Chapter 98 Pathophysiology of Ocular Trauma
Introduction
Ocular trauma is a major cause of ocular morbidity and a leading cause of monocular visual loss. It is estimated that over 2 million eye injuries occur in the USA each year.1 Children and young adults are particularly at risk, and consequently, society suffers significant socioeconomic loss, as well as human loss, from these common injuries. In 1990, national projections estimated that more than 227 000 hospital days and US$175–200 million in hospital charges alone result from ocular injuries every year.2
A classification of ocular trauma with standardized terminology was developed by Kuhn and associates.3 The International Society of Ocular Trauma subsequently used this terminology to develop a classification system for mechanical injuries of the eye (Box 98.1). This system has proved useful for describing ocular trauma without miscommunication and for facilitating the delivery of optimal patient care.
Different types of ocular injuries have different pathophysiologic and therapeutic ramifications; therefore, knowledge of the initial mechanism of injury to the macula or optic nerve is critical for determining visual prognosis.4–6 Subsequent wound healing responses leading to intraocular proliferation, traction retinal detachment and posttraumatic proliferative vitreoretinopathy (PVR) can play major roles in determining the final visual outcome.4,6–9
Before the use of vitrectomy, wound closure often left vitreous incarcerated in the lips of the scleral or corneal edges of the wound, providing a scaffold for future proliferation. In the first large prospective study, conducted between 1952 and 1970, only 6% of patients with open-globe injury gained visual acuity of 5/200 or better.10 Our improved understanding of wound healing and the advent of vitrectomy techniques in the 1970s permitted more successful repair of posterior segment wounds and resulted in a marked decrease in enucleations.8,9,11,12
This chapter reviews the pathophysiology and the therapeutic aspects related to open-globe injury involving the posterior segment. (See also Chapter 91, Traumatic chorioretinopathies; Chapter 97, Pathogenesis of proliferative vitreoretinopathy, and Chapter 110, Surgery for ocular trauma.)
Anatomic change
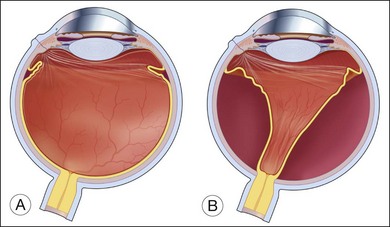
While the direct damage to the ocular tissue depends upon the nature of trauma, the integrity of the vitreoretinal barrier is commonly disrupted by ocular trauma. The vitreous is normally attached to all contiguous structures, including the posterior lens capsule, the pars plana of the ciliary body, the retina, and the optic disc; but the strength of this attachment varies. The vitreous is most firmly attached at its base and is relatively firmly attached to the lens, fovea–parafoveal area, margin of the optic nerve head, and along major retinal blood vessels.13 Weakening of the vitreoretinal interface induced by mechanical force can lead to acute posterior vitreous detachment (PVD) from the retina. When the detachment reaches a point of firmer attachment to the retina, typically the vitreous base, it exerts traction on the retina. The abrupt PVD and/or prolapse and incarceration of the vitreous through the penetrating wound predispose the eye to vitreous traction on the retina and tractional retinal detachment (Fig. 98.1).14–18
Histopathologic findings
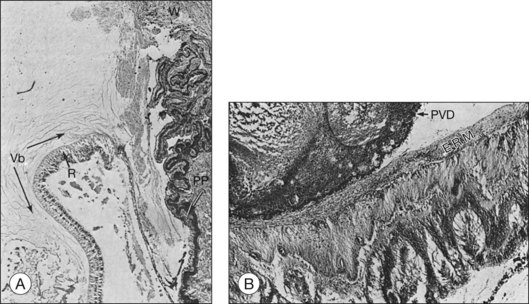
Histopathological evaluation of human eyes enucleated after penetrating trauma has shown that healing of limbal and sclera wounds was more rapid than healing of corneal wounds.17 As early as 4 days after injury, fibroblastic proliferation had occurred from the episclera, and by 1 week, from the stroma of the ciliary body and choroid. At 2 weeks, a mass of vascularized fibrous tissue joined the wound edges; and by 4–6 weeks, a dense fibrous scar had formed. This fibrous ingrowth in limbal or scleral wounds occurred in relation to vitreous incarceration and damage to the lens and/or vitreous hemorrhage. Condensation of vitreous fibrils over the vitreous base was followed by PVD. Typically, the vitreous remained attached anteriorly, and the condensed vitreous fibrils remained attached to the peripheral retina, frequently radiating from a limbal or scleral wound associated with fibroblasts (Fig. 98.2). Retinal detachment was present in most eyes. Although retinal tears were found in a few eyes, it was impossible to exclude a rhegmatogenous component since not all eyes were serially sectioned. Retinal hemorrhage and choroidal hemorrhage were common within the first 2 months and 2 weeks of injury, respectively. Epiretinal membranes were present over both the peripheral and posterior retina by 6 weeks after injury. Subretinal membranes were delicate, branching, and dendritic in appearance 1 and 2 weeks after injury and were thickened and attached to folds in the retina in later phases. Intraocular inflammatory infiltrate, mainly monocytes, was prominent in the anterior chamber or vitreous. Almost all eyes contained some macrophages, either lining the PVD or accumulated in areas of subretinal hemorrhage. Fibroblastic proliferation within the vitreous was present in the area of the wound, resulting in a cyclitic membrane in the early weeks after injury and containing fibroblast-like cells 2 months after injury.
Experimental models
Human specimens obtained from surgery, such as periretinal membranes, vitreous aspirates, and enucleated eyes, provide information about the pathophysiology of open-globe injury.17,18 However, since these specimens often represent only the advanced stages of disease and since they encompass the secondary effects of retinal detachment and PVR, their contributions to our understanding of the complex mechanism of trauma-induced injury are somewhat limited. Therefore, animal models that reproduce various types of ocular trauma have played an important role in our understanding of pathogenesis.
Cleary and Ryan developed penetrating injury models in rabbits and rhesus monkeys using a standard technique.19–24 A knife wound was made using a stab incision through the pars plana and then lengthened to 8 mm with scissors. Vitreous gel prolapsed through the wound and the vitreous face was ruptured in a manner similar to that encountered in the perforated human eye. The wound was then carefully closed and 0.5 cc of autologous blood was injected into the midvitreous. With this standardized method, traction retinal detachment was achieved with remarkable reproducibility. During the second week after injury, the blood changed to a contracted clot and the posterior vitreous detached. As early as 4 weeks after the injury, fibrous tissue grew from the wound into the vitreous, the blood clot formed fibrous tissue, and the posterior vitreous detached. Epiretinal membranes became visible around this time and progressed for up to 15 weeks. The retinal detachment typically occurred between 6 and 11 weeks after the injury. The configuration of the retinal detachment was indicative of the key processes involved. When the vitreous detached posteriorly, the anteroperipheral portion of the vitreous remained firmly attached to the peripheral retina in the area of the vitreous base. Subsequently, the peripheral retina was dragged forward toward the pars plana through its entire circumference, forming a funnel-shaped configuration with full-thickness folds.19,20,25 The presence of intravitreal blood was a potent stimulant to the development of this cascade of the wound healing process. Some 73% of 25 monkey eyes with intravitreal blood injections developed tractional retinal detachments as opposed to only 24% of eyes that received only balanced salt solution injections.19
Penetrating injuries in human eyes are often accompanied by contusions. An animal model used to study this combination employed pigs because pig sclera is sturdy enough to withstand a blunt pellet injury.26–28 An airgun delivered a pellet to the limbus of a pig eye with standardized force at impact. An 8-mm incision was then made in the pars plana as previously described. The main features were the development of intravitreal proliferation and traction retinal detachment. Additionally, subretinal hemorrhage was frequently associated, leading to subretinal fibrous membrane formation.
Animal models are useful in reproducing the findings observed in ocular trauma in humans; and furthermore, these models are valuable for evaluating surgical techniques and therapeutic drugs.29,30 For instance, the morphology of the wound and the efficacy of vitrectomy have been studied in this animal model of a contusion injury. Because of initial uveal engorgement and inflammatory swelling, early surgical intervention was hazardous. The findings support the clinical impression that vitrectomy in traumatized eyes with a substantial contusive component is best delayed for 1 or 2 weeks.26,27 (This is discussed in more detail in Chapter 110, Surgery for ocular trauma.)
A human RPE culture system is another useful means of investigating the RPE cell’s behavioral patterns, such as the migration, proliferation, and alteration of its phenotype, and the growth factors and cytokine-secreting patterns in order to understand the process of PVR.31–36
Wound healing and traumatic proliferative vitreoretinopathy
Wound healing in the eye is similar to that in other bodily tissues, consisting of three phases: exudation/inflammation, proliferation, and regeneration.37 The typical wound healing response in the anatomical setting of the eye and the vitreoretinal relationship explain the development of traction retinal detachment after penetrating ocular injury.
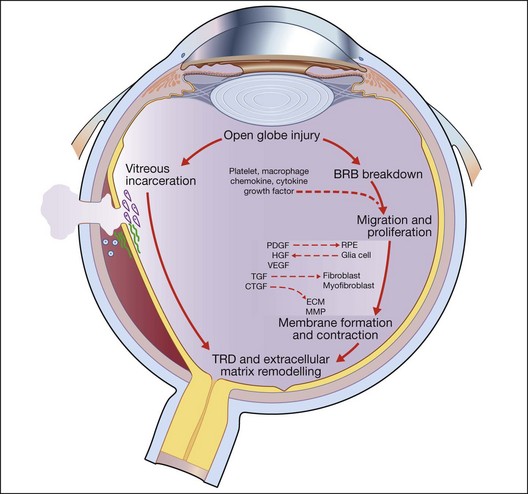
Open-globe injury results in a breakdown of the blood–retinal barrier and allows the entry of a variety of cells into the intraocular milieu, causing the expression of a variety of chemokines, inflammatory cytokines, and growth factors that affect the adjacent RPE, fibroblasts, and glial cells. In response, these previously resting cells undergo proliferation and migration as they change their pattern of gene expression, resulting in alterations of their own cytokine, extracellular matrix, and receptor profiles. Some cells – myofibroblasts, for example – proliferate and produce strong contractile forces that oppose the physiological forces that normally keep the retina attached and a tractional retinal detachment occurs. Following the natural course, proliferation is accompanied by a progressive accumulation of extracellular collagen and by a decrease in inflammation and inflammatory mediators.18,20 This wound healing process is central to the final common pathway that leads to traction retinal detachment and posttraumatic PVR in open globe injury (Fig. 98.3).
Cellular constituents
Epiretinal membranes removed during vitreous surgery for PVR after injury have been analyzed to gain an understanding of the origin and characteristics of their cellular constituents.38–40 Depending on the nature of the injury and the stage of the response, these membranes contain variable numbers of cells that are phenotypically identified as inflammatory cells, RPE cells, glial cells, fibroblasts, and myofibroblasts.
Inflammatory cells are among the earliest cell types to appear in the wound healing response. They may be attracted to the wound by chemokines upregulated in traumatized retinal tissue, by breakdown of the blood–retinal barrier, or as a response to intraocular blood.31,41–45 The cytokine products of these cells may be critical for the activation of other retinal cell types, further recruitment of inflammatory cells, and formation of collagen. Macrophages are a constant feature of experimental traction retinal detachment.42,43 In the primate model of posterior penetrating injury, macrophages are present before the invasion and proliferation of fibroblast or RPE cells.19,20 The finding of cellular and humoral immune responses to retinal antigens following retinal detachment and experimental PVR has suggested the possibility of an autoimmune component in PVR, although the evidence for a pathogenic role of such a response is incomplete.45–48
The RPE cell is central to the pathophysiological response seen in posttraumatic PVR. The RPE cells have critical characteristics, including migration and proliferation.32,33,38 The posttraumatic stimuli that are specifically responsible for RPE changes are not completely understood. RPE cell growth appears to be regulated by both paracrine and autocrine stimulation. RPE cells were shown to proliferate and form multilayered colonies of dedifferentiated RPE cells within 24 hours of retinal detachment in cats.49 They were also consistently found in the membranes of animal models of penetrating injury.20,22,26 Additionally, cultured RPE cells, just like RPE cells under the detached retina, produce mitogenic and chemotactic growth factors, such as platelet-derived growth factor (PDGF) and hepatocyte growth factor (HGF), and possess receptors for each of these growth factors. RPE cells respond not only to growth factors from RPE cells (autocrine) but also to those from surrounding tissue or from serum (paracrine), resulting in recruitment of additional RPE cells, and thus augmenting the process.34–36,50–56 In response to these stimulations, RPE cells may alter their phenotype to cells with a macrophage, fibroblast, or myofibroblast morphology.32,33,38 This fibroblastic RPE may synthesize and remodel the matrix on the retinal surface, contributing to the formation of the membrane. It was demonstrated that the proportion of RPE in human membranes varies according to the age of the membrane. The number of RPE cells is greater in early (<4 months) specimens and declines progressively as the membranes mature with more advanced extracellular matrices.57
Glial cells, identified by their typical morphologic characteristics and immunoreactivity to glial fibrillary acidic protein (GFAP), were found in neurosensory retina and membrane from full-thickness retinectomy specimens obtained at surgery for PVR, with increased expression correlated to the severity of degeneration after trauma.57–60 Glial cells appear to be involved in PVR formation through migration onto the surface of the retina and may be involved in remodeling of intraretinal synapses, possibly contributing to visual recovery after retinal injury.
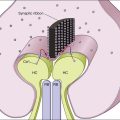
Fibroblastic proliferation is critical to the progression of post-traumatic proliferative response. Although fibroblasts are typically derived from RPE and glial cells, the immunohistochemical markers for these cells are missing in some fibroblasts, making their derivation uncertain.38,61,62 In studies using an animal model of globe perforation with long posterior wounds and injection of intravitreal blood, where membranes extended from the wounds and β-galactosidase labeled Tenon fibroblasts were identified in the vitreous and membrane, it was determined that at least some of the fibroblasts may have originated from Tenon’s layer at the wound edge (Fig. 98.4).20,24,25,63,64
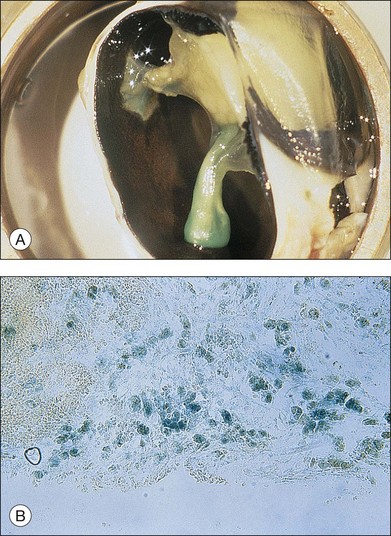
Myofibroblasts are an important component of wound contraction in granulation tissue. Similarly, although the origin of these cells is unknown, RPE cells, fibroblasts, and other cell types have been implicated.38,61,62 Ultrastructural analysis showing that myofibroblasts contain myofibrils and smooth muscle actin suggests that these myofibroblasts may produce contractile forces that cause contraction of the vitreous, retina, and membrane in PVR.65–67 This force of cellular traction may be transmitted directly by cellular attachment to the adjacent tissue or indirectly through traction on collagen fibrils that are attached to the tissue. Experimental work suggested an alternative mechanism of membrane contraction involving the interaction of RPE cells and collagen.14 Collagen fibers are pulled by the RPE cells by alternating extension and retraction of their lamellipodia. Collagen is piled up adjacent to the RPE cell with subsequent tissue shortening.
Growth factors
Several growth factors appear to play critical roles, including platelet-derived growth factor (PDGF), hepatocyte growth factor (HGF), transforming growth factor-β (TGF-β), connective tissue growth factor (CTGF) and vascular endothelial growth factor (VEGF). The association of these growth factors with cells in the membranes suggests that they are produced locally during the wound healing response.31,34–36,55,56,68
Platelet-derived growth factor may be released from platelet α-granules after tissue damage or from endogenous retinal cells such as RPE cells. As a potent mitogenic and chemotactic stimulant for RPE and glial cells, PDGF receptor (PDGFR) signaling seems to play a central role in the development of PVR.36,69 Analysis of epiretinal membranes from patients with PVR shows that PDGFRs are activated in these membranes.70 Experimental studies using an organ culture model showed that RPE cell-mediated retinal contraction can be inhibited by neutralizing antibodies against PDGF; and studies using a mouse fibroblast-induced model of PVR showed that PVR was diminished when fibroblasts isolated from PDGF-receptor knockout mice were used.71,72 However, recent evidence suggests that PDGF itself may not be an appropriate therapeutic target. Neutralizing antibodies do not effectively attenuate experimental PVR using cells with intact PDGFR, and other growth factors outside the PDGF family can activate PDGFRα without engaging its ligand-binding domain.73 Indirect activation of PDGFRα by non-PDGF family growth factors appears to promote PVR by chronic activation of Akt and suppression of p53.74
Hepatocyte growth factor is also mitogenic and chemotactic for RPE cells and was found in the vitreous of patients with PVR.34,50,51,53 Experimentally, cultured human RPE cells responded to HGF by epithelial-to-mesenchymal shape change and by cell migration response that increased with increasing concentrations of HGF.34,50,51,53 Activation of mitogen-activated protein kinases (MAPK) is a component of the HGF-induced RPE change.54 This response was reduced in the presence of neutralizing antibody.51 In vivo studies in rabbits have shown that overexpression of HGF in RPE leads to subretinal proliferation of RPE.55
Transforming growth factor-β is over expressed in the vitreous of patients with PVR, and the contractile effect of hyalocyte-containing collagen gels correlates with vitreal concentration of activated TGF-β2.75 Similarly, there is strong immunoreactivity for CTGF in human PVR membranes, and CTGF N-terminal fragment accumulates in the vitreous of patients with PVR.76 CTGF functions as a downstream mediator of TGF-β action on fibroblasts and RPE where it stimulates cell migration and cell matrix deposition.34,76 In vivo studies have shown that CTGF promotes the development of highly fibrotic PVR membranes in rabbits.76 Recently, CTGF has been shown to promote the profibrotic activities of TGF-β by regulating fibronectin-extra domain A (EDA) through protein–protein interactions between CTGF and both TGF-β and TGF-β receptor II.77
Vascular endothelial growth factor also has been localized to PVR membranes. However, the relatively low incidence of neovascularization in posttraumatic PVR membrane may suggest that VEGF may act as chemotactic factor for RPE, a cell type that also expresses VEGF receptors.29
In addition to various growth factors, proinflammatory cytokines such as tumor necrosis factor-α and interleukin appear to have dramatic effects on many cell types, including RPE, and may alter cellular function by stimulating proliferation, migration, and expression of cell surface integrin and cell adhesion molecule, as well as extracellular matrix production and invasiveness.35,78,79 It was also shown that infiltrating macrophages or resident RPE or glial cells may be the source of these cytokines.35,68 A strong association has been found between a polymorphism in the tumor necrosis factor locus and PVR in a case-controlled study.80
The stage specificity of growth factor expression during PVR development has been described, where PDGF-AA is expressed uniformly throughout all stages of PVR, while HGF expression peaks during mid stage, and CTGF expression is highest during late stage PVR. These results may be applicable to a stage-specific therapeutic approach for PVR.81
Extracellular matrix
In addition to cellular response, extracellular matrices (ECM) are important components of human PVR membranes, similar to the wound healing process in other organs. Initially, the formation of a fibrin-rich membrane can be observed.82,83 Intraocular fibrin may provide a structure for the formation of complex membranes by stimulating the migration of RPE cells along the fibrin sheets.84 Subsequently, membrane is characterized by the presence of interstitial collagens I and III and fibronectin within the membranes.57,78,85,86 The collagens and fibronectin may be derived from RPE, glial cells, or macrophages, although the most consistent association is with RPE. Similar patterns of ECM expression are found in experimental PVR produced by injection of fibroblasts in the rabbit.87 Provisional ECM components (collagen I, fibronectin) in PVR membranes may play important roles in the progression of the wound healing response by stimulating the RPE and glial cells through activation of integrin receptors, resulting in altered cell behavior, including chemotaxis and migration.79,88
The wound healing process of PVR involves the unbalanced action of matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the degradation and contraction of extracellular matrix.89–92 MMP-9 and MMP-2 are the major MMPs that have been detected in vitreous samples and proliferative membranes of PVR patients.90,91,93 In in vitro models of collagen contraction mediated by the retinal pigment epithelium, synthetic MMP inhibitors have shown an anticontraction effect in a dose-dependent manner.94
Special conditions
Traumatic endophthalmitis
Endophthalmitis is a particularly devastating complication of open-globe injury, affecting from 3.1% to 30% of eyes with these injuries in the absence of intraocular foreign body (IOFB).95,96 Approximately 75% of all posttraumatic culture-positive endophthalmitis cases are infected by Gram-positive organisms, with Bacillus species causing about 20% of the infections.97 Risk factors associated with the development of endophthalmitis include the presence of an IOFB, lens rupture, delayed timing of primary repair, age >50 years, female gender, large wound size, location of wound, ocular tissue prolapse, placement of a primary intraocular lens, and rural location.95,96,98–100
The incidence of infectious endophthalmitis after penetrating injury with IOFB was reported higher, varying from 1.3% to 60%.95,100–102 A delay in IOFB removal or primary repair of the wound more than 24 hours after the injury have been associated with the increased risk of endophthalmitis103 (see also Chapter 87, Endogenous ophthalmitis).
Intraocular foreign body
Preoperative retinal detachment, the location and the size of IOFB, and scleral or corneoscleral entry wound are predictive of a postoperative retinal detachment.97,101,104 IOFBs also have a related higher risk of endophthalmitis, which increases dramatically when the IOFB is of a non-metallic material.101 A chronically retained iron IOFB results in an extinguished electroretinogram and blindness, referred to as ocular siderosis, when there is progressive deposition of iron in the ocular tissues. Collections of dense ferritin particles are seen in the cytoplasm and organelles of ocular cells, and it has been hypothesized that these large accumulations cause physical damage that kills retinal cells.105 Copper IOFBs are of particular concern because they can rapidly elicit a sterile endophthalmitis-like reaction with hypopyon and retinal detachment. Ionization of copper causes changes in the neurosensory retina that, if left untreated, can lead to loss of vision within a few hours.106
Post-traumatic infection and the presence of an IOFB are likely to increase the risk of PVR. Definitive treatment involves vitrectomy, removal of the IOFB, and intravitreal as well as systemic antibiotic therapy.107
Combat ocular injury
The main mechanism of open-globe injury in the military population is secondary to blast explosions, e.g. from improvised explosive devices (IEDs). Blast-associated injuries have been demonstrated to result in poor functional outcomes despite surgical intervention because of the surgical complexities and extensive blunt ocular concussive damage.108,109
Intraocular foreign bodies in combat-related ocular injury may have a reduced risk of endophthalmitis since they are usually propelled with high velocity from the explosion and may attain high temperatures, leading to self-sterilization. Predictably, visual outcomes were substantially associated with the depth of penetration of the IOFBs rather than with the timing of removal.109
Therapeutic aspects
Surgical approach
Throughout the literature on the pathophysiology of ocular trauma and related wound healing processes, vitrectomy is shown to be beneficial in that it removes the blood, vitreous scaffolds, and other stimuli for PVR see Chapter 110, Surgery for ocular trauma, for a description of trauma principles and treatment techniques. Although progress in instrumentation and surgical techniques has provided dramatic improvements in anatomical repair, including repairing the laceration, reattaching the retina, and removing IOFBs after open-globe injury, a variety of late complications may develop, including new or recurrent retinal detachment and progressive proliferative membranes.110,111 Therefore, pharmacologic approaches have been studied to modify the wound healing process by inhibiting the development of PVR.
Pharmacologic approach
Corticosteroids reduce intraocular inflammation and adversely affect wound healing.112 Machemer and Tano et al. suggested intravitreal application of steroids to suppress inflammation locally and to reduce proliferation of cells in patients with aggressive PVR.113,114 Since soluble cortisone is washed out of the eye within approximately 24 hours after a single intravitreal injection, the use of crystalline triamcinolone has been described for the treatment of PVR. As part of its wide utilization for a number of other retinal diseases, intravitreal triamcinolone is now commonly used in PVR.115 However, posttraumatic PVR is a long-term complication that may require drug release over months, and long-term injections of triamcinolone are associated with complications, such as elevated intraocular pressure, cataract formation, and endophthalmitis.116 Dexamethasone intravitreal implant is a potential approach, but it needs to be further studied for the treatment of PVR.117
Anti-proliferative drugs have been considered for the treatment of PVR, and a wide spectrum of drugs such as 5-fluorouracil, daunomycin, cyclosporine, mitomycin C, hypericin and Taxol have been tested in experimental models or human clinical trials to decrease uncontrolled mitogenic activity of the cells at the vitreoretinal interface.30,118–123 However, none of these drugs was entirely satisfactory because their inhibitory effects were transient, and most have a narrow therapeutic window.124,125
Agents that block growth factors and their signaling also have been considered to modify the wound healing process. Targeting PDGF, HGF, and protein kinase C through approaches such as direct binding blockers, receptor blockers, and gene therapy seem to have potential in experimental models but further tests in humans are required prior to clinical use.126–129 Various MMP inhibitors have been suggested to reduce the severity of PVR because cell-mediated collagen contraction and cell migration and invasion in PVR are mediated by MMPs, and these inhibitors may be adjunctive to other pharmacologic approaches.89–91,94
Although these therapies would likely be of no use in the treatment of an established membrane, their possible use in the prevention of intraocular proliferation in specific high-risk populations with ocular trauma is being explored. Recent advances in drug delivery to the vitreous and retina including injectable particles and implantable devices may be even more helpful for these pharmacologic approaches for the treatment of PVR.130
1 McGwin G, Jr., Xie A, Owsley C. Rate of eye injury in the United States. Arch Ophthalmol. 2005;123:970–976.
2 Tielsch JM, Parver LM. Determinants of hospital charges and length of stay for ocular trauma. Ophthalmology. 1990;97:231–237.
3 Kuhn F, Morris R, Witherspoon D, et al. A standard classification of ocular trauma. Ophthalmology. 1996;103:240–243.
4 de Bustros S, Michels RG, Glaser BM. Evolving concepts in the management of posterior segment penetrating ocular injuries. Retina. 1990;10:S72–S75.
5 Fuller DG, Hutton WL. Prediction of postoperative vision in eyes with severe trauma. Retina. 1990;10:S20–S34.
6 Shock JP, Adams D. Long-term visual acuity results after penetrating and perforating ocular injuries. Am J Ophthalmol. 1985;100:714–718.
7 Esmaeli B, Elner SG, Schork MA, et al. Visual outcome and ocular survival after penetrating trauma. A clinicopathologic study. Ophthalmology. 1995;102:393–400.
8 Pieramici DJ, MacCumber MW, Humayun MU, et al. Open-globe injury. Update on types of injuries and visual results. Ophthalmology. 1996;103:1798–1803.
9 Soheilian M, Peyman GA, Wafapoor H, et al. Surgical management of traumatic retinal detachment with perfluorocarbon liquid. The Vitreon Study Group. Int Ophthalmol. 1996;20:241–249. –1997
10 Cherry PMH. Rupture of the globe. Arch Ophthalmol. 1972;88:498–507.
11 Moon C, Lee J, Sohn J, et al. The result of consecutive vitrectomy in penetrating ocular injury. J Korean Ophthalmol Soc. 1996;37:1937–1945.
12 Spiegel D, Nasemann J, Nawrocki J, et al. Severe ocular trauma managed with primary pars plana vitrectomy and silicone oil. Retina. 1997;17:275–285.
13 Fine BS, Tousimis AJ. The structure of the vitreous body and the suspensory ligaments of the lens. Arch Ophthalmol. 1961;65:95–110.
14 Glaser BM, Cardin A, Biscoe B. Proliferative vitreoretinopathy. The mechanism of development of vitreoretinal traction. Ophthalmology. 1987;94:327–332.
15 Hsu HT, Patterson R, Ryan SJ. Traumatic posterior vitreous detachment: scanning electron microscopy of an experimental model in the monkey eye. Scan Electron Microsc. 1984;Pt 3:1361–1368.
16 Matsumoto B, Blanks JC, Ryan SJ. Topographic variations in the rabbit and primate internal limiting membrane. Invest Ophthalmol Vis Sci. 1984;25:71–82.
17 Winthrop SR, Cleary PE, Minckler DS, et al. Penetrating eye injuries: a histopathological review. Br J Ophthalmol. 1980;64:809–817.
18 Punnonen E. Pathological findings in eyes enucleated because of perforating injury. Acta Ophthalmol (Copenh). 1990;68:265–269.
19 Cleary PE, Ryan SJ. Method of production and natural history of experimental posterior penetrating eye injury in the rhesus monkey. Am J Ophthalmol. 1979;88:212–220.
20 Cleary PE, Ryan SJ. Histology of wound, vitreous, and retina in experimental posterior penetrating eye injury in the rhesus monkey. Am J Ophthalmol. 1979;88:221–231.
21 Cleary PE, Ryan SJ. Experimental posterior penetrating eye injury in the rabbit. I. Method of production and natural history. Br J Ophthalmol. 1979;63:306–311.
22 Cleary PE, Ryan SJ. Experimental posterior penetrating eye injury in the rabbit. II. Histology of wound, vitreous, and retina. Br J Ophthalmol. 1979;63:312–321.
23 Cleary PE, Jarus G, Ryan SJ. Experimental posterior penetrating eye injury in the rhesus monkey: vitreous-lens admixture. Br J Ophthalmol. 1980;64:801–808.
24 Cleary PE, Minckler DS, Ryan SJ. Ultrastructure of traction retinal detachment in rhesus monkey eyes after a posterior penetrating ocular injury. Am J Ophthalmol. 1980;90:829–845.
25 Hsu HT, Ryan SJ. Natural history of penetrating ocular injury with retinal laceration in the monkey. Graefes Arch Clin Exp Ophthalmol. 1986;224:1–6.
26 Gregor Z, Ryan SJ. Combined posterior contusion and penetrating injury in the pig eye. II. Histological features. Br J Ophthalmol. 1982;66:799–804.
27 Gregor Z, Ryan SJ. Combined posterior contusion and penetrating injury in the pig eye: III. A controlled treatment trial of vitrectomy. Br J Ophthalmol. 1983;67:282–285.
28 Gregor Z, Ryan SJ. Combined posterior contusion and penetrating injury in the pig eye. I. A natural history study. Br J Ophthalmol. 1982;66:793–798.
29 Chen YS, Vinores SA, Campochiaro PA. Simultaneous expression of vascular endothelial growth factor (VEGF) and its receptors in epiretinal membrane. ARVO abstract. 1996:577.
30 Wiedemann P, Sorgente N, Ryan SJ. Proliferative vitreoretinopathy: the rabbit cell injection model for screening of antiproliferative drugs. J Pharmacol Methods. 1984;12:69–78.
31 Kruger EF, Nguyen QD, Ramos-Lopez M, et al. Proliferative vitreoretinopathy after trauma. Int Ophthalmol Clin. 2002;42:129–143.
32 Abe T, Durlu YK, Tamai M. The properties of retinal pigment epithelial cells in proliferative vitreoretinopathy compared with cultured retinal pigment epithelial cells. Exp Eye Res. 1996;63:201–210.
33 Campochiaro PA, Jerdan JA, Glaser BM, et al. Vitreous aspirates from patients with proliferative vitreoretinopathy stimulate retinal pigment epithelial cell migration. Arch Ophthalmol. 1985;103:1403–1405.
34 Hinton DR, He S, Jin ML, et al. Novel growth factors involved in the pathogenesis of proliferative vitreoretinopathy. Eye. 2002;16:422–428.
35 Limb GA, Earley O, Jones SE, et al. Expression of mRNA coding for TNF alpha, IL-1 beta and IL-6 by cells infiltrating retinal membranes. Graefes Arch Clin Exp Ophthalmol. 1994;232:646–651.
36 Campochiaro PA, Glaser BM. Platelet-derived growth factor is chemotactic for human retinal pigment epithelial cells. Arch Ophthalmol. 1985;103:576–579.
37 Irvin TT. The healing wound. In: Bucknall TE, Ellis H. Wound healing for surgeons. London: Bailliere Tindall; 1984:3–28.
38 Kampik A, Kenyon KR, Michels RG, et al. Epiretinal and vitreous membranes. Comparative study of 56 cases. Arch Ophthalmol. 1981;99:1445–1454.
39 Schwartz D, de Ia Cruz ZC, Green WR, et al. Proliferative vitreoretinopathy. Ultrastructural study of 20 retroretinal membranes removed by vitreous surgery. Retina. 1988;8:275–281.
40 Rentsch FJ. The ultrastructure of preretinal macular fibrosis. Graefes Arch Clin Exp Ophthalmol. 1977;203:321–337.
41 Topping TM, Abrams GW, Machemer R. Experimental double-perforating injury of the posterior segment in rabbit eyes: the natural history of intraocular proliferation. Arch Ophthalmol. 1979;97:735–742.
42 Leibovich SJ, Ross R. The role of macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol. 1975;78:71–100.
43 Charteris DG, Hiscott P, Robey HL, et al. Inflammatory cells in proliferative vitreoretinopathy subretinal membranes. Ophthalmology. 1993;100:43–46.
44 Baudouin C, Fredi-Reygrobellet D, Brignole F, et al. Growth factors in vitreous and subretinal fluid cells from patients with proliferative vitreoretinopathy. Ophthalmic Res. 1993;25:52–59.
45 Charteris DG, Hiscott P, Grierson I, et al. Proliferative vitreoretinopathy. Lymphocytes in epiretinal membranes. Ophthalmology. 1992;99:1364–1367.
46 Grisanti S, Heimann K, Wiedemann P. Immune response to specific molecules of the retina in proliferative vitreoretinal disorders. Graefes Arch Clin Exp Ophthalmol. 1994;232:302–307.
47 Baudouin C, Fredi-Reygrobeilet D, Gordon WC, et al. Immunohistologic study of epiretinal membranes in proliferative vitreoretinopathy. Am J Ophthalmol. 1990;110:593–598.
48 Grisanti S, Wiedemann P, Weller M, et al. The significance of complement in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1991;32:2711–2717.
49 Anderson DH, Stern WH, Fisher SK, et al. The onset of pigment epithelial proliferation after retinal detachment. Invest Ophthalmol Vis Sci. 1981;21:10–16.
50 Grieson I, Heathcote L, Hiscott P, et al. Hepatocyte growth factor/scatter factor in the eye. Prog Retin Eye Res. 2000;19:779–802.
51 Briggs MC, Grierson I, Hiscott P, et al. Active scatter factor (HGF/SF) in proliferative vitreoretinal disease. Invest Ophthalmol Vis Sci. 2000;41:3085–3094.
52 Lashikari K, Rahimi N, Kazlauskas A. Hepatocyte growth factor receptor in human RPE cells: implications in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1999;40:149–156.
53 Jin M, Chen Y, He S, et al. Hepatocyte growth factor and its role in the pathogenesis of retinal detachment. Invest Ophthalmol Vis Sci. 2004;45:323–329.
54 Liou GI, Matragoon S, Samuel S, et al. MAP kinase and beta-catenin signaling in HGF induced RPE migration. Mol Vis. 2002;8:483–493.
55 Robbins SG, Mixon RN, Wilson DJ, et al. Platelet-derived growth factor ligands and receptors immunolocalized in proliferative retinal diseases. Invest Ophthalmol Vis Sci. 1994;35:3649–3663.
56 Westra I, Robbins SG, Wilson DJ, et al. Time course of growth factor staining in a rabbit model of traumatic retinal detachment. Graefes Arch Clin Exp Ophthalmol. 1995;233:573–581.
57 Morino I, Hiscott P, McKechnie N, et al. Variation in epiretinal membrane components with clinical duration of the proliferative tissue. Br J Ophthalmol. 1990;74:393–399.
58 Jerdan JA, Pepose JS, Michels RG, et al. Proliferative vitreoretinopathy membranes. An immunohistochemical study. Ophthalmology. 1989;96:801–810.
59 Sethi CS, Lewis GP, Fisher SK, et al. Glial remodeling and neural plasticity in human retinal detachment with proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2005;46:329–342.
60 Charteris DG, Downie J, Aylward GW, et al. Intraretinal and periretinal pathology in anterior proliferative vitreoretinpathy. Graefes Arch Clin Exp Ophthalmol. 2007;245:93–100.
61 Gabbiani G. The cellular derivation and the life span of the myofibroblast. Pathol Res Pract. 1996;192:708–711.
62 Hui YN, Sorgente N, Ryan SJ. Ultrastructures of the glial epiretinal membrane induced by activated macrophages. Clin Med J (Engl). 1992;105:577–581.
63 Hsu HT, Ryan SJ. Experimental retinal detachment in the rabbit. Penetrating ocular injury with retinal laceration. Retina. 1986;6:66–69.
64 Santos RO, Murata T, Cui JZ, et al. The role of tenon fibroblasts in the pathogenesis of proliferative vitreo-retinopathy due to perforating eye injury. ARVO abstract. 1998:517.
65 Desmoulière A. Factors influencing myofibroblast differentiation during wound healing and fibrosis. Cell Biol Int. 1995;19:471–476.
66 Jester JV, Petroll WM, Barry PA, et al. Expression of alpha-smooth muscle (alpha-SM) actin during corneal stromal wound healing. Invest Ophthalmol Vis Sci. 1995;36:809–819.
67 Sakamoto T, Hinton DR, Sakamoto H, et al. Collagen gel contraction induced by retinal pigment epithelial cells and choroidal fibroblasts involves the protein kinase C pathway. Curr Eye Res. 1994;13:451–459.
68 Limb GA, Alam A, Earley O, et al. Distribution of cytokine proteins within epiretinal membranes in proliferative vitreoretinopathy. Curr Eye Res. 1994;13:791–798.
69 Harvey AK, Roberge F, Hjelmeland LM. Chemotaxis of rat retinal glia to growth factors found in repairing wounds. Invest Ophthalmol Vis Sci. 1987;28:1092–1099.
70 Cui J, Lei H, Samad A, et al. PDGF receptors are activated in human epiretinal membranes. Exp Eye Res. 2009;88:438–444.
71 Carrington L, McLeod D, Boulton M. IL-10 and antibodies to TGF-β2 and PDGF inhibit RPE-mediated retinal contraction. Invest Ophthalmol Vis Sci. 2000;41:1210–1216.
72 Andrews A, Balciunaite E, Leong FL, et al. Platelet-derived growth factor plays a key role in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1999;40:2683–2689.
73 Lei H, Velez G, Hovland P, et al. Growth factors outside the PDGF family drive experimental PVR. Invest Ophthalmol Vis Sci. 2009;50:3394–3403.
74 Lei H, Velez G, Kazlauskas A. Pathological signaling via platelet-derived growth factor receptor {alpha} involves chronic activation of Akt and suppression of p53. Mol Cell Biol. 2011;31:1788–1799.
75 Kita T, Hata Y, Arita R, et al. Role of TGF-beta in proliferative vitreoretinal diseases and ROCK as a therapeutic target. Proc Natl Acad Sci U S A. 2008;105:17504–17509.
76 He S, Chen Y, Khankan R, et al. Connective tissue growth factor as a mediator of intraocular fibrosis. Invest Ophthalmol Vis Sci. 2008;49:4078–4088.
77 Khankan R, Oliver N, He S, et al. Regulation of fibronectin-EDA through CTGF domain-specific interactions with TGF-beta2 and its receptor TGF-betaRII. Invest Ophthalmol Vis Sci. 2011;52:5068–5078.
78 Casaroli Marano RP, Vilaró S. The role of fibronectin, laminin, vitronectin and their receptors on cellular adhesion in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1994;35:2791–2803.
79 Hunt RC, Pakalnis VA, Choudhury P, et al. Cytokines and serum cause alpha-2 beta-1 integrin-mediated contraction of collagen gels by cultured retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1994;35:955–963.
80 Rojas J, Fernandez I, Pastor JC, et al. A strong genetic association between the tumor necrosis factor locus and proliferative vitreoretinopathy: the retina 4 project. Ophthalmology. 2010;117:2417–2423.
81 Cui JZ, Chiu A, Maberley D, et al. Stage specificity of novel growth factor expression during development of proliferative vitreoretinopathy. Eye. 2007;21:200–208.
82 Jaffe GJ, Schwartz D, Han DP, et al. Risk factors for postvitrectomy fibrin formation. Am J Ophthalmol. 1990;109:661–667.
83 Johnson RN, Blankenship G. A prospective, randomized, clinical trial of heparin therapy for postoperative intraocular fibrin. Ophthalmology. 1988;95:312–317.
84 Vidaurri-Leal JS, Glaser BM. Effect of fibrin on morphologic characteristics of retinal pigment epithelial cells. Arch Ophthalmol. 1984;102:1376–1379.
85 Scheiffarth OF, Kampik A, Günther H, et al. Proteins of the extracellular matrix in vitreoretinal membranes. Graefes Arch Clin Exp Ophthalmol. 1988;226:357–361.
86 Tervo K, Latvala T, Suomalainen VP, et al. Cellular fibronectin and tenascin in experimental perforating scleral wounds with incarceration of the vitreous. Graefes Arch Clin Exp Ophthalmol. 1995;233:168–172.
87 Spee C, Soriano D, Kohen L, et al. ECM production during the time course of PVR in a rabbit model. ARVO abstract. 1994:3651.
88 Robbins SG, Brem RB, Wilson DJ, et al. Immunolocalization of integrins in proliferative retinal membranes. Invest Ophthalmol Vis Sci. 1994;35:3475–3485.
89 Hiscott P, Sheridan C, Magee RM, et al. Matrix and the retinal pigment epithelium in proliferative retinal disease. Prog Retin Eye Res. 1999;18:167–190.
90 Webster L, Chignell AH, Limb GA. Predominance of MMP-1 and MMP-2 in epiretinal and subretinal membranes of proliferative vitreoretinopathy. Exp Eye Res. 1999;68:91–98.
91 Kon CH, Occleston NL, Charteris D, et al. A prospective study of matrix metalloproteinases in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1998;39:1524–1529.
92 Sheridan CM, Occleston NL, Hiscott P, et al. Matrix metalloproteinases: a role in the contraction of vitreo-retinal scar tissue. Am J Pathol. 2001;159:1555–1566.
93 Symeonidis C, Papakonstantinou E, Androudi S, et al. Interleukin-6 and the matrix metalloproteinase response in the vitreous during proliferative vitreoretinopathy. Cytokine. 2011;54:212–217.
94 Ozerdem U, Mach-Hofacre B, Keefe K, et al. The effect of prinomastat (AG3340), a synthetic inhibitor of matrix metalloproteinases, on posttraumatic proliferative vitreoretinopathy. Ophthalmic Res. 2001;33:20–23.
95 Brinton GS, Topping TM, Hyndiuk RA, et al. Posttraumatic endophthalmitis. Arch Ophthalmol. 1984;102:547–550.
96 Essex RW, Yi Q, Charles PG, et al. Post-traumatic endophthalmitis. Ophthalmology. 2004;111:2015–2022.
97 Alfaro DV, Roth D, Liggett PE. Posttraumatic endophthalmitis. Causative organisms, treatment, and prevention. Retina. 1994;14:206–211.
98 Andreoli CM, Andreoli MT, Kloek CE, et al. Low rate of endophthalmitis in a large series of open globe injuries. Am J Ophthalmol. 2009;147:601–608.e2.
99 Boldt HC, Pulido JS, Blodi CF, et al. Rural endophthalmitis. Ophthalmology. 1989;96:1722–1726.
100 Thompson JT, Parver LM, Enger CL, et al. Infectious endophthalmitis after penetrating injuries with retained intraocular foreign bodies. National Eye Trauma System. Ophthalmology. 1993;100:1468–1474.
101 Jonas JB, Knorr HLJ, Budde WM. Prognostic factors in ocular injuries caused by intraocular or retrobulbar foreign bodies. Ophthalmology. 2000;107:823–828.
102 Cebulla CM, Flynn HW, Jr. Endophthalmitis after open globe injuries. Am J Ophthalmol. 2009;147:567–568.
103 Wani VB, Al-Ajmi M, Thalib L, et al. Vitrectomy for posterior segment intraocular foreign bodies: visual results and prognostic factors. Retina. 2003;23:654–660.
104 Parrish CM, O’Day DM. Traumatic endophthalmitis. Int Ophthalmol Clin. 1987;27:112–119.
105 Tawara A. Transformation and cytotoxicity of iron in siderosis bulbi. Invest Ophthalmol Vis Sci. 1986;27:226–236.
106 Mester V, Kuhn F. Intraocular foreign bodies. Ophthalmol Clin North Am. 2002;15:235–242.
107 Jonas J, Budde WM. Early versus late removal of retained intraocular foreign bodies. Retina. 1999;19:193–197.
108 Bajaire B, Oudovitchenko E, Morales E. Vitreoretinal surgery of the posterior segment for explosive trauma in terrorist warfare. Graefes Arch Clin Exp Ophthalmol. 2006;244:991–995.
109 Colyer MH, Chun DW, Bowker KS, et al. Perforating globe injuries during operation Iraqi Freedom. Ophthalmology. 2008;115:2087–2093.
110 Cox MS, Freeman HM. Retinal detachment due to ocular penetration. I. Clinical characteristics and surgical results. Arch Ophthalmol. 1978;96:1354–1361.
111 Chiquet C, Gain P, Zech JC, et al. [Risk factors for secondary retinal detachment after extraction of intraocular foreign bodies.]. Can J Ophthalmol. 2002;37:168–176.
112 Pessa ME, Bland KI, Copelend EM, III. Growth factors and determinants of wound repair. J Surg Res. 1987;42:207–217.
113 Machemer R. Proliferative vitreoretinopathy (PVR) :a personal account of its pathogenesis and treatment. Proctor lecture. Invest Ophthalmol Vis Sci. 1988;29:1771–1783.
114 Tano Y, Sugita G, Abrams G, et al. Inhibition of intraocular proliferation with intravitreal corticosteroids. Am J Ophthalmol. 1980;89:131–136.
115 Jonas JB, Hayler JK, Panda-Jonas S. Intravitreal injection of crystalline cortisone as adjunctive treatment of proliferative vitreoretinopathy. Br J Ophthalmol. 2000;84:1064–1067.
116 Ozkiriş A, Erkiliç K. Complications of intravitreal injection of triamcinolone acetonide. Can J Ophthalmol. 2005;40:63–68.
117 Haller JA, Bandello F, Belfort R, Jr., et al. Randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with macular edema due to retinal vein occlusion. Ophthalmology. 2010;117:1134–1146.
118 van Bockxmeer FM, Martin CE, Thompson DE, et al. Taxol for the treatment of proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1985;26:1140–1147.
119 Harris MS, Sakamoto T, Kimura H, et al. Hypericin inhibits cell growth and induces apoptosis in retinal pigment epithelial cells: possible involvement of protein kinase C. Curr Eye Res. 1996;15:255–262.
120 Murphy TL, Sakamoto T, Hinton DR, et al. Migration of retinal pigment epithelium cells in vitro is regulated by protein kinase C. Exp Eye Res. 1995;60:683–695.
121 Tahara YR, Sakamoto TR, Oshima YR, et al. The antidepressant hypericin inhibits progression of experimental proliferative vitreoretinopathy. Curr Eye Res. 1999;19:323–329.
122 Wiedemann P, Hilgers RD, Bauer P, et al. Adjunctive daunorubicin in the treatment of proliferative vitreoretinopathy: results of a multicenter clinical trial. Daunomycin Study Group. Am J Ophthalmol. 1998;126:550–559.
123 Asaria RH, Kon CH, Bunce C, et al. Adjuvant 5-fluorouracil and heparin prevents proliferative vitreoretinopathy. Results from a randomized, double-blind, controlled clinical trial. Ophthalmology. 2001;108:1179–1183.
124 Steinhorst UH, Hatchell DL, Chen EP, et al. Ocular toxicity of daunomycin: effects of subdivided doses on the rabbit retina after vitreous gas compression. Graefes Arch Clin Exp Ophthalmol. 1993;231:591–594.
125 Penha FM, Rodrigues EB, Maia M, et al. Retinal and ocular toxicity in ocular application of drugs and chemicals-part II: retinal toxicity of current and new drugs. Ophthalmic Res.. 2010;44:205–224.
126 Ikuno Y, Leong FL, Kazlauskas A. Attenuation of experimental proliferative vitreoretinopathy by inhibiting the platelet-derived growth factor receptor. Invest Ophthalmol Vis Sci. 2000;41:3107–3116.
127 Ikuno Y, Kazlauskas A. An in vivo gene therapy approach for experimental proliferative vitreoretinopathy using the truncated platelet-derived growth factor alpha receptor. Invest Ophthalmol Vis Sci. 2002;43:2406–2411.
128 Date K, Matsumoto K, Shimura H, et al. HGF/NK4 is a specific antagonist for pleiotrophic actions of hepatocyte growth factor. FEBS Lett. 1997;420:1–6.
129 Hinton DR, He S, Graf K, et al. Mitogen-activated protein kinase activation mediates PDGF-directed migration of RPE cells. Exp Cell Res. 1998;239:11–15.
130 Guidetti B, Azéma J, Malet-Martino M, et al. Delivery systems for the treatment of proliferative vitreoretinopathy: materials, devices and colloidal carriers. Curr Drug Deliv. 2008;5:7–19.