Chapter 203 Peripheral Nerve Tumors of the Extremities
In the United States, it is estimated that annually six out of every 1 million people undergo surgery for peripheral nerve tumors.1 The neurosurgeon is often consulted to diagnose and manage tumors involving the peripheral nerves of the extremities. Intraneural tumors arise from components of the nerve sheath (Schwann cells, perineural cells, fibroblasts), neurons, or invading metastases and cause expansion of the nerve. Extraneural tumors arise from adjacent structures and can compress or infiltrate the nerve. The majority of tumors involving the peripheral nervous system are benign tumors of the nerve sheath (schwannoma or neurofibroma). Tumors can arise sporadically or be associated with genetic syndromes that produce multiple tumors such as neurofibromatosis or schwannomatosis.
Presentation
Patients with a peripheral nerve tumor usually present to clinic with a visible or palpable mass (may be symptomatic or asymptomatic), sensory alterations, pain, weakness, or autonomic dysfunction, or the tumor may be an incidental finding (Table 203-1). Many peripheral nerve tumors are first noticed as asymptomatic lumps. There may be swelling or a history of swelling around the lump. Sensory alterations might consist of anesthesia, hypoesthesia, hyperesthesia, paresthesias, or dysesthesia in the distribution of the affected nerve. Stimulation of the skin with innocuous mechanical or thermal stimuli might elicit pain (allodynia) or patients might have a heightened response to painful stimuli (hyperalgesia). The area surrounding the tumor might spontaneously ache or be tender to palpation. Palpation of the mass might elicit paresthesias or dysesthias. Involvement by the tumor of a motor or mixed nerve might produce frank weakness or, more commonly, subtle functional impairment with fine motor tasks. Autonomic dysfunction can manifest as alterations in the coloration, texture, or temperature of the skin or in the amount of sweat. On occasion, peripheral nerve tumors cause complex regional pain syndrome.
TABLE 203-1 Common Presenting Symptoms of an Extremity Peripheral Nerve Tumor
| Visible or palpable mass |
| Pain |
| Altered sensation (anesthesia, hypoesthesia, hyperesthesia, paresthesia, or dysesthesia) |
| Weakness |
| Autonomic dysfunction (sweating, discoloration, edema, temperature changes) |
| Incidental finding on imaging studies |
History and Examination
A thorough physical examination should be performed on all patients with peripheral nerve tumors. Patients should be examined for diagnostic signs of NF1 (Table 203-2). The neurologic examination includes testing the cranial nerves as well as the motor and sensory function and deep tendon reflexes in all four extremities. A systematic and reproducible examination permits detection of subtle clinical deficits due to the tumor, pathology at higher levels of the neuraxis, or previously undetected tumors elsewhere in the body. Changes in motor function are a late finding; often examination detects only subtle sensory changes or no deficits at all. Discoloration or atrophy of the skin, abnormal sweating, temperature changes, nailbed atrophy, or distal swelling may be findings of autonomic dysfunction.
TABLE 203-2 National Institutes of Health Criteria for Neurofibromatosis Type 155
Imaging
Historically, the diagnosis of peripheral nerve tumors was based on history, physical examination, and electrophysiologic studies. Although soft tissue masses are not visible on plain film radiographs, these images can show secondary skeletal changes due to tumor mass effect or calcifications intrinsic to the tumor. Computed tomography (CT) also can demonstrate secondary skeletal changes and intrinsic calcifications and offers improved resolution of soft tissues. CT is the imaging modality of choice for patients with contraindications to MRI. Since the 1980s, MRI has emerged as the gold standard for imaging peripheral nerve tumors.2 MRI permits detailed visualization of soft tissue at high resolution and can demonstrate the presence of a mass, the anatomy of involved peripheral nerves, and their relationships to adjacent structures such as major blood vessels and joints. The advent of high-resolution MR neurography (MRN) combined with the expansion of the number of neuroradiologists experienced with performing and interpreting these studies has significantly affected the field since the turn of the 21st century.3–5 MRN offers improved contrast and resolution that facilitates determining the precise location, size, shape, and resectability of peripheral nerve masses.5
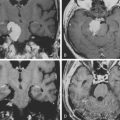
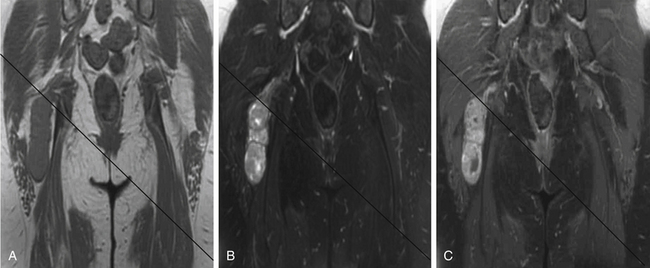
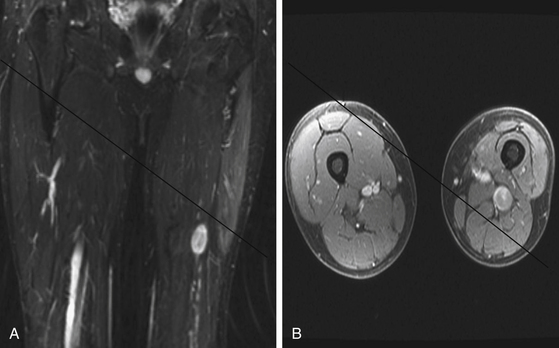
A combination of T1, T2, STIR (short tau inversion recovery), and gadolinium-enhanced scans provide clues regarding the nature of the tumor and can help distinguish benign and malignant lesions. MRI provides crucial information for preoperative planning and aids in determining the specific pathology of the tumor. Further information can be obtained regarding cellular composition and areas of hemorrhage or cysts. For example, benign nerve sheath tumors are often isointense or slightly hyperintense on T1, hyperintense on T2, and heterogeneously enhancing (Figs. 203-1 and 203-2). The majority of neurofibromas, and some schwannomas, display increased central and decreased peripheral T1 signal intensity (the “target sign”) and an opposite pattern of increased peripheral and decreased central T2 signal intensity.6 Patients with NF1 or schwannomatosis often have multiple tumors throughout their bodies and can have multiple discrete nodules on an affected nerve or develop large plexiform tumors (Fig. 203-3).
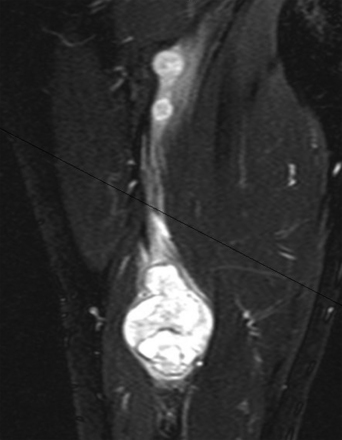
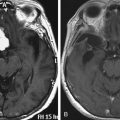
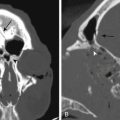
The capacity to accurately assess tumor grade or malignancy based on MRI remains limited. In general, malignant tumors heterogeneously enhance, are less discrete, and are more likely to have areas of necrosis or hemorrhage (Fig. 203-4) However, benign tumors can also have all of these characteristics. A rapid increase in size of a tumor on serial MRI can suggest malignancy, but small increases in tumor volume are difficult to detect.
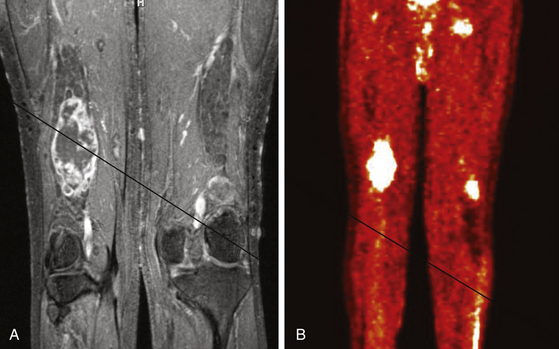
FIGURE 203-4 Magnetic resonance image (MRI) and 18FDG-PET (18fluorodeoxyglucose positron emission tomography) of a malignant peripheral nerve sheath tumor (MPNST) of the sciatic nerve. A, Coronal T1-weighted post-gadolinium image demonstrates a large, infiltrative, heterogeneously enhancing tumor of the right sciatic nerve with areas of necrosis. B, Coronal FDG-PET scan demonstrating marked uptake within the right sciatic nerve tumor.
Metabolic imaging using with 18fluorodeoxyglucose (18FDG) positron emission tomography (PET) often adds valuable information regarding the grade of a tumor (see Fig. 203-4). Ferner and colleagues introduced the use of 18FDG-PET to detect malignant transformation of benign plexiform neurofibromas to MPNSTs.7 Several small series have reported sensitivity ranging from 89% to 100% and specificity ranging from 83% to 95% depending on the time of scanning after injection and the cutoffs used for standard uptake values.8–10 The addition of CT anatomic imaging to 18FGD PET can facilitate targeting biopsies to metabolic hot spots, to further augment diagnostic sensitivity.11
Benign Peripheral Nerve Tumors
The majority of peripheral nerve tumors are benign. Intraneural tumors can arise from the nerve sheath (schwannoma, neurofibroma, intraneural perineuroma) or other intrinsic structures of the nerve (desmoid tumor, intraneural ganglia, lipomatous tumors, ganglioneuroma, metastases, and others). Extraneural tumors represent a diverse population of entities that can arise from or metastasize to adjacent soft tissue, bones, or joints and compress or invade peripheral nerves. This section focuses on schwannomas and neurofibromas.
Schwannoma
Schwannomas (neurilemmomas) are benign tumors that arise from the nerve sheath. They are typically solitary and slow growing. A schwannoma can arise from any peripheral nerve with a Schwann cell, including distal portions of cranial nerves. Sporadic schwannomas can occur at any age but are most common in the third to sixth decades of life.12
Bilateral acoustic schwannomas are pathognomonic for the diagnosis of NF2, but there is no association between peripheral schwannoma and NF1.13 Since the turn of the 21st century the diagnosis of schwannomatosis has gained acceptance for patients with multiple peripheral nerve schwannomas in the absence of acoustic schwannomas. The specific diagnostic criteria for schwannomatosis are listed in Table 203-3. The annual incidence of new diagnoses has been estimated to be 1 in 1,808,300 in a Finnish population-based study, which would correlate to a birthrate of about 1 in 80,000.14 Schwannomatosis usually occurs sporadically, but cases of autosomal dominant transmission have been reported.15 The tumors that develop may be conventional globular schwannomas or plexiform and arise throughout the body or may be limited to a single segment or nerve. It is currently unknown whether the diagnosis of schwannomatosis alters the risk of malignant transformation of a schwannoma.
| Definite Schwannomatosis |
Two or more pathologically proved schwannomas, without symptoms of cranial nerve
VIII dysfunction at >30 years of age.
Two or more pathologically proved schwannomas in an anatomically limited distribution (single limb or segment of the spine) without symptoms of cranial nerve VIII dysfunction at any age
Pathology
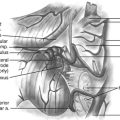
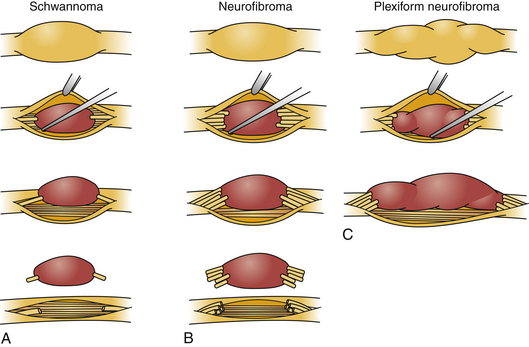
Schwannomas can be broadly classified into four subcategories: conventional schwannoma (the focus of this section); cellular schwannoma, a benign tumor that histologically resembles MPNST; plexiform schwannoma, which simulates plexiform neurofibroma or in its cellular form, MPNST; melanocytic schwannoma, which is often mistaken for a melanoma.16 Conventional schwannomas are usually globoid and homogeneously firm, but they can have ballotable cystic areas. On gross examination the tumor has a well-defined capsule, appears shiny, and is tan-yellow. Histologically, they are distinguished from other nerve sheath tumors by being composed exclusively of Schwann cells; perineural cells and fibroblasts are not present.17 Diagnostic features include a fibrous capsule, hyaline vessels, cellular (Antoni A) and loose-textured (Antoni B) areas, and Verocay bodies (opposing rows of spindle nuclei separated by anucleate rows of eosinophilic processes).16 Unlike neurofibromas, schwannomas arise within the nerve sheath and displace the fascicles as opposed to enveloping them (Fig. 203-5). However, often a small fascicle entering or exiting the proximal and distal poles of the tumor can be identified.
Molecular Biology
Mutations in the NF2 gene at the 22q12.2 locus are found in sporadic schwannomas and those in patients with NF2.18 The schwannomas that develop in patients with schwannomatosis show a multitude of truncations in the NF2 gene, and multiple tumors from an individual patient can each have unique mutations.15,19 The NF2 tumor suppressor gene codes for merlin (schwannomin), a cell membrane protein that functions in intercellular signaling and helps mediate growth arrest. The loss of both wild type NF2 alleles (two-hit hypothesis of tumorigenesis) results in marked reduction in merlin protein and tumor development.20
Management
The natural history of a schwannoma is to be slow growing, and thus intervention usually is not warranted until symptoms are present. The indications for surgical resection of benign peripheral nerve tumors are listed in Table 203-4. Unless the tumor is massive, or prior surgery has been performed, operative excision can often be accomplished with little or no injury to the parent nerve. Recurrences after removal are infrequent. Although radiosurgery is commonly used to treat acoustic schwannomas, there is only one reported case of stereotactic radiosurgery for a peripheral nerve schwannoma.21 Reasons for this discrepancy include the significantly lower morbidity associated with surgical resection of peripheral nerve schwannomas compared to acoustic schwannomas and the concern that radiation might induce malignant transformation.
TABLE 203-4 Indications for Surgical Intervention for Benign Peripheral Nerve Tumors
| Neurologic symptoms |
| Pain |
| Concern for malignancy |
| Cosmesis |
| To obtain diagnosis of pathology |
Tumors with imaging findings characteristic of benign peripheral nerve tumors do not require biopsy unless unusual clinical features exist. Biopsy is indicated when there is a question of diagnosis and opting for serial MRI might place the patient at risk. Fine-needle aspiration, used in conjunction with needle core biopsy, has been used to distinguish a large neurofibroma from a neurogenic sarcoma,22 but we have seen several cases in which needle biopsy failed to diagnose malignancy owing to sampling error. In addition, percutaneous biopsies risk injury to nerve fascicles coursing over the surface of the tumor.
Surgical Technique
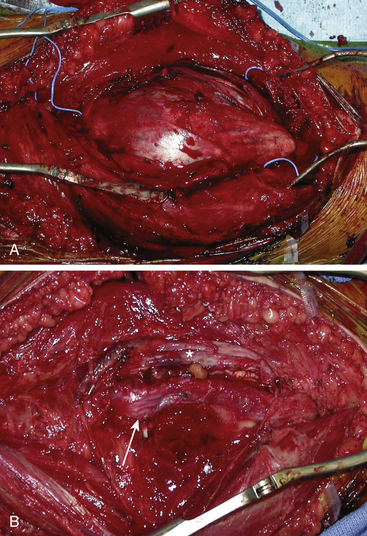
In some cases the tumor is enveloped with fascicles, and the appearance on first inspection suggests that the tumor cannot be resected. An internal neurolysis with patient separation of the fascicles will reveal that the tumor can be separated from the fascicles (Fig. 203-6). Loupe magnification or an operating microscope is required to identify the fascicles coursing over the tumor. A nerve stimulator is used to help identify an afascicular entry point. An afascicular dissection plane is established by gently separating and elevating the fascicles from the tumor capsule. For very large tumors, dissection may be facilitated by entering the capsule and debulking the tumor from within using an ultrasonic aspirator. On the rare occasion when a significant fascicle is sacrificed, nerve grafting should be considered.
Neurofibroma
Neurofibromas are benign tumors that arise from the nerve sheath. They can arise sporadically as solitary tumors but are usually part of NF1 (von Recklinghausen’s disease). NF1 is one of the most common autosomal dominant disorders of the nervous system, occurring with an estimated incidence of 1 in 2500 to 3000 persons independent of ethnicity, race, and gender.23 The diagnostic criteria for NF1 are listed in Table 203-2. The majority of cases of NF1 are diagnosed in children.
Pathology
Neurofibromas can be broadly classified into five subcategories. Localized cutaneous neurofibromas grow as hyperpigmented nodular lesions of the skin and are the most common. Localized intraneural neurofibromas grow within peripheral nerves, envelope nerve fascicles, and cause focal fusiform dilation. Plexiform neurofibromas are multinodular tumors that arise in peripheral nerves or at the level of nerve plexuses and cause longitudinal sausage-like enlargement of the parent nerve(s). They are rarely seen in patients without the diagnosis of NF1. Diffuse neurofibromas involve skin and subcutaneous tissue but are more extensive than localized cutaneous neurofibromas. Massive soft tissue neurofibromas infiltrate the soft tissue of an extremity and can cause elephantiasis neuromatosa, gigantism, or disfiguring soft tissue masses, and can have underlying intraneural or plexiform neurofibromas.24
The microscopic appearance of neurofibromas varies slightly across the different subcategories. Intraneural neurofibromas likely arise from NF1-deficient Schwann cells25 and are composed of a mixture of Schwann cells, fibroblasts, perineural cells, and mast cells. A crucial histologic feature is that, unlike schwannomas, the tumor involves the cross section of the nerve fascicle, resulting in a lack of surgical cleavage plane between normal nerve fibers and tumor (see Fig. 203-5). Proliferating cells spread along the nerve fibers and expand the fascicle. Spared nerve fibers are often centrally situated within the nerve. It is common to observe coarse collagen bundles and mucin pools. Plexiform neurofibromas consist of multiple intraneural fibromas that appear to form a tangled mass.
Molecular Biology
Patients with NF1 are born with a heterozygous mutation in the NF1 gene located 17q11.2 locus.26–28 Sporadic neurofibromas represent somatic mutations to both copies of the NF1 gene. Tumorigenesis results from loss of the normal allele in NF1+/− Schwann cells. The NF1 gene codes for neurofibromin, an intracellular signaling protein that contributes to inactivating the Ras-GTP signaling cascade and positively regulating cyclic adenosine monophosphate (cAMP) levels.29 The absence of functional neurofibromin results in increased cell proliferation and survival.30
Management
The management of neurofibromas reflects the severity of the symptoms. Localized intraneural neurofibromas usually occur as slowly enlarging, painless, soft, mobile masses. Plexiform neurofibromas should be monitored closely for changes in size or the development of symptoms. As the tumors grow, symptoms arise from axonal compression with neurologic deficits (including pain) or secondary to compression of neighboring tissues. The indications for surgical intervention are the same as for schwannomas (see Table 203-4). In contrast to schwannomatosis, because of the elevated risk of malignant transformation in NF1, percutaneous biopsy is used when there is rapid tumor growth or rapid onset of pain.
Surgical Technique
Once the region of the nerve containing the tumor is exposed, magnification is employed. The actual tumor might not be visible on the surface of the nerve, and an internal neurolysis is required. As the epineurium is opened, the tumor can be identified. Often there is a pseudocapsule over the tumor that needs to be opened. Once the dissection is carried out to the correct plane, some uninvolved fascicles can usually be identified and protected. Electrical stimulation is used to assess the function of fascicles enlarged by tumor. Unlike the fascicle entering a schwannoma, those entering a neurofibroma are often still functioning. Removal of the tumor inevitably requires sacrifice of one or more fascicles (see Fig. 203-5). The tumor then can be gently dissected away from the remaining uninvolved fascicles.
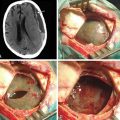
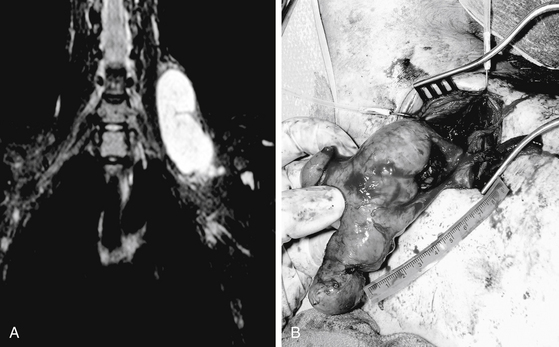
The goal of surgical intervention for extensive plexiform neurofibromas is often to debulk the tumor and alleviate the symptoms caused by neural compression (Fig. 203-7, and see Fig. 203-5). Closure of the wound is achieved in layers with care not to entrap the nerve. If a nerve graft was required, a splint is employed for 2 to 4 weeks.
Other Benign Tumors
Intraneural perineurioma is a benign peripheral nerve tumor arising from perineurial cells. Other names for this tumor include hypertrophic (mono)neuropathy, interstitial hypertrophic neuropathy, pseudo-onion bulb (mono)neuropathy, hypertrophic neurofibroma, and intraneural neurofibroma.31 Intraneural perineurioma typically occurs in adolescents or young adults as a slow-growing, painless mononeuropathy that characteristically causes progressive loss of motor function and, uncommonly, a sensory deficit.32 This tumor produces discrete fusiform dilation of the involved nerve, with tumor enveloping and dilating the fascicles. Immunohistologic examination typically shows pseudo–onion bulbs of proliferating perineurial cells that are epithelial membrane antigen (EMA) positive and S100 protein negative. Molecular analysis of tumor tissue often demonstrates abnormalities on the long arm of chromosome 22.33 Surgical intervention is indicated for symptomatic perineuriomas and may consist of neurolysis, resection, and/or grafting. Local recurrence is very rare, and there are no reported cases of metastasis.
Intraneural ganglion cysts are benign soft tissue masses involving peripheral nerves at locations in proximity to joints. The most common sites are the ulnar nerve in the cubital tunnel and peroneal nerve at the knee. Patients present with palpable masses near the joint that may be associated with local tenderness, radiating pain, or neurologic dysfunction. The pathogenesis remains controversial. Proposed theories include a role for embryonal rests,34 cystic degeneration of a tumor,35 organization of a hematoma,36 or focal degeneration of epi- or perineural tissue.37 The most comprehensive theory of pathogenesis has been proposed by Spinner and colleagues.38 The unified (articular) theory proposes that intraneural ganglia are derived from degenerative synovial joints. Joint fluid penetrates through a capsular rent and dissects within the epineurium of nerves by way of articular branches, typically causing intrinsic compression of nerve. The size, shape, and direction of propagation of these intraneural cysts are influenced by pressure fluxes. Surgical resection is indicated for symptomatic lesions, but historically ganglion cysts have a high recurrence rate. Surgical resection involves dissecting the cyst and its articular branch, performing longitudinal epineurotomy to decompress the intraneural portion of the cyst, disconnecting the articular branch near the joint, and disarticulating the joint (for cysts involving the tibiofibular joint and peroneal nerve).39
Benign fatty tumors involving peripheral nerve are encountered on occasion. Lipomatous tumors fall into four general classes: well-encapsulated intraneural lipomas, diffusely infiltrating fibrofatty tumors (lipofibromatous hamartomas), macrodystrophia lipomatosa (an infiltrating fibrofatty lesion with associated focal macrodactyly), and extraneural soft tissue lipomas that can compress peripheral nerves.40 Intraneural lipomas arise from normally occurring intraneural adipose cells and have a fine capsule. Lipofibromatous hamartomas are composed of fibrous fatty tissue intimately associated with neural elements. Surgical intervention is indicated for symptomatic fatty tumors. Intraneural lipomas can usually be safely resected. Lipofibromatous hamartomas may be extensive, and resection may be very challenging, with a high risk for complications; neurolysis is often a safer and beneficial option.
Malignant Peripheral Nerve Sheath Tumors
MPNSTs are rare neurogenic sarcomas of ectomesenchymal origin that account for approximately 3% to 10% of soft tissue sarcomas. The World Health Organization (WHO) coined the term malignant peripheral nerve sheath tumor to replace previous heterogeneous and often confusing terminology, such as malignant schwannoma, malignant neurilemmoma, and neurofibrosarcoma, used to describe tumors of neurogenic origin and similar biologic behavior. Both sporadic and NF1-associated MPNSTs arise in neurofibromas, in normal peripheral nerves, or rarely in other tumors, including schwannoma, ganglioneuroma, and pheochromocytoma.41 Although rare in the general population (incidence about 0.001%), MPNSTs arise in 5% to 42% of patients with NF1.42–44
Pathology
MPNSTs are typically firm, fusiform tumors intimately adherent to the nerve. Histologically, there is great variability among MPNSTs, which often provides a diagnostic challenge. Common histologic findings include highly cellular and mitotically active spindle cells (more than 4 mitoses per 10 high power fields), abrupt variations in cellularity, increased perivascular cellularity, herringbone growth patterns, nuclear enlargement, hyperchromasia, and necrosis.45 Histologic variants of MPNST include epithelioid, glandular, and rhabdomyosarcomatous. Epithelioid MPNSTs are composed of malignant epithelioid cells. Glandular MPNST demonstrate cytokeratin-positive glands, often associated with mucin. Rhabdomyosarcomatous MPNSTs show malignant cells resembling embryonal rhabdomyosarcoma. Although there is no consensus on MPNST grading, they may be classified high, intermediate, or low based on the degree of cellularity, nuclear pleomorphism, anaplasia, mitotic rate, microvascular proliferation, and degree of necrosis and invasion.46
Molecular Biology
The loss of both NF1 alleles is found in the majority of MPNSTs. However, the loss of NF1 alone in both sporadic and NF1 tumors does not result in MPNST formation; additional genetic hits are required for malignant transformation.47 Most MPNSTs, but not neurofibromas, have alterations of the INK4A gene, which encodes two growth inhibitors, p16INK4A and p19ARF.47,48 Alterations in the Ras and mTOR signaling pathways have been demonstrated in MPNSTs.49 Overexpression of p53 is seen in many types of human tumors, including MPNSTs (but not benign peripheral nerve sheath tumors).50 Interestingly, TP53 tumor suppressor gene mutations and p53 overexpression are more common in sporadic than in NF1-related tumors.47
Management
Patients are often referred after a fine needle or core biopsy. In our experience the benefit of these biopsy techniques is limited because they can provide nondiagnostic samples or missed diagnosis due to a sampling error in a heterogeneous tumor, or they can produce new neural deficits. Open biopsy and surgical resection is the mainstay for management of MPNST. If there is suspicion of malignancy from prior needle biopsy or intraoperative findings and frozen section then the nerve or element of the plexus should be completely resected with negative margins. Inevitably, a neurologic deficit results from resection of the involved nerve. Nerve grafting has a low success rate because proximal nerves are not amenable to re-innervation with grafting, the natural history of the disease may be too short for effective reinnervation, and postoperative irradiation of the tumor, and thus the graft site invariably kills the graft.51
The ability to achieve tumor-free margins provides improved prognosis.52 A curative resection consists of complete tumor resection with 2 cm of tumor-free margins in all directions.42 For most MPNSTs in the extremities, we advocate limb-sparing radical resection. MPNSTs that arise at the level of the plexus or farther proximal present additional challenges because radical resection produces significant morbidity because it would require sacrifice of adjacent nerves and the major extremity vessels. For proximal MPNSTs, maximal safe resection combined with implantation of radioactive seeds is an option that can provide adequate local control. Overall, limb-sparing radical resection provides local control in 85% to 90% of soft tissue sarcomas.53
Adjuvant therapy options including radiation and chemotherapy may accompany surgical intervention but remain a topic of controversy. Radiation has been shown to reduce local recurrences and is advocated for MPNSTs even if adequate surgical margins are obtained. MPNSTs are poorly responsive to chemotherapy. Adjuvant chemotherapy may be used as a palliative measure when systemic metastasis has occurred.42
The prognosis for patients with MPNST remains poor. The 10-year survival rate after definitive surgery is less than 50%. Negative prognostic factors include tumor size (larger than 5 cm), tumor grade, positive margins, and proximal location. NF1 does not appear to predict worse prognosis even though patients with NF1 are typically younger, have larger tumors, and have longer duration of preoperative symptoms.54
Angelov L., Davis A., O’Sullivan B., et al. Neurogenic sarcomas: experience at the University of Toronto. Neurosurgery. 1998;43(1):56-64.
Anghileri M., Miceli R., Fiore M., et al. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer. 2006;107(5):1065-1074.
Boyanton B.L.Jr., Jones J.K., Shenaq S.M., et al. Intraneural perineurioma: a systematic review with illustrative cases. Arch Pathol Lab Med. 2007;131(9):1382-1392.
Birindelli S., Perrone F., Oggionni M., et al. Rb and TP53 pathway alterations in sporadic and NF1-related malignant peripheral nerve sheath tumors. Lab Invest. 2001;81(6):833-844.
Carli M., Ferrari A., Mattke A., et al. Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J Clin Oncol. 2005;23(33):8422-8430.
Ferner R.E., Lucas J.D., O’Doherty M.J., et al. Evaluation of 18fluorodeoxyglucose positron emission tomography (18FDG PET) in the detection of malignant peripheral nerve sheath tumours arising from within plexiform neurofibromas in neurofibromatosis 1. J Neurol Neurosurg Psychiatry. 2000;68(3):353-357.
Gachiani J., Kim D., Nelson A., Kline D. Surgical management of malignant peripheral nerve sheath tumors. Neurosurg Focus. 2007;22(6):E13.
Kourea H.P., Orlow I., Scheithauer B.W., et al. Deletions of the INK4A gene occur in malignant peripheral nerve sheath tumors but not in neurofibromas. Am J Pathol. 1999;155(6):1855-1860.
Kurtkaya-Yapicier O., Scheithauer B., Woodruff J.M. The pathobiologic spectrum of Schwannomas. Histol Histopathol. 2003;18(3):925-934.
Kwok K., Slimp J., Born D., et al. The evaluation and management of benign peripheral nerve tumors and masses. In: Berger M., Prados M. Textbook of Neuro-Oncology. Philadelphia: Elsevier Saunders; 2005:535-563.
Lim M., Adler J.R. CyberKnife radiosurgery for extremity schwannomas: technical note and case report. Stereotact Funct Neurosurg. 2006;84(2-3):60-63.
Louis D.N., Ramesh V., Gusella J.F. Neuropathology and molecular genetics of neurofibromatosis 2 and related tumors. Brain Pathol. 1995;5(2):163-172.
MacCollin M., Chiocca E.A., Evans D.G., et al. Diagnostic criteria for schwannomatosis. Neurology. 2005;64(11):1838-1845.
National Institutes of Health: Consensus Development Conference Statement: neurofibromatosis. Bethesda, Md., USA, July 13-15, 1987. Neurofibromatosis. 1988;1(3):172-178.
Resnick J.M., Fanning C.V., Caraway N.P., et al. Percutaneous needle biopsy diagnosis of benign neurogenic neoplasms. Diagn Cytopathol. 1997;16(1):17-25.
Scheithauer B.W., Giannini C., Woodruff J.M. Pathology and Genetics of Tumours of the Nervous System. Lyon, France: IARC Press; 2000. 169-171
Scheithauer B.W., Woodruff J.M., Erlandson R.A. Primary malignant tumors of the peripheral nerve. In: Rosai J., Sobin L. Tumors of the Peripheral Nervous System. Washington DC: Armed Forces Institute of Pathology; 1999:303-372.
Singh T., Kliot M. Imaging of peripheral nerve tumors. Neurosurg Focus. 2007;22(6):E6.
Skovronsky D.M., Oberholtzer J.C. Pathologic classification of peripheral nerve tumors. Neurosurg Clin N Am. 2004;15(2):157-166.
Spinner R.J., Atkinson J.L., Tiel R.L. Peroneal intraneural ganglia: the importance of the articular branch. A unifying theory. J Neurosurg. 2003;99(2):330-343.
Spinner R.J., Desy N.M., Rock M.G., Amrami K.K. Peroneal intraneural ganglia. Part I. Techniques for successful diagnosis and treatment. Neurosurg. 2007;22(6):E16.
Terzis J.K., Daniel R.K., Williams H.B., Spencer P.S. Benign fatty tumors of the peripheral nerves. Ann Plast Surg. 1978;1(2):193-216.
Wallace M.R., Marchuk D.A., Andersen L.B., et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249(4965):181-186.
Woodruff J.M. Pathology of tumors of the peripheral nerve sheath in type 1 neurofibromatosis. Am J Med Genet. 1999;89(1):23-30.
Woodruff J.M., Erlandson R.A., Scheithauer B.W. Nerve sheath tumors. Am J Surg Pathol. 1995;19(5):608-611.
1. Kwok K., Slimp J., Born D., et al. The evaluation and management of benign peripheral nerve tumors and masses. In: Berger M., Prados M. Textbook of Neuro-Oncology. Philadelphia: Elsevier Saunders; 2005:535-563.
2. Kransdorf M.J., Jelinek J.S., Moser R.P.Jr., et al. Soft-tissue masses: diagnosis using MR imaging. AJR Am J Roentgenol. 1989;153(3):541-547.
3. Ferrari A., Miceli R., Meazza C., et al. Soft tissue sarcomas of childhood and adolescence: the prognostic role of tumor size in relation to patient body size. J Clin Oncol. 2009;20(27(3)):371-376.
4. Grant G.A., Britz G.W., Goodkin R., et al. The utility of magnetic resonance imaging in evaluating peripheral nerve disorders. Muscle Nerve. 2002;25(3):314-331.
5. Singh T., Kliot M. Imaging of peripheral nerve tumors. Neurosurg Focus. 2007;22(6):E6.
6. Suh J.S., Abenoza P., Galloway H.R., et al. Peripheral (extracranial) nerve tumors: correlation of MR imaging and histologic findings. Radiology. 1992;183(2):341-346.
7. Ferner R.E., Lucas J.D., O’Doherty M.J., et al. Evaluation of 18fluorodeoxyglucose positron emission tomography (18FDG PET) in the detection of malignant peripheral nerve sheath tumours arising from within plexiform neurofibromas in neurofibromatosis 1. J Neurol Neurosurg Psychiatry. 2000;68(3):353-357.
8. Cardona S., Schwarzbach M., Hinz U., et al. Evaluation of F18-deoxyglucose positron emission tomography (FDG-PET) to assess the nature of neurogenic tumours. Eur J Surg Oncol. 2003;29(6):536-541.
9. Ferner R.E., Golding J.F., Smith M., et al. [18F]2-fluoro-2-deoxy-d-glucose positron emission tomography (FDG PET) as a diagnostic tool for neurofibromatosis 1 (NF1) associated malignant peripheral nerve sheath tumours (MPNSTs): a long-term clinical study. Ann Oncol. 2008;19(2):390-394.
10. Benz M.R., Czernin J., Dry S.M., et al. Quantitative F18-fluorodeoxyglucose positron emission tomography accurately characterizes peripheral nerve sheath tumors as malignant or benign. Cancer. 2010;116(2):451-458.
11. Karabatsou K., Kiehl T.R., Wilson D.M., et al. Potential role of 18fluorodeoxyglucose-positron emission tomography/computed tomography in differentiating benign neurofibroma from malignant peripheral nerve sheath tumor associated with neurofibromatosis 1. Neurosurgery. 2009;65(suppl 4):160-170.
12. Woodruff J.M., Erlandson R.A., Scheithauer B.W. Nerve sheath tumors. Am J Surg Pathol. 1995;19(5):608-611.
13. Martuza R.L., Eldridge R. Neurofibromatosis 2 (bilateral acoustic neurofibromatosis). N Engl J Med. 1988;318(11):684-688.
14. Antinheimo J., Sankila R., Carpen O., et al. Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology. 2000;54(1):71-76.
15. Jacoby L.B., Jones D., Davis K., et al. Molecular analysis of the NF2 tumor-suppressor gene in schwannomatosis. Am J Hum Genet. 1997;61(6):1293-1302.
16. Kurtkaya-Yapicier O., Scheithauer B., Woodruff J.M. The pathobiologic spectrum of Schwannomas. Histol Histopathol. 2003;18(3):925-934.
17. Enzinger F.M., Weiss S. Enzinger and Weiss’s Soft Tissue Tumors. St. Louis: Mosby; 2001.
18. Wolff R.K., Frazer K.A., Jackler R.K., et al. Analysis of chromosome 22 deletions in neurofibromatosis type 2-related tumors. Am J Hum Genet. 1992;51(3):478-485.
19. Kaufman D.L., Heinrich B.S., Willett C., et al. Somatic instability of the NF2 gene in schwannomatosis. Arch Neurol. 2003;60(9):1317-1320.
20. Louis D.N., Ramesh V., Gusella J.F. Neuropathology and molecular genetics of neurofibromatosis 2 and related tumors. Brain Pathol. 1995;5(2):163-172.
21. Lim M., Adler J.R. CyberKnife radiosurgery for extremity schwannomas: technical note and case report. Stereotact Funct Neurosurg. 2006;84(2-3):60-63.
22. Resnick J.M., Fanning C.V., Caraway N.P., et al. Percutaneous needle biopsy diagnosis of benign neurogenic neoplasms. Diagn Cytopathol. 1997;16(1):17-25.
23. Huson S.M., Harper P.S., Compston D.A. Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain. 1988;111(Pt 6):1355-1381.
24. Woodruff J.M. Pathology of tumors of the peripheral nerve sheath in type 1 neurofibromatosis. Am J Med Genet. 1999;89(1):23-30.
25. Muir D., Neubauer D., Lim I.T., et al. Tumorigenic properties of neurofibromin-deficient neurofibroma Schwann cells. Am J Pathol. 2001;158(2):501-513.
26. Wallace M.R., Marchuk D.A., Andersen L.B., et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249(4965):181-186.
27. Upadhyaya M., Han S., Consoli C., et al. Characterization of the somatic mutational spectrum of the neurofibromatosis type 1 (NF1) gene in neurofibromatosis patients with benign and malignant tumors. Hum Mutat. 2004;23(2):134-146.
28. Cawthon R.M., Weiss R., Xu G.F., et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62(1):193-201.
29. Dasgupta B., Dugan L.L., Gutmann D.H. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase–activating polypeptide-mediated signaling in astrocytes. J Neurosci. 2003;23(26):8949-8954.
30. Viskochil D. Genetics of neurofibromatosis 1 and the NF1 gene. J Child Neurol. 2002;17(8):562-570.
31. Boyanton B.L.Jr., Jones J.K., Shenaq S.M., et al. Intraneural perineurioma: a systematic review with illustrative cases. Arch Pathol Lab Med. 2007;131(9):1382-1392.
32. Scheithauer B.W., Giannini C., Woodruff J.M. Pathology and Genetics of Tumours of the Nervous System. Lyon, France: IARC Press; 2000. :169-171
33. Emory T.S., Scheithauer B.W., Hirose T., et al. Intraneural perineurioma. A clonal neoplasm associated with abnormalities of chromosome 22. Am J Clin Pathol. 1995;103(6):696-704.
34. Brooks D.M. Nerve compression by simple ganglia. J Bone Joint Surg Br. 1952;34(3):391-400.
35. Jenkins S.A. Solitary tumours of peripheral nerve trunks. J Bone Joint Surg Br. 1952;34(3):401-411.
36. Gurdjian E.S., Larsen R.D., Lindner D.W. Intraneural cyst of the peroneal and ulnar nerves. Report of two cases. J Neurosurg. 1965;23(1):76-78.
37. Carp L., Stout A.P. A study of ganglion. With special reference to treatment. Surg Gynecol Obstet. 1928;47:460-468.
38. Spinner R.J., Atkinson J.L., Tiel R.L. Peroneal intraneural ganglia: the importance of the articular branch. A unifying theory. J Neurosurg. 2003;99(2):330-343.
39. Spinner R.J., Desy N.M., Rock M.G., Amrami K.K. Peroneal intraneural ganglia. Techniques for successful diagnosis and treatment. Neurosurg. 2007;22(6):E16.
40. Terzis J.K., Daniel R.K., Williams H.B., Spencer P.S. Benign fatty tumors of the peripheral nerves. Ann Plast Surg. 1978;1(2):193-216.
41. Scheithauer B.W., Woodruff J.M., Erlandson R.A. Primary malignant tumors of the peripheral nerve. In: Rosai J., Sobin L. Tumors of the Peripheral Nervous System. Washington DC: Armed Forces Institute of Pathology; 1999:303-372.
42. Angelov L., Davis A., O’Sullivan B., et al. Neurogenic sarcomas: experience at the University of Toronto. Neurosurgery. 1998;43(1):56-64.
43. Ducatman B.S., Scheithauer B.W., Piepgras D.G., et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57(10):2006-2021.
44. Kar M, Deo SV, Shukla NK et al. Malignant peripheral nerve sheath tumors (MPNST) clinicopathological study and treatment outcome of twenty-four cases. World J Surg Oncol. 4:55, 1006.
45. Skovronsky D.M., Oberholtzer J.C. Pathologic classification of peripheral nerve tumors. Neurosurg Clin N Am. 2004;15(2):157-166.
46. Baehring J.M., Betensky R.A., Batchelor T.T. Malignant peripheral nerve sheath tumor: the clinical spectrum and outcome of treatment. Neurology. 2003;61(5):696-698.
47. Birindelli S., Perrone F., Oggionni M., et al. Rb and TP53 pathway alterations in sporadic and NF1-related malignant peripheral nerve sheath tumors. Lab Invest. 2001;81(6):833-844.
48. Kourea H.P., Orlow I., Scheithauer B.W., et al. Deletions of the INK4A gene occur in malignant peripheral nerve sheath tumors but not in neurofibromas. Am J Pathol. 1999;155(6):1855-1860.
49. Johannessen C.M., Johnson B.W., Williams S.M., et al. TORC1 is essential for NF1-associated malignancies. Curr Biol. 2008;18(1):56-62.
50. Verdijk R.M., den Bakker M.A., Dubbink H.J., et al. TP53 mutation analysis of malignant peripheral nerve sheath tumors. J Neuropathol Exp Neurol. 2010;69(1):16-26.
51. Gachiani J., Kim D., Nelson A., Kline D. Surgical management of malignant peripheral nerve sheath tumors. Neurosurg Focus. 2007;22(6):E13.
52. Carli M., Ferrari A., Mattke A., et al. Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J Clin Oncol. 2005;23(33):8422-8430.
53. Frustaci S., Gherlinzoni F., De P.A., et al. Adjuvant chemotherapy for adult soft tissue sarcomas of the extremities and girdles: results of the Italian randomized cooperative trial. J Clin Oncol. 2001;19(5):1238-1247.
54. Anghileri M., Miceli R., Fiore M., et al. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer. 2006;107(5):1065-1074.
55. National Institutes of Health: Consensus Development Conference Statement: neurofibromatosis. Bethesda, Md., USA, July 13-15, 1987. Neurofibromatosis. 1988;1(3):172-178.
56. MacCollin M., Chiocca E.A., Evans D.G., et al. Diagnostic criteria for schwannomatosis. Neurology. 2005;64(11):1838-1845.