Chapter 97 Pathogenesis of Proliferative Vitreoretinopathy
Introduction
PVR is reported to have an incidence of 5–10% of all rhegmatogenous retinal detachments (RRD),1 and it is believed to be the most common cause of ultimate failure of a surgery for RRD. A high rate of PVR has also been reported following posterior segment procedures or disorders such as giant retinal tears, macular relocation surgery, endoresection of tumors and chip implantation. In human immunodeficiency, virus-infected patients PVR has been developed in 29% of cytomegalovirus retinitis-related retinal detachment.2 In children, postoperative PVR occurs in higher incidence and is characterized by a rapid and aggressive development.3 PVR is also a common complication following a variety of ocular injuries with the highest frequency after perforating and penetrating wounds, in which the incidence is 10–45%.
Soon after the development of vitrectomy, PVR became a specific indication for vitreous surgery. The changing understanding of PVR pathogenesis is reflected in the different classification systems. In the beginning, it was assumed to be primarily due to changes in the vitreous gel (“massive vitreous retraction”, “massive preretinal retraction”). Then the involvement of cells was recognized, and the condition was re-termed “massive periretinal proliferation.”4 Later, a distinct biomicroscopic classification was developed in an attempt to categorize the severity of the disease (Retina Society Classification 1983). A major problem of the latter system was that it did not reflect prognosis and surgical difficulty. The Cologne classification5 and the Silicone Oil Study classification6 took these aspects into account by separating anterior and posterior PVR. The updated Retina Society classification is a compromise, which describes severity (A, B, C), localization, updated grade, and updated contraction type.7 However, this classification still does not consider many aspects of the disorder. Therefore, a classification that provides additional relevant clinical information, such as the evolutionary stages of the disease (biologic activity) and the degree of surgical difficulty is desirable.8
Clinically, early stages of PVR are characterized by an increased reflectance and a cellophane appearance of the inner retinal surface. Additionally, tortuosity of both small and larger vessels is regularly observed. The pathological hallmarks of the advanced PVR include periretinal membrane formation, causing development of surface wrinkling and single or multifocal star-folds (Fig. 97.1). In the final stages, multidirectional tractional forces produced by posterior and/or anterior PVR form a narrow or closed funnel of the detached retina.9
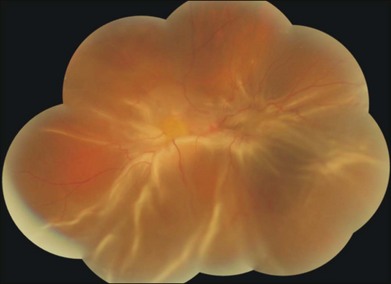
Fig. 97.1 Human proliferative vitreoretinopathy showing typical surface wrinkling and multifocal star-folds.
A retinal break is a prerequisite for the development of PVR. Almost all risk factors for the PVR are associated with intravitreal dispersion of retinal pigment epithelial (RPE) cells or the breakdown of the blood–retinal barrier (BRB).10,11 The size of breaks, the extent of detachment, the presence of preoperative inflammation or low-grade PVR, and iatrogenic complications are important factors in the pathogenesis of severe PVR after the surgery for retinal detachment.12,13
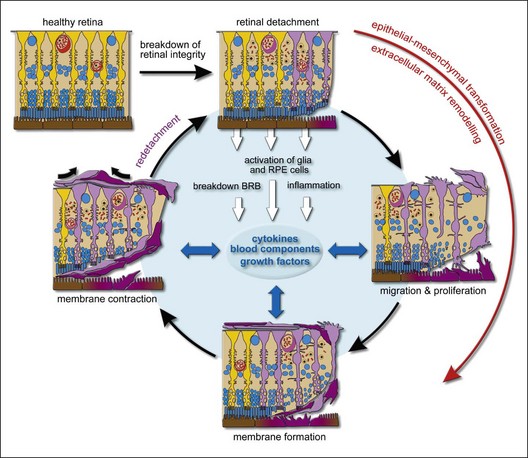
Several investigators have underlined the parallels between PVR and the general wound-healing process.14,15 PVR develops in a sequence of three overlapping phases: inflammation, cellular proliferation, and extracellular matrix remodeling. The time course of PVR development is poorly investigated. Data which were accumulated from clinical analysis could reveal that on average it takes 4–8 weeks for PVR development after surgery.8,16 Detailed PVR chronobiology was evaluated in the experimental animal model of PVR induced by macrophages. The inflammation phase is initiated immediately in response to macrophage injection. Cellular proliferation can be detected as early as days 4 ± 7 and peaks on days 10 ± 14. The scarring-induced retinal detachment occurs during the 2nd and 3rd weeks after macrophage injection.17
Cellular basis of PVR
Composition of membranes
The composition of the membranes changes with time, from early cellular to late paucicellular and rather fibrotic membranes.18 In fibroproliferative membranes, glial cells, RPE cells, and fibrocytes transdifferentiate into contractile myofibrocytes,19 which generate tractional forces through the expression of the contractile protein α-smooth-muscle actin. Fibroblastic transdifferentiation of cells causes a reduction in cell-specific proteins such as glial fibrillary acidic protein (GFAP; glial cells) and cytokeratins (RPE cells), while proteins involved in motility and proliferation such as α-smooth-muscle actin (which is normally not expressed by the cells) are upregulated.19–21 This cellular transdifferentiation may be the reason for the paradox that relatively few glial and pigment epithelial cells can be detected in periretinal membranes with the commonly used immunocytochemical markers.
RPE cells
EMT is an important process during normal development in which epithelial cells transform to mesenchymal cells, and show alterations in polarized organization, intercellular adhesion and become migratory and invasive. The critical role of EMT has also been recognized in numerous pathological conditions including tumor progression and fibrosis. EMT appears to play a critical role in PVR. During retinal detachment, RPE cells gain access through retinal breaks into the vitreous cavity, adhere to the detached retina, and undergo EMT to a fibrocyte phenotype. Studies on EMT show that various factors such as cytokines, modified growth factor signaling, loss of normal matrix adhesion, and changes in cell–cell adhesion profile may initiate this process. Recent studies suggest that disruption and loss of cell–cell contacts initiate EMT and proliferation of RPE cells.22,23 During EMT, RPE cells lose their epithelial phenotypes and acquire mesenchymal, fibroblast-like properties. Characteristic features of this process are the upregulation of α-smooth-muscle actin and vimentin, the loss of the zona occludens protein ZO-1, the reduced intercellular adhesion and the increased motility and enhanced migration24 which is supported by the enhanced matrix metalloproteinases (MMPs) activity.
RPE cell migration is a complex process that includes changes in cell attachment, spreading, and cytoskeletal reorganization, and it is regulated by cell matrix, matrix-dependent enzymes, cytokines, and growth factors. RPE cell migration is also mediated by cell membrane-associated signaling.25
Glial cells
Whereas the role of RPE cells has been widely discussed in the literature over the last 30 years, the impact of glial cells on the pathogenesis of PVR was frequently overlooked. Glial cells and, in particular, Müller cells, play a central role in retinal physiology. They support the neuronal activity, the integrity of the BRB, and maintain the ionic and osmotic homeostasis. Virtually all retinal diseases are associated with reactive gliosis, which is characterized by cellular hypertrophy and upregulation of the intermediate filaments vimentin and GFAP. Moreover, Müller cell gliosis is associated with proliferation and PVR is a key example of massive and long-lasting cellular proliferation.26 Reactive gliosis is observed within minutes after experimental retinal detachment and proceeds so long as the retina remains detached.27 In the course of PVR, Müller cells proliferate and migrate out of the retina where they are a constant part of fibrocellular membranes. Fibroproliferative membranes are focally connected to the retina via hypertrophied processes of Müller cells, rising from the retinal tissue into the membranes.1,28,29
The proliferation of Müller cells is accompanied by alterations in their membrane conductance. Müller cells of patients with PVR, and those from animal models of PVR, display a severe downregulation of the potassium conductance mediated by inwardly rectifying potassium (Kir) channels.30–33 The downregulation of functional Kir channels causes a depolarization of the cells which is a prerequisite for the reentry of the cells into the proliferation cycle.34 Moreover, the impaired retinal potassium homeostasis may favor neuronal hyperexcitation and glutamate toxicity. Therefore, the depolarization of Müller cells may contribute to an impairment of the regular glial–neuronal interactions in the retina and, thus, to the neuronal degeneration observed in PVR.35 Furthermore, activated Müller cells may act as modulators of immune and inflammatory processes, for example by producing proinflammatory cytokines.36 Reactive gliosis is a limiting factor in the recovery of vision after retinal reattachment.29 In the healthy retina, Müller cells serve as soft, compliant embedding for neurons and also as a soft substrate required for neurite growth and facilitating neuronal plasticity.37 The hypertrophied and proliferating glial cells fill the spaces left by dying neurons and degenerated axons and generate so-called glial scars. Reactive gliosis is associated with an increased stiffness of Müller cells.38 Such rigid glial scars may constitute a mechanical obstacle for regenerative axon growth. Therefore, attempts to reduce Müller cell gliosis may be a promising tool to inhibit retinal degeneration and to support neuroregeneration after retinal reattachment.29,35
Blood-borne cells
Inflammation is an important step in the pathogenesis of PVR and is associated with influx of blood-derived cells into the retinal tissue and vitreous. Blood-borne cells such as macrophages and fibrocytes are frequently found in PVR membranes. Macrophage-like cells might be also derived from RPE cells which have undergone EMT. Circulating fibrocytes and macrophages may function as precursors of myofibroblasts in PVR membranes and may contribute to the fibrocellular membranes directly.39,40 Hyalocytes (cells in the cortical vitreous) belong to the monocyte/macrophage lineage and play a significant role in the synthesis of ECM, modulation of immune reaction, and modulation of inflammation. The strong contractile properties of hyalocytes suggest also a critical role in the genesis of PVR.41
Stimulation of cellular proliferation and migration
There is a great deal of evidence supporting the idea that soluble factors including growth factors and cytokines play a central role in the pathogenesis of PVR, inducing key cellular responses such as chemotaxis, proliferation, migration, and extracellular matrix remodeling.42 Growth factors are proteins that bind to receptors on the cell surface, with the primary result of activating cellular proliferation and/or differentiation. Cytokines specifically denote signaling proteins that are used extensively in cellular communication. They often exhibit growth factor activity and can exert autocrine, paracrine and endocrine effects as do the hormones. Interleukins are cytokines specifically directed to leucocytes. A number of soluble factors contribute to PVR, but the evidence accumulated so far is greatest for the factors below.
Blood components
A breakdown of the blood–retinal barrier is a characteristic and essential event in the pathogenesis of PVR. Serum, which enters the retina and vitreous seems to play an important role in the transmission of mitogenic signals to both Müller cells and RPE cells. Thrombin, a serum component, stimulates the proliferation of retinal glial cells and RPE cells.43,44 Fibronectin stimulates the migration of glial cells43 and plays an important role in ECM remodeling (see below). The vitreous of experimental PVR animals and retinal surgery patients contains the serum-derived protease plasmin. It is responsible for the generation of active platelet-derived growth factor-C (PDGF-C), which is the major PDGF isoform involved in the development of PVR45 (see below).
Platelet-derived growth factor (PDGF)
PDGF, a wound-healing cytokine, seems to play a critical role in the development of PVR. Both PDGF and PDGF receptor (PDGFR) are present in PVR membranes.46 Vitreal PDGF could be detected in eight out of nine PVR patients, but only in one out of 16 patients with other vitreoretinal diseases.45 RPE and retinal glial cells produce PDGFs and possess their receptors.46 PDGF is a potent chemoattractant and mitogen for many cell types, including RPE and glial cells.47,48
Among the four PDGF family members, PDGF-C was the predominant isoform in PVR specimens.45 Plasmin is suggested to be the major PDGF-C processing protease.49 Analysis of epiretinal PVR membranes indicated that a greater percentage of the PDGFRα receptors is activated as compared to PDGFRβ and that both retinal pigment epithelial and glial cells express activated PDGFRα receptors.50 Abundant binding of PDGF-C to PDGFRα explains why PDGFRα activation is more important in the pathogenesis of PVR than PDGFRβ activation.45 In a rabbit model of PVR, delivery of retrovirus containing a dominant negative PDGFRα suppresses the development of retinal detachment.51 Furthermore, oral administration of tyrosine kinase inhibitors, which block signaling through PDGF receptors, reduce PDGF-induced epiretinal membrane formation and retinal detachment in transgenic mice.52 It has been shown recently that PDGFRα can also be activated by other growth factors besides PDGF.53 This indirect activation of PDGFRα involves the formation of intracellular reactive oxygen species (ROS). Initial studies have shown that vitreal injection of the antioxidant N-acetyl cysteine prevents the development of experimental PVR.54
Transforming growth factor-β
TGF-β is implicated in the tissue contraction in different fibrous diseases including proliferative eye diseases such as proliferative diabetic retinopathy and PVR.55 The vitreous of PVR patients contains high levels of TGF-β.56 TGF-β was also localized to subretinal strands of PVR patients.57 Müller cells are one source of TGF-β in the retina.58 Experimental retinal detachment increases the expression of TGF-β and TGF receptor II in Müller cells and in hypertrophied Müller cells processes that contribute to the formation of periretinal membranes.59 TGF-β-treated RPE cells differentiate along a myofibroblast pathway.24 Furthermore, gene transfer of a soluble TGF-β type II receptor (which traps TGF-β) suppresses the progression of experimental PVR.60 These experimental and clinical studies indicate that inhibition of TGF-β signaling could be a therapeutic approach to prevent the progression of PVR. Intravitreal injection of the cicatricial contraction inhibitor simvastatin could prevent PVR progression in a dose-dependent manner.61 TGF-β likely mediates cicatricial contraction via activation of the Rho-kinase (ROCK) pathway; this pathway is also activated by various other growth factors implicated in cell adhesion, proliferation and migration. It makes it an interesting therapeutic target for treating PVR.62,63
Monocyte chemotactic protein-1 (MCP1)
The chemokine MCP-1 has a role during the initial stage of PVR. In experimental retinal detachment, the expression of MCP-1 increases within one hour, and the MCP-1 protein level increases within six hours of detachment.64 MCP-1 stimulates the migration of RPE cells; this effect is inhibited by dexamethasone.65 MCP-1 was detected in a substantial percent of vitreous samples of patients with PVR.66 Its expression level in PVR patients was much higher than in patients with rhegmatogenous retinal detachment, macular hole or idiopathic epimacular membrane.
Basic fibroblast growth factor (bFGF)
Many observations show that bFGF may be a putative target for PVR therapy. bFGF levels are raised in PVR vitreous compared to non-PVR vitreous.67 Fibroproliferative membranes contain bFGF.68 The vitreous bFGF expression levels in PVR-D patients were notably higher than those in PVR-C, vitreous hemorrhage and control group.69 A blockade of integrin receptors inhibits RPE cell attachment, migration and invasion stimulated by bFGF.70
Hepatocyte growth factor (HGF)
HGF (also known as scatter factor) is a multipotential cytokine that plays a role in scattering of retinal cells, chemotaxis and EMT.71,72 Increased expression level of HGF has been shown in vitreous and in epiretinal membranes of patients with PVR.73,74 In epiretinal membranes of patients with PVR the expression of HGF and its receptor, the c-Met tyrosine kinase, can be localized in various cell types, including glial and RPE cells.46,75
Connective tissue growth factor (CTGF)
Both in vivo and in vitro studies demonstrate that CTGF is a crucial factor in the pathogenesis of PVR.76 CTGF is up-regulated in a model of RPE monolayer scrape wounding in vitro. Recombinant human CTGF protein could stimulate RPE migration in a dose-dependent manner; this effect is suppressed by dexamethasone.77 CTGF is expressed in fibroproliferative membranes and vitreous samples78 from patients with PVR.74 During the development of PVR the expression of CTGF in epiretinal membranes increases from early to late stage, and localizes to RPE cells in early stage and to glial cells in the late stage of PVR.46
Epidermal growth factor (EGF)
EGF may activate RPE cells in PVR.79 EGF induces RPE proliferation and migration through the EGF–EGFR–MAPK signal pathway in a concentration-dependent manner.80 EGF could also promote integrin-α5 expression, which subsequently activates RPE cells.81 In addition, EMT of RPE cells could be induced by EGF.24
Vascular endothelial growth factor (VEGF)
VEGF is a regulatory mediator of cellular proliferation and vascular permeability. Anti-VEGF therapy is widely used to treat retinal or choroidal neovascular disorders, such as diabetic retinopathy, retinopathy of prematurity, and age-related macular degeneration. The VEGF concentration in the vitreous of PVR patients is significantly higher when compared with retinal detachment and macular hole.82 VEGF is also expressed in PVR membranes.83 Chronic PVR is often associated with neovascularization in the far periphery of the retina, the ciliary body, and the anterior segment, and VEGF is likely implicated in the induction of neovascularization. As an autocrine and paracrine stimulator, VEGF secreted by RPE, glial and other cells may contribute to the progression of fibrovascular membrane formation.
Cytokines
It has been suggested that cytokine-mediated pathways of inflammation play an important role in the development of PVR. Inflammatory cells, RPE cells and retinal glial cells are possible sources of IL-6, IL-1β, TNF-α and interferon gamma (INF-γ) in the PVR vitreous.33 Cytokines in the vitreous84 may stimulate important cellular processes, including migration, proliferation, and production of extracellular matrix. In the early stage and proliferative phase of experimental PVR, cytokines are present in large amounts in the vitreous; the cytokine levels decrease to normal in the remodeling (scarring) phase.85
Extracellular matrix remodeling
The ECM is composed of a dynamic and complex array of collagens, glycoproteins, glycosaminoglycans and proteoglycans. All these molecules form the substrate surrounding the cells and tissues to provide a mechanical and structural support and modulate autocrine signaling loops and the recruitment of cells.86 Moreover, the ECM interacts with the cytoskeleton and with cytokines to transmit biological signals which affect the cell behavior, development, migration, proliferation, transdifferentiation, contraction, and remodeling. Integrins and proteoglycans on the cell surface are the main adhesion receptors that promote signal transduction.87 Bruch’s membrane and the interphotoreceptor matrix are specialized types of ECM that surround the RPE and photoreceptors. At early stages of rhegmatogenous retinal detachment and PVR, the neural retina layer detaches from the RPE layer, resulting in a disintegration of the interphotoreceptor matrix.88 Retinal detachment causes a dissociation of RPE cells from Bruch’s membrane and a dispersion of the cells into the vitreal cavity (grade A PVR). The time-dependent increase in the extracellular matrix content of PVR membranes suggests that components of the ECM stimulate its production in an autocrine/paracrine manner.8
The wound-healing process is a complex and dynamic process of restoring tissue structures and proper organ function. But it can be detrimental for tissue function when it becomes excessive such as in PVR. ECM components play a central role in all phases of PVR. ECM remodeling with fibrocellular membrane contraction is a culmination of this process. During the remodeling phase, transdifferentiated RPE, glial, and blood-borne cells can generate tractional forces through contraction of vitreal and newly synthesized ECM resulting in retinal surface wrinkling, star-fold formation, and tractional retinal detachment. The synthesis and deposition of ECM components on both sides of the retina is a key event of remodelling phase. Structural proteins, adhesive proteins and anti-adhesive proteins are three components of the extracellular matrix in PVR membranes.89 Collagen (which includes various types) is one of the major structural proteins and components of PVR membranes8 may be produced by RPE, glial and fibroblast-like cells.90–92 The expression of fibronectin, an adhesion protein, in the normal retina is low; under pathological conditions, its concentration in the vitreous humor increases quickly.93 Fibronectin promotes adhesion between cells and ECM and might stimulate RPE cells to migrate toward membranes.94 In addition, fibronectin amplifies the EMT of RPE cells induced by TGF-β.95 TGF-β also induces the synthesis of ECM components such as collagens and fibronectin.63,90,96 The function of anti-adhesive proteins is contrary to adhesive proteins. Anti-adhesive thrombospondin in PVR membranes could facilitate the activated cells to detach from the ECM and to migrate into the wounded area.97 The balance between MMPs and tissue inhibitors of MMPs (TIMPs) regulates the turnover of the extracellular matrix and the tissue remodeling associated with PVR. Cellular components of PVR membranes, including RPE cells, glial cells, and fibroblasts synthesize MMPs.98 The expression of MMP-2 and MMP-9 is upregulated dramatically in PVR membranes.94 In addition, MMP activity may be required for ECM contraction.99
Myofibroblasts play a key role in tissue remodeling. They participate in a variety of phenomena and originate from different cellular types suggesting that the term myofibroblast describes a functional status rather than fixed cell type.100 The transformation of RPE, glial and blood-borne cells to myofibroblasts is characterized by expression of α-smooth-muscle actin, a contractive component which is thought to be essential for tractional force generation.100 Transmembrane integrins at the myofibroblast surface link bundles of actin microfilaments with extracellular matrix. Through this mechanotransduction system, the contractile force generated by stress fibers containing α-smooth-muscle actin can be transmitted to the surrounding ECM.101 Cytokines and the components of ECM modulate the contractile activities of myofibroblasts. Blood serum components and the members of PDGF, IGF and (TGF)-β families are considered to be the major mediators of ECM contraction in the PVR process.19,102
In an experimental animal model, intravitreal injections of RPE cells or mixtures of RPE and glial cells show a progressive contractive response.103 Transdifferentiated RPE cells can attach to individual strands of collagen and pull collagen by alternating extension and retraction of the lamellipodia.104 Vitreous cortex which consists of densely packed collagen fibrils may form a scaffold for fibrocellular proliferation and is an active player in the remodeling process. The studies of cell-mediated gel contraction demonstrate that vitreous samples from PVR patients contain sufficient amounts of biologically active factors to induce extracellular matrix contraction.102,105 Clinical data also support the role of vitreous as PVR-stimulated milieu. Thus tamponade with heavy silicone oil can displace the PVR milieu from the lower part to the upper retina resulting in upper PVR.106 Removal of the vitreous (scaffold and PVR-stimulated milieu) is a rational treatment target for PVR.
Biomarkers
Intensive development of molecular biology techniques has permitted identification of a wide variety of cellular and biological characteristics of PVR, and the search for predictive molecular risk factors (biomarkers) of PVR susceptibility was intensified in recent years. To find proteins that are specifically involved in distinct pathophysiological events of PVR, vitreous proteomes were investigated. More than 100 PVR-specific proteins have been identified, including proteins involved in metabolic dysfunction, immune responses, and cytoskeleton remodeling. Significant differences were detected in the composition of vitreous proteins between control and PVR samples. While the cytoskeletal and metabolic proteins, such as enolases, are downregulated in severe PVR, complement components, the serine proteinase inhibitor (serpin), and proteins involved in cell proliferation are upregulated.107,108 The activity of the enzyme lysyl oxidase, which contributes to extracellular matrix integrity, is decreased in PVR vitreous.109 In addition, the vitreous of PVR patients contains increased levels of the MMP-2 and -9; the vitreal content of MMPs (MMP-1, -2, -3, -8, -9 and TIMP-1) correlates with the grade of PVR.110 The elevation of the vitreal content of inflammation-associated proteins such as α1-antitrypsin, apolipoprotein A-IV, serum albumin, and transferrin is consistent with the significance of inflammation in the pathogenesis of PVR.111 This suggests that steroids such as triamcinolone acetonide might be helpful as adjuvant therapy of PVR. Kininogen 1 (essential for assembly of the kallikrein–kinin system) is one of the relatively high-abundant proteins in the vitreous and serum of PVR patients and may thus represent a serum biomarker of PVR.108 Attempts to assess the genetic contribution to PVR have also been made.112 Interestingly, a strong genetic association between the tumor necrosis factor locus and PVR was observed which underlines the importance of inflammation in the pathogenesis of PVR.113 It can be concluded that PVR is a complex syndrome, which involves impairment of metabolic function, remodeling of the cytoskeleton and extracellular matrix, immune responses, and inflammation.
Conclusion
Despite the substantial progress that has been made during the past 30 years, the pathogenesis of PVR is incompletely understood. At present PVR can be considered as an excessive vitreoretinal wound healing process characterized by the phases of inflammation, proliferation, and remodeling (Fig. 97.2). Once the vicious cycle of detachment and “storm of cytokines” has started a permanent functional failure due to structural retinal changes is initiated. Multiple different cellular and soluble factors are involved in the development of PVR, including growth and inflammatory factors, serum, fibrin, MMPs. Therefore, therapeutic options based on the inhibition of one factor or phenomenon may be regarded with scepticism. Further research to understand the relative importance of distinct factors in the formation and progression of PVR may lead to more effective therapeutic approaches for the treatment of PVR. Until then surgery is the treatment of choice. Removal of activated cells and membranes and complete vitreous removal is a rational treatment goal achieved by surgery. In the future combined analysis of clinical risk factors and biomarkers should improve the identification of patients at high risk of PVR formation and may allow targeted application of appropriate adjunctive therapy.
1 Charteris DG, Sethi CS, Lewis GP, et al. Proliferative vitreoretinopathy-developments in adjunctive treatment and retinal pathology. Eye (Lond). 2002;16:369–374.
2 Kunavisarut P, Bijlsma WR, Pathanapitoon K, et al. Proliferative vitreoretinopathy in human immunodeficiency virus-infected patients in the era of highly active antiretroviral therapy. Am J Ophthalmol. 2010;150:218–222.
3 Scott IU, Flynn HW, Jr., Azen SP, et al. Silicone oil in the repair of pediatric complex retinal detachments: a prospective, observational, multicenter study. Ophthalmology. 1999;106:1399–1407. discussion 407–8
4 Laqua H, Machemer R. Glial cell proliferation in retinal detachment (massive periretinal proliferation). Am J Ophthalmol. 1975;80:602–618.
5 Heimann K, Wiedemann P. Proliferative vitreoretinopathy. Heidelberg: Kaden Verlag, 1989.
6 Lean JS, Stern WH, Irvine AR, et al. Classification of proliferative vitreoretinopathy used in the silicone study. The Silicone Study Group. Ophthalmology. 1989;96:765–771.
7 Machemer R, Aaberg TM, Freeman HM, et al. An updated classification of retinal detachment with proliferative vitreoretinopathy. Am J Ophthalmol. 1991;112:159–165.
8 Pastor JC, de la Rua ER, Martin F. Proliferative vitreoretinopathy: risk factors and pathobiology. Prog Retin Eye Res. 2002;21:127–144.
9 Iandiev I, Bringmann A, Wiedemann P. [Proliferative vitreoretinopathy–pathogenesis and therapy]. Klin Monbl Augenheilkd. 2010;227:168–174.
10 Nagasaki H, Shinagawa K. Risk factors for proliferative vitreoretinopathy. Curr Opin Ophthalmol. 1995;6:70–75.
11 Nagasaki H, Shinagawa K, Mochizuki M. Risk factors for proliferative vitreoretinopathy. Prog Retin Eye Res. 1998;17:77–98.
12 Girard P, Mimoun G, Karpouzas I, et al. Clinical risk factors for proliferative vitreoretinopathy after retinal detachment surgery. Retina. 1994;14:417–424.
13 Wickham L, Ho-Yen GO, Bunce C, et al. Surgical failure following primary retinal detachment surgery by vitrectomy: risk factors and functional outcomes. Br J Ophthalmol. 2010;95:1234–1238.
14 Weller M, Wiedemann P, Heimann K. Proliferative vitreoretinopathy–is it anything more than wound healing at the wrong place? Int Ophthalmol. 1990;14:105–117.
15 Kirchhof B, Sorgente N. Pathogenesis of proliferative vitreoretinopathy. Modulation of retinal pigment epithelial cell functions by vitreous and macrophages. Dev Ophthalmol. 1989;16:1–153.
16 Mietz H, Heimann K. Onset and recurrence of proliferative vitreoretinopathy in various vitreoretinal disease. Br J Ophthalmol. 1995;79:874–877.
17 Hui YN, Hu D. Prevention of experimental proliferative vitreoretinopathy with daunomycin and triamcinolone based on the time course of the disease. Graefes Arch Clin Exp Ophthalmol. 1999;237:601–605.
18 Hiscott PS, Grierson I, McLeod D. Natural history of fibrocellular epiretinal membranes: a quantitative, autoradiographic, and immunohistochemical study. Br J Ophthalmol. 1985;69:810–823.
19 Guidry C. The role of Muller cells in fibrocontractive retinal disorders. Prog Retin Eye Res. 2005;24:75–86.
20 Sramek SJ, Wallow IH, Stevens TS, et al. Immunostaining of preretinal membranes for actin, fibronectin, and glial fibrillary acidic protein. Ophthalmology. 1989;96:835–841.
21 McGillem GS, Dacheux RF. Rabbit retinal Muller cells undergo antigenic changes in response to experimentally induced proliferative vitreoretinopathy. Exp Eye Res. 1999;68:617–627.
22 Tamiya S, Liu L, Kaplan HJ. Epithelial-mesenchymal transition and proliferation of retinal pigment epithelial cells initiated upon loss of cell-cell contact. Invest Ophthalmol Vis Sci. 2010;51:2755–2763.
23 Pratt CH, Vadigepalli R, Chakravarthula P, et al. Transcriptional regulatory network analysis during epithelial-mesenchymal transformation of retinal pigment epithelium. Mol Vis. 2008;14:1414–1428.
24 Lee H, O’Meara SJ, O’Brien C, et al. The role of gremlin, a BMP antagonist, and epithelial-to-mesenchymal transition in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2007;48:4291–4299.
25 He S, Kumar SR, Zhou P, et al. Soluble EphB4 inhibition of PDGF-induced RPE migration in vitro. Invest Ophthalmol Vis Sci. 2010;51:543–552.
26 Bringmann A, Pannicke T, Grosche J, et al. Muller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25:397–424.
27 Geller SF, Lewis GP, Fisher SK. FGFR1, signaling, and AP-1 expression after retinal detachment: reactive Muller and RPE cells. Invest Ophthalmol Vis Sci. 2001;42:1363–1369.
28 Charteris DG, Downie J, Aylward GW, et al. Intraretinal and periretinal pathology in anterior proliferative vitreoretinopathy. Graefes Arch Clin Exp Ophthalmol. 2007;245:93–100.
29 Fisher SK, Lewis GP. Muller cell and neuronal remodeling in retinal detachment and reattachment and their potential consequences for visual recovery: a review and reconsideration of recent data. Vision Res. 2003;43:887–897.
30 Bringmann A, Francke M, Pannicke T, et al. Human Muller glial cells: altered potassium channel activity in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1999;40:3316–3323.
31 Francke M, Pannicke T, Biedermann B, et al. Loss of inwardly rectifying potassium currents by human retinal glial cells in diseases of the eye. Glia. 1997;20:210–218.
32 Francke M, Faude F, Pannicke T, et al. Electrophysiology of rabbit Muller (glial) cells in experimental retinal detachment and PVR. Invest Ophthalmol Vis Sci. 2001;42:1072–1079.
33 Bringmann A, Wiedemann P. Involvement of Muller glial cells in epiretinal membrane formation. Graefes Arch Clin Exp Ophthalmol. 2009;247:865–883.
34 Bringmann A, Francke M, Pannicke T, et al. Role of glial K(+) channels in ontogeny and gliosis: a hypothesis based upon studies on Muller cells. Glia. 2000;29:35–44.
35 Francke M, Faude F, Pannicke T, et al. Glial cell-mediated spread of retinal degeneration during detachment: a hypothesis based upon studies in rabbits. Vision Res. 2005;45:2256–2267.
36 Drescher KM, Whittum-Hudson JA. Evidence for induction of interferon-alpha and interferon-beta in retinal glial cells of Muller. Virology. 1997;234:309–316.
37 Lu YB, Franze K, Seifert G, et al. Viscoelastic properties of individual glial cells and neurons in the CNS. Proc Natl Acad Sci USA. 2006;103:17759–17764.
38 Lu YB, Iandiev I, Hollborn M, et al. Reactive glial cells: increased stiffness correlates with increased intermediate filament expression. FASEB J. 2011;25:624–631.
39 Abu El-Asrar AM, Struyf S, Van Damme J, et al. Circulating fibrocytes contribute to the myofibroblast population in proliferative vitreoretinopathy epiretinal membranes. Br J Ophthalmol. 2008;92:699–704.
40 Lin ML, Li YP, Li ZR, et al. Macrophages acquire fibroblast characteristics in a rat model of proliferative vitreoretinopathy. Ophthalmic Res. 2011;45:180–190.
41 Sakamoto T, Ishibashi T. Hyalocytes: essential cells of the vitreous cavity in vitreoretinal pathophysiology? Retina. 2011;31:222–228.
42 Wiedemann P. Growth factors in retinal diseases: proliferative vitreoretinopathy, proliferative diabetic retinopathy, and retinal degeneration. Surv Ophthalmol. 1992;36:373–384.
43 Puro DG, Mano T, Chan CC, et al. Thrombin stimulates the proliferation of human retinal glial cells. Graefes Arch Clin Exp Ophthalmol. 1990;228:169–173.
44 Parrales A, Palma-Nicolas JP, Lopez E, et al. Thrombin stimulates RPE cell proliferation by promoting c-Fos-mediated cyclin D1 expression. J Cell Physiol. 2010;222:302–312.
45 Lei H, Hovland P, Velez G, et al. A potential role for PDGF-C in experimental and clinical proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2007;48:2335–2342.
46 Cui JZ, Chiu A, Maberley D, et al. Stage specificity of novel growth factor expression during development of proliferative vitreoretinopathy. Eye (Lond). 2007;21:200–208.
47 Li R, Maminishkis A, Wang FE, et al. PDGF-C and -D induced proliferation/migration of human RPE is abolished by inflammatory cytokines. Invest Ophthalmol Vis Sci. 2007;48:5722–5732.
48 Moon SW, Chung EJ, Jung SA, et al. PDGF stimulation of Muller cell proliferation: Contributions of c-JNK and the PI3K/Akt pathway. Biochem Biophys Res Commun. 2009;388:167–171.
49 Lei H, Velez G, Hovland P, et al. Plasmin is the major protease responsible for processing PDGF-C in the vitreous of patients with proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2008;49:42–48.
50 Cui J, Lei H, Samad A, et al. PDGF receptors are activated in human epiretinal membranes. Exp Eye Res. 2009;88:438–444.
51 Ikuno Y, Kazlauskas A. An in vivo gene therapy approach for experimental proliferative vitreoretinopathy using the truncated platelet-derived growth factor alpha receptor. Invest Ophthalmol Vis Sci. 2002;43:2406–2411.
52 Saishin Y, Takahashi K, Seo MS, et al. The kinase inhibitor PKC412 suppresses epiretinal membrane formation and retinal detachment in mice with proliferative retinopathies. Invest Ophthalmol Vis Sci. 2003;44:3656–3662.
53 Lei H, Velez G, Hovland P, et al. Growth factors outside the PDGF family drive experimental PVR. Invest Ophthalmol Vis Sci. 2009;50:3394–3403.
54 Lei H, Velez G, Cui J, et al. N-acetylcysteine suppresses retinal detachment in an experimental model of proliferative vitreoretinopathy. Am J Pathol. 2010;177:132–140.
55 Saika S, Yamanaka O, Okada Y, et al. TGF beta in fibroproliferative diseases in the eye. Front Biosci (Schol Ed). 2009;1:376–390.
56 Kita T, Hata Y, Kano K, et al. Transforming growth factor-beta2 and connective tissue growth factor in proliferative vitreoretinal diseases: possible involvement of hyalocytes and therapeutic potential of Rho kinase inhibitor. Diabetes. 2007;56:231–238.
57 Winkler J, Hoerauf H. TGF-β and RPE-derived cells in taut subretinal strands from patients with proliferative vitreoretinopathy. Eur J Ophthalmol. 2011;21:422–426.
58 Anderson DH, Guerin CJ, Hageman GS, et al. Distribution of transforming growth factor-beta isoforms in the mammalian retina. J Neurosci Res. 1995;42:63–79.
59 Guerin CJ, Hu L, Scicli G, et al. Transforming growth factor beta in experimentally detached retina and periretinal membranes. Exp Eye Res. 2001;73:753–764.
60 Oshima Y, Sakamoto T, Hisatomi T, et al. Gene transfer of soluble TGF-beta type II receptor inhibits experimental proliferative vitreoretinopathy. Gene Ther. 2002;9:1214–1220.
61 Kawahara S, Hata Y, Kita T, et al. Potent inhibition of cicatricial contraction in proliferative vitreoretinal diseases by statins. Diabetes. 2008;57:2784–2793.
62 Kita T, Hata Y, Arita R, et al. Role of TGF-beta in proliferative vitreoretinal diseases and ROCK as a therapeutic target. Proc Natl Acad Sci USA. 2008;105:17504–17509.
63 Itoh Y, Kimoto K, Imaizumi M, et al. Inhibition of RhoA/Rho-kinase pathway suppresses the expression of type I collagen induced by TGF-beta2 in human retinal pigment epithelial cells. Exp Eye Res. 2007;84:464–472.
64 Nakazawa T, Matsubara A, Noda K, et al. Characterization of cytokine responses to retinal detachment in rats. Mol Vis. 2006;12:867–878.
65 Han QH, Hui YN, Du HJ, et al. Migration of retinal pigment epithelial cells in vitro modulated by monocyte chemotactic protein-1: enhancement and inhibition. Graefes Arch Clin Exp Ophthalmol. 2001;239:531–538.
66 Abu el-Asrar AM, Van Damme J, Put W, et al. Monocyte chemotactic protein-1 in proliferative vitreoretinal disorders. Am J Ophthalmol. 1997;123:599–606.
67 Asaria RH, Kon CH, Bunce C, et al. Silicone oil concentrates fibrogenic growth factors in the retro-oil fluid. Br J Ophthalmol. 2004;88:1439–1442.
68 Hueber A, Wiedemann P, Esser P, et al. Basic fibroblast growth factor mRNA, bFGF peptide and FGF receptor in epiretinal membranes of intraocular proliferative disorders (PVR and PDR). Int Ophthalmol. 1996;20:345–350.
69 Liang X, Li C, Gao R, et al. Quantitative study of basic fibroblast growth factor in vitreous with proliferative vitreoretinopathy. Yan Ke Xue Bao. 2000;16:7–10.
70 Hoffmann S, He S, Jin M, et al. A selective cyclic integrin antagonist blocks the integrin receptors alphavbeta3 and alphavbeta5 and inhibits retinal pigment epithelium cell attachment, migration and invasion. BMC Ophthalmol. 2005;5:16.
71 Briggs MC, Grierson I, Hiscott P, et al. Active scatter factor (HGF/SF) in proliferative vitreoretinal disease. Invest Ophthalmol Vis Sci. 2000;41:3085–3094.
72 Lashkari K, Rahimi N, Kazlauskas A. Hepatocyte growth factor receptor in human RPE cells: implications in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1999;40:149–156.
73 Grierson I, Heathcote L, Hiscott P, et al. Hepatocyte growth factor/scatter factor in the eye. Prog Retin Eye Res. 2000;19:779–802.
74 Hinton DR, He S, Jin ML, et al. Novel growth factors involved in the pathogenesis of proliferative vitreoretinopathy. Eye (Lond). 2002;16:422–428.
75 Hollborn M, Krausse C, Iandiev I, et al. Glial cell expression of hepatocyte growth factor in vitreoretinal proliferative disease. Lab Invest. 2004;84:963–972.
76 He S, Chen Y, Khankan R, et al. Connective tissue growth factor as a mediator of intraocular fibrosis. Invest Ophthalmol Vis Sci. 2008;49:4078–4088.
77 Guo CM, Wang YS, Hu D, et al. Modulation of migration and Ca2+ signaling in retinal pigment epithelium cells by recombinant human CTGF. Curr Eye Res. 2009;34:852–862.
78 Kita T, Hata Y, Miura M, et al. Functional characteristics of connective tissue growth factor on vitreoretinal cells. Diabetes. 2007;56:1421–1428.
79 Liang CM, Tai MC, Chang YH, et al. Glucosamine inhibits epidermal growth factor-induced proliferation and cell-cycle progression in retinal pigment epithelial cells. Mol Vis. 2010;16:2559–2571.
80 Yan F, Hui YN, Li YJ, et al. Epidermal growth factor receptor in cultured human retinal pigment epithelial cells. Ophthalmologica. 2007;221:244–250.
81 Chen Z, Chen CZ, Gong WR, et al. Integrin-alpha5 mediates epidermal growth factor-induced retinal pigment epithelial cell proliferation and migration. Pathobiology. 2010;77:88–95.
82 Ogata N, Nishikawa M, Nishimura T, et al. Inverse levels of pigment epithelium-derived factor and vascular endothelial growth factor in the vitreous of eyes with rhegmatogenous retinal detachment and proliferative vitreoretinopathy. Am J Ophthalmol. 2002;133:851–852.
83 Armstrong D, Augustin AJ, Spengler R, et al. Detection of vascular endothelial growth factor and tumor necrosis factor alpha in epiretinal membranes of proliferative diabetic retinopathy, proliferative vitreoretinopathy and macular pucker. Ophthalmologica. 1998;212:410–414.
84 Kon CH, Occleston NL, Aylward GW, et al. Expression of vitreous cytokines in proliferative vitreoretinopathy: a prospective study. Invest Ophthalmol Vis Sci. 1999;40:705–712.
85 Hui Y, Shi Y, Zhang X, et al. [TNF-alpha, IL-8 and IL-6 in the early inflammatory stage of experimental PVR model induced by macrophages]. Zhonghua Yan Ke Za Zhi. 1999;35:140–143.
86 Kim SH, Turnbull JE, Guimond S. Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol. 2011;209:139–151.
87 Byron A, Morgan MR, Humphries MJ. Adhesion signalling complexes. Curr Biol. 2010;20:R1063–R1067.
88 Hou X, Han QH, Hu D, et al. Mechanical force enhances MMP-2 activation via p38 signaling pathway in human retinal pigment epithelial cells. Graefes Arch Clin Exp Ophthalmol. 2009;247:1477–1486.
89 Wang LH, Li GL. [Progress in studies on effects of extracellular matrix in occurrence of proliferative vitreoretinopathy]. Zhonghua Yan Ke Za Zhi. 2008;44:759–763.
90 Hollborn M, Reichenbach A, Wiedemann P, et al. Contrary effects of cytokines on mRNAs of cell cycle- and ECM-related proteins in hRPE cells in vitro. Curr Eye Res. 2004;28:215–223.
91 Jerdan JA, Pepose JS, Michels RG, et al. Proliferative vitreoretinopathy membranes. An immunohistochemical study. Ophthalmology. 1989;96:801–810.
92 Hollborn M, Faude F, Wiedemann P, et al. Elevated proto-oncogene and collagen mRNA expression in PVR retinas. Graefes Arch Clin Exp Ophthalmol. 2003;241:439–446.
93 George B, Chen S, Chaudhary V, et al. Extracellular matrix proteins in epiretinal membranes and in diabetic retinopathy. Curr Eye Res. 2009;34:134–144.
94 Ioachim E, Stefaniotou M, Gorezis S, et al. Immunohistochemical study of extracellular matrix components in epiretinal membranes of vitreoproliferative retinopathy and proliferative diabetic retinopathy. Eur J Ophthalmol. 2005;15:384–391.
95 Gamulescu MA, Chen Y, He S, et al. Transforming growth factor beta2-induced myofibroblastic differentiation of human retinal pigment epithelial cells: regulation by extracellular matrix proteins and hepatocyte growth factor. Exp Eye Res. 2006;83:212–222.
96 Khankan R, Oliver N, He S, et al. Regulation of fibronectin-EDA through CTGF domain-specific interactions with TGFβ2 and its Receptor TGFβRII. Invest Ophthalmol Vis Sci. 2011;52:5068–5078.
97 Hiscott P, Paraoan L, Choudhary A, et al. Thrombospondin 1, thrombospondin 2 and the eye. Prog Retin Eye Res. 2006;25:1–18.
98 Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–839.
99 Sheridan CM, Occleston NL, Hiscott P, et al. Matrix metalloproteinases: a role in the contraction of vitreo-retinal scar tissue. Am J Pathol. 2001;159:1555–1566.
100 Hinz B, Gabbiani G. Fibrosis: recent advances in myofibroblast biology and new therapeutic perspectives. F1000 Biol Rep. 2010;2:78.
101 Tomasek JJ, Gabbiani G, Hinz B, et al. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363.
102 Kita T. [Molecular mechanisms of preretinal membrane contraction in proliferative vitreoretinal diseases and ROCK as a therapeutic target]. Nippon Ganka Gakkai Zasshi. 2010;114:927–934.
103 Peters MA, Burke JM, Clowry M, et al. Development of traction retinal detachments following intravitreal injections of retinal Muller and pigment epithelial cells. Graefes Arch Clin Exp Ophthalmol. 1986;224:554–563.
104 Glaser BM, Cardin A, Biscoe B. Proliferative vitreoretinopathy. The mechanism of development of vitreoretinal traction. Ophthalmology. 1987;94:327–332.
105 Beutel J, Luke M, Bartz-Schmidt KU, et al. [Vitreal-induced RPE cell traction. Investigation of pathological vitreous samples in an in vitro contraction model]. Ophthalmologe. 2009;106:893–898.
106 Joussen AM, Lux A, Kirchhof B. Shifting of the proliferative vitreoretinopathy milieu after tamponade with heavy silicone oil in eyes prone to proliferative vitreoretinopathy and bleeding. Br J Ophthalmol. 2009;93:128–129.
107 Grisanti S, Wiedemann P, Weller M, et al. The significance of complement in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1991;32:2711–2717.
108 Yu J, Liu F, Cui SJ, et al. Vitreous proteomic analysis of proliferative vitreoretinopathy. Proteomics. 2008;8:3667–3678.
109 Coral K, Angayarkanni N, Madhavan J, et al. Lysyl oxidase activity in the ocular tissues and the role of LOX in proliferative diabetic retinopathy and rhegmatogenous retinal detachment. Invest Ophthalmol Vis Sci. 2008;49:4746–4752.
110 Symeonidis C, Papakonstantinou E, Souliou E, et al. Correlation of matrix metalloproteinase levels with the grade of proliferative vitreoretinopathy in the subretinal fluid and vitreous during rhegmatogenous retinal detachment. Acta Ophthalmol. 2011;89:339–345.
111 Shitama T, Hayashi H, Noge S, et al. Proteome Profiling of Vitreoretinal Diseases by Cluster Analysis. Proteomics Clin Appl. 2008;2:1265–1280.
112 Sanabria Ruiz-Colmenares MR, Pastor Jimeno JC, Garrote Adrados JA, et al. Cytokine gene polymorphisms in retinal detachment patients with and without proliferative vitreoretinopathy: a preliminary study. Acta Ophthalmol Scand. 2006;84:309–313.
113 Rojas J, Fernandez I, Pastor JC, et al. A strong genetic association between the tumor necrosis factor locus and proliferative vitreoretinopathy: The Retina 4 Project. Ophthalmology. 2010;117:2417. e2–23.e2