CHAPTER 383 Pathobiology of True Arteriovenous Malformations
Harvey Cushing and Walter Dandy are credited with the modern conceptualization of cerebrovascular malformations. McCormick in 19662 and Russell and Rubenstein3 described four types of vascular malformations, and this is now accepted as the current nomenclature. Cerebrovascular malformations are classified according to their histopathologic features as arteriovenous malformation (AVM), venous angioma, cavernous malformation (CM), and capillary telangiectasia. The focus of this chapter is on true AVMs. CMs, including their genetics, and venous angiomas are discussed in detail in Part 7. A possible fifth category is a direct fistula, or arteriovenous fistula (AVF). These conditions are regarded as acquired lesions involving single or multiple dilated arterioles that connect directly to a vein without a nidus. They are high-flow, high-pressure lesions that have a low incidence of hemorrhage (examples include vein of Galen aneurysmal malformation, dural AVF [DAVF], and carotid cavernous fistula). DAVFs are not discussed in this chapter because they are considered acquired lesions but are covered in Part 6. Later chapters provide a comprehensive analysis of the surgical principles and techniques related to AVMs.
Arteriovenous Malformations
True AVMs are abnormalities of the intracranial vessels in which the arterial and venous systems are connected without an intervening capillary bed.4
Pathology, Pathogenesis, and Pathophysiology
AVMs are high-flow cerebrovascular lesions that consist of a tangle of abnormal blood vessels. Three morphologic features are typical of these lesions: feeding arteries, draining veins, and a dysplastic vascular nidus composed of a tangle of abnormal vessels that acts as a shunt from the arterial to the venous system.5 Gross pathologic features include the absence of a capillary bed and the presence of small feeding arteries composed of variable amounts of smooth muscle and elastic laminae, with single or multiple direct arteriovenous (AV) connections. The lack of a capillary bed results in low resistance and, with the AV connections, permits high-flow AV shunting. Although the feeding vessels and draining veins themselves may not be congenitally abnormal, their communication through the nidus subsequently leads to arterial dilation and venous arterialization (Fig. 383-1).
This chronic high-flow shunt produces secondary structural changes in the feeding and draining vessels, dilation of the feeding arteries, and dilation and thickening of the draining veins.5 Smooth muscle hyperplasia associated with fibroblasts and connective tissue elements known as fibromuscular cushions develops in the feeding arteries.
The nidus (Latin = nest) was described by Cushing and Bailey as a “snarl” based on its gross appearance.1 The nidus is a conglomeration of numerous AV shunts without interposed brain tissue and no capillary bed. Microscopically, the nidus has thin collagenous walls in the venous elements with muscular elastic walls in the feeding arteries.
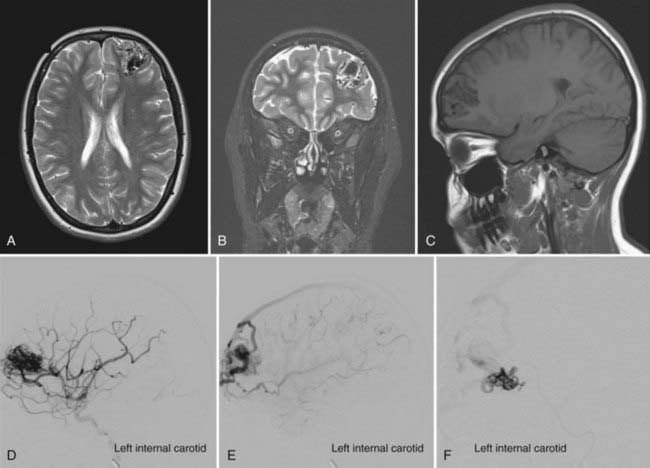
Aneurysms are associated with 2.3% to 16.7% of AVMs (Fig. 383-2).6,7 The pathophysiology is not known definitively, but it is believed to be secondary to a high-flow vasculopathy.8–10 AVM-associated aneurysms are classified by location as either flow related (85%) or unrelated (15%). The latter are located on remote vessels that bear no relationship to the supply of the AVM.11,12 Flow-related aneurysms occur along vessels that supply the AVM and are classified as being either proximal (from arteries on the circle of Willis or the proximal feeding vessels up to a primary bifurcation) or distal (from the feeding vessel distal to its origin from the parent artery at its primary bifurcation). Intranidal aneurysms occur within the nidus of the AVM and show early filling during angiography.11 They account for 5.5% of flow-related aneurysms.11
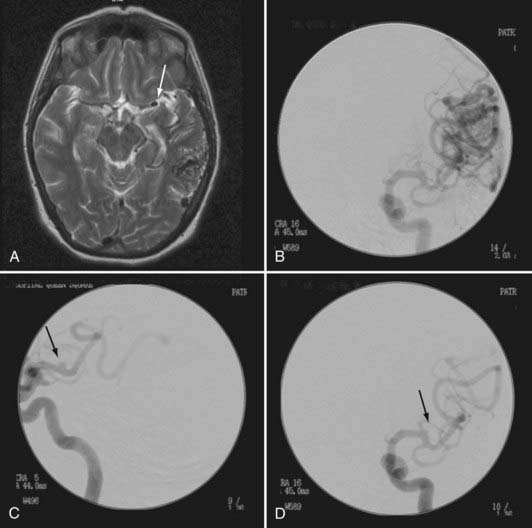
Varix formation may be associated with AVMs, especially those with a paucity of venous drainage. The pathophysiology of this venopathy is unclear but is believed to be due to secondary angiomorphologic changes induced by high flow downstream of the AV shunt of the nidus.13 Valavanis and Yasargil associated this condition with an increased risk for hemorrhage.13
The perinidal capillary network may be a cause of recurrence of surgically resected AVMs. Dilated capillaries (10 to 25 times larger than normal capillaries) form a ring (1 to 7 mm) around the nidus. These capillaries are connected to the nidus, to the feeding arteries/draining veins, and to surrounding normal brain vessels.14,15 Unlike CMs, intervening neural parenchyma may be present within the compact network of dysplastic vascular channels that forms the nidus. The parenchymal elements tend to be gliotic, hemosiderin stained, and nonfunctional. Vascular or interstitial calcification is frequently a feature. A type of proliferative or diffuse AVM without a focal nidus is often seen in pediatric patients and has been referred to by Lasjaunias and colleagues as cerebral proliferative angiopathy.16
Etiology
AVMs may be classified as being sporadic or syndromic in origin. Sporadic AVMs are by far the most common, with a global prevalence of 0.04% to 0.52%.17,18 Although sporadic, there is increasing evidence that they develop as the result of upregulation or downregulation of multiple homeobox genes that are involved in angiogenesis, such as Hox D319 and Hox B3.20 Syndromic AVMs account for approximately 2% of cases. Multiple familial AVMs are seen in hereditary hemorrhagic telangiectasia (HHT).21 Cerebrofacial arteriovenous metameric syndromes (CAMSs) involve the face and brain.22
Three percent to 20% of sporadic AVMs are diagnosed in children,23 and AVMs are the most common cause of spontaneous brain hemorrhage in children (excluding the neonatal period).24,25 AVMs are prone to apoplectic hemorrhage by rupture of nidal vessels or associated aneurysms or by obstruction to venous outflow. Bleeding is typically from rupture of a draining vein in association with dilation, kinking, and thrombosis or from rupture of flow-related aneurysms, which are more prevalent than in adults.26 Large AVMs generate an arterial steal phenomenon, and older children may exhibit progressive neurological deterioration or chronic epilepsy (or both).27 If sufficient AV shunting is present, they may initially be manifested as congestive cardiac failure in neonates and infants.
Staging, Grading, or Classification Criteria
The Spetzler-Martin scale classifies AVMs according to size, location in relation to functionally eloquent cortex, and type of venous drainage.28 Multiple brain AVMs are typically seen as part of HHT in association with pulmonary and hepatic AVMs and cutaneous and mucosal telangiectases21 or CAMSs.22 A typical example is Wyburn-Mason syndrome, in which an AVM, usually of the diencephalon/optic pathway or midbrain/thalamus, occurs in conjunction with retinal vascular malformations seen on funduscopy (Fig. 383-3). Other CAMSs have been described that link facial development with embryologic brain development.22 Hence, in CAMS-1, prosencephalic AVMs affecting the hypothalamus/hypophysis are seen in association with a facial AVM of the nose. In CAMS-2, AVMs affecting the lateral prosencephalon (occipital lobe, thalamus) are seen in association with facial AVMs of the maxilla. In CAMS-3, AVMs of the rhombencephalon (cerebellum, pons) are seen with facial AVMs of the mandible.29,30
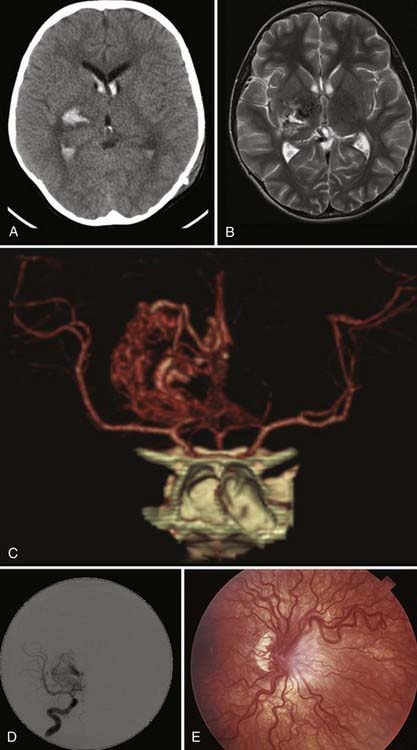
AVMs occur throughout the neuraxis, and most come to attention between the ages of 20 and 50 years because of symptoms. They are definitively identified by angiography,31 which shows not only the nidus of tangled vessels but also early venous filling secondary to direct arterial-to-venous shunting within the lesion. Clinically, they are primarily manifested as hemorrhage, which is seen in approximately 65% of symptomatic lesions17; 15% to 35% have seizures as the initial symptom,32 and the remainder are manifested as headache or progressive neurologic deficits. Although a hemorrhagic manifestation is also common with CMs, the extent of hemorrhage in AVMs is frequently more dramatic and disabling because of the high-flow nature of these lesions. Consequently, mortality rates associated with AVM hemorrhage are approximately 6% to 29%.17,33,34
Venous Angioma
Synonym: developmental venous anomaly
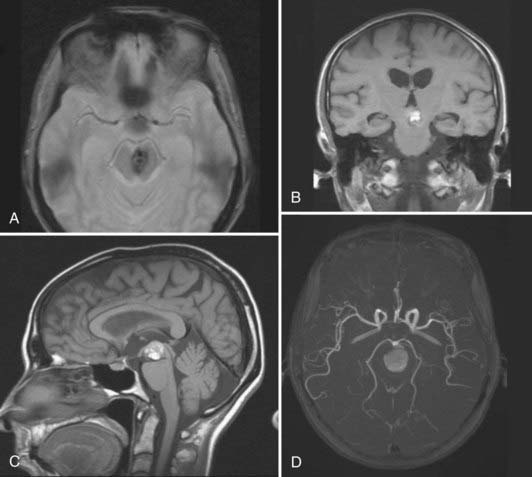
Venous angiomas are the most common intracranial vascular malformation.35–37 According to one hypothesis, an intrauterine ischemic event occurs during the formation of medullary veins and results in collateral venous drainage.35 The pathologic characteristics of these lesions consist of anomalous veins separated by normal brain tissue. Histologic section shows that the walls of the veins are thickened and hyalinized and usually lack elastic tissue and smooth muscle.4
Cavernous Malformation
CMs are well-circumscribed, multilobulated, angiographically occult vascular malformations.
Pathology, Pathogenesis, and Pathophysiology
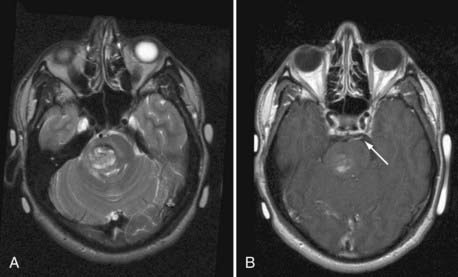
CMs are composed of sinusoidal vascular channels (or caverns) lined by a single layer of endothelium. The caverns are separated by a collagenous stroma that is devoid of elastin, smooth muscle, or other mature vascular wall elements. Lack of intervening brain parenchyma is a pathologic hallmark. Macroscopically, CMs are often referred to as having a “mulberry” appearance.2,4 Within the cavernoma, hyalinization, thrombosis with varying degrees of organization, calcification, cyst formation, and cholesterol deposition are common. The surrounding brain parenchyma exhibits evidence of previous microhemorrhage, hemosiderin staining, and hemosiderin-laden macrophages. A surrounding parenchymal gliomatous reaction is characteristic and may form a capsule around the lesion.38,39 Based on the magnetic resonance imaging (MRI) appearance (Figs. 383-4 and 383-5) of CMs, they have been classified into four types reflecting their dynamic nature and propensity for hemorrhage.40
Capillary Telangiectasia
Capillary telangiectases, also known as capillary malformations, are vascular malformations composed of dilated capillaries with normal intervening neural tissue. They are the second most common vascular malformation affecting the brain. Their estimated prevalence from autopsy studies is 0.3%.41 They account for 12.4% of angiographically occult vascular malformations but for only 2.6% of symptomatic lesions.42 Capillary telangiectases are not detectable with conventional cerebral angiography or contrast-enhanced computed tomography (CT).43 Lee and coworkers in 1997 characterized the MRI appearance of histopathologically unproven lesions thought to be capillary telangiectases on clinical grounds.43 They reported that they tend to be small, homogeneously enhancing lesions. They are hypointense to isointense on T1-weighted imaging and isointense to hyperintense on T2-weighted and proton density–weighted imaging and are seen most reliably on susceptibility-sensitive or gradient echo sequences. However, they can be difficult to appreciate and are likely to be under-recognized on CT or MRI. Asymptomatic lesions are identified rarely, but when they are, surgery is not recommended. Although several case reports have implicated capillary telangiectases as a cause of hemorrhage, this has been proved histopathologically in just a few cases.44,45
Pathology, Pathogenesis, and Pathophysiology
Blackwood in 1941 provided the first classic pathologic description.46 On gross examination, the cut surface of the brain has small areas resembling pink or brownish petechial hemorrhages. Microscopically, they resemble small tufts of capillaries. There is no smooth muscle and an absence of elastic fibers, and there are no feeding or draining vessels. Normal brain parenchyma is present between the dilated capillaries. Mild gliosis can surround the parenchyma; however, hemosiderin and other evidence of previous hemorrhage are unusual. “Hemangioma calcificans” is a rare calcified variant of capillary telangiectasia.47 The pathogenesis of these lesions is unknown.
Mixed Lesions: True Arteriovenous Malformations with Other Vascular Malformations
Mixed lesions that include a component of a true AVM are relatively uncommon.
Arteriovenous Malformations and Capillary Telangiectasia
Chang and coauthors reported a patient in whom capillary telangiectasia developed and became symptomatic approximately 1 month after resection of an adjacent AVM.48 They postulated that growth of the capillary telangiectasia may have been caused by the decompressive effect or changes in hemodynamics associated with resection of the AVM or by the release of angiogenic factors after surgery. This is the only report of such an association.
Arteriovenous Malformations and Developmental Venous Anomalies
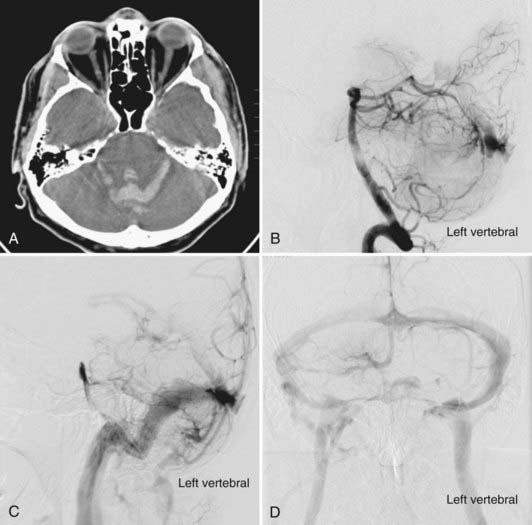
The coexistence of developmental venous anomalies (DVAs) and AVMs is well recognized despite being rare. The earliest description may have been that of Hirata and colleagues in 1986.49 This patient did not have an AVM nidus, however, and it may have been an AVF. Awad and associates reported a series of three lesions that clearly had both AVM and DVA components.50 The signs and symptoms in these three patients were thought to be caused by the AVM, not the DVA. These combined lesions preserve the appearance of the DVA on angiography with the exception of AV shunting during the arterial phase of the injection. Consequently, the venous lesion is identified earlier (Fig. 383-6). Frequently, it is difficult to appreciate the DVA separate from the AVM on MRI, and conventional cerebral angiography is more sensitive. Meyer and coworkers emphasized the pitfall of obliterating the DVA when approaching the AVM component in patients with symptomatic hemorrhage.51 From a pathophysiologic perspective, the association of an AVM with a DVA is perhaps the simplest of all the mixed malformations to explain. Mullan and colleagues reviewed four such patients and discussed the pathophysiology in the context of embryologic development of the cerebral venous system.52 They suggested that the AVM might form in a fashion similar to a DAVF: thrombosis of the star cluster would allow the DVA to become arterialized and form the basis of an AVM. Nussbaum and coauthors described the association of small AVMs with a DVA in the posterior fossa.53 They observed that venous hypertension may increase directly, through erythrocyte diapedesis, or indirectly by tissue ischemia causing an increase in the levels of angiogenic factors, an explanation previously suggested by Wilson.54 This venous hypertension may become chronic at the site of drainage of the DVA, with an acute increase reflecting a rise in intracranial venous pressure or occurring as a result of acute thrombosis. Comey and collaborators55 cited Dillon’s unpublished series of patients in whom so-called collector vein stenosis was suggested as a further mechanism for venous hypertension. Wilson speculated that venous hypertension may force previously diminutive AV shunts open, which could then enlarge over time.54 This supports the development of AVFs but not necessarily AVMs. In support of these theories, the literature has described areas of signal abnormality and enhancement surrounding some DVAs associated with CMs. This finding may indicate that some patients have legitimate hypertension and disruption of the blood-brain barrier,55 which is indicated by contrast enhancement. This concept is supported by a case reported by Ciricillo and coworkers.56 An angiographically occult AVM in association with a DVA was resected from a child in whom new angiographically occult lesions later developed that were presumed to be CMs. More recently, Im and coauthors reported a series of 15 atypical DVAs with arterialization but without a distinct nidus that behaved clinically as AVMs.57 The most interesting question that this article raises is whether these lesions are separate types of vascular malformations or simply part of a continuum of these lesions.58 The answer at present is unknown, and any theories will ultimately be proved or disproved by histochemical analysis.56
Arteriovenous Malformations and Cavernous Malformations
AVMs have been described in association with CMs. Garner and coworkers reported one such patient,59 and Awad50 and coauthors reported three. The latter group suggested that the AVM portion of the lesion tends to be angiographically and radiographically occult, thus making preoperative diagnosis extremely difficult. With respect to pathogenesis, smooth muscle may develop in the wall of the CM as a reaction to angiogenic factors. An alternative theory suggests that microhemorrhage from an occult AVM precipitates development of the CM.
Genetics of Arteriovenous Malformations
Considerable progress continues to be made in understanding the genetics of cerebrovascular malformations.60 Certain rare familial or congenital syndromes include these malformations among their constellations of abnormalities. Recognition of familial clustering in a subset of patients with cerebrovascular malformations has led to linkage analysis studies investigating the underlying genetic basis of these lesions.61 Using a positional cloning strategy, investigators identified two cerebrovascular malformation genes in patients with HHT, a syndrome that features cerebral AVMs. This suggests that a genetic defect underlies at least some vascular malformations. In addition to enhancing presymptomatic screening, identification of the genes responsible may result in better understanding of the pathogenesis of these lesions and, ultimately, in novel treatments.
AVMs account for the vast majority of symptomatic vascular malformations and are considered to be congenital developmental anomalies of blood vessels that arise during development of the embryonic cerebral circulation.4 They result from a persistent direct connection between the arterial and venous portions of the primitive vascular plexus in the embryo before the fourth week of gestation62; although there is evidence of a genetic basis for some categories of AVMs, their exact genetic origins have not been fully identified. Three general categories of cerebral AVMs are recognized: AVMs as part of an inherited disease in which a genetic factor has clearly been identified (e.g., HHT), familial cases of AVMs without a known genetic cause, and AVMs as part of a developmental neurocutaneous disorder (e.g., Wyburn-Mason syndrome). This section focuses on the genetic aspects of AVMs, with particular attention paid to familial and hereditary syndromes featuring AVMs.
Hereditary Syndromes Featuring Cerebral Arteriovenous Malformations
HHT, also known as Rendu-Osler-Weber syndrome, is a rare autosomal dominant vascular dysplasia characterized by vascular malformations in multiple organ systems. The nasal, mucocutaneous, pulmonary, cerebral, gastrointestinal, and hepatic vascular beds are most commonly affected.63 The clinical diagnosis of HHT is generally made according to the established Curaçao criteria.64 An individual is considered to have HHT if three of the following four criteria are met: recurrent spontaneous epistaxis; mucocutaneous telangiectasia; visceral involvement such as pulmonary and cerebral/spinal AVMs, gastrointestinal bleeding, or intrahepatic shunting; and a family history of HHT. The vascular malformations affecting the brain are primarily AVMs. Symptomatic cerebral AVMs are present in up to 5% of patients with HHT.63,65 Screening for asymptomatic lesions reveals a higher incidence of nearly 13%.66,67 Families with this uncommon angiodysplastic disorder often have multiple cerebral AVMs, with up to a third of HHT patients harboring AVMs having multiple lesions.65 HHT is known to have age-dependent penetrance (almost complete by 40 years of age).
HHT is a genetically and clinically heterogeneous disorder. Linkage analysis has mapped the genes underlying this syndrome to regions 9q33-q34.168,69 on chromosome 9 (HHT type 1 [HHT1]) and 12q11-q1470,71 on chromosome 12 (HHT2). HHT1 (Online Mendelian Inheritance in Man [OMIM] 187300) is caused by mutations in the ENG (endoglin) gene72,73 and is associated with a higher prevalence of pulmonary and cerebral AVMs. HHT2 (OMIM 600376) is caused by mutations in the ACVRL1 (activin receptor–like kinase 1 or ALK-1) gene74 and is associated with a milder pattern of disease expression and later age at onset. About 20% of HHT families remain unclassified after mutation analysis of these two genes, thus suggesting that other genes may be implicated.75 Indeed, in 2005 Cole and coworkers identified a new locus for HHT (HHT3) mapped to chromosome 5q31.3-32, although the causative gene remains unidentified.76 Mutations affecting the two genes endoglin (HHT1) and ALK-1 (HHT2) can partially explain the phenotypic heterogeneity of HHT, such as disease severity, variation in organ involvement, and age at onset. Distinct genotype-phenotype correlations do exist; for example, HHT1 is associated with a higher incidence of pulmonary and cerebral AVMs. Distinct mutations within the ALK-1 locus itself also appear to be linked to specific phenotypic traits77; however, the severity and type of clinical manifestations can differ even among family members carrying the same mutation,78 which suggests that epigenetic factors, such as the environment, blood pressure, or hormonal factors, may influence the clinical manifestations. Modifier genes may act to alter expression of the clinical characteristics.79 This is supported by murine models of HHT that show different phenotypic traits based on the background strain,80 thus suggesting that the genetic background modifies clinical expression of the disease. A subset of patients with a combined syndrome of juvenile polyposis and HHT (OMIM 175050) harbor mutations in the MADH4 gene.81
Linkage analysis in 1994 first mapped HHT to chromosome 9q33-q34.1,68,69 where endoglin was previously mapped in 1993.73 Subsequent testing confirmed endoglin as the disease-associated gene for HHT1.72 Endoglin is a receptor for transforming growth factor-β (TGF-β), specifically, the type III TGF-β receptor, and endoglin is the most abundant TGF-β binding protein in endothelial cells (ECs).60 Subsequently, mutations in another gene at the 12q locus were identified that affect ALK-1.74 ALK-1 is a member of the type I TGF-β receptor family
The TGF-β superfamily of proteins is involved in many functions, including vasculogenesis, wound repair, and angiogenesis. TGF-β itself is a potent angiogenic factor that plays important roles in tissue repair, growth, and differentiation. Endoglin, a type III TGF-β receptor, is a membrane glycoprotein with a molecular weight of 180 kD.60 By associating with the TGF-β type II and type I receptors (ALK-5 and ALK-1) at the cell surface, endoglin acts as a component of the type I TGF-β receptor complex, which binds TGF-β isoforms.82,83 Its expression is limited primarily to ECs and activated monocytes. Knockout mice lacking endoglin die during gestation secondary to defective vascular development, thus suggesting that endoglin is critical for vascular development. Mutated forms of endoglin may act as a dominant negative protein.84 However, studies of ECs and monocytes in both cerebral and pulmonary AVMs from patients with HHT1 have shown that normal endoglin dimers are still expressed at the cell surface but are reduced by 50% in comparison to normal individuals. No mutant endoglin was expressed on the cell surface but instead was found only as an intracellular homodimer.85 Such studies suggest that the mutant form of endoglin is not secreted to form heterodimers at the cell surface and is therefore unlikely to act as a dominant negative protein. This supports a model of haploinsufficiency,75 namely, that reduced levels of functional endoglin are the mechanism by which vascular abnormalities occur in HHT1. Because there is still some normal endoglin expression, the AVMs cannot be attributed to loss of heterozygosity and complete loss of endoglin expression.86
Like endoglin, ALK-1, one of the type I TGF-β receptors, is expressed exclusively on ECs. Mutations of the ALK-1 gene in HHT2 patients indicate that ALK-1 also plays an important role in vascular development.87 Mice lacking ALK-1 die during gestation, similar to endoglin knockout mice. They exhibit vascular abnormalities, including hyperdilation of vessels and abnormal fusion of capillary structures.88 The mutations that have been identified in patients with HHT2 appear to result in disruption of the activity of translated protein or instability of mutant mRNA. ALK-1 levels are reduced in ECs from patients with HHT2.89 Therefore, reduced expression of ALK-1 appears to be the primary defect in HHT2, as with endoglin in HHT1. In fact, most mutations of endoglin and ALK-1 identified to date in patients with HHT create null alleles that lead to reduced message or protein levels. This supports haploinsufficiency as the predominant mechanism, with inheritance of a mutation leading to reduced levels of the protein on the surface of vascular endothelium and predisposing affected individuals to the development of HHT-associated vascular lesions.90 The precise mechanism by which endoglin and ALK-1 mutations result in vascular abnormalities is unknown, but both proteins play a role in TGF-β signaling. Binding of TGF-β to type II TGF-β receptors, which is accelerated in the presence of endoglin, results in phosphorylation of the TGF-β type I receptors ALK-I and ALK-5. Phosphorylated ALK-1 and ALK-5 activate downstream proteins, known as Smad proteins, that enter the nucleus and regulate gene transcription involved in angiogenesis.91 Mutations in both endoglin and ALK-1 could result in alterations in this TGF-β signaling cascade. Because TGF-β is one of the factors critical for vascular homeostasis, disruption of its effects by altering the receptors or signaling pathways could result in abnormal vessel function. ECs with an inability to respond to TGF-β appropriately may form abnormal blood vessels, the basis for lesions such as AVMs. The existence of these genetic defects has not been studied in the rare instances of familial AVMs or investigated as the cause of sporadic AVMs; however, analysis of endoglin expression in sporadic AVMs has revealed no reduction or mutant forms, thus suggesting that endoglin deficiency may not be directly involved in the generation of sporadic AVMs.92
Familial Cerebral Arteriovenous Malformations
Genetically inherited angiodysplastic syndromes such as HHT provide the strongest evidence for the underlying genetic basis of some AVMs. Isolated familial predisposition in the absence of an angiodysplastic syndrome is exceedingly rare. Existing reports show several members and relatives of the same family harboring AVMs, but without clear demonstration of a genetic basis. Only about 20 cases of familial AVMs have been documented. Some authors have suggested an autosomal dominant mode of transmission with variable penetrance93–95 or an X-linked recessive pattern of inheritance.96 The clinical manifestations and location of familial AVMs do not appear to differ significantly from those of nonfamilial lesions. The age at diagnosis tends to be younger, and a higher proportion of such patients appear to have multiple AVMs.
Congenital Syndromes Featuring Arteriovenous Malformations
AVMs are considered to be congenital lesions that arise during a critical period of development of the brain vasculature. Congenital syndromes featuring AVMs or other vascular malformations suggest an underlying defect early in vascular development. To date, the involvement of genetic factors in such nonhereditary syndromes has not been clearly demonstrated. These are groups of rare syndromes that feature cerebrovascular abnormalities in conjunction with cutaneous vascular malformations. Wyburn-Mason syndrome, otherwise referred to as unilateral retinocephalic vascular malformation, consists of cutaneous vascular nevi, retinal and optic nerve vascular malformations, and ipsilateral or bilateral mesencephalic cerebral AVMs.97,98 Although the genetic defect is unknown, an association with the gene mutations responsible for HHT has been postulated.99
Advances in genetic techniques have allowed the identification of specific genetic defects underlying some forms of familial cerebrovascular malformations (i.e., familial CMs) and AVMs associated with the angiodysplastic syndrome HHT. This knowledge provides insight into the potential pathogenesis of these lesions. The reduced expression of endoglin and ALK-1 that is a result of mutations causing HHT raises the possibility of new therapeutic options, such as in vivo gene transfer targeting endoglin or ALK-1 expression in the vasculature.100 However, it is noteworthy that the genetic defects found to underlie these vascular malformations do not account for all cases, which suggests that multiple different gene defects may result in molecular changes that converge to give rise to the same lesion. Similarly, the effect of environmental factors cannot be ignored because certain genetic defects may result only in a predisposition to the development of certain malformations.
The Biology of Vasculogenesis and Angiogenesis
Vasculogenesis refers to the formation of primitive blood vessels inside the embryo (the primitive heart and primary vascular plexus) and its surrounding membranes (the extraembryonic yolk sac). Streeter detailed the development of the vascular system of the brain.101 Hematopoietic cells and ECs form the primitive vascular plexus by expansion, differentiation, and interconnection of mesoderm-derived stem cells (i.e., hemangioblasts) that aggregate and form blood islands.102–104 ECs fuse together to form the extraembryonic vascular network, which grows toward the embryo. The first intraembryonic angioblasts appear at the single-somite stage (during the third week of embryonic life), and interconnection with their extraembryonic counterparts is established at the two-somite stage.105 After circulation is initiated (at the end of the fourth gestational week), vascular smooth muscle cells and pericytes are recruited to the endothelial tubes to form increasingly mature vessels. In AVMs, direct connections between the future arterial and venous sides of the primitive vascular plexus persist, in addition to failure of development of an interposed capillary network, and AVMs are thought to develop during this embryonic stage.
Hemodynamic Effects of Arteriovenous Malformations
Acting as AV shunts, AVMs exert significant effects on cerebral hemodynamics. Generally, the presence of an AV shunt in the peripheral circulation results in venous hypertension downstream and arterial hypotension upstream. Such changes can trigger vascular remodeling, the process of which can further affect local hemodynamics. Whether acquired or congenital, ongoing vascular remodeling and angiogenesis presumably triggered by aberrant local hemodynamics have been considered to be a critical component of their pathophysiology.106–108 Clinical studies investigating the physiologic effects of AV shunts in the cerebral circulation have largely focused on AVMs. The hemodynamic effects of AV shunting are predictable from classic biomechanical and fluid dynamic theory.109–112 Rapid shunt flow results in an increased total amount of bulk flow through AV shunts. This increased flow results in cerebral arterial hypotension along the path of the shunt.113,114 Patients with AVMs have a progressive decrease in arterial pressure that proceeds from the circle of Willis to the AVM nidus. In patients with large, high-flow AV shunts, there may be normal brain regions in which cerebral arterial hypotension is below the range of normal autoregulation. Despite significant cerebral arterial hypotension, the majority of patients are free of ischemic symptoms. Hypotensive normal brain regions can, for the most part, be demonstrated to have relatively normal levels of tissue perfusion, thus implying some adaptive change in total cerebrovascular resistance.115,116
Active Vascular Remodeling and Angiogenesis in Arteriovenous Malformations
There is a growing body of clinical and experimental evidence suggesting that AVMs can undergo significant vascular remodeling and angiogenesis in adult life. The variable nature of the clinical course of AVMs, especially with respect to their propensity for growth, regression, and spontaneous hemorrhage,117 strongly suggests that active vascular changes are taking place in the majority of patients. A review of studies that examined interval angiograms from a total of 106 patients with a mean follow-up time of 8.4 years showed that more than half of the AVMs increased in size; approximately a fifth of the AVMs decreased in size or vanished. These clinical observations suggest that AVMs can spontaneously regress or undergo extensive regrowth, either de novo or from a retained fragment, after surgical extirpation or complete obliteration by radiosurgery. Histopathologic studies have presented further evidence to support the notion of active angiogenesis and vascular remodeling in AVMs. Hatva and coworkers examined EC proliferation rates in nine adult AVM specimens by using the Ki-67 index and compared them with a single control cortical sample from an 11-year-old patient.118 Although statistical details such as standard deviation or range were not described, the Ki-67 index of AVM ECs was higher than that of control brain (2.5% versus 0.5%). They presumably included ECs from both the nidal vessels and capillaries in adjacent brain tissue for calculation of the Ki-67 index. A similar study using a much larger number of specimens (37 AVMs and 5 controls) to compensate for possible heterogeneity in the angioarchitecture of AVMs noted an approximate sevenfold increase in nonresting ECs in AVMs in comparison to control brain specimens, findings suggesting the presence of active angiogenesis in AVMs. The EC turnover rate in AVMs appears to be somewhere between that of normal quiescent cerebrovascular ECs and ECs in progressing tumors. Angiogenesis and vascular remodeling are controlled by the orchestration of a number of angiogenesis-related factors.119,120 The concerted effects of key angiogenic factors may be maintaining the angiogenic phenotype of vascular malformations.
Vascular Endothelial Cell Growth Factor
Vascular endothelial cell growth factor (VEGF) type A is a potent EC mitogen and morphogen that drives angiogenesis in a wide variety of tissues and lesions. VEGF appears to have at least five isoforms expressed in vascular tissues along with two main vascular receptors with different functions121: VEGF-R1 (also called flt-1) and VEGF-R2 (also called flk-1 or KDR). A number of observational studies have shown increased expression of VEGF in AVMs at both the protein and mRNA level.122,123 These reports showed increased VEGF expression in the vascular wall of AVMs, thus suggesting increased endothelial mitogenic activity in AVMs that can maintain higher angiogenic activity. Increased expression of VEGF may be associated with the clinical behavior (i.e., recurrence) of AVMs. Kader and coauthors reported a series of five pediatric patients who had undergone resection of their AVMs and had a negative postoperative control angiogram but in whom the AVMs eventually regrew.124 Whether this regrowth was de novo or from a retained fragment is unclear, but the angiogenic potential in these patients seems unequivocal. Subsequent studies found a high degree of astrocytic VEGF expression in AVMs from the initial operation in children with recurrent AVMs than in nonrecurrent AVMs.125
Hypoxia-inducible factor-1 (HIF-1) is a major upstream activator of VEGF.126 Regulation of HIF-1 is primarily by destruction of the α subunit.127,128 Although decreased O2 delivery is the best known stimulus, recent evidence points to a direct mechanical effect leading to activation of HIF-1α. This effect has recently been described in chondrocytes subject to joint motion.129 In the heart, HIF-1α is increased not only in ischemic myocardium but also in remote nonischemic regions,130 which suggests that left ventricular dysfunction is a mechanical trigger. Mechanical deformation of vascular smooth muscle cells can cause activation of HIF-1α.131 HIF-1α is a target of intensive efforts to develop pharmacologic blockers because of its involvement in a wide range of disease states.132 An immunohistochemical study showed that HIF-1α was expressed in a majority of both embolized and nonembolized AVM samples.133 Although HIF-1α appeared to be expressed slightly more frequently in embolized AVMs than in nonembolized AVMs, future studies using more quantitative methods will be needed to verify this potential difference in HIF-1α between embolized AVMs and nonembolized AVMs. The increased HIF-1α in AVMs may be responsible for the increased VEGF expression observed in multiple studies. The mechanisms underlying the increased HIF-1α expression in AVMs are not well understood. Although hypoxia is a major stimulant of HIF-1α expression, AVM blood vessels are generally exposed to abnormally high blood flow. Unless AVMs are complicated by hemorrhage or embolization treatment, AVM tissues are likely to be free of ischemia or hypoxia. Mechanical stress on cells inside the vascular wall induced by abnormally high blood flow may be triggering expression of HIF-1α, as previously suggested in other pathologic states.
Angiopoietin and Tie-2
Angiopoietins (Ang-1 and Ang-2) and their receptor Tie-2 play a critical role in angiogenesis and vascular stability.134–138 Ang-1, an agonist for the Tie-2 receptor, promotes interaction between ECs and peri-EC support cells to stabilize vessels.134,135
Ang-2 is an antagonist for the Tie-2 receptor that acts to destabilize these attachments by preventing stimulation of Tie-2 by Ang-1. Ang-1 and Ang-2 are both approximately 75-kD secreted proteins with considerable sequence homology; both bind to Tie-2 with similar affinity, and neither bind to Tie-1. Ang-1 is widely expressed in embryos and adults, whereas Ang-2 seems to be present in tissues undergoing active vascular remodeling, such as the ovary, uterus, and placenta.136 Although it lacks mitogenic effects on ECs, Ang-1 induces phosphorylation of Tie-2 in cultured ECs, but Ang-2 does not. Ang-2 competitively inhibits the Ang-1–induced kinase activity of Tie-2 and functions as an Ang-1 antagonist. Based on the data available, the Ang-1/Tie-2 circuit allows or mediates vessel maturation from simple EC tubes into complete vascular structures (i.e., a construction phase).120 When expression of Ang-2 blocks the Ang-1 signal, vascular deconstruction ensues (e.g., loosening of the tight complex of EC/support cells). Exposing ECs to a milieu containing VEGF and other growth factors can result in reinstitution of the construction phase. It has been suggested that Ang-2 may have a more biologically active role than just being an antagonist of Ang-1.136,139,140 For example, a high concentration of Ang-2 can induce phosphorylation of Tie-2 and exert an antiapoptotic effect.139 The presence of VEGF appears to influence the actions of Ang-2. If VEGF is present, the proangiogenic state is enhanced. In the absence of VEGF, however, vessels undergo involution and regression.120 Tie-2 is known to be upregulated and activated during other angiogenic processes such as wound healing,141 tumor growth,142–144 and the female reproductive cycle.141 Tie-2 has been shown to be expressed and phosphorylated in a number of quiescent adult tissues as well.141 Tie-2 is mutated in a familial venous malformation (VM) syndrome.145,146 The VMs in affected individuals consist of dilated vessels that often lack or have a reduced smooth muscle layer. Abnormal VM Tie-2 stimulation results in the activation of a different subset of signaling proteins and subsequently leads to altered biologic functions, (i.e., the “activating mutation” does not necessarily lead to gain of normal function).
The vascular phenotypes observed with overexpression of Ang-2 or homozygous disruption of the Tie-2 gene are strikingly similar to AVM vessels.134,137,138 They display abnormally dilated vessels that lack mature peri-EC support structure. There appears to be an abnormal balance in the angiopoietin–Tie-2 system in AVMs. In AVM tissues, Ang-2 (antagonist) is markedly increased and Ang-1 (agonist) is decreased in comparison to control brain tissues.147 Studies analyzing homogenized AVM tissue have shown markedly decreased Tie-2 expression at both the mRNA and protein levels.147,148 This finding was further confirmed by another independent study using the gene microarray technique to compare AVM tissues and superficial temporal artery samples.149 However, an immunohistochemistry study showed Tie-2 expression in only 2 of 185 blood vessels from AVMs and in none of 176 control blood vessels.150 The lack of expression of Tie-2 in the control blood vessels in this immunohistochemical study is rather surprising because it is well known that Tie-2 is constitutively expressed in normal blood vessels in a wide variety of tissues and has been used as an EC marker. The contradictions among these studies underscore a fundamental difficulty in using the immunohistochemical technique to quantitatively or semiquantitatively assess expression of angiogenic factors. The reduced levels of Tie-2 mRNA observed in Ang-1 null mice suggest that Tie-2 levels may be a function of ligands.134,151 Ang-1 stimulation of Tie-2 may be required to maintain baseline expression of Tie-2. The angiopoietin–Tie-2 system appears to have an autoregulatory feedback system that may be regulating the overall activity of the Tie-2 system in both physiologic and pathologic conditions.152 Dysregulation of the feedback system in the angiopoietin–Tie-2 system may be associated with the pathologic angiogenesis observed in AVMs.152 The abnormal balance of angiopoietins in AVMs—decreased levels of Ang-1 along with increased levels of Ang-2—would be expected to result in an overall decrease in Tie-2 signaling, which would lead to vascular instability. Furthermore, decreased levels of Tie-2, if it exists, will further impair vascular stability in AVMs. This may, in part, explain the aberrant vascular phenotype in AVMs (i.e., dilated vessels with a relative lack of mature peri-EC support).
Matrix Metalloproteinases
Matrix metalloproteinases (MMPs), a family of proteolytic enzymes, degrade extracellular matrix proteins, cell surface molecules, and other pericellular substances.153 By degrading the vascular extracellular matrix, MMPs can create a microenvironment that facilitates angiogenesis and vascular remodeling. MMP-9 and MMP-2, which have the capability of degrading gelatin, have been extensively studied in physiologic and pathologic angiogenesis and vascular remodeling. MMP-9, also known as gelatinase B, degrades components of vascular extracellular matrices, including type IV and V collagen, fibronectin, and elastin.153 High levels of MMP-9 expression are detected in structurally unstable vasculature, including cerebral aneurysms,154,155 abdominal aortic aneurysms,156–158 and atherosclerotic carotid artery.159 Excessive degradation of the vascular matrix may contribute to the destabilization of vessels and lead to weakening of the vessel wall and vessel rupture.160 An abnormal expression pattern of MMP-9 and tissue inhibitors of metalloproteinases (TIMPs) has been observed in AVMs.161 There is markedly increased MMP-9 activity in AVMs in comparison to control brain samples. MMP-9 is expressed in the EC/peri-EC layer of AVMs. The increased activity of MMP-9 can be expected to cause degradation of the vascular matrix, thereby impairing the structural stability of AVM vessels. There is increasing interest in using MMP inhibitors in treating vascular diseases, including abdominal aortic aneurysms. It has been proposed that pharmacologic inhibition of MMPs may stabilize the unstable blood vessels and prevent complications such as vessel rupture.162 At subantimicrobial doses, doxycycline is an inhibitor of MMPs. In patients with abdominal aortic aneurysm, treatment with doxycycline for 1 week before the repair surgery resulted in decreased MMP-9 and MMP-2 in the wall of the aneurysms.158 Similar results have been reported in patients with atherosclerotic carotid plaque who received doxycycline for 2 to 8 weeks.163 Doxycycline can inhibit MMP-9 activity in mouse brain hyperstimulated with adenovirally transduced VEGF.164 Because MMP-9 appears to be highly expressed in the nidal vessels, these proteases or related ones may serve as potential pharmacologic targets to modify the clinical behavior of AVMs.
Concerted Effects of Angiogenic Factors
Active angiogenesis and vascular remodeling are controlled by the orchestration of a number of angiogenesis-related factors.119,120 Numerous studies have investigated various angiogenic factors, but the results need to be interpreted with caution because some of the studies used less quantitative methods such as immunohistochemistry. The concerted effect of key angiogenic factors may be maintenance of the angiogenic phenotype of AVMs and thereby determination of the clinical course. For example, in addition to serving as a major proteolytic factor during angiogenesis, MMP-9 appears to initiate and sustain angiogenesis during carcinogenesis by increasing the bioavailability of VEGF.165 Ang-2 appears to increase MMP-9 expression in the presence of VEGF.166 The increased expression of Ang-2 and VEGF in AVMs may be contributing to the increased MMP-9 activity, and an intricate balance among these factors may be determining the angiogenic potential of AVMs. There is increasing interest in studying the expression profile of a large number of genes and their products. Two independent studies used the gene microarray technique to characterize the gene expression pattern in intracranial vascular malformations. Conventional molecular assay techniques, such as Western blot, Northern blot, immunohistochemistry, and in situ hybridization, are designed to measure the expression of a single gene or its product. However, these techniques are not suitable for dissection of multiple interacting molecular pathways or screening of potential molecular abnormalities, where the list of candidate genes or products can be extensive. The gene microarray technique has become a powerful molecular tool to analyze the gene expression profile by measuring expression levels of more than 1000 genes.167 It has been used to identify a disease-specific gene expression profile or screen for potential therapeutic targets.168 There have been two studies that analyzed the gene expression profile of AVM samples with the gene microarray technique. Shenkar and coworkers compared the gene expression profile of AVMs with superficial temporal artery samples.149 They found a number of genes related to angiogenesis and structural components of the vascular wall to be differentially expressed between AVMs and superficial temporal arteries. Hashimoto’s group compared gene expression profile between AVM and control brain samples and confirmed the main findings from the gene microarray by performing Western blot and immunohistochemistry.169 Out of 12,625 gene probes assayed, 1781 gene probes showed differential expression between AVMs and controls. AVM samples had a gene expression pattern that was distinct from those of control brain samples. Although these studies are still preliminary, they clearly show the potential of the gene microarray technique as a screening tool for studying multiple pathways simultaneously in AVM specimens. Findings from these studies are expected to lead to analysis of new candidate genes or proteins in AVMs.
Hypothetical Events Leading to the Development of Vascular Malformations
Despite the recent progress in identifying the abnormal expression profile of various angiogenesis-related factors in AVM tissues, events that lead to AVM development and formation are not well understood. Although the extradural fistula models are reasonable approximations of DAVF, there has been a lack of studies delineating the mechanistic steps between venous hypertension and angiogenic activity. Because there is no good animal AVM model that mimics the syndrome of recurrent intracranial hemorrhage, researchers have been forced to focus on observational studies of human tissue. However, careful observation with clinical correlation of a large number of samples can provide important insight into the pathophysiology of AVMs. Such studies coupled with findings from experimental angiogenesis models in the brain170,171 can generate new hypotheses. Hashimoto and Young proposed a speculative and simplified conceptual synthesis of experimental observations relevant to the pathogenesis of AVMs.172 The inciting event is unknown at present, but there are some reasonable candidates. For example, there are many conditions that might result in a localized area of microvascular thrombosis, such as minor cranial trauma either prenatally or postnatally or a state of relative thrombophilia, such as in DAVF. Altered protein function, perhaps even subtle and derived from some combination of polymorphic genes, might take otherwise innocuous events and conspire to create the circumstances for development of the human AVM phenotype. Not all gene alterations need to be present in a given AVM patient; multiple alterations could converge on a single set of pathways. The inciting event leads to a region of venous hypertension that stimulates elaboration of HIF-1. Downstream from HIF-1, VEGF stimulation begins a process of focal angiogenesis (two other important sources of VEGF are release from leukocytes/macrophages and MMP-9–mediated release from the extracellular matrix). The resultant angiogenesis stimulates multiple cell types to secrete inflammatory cytokines such as interleukin-6 (IL-6), which in turn could attract leukocytes and macrophages; IL-6 and leukocytes may further contribute to MMP expression and activity by accelerating EC proliferation, migration, and microvessel sprouting. IL-6 may enhance angiogenesis by either stimulation of VEGF or activation of MMPs. The angiogenic process might be turned pathologic in the presence of some predisposing genetic alteration in TGF-β or Tie-2/angiopoietin signaling function, as suggested by familial genetic disorders. Development of a vascular malformation might become self-sustaining through the opening of AV shunts and increased flow. High flow increases endothelial shear and activates nitric oxide (NO) synthase and NO production, thereby further driving VEGF stimulation. Downstream activation of MMPs ultimately destabilizes the vascular walls and sets the stage for rupture (injection of MMP into the brain was one of the original models for mimicking intracranial hemorrhage).173
Acknowledgment
The authors thank Dr. Roxana Gunny, F.R.C.R., and Dr. Stefan Brew, F.R.C.R., for providing images.
Awad IA, Robinson JRJr, Mohanty S, et al. Mixed vascular malformations of the brain: clinical and pathogenetic considerations. Neurosurgery. 1993;33:179-188.
Bhattacharya JJ, Luo CB, Shu DC, et al. Wyburn-Mason or Bonnet-Dechame-Blanc as cerebrofacial arteriovenous metameric syndromes (CAMS). A new concept and a new classification. Intervent Neuroradiol. 2001;7:5-17.
Bland LI, Lapham LW, Ketonen L, et al. Acute cerebellar hemorrhage secondary to capillary telangiectasia in an infant: A case report. Arch Neurol. 1994;51:1151-1154.
Boudreau NJ, Varner JA. The homeobox factor Hox D3 promotes integrin α5β1 expression and function during angiogenesis. J Biol Chem. 2004;279:4862-4868.
Gao E, Young WL, Ornstein E, et al. A theoretical model of cerebral hemodynamics: application to the study of arteriovenous malformations. J Cereb Blood Flow Metab. 1997;17:905-918.
Gault J, Sarin H, et al. Pathobiology of human cerebrovascular malformations: basic mechanisms and clinical relevance. Neurosurgery. 2004;55:1-17.
Hashimoto T, Emala CW, Joshi S, et al. Abnormal pattern of tie-2 and vascular endothelial growth factor receptor expression in human cerebral arteriovenous malformations. Neurosurgery. 2000;47:910-918. discussion 918-919
Hashimoto T, Lawton MT, Wen G, et al. Gene microarray analysis of human brain arteriovenous malformations. Neurosurgery. 2004;54:410-425.
Kader A, Young WL, Pile-Spellman J, et al. The influence of hemodynamic and anatomic factors on hemorrhage from cerebral arteriovenous malformations. Neurosurgery. 1994;34:801-808.
Kilic T, Pamir MN, Kullu S, et al. Expression of structural proteins and angiogenic factors in cerebrovascular anomalies. Neurosurgery. 2000;46:1179-1191. discussion 1191-1192
Lawton MT, Jacobowitz R, Spetzler RF. Redefined role of angiogenesis in the pathogenesis of dural arteriovenous malformations. J Neurosurg. 1997;87:267-274.
Martin NA, Vinters HV. Arteriovenous malformations. In: Carter LP, Spetzler RF, Hamilton MG, editors. Neurovascular Surgery. New York: McGraw-Hill; 1995:875-903.
McCormick WF. The pathology of vascular (“arteriovenous”) malformations. J Neurosurg. 1966;24:807-816.
Miyasaka K, Wolpert SM, Prager RJ. The association of cerebral aneurysms, infundibula and intracranial arteriovenous malformations. Stroke. 1982;13:196-203.
Mullan S, Mojtahedi S, Johnson DL, et al. Embryological basis of some aspects of cerebral vascular fistulas and malformations. J Neurosurg. 1996;85:1-8.
Nussbaum ES, Heros RC, Madison MT, et al. The pathogenesis of arteriovenous malformations: insights provided by a case of multiple arteriovenous malformations developing in relation to a developmental venous anomaly. Neurosurgery. 1998;43:347-352.
Putman C, Chaloupka J, Fulbright R, et al. Exceptional multiplicity of cerebral arteriovenous malformations associated with hereditary hemorrhagic telangiectasia (Osler Weber Rendu). AJNR Am J Neuroradiol. 1996;17:1733-1742.
Russell DS, Rubenstein LJ. Pathology of Tumours of the Nervous System, 3rd ed. Baltimore: Williams & Wilkins; 1971.
Saito Y, Kobayashi N. Cerebral venous angiomas: clinical evaluation and possible etiology. Radiology. 1981;139:87-94.
Shenkar R, Elliott JP, Diener K, et al. Differential gene expression in human cerebrovascular malformations. Neurosurgery. 2003;52:465-478.
Smith ER, Butler WE, Ogilvy CS. Surgical approaches to vascular anomalies of the child’s brain. Curr Opin Neurol. 2002;15:165-171.
Tu J, Stoodley MA, Morgan MK, et al. Ultrastructure of perinidal capillaries in cerebral arteriovenous malformations. Neurosurgery. 2006;58:961-970.
Valavanis A, Yasargil M. The endovascular treatment of brain arteriovenous malformations. Adv Tech Stand Neurosurg. 1998;24:131-214.
Wilson CB. Cryptic vascular malformations. Clin Neurosurg. 1992;38:49-84.
Zabramski JM, Wascher TM, Spetzler RF, et al. The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg. 1994;80:422-432.
1 Cushing H, Bailey P. Tumours Arising from the Blood Vessels of the Brain: Angiomatous Malformations and Hemangioblastomas, Vol. 3. Springfield, IL: Charles C Thomas. 1928. 19-34
2 McCormick WF. The pathology of vascular (“arteriovenous”) malformations. J Neurosurg. 1966;24:807-816.
3 Russell DS, Rubenstein LJ. Pathology of Tumours of the Nervous System, 3rd ed. Baltimore: Williams & Wilkins; 1971.
4 Kalimo H, Kase M, Haltia M. Vascular diseases. In: Graham D, Lantos P, editors. Greenfield’s Neuropathology. 6th ed. New York: Oxford University Press; 1997:345-347.
5 Martin NA, Vinters HV. Arteriovenous malformations. In: Carter LP, Spetzler RF, Hamilton MG, editors. Neurovascular Surgery. New York: McGraw-Hill; 1995:875-903.
6 Patterson JH, McKissock WA. A clinical survey of intracranial angiomas with special reference to their mode of progression and surgical treatment: a report of 110 cases. Brain. 1956;79:233-266.
7 Miyasaka K, Wolpert SM, Prager RJ. The association of cerebral aneurysms, infundibula and intracranial arteriovenous malformations. Stroke. 1982;13:196-203.
8 Kader A, Young WL, Pile-Spellman J, et al. The influence of hemodynamic and anatomic factors on hemorrhage from cerebral arteriovenous malformations. Neurosurgery. 1994;34:801-808.
9 Gao E, Young WL, Pile-Spellman J, et al. Cerebral arteriovenous malformation feeding artery aneurysms: a theoretical model of intravascular pressure changes after treatment. Neurosurgery. 1997;41:1345-1358.
10 Kondziolka D, Nixon BJ, Lasjaunias P, et al. Cerebral arteriovenous malformations with associated arterial aneurysms: hemodynamic and therapeutic considerations. Can J Neurol Sci. 1988;15:130-134.
11 Redekop G, TerBrugge K, Montanera W, et al. Arterial aneurysms associated with cerebral arteriovenous malformations: classification, incidence, and risk of hemorrhage. J Neurosurg. 1998;89:539-546.
12 Thompson RC, Steinberg GK, Levy RP, et al. The management of patients with arteriovenous malformations and associated intracranial aneurysms. Neurosurgery. 1998;43:202-212.
13 Valavanis A, Yasargil M. The endovascular treatment of brain arteriovenous malformations. Adv Tech Stand Neurosurg. 1998;24:131-214.
14 Tu J, Stoodley MA, Morgan MK, et al. Ultrastructure of perinidal capillaries in cerebral arteriovenous malformations. Neurosurgery. 2006;58:961-970.
15 Sato S, Kodama N, Sasaki T, et al. Perinidal dilated capillary networks in cerebral arteriovenous malformations. Neurosurgery. 2004;54:163-168. discussion 168-170
16 Lasjaunias P, Landrieu P, Rodesch G, et al. Cerebral proliferative angiopathy: clinical and angiographic description of an entity different from cerebral AVMs. Stroke. 2008;39:878-885.
17 Brown RDJr, Wiebers DO, Torner JC, et al. Frequency of intracranial hemorrhage as a presenting symptom and subtype analysis: a population-based study of intracranial vascular malformations in Olmsted Country, Minnesota. J Neurosurg. 1996;85:29-32.
18 Arteriovenous Malformation Study Group. Arteriovenous malformations of the brain in adults. N Engl J Med. 1999;340:1812-1818.
19 Boudreau NJ, Varner JA. The homeobox factor Hox D3 promotes integrin α5β1 expression and function during angiogenesis. J Biol Chem. 2004;279:4862-4868.
20 Myers C, Charboneau A, Boudreau NJ. Homeobox B3 promotes capillary morphogenesis and angiogenesis. J Cell Biol. 2000;148:343-351.
21 Putman C, Chaloupka J, Fulbright R, et al. Exceptional multiplicity of cerebral arteriovenous malformations associated with hereditary hemorrhagic telangiectasia (Osler Weber Rendu). AJNR Am J Neuroradiol. 1996;17:1733-1742.
22 Bhattacharya JJ, Luo CB, Shu DC, et al. Wyburn-Mason or Bonnet-Dechame-Blanc as cerebrofacial arteriovenous metameric syndromes (CAMS). A new concept and a new classification. Intervent Neuroradiol. 2001;7:5-17.
23 Bristol RE, Albuquerque FC, Spetzler RF, et al. Surgical management of arteriovenous malformations in children. J Neurosurg. 2006;105(2 suppl):88-93.
24 Celli P, Ferrante L, Palma L, et al. Cerebral arteriovenous malformations in children. Clinical features and outcome of treatment in children and in adults. Surg Neurol. 1984;22:43-49.
25 Smith ER, Butler WE, Ogilvy CS. Surgical approaches to vascular anomalies of the child’s brain. Curr Opin Neurol. 2002;15:165-171.
26 Humphreys RP, Hoffman HJ, Drake JM, et al. Choices in the 1990s for the management of pediatric cerebral arteriovenous malformations. Pediatr Neurosurg. 1996;25:277-285.
27 Han PP, Ponce FA, Spetzler RF. Intention-to-treat analysis of Spetzler-Martin grades IV and V arteriovenous malformations: natural history and treatment paradigm. J Neurosurg. 2003;98:3-7.
28 Spetzler RF, Martin NA. A proposed grading system for arteriovenous malformations. J Neurosurg. 1986;65:476-483.
29 Wong IYC, Batista LL, Alvarez H, et al. Craniofacial arteriovenous metameric syndrome (CAMS) 3—a transitional pattern between CAM 1 and 2 and spinal arteriovenous metameric syndromes. Neuroradiology. 2003;45:611-615.
30 Kang HS, Han MH, Kwon BJ, et al. Cerebellopontine vascular malformation: a rare type of cerebrofacial arteriovenous metameric syndrome. J Neurosurg. 2005;102:156-160.
31 Osborn A. Diagnostic Radiology. Chicago: CV Mosby; 1994.
32 Morello G, Borghi G. Cerebral angiomas: a report of 154 personal cases and a comparison between the results of surgical excision and conservative management. Acta Neurochir (Wien). 1973;28:135-155.
33 Wilkins R. The natural history of intracranial vascular malformations: a review. Neurosurgery. 1985;16:421-430.
34 Graf C, Perret G, Torner J. Bleeding from cerebral arteriovenous malformations as part of their natural history. J Neurosurg. 1983;58:331-337.
35 Saito Y, Kobayashi N. Cerebral venous angiomas: clinical evaluation and possible etiology. Radiology. 1981;139:87-94.
36 Lasjaunias P, Burrows P, Planet C. Developmental venous anomalies (DVA): The so-called venous angioma. Neurosurg Rev. 1994;9:232-242.
37 Wolf A, Brock S. The pathology of cerebral angiomas: a study of nine cases. Bull Neurol Inst N Y. 1935;4:144-176.
38 Johnson P, Wascher T, Golfinos J, et al. Definition and pathological features. In: Awad I, Barrow D, editors. Cavernous Malformations. Park Ridge, IL: American Association of Neurological Surgeons; 1993:1-11.
39 Ramina R, Ingunza W, Vonofakos D. Cystic cerebral cavernous angioma with dense calcification: case report. J Neurosurg. 1980;52:259-262.
40 Zabramski JM, Wascher TM, Spetzler RF, et al. The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg. 1994;80:422-432.
41 Sarwar M, McCormick WF. Intracerebral venous angioma: case report and review. Arch Neurol. 1978;35:323-325.
42 Rigamonti D, Hsu FPK, Huhn S. Angiographically occult vascular malformations. In: Carter LP, Spetzler RF, Hamilton MG, editors. Neurovascular Surgery. New York: McGraw-Hill; 1995:521-540.
43 Lee RR, Becher MW, Benson ML, et al. Brain capillary telangiectasia: MR imaging appearance and clinicohistopathologic findings. Radiology. 1997;205:797-805.
44 Bland LI, Lapham LW, Ketonen L, et al. Acute cerebellar hemorrhage secondary to capillary telangiectasia in an infant: a case report. Arch Neurol. 1994;51:1151-1154.
45 Thrash AM. Vascular malformations of the cerebellum. Arch Pathol. 1963;75:65-69.
46 Blackwood W. Two cases of benign cerebral telangiectasis. J Pathol Bacteriol. 1941;52:209-212.
47 Vaquero J, Manrique M, Oya S, et al. Calcified telangiectatic hamartomas of the brain. Surg Neurol. 1980;13:453-457.
48 Chang SD, Steinberg GK, Rosario M, et al. Mixed arteriovenous malformation and capillary telangiectasia: A rare subset of mixed vascular malformations: case report. J Neurosurg. 1997;86:699-703.
49 Hirata Y, Matsukado Y, Nagahiro S, et al. Intracerebral venous angioma with arterial blood supply: a mixed angioma. Surg Neurol. 1986;25:227-232.
50 Awad IA, Robinson JRJr, Mohanty S, et al. Mixed vascular malformations of the brain: clinical and pathogenetic considerations. Neurosurgery. 1993;33:179-188.
51 Meyer B, Stangl AP, Schramm J. Association of venous and true arteriovenous malformation: A rare entity among mixed vascular malformations of the brain: case report. J Neurosurg. 1995;83:141-144.
52 Mullan S, Mojtahedi S, Johnson DL, et al. Cerebral venous malformation arteriovenous malformation transition forms. J Neurosurg. 1996;85:9-13.
53 Nussbaum ES, Heros RC, Madison MT, et al. The pathogenesis of arteriovenous malformations: insights provided by a case of multiple arteriovenous malformations developing in relation to a developmental venous anomaly. Neurosurgery. 1998;43:347-352.
54 Wilson CB. Cryptic vascular malformations. Clin Neurosurg. 1992;38:49-84.
55 Comey CH, Kondziolka D, Yonas H. Regional parenchymal enhancement with mixed cavernous/venous malformations of the brain: case report. J Neurosurg. 1997;86:155-158.
56 Ciricillo SF, Dillon WP, Fink ME, et al. Progression of multiple cryptic vascular malformations associated with anomalous venous drainage: case report. J Neurosurg. 1994;81:477-481.
57 Im SH, Han MH, Kwon BJ, et al. Venous-predominant parenchymal arteriovenous malformation: a rare subtype with a venous drainage pattern mimicking developmental venous anomaly. J Neurosurg. 2008;108:1142-1147.
58 Wolfe SQ, Heros RC. Developmental venous anomalies with arterial supply [editorial]. J Neurosurg. 2008;108:1139-1141.
59 Garner TB, Del Curling OJr, Kelly DLJr, et al. The natural history of intracranial venous angiomas. J Neurosurg. 1991;75:715-722.
60 Amin-Hanjani S. The genetics of cerebrovascular malformations. Semin Cerebrovasc Dis Stroke. 2002;2:73-81.
61 Gault J, Sarin H, Awadallah NA, et al. Pathobiology of human cerebrovascular malformations: basic mechanisms and clinical relevance. Neurosurgery. 2004;55:1-17.
62 Mullan S, Mojtahedi S, Johnson DL, et al. Embryological basis of some aspects of cerebral vascular fistulas and malformations. J Neurosurg. 1996;85:1-8.
63 Guttmacher AE, Marchuk DA, White RIJr. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;333:918-924.
64 Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary haemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000;91:66-67.
65 Maher CO, Piepgras DG, Brown RDJr, et al. Cerebrovascular manifestations in 321 cases of hereditary hemorrhagic telangiectasia. Stroke. 2001;32:877-882.
66 Haitjema T, Disch F, Overtoom TT, et al. Screening family members of patients with hereditary hemorrhagic telangiectasia. Am J Med. 1995;99:519-524.
67 Willemse RB, Mager JJ, Westermann CJ, et al. Bleeding risk of cerebrovascular malformations in hereditary hemorrhagic telangiectasia. J Neurosurg. 2000;92:779-784.
68 McDonald MT, Papenberg KA, Ghosh S, et al. A disease locus for hereditary haemorrhagic telangiectasia maps to chromosome 9q3334. Nat Genet. 1994;6:197-204.
69 Shovlin CL, Hughes JM, Tuddenham EG, et al. A gene for hereditary haemorrhagic telangiectasia maps to chromosome 9q3. Nat Genet. 1994;6:205-209.
70 Vincent P, Plauchu H, Hazan J, et al. A third locus for hereditary haemorrhagic telangiectasia maps to chromosome 12q. Hum Mol Genet. 1995;4:945-949.
71 Johnson DW, Berg JN, Gallione CJ, et al. A second locus for hereditary hemorrhagic telangiectasia maps to chromosome 12. Genome Res. 1995;5:21-28.
72 McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345-351.
73 Fernandez-Riuz E, St-Jacques S, Bellon T, et al. Assignment of the human endoglin gene (END) to 9q34Rqter. Cytogenet Cell Genet. 1993;64:204-207.
74 Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189-195.
75 Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. Med Genet. 2006;43:97-110.
76 Cole S, Begbie M, Wallace G, et al. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005;42:577-582.
77 Kjeldsen AD, Brusgaard K, Poulsen L, et al. Mutations in the ALK-I gene and the phenotype of hereditary hemorrhagic telangiectasia in two large Danish families. Am J Med Genet. 2001;98:298-302.
78 McDonald JE, Miller FJ, Hallam SE, et al. Clinical manifestations in a large hereditary hemorrhagic telangiectasia (HHT) type 2 kindred. Am J Med Genet. 2000;93:320-327.
79 Bourdeau A, Faughnan ME, McDonald ML, et al. Potential role of modifier genes influencing transforming growth factor-βl levels in the development of vascular defects in endoglin heterozygous mice with hereditary hemorrhagic telangiectasia. Am J Pathol. 2001;158:2011-2020.
80 Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104:1343-1351.
81 Gallione CJ, Repetto GM, Legius E, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004;363:852-859.
82 Cheifetz S, Bellon T, Cales C, et al. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J Biol Chem. 267, 1992. 19027–19GG030
83 Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J Biol Chem. 1999;274:584-594.
84 Lux A, Gallione CJ, Marchuk DA. Expression analysis of endoglin missense and truncation mutations: insights into protein structure and disease mechanisms. Hum Mol Genet. 2000;9:745-755.
85 Pece N, Vera S, Cymerman U, et al. Mutant endoglin in hereditary hemorrhagic telangiectasia type 1 is transiently expressed intracellularly and is not a dominant negative. J Clin Invest. 1997;100:2568-2579.
86 Bourdeau A, Cymerman U, Paquet ME, et al. Endoglin expression is reduced in normal vessels but still detectable in arteriovenous malformations of patients with hereditary hemorrhagic telangiectasia type 1. Am J Pathol. 2000;156:911-923.
87 Berg JN, Gallione CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet. 1997;61:60-67.
88 Oh SP, Seki T, Goss KA, et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A. 2000;97:2626-2631.
89 Abdalla SA, Pece-Barbara N, Vera S, et al. Analysis of ALK-1 and endoglin in newborns from families with hereditary hemorrhagic telangiectasia type 2. Hum Mol Genet. 2000;9:1227-1237.
90 Pece-Barbara N, Cymerman U, Vera S, et al. Expression analysis of four endoglin missense mutations suggests that haploinsufficiency is the predominant mechanism for hereditary hemorrhagic telangiectasia type 1. Hum Mol Genet. 1999;8:2171-2181.
91 Wrana JL. Regulation of Smad activity. Cell. 2000;100:189-192.
92 Matsubara S, Bourdeau A, terBrugge KG, et al. Analysis of endoglin expression in normal brain tissue and in cerebral arteriovenous malformations. Stroke. 2000;31:2653-2660.
93 Snead OCIII, Acker JD, Morawetz R. Familial arteriovenous malformation. Ann Neurol. 1979;5:585-587.
94 Aberfeld DC, Rao KR. Familial arteriovenous malformation of the brain. Neurology. 1981;31:184-186.
95 Boyd MC, Steinbok P, Paty DW. Familial arteriovenous malformations. Report of four cases in one family. J Neurosurg. 1985;62:597-599.
96 Brilli RJ, Sacchetti A, Neff S. Familial arteriovenous malformations in children. Pediatr Emerg Care. 1995;11:376-378.
97 Kikuchi K, Kowada M, Sakamoto T, et al. Wyburn-Mason syndrome: report of a rare case with computed tomography and angiographic evaluations. J Comput Tomogr. 1988;12:111-115.
98 Patel U, Gupta SC. Wyburn-Mason syndrome. A case report and review of the literature. Neuroradiology. 1990;31:544-546.
99 Jessurun GA, Kamphuis DJ, van der Zande FH, et al. Cerebral arteriovenous malformations in The Netherlands Antilles. High prevalence of hereditary hemorrhagic telangiectasia–related single and multiple cerebral arteriovenous malformations. Clin Neurol Neurosurg. 1993;95:193-198.
100 Velasco B, Ramirez JR, Relloso M, et al. Vascular gene transfer driven by endoglin and ICAM-2 endothelial-specific promoters. Gene Ther. 2001;8:897-904.
101 Streeter GL. The developmental alterations in the vascular system of the brain of human embryo. Contrib Embryol. 1918;8:5-38.
102 Folkman J, D’Amore PA. Blood vessel formation: what is its molecular basis? Cell. 1996;87:1153-1155.
103 Patan S. Vasculogenesis and angiogenesis as mechanisms of vascular network formation, growth and remodeling. J Neurooncol. 2000;50:1-15.
104 Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73-91.
105 Yamada S. Arteriovenous Malformations in Functional Areas of the Brain. Armonk, NY: Futura; 1999.
106 Hashimoto T, Mesa-Tejada R, Quick CM, et al. Evidence of increased endothelial cell turnover in brain arteriovenous malformations. Neurosurgery. 2001;49:124-131. discussion 131-132
107 Uranishi R, Nakase H, Sakaki T. Expression of angiogenic growth factors in dural arteriovenous fistula. J Neurosurg. 1999;91:781-786.
108 Lawton MT, Jacobowitz R, Spetzler RF. Redefined role of angiogenesis in the pathogenesis of dural arteriovenous malformations. J Neurosurg. 1997;87:267-274.
109 Gao E, Young WL, Ornstein E, et al. A theoretical model of cerebral hemodynamics: application to the study of arteriovenous malformations. J Cereb Blood Flow Metab. 1997;17:905-918.
110 Gao E, Young WL, Pile-Spellman J, et al. Cerebral arteriovenous malformation feeding artery aneurysms: a theoretical model of intravascular pressure changes after treatment. Neurosurgery. 1997;41:1345-1358.
111 Quick CM, Young WL, Leonard EF, et al. Model of structural and functional adaptation of small conductance vessels to arterial hypotension. Am J Physiol Heart Circ Physiol. 2000;279:H1645-H1653.
112 Quick CM, Leonard EF, Young WL. Adaptation of cerebral circulation to brain arteriovenous malformations increases feeding artery pressure and decreases regional hypotension. Neurosurgery. 2002;50:167-173. discussion 173-175
113 Fogarty-Mack P, Pile-Spellman J, Hacein-Bey L, et al. The effect of arteriovenous malformations on the distribution of intracerebral arterial pressures. AJNR Am J Neuroradiol. 1996;17:1443-1449.
114 Duong DH, Young WL, Vang MC, et al. Feeding artery pressure and venous drainage pattern are primary determinants of hemorrhage from cerebral arteriovenous malformations. Stroke. 1998;29:1167-1176.
115 Young WL, Kader A, Ornstein E, et al. Cerebral hyperemia after arteriovenous malformation resection is related to “breakthrough” complications but not to feeding artery pressure. The Columbia University Arteriovenous Malformation Study Project. Neurosurgery. 1996;38:1085-1093. discussion 1093-1095
116 Hacein-Bey L, Nour R, Pile-Spellman J, et al. Adaptive changes in autoregulation to chronic cerebral hypotension with arteriovenous malformations: an acetazolamide enhanced single-photon emission CT study. AJNR Am J Neuroradiol. 1995;16:1865-1874.
117 Arteriovenous Malformation Study Group. Current concepts: arteriovenous malformations of the brain in adults. N Engl J Med. 1999;340:1812-1818.
118 Hatva E, Jaaskelainen J, Hirvonen H, et al. Tie endothelial cell–specific receptor tyrosine kinase is upregulated in the vasculature of arteriovenous malformations. J Neuropathol Exp Neurol. 1996;55:1124-1133.
119 Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401-410.
120 Hanahan D. Signaling vascular morphogenesis and maintenance. Science. 1997;277:48-50.
121 Gale NW, Yancopoulos GD. Growth factors acting via endothelial cell–specific receptor tyrosine kinases: VEGFs, angiopoietins, and ephrins in vascular development. Genes Dev. 1999;13:1055-1066.
122 Rothbart D, Awad IA, Lee J, et al. Expression of angiogenic factors and structural proteins in central nervous system vascular malformations. Neurosurgery. 1996;38:915-924. discussion 924-925
123 Kilic T, Pamir MN, Kullu S, et al. Expression of structural proteins and angiogenic factors in cerebrovascular anomalies. Neurosurgery. 2000;46:1179-1191. discussion 1191-1192
124 Kader A, Goodrich JT, Sonstein WJ, et al. Recurrent cerebral arteriovenous malformations after negative postoperative angiograms [see comments of A Patil and AJ Fox, with response, J Neurosurg 86:170-171, 1997]. J Neurosurg. 1996;85:14-18.
125 Sonstein WJ, Kader A, Michelsen WJ, et al. Expression of vascular endothelial growth factor in pediatric and adult cerebral arteriovenous malformations: an immunocytochemical study. J Neurosurg. 1996;85:838-845.
126 Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol. 2002;64:993-998.
127 Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468-472.
128 Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464-468.
129 Pufe T, Lemke A, Kurz B, et al. Mechanical overload induces VEGF in cartilage discs via hypoxia-inducible factor. Am J Pathol. 2004;164:185-192.
130 Kim CH, Cho YS, Chun YS, et al. Early expression of myocardial HIF-1α in response to mechanical stresses: regulation by stretch-activated channels and the phosphatidylinositol 3-kinase signaling pathway. Circ Res. 2002;90:E25-E33.
131 Chang H, Shyu KG, Wang BW, et al. Regulation of hypoxia-inducible factor-1α by cyclical mechanical stretch in rat vascular smooth muscle cells. Clin Sci (Lond). 2003;105:447-456.
132 Giaccia A, Siim BG, Johnson RS. HIF-1 as a target for drug development. Nat Rev Drug Discov. 2003;2:803-811.
133 Sure U, Battenberg E, Dempfle A, et al. Hypoxia-inducible factor and vascular endothelial growth factor are expressed more frequently in embolized than in nonembolized cerebral arteriovenous malformations. Neurosurgery. 2004;55:663-669. discussion 669-670
134 Suri C, Jones PF, Patan S, et al. Requisite role of angiopoietin-1, a ligand for the tie2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171-1180.
135 Davis S, Aldrich TH, Jones PF, et al. Isolation of angiopoietin-1, a ligand for the tie2 receptor, by secretion-trap expression cloning. Cell. 1996;87:1161-1169.
136 Maisonpierre PC, Suri C, Jones PF, et al. Angiopoietin-2, a natural antagonist for tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55-60.
137 Dumont DJ, Gradwohl G, Fong G-H, et al. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev. 1994;8:1897-1909.
138 Sato TN, Tozawa Y, Deutsch U, et al. Distinct roles of the receptor tyrosine kinase tie-1 and tie2 in blood vessel formation [letter]. Nature. 1995;376:70-74.
139 Kim I, Kim JH, Moon SO, et al. Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3-kinase/akt signal transduction pathway. Oncogene. 2000;19:4549-4552.
140 Teichert-Kuliszewska K, Maisonpierre PC, Jones N, et al. Biological action of angiopoietin-2 in a fibrin matrix model of angiogenesis is associated with activation of tie2. Cardiovasc Res. 2001;49:659-670.
141 Wong AL, Haroon ZA, Werner S, et al. Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ Res. 1997;81:567-574.
142 Peters KG, Coogan A, Berry D, et al. Expression of tie2/tek in breast tumour vasculature provides a new marker for evaluation of tumour angiogenesis. Br J Cancer. 1998;77:51-56.
143 Lin P, Polverini P, Dewhirst M, et al. Inhibition of tumor angiogenesis using a soluble receptor establishes a role for tie2 in pathologic vascular growth. J Clin Invest. 1997;100:2072-2078.
144 Lin P, Buxton JA, Acheson A, et al. Antiangiogenic gene therapy targeting the endothelium-specific receptor tyrosine kinase tie2. Proc Natl Acad Sci U S A. 1998;95:8829-8834.
145 Vikkula M, Boon LM, Carraway KL, et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase tie2. Cell. 1996;87:1181-1190.
146 Calvert JT, Riney TJ, Kontos CD, et al. Allelic and locus heterogeneity in inherited venous malformations. Hum Mol Genet. 1999;8:1279-1289.
147 Hashimoto T, Lam T, Boudreau NJ, et al. Abnormal balance in the angiopoietin-tie2 system in human brain arteriovenous malformations. Circ Res. 2001;89:111-113.
148 Hashimoto T, Emala CW, Joshi S, et al. Abnormal pattern of tie-2 and vascular endothelial growth factor receptor expression in human cerebral arteriovenous malformations. Neurosurgery. 2000;47:910-918. discussion 918-919
149 Shenkar R, Elliott JP, Diener K, et al. Differential gene expression in human cerebrovascular malformations. Neurosurgery. 2003;52:465-478.
150 Uranishi R, Baev NI, Ng PY, et al. Expression of endothelial cell angiogenesis receptors in human cerebrovascular malformations. Neurosurgery. 2001;48:359-367. discussion 367-368
151 Folkman J, D’Amore PA. Blood vessel formation: what is its molecular basis? (minireview). Cell. 1996;87:1153-1155.
152 Hashimoto T, Wu Y, Boudreau NJ, et al. Regulation of tie2 expression by angiopoietin: potential feedback system. Endothelium. 2004;11:207-210.
153 Sternlicht M, Bergers G. Matrix metalloproteinases as emerging targets in anticancer therapy: status and prospects. Emerg Ther Targets. 2000;4:609-633.
154 Gaetani P, Rodriguez Y, Baena R, et al. Metalloproteases and intracranial vascular lesions. Neurol Res. 1999;21:385-390.
155 Todor DR, Lewis I, Bruno G, et al. Identification of a serum gelatinase associated with the occurrence of cerebral aneurysms as pro-matrix metalloproteinase-2. Stroke. 1998;29:1580-1583.
156 Knox JB, Sukhova GK, Whittemore AD, et al. Evidence for altered balance between matrix metalloproteinases and their inhibitors in human aortic diseases. Circulation. 1997;95:205-212.
157 Goodall S, Crowther M, Hemingway DM, et al. Ubiquitous elevation of matrix metalloproteinase-2 expression in the vasculature of patients with abdominal aneurysms. Circulation. 2001;104:304-309.
158 Curci JA, Mao D, Bohner DG, et al. Preoperative treatment with doxycycline reduces aortic wall expression and activation of matrix metalloproteinases in patients with abdominal aortic aneurysms. J Vasc Surg. 2000;31:325-342.
159 Loftus IM, Naylor AR, Goodall S, et al. Increased matrix metalloproteinase-9 activity in unstable carotid plaques. A potential role in acute plaque disruption. Stroke. 2000;31:40-47.
160 Chyatte D, Lewis I. Gelatinase activity and the occurrence of cerebral aneurysms. Stroke. 1997;28:799-804.
161 Hashimoto T, Wen G, Lawton MT, et al. Abnormal expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in brain arteriovenous malformations. Stroke. 2003;34:925-931.
162 Rosenberg GA. Growth and bleeding in BAVM: another role for MMPS. Stroke. 2003;34:925-931.
163 Axisa B, Loftus IM, Naylor AR, et al. Prospective, randomized, double-blind trial investigating the effect of doxycycline on matrix metalloproteinase expression within atherosclerotic carotid plaques. Stroke. 2002;33:2858-2864.
164 Lee CZ, Xu B, Hashimoto T, et al. Doxycycline suppresses cerebral matrix metalloproteinase-9 and angiogenesis induced by focal hyperstimulation of vascular endothelial growth factor in a mouse model. Stroke. 2004;35:1715-1719.
165 Bergers G, Brekken R, McMahon G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737-744.
166 Etoh T, Inoue H, Tanaka S, et al. Angiopoietin-2 is related to tumor angiogenesis in gastric carcinoma: possible in vivo regulation via induction of proteases. Cancer Res. 2001;61:2145-2153.
167 Rutka JT, Taylor M, Mainprize T, et al. Molecular biology and neurosurgery in the third millennium. Neurosurgery. 2000;46:1034-1051.
168 Geraci MW, Moore M, Gesell T, et al. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ Res. 2001;88:555-562.
169 Hashimoto T, Lawton MT, Wen G, et al. Gene microarray analysis of human brain arteriovenous malformations. Neurosurgery. 2004;54:410-425.
170 Yang GY, Xu B, Hashimoto T, et al. Induction of focal angiogenesis through adenoviral vector mediated vascular endothelial cell growth factor gene transfer in the mature mouse brain. Angiogenesis. 2003;6:151-158.
171 Xu B, Wu YQ, Huey M, et al. Vascular endothelial growth factor induces abnormal microvasculature in the endoglin heterozygous mouse brain. J Cereb Blood Flow Metab. 2004;24:237-244.
172 Hashimoto T, Young WL. Roles of angiogenesis and vascular remodeling in brain vascular malformations. Semin Cerebrovasc Dis Stroke. 2004;4:217-225.
173 Rosenberg GA, Mun-Bryce S, Wesley M, et al. Collagenase-induced intracerebral hemorrhage in rats. Stroke. 1990;21:801-807.