S2 spinal nerve ventral rami. The medial trunk of the sciatic nerve (which forms the tibial nerve) derives from the ventral branches of the same ventral rami (L4-S2).
LUMBOSACRAL PLEXOPATHIES
Plexopathies are typically recognized when motor, sensory, and if applicable, reflex deficits occur in multiple nerve and segmental distributions confined to one extremity. If localization within the lumbosacral plexus can be accomplished, designation as a lumbar plexopathy, a sacral plexopathy, a lumbosacral trunk lesion, or a panplexopathy is the best localization that can be expected. Although lumbar plexopathies may be bilateral, usually occurring in a stepwise and chronologically dissociated manner, sacral plexopathies are more likely to behave in this manner due to their closer anatomic proximity. The differential diagnosis of plexopathy includes disorders of the conus medullaris and cauda equina (polyradiculopathy). If there is a paucity of pain and sensory involvement, motor neuron disease should be considered as well.
The causes of lumbosacral plexopathies are listed in Table 459-10. Diabetic radiculopathy (discussed above) is a fairly common cause of painful leg weakness. Lumbosacral plexopathies are a well-recognized complication of retroperitoneal hemorrhage. Various primary and metastatic malignancies can affect the lumbosacral plexus as well; these include carcinoma of the cervix, endometrium, and ovary; osteosarcoma; testicular cancer; multiple myeloma; lymphoma; acute myelogenous leukemia; colon cancer; squamous cell carcinoma of the rectum; adenocarcinoma of unknown origin; and intraneural spread of prostate cancer.
|
LUMBOSACRAL PLEXOPATHIES: ETIOLOGIES |
RECURRENT NEOPLASTIC DISEASE OR RADIATION-INDUCED PLEXOPATHY
The treatment for various malignancies is often radiation therapy, the field of which may include parts of the brachial plexus. It can be difficult in such situations to determine if a new brachial or lumbosacral plexopathy is related to tumor within the plexus or from radiation-induced nerve damage. Radiation can be associated with microvascular abnormalities and fibrosis of surrounding tissues, which can damage the axons and the Schwann cells. Radiation-induced plexopathy can develop months or years following therapy and is dose dependent.
Tumor invasion is usually painful and more commonly affects the lower trunk, whereas radiation injury is often painless and affects the upper trunk. Imaging studies such as MRI and CT scans are useful but can be misleading with small microscopic invasion of the plexus. EMG can be informative if myokymic discharges are appreciated, as this finding strongly suggests radiation-induced damage.
EVALUATION AND TREATMENT OF PLEXOPATHIES
Most patients with plexopathies will undergo both imaging with MRI and EDx evaluations. Severe pain from acute idiopathic lumbosacral plexopathy may respond to a short course of glucocorticoids.
460 |
Guillain-Barré Syndrome and Other Immune-Mediated Neuropathies |
GUILLAIN-BARRÉ SYNDROME
Guillain-Barré syndrome (GBS) is an acute, frequently severe, and fulminant polyradiculoneuropathy that is autoimmune in nature. It occurs year-round at a rate of between 1 and 4 cases per 100,000 annually; in the United States, ~5000–6000 cases occur per year. Males are at slightly higher risk for GBS than females, and in Western countries, adults are more frequently affected than children.
Clinical Manifestations GBS manifests as a rapidly evolving areflexic motor paralysis with or without sensory disturbance. The usual pattern is an ascending paralysis that may be first noticed as rubbery legs. Weakness typically evolves over hours to a few days and is frequently accompanied by tingling dysesthesias in the extremities. The legs are usually more affected than the arms, and facial diparesis is present in 50% of affected individuals. The lower cranial nerves are also frequently involved, causing bulbar weakness with difficulty handling secretions and maintaining an airway; the diagnosis in these patients may initially be mistaken for brainstem ischemia. Pain in the neck, shoulder, back, or diffusely over the spine is also common in the early stages of GBS, occurring in ~50% of patients. Most patients require hospitalization, and in different series, up to 30% require ventilatory assistance at some time during the illness. The need for mechanical ventilation is associated with more severe weakness on admission, a rapid tempo of progression, and the presence of facial and/or bulbar weakness during the first week of symptoms. Fever and constitutional symptoms are absent at the onset and, if present, cast doubt on the diagnosis. Deep tendon reflexes attenuate or disappear within the first few days of onset. Cutaneous sensory deficits (e.g., loss of pain and temperature sensation) are usually relatively mild, but functions subserved by large sensory fibers, such as deep tendon reflexes and proprioception, are more severely affected. Bladder dysfunction may occur in severe cases but is usually transient. If bladder dysfunction is a prominent feature and comes early in the course, diagnostic possibilities other than GBS should be considered, particularly spinal cord disease. Once clinical worsening stops and the patient reaches a plateau (almost always within 4 weeks of onset), further progression is unlikely.
Autonomic involvement is common and may occur even in patients whose GBS is otherwise mild. The usual manifestations are loss of vasomotor control with wide fluctuation in blood pressure, postural hypotension, and cardiac dysrhythmias. These features require close monitoring and management and can be fatal. Pain is another common feature of GBS; in addition to the acute pain described above, a deep aching pain may be present in weakened muscles that patients liken to having overexercised the previous day. Other pains in GBS include dysesthetic pain in the extremities as a manifestation of sensory nerve fiber involvement. These pains are self-limited and often respond to standard analgesics (Chap. 18).
Several subtypes of GBS are recognized, as determined primarily by electrodiagnostic (Edx) and pathologic distinctions (Table 460-1). The most common variant is acute inflammatory demyelinating polyneuropathy (AIDP). Additionally, there are two axonal variants, which are often clinically severe—the acute motor axonal neuropathy (AMAN) and acute motor sensory axonal neuropathy (AMSAN) subtypes. In addition, a range of limited or regional GBS syndromes are also encountered. Notable among these is the Miller Fisher syndrome (MFS), which presents as rapidly evolving ataxia and areflexia of limbs without weakness, and ophthalmoplegia, often with pupillary paralysis. The MFS variant accounts for ~5% of all cases and is strongly associated with antibodies to the ganglioside GQ1b (see “Immunopathogenesis,” below). Other regional variants of GBS include (1) pure sensory forms; (2) ophthalmoplegia with anti-GQ1b antibodies as part of severe motor-sensory GBS; (3) GBS with severe bulbar and facial paralysis, sometimes associated with antecedent cytomegalovirus (CMV) infection and anti-GM2 antibodies; and (4) acute pandysautonomia (Chap. 454).
|
SUBTYPES OF GUILLAIN-BARRÉ SYNDROME (GBS) |

Antecedent Events Approximately 70% of cases of GBS occur 1–3 weeks after an acute infectious process, usually respiratory or gastrointestinal. Culture and seroepidemiologic techniques show that 20–30% of all cases occurring in North America, Europe, and Australia are preceded by infection or reinfection with Campylobacter jejuni. A similar proportion is preceded by a human herpes virus infection, often CMV or Epstein-Barr virus. Other viruses (e.g., HIV, hepatitis E) and also Mycoplasma pneumoniae have been identified as agents involved in antecedent infections, as have recent immunizations. The swine influenza vaccine, administered widely in the United States in 1976, is the most notable example. Influenza vaccines in use from 1992 to 1994, however, resulted in only one additional case of GBS per million persons vaccinated, and the more recent seasonal influenza vaccines appear to confer a GBS risk of <1 per million. Epidemiologic studies looking at H1N1 vaccination demonstrated at most only a slight increased risk of GBS. Meningococcal vaccinations (Menactra) does not appear to carry an increased risk. Older-type rabies vaccine, prepared in nervous system tissue, is implicated as a trigger of GBS in developing countries where it is still used; the mechanism is presumably immunization against neural antigens. GBS also occurs more frequently than can be attributed to chance alone in patients with lymphoma (including Hodgkin’s disease), in HIV-seropositive individuals, and in patients with systemic lupus erythematosus (SLE). C. jejuni has also been implicated in summer outbreaks of AMAN among children and young adults exposed to chickens in rural China.
Immunopathogenesis Several lines of evidence support an autoimmune basis for acute inflammatory demyelinating polyneuropathy (AIDP), the most common and best-studied type of GBS; the concept extends to all of the subtypes of GBS (Table 460-1).
It is likely that both cellular and humoral immune mechanisms contribute to tissue damage in AIDP. T cell activation is suggested by the finding that elevated levels of cytokines and cytokine receptors are present in serum (interleukin [IL] 2, soluble IL-2 receptor) and in cerebrospinal fluid (CSF) (IL-6, tumor necrosis factor α, interferon γ). AIDP is also closely analogous to an experimental T cell–mediated immunopathy designated experimental allergic neuritis (EAN). EAN is induced in laboratory animals by immune sensitization against protein fragments derived from peripheral nerve proteins, and in particular against the P2 protein. Based on analogy to EAN, it was initially thought that AIDP was likely to be primarily a T cell–mediated disorder; however, abundant data now suggest that autoantibodies directed against nonprotein determinants may be central to many cases.
Circumstantial evidence suggests that all GBS results from immune responses to nonself antigens (infectious agents, vaccines) that misdirect to host nerve tissue through a resemblance-of-epitope (molecular mimicry) mechanism (Fig. 460-1). The neural targets are likely to be glycoconjugates, specifically gangliosides (Table 460-2; Fig. 460-2). Gangliosides are complex glycosphingolipids that contain one or more sialic acid residues; various gangliosides participate in cell-cell interactions (including those between axons and glia), modulation of receptors, and regulation of growth. They are typically exposed on the plasma membrane of cells, rendering them susceptible to an antibody-mediated attack. Gangliosides and other glycoconjugates are present in large quantity in human nervous tissues and in key sites, such as nodes of Ranvier. Antiganglioside antibodies, most frequently to GM1, are common in GBS (20–50% of cases), particularly in AMAN and AMSAN and in those cases preceded by C. jejuni infection. Furthermore, isolates of C. jejuni from stool cultures of patients with GBS have surface glycolipid structures that antigenically cross react with gangliosides, including GM1, concentrated in human nerves. Sialic acid residues from pathogenic C. jejuni strains can also trigger activation of dendritic cells via signaling through a toll-like receptor (TLR4), promoting B cell differentiation and further amplifying humoral autoimmunity. Another line of evidence is derived from experience in Europe with parenteral use of purified bovine brain gangliosides for treatment of various neuropathic disorders. Between 5 and 15 days after injection, some recipients developed acute motor axonal GBS with high titers of anti-GM1 antibodies that recognized epitopes at nodes of Ranvier and motor endplates. Experimentally, anti-GM1 antibodies can trigger complement-mediated injury at paranodal axon-glial junctions, disrupting the clustering of sodium channels and likely contributing to conduction block (see “Pathophysiology,” below).
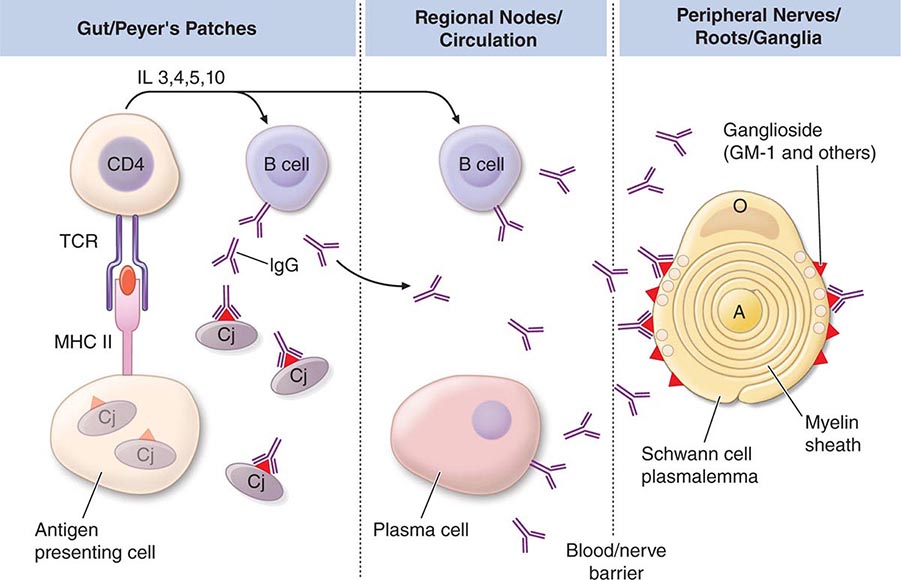
FIGURE 460-1 Postulated immunopathogenesis of Guillain-Barré syndrome (GBS) associated with Campylobacter jejuni infection. B cells recognize glycoconjugates on C. jejuni (Cj) (triangles) that cross-react with ganglioside present on Schwann cell surface and subjacent peripheral nerve myelin. Some B cells, activated via a T cell–independent mechanism, secrete primarily IgM (not shown). Other B cells (upper left side) are activated via a partially T cell–dependent route and secrete primarily IgG; T cell help is provided by CD4 cells activated locally by fragments of Cj proteins that are presented on the surface of antigen-presenting cells (APCs). A critical event in the development of GBS is the escape of activated B cells from Peyer’s patches into regional lymph nodes. Activated T cells probably also function to assist in opening of the blood-nerve barrier, facilitating penetration of pathogenic autoantibodies. The earliest changes in myelin (right) consist of edema between myelin lamellae and vesicular disruption (shown as circular blebs) of the outermost myelin layers. These effects are associated with activation of the C5b-C9 membrane attack complex and probably mediated by calcium entry; it is possible that the macrophage cytokine tumor necrosis factor (TNF) also participates in myelin damage. A, axon; B, B cell; MHC II, class II major histocompatibility complex molecule; O, oligodendrocyte; TCR, T cell receptor.
|
PRINCIPAL ANTIGLYCOLIPID ANTIBODIES IMPLICATED IN IMMUNE NEUROPATHIES |
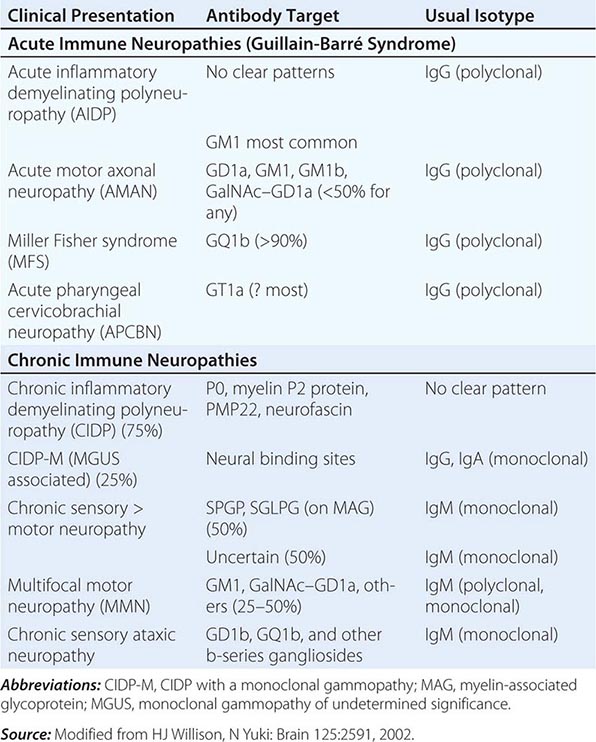
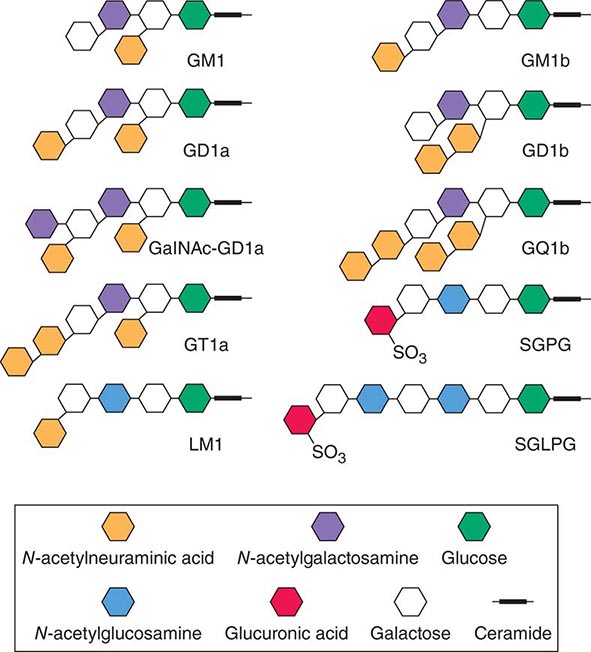
FIGURE 460-2 Glycolipids implicated as antigens in immune-mediated neuropathies. (Modified from HJ Willison, N Yuki: Brain 125: 2591, 2002.)
Anti-GQ1b IgG antibodies are found in >90% of patients with MFS (Table 460-2; Fig. 460-2), and titers of IgG are highest early in the course. Anti-GQ1b antibodies are not found in other forms of GBS unless there is extraocular motor nerve involvement. A possible explanation for this association is that extraocular motor nerves are enriched in GQ1b gangliosides in comparison to limb nerves. In addition, a monoclonal anti-GQ1b antibody raised against C. jejuni isolated from a patient with MFS blocked neuromuscular transmission experimentally.
Taken together, these observations provide strong but still inconclusive evidence that autoantibodies play an important pathogenic role in GBS. Although antiganglioside antibodies have been studied most intensively, other antigenic targets may also be important. One report identified IgG antibodies against Schwann cells and neurons (nerve growth cone region) in some GBS cases. Proof that these antibodies are pathogenic requires that they be capable of mediating disease following direct passive transfer to naïve hosts; this has not yet been demonstrated, although one case of possible maternal-fetal transplacental transfer of GBS has been described.
In AIDP, an early step in the induction of tissue damage appears to be complement deposition along the outer surface of the Schwann cell. Activation of complement initiates a characteristic vesicular disintegration of the myelin sheath and also leads to recruitment of activated macrophages, which participate in damage to myelin and axons. In AMAN, the pattern is different in that complement is deposited along with IgG at the nodes of Ranvier along large motor axons. Interestingly, in cases of AMAN, antibodies against GD1a appear to have a fine specificity that favors binding to motor rather than sensory nerve roots, even though this ganglioside is expressed on both fiber types.
Pathophysiology In the demyelinating forms of GBS, the basis for flaccid paralysis and sensory disturbance is conduction block. This finding, demonstrable electrophysiologically, implies that the axonal connections remain intact. Hence, recovery can take place rapidly as remyelination occurs. In severe cases of demyelinating GBS, secondary axonal degeneration usually occurs; its extent can be estimated electrophysiologically. More secondary axonal degeneration correlates with a slower rate of recovery and a greater degree of residual disability. When a severe primary axonal pattern is encountered electrophysiologically, the implication is that axons have degenerated and become disconnected from their targets, specifically the neuromuscular junctions, and must therefore regenerate for recovery to take place. In motor axonal cases in which recovery is rapid, the lesion is thought to be localized to preterminal motor branches, allowing regeneration and reinnervation to take place quickly. Alternatively, in mild cases, collateral sprouting and reinnervation from surviving motor axons near the neuromuscular junction may begin to reestablish physiologic continuity with muscle cells over a period of several months.
Laboratory Features CSF findings are distinctive, consisting of an elevated CSF protein level (1–10 g/L [100–1000 mg/dL]) without accompanying pleocytosis. The CSF is often normal when symptoms have been present for ≤48 h; by the end of the first week, the level of protein is usually elevated. A transient increase in the CSF white cell count (10–100/μL) occurs on occasion in otherwise typical GBS; however, a sustained CSF pleocytosis suggests an alternative diagnosis (viral myelitis) or a concurrent diagnosis such as unrecognized HIV infection, leukemia or lymphoma with infiltration of nerves, or neurosarcoidosis. Edx features are mild or absent in the early stages of GBS and lag behind the clinical evolution. In AIDP, the earliest features are prolonged F-wave latencies, prolonged distal latencies, and reduced amplitudes of compound muscle action potentials (CMAPs), probably owing to the predilection for involvement of nerve roots and distal motor nerve terminals early in the course. Later, slowing of conduction velocity, conduction block, and temporal dispersion may be appreciated (Table 460-1). Occasionally, sensory nerve action potentials (SNAPs) may be normal in the feet (e.g., sural nerve) when abnormal in the arms. This is also a sign that the patient does not have one of the more typical “length-dependent” polyneuropathies. In cases with primary axonal pathology, the principal Edx finding is reduced amplitude of CMAPs (and also SNAPS with AMSAN) without conduction slowing or prolongation of distal latencies.
Diagnosis GBS is a descriptive entity. The diagnosis of AIDP is made by recognizing the pattern of rapidly evolving paralysis with areflexia, absence of fever or other systemic symptoms, and characteristic antecedent events. In 2011, the Brighton Collaboration developed a new set of case definitions for GBS in response to needs of epidemiologic studies of vaccination and assessing risks of GBS (Table 460-3). These criteria have subsequently been validated. Other disorders that may enter into the differential diagnosis include acute myelopathies (especially with prolonged back pain and sphincter disturbances); diphtheria (early oropharyngeal disturbances); Lyme polyradiculitis and other tick-borne paralyses; porphyria (abdominal pain, seizures, psychosis); vasculitic neuropathy (check erythrocyte sedimentation rate, described below); poliomyelitis (fever and meningismus common); West Nile virus; CMV polyradiculitis (in immunocompromised patients); critical illness neuropathy or myopathy; neuromuscular junction disorders such as myasthenia gravis and botulism (pupillary reactivity lost early); poisonings with organophosphates, thallium, or arsenic; paralytic shellfish poisoning; or severe hypophosphatemia (rare). Laboratory tests are helpful primarily to exclude mimics of GBS. Edx features may be minimal, and the CSF protein level may not rise until the end of the first week. If the diagnosis is strongly suspected, treatment should be initiated without waiting for evolution of the characteristic Edx and CSF findings to occur. GBS patients with risk factors for HIV or with CSF pleocytosis should have a serologic test for HIV.
|
BRIGHTON CRITERIA FOR DIAGNOSIS OF GUILLAIN-BARRÉ SYNDROME (GBS) AND MILLER FISHER SYNDROME |
Abbreviation: CSF, cerebrospinal fluid.
Source: From JJ Sejvar et al: Guillain-Barré syndrome and Fisher syndrome: case definitions and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine 29:599, 2011. Validation study published by C Fokke et al: Diagnosis of Guillain-Barré syndrome and validation of Brighton criteria. Brain 137:33, 2014.
Prognosis and Recovery Approximately 85% of patients with GBS achieve a full functional recovery within several months to a year, although minor findings on examination (such as areflexia) may persist and patients often complain of continued symptoms, including fatigue. The mortality rate is <5% in optimal settings; death usually results from secondary pulmonary complications. The outlook is worst in patients with severe proximal motor and sensory axonal damage. Such axonal damage may be either primary or secondary in nature (see “Pathophysiology,” above), but in either case successful regeneration cannot occur. Other factors that worsen the outlook for recovery are advanced age, a fulminant or severe attack, and a delay in the onset of treatment. Between 5 and 10% of patients with typical GBS have one or more late relapses; such cases are then classified as chronic inflammatory demyelinating polyneuropathy (CIDP).
CHRONIC INFLAMMATORY DEMYELINATING POLYNEUROPATHY
CIDP is distinguished from GBS by its chronic course. In other respects, this neuropathy shares many features with the common demyelinating form of GBS, including elevated CSF protein levels and the Edx findings of acquired demyelination. Most cases occur in adults, and males are affected slightly more often than females. The incidence of CIDP is lower than that of GBS, but due to the protracted course, the prevalence is greater.
Clinical Manifestations Onset is usually gradual over a few months or longer, but in a few cases, the initial attack is indistinguishable from that of GBS. An acute-onset form of CIDP should be considered when GBS deteriorates >9 weeks after onset or relapses at least three times. Symptoms are both motor and sensory in most cases. Weakness of the limbs is usually symmetric but can be strikingly asymmetric in multifocal acquired demyelinating sensory and motor (MADSAM) neuropathy variant (Lewis-Sumner syndrome) in which discrete peripheral nerves are involved. There is considerable variability from case to case. Some patients experience a chronic progressive course, whereas others, usually younger patients, have a relapsing and remitting course. Some have only motor findings, and a small proportion present with a relatively pure syndrome of sensory ataxia. Tremor occurs in ~10% and may become more prominent during periods of subacute worsening or improvement. A small proportion have cranial nerve findings, including external ophthalmoplegia. CIDP tends to ameliorate over time with treatment; the result is that many years after onset, nearly 75% of patients have reasonable functional status. Death from CIDP is uncommon.
Diagnosis The diagnosis rests on characteristic clinical, CSF, and electrophysiologic findings. The CSF is usually acellular with an elevated protein level, sometimes several times normal. As with GBS, a CSF pleocytosis should lead to the consideration of HIV infection, leukemia or lymphoma, and neurosarcoidosis. Edx findings reveal variable degrees of conduction slowing, prolonged distal latencies, distal and temporal dispersion of CMAPs, and conduction block as the principal features. In particular, the presence of conduction block is a certain sign of an acquired demyelinating process. Evidence of axonal loss, presumably secondary to demyelination, is present in >50% of patients. Serum protein electrophoresis with immunofixation is indicated to search for monoclonal gammopathy and associated conditions (see “Monoclonal Gammopathy of Undetermined Significance,” below). In all patients with presumptive CIDP, it is also reasonable to exclude vasculitis, collagen vascular disease (especially SLE), chronic hepatitis, HIV infection, amyloidosis, and diabetes mellitus. Other associated conditions include inflammatory bowel disease and lymphoma.
Pathogenesis Although there is evidence of immune activation in CIDP, the precise mechanisms of pathogenesis are unknown. Biopsy typically reveals little inflammation and onion-bulb changes (imbricated layers of attenuated Schwann cell processes surrounding an axon) that result from recurrent demyelination and remyelination (Fig. 460-1). The response to therapy suggests that CIDP is immune-mediated; CIDP responds to glucocorticoids, whereas GBS does not. Passive transfer of demyelination into experimental animals has been accomplished using IgG purified from the serum of some patients with CIDP, lending support for a humoral autoimmune pathogenesis. A minority of patients have serum antibodies against P0, myelin P2 protein, PMP22, or neurofascin. It is also of interest that a CIDP-like illness developed spontaneously in the nonobese diabetic (NOD) mouse when the immune co-stimulatory molecule B7-2 (CD86) was genetically deleted; this suggests that CIDP can result from altered triggering of T cells by antigen-presenting cells.
Approximately 25% of patients with clinical features of CIDP also have a monoclonal gammopathy of undetermined significance (MGUS). Cases associated with monoclonal IgA or IgG kappa usually respond to treatment as favorably as cases without a monoclonal gammopathy. Patients with IgM monoclonal gammopathy and antibodies directed against myelin-associated glycoprotein (MAG) have a distinct polyneuropathy, tend to have more sensory findings and a more protracted course, and usually have a less satisfactory response to treatment.
MULTIFOCAL MOTOR NEUROPATHY
Multifocal motor neuropathy (MMN) is a distinctive but uncommon neuropathy that presents as slowly progressive motor weakness and atrophy evolving over years in the distribution of selected nerve trunks, associated with sites of persistent focal motor conduction block in the same nerve trunks. Sensory fibers are relatively spared. The arms are affected more frequently than the legs, and >75% of all patients are male. Some cases have been confused with lower motor neuron forms of amyotrophic lateral sclerosis (Chap. 452). Less than 50% of patients present with high titers of polyclonal IgM antibody to the ganglioside GM1. It is uncertain how this finding relates to the discrete foci of persistent motor conduction block, but high concentrations of GM1 gangliosides are normal constituents of nodes of Ranvier in peripheral nerve fibers. Pathology reveals demyelination and mild inflammatory changes at the sites of conduction block.
Most patients with MMN respond to high-dose IVIg (dosages as for CIDP, above); periodic re-treatment is required (usually at least monthly) to maintain the benefit. Some refractory patients have responded to rituximab or cyclophosphamide. Glucocorticoids and PE are not effective.
NEUROPATHIES WITH MONOCLONAL GAMMOPATHY
MULTIPLE MYELOMA
Clinically overt polyneuropathy occurs in ~5% of patients with the commonly encountered type of multiple myeloma, which exhibits either lytic or diffuse osteoporotic bone lesions. These neuropathies are sensorimotor, are usually mild and slowly progressive but may be severe, and generally do not reverse with successful suppression of the myeloma. In most cases, Edx and pathologic features are consistent with a process of axonal degeneration.
In contrast, myeloma with osteosclerotic features, although representing only 3% of all myelomas, is associated with polyneuropathy in one-half of cases. These neuropathies, which may also occur with solitary plasmacytoma, are distinct because they (1) are usually demyelinating in nature and resemble CIDP; (2) often respond to radiation therapy or removal of the primary lesion; (3) are associated with different monoclonal proteins and light chains (almost always lambda as opposed to primarily kappa in the lytic type of multiple myeloma); (4) are typically refractory to standard treatments of CIDP; and (5) may occur in association with other systemic findings including thickening of the skin, hyperpigmentation, hypertrichosis, organomegaly, endocrinopathy, anasarca, and clubbing of fingers. These are features of the POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes). Levels of vascular endothelial growth factor (VEGF) are increased in the serum, and this factor is felt to somehow play a pathogenic role in this syndrome. Treatment of the neuropathy is best directed at the osteosclerotic myeloma using surgery, radiotherapy, chemotherapy, or autologous peripheral blood stem cell transplantation.
Neuropathies are also encountered in other systemic conditions with gammopathy, including Waldenström’s macroglobulinemia, primary systemic amyloidosis, and cryoglobulinemic states (mixed essential cryoglobulinemia, some cases of hepatitis C).
MONOCLONAL GAMMOPATHY OF UNDETERMINED SIGNIFICANCE
Chronic polyneuropathies occurring in association with MGUS are usually associated with the immunoglobulin isotypes IgG, IgA, and IgM. Most patients present with isolated sensory symptoms in their distal extremities and have Edx features of an axonal sensory or sensorimotor polyneuropathy. These patients otherwise resemble idiopathic sensory polyneuropathy, and the MGUS might just be coincidental. They usually do not respond to immunotherapies designed to reduce the concentration of the monoclonal protein. Some patients, however, present with generalized weakness and sensory loss and Edx studies indistinguishable from CIDP without monoclonal gammopathy (see “Chronic Inflammatory Demyelinating Polyneuropathy,” above), and their response to immunosuppressive agents is also similar. An exception is the syndrome of IgM kappa monoclonal gammopathy associated with an indolent, longstanding, sometimes static sensory neuropathy, frequently with tremor and sensory ataxia. Most patients are male and older than age 50 years. In the majority, the monoclonal IgM immunoglobulin binds to a normal peripheral nerve constituent, MAG, found in the paranodal regions of Schwann cells. Binding appears to be specific for a polysaccharide epitope that is also found in other normal peripheral nerve myelin glycoproteins, P0 and PMP22, and also in other normal nerve-related glycosphingolipids (Fig. 460-1). In the MAG-positive cases, IgM paraprotein is incorporated into the myelin sheaths of affected patients and widens the spacing of the myelin lamellae, thus producing a distinctive ultrastructural pattern. Demyelination and remyelination are the hallmarks of the lesions, but axonal loss develops over time. These anti-MAG polyneuropathies are typical refractory to immunotherapy. In a small proportion of patients (30% at 10 years), MGUS will in time evolve into frankly malignant conditions such as multiple myeloma or lymphoma.
VASCULITIC NEUROPATHY
Peripheral nerve involvement is common in polyarteritis nodosa (PAN), appearing in half of all cases clinically and in 100% of cases at postmortem studies (Chap. 385). The most common pattern is multifocal (asymmetric) motor-sensory neuropathy (mononeuropathy multiplex) due to ischemic lesions of nerve trunks and roots; however, some cases of vasculitic neuropathy present as a distal, symmetric sensorimotor polyneuropathy. Symptoms of neuropathy are a common presenting complaint in patients with PAN. The Edx findings are those of an axonal process. Small- to medium-sized arteries of the vasa nervorum, particularly the epineural vessels, are affected in PAN, resulting in a widespread ischemic neuropathy. A high frequency of neuropathy occurs in allergic angiitis and granulomatosis (Churg-Strauss syndrome [CSS]).
Systemic vasculitis should always be considered when a subacute or chronically evolving mononeuropathy multiplex occurs in conjunction with constitutional symptoms (fever, anorexia, weight loss, loss of energy, malaise, and nonspecific pains). Diagnosis of suspected vasculitic neuropathy is made by a combined nerve and muscle biopsy, with serial section or skip-serial techniques.
Approximately one-third of biopsy-proven cases of vasculitic neuropathy are “nonsystemic” in that the vasculitis appears to affect only peripheral nerves. Constitutional symptoms are absent, and the course is more indolent than that of PAN. The erythrocyte sedimentation rate may be elevated, but other tests for systemic disease are negative. Nevertheless, clinically silent involvement of other organs is likely, and vasculitis is frequently found in muscle biopsied at the same time as nerve.
Vasculitic neuropathy may also be seen as part of the vasculitis syndrome occurring in the course of other connective tissue disorders (Chap. 385). The most frequent is rheumatoid arthritis, but ischemic neuropathy due to involvement of vasa nervorum may also occur in mixed cryoglobulinemia, Sjögren’s syndrome, granulomatosis with polyangiitis (Wegener’s), hypersensitivity angiitis, systemic lupus erythematosus, and progressive systemic sclerosis.
Some vasculitides are associated with antineutrophil cytoplasmic antibodies (ANCAs), which in turn, are subclassified as cytoplasmic (cANCA) or perinuclear (pANCA). cANCAs are directed against proteinase 3 (PR3), whereas pANCAs target myeloperoxidase (MPO). PR3/cANCAs are associated with granulomatosis with polyangiitis (Wegener’s), whereas MPO/pANCAs are typically associated with microscopic polyangiitis, CSS, and less commonly PAN. Of note, MPO/pANCA has also been seen in minocycline-induced vasculitis.
Management of these neuropathies, including the “nonsystemic” vasculitic neuropathy, consists of treatment of the underlying condition as well as the aggressive use of glucocorticoids and cyclophosphamide. Use of these immunosuppressive agents has resulted in dramatic improvements in outcome, with 5-year survival rates now greater than 80%. Recent clinical trials found that the combination of rituximab and glucocorticoids is not inferior to cyclophosphamide and glucocorticoids. Thus, combination therapy with glucocorticoids and rituximab is increasingly recommended as the standard initial treatment, particularly for ANCA-associated vasculitis.
ANTI-Hu PARANEOPLASTIC NEUROPATHY (CHAP. 122)
This uncommon immune-mediated disorder manifests as a sensory neuronopathy (i.e., selective damage to sensory nerve bodies in dorsal root ganglia). The onset is often asymmetric with dysesthesias and sensory loss in the limbs that soon progress to affect all limbs, the torso, and the face. Marked sensory ataxia, pseudoathetosis, and inability to walk, stand, or even sit unsupported are frequent features and are secondary to the extensive deafferentation. Subacute sensory neuronopathy may be idiopathic, but more than half of cases are paraneoplastic, primarily related to lung cancer, and most of those are small-cell lung cancer (SCLC). Diagnosis of the underlying SCLC requires awareness of the association, testing for the paraneoplastic antibody, and often positron emission tomography (PET) scanning for the tumor. The target antigens are a family of RNA-binding proteins (HuD, HuC, and Hel-N1) that in normal tissues are only expressed by neurons. The same proteins are usually expressed by SCLC, triggering in some patients an immune response characterized by antibodies and cytotoxic T cells that cross-react with the Hu proteins of the dorsal root ganglion neurons, resulting in immune-mediated neuronal destruction. An encephalomyelitis may accompany the sensory neuronopathy and presumably has the same pathogenesis. Neurologic symptoms usually precede, by ≤6 months, the identification of SCLC. The sensory neuronopathy runs its course in a few weeks or months and stabilizes, leaving the patient disabled. Most cases are unresponsive to treatment with glucocorticoids, IVIg, PE, or immunosuppressant drugs.
461 |
Myasthenia Gravis and Other Diseases of the Neuromuscular Junction |
Myasthenia gravis (MG) is a neuromuscular disorder characterized by weakness and fatigability of skeletal muscles. The underlying defect is a decrease in the number of available acetylcholine receptors (AChRs) at neuromuscular junctions due to an antibody-mediated autoimmune attack. Treatment now available for MG is highly effective, although a specific cure has remained elusive.
PATHOPHYSIOLOGY
At the neuromuscular junction (Fig. 461-1, Video 461-1), acetylcholine (ACh) is synthesized in the motor nerve terminal and stored in vesicles (quanta). When an action potential travels down a motor nerve and reaches the nerve terminal, ACh from 150 to 200 vesicles is released and combines with AChRs that are densely packed at the peaks of postsynaptic folds. The AChR consists of five subunits (2α, 1β, 1δ, and 1γ or ε) arranged around a central pore. When ACh combines with the binding sites on the α subunits of the AChR, the channel in the AChR opens, permitting the rapid entry of cations, chiefly sodium, which produces depolarization at the end-plate region of the muscle fiber. If the depolarization is sufficiently large, it initiates an action potential that is propagated along the muscle fiber, triggering muscle contraction. This process is rapidly terminated by hydrolysis of ACh by acetylcholinesterase (AChE), which is present within the synaptic folds, and by diffusion of ACh away from the receptor.
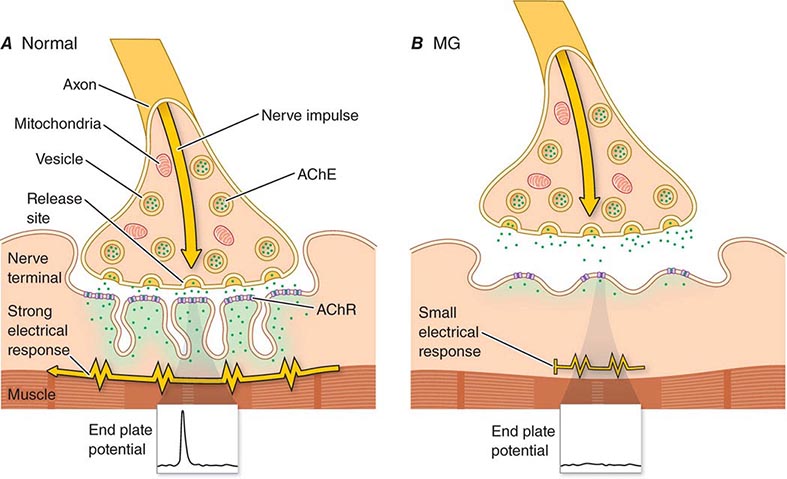
FIGURE 461-1 Diagrams of (A) normal and (B) myasthenic neuromuscular junctions. AChE, acetylcholinesterase. See text for description of normal neuromuscular transmission. The myasthenia gravis (MG) junction demonstrates a normal nerve terminal; a reduced number of acetylcholine receptors (AChRs) (stippling); flattened, simplified postsynaptic folds; and a widened synaptic space. See Video 461-1 also. (Modified from DB Drachman: N Engl J Med 330:1797, 1994; with permission.)
In MG, the fundamental defect is a decrease in the number of available AChRs at the postsynaptic muscle membrane. In addition, the postsynaptic folds are flattened, or “simplified.” These changes result in decreased efficiency of neuromuscular transmission. Therefore, although ACh is released normally, it produces small end-plate potentials that may fail to trigger muscle action potentials. Failure of transmission at many neuromuscular junctions results in weakness of muscle contraction.
The amount of ACh released per impulse normally declines on repeated activity (termed presynaptic rundown). In the myasthenic patient, the decreased efficiency of neuromuscular transmission combined with the normal rundown results in the activation of fewer and fewer muscle fibers by successive nerve impulses and hence increasing weakness, or myasthenic fatigue. This mechanism also accounts for the decremental response to repetitive nerve stimulation seen on electrodiagnostic testing.
The neuromuscular abnormalities in MG are caused by an autoimmune response mediated by specific anti-AChR antibodies. The anti-AChR antibodies reduce the number of available AChRs at neuromuscular junctions by three distinct mechanisms: (1) accelerated turnover of AChRs by a mechanism involving cross-linking and rapid endocytosis of the receptors; (2) damage to the postsynaptic muscle membrane by the antibody in collaboration with complement; and (3) blockade of the active site of the AChR, i.e., the site that normally binds ACh. An immune response to muscle-specific kinase (MuSK), a protein involved in AChR clustering at neuromuscular junctions, can also result in MG, with reduction of AChRs demonstrated experimentally. Anti-MuSK antibody occurs in about 40% of patients without AChR antibody. A small proportion of patients whose sera are negative for both AChR and MuSK antibodies have antibodies to another protein at the neuromuscular junction—low-density lipoprotein receptor-related protein 4 (lrp4)—that is important for clustering of AChRs. The pathogenic antibodies are IgG and are T cell dependent. Thus, immunotherapeutic strategies directed against either the antibody-producing B cells or helper T cells are effective in this antibody-mediated disease.
How the autoimmune response is initiated and maintained in MG is not completely understood, but the thymus appears to play a role in this process. The thymus is abnormal in ~75% of patients with AChR antibody–positive MG; in ~65% the thymus is “hyperplastic,” with the presence of active germinal centers detected histologically, although the hyperplastic thymus is not necessarily enlarged. An additional 10% of patients have thymic tumors (thymomas). Muscle-like cells within the thymus (myoid cells), which express AChRs on their surface, may serve as a source of autoantigen and trigger the autoimmune reaction within the thymus gland.
CLINICAL FEATURES
MG is not rare, having a prevalence as high as 2–7 in 10,000. It affects individuals in all age groups, but peaks of incidence occur in women in their twenties and thirties and in men in their fifties and sixties. Overall, women are affected more frequently than men, in a ratio of ~3:2. The cardinal features are weakness and fatigability of muscles. The weakness increases during repeated use (fatigue) or late in the day and may improve following rest or sleep. The course of MG is often variable. Exacerbations and remissions may occur, particularly during the first few years after the onset of the disease. Remissions are rarely complete or permanent. Unrelated infections or systemic disorders can lead to increased myasthenic weakness and may precipitate “crisis” (see below).
The distribution of muscle weakness often has a characteristic pattern. The cranial muscles, particularly the lids and extraocular muscles, are typically involved early in the course of MG; diplopia and ptosis are common initial complaints. Facial weakness produces a “snarling” expression when the patient attempts to smile. Weakness in chewing is most noticeable after prolonged effort, as in chewing meat. Speech may have a nasal timbre caused by weakness of the palate or a dysarthric “mushy” quality due to tongue weakness. Difficulty in swallowing may occur as a result of weakness of the palate, tongue, or pharynx, giving rise to nasal regurgitation or aspiration of liquids or food. Bulbar weakness is especially prominent in MuSK antibody–positive MG. In ~85% of patients, the weakness becomes generalized, affecting the limb muscles as well. If weakness remains restricted to the extraocular muscles for 3 years, it is likely that it will not become generalized, and these patients are said to have ocular MG. The limb weakness in MG is often proximal and may be asymmetric. Despite the muscle weakness, deep tendon reflexes are preserved. If weakness of respiration becomes so severe as to require respiratory assistance, the patient is said to be in crisis.
DIAGNOSIS AND EVALUATION
(Table 461-1) The diagnosis is suspected on the basis of weakness and fatigability in the typical distribution described above, without loss of reflexes or impairment of sensation or other neurologic function. The suspected diagnosis should always be confirmed definitively before treatment is undertaken; this is essential because (1) other treatable conditions may closely resemble MG and (2) the treatment of MG may involve surgery and the prolonged use of drugs with potentially adverse side effects.
|
DIAGNOSIS OF MYASTHENIA GRAVIS (MG) |
Abbreviations: AChR, acetylcholine receptor; CT, computed tomography; MRI, magnetic resonance imaging; MuSK, muscle-specific tyrosine kinase.
Antibodies to AChR, MuSK, or lpr4 As noted above, anti-AChR antibodies are detectable in the serum of ~85% of all myasthenic patients but in only about 50% of patients with weakness confined to the ocular muscles. The presence of anti-AChR antibodies is virtually diagnostic of MG, but a negative test does not exclude the disease. The measured level of anti-AChR antibody does not correspond well with the severity of MG in different patients. However, in an individual patient, a treatment-induced fall in the antibody level often correlates with clinical improvement, whereas a rise in the level may occur with exacerbations. Antibodies to MuSK have been found to be present in ~40% of AChR antibody–negative patients with generalized MG, and their presence is a useful diagnostic test in these patients. MuSK antibodies are rarely present in AChR antibody–positive patients or in patients with MG limited to ocular muscles. These antibodies may interfere with clustering of AChRs at neuromuscular junctions, as MuSK is known to do during early development. A small proportion of MG patients without antibodies to AChR or MuSK may have antibodies to lrp4, although a test for lrp4 antibodies is not yet commercially available. Finally, antibodies against agrin have recently been found in some patients with MG. Agrin is a protein derived from motor nerves that normally binds to lrp4 and thus may also interfere with clustering of AChRs at neuromuscular junctions. There may well be other—as yet undefined—antibodies that impair neuromuscular transmission.
Electrodiagnostic Testing Repetitive nerve stimulation may provide helpful diagnostic evidence of MG. Anti-AChE medication is stopped 6–24 h before testing. It is best to test weak muscles or proximal muscle groups. Electric shocks are delivered at a rate of two or three per second to the appropriate nerves, and action potentials are recorded from the muscles. In normal individuals, the amplitude of the evoked muscle action potentials does not change at these rates of stimulation. However, in myasthenic patients, there is a rapid reduction of >10–15% in the amplitude of the evoked responses.
Anticholinesterase Test Drugs that inhibit the enzyme AChE allow ACh to interact repeatedly with the limited number of AChRs in MG, producing improvement in muscle strength. Edrophonium is used most commonly for diagnostic testing because of the rapid onset (30 s) and short duration (~5 min) of its effect. An objective end point must be selected to evaluate the effect of edrophonium, such as weakness of extraocular muscles, impairment of speech, or the length of time that the patient can maintain the arms in forward abduction. An initial IV dose of 2 mg of edrophonium is given. If definite improvement occurs, the test is considered positive and is terminated. If there is no change, the patient is given an additional 8 mg IV. The dose is administered in two parts because some patients react to edrophonium with side effects such as nausea, diarrhea, salivation, fasciculations, and rarely with severe symptoms of syncope or bradycardia. Atropine (0.6 mg) should be drawn up in a syringe and ready for IV administration if these symptoms become troublesome. The edrophonium test is now reserved for patients with clinical findings that are suggestive of MG but who have negative antibody and electrodiagnostic test results. False-positive tests occur in occasional patients with other neurologic disorders, such as amyotrophic lateral sclerosis, and in placebo-reactors. False-negative or equivocal tests may also occur. In some cases, it is helpful to use a longer-acting drug such as neostigmine (15 mg PO), because this permits more time for detailed evaluation of strength.
Inherited Myasthenic Syndromes The congenital myasthenic syndromes (CMS) comprise a heterogeneous group of disorders of the neuromuscular junction that are not autoimmune but rather are due to genetic mutations in which virtually any component of the neuromuscular junction may be affected. Alterations in function of the presynaptic nerve terminal, in the various subunits of the AChR, AChE, or the other molecules involved in end-plate development or maintenance, have been identified in the different forms of CMS. These disorders share many of the clinical features of autoimmune MG, including weakness and fatigability of skeletal muscles, in some cases involving extraocular muscles (EOMs), lids, and proximal muscles, similar to the distribution in autoimmune MG. CMS should be suspected when symptoms of myasthenia have begun in infancy or childhood and AChR antibody tests are consistently negative. By far the most common genetic defects occur in the AChR or other postsynaptic molecules (67% in the Mayo Clinic series of 350 CMS patients), with about equal frequencies of abnormalities in AChE (13%) and the various maintenance molecules (DOK7, GFPT, etc.; ~14%). In the forms that involve the AChR, a wide variety of mutations have been identified in each of the subunits, but the ε subunit is affected in ~75% of these cases. In most of the recessively inherited forms of CMS, the mutations are heteroallelic; that is, different mutations affecting each of the two alleles are present. Features of the four most common forms of CMS are summarized in Table 461-2. Although clinical features and electrodiagnostic and pharmacologic tests may suggest the correct diagnosis, molecular analysis is required for precise elucidation of the defect; this may lead to helpful treatment as well as genetic counseling.
|
THE CONGENITAL MYASTHENIC SYNDROMES |
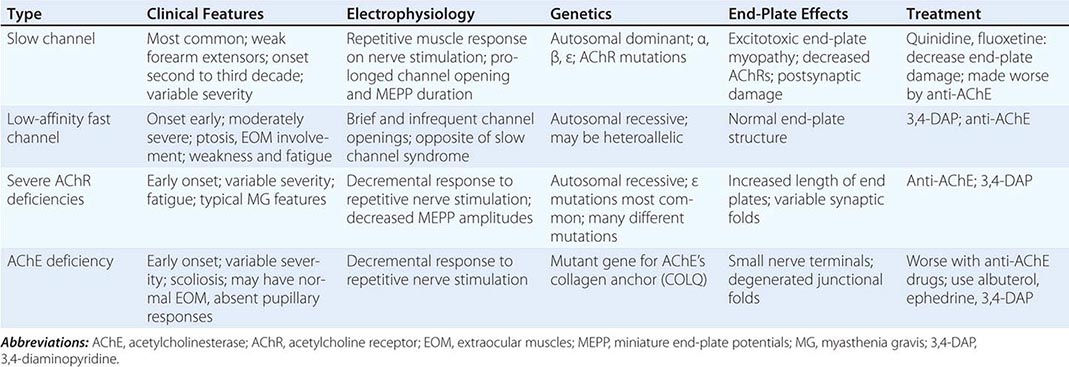
Differential Diagnosis Other conditions that cause weakness of the cranial and/or somatic musculature include the nonautoimmune CMS discussed above, drug-induced myasthenia, Lambert-Eaton myasthenic syndrome (LEMS), neurasthenia, hyperthyroidism (Graves’ disease), botulism, intracranial mass lesions, oculopharyngeal dystrophy, and mitochondrial myopathy (Kearns-Sayre syndrome, progressive external ophthalmoplegia). Treatment with penicillamine (used for scleroderma or rheumatoid arthritis) may result in true autoimmune MG, but the weakness is usually mild, and recovery occurs within weeks or months after discontinuing its use. Aminoglycoside antibiotics or procainamide can cause exacerbation of weakness in myasthenic patients; very large doses can cause neuromuscular weakness in normal individuals.
LEMS is a presynaptic disorder of the neuromuscular junction that can cause weakness similar to that of MG. The proximal muscles of the lower limbs are most commonly affected, but other muscles may be involved as well. Cranial nerve findings, including ptosis of the eyelids and diplopia, occur in up to 70% of patients and resemble features of MG. However, the two conditions are usually readily distinguished, because patients with LEMS have depressed or absent reflexes and experience autonomic changes such as dry mouth and impotence. Nerve stimulation produces an initial low-amplitude response and, at low rates of repetitive stimulation (2–3 Hz), decremental responses like those of MG; however, at high rates (50 Hz), or following exercise, incremental responses occur. LEMS is caused by autoantibodies directed against P/Q-type calcium channels at the motor nerve terminals, which can be detected in ~85% of LEMS patients by radioimmunoassay. These autoantibodies result in impaired release of ACh from nerve terminals. Many patients with LEMS have an associated malignancy, most commonly small-cell carcinoma of the lung, which may express calcium channels that stimulate the autoimmune response. The diagnosis of LEMS may signal the presence of a tumor long before it would otherwise be detected, permitting early removal. Treatment of LEMS involves plasmapheresis and immunosuppression, as for MG. 3,4-Diaminopyridine (3,4-DAP) and pyridostigmine may also be symptomatically helpful. 3,4-DAP acts by blocking potassium channels, which results in prolonged depolarization of the motor nerve terminals and thus enhances ACh release. Pyridostigmine prolongs the action of ACh, allowing repeated interactions with AChRs.
Botulism (Chap. 178) is due to potent bacterial toxins produced by any of eight different strains of Clostridium botulinum. The toxins enzymatically cleave specific proteins essential for the release of ACh from the motor nerve terminal, thereby interfering with neuromuscular transmission. Most commonly, botulism is caused by ingestion of improperly prepared food containing toxin. Rarely, the nearly ubiquitous spores of C. botulinum may germinate in wounds. In infants, the spores may germinate in the gastrointestinal (GI) tract and release toxin, causing muscle weakness. Patients present with myasthenia-like bulbar weakness (e.g., diplopia, dysarthria, dysphagia) and lack sensory symptoms and signs. Weakness may generalize to the limbs and may result in respiratory failure. Reflexes are present early, but they may be diminished as the disease progresses. Mentation is normal. Autonomic findings include paralytic ileus, constipation, urinary retention, dilated or poorly reactive pupils, and dry mouth. The demonstration of toxin in serum by bioassay is definitive, but the results usually take a relatively long time to be completed and may be negative. Nerve stimulation studies reveal findings of presynaptic neuromuscular blockade with reduced compound muscle action potentials (CMAPs) that increase in amplitude following high-frequency repetitive stimulation. Treatment includes ventilatory support and aggressive inpatient supportive care (e.g., nutrition, deep vein thrombosis prophylaxis) as needed. Antitoxin should be given as early as possible to be effective and can be obtained through the Centers for Disease Control and Prevention. A preventive vaccine is available for laboratory workers or other highly exposed individuals.
Neurasthenia is the historic term for a myasthenia-like fatigue syndrome without an organic basis. These patients may present with subjective symptoms of weakness and fatigue, but muscle testing usually reveals the “give-away weakness” characteristic of nonorganic disorders; the complaint of fatigue in these patients means tiredness or apathy rather than decreasing muscle power on repeated effort. Hyperthyroidism is readily diagnosed or excluded by tests of thyroid function, which should be carried out routinely in patients with suspected MG. Abnormalities of thyroid function (hyper- or hypothyroidism) may increase myasthenic weakness. Diplopia resembling that in MG may occasionally be due to an intracranial mass lesion that compresses nerves to the EOMs (e.g., sphenoid ridge meningioma), but magnetic resonance imaging (MRI) of the head and orbits usually reveals the lesion.
Progressive external ophthalmoplegia is a rare condition resulting in weakness of the EOMs, which may be accompanied by weakness of the proximal muscles of the limbs and other systemic features. Most patients with this condition have mitochondrial disorders that can be detected on muscle biopsy (Chap. 462e).
Search for Associated Conditions (Table 461-3) Myasthenic patients have an increased incidence of several associated disorders. Thymic abnormalities occur in ~75% of AChR antibody–positive patients, as noted above. Neoplastic change (thymoma) may produce enlargement of the thymus, which is detected by computed tomography (CT) scanning of the anterior mediastinum. A thymic shadow on CT scan may normally be present through young adulthood, but enlargement of the thymus in a patient age >40 years is highly suspicious of thymoma. Hyperthyroidism occurs in 3–8% of patients and may aggravate the myasthenic weakness. Thyroid function tests should be obtained in all patients with suspected MG. Because of the association of MG with other autoimmune disorders, blood tests for rheumatoid factor and antinuclear antibodies should also be carried out. Chronic infection of any kind can exacerbate MG and should be sought carefully. Finally, measurements of ventilatory function are valuable because of the frequency and seriousness of respiratory impairment in myasthenic patients.
|
DISORDERS ASSOCIATED WITH MYASTHENIA GRAVIS AND RECOMMENDED LABORATORY TESTS |
Abbreviations: CT, computed tomography; MRI, magnetic resonance imaging; PPD, purified protein derivative.
Because of the side effects of glucocorticoids and other immunosuppressive agents used in the treatment of MG, a thorough medical investigation should be undertaken, searching specifically for evidence of chronic or latent infection (such as tuberculosis or hepatitis), hypertension, diabetes, renal disease, and glaucoma.
|
TREATMENT |
MYASTHENIA GRAVIS |
The prognosis has improved strikingly as a result of advances in treatment. Nearly all myasthenic patients can be returned to full productive lives with proper therapy. The most useful treatments for MG include anticholinesterase medications, immunosuppressive agents, thymectomy, and plasmapheresis or intravenous immunoglobulin (IVIg) (Fig. 461-2).
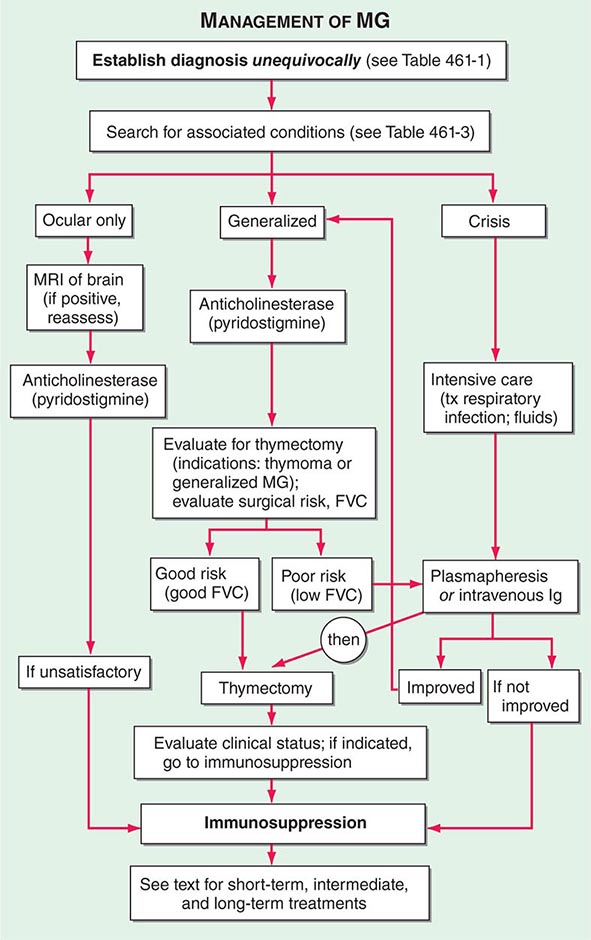
FIGURE 461-2 Algorithm for the management of myasthenia gravis. FVC, forced vital capacity; MRI, magnetic resonance imaging.
ANTICHOLINESTERASE MEDICATIONS
Anticholinesterase medication produces at least partial improvement in most myasthenic patients, although improvement is complete in only a few. Patients with anti-MuSK MG generally obtain less benefit from anticholinesterase agents than those with AChR antibodies. Pyridostigmine is the most widely used anticholinesterase drug. The beneficial action of oral pyridostigmine begins within 15–30 min and lasts for 3–4 h, but individual responses vary. Treatment is begun with a moderate dose, e.g., 30–60 mg three to four times daily. The frequency and amount of the dose should be tailored to the patient’s individual requirements throughout the day. For example, patients with weakness in chewing and swallowing may benefit by taking the medication before meals so that peak strength coincides with mealtimes. Long-acting pyridostigmine may occasionally be useful to get the patient through the night but should not be used for daytime medication because of variable absorption. The maximum useful dose of pyridostigmine rarely exceeds 120 mg every 4–6 h during daytime. Overdosage with anticholinesterase medication may cause increased weakness and other side effects. In some patients, muscarinic side effects of the anticholinesterase medication (diarrhea, abdominal cramps, salivation, nausea) may limit the dose tolerated. Atropine/diphenoxylate or loperamide is useful for the treatment of GI symptoms.
THYMECTOMY
Two separate issues should be distinguished: (1) surgical removal of thymoma, and (2) thymectomy as a treatment for MG. Surgical removal of a thymoma is necessary because of the possibility of local tumor spread, although most thymomas are histologically benign. In the absence of a tumor, the available evidence suggests that up to 85% of patients experience improvement after thymectomy; of these, ~35% achieve drug-free remission. However, the improvement is typically delayed for months to years. The advantage of thymectomy is that it offers the possibility of long-term benefit, in some cases diminishing or eliminating the need for continuing medical treatment. Review of the published studies showed that following thymectomy, MG patients were 1.7 times as likely to improve and twice as likely to attain remission as those who did not have surgical thymectomy. In view of these potential benefits and of the negligible risk in skilled hands, thymectomy has gained widespread acceptance in the treatment of MG. It is the consensus that thymectomy should be carried out in all patients with generalized MG who are between the ages of puberty and at least 55 years. Whether thymectomy should be recommended in children, in adults >55 years of age, and in patients with weakness limited to the ocular muscles is still a matter of debate. There is also evidence that patients with MuSK antibody–positive MG respond less well to thymectomy than those with AChR antibody. Thymectomy must be carried out in a hospital where it is performed regularly and where the staff is experienced in the pre- and postoperative management, anesthesia, and surgical techniques of total thymectomy. Thymectomy should never be carried out as an emergency procedure, but only when the patient is adequately prepared. If necessary, treatment with IVIg or plasmapheresis may be used before surgery, but it is helpful to try to avoid immunosuppressive agents because of the risk of infection.
Immunosuppression using one or more of the available agents is effective in nearly all patients with MG. The choice of drugs or other immunomodulatory treatments should be guided by the relative benefits and risks for the individual patient and the urgency of treatment. It is helpful to develop a treatment plan based on short-term, intermediate-term, and long-term objectives. For example, if immediate improvement is essential either because of the severity of weakness or because of the patient’s need to return to activity as soon as possible, IVIg should be administered or plasmapheresis should be undertaken. For the intermediate term, glucocorticoids and cyclosporine or tacrolimus generally produce clinical improvement within a period of 1–3 months. The beneficial effects of azathioprine and mycophenolate mofetil usually begin after many months (as long as a year), but these drugs have advantages for the long-term treatment of patients with MG. There is a growing body of evidence that rituximab is effective in many MG patients, especially those with MuSK antibody.
Glucocorticoid Therapy Glucocorticoids, when used properly, produce improvement in myasthenic weakness in the great majority of patients. To minimize adverse side effects, prednisone should be given in a single dose rather than in divided doses throughout the day. The initial dose should be relatively low (15–25 mg/d) to avoid the early weakening that occurs in up to one-third of patients treated initially with a high-dose regimen. The dose is increased stepwise, as tolerated by the patient (usually by 5 mg/d at 2- to 3-day intervals), until there is marked clinical improvement or a dose of 50–60 mg/d is reached. This dose is maintained for 1–3 months and then is gradually modified to an alternate-day regimen over the course of an additional 1–3 months; the goal is to reduce the dose on the “off day” to zero or to a minimal level. Generally, patients begin to improve within a few weeks after reaching the maximum dose, and improvement continues to progress for months or years. The prednisone dosage may gradually be reduced, but usually months or years may be needed to determine the minimum effective dose, and close monitoring is required. Few patients are able to do without immunosuppressive agents entirely. Patients on long-term glucocorticoid therapy must be followed carefully to prevent or treat adverse side effects. The most common errors in glucocorticoid treatment of myasthenic patients include (1) insufficient persistence—improvement may be delayed and gradual; (2) tapering the dosage too early, too rapidly, or excessively; and (3) lack of attention to prevention and treatment of side effects.
The management of patients treated with glucocorticoids is discussed in Chap. 406.
Other Immunosuppressive Drugs Mycophenolate mofetil, azathioprine, cyclosporine, tacrolimus, rituximab, and occasionally cyclophosphamide are effective in many patients, either alone or in various combinations.
Mycophenolate mofetil has become one of the most widely used drugs in the treatment of MG because of its effectiveness and relative lack of side effects. A dose of 1–1.5 g bid is recommended. Its mechanism of action involves inhibition of purine synthesis by the de novo pathway. Since lymphocytes have only the de novo pathway, but lack the alternative salvage pathway that is present in all other cells, mycophenolate inhibits proliferation of lymphocytes but not proliferation of other cells. It does not kill or eliminate preexisting autoreactive lymphocytes, and therefore clinical improvement may be delayed for many months to a year, until the preexisting autoreactive lymphocytes die spontaneously. The advantage of mycophenolate lies in its relative lack of adverse side effects, with only occasional production of GI symptoms, rare development of leukopenia, and very small risks of malignancy or progressive multifocal leukoencephalopathy inherent in nearly all immunosuppressive treatments. Although two published studies did not show positive outcomes, most experts attribute the negative results to flaws in the trial designs, and mycophenolate is widely used for long-term treatment of myasthenic patients.
Until recently, azathioprine has been the most commonly used immunosuppressive agent for MG because of its relative safety in most patients and long track record. Its therapeutic effect may add to that of glucocorticoids and/or allow the glucocorticoid dose to be reduced. However, up to 10% of patients are unable to tolerate azathioprine because of idiosyncratic reactions consisting of flu-like symptoms of fever and malaise, bone marrow suppression, or abnormalities of liver function. An initial dose of 50 mg/d should be used for several days to test for these side effects. If this dose is tolerated, it is increased gradually to about 2–3 mg/kg of total body weight, or until the white blood count falls to 3000–4000/μL. The beneficial effect of azathioprine takes 3–6 months to begin and even longer to peak. In patients taking azathioprine, allopurinol should never be used to treat hyperuricemia. Because the two drugs share a common degradation pathway; the result may be severe bone marrow suppression due to increased effects of the azathioprine.
The calcineurin inhibitors cyclosporine and tacrolimus (FK506) are approximately as effective as azathioprine and are being used increasingly in the management of MG. Their beneficial effect appears more rapidly than that of azathioprine. Either drug may be used alone, but they are usually used as an adjunct to glucocorticoids to permit reduction of the glucocorticoid dose. The usual dose of cyclosporine is 4–5 mg/kg per day, and the average dose of tacrolimus is 0.07–0.1 mg/kg per day, given in two equally divided doses (to minimize side effects). Side effects of these drugs include hypertension and nephrotoxicity, which must be closely monitored. “Trough” blood levels are measured 12 h after the evening dose. The therapeutic range for the trough level of cyclosporine is 150–200 ng/L, and for tacrolimus, it is 5–15 ng/L.
Rituximab (Rituxan) is a monoclonal antibody that binds to the CD20 molecule on B lymphocytes. It has been widely used for the treatment of B cell lymphomas and has also proven successful in the treatment of several autoimmune diseases including rheumatoid arthritis, pemphigus, and some IgM-related neuropathies. There is now an extensive literature on the benefit of rituximab in MG. It is particularly effective in MuSK antibody–positive MG, although some patients with AChR antibody MG respond to it as well. The usual dose is 375 mg/m2, given IV in 4 weekly infusions, or 1 g, given IV on two occasions 2 weeks apart.
For the occasional patient with MG that is genuinely refractory to optimal treatment with conventional immunosuppressive agents, a course of high-dose cyclophosphamide may induce long-lasting benefit by “rebooting” the immune system. At high doses, cyclophosphamide eliminates mature lymphocytes but spares hematopoietic precursors (stem cells), because they express the enzyme aldehyde dehydrogenase, which hydrolyzes cyclophosphamide. At present, this procedure is reserved for refractory patients and should be administered only in a facility fully familiar with this approach. Maintenance immunotherapy after rebooting is usually required to sustain the beneficial effect.
PLASMAPHERESIS AND INTRAVENOUS IMMUNOGLOBULIN
Plasmapheresis has been used therapeutically in MG. Plasma, which contains the pathogenic antibodies, is mechanically separated from the blood cells, which are returned to the patient. A course of five exchanges (3–4 L per exchange) is generally administered over a 10- to 14-day period. Plasmapheresis produces a short-term reduction in anti-AChR antibodies, with clinical improvement in many patients. It is useful as a temporary expedient in seriously affected patients or to improve the patient’s condition prior to surgery (e.g., thymectomy).
The indications for the use of IVIg are the same as those for plasma exchange: to produce rapid improvement to help the patient through a difficult period of myasthenic weakness or prior to surgery. This treatment has the advantages of not requiring special equipment or large-bore venous access. The usual dose is 2 g/kg, which is typically administered over 5 days (400 mg/kg per day). If tolerated, the total dose of IVIg can be given over a 3- to 4-day period. Improvement occurs in ~70% of patients, beginning during treatment, or within a week, and continuing for weeks to months. The mechanism of action of IVIg is not known; the treatment has no consistent effect on the measurable amount of circulating AChR antibody. Adverse reactions are generally not serious but may include headache, fluid overload, and rarely aseptic meningitis or renal failure. IVIg should rarely be used as a long-term treatment in place of rationally managed immunosuppressive therapy. Unfortunately, there is a tendency for physicians unfamiliar with immunosuppressive treatments to rely on repeated IVIg infusions, which usually produce only intermittent benefit, do not reduce the underlying autoimmune response, and are very costly. The intermediate and long-term treatment of myasthenic patients requires other methods of therapy outlined earlier in this chapter.
MANAGEMENT OF MYASTHENIC CRISIS
Myasthenic crisis is defined as an exacerbation of weakness sufficient to endanger life; it usually consists of respiratory failure caused by diaphragmatic and intercostal muscle weakness. Crisis rarely occurs in properly managed patients. Treatment should be carried out in intensive care units staffed with teams experienced in the management of MG, respiratory insufficiency, infectious disease, and fluid and electrolyte therapy. The possibility that deterioration could be due to excessive anticholinesterase medication (“cholinergic crisis”) is best excluded by temporarily stopping anticholinesterase drugs. The most common cause of crisis is intercurrent infection. This should be treated immediately, because the mechanical and immunologic defenses of the patient can be assumed to be compromised. The myasthenic patient with fever and early infection should be treated like other immunocompromised patients. Early and effective antibiotic therapy, respiratory assistance (preferably noninvasive, using bilevel positive airway pressure), and pulmonary physiotherapy are essentials of the treatment program. As discussed above, plasmapheresis or IVIg is frequently helpful in hastening recovery.
DRUGS TO AVOID IN MYASTHENIC PATIENTS
Many drugs have been reported to exacerbate weakness in patients with MG (Table 461-4), but not all patients react adversely to all of these. Conversely, not all “safe” drugs can be used with impunity in patients with MG. As a rule, the listed drugs should be avoided whenever possible, and myasthenic patients should be followed closely when any new drug is introduced.
|
DRUGS WITH INTERACTIONS IN MYASTHENIA GRAVIS (MG) |
PATIENT ASSESSMENT
To evaluate the effectiveness of treatment as well as drug-induced side effects, it is important to assess the patient’s clinical status systematically at baseline and on repeated interval examinations. Because of the variability of symptoms of MG, the interval history and physical findings on examination must be taken into account. The most useful clinical tests include forward arm abduction time (up to a full 5 min), spirometry with determination of forced vital capacity, range of eye movements, and time to development of ptosis on upward gaze. Manual muscle testing or, preferably, quantitative dynamometry of limb muscles, especially proximal muscles, is also important. An interval form can provide a succinct summary of the patient’s status and a guide to treatment results; an abbreviated form is shown in Fig. 461-3. A progressive reduction in the patient’s AChR antibody level also provides clinically valuable confirmation of the effectiveness of treatment; conversely, a rise in AChR antibody levels during tapering of immunosuppressive medication may predict clinical exacerbation. For reliable quantitative measurement of AChR antibody levels, it is best to compare antibody levels from prior frozen serum aliquots with current serum samples in simultaneously run assays.
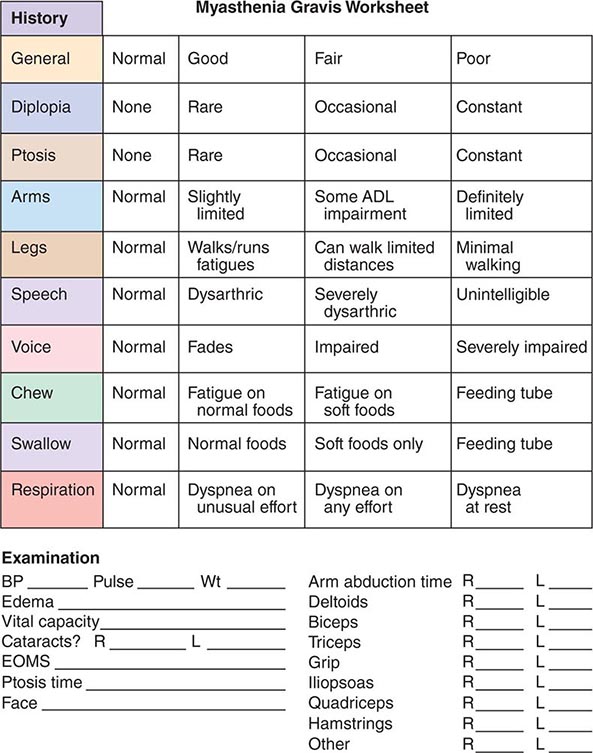
FIGURE 461-3 Abbreviated interval assessment form for use in evaluating treatment for myasthenia gravis.