CHAPTER 361 Pathobiology of Intracranial Aneurysms
Aneurysms are classified according to their shape and pathogenesis. Saccular aneurysms are berry-shaped or multilobed outpouchings on a blood vessel and are generally associated with cerebral artery bifurcations in the circle of Willis. Fusiform aneurysms are blood vessel dilations that usually result from dissections when blood ruptures into the wall of an artery. Infectious aneurysms arise from focal necrosis of an arterial wall after bacterial or fungal infection. Aneurysms are solitary lesions in about 70% to 75% of patients, whereas multiple lesions occur in about 25% to 30%.1 The majority of aneurysms develop spontaneously, but a small fraction (1% to 2%) are associated with trauma,2 infection,3 or tumor.4
About 85% of cases of subarachnoid hemorrhage are caused by rupture of saccular aneurysms.5 The most frequent sites for saccular aneurysms related to the circle of Willis are the internal carotid artery, anterior communicating artery, and middle cerebral artery.6 When subarachnoid hemorrhage occurs, the subarachnoid space is filled with arterial blood, and nerve fibers within the leptomeninges are stimulated, which can potentially cause intense pain and headache. Arteries located in the subarachnoid space may undergo spasm and result in poor cerebral perfusion and ischemia, which in turn may cause infarction. Flow of cerebrospinal fluid may be impeded as blood becomes organized into scar tissue that blocks the subarachnoid space. When drainage of cerebrospinal fluid is impeded, hydrocephalus with dilation of the cerebral ventricles occurs.7 The reported mortality of patients in whom subarachnoid hemorrhage is diagnosed is about 40% within the first 30 days. Among those who survive, less than 25% have a good functional outcome.8
Pathobiology of Saccular Intracranial Aneurysms
Saccular intracranial aneurysms are thought to arise as a result of disrupted balance between local hemodynamic stress and arterial wall strength.9–11 An understanding of the biologic changes that occur at the tissue, cellular, and molecular levels associated with aneurysm growth and rupture and their impact on the health of the patient is important in developing strategies for prevention, diagnosis, and treatment. Most of our knowledge of the pathology of intracranial aneurysms is based on histologic study of specimens obtained during surgery or autopsy. Such studies indicate that the structural integrity of aneurysm walls is compromised by a number of factors, such as changes in the matrix, hypertension, atherosclerosis, aging, gender, and genetic factors. Current evidence on the pathogenesis of intracranial aneurysms suggests that arterial wall proteolysis by matrix metalloproteinases, apoptosis, and chronic inflammation play a key role in disease progression.
Histology of Normal Intracranial Arteries
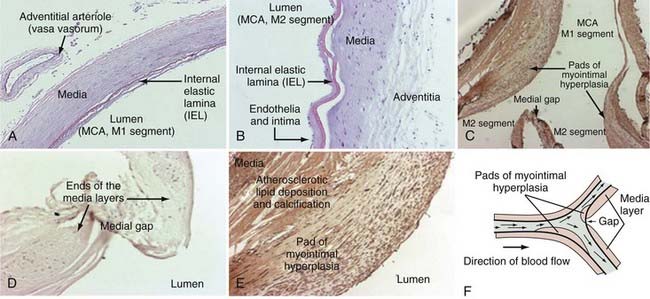
Intracranial arteries vary in size from fine communicating arteries (0.25 mm) to the basilar and internal carotid arteries (3 to 5 mm). Like all arteries, intracranial arteries are composed of three distinct microscopic layers: tunica intima, tunica media, and tunica adventitia, commonly referred to as the intima, media, and adventitia (Fig. 361-1A and B). The innermost layer is the intima, which is composed of a monolayer of endothelial cells on the luminal side in direct contact with blood flow, followed by a subendothelial connective tissue layer. A membrane of elastic fibers called the internal elastic lamina separates the intima from the media. The media is composed mainly of concentric sheets of smooth muscle cells and collagen fibers.12,13 Medial collagen, apparently predominantly type III, can be stretched despite its highly aligned arrangement.12 The outermost layer is the adventitia, which consists primarily of a complex network of type I collagen fibers, some elastin, nerves, fibroblasts, and the vasa vasorum. Adventitial collagen fibers adjacent to the media are oriented in a circumferential fashion and gradually change to a longitudinal orientation toward the outer layer.14–16 Adventitial collagen fibrils, 50 to 100 nm in diameter,17,18 have a wavy appearance and are unstretched at physiologic pressure levels.16,19 In most arteries, the adventitia is separated from the media by another layer of elastic fibers called the external elastic lamina. However, this structure is not present in intracranial arteries.
A substantial portion of the connective tissue in the arterial wall is filled with extracellular matrix, a complex and dynamic meshwork of proteins and proteoglycans that provide structural support to the vessel wall, as well as participate in various biologic activities such as cell proliferation, differentiation, and migration.20 Synthesized and secreted by resident cells, extracellular matrix molecules become highly organized to adapt to the functional requirements of the particular tissue. Examples of extracellular matrix components are collagen, elastin, fibronectin, laminin, nidogen/entactins, perlecan, and syndecans.
About 80% to 90% of the total arterial collagen, composed of collagen types I and III, consists of fibrillar collagen, which provides tensile strength to blood vessels. The defining structural feature of collagen is a triple-stranded helical structure (designated procollagen) of three polypeptide chains, referred to as α chains. Once secreted, propolypeptides of procollagen are removed by proteolytic enzymes. This allows the formation of higher order polymers called collagen fibrils (10 to 30 nm in diameter), which often aggregate into even larger bundles called collagen fibers (500 to 3000 nm in diameter). The remaining 10% to 20% of arterial collagen (types IV, V, VI, and VIII)21 is composed of network-forming collagen that gives rise to net-like structures, such as in the basement membrane.
Elastin is the core component of elastic fibers and it provides resilience to blood vessels. On secretion, elastin precursor molecules become highly cross-linked to one another and form networks of elastin fibers and sheets. Microfibrils, composed of glycoproteins, are thought to provide scaffolding for elastin molecules and are subsequently displaced as elastin is deposited into the growing fiber. Elastin makes up about 90% of elastic fibers, with microfibrillar glycoproteins accounting for the remaining 10%.22
Fibronectin is a multifunctional glycoprotein that provides stability to vessel walls and is involved in intercellular contact and repair mechanisms.23,24 Fibronectins connect cells with collagen fibers, thereby allowing cells to move through the extracellular matrix. They bind collagen and cell surface receptors called integrins, which causes reorganization of the cell cytoskeleton and cell movement. Fibronectins are secreted in an inactive form. Binding to integrins allows them to dimerize and become active. Fibronectins also help at the site of tissue injury by binding to platelets during blood clotting and facilitating movement of cells to the affected area during wound healing.
Laminins, a family of heterotrimeric glycoproteins that are usually found in basement membranes, function in migration, adhesion, and cell signaling.25,26 They are formed from the assembly of three subunits (α, β, and γ). Several laminin isoforms are derived from the combination of five α, four β, and six γ subunits. Vascular endothelial cells have been shown to produce the laminin-8 and laminin-10 isoforms.25,27,28
In 1930, Forbus described the presence of discontinuities in the smooth muscle layer of arterial vessels, especially at the apex of vessel bifurcations. For many years, these medial gaps were believed to represent congenital medial defects that contribute to the development of intracranial aneurysms.29 In 1983, Stehbens demonstrated that the so-called medial defects could be visualized under light microscopy and that they are actually physiologic intersections of two muscle layers rather than defects.30 This finding is in agreement with more recent studies showing that highly organized tendon-like collagen is consistently found in the region of the medial gap, which would provide strength and stability to the vessel, and is inconsistent with the concept of defect.17 Accordingly, experimentally induced intracranial aneurysms in rats were shown not to originate at the site of these medial gaps.31 The histologic characteristics of these medial gaps are shown in Figure 361-1.32
Histology of Intracranial Arteries from Patients with Aneurysms
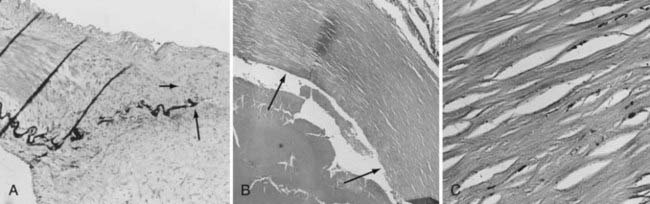
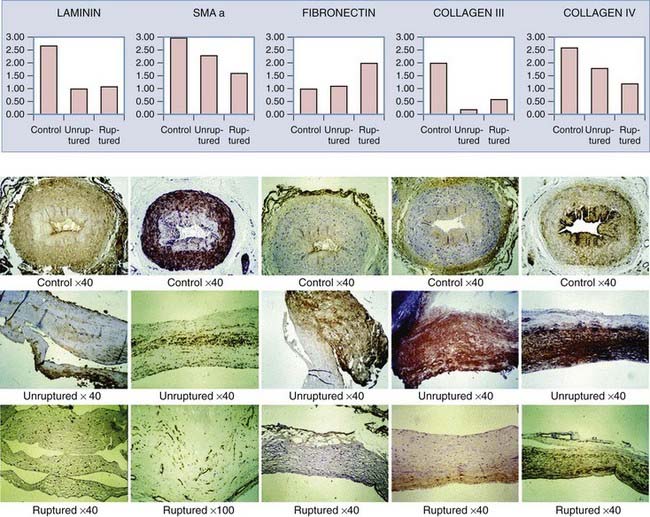
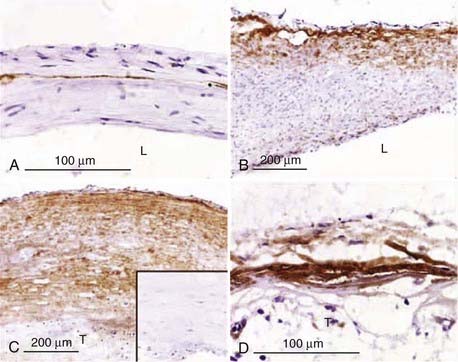
In contrast to the highly ordered structure of a normal arterial wall, aneurysms have a disorganized wall structure with less distinct layers. A common feature of intracranial aneurysm walls is loss or rupture of the internal elastic lamina (Fig. 361-2A).33–36 Other characteristics include intimal thickening resembling myointimal hyperplasia, muscle fiber disarray, depletion of cellular components (Fig. 361-2B and C), and irregularities in the luminal surface (Fig. 361-3).33,34,36–42 Atherosclerotic lesions have also been identified in some aneurysms.38,43–45 Immunohistochemical studies on 18 intracranial aneurysm specimens showed intermingling of collagen type I and fibronectin throughout the aneurysm wall. This finding is in contrast to parent and normal arteries, in which collagen and fibronectin are limited to the adventitia and media, respectively.46 Expression of laminin, collagen types III and IV, and the angiogenic growth factor transforming growth factor-α has been found to be decreased in some aneurysm samples when compared with normal vessels (Fig. 361-4).47
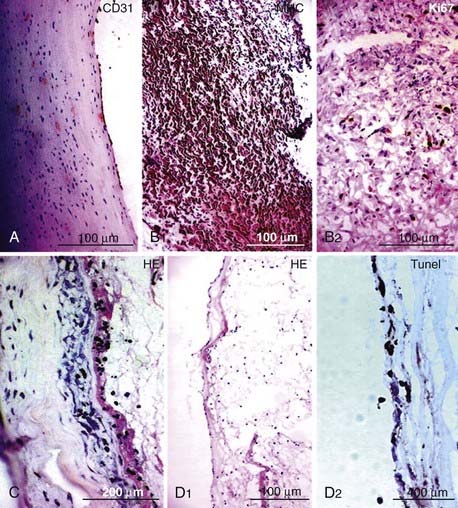
Changes in the medial smooth muscle cell phenotype have also been reported in intracranial aneurysms. In control cerebral arteries, α-smooth muscle actin, desmin, and the smooth muscle myosin heavy-chain (MHC) isoforms SM1 and SM2 were strongly expressed, whereas another MHC isoform, SMemb, was weakly expressed. In aneurysm walls, no expression of desmin was observed. In some aneurysms, SMemb expression was higher or SM2 expression was lower than in control arteries. Desmin is the major intermediate filament of skeletal, cardiac, and smooth muscle tissue48 and is believed to be important for strengthening and maintaining the integrity of smooth muscle cells.49 These observations suggest that a phenotypic switch in smooth muscle cells is associated with vascular remodeling in the aneurysm wall.50
Histologic differences have been noted between ruptured and unruptured aneurysms. A denser expression of fibronectin has been observed in ruptured aneurysms than in unruptured aneurysms.47 Ruptured walls appear to have more pronounced endothelial damage and loss of wall density.41,51–53 In a study involving 44 ruptured and 27 unruptured aneurysms, Kataoka and colleagues found that unruptured aneurysms had thick walls resembling myointimal hyperplasia whereas ruptured aneurysms had thin, hyalinized, and hypocellular walls.41 In a series of 66 intracranial aneurysm samples, Frosen and associates identified four histologic aneurysm wall types and described morphologic changes in the aneurysm walls that correlated with rupture. These wall types were endothelialized wall with linearly organized smooth muscle cells (42% ruptured) (Fig. 361-5A), thickened wall with disorganized smooth muscle cells (55% ruptured) (Fig. 361-5B), hypocellular wall with myointimal hyperplasia (64% ruptured) (Fig. 361-5C), and thin, hyalinized, and hypocellular wall (100% ruptured) (Fig. 361-5D).51
Structural and biochemical changes have also been noted in the nonaneurysmal tissue of aneurysm patients. In a study of the intracranial arteries of 70 normal individuals and 35 patients with intracranial aneurysms who died of aneurysm rupture, a dense, uniformly distributed network of reticular fibers surrounding the smooth muscle cells was observed in the media of normal individuals. This pattern did not appear to change with age, at least until the age of 50. In contrast, a decreased number of reticular fibers was observed in the arteries of aneurysm patients, all of whom died before the age of 50.54 Similar results were obtained in two other independent investigations.55,56 Aside from decreased numbers, irregular distribution and shorter reticular fiber length were also noted.55,56 Biochemical studies have shown that the skin fibroblasts and intracranial and temporal arteries of some aneurysm patients are deficient in collagen type III relative to collagen type I.57–60 These observations led to the hypothesis that “intrinsic” abnormalities in the walls of intracranial arteries lead to conditions that favor the formation and rupture of intracranial arteries.
Mechanisms of Aneurysm Wall Degeneration and Rupture
In normal arteries, myointimal hyperplasia is an adaptive mechanism in response to hemodynamic stress or mechanical injury. Pads of myointimal hyperplasia are formed when endothelial damage from injury or stress induces a change in the phenotype of medial smooth muscle cells that promotes their migration to and proliferation at the damaged endothelial lining.61 The histopathologic resemblance of unruptured intracranial aneurysm walls to myointimal hyperplasia suggests that the aneurysm wall reacts similarly to hemodynamic stress by increasing cell proliferation and matrix synthesis. However, it is still unclear whether the myointimal hyperplasia in aneurysms is an adaptive mechanism of the vascular wall to compensate for excessive stress or whether it contributes to weakening of the wall.
Histologic studies suggest that intracranial aneurysm walls rupture as a consequence of matrix degeneration and decellularization, which may be the result of direct vascular stress or injury or correspond to defective homeostatic maintenance or repair mechanisms.62,63 Although it is still unclear how risk factors for subarachnoid hemorrhage, such as smoking, hypertension, female gender, previous subarachnoid hemorrhage, and age, contribute to rupture, data suggest that proteolysis, programmed cell death, inflammation, and hemodynamic stress are events that are associated with aneurysm wall degeneration and rupture.
Proteolysis and Intracranial Aneurysms
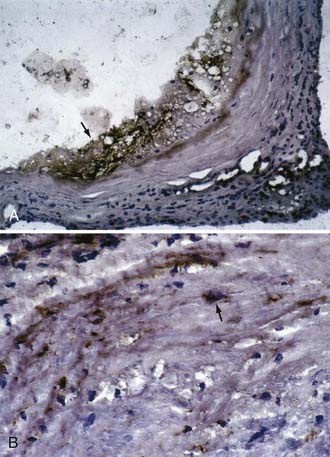
Extracellular matrix components are constantly being synthesized and degraded. When compared with normal intracranial arteries, aneurysm tissue, especially ruptured aneurysm tissue, displays increased activity or expression (or both) of matrix-degrading proteases that regulate remodeling of the arterial wall. Prominent in aneurysm walls are matrix metalloproteinase-2 (MMP-2) and MMP-9, which are classified as gelatinases and are capable of degrading both elastin and denatured collagen. In a series of 23 patients with intracranial aneurysms, Bruno and associates found focal areas of gelatinase activity attributable to MMP-2 and MMP-9 in both ruptured and unruptured aneurysms. Increased protease activity correlated with increased expression of MMP-2, plasmin (Fig. 361-6), and membrane-type MMP,11 with the latter two being involved in activation of MMP-2. Increased serum levels of elastase have also been observed in aneurysm patients.64,65 Furthermore, cathepsin D–expressing cells have been found in aneurysm walls in regions where the collagen layer was degraded. Cathepsin D is an endopeptidase that can also digest extracellular matrix proteins.
Data suggest that increased proteolysis may also contribute to rupture of intracranial aneurysms. In a series of 30 aneurysm patients, Jin and colleagues found that mRNA levels of MMP-2 and MMP-9 were higher in ruptured aneurysms than in unruptured aneurysms. Levels of MMP-2 and MMP-9 were elevated relative to their inhibitors (called tissue inhibitors of MMPs [TIMPs]) in ruptured aneurysms.66 Patients with ruptured aneurysms had higher serum MMP-2 and MMP-9 levels than did those with unruptured aneurysms.66 Upregulation of elastase activity has also been observed in the arterial walls of ruptured aneurysms in comparison to unruptured aneurysms.64
Apoptosis and Intracranial Aneurysms
Very few cells undergo programmed cell death (apoptosis) in normal arterial wall. In contrast, many apoptotic cells are found in intracranial aneurysm walls, especially in ruptured aneurysms.51,53 Experimental and clinical studies have shown that the decreased number of smooth muscle cells in aneurysm walls is primarily due to apoptosis.67,68 Higher apoptosis levels were also found in the meningeal and superficial temporal arteries in patients with ruptured aneurysms than in patients with unruptured lesions.53 In a series of five ruptured aneurysms, Hara and coworkers found that many apoptotic cells were localized in the neck and dome of aneurysms, close to the rupture point, whereas no apoptosis was detected in control arteries.67 These results suggest that apoptosis plays an important role in the development and rupture of intracranial aneurysms.
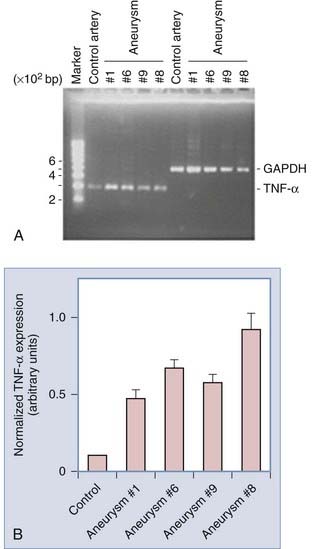
The events that trigger apoptosis in aneurysm walls is unclear. It had been proposed that apoptosis is induced by cytokines released by inflammatory cells that infiltrate aneurysm tissues. Jayaraman and associates showed that mRNA and protein expression of the proinflammatory cytokine tumor necrosis factor-α (TNF-α) and its proapoptotic downstream target Fas-associated death domain protein are increased in ruptured aneurysms (Fig. 361-7),69 thus suggesting a role for TNF-α in promoting inflammation and subsequent apoptosis in aneurysm walls.
A role for the c-Jun amino-terminal kinase (JNK) pathway in apoptosis in intracranial aneurysms has also been proposed. Inflammatory signals are capable of stimulating JNK (a member of the mitogen-activated protein kinase family) to initiate apoptosis via phosphorylation of the c-Jun transcription factor. In a series of 12 intracranial aneurysms and 5 control vessels, Takagi and coworkers found that phosphorylation of JNK and its target c-Jun was increased in aneurysm tissues (Fig. 361-8) in regions where apoptosis in smooth muscle cells was also increased.70 In a series of 12 ruptured and 12 unruptured aneurysms, Laaksamo and associates found that p54 JNK phosphorylation was 1.5-fold higher in ruptured aneurysms than in unruptured aneurysms. Furthermore, phosphorylation of p54 JNK and the protein levels of both JNK and c-Jun were found to correlate with aneurysm size.71
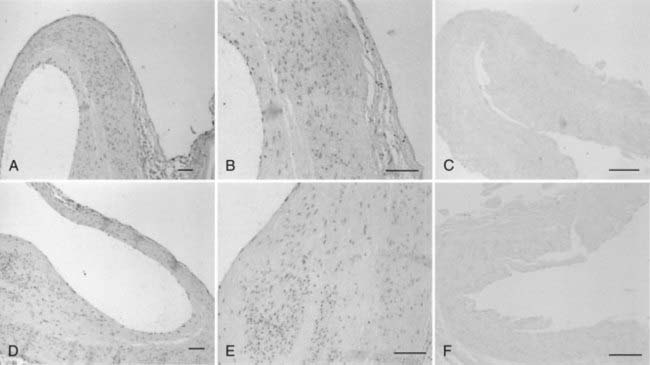
Inflammation and Intracranial Aneurysms
The presence of macrophages, T cells, B cells, immunoglobulin antibodies, and activated complement in intracranial aneurysm walls, especially in ruptured aneurysms, indicates that active innate and adaptive immunologic reactions are associated with aneurysm formation and rupture (Fig. 361-9).41,45,51,52,72 Upregulated expression of the proinflammatory chemokine monocyte chemoattractant protein-1 (MCP-1) in the aneurysm wall has also been observed. Even though MCP-1 mRNA was not detectable in normal arteries, it was often observed in the intima of aneurysmal walls and appeared to be expressed in the cytoplasm of fibroblast cells that assembled with monocyte-like cells.42 The extent of inflammatory cell infiltration and complement activation appeared to be higher in ruptured than unruptured aneurysms.51,52
Further evidence of the development of active immune reactions in intracranial aneurysm specimens was demonstrated in a microarray study by Krischek and colleagues in which the global transcription profiles of six ruptured and four unruptured aneurysm tissues were compared with the profiles of four control arteries. Genes involved in antigen presentation, such as major histocompatibility complex class II genes, emerged as those with the highest upregulation in the aneurysm samples.73
Hemodynamic Stress and Inflammation and Intracranial Aneurysms
Intracranial aneurysms have been induced by hypertension and carotid ligation in animal models.68,74–86 The aneurysmal changes, detected about 3 months after surgery, were very similar to changes in human saccular aneurysms. Such changes include fragmentation of the internal elastic lamina, thinning of the smooth muscle cell layer, degeneration of adventitial tissue, de-endothelialization, macrophage infiltration, and apoptosis. These data suggest that hemodynamic stress leads to arterial wall degeneration and eventually to aneurysm formation.
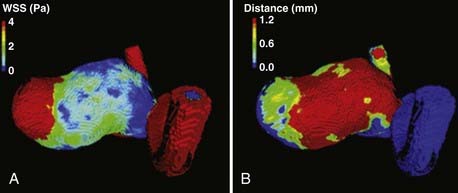
Although the intracranial aneurysms in these models seem to form at areas of high hemodynamic stress such as at arterial bifurcations, studies on intra-aneurysmal hemodynamic flow in human intracranial aneurysms show that aneurysm growth is associated with low shear stress. By monitoring aneurysm size with time-averaged shear stress in 7 aneurysm patient models, Boussel and associates found that growth probably occurs in aneurysms in which the endothelial cells are exposed to abnormally low wall shear stress (Fig. 361-10).87 In a study measuring intra-aneurysmal flow in 3 aneurysm models, Yamaguchi and coauthors have shown that aneurysms with higher aspect ratios (dome/neck measurement, known to correlate with rupture) are associated with low wall shear stress.88 In another study in which wall shear distribution in 8 ruptured aneurysms and 18 unruptured internal carotid aneurysms was retrospectively analyzed, Jou and colleagues found that in ruptured aneurysms, a greater proportion of the aneurysm was exposed to low wall shear stress.89
Atherosclerosis and Inflammation and Intracranial Aneurysms
Immune and adaptive immune responses are activated in atherosclerotic lesions, including infiltration of macrophages, T cells,90–92 B cells,93 mast cells,94,95 and natural killer cells96; binding of antibodies97; and activation of complement.98,99 Macrophages induce apoptosis and proteolysis by secreting inflammatory cytokines such as TNF-α and matrix metalloproteinases, respectively. Macrophages also induce the production of growth factors and more cytokines by activating T cells and B cells.100 The inflammation in atherosclerosis is thought to be triggered by the accumulation of lipids and oxidative or enzymatic modification.101 Modified lipids, such as low-density lipoprotein, form neoepitopes that are recognized by and activate the immune system. Oxidative stress and free radicals in atherosclerotic walls may also activate the immune response.
Several studies support the hypothesis that aneurysm formation and atherosclerosis might be related. In a histologic examination of atherosclerotic saccular aneurysms, Kosierkiewicz and coworkers found that aneurysm size is related to the progression of atherosclerotic plaque. In small aneurysms, atherosclerotic lesions were characterized by intimal thickening with only minimal inflammatory cell infiltration. In large aneurysms, atherosclerotic lesions were more advanced and exhibited more extensive macrophage, lymphocyte, and natural killer cell infiltration.45 In a study involving 50 patients with intracranial aneurysms and no significant atheromatous disease, Bolger and associates found that elevated serum levels of lipoprotein (a) (Lp[a]), an independent risk factor for atherosclerosis, were also detected in aneurysm patients, whose average serum Lp(a) levels were twofold higher than in healthy normal controls.102 These data suggest that Lp(a) is also associated with the presence of intracranial aneurysms. In a study investigating the wall shear stress distribution between areas with and without atherosclerotic changes in a single, large middle cerebral artery aneurysm, Tateshima and colleagues showed that low wall shear stress correlated with atherosclerotic areas.103
Despite considerable resemblance to atherosclerosis, some studies suggest that aneurysm and atherosclerosis might be two separate processes. In an autopsy study of six intracranial aneurysms, Hassler detected slight atherosclerotic changes near the vicinity of the aneurysms. However, no apparent relationship between the presence of atherosclerosis and aneurysms was found.37 Although an association between elevated Lp(a) levels and intracranial aneurysm formation was found (described earlier), examination of Lp(a) expression in intracranial aneurysm samples and control arteries with and without atherosclerosis showed that Lp(a) expression in aneurysms may occur independently of atherosclerosis.43
Conclusion
Familial aggregation of intracranial aneurysms, the strongest risk factor for aneurysmal subarachnoid hemorrhage, strongly suggests a genetic role in the development of this disease (the topic of another chapter). The importance of identifying causative genetic mutations to provide a deeper understanding of the pathogenesis of aneurysm formation has been appreciated and has been a focus of research in the past decade. With the use of molecular genetic techniques such as linkage and association studies, several intracranial aneurysm susceptibility loci have been reported. However, no single disease-causing genetic variant has been identified in these regions. Recently, using a genome-wide linkage approach in a single, large white family, we identified a new susceptibility region in chromosome 13q and are currently examining genes in this interval.104 In addition, we have noted mutations in the type 3 receptors for transforming growth factor in a small subset of familial aneurysm patients.105 Knowledge of genetic determinants may provide diagnostic tools for identifying individuals at increased risk for aneurysm formation, insight on aneurysm pathobiology, and clues on how to prevent or stop aneurysm growth.
Boussel L, Rayz V, McCulloch C, et al. Aneurysm growth occurs at region of low wall shear stress: patient-specific correlation of hemodynamics and growth in a longitudinal study. Stroke. 2008;39:2997.
Caird J, Napoli C, Taggart C, et al. Matrix metalloproteinases 2 and 9 in human atherosclerotic and non-atherosclerotic cerebral aneurysms. Eur J Neurol. 2006;13:1098.
Draghia F, Draghia AC, Onicescu D. Electron microscopic study of the arterial wall in the cerebral aneurysms. Rom J Morphol Embryol. 2008;49:101.
Forbus W. On the origin of miliary aneurysms of the superficial cerebral arteries. Bull Johns Hopkins Hosp. 1930;47:239.
Frosen J. The Pathobiology of Saccular Cerebral Artery Aneurysm Rupture and Repair. Helsinki: University of Helsinki; 2006.
Frosen J, Piippo A, Paetau A, et al. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke. 2004;35:2287.
Hegedus K. Some observations on reticular fibers in the media of the major cerebral arteries. A comparative study of patients without vascular diseases and those with ruptured berry aneurysms. Surg Neurol. 1984;22:301.
Inci S, Spetzler RF. Intracranial aneurysms and arterial hypertension: a review and hypothesis. Surg Neurol. 2000;53:530.
Jamous MA, Nagahiro S, Kitazato KT, et al. Endothelial injury and inflammatory response induced by hemodynamic changes preceding intracranial aneurysm formation: experimental study in rats. J Neurosurg. 2007;107:405.
Jayaraman T, Berenstein V, Li X, et al. Tumor necrosis factor alpha is a key modulator of inflammation in cerebral aneurysms. Neurosurgery. 2005;57:558.
Jin D, Sheng J, Yang X, et al. Matrix metalloproteinases and tissue inhibitors of metalloproteinases expression in human cerebral ruptured and unruptured aneurysm. Surg Neurol. 2007;68(suppl 2):S11.
Kataoka K, Taneda M, Asai T, et al. Structural fragility and inflammatory response of ruptured cerebral aneurysms. A comparative study between ruptured and unruptured cerebral aneurysms. Stroke. 1999;30:1396.
Kilic T, Sohrabifar M, Kurtkaya O, et al. Expression of structural proteins and angiogenic factors in normal arterial and unruptured and ruptured aneurysm walls. Neurosurgery. 2005;57:997.
Kosierkiewicz TA, Factor SM, Dickson DW. Immunocytochemical studies of atherosclerotic lesions of cerebral berry aneurysms. J Neuropathol Exp Neurol. 1994;53:399.
Krex D, Schackert HK, Schackert G. Genesis of cerebral aneurysms—an update. Acta Neurochir (Wien). 2001;143:429.
Krischek B, Kasuya H, Tajima A, et al. Network-based gene expression analysis of intracranial aneurysm tissue reveals role of antigen presenting cells. Neuroscience. 2008;154:1398.
Laaksamo E, Tulamo R, Baumann M, et al. Involvement of mitogen-activated protein kinase signaling in growth and rupture of human intracranial aneurysms. Stroke. 2008;39:886.
Nakajima N, Nagahiro S, Sano T, et al. Phenotypic modulation of smooth muscle cells in human cerebral aneurysmal walls. Acta Neuropathol. 2000;100:475.
Pentimalli L, Modesti A, Vignati A, et al. Role of apoptosis in intracranial aneurysm rupture. J Neurosurg. 2004;101:1018.
Ruigrok YM, Rinkel GJ, Wijmenga C. Genetics of intracranial aneurysms. Lancet Neurol. 2005;4:179.
Schlote W, Gaus C. Histologic aspects from ruptured and nonruptured aneurysms. Neurol Res. 1994;16:59.
Takagi Y, Ishikawa M, Nozaki K, et al. Increased expression of phosphorylated c-Jun amino-terminal kinase and phosphorylated c-Jun in human cerebral aneurysms: role of the c-Jun amino-terminal kinase/c-Jun pathway in apoptosis of vascular walls. Neurosurgery. 2002;51:997.
Tulamo R, Frosen J, Junnikkala S, et al. Complement activation associates with saccular cerebral artery aneurysm wall degeneration and rupture. Neurosurgery. 2006;59:1069.
van den Berg JS, Limburg M, Pals G, et al. Some patients with intracranial aneurysms have a reduced type III/type I collagen ratio. A case-control study. Neurology. 1997;49:1546.
Zhang B, Fugleholm K, Day LB, et al. Molecular pathogenesis of subarachnoid haemorrhage. Int J Biochem Cell Biol. 2003;35:1341.
1 Rinne J, Hernesniemi J, Puranen M, et al. Multiple intracranial aneurysms in a defined population: prospective angiographic and clinical study. Neurosurgery. 1994;35:803.
2 Benoit BG, Wortzman G. Traumatic cerebral aneurysms. Clinical features and natural history. J Neurol Neurosurg Psychiatry. 1973;36:127.
3 Clare CE, Barrow DL. Infectious intracranial aneurysms. Neurosurg Clin N Am. 1992;3:551.
4 Damasio H, Seabra-Gomes R, da Silva JP, et al. Multiple cerebral aneurysms and cardiac myxoma. Arch Neurol. 1975;32:269.
5 Kassell NF, Torner JC, Jane JA, et al. The International Cooperative Study on the Timing of Aneurysm Surgery. Part 2: surgical results. J Neurosurg. 1990;73:37.
6 Clarke G, Mendelow AD, Mitchell P. Predicting the risk of rupture of intracranial aneurysms based on anatomical location. Acta Neurochir (Wien). 2005;147:259.
7 Heros RC. Acute hydrocephalus after subarachnoid hemorrhage. Stroke. 1989;20:715.
8 Zhang B, Fugleholm K, Day LB, et al. Molecular pathogenesis of subarachnoid haemorrhage. Int J Biochem Cell Biol. 2003;35:1341.
9 Stehbens WE. Etiology of intracranial berry aneurysms. J Neurosurg. 1989;70:823.
10 Inci S, Spetzler RF. Intracranial aneurysms and arterial hypertension: a review and hypothesis. Surg Neurol. 2000;53:530.
11 Bruno G, Todor R, Lewis I, et al. Vascular extracellular matrix remodeling in cerebral aneurysms. J Neurosurg. 1998;89:431.
12 Canham PB, Talman EA, Finlay HM, et al. Medial collagen organization in human arteries of the heart and brain by polarized light microscopy. Connect Tissue Res. 1991;26:121.
13 Walmsley JG, Campling MR, Chertkow HM. Interrelationships among wall structure, smooth muscle orientation, and contraction in human major cerebral arteries. Stroke. 1983;14:781.
14 Finlay HM, McCullough L, Canham PB. Three-dimensional collagen organization of human brain arteries at different transmural pressures. J Vasc Res. 1995;32:301.
15 Rowe AJ, Finlay HM, Canham PB. Collagen biomechanics in cerebral arteries and bifurcations assessed by polarizing microscopy. J Vasc Res. 2003;40:406.
16 Smith JF, Canham PB, Starkey J. Orientation of collagen in the tunica adventitia of the human cerebral artery measured with polarized light and the universal stage. J Ultrastruct Res. 1981;77:133.
17 Finlay HM, Whittaker P, Canham PB. Collagen organization in the branching region of human brain arteries. Stroke. 1998;29:1595.
18 Merrilees MJ, Tiang KM, Scott L. Changes in collagen fibril diameters across artery walls including a correlation with glycosaminoglycan content. Connect Tissue Res. 1987;16:237.
19 Canham PB, Whittaker P, Barwick SE, et al. Effect of pressure on circumferential order of adventitial collagen in human brain arteries. Can J Physiol Pharmacol. 1992;70:296.
20 Krex D, Schackert HK, Schackert G. Genesis of cerebral aneurysms—an update. Acta Neurochir (Wien). 2001;143:429.
21 Mayne R. Collagenous proteins of blood vessels. Arteriosclerosis. 1986;6:585.
22 Ruigrok YM, Rinkel GJ, Wijmenga C. Genetics of intracranial aneurysms. Lancet Neurol. 2005;4:179.
23 Hynes RO, Yamada KM. Fibronectins: multifunctional modular glycoproteins. J Cell Biol. 1982;95:369.
24 Hedin U, Bottger BA, Forsberg E, et al. Diverse effects of fibronectin and laminin on phenotypic properties of cultured arterial smooth muscle cells. J Cell Biol. 1988;107:307.
25 Patarroyo M, Tryggvason K, Virtanen I. Laminin isoforms in tumor invasion, angiogenesis and metastasis. Semin Cancer Biol. 2002;12:197.
26 Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Dev Dyn. 2000;218:213.
27 Frieser M, Nockel H, Pausch F, et al. Cloning of the mouse laminin alpha 4 cDNA. Expression in a subset of endothelium. Eur J Biochem. 1997;246:727.
28 Sorokin LM, Pausch F, Frieser M, et al. Developmental regulation of the laminin alpha5 chain suggests a role in epithelial and endothelial cell maturation. Dev Biol. 1997;189:285.
29 Forbus W. On the origin of miliary aneurysms of the superficial cerebral arteries. Bull Johns Hopkins Hosp. 1930;47:239.
30 Stehbens W. The pathology of intracranial arterial aneurysms and their complications. In: Fox J, editor. Intracranial Aneurysms. New York: Springer; 1983:272.
31 Futami K, Yamashita J, Higashi S. Do cerebral aneurysms originate at the site of medial defects? Microscopic examinations of experimental aneurysms at the fenestration of the anterior cerebral artery in rats. Surg Neurol. 1998;50:141.
32 Frosen J. The Pathobiology of Saccular Cerebral Artery Aneurysm Rupture and Repair. Helsinki: University of Helsinki; 2006.
33 Draghia F, Draghia AC, Onicescu D. Electron microscopic study of the arterial wall in the cerebral aneurysms. Rom J Morphol Embryol. 2008;49:101.
34 Stehbens WE. Ultrastructure of aneurysms. Arch Neurol. 1975;32:798.
35 Scanarini M, Mingrino S, Giordano R, et al. Histological and ultrastructural study of intracranial saccular aneurysmal wall. Acta Neurochir (Wien). 1978;43:171.
36 Nakayama Y, Tanaka A, Kumate S, et al. Giant fusiform aneurysm of the basilar artery: consideration of its pathogenesis. Surg Neurol. 1999;51:140.
37 Hassler O. Scanning electron microscopy of saccular intracranial aneurysms. Am J Pathol. 1972;68:511.
38 Schlote W, Gaus C. Histologic aspects from ruptured and nonruptured aneurysms. Neurol Res. 1994;16:59.
39 Shokunbi MT, Vinters HV, Kaufmann JC. Fusiform intracranial aneurysms. Clinicopathologic features. Surg Neurol. 1988;29:263.
40 Abruzzo T, Shengelaia GG, Dawson RC3rd, et al. Histologic and morphologic comparison of experimental aneurysms with human intracranial aneurysms. AJNR Am J Neuroradiol. 1998;19:1309.
41 Kataoka K, Taneda M, Asai T, et al. Structural fragility and inflammatory response of ruptured cerebral aneurysms. A comparative study between ruptured and unruptured cerebral aneurysms. Stroke. 1999;30:1396.
42 Cao Y, Zhao J, Wang S, et al. Monocyte chemoattractant protein-1 mRNA in human intracranial aneurysm walls. Zhonghua Yu Fang Yi Xue Za Zhi. 2002;36:519.
43 Caird J, Burke M, Roberts G, et al. Apolipoprotein(a) expression in intracranial aneurysms. Neurosurgery. 2003;52:854.
44 Caird J, Napoli C, Taggart C, et al. Matrix metalloproteinases 2 and 9 in human atherosclerotic and non-atherosclerotic cerebral aneurysms. Eur J Neurol. 2006;13:1098.
45 Kosierkiewicz TA, Factor SM, Dickson DW. Immunocytochemical studies of atherosclerotic lesions of cerebral berry aneurysms. J Neuropathol Exp Neurol. 1994;53:399.
46 Austin G, Fisher S, Dickson D, et al. The significance of the extracellular matrix in intracranial aneurysms. Ann Clin Lab Sci. 1993;23:97.
47 Kilic T, Sohrabifar M, Kurtkaya O, et al. Expression of structural proteins and angiogenic factors in normal arterial and unruptured and ruptured aneurysm walls. Neurosurgery. 2005;57:997.
48 Small JV, Sobieszek A. Studies on the function and composition of the 10-nm (100-Å) filaments of vertebrate smooth muscle. J Cell Sci. 1977;23:243.
49 Li Z, Colucci-Guyon E, Pincon-Raymond M, et al. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev Biol. 1996;175:362.
50 Nakajima N, Nagahiro S, Sano T, et al. Phenotypic modulation of smooth muscle cells in human cerebral aneurysmal walls. Acta Neuropathol. 2000;100:475.
51 Frosen J, Piippo A, Paetau A, et al. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke. 2004;35:2287.
52 Tulamo R, Frosen J, Junnikkala S, et al. Complement activation associates with saccular cerebral artery aneurysm wall degeneration and rupture. Neurosurgery. 2006;59:1069.
53 Pentimalli L, Modesti A, Vignati A, et al. Role of apoptosis in intracranial aneurysm rupture. J Neurosurg. 2004;101:1018.
54 Hegedus K. Some observations on reticular fibers in the media of the major cerebral arteries. A comparative study of patients without vascular diseases and those with ruptured berry aneurysms. Surg Neurol. 1984;22:301.
55 Chyatte D, Reilly J, Tilson MD. Morphometric analysis of reticular and elastin fibers in the cerebral arteries of patients with intracranial aneurysms. Neurosurgery. 1990;26:939.
56 Ostergaard JR, Reske-Nielsen E, Oxlund H. Histological and morphometric observations on the reticular fibers in the arterial beds of patients with ruptured intracranial saccular aneurysms. Neurosurgery. 1987;20:554.
57 van den Berg JS, Limburg M, Pals G, et al. Some patients with intracranial aneurysms have a reduced type III/type I collagen ratio. A case-control study. Neurology. 1997;49:1546.
58 Neil-Dwyer G, Bartlett JR, Nicholls AC, et al. Collagen deficiency and ruptured cerebral aneurysms. A clinical and biochemical study. J Neurosurg. 1983;59:16.
59 Ostergaard JR, Oxlund H. Collagen type III deficiency in patients with rupture of intracranial saccular aneurysms. J Neurosurg. 1987;67:690.
60 Pope FM, Nicholls AC, Narcisi P, et al. Some patients with cerebral aneurysms are deficient in type III collagen. Lancet. 1981;1:973.
61 Kraiss LC, Clowes AW. Response of the arterial wall to injury and intimal hyperplasia. In: San S, editor. The Basic Science of Vascular Disease. New York: Futura; 1997:289.
62 de la Monte SM, Moore GW, Monk MA, et al. Risk factors for the development and rupture of intracranial berry aneurysms. Am J Med. 1985;78:957.
63 Ferguson GG. Physical factors in the initiation, growth, and rupture of human intracranial saccular aneurysms. J Neurosurg. 1972;37:666.
64 Connolly ESJr, Fiore AJ, Winfree CJ, et al. Elastin degradation in the superficial temporal arteries of patients with intracranial aneurysms reflects changes in plasma elastase. Neurosurgery. 1997;40:903.
65 Baker CJ, Fiore A, Connolly ESJr, et al. Serum elastase and alpha-1-antitrypsin levels in patients with ruptured and unruptured cerebral aneurysms. Neurosurgery. 1995;37:56.
66 Jin D, Sheng J, Yang X, et al. Matrix metalloproteinases and tissue inhibitors of metalloproteinases expression in human cerebral ruptured and unruptured aneurysm. Surg Neurol. 2007;68(suppl 2):S11.
67 Hara A, Yoshimi N, Mori H. Evidence for apoptosis in human intracranial aneurysms. Neurol Res. 1998;20:127.
68 Kondo S, Hashimoto N, Kikuchi H, et al. Apoptosis of medial smooth muscle cells in the development of saccular cerebral aneurysms in rats. Stroke. 1998;29:181.
69 Jayaraman T, Berenstein V, Li X, et al. Tumor necrosis factor alpha is a key modulator of inflammation in cerebral aneurysms. Neurosurgery. 2005;57:558.
70 Takagi Y, Ishikawa M, Nozaki K, et al. Increased expression of phosphorylated c-Jun amino-terminal kinase and phosphorylated c-Jun in human cerebral aneurysms: role of the c-Jun amino-terminal kinase/c-Jun pathway in apoptosis of vascular walls. Neurosurgery. 2002;51:997.
71 Laaksamo E, Tulamo R, Baumann M, et al. Involvement of mitogen-activated protein kinase signaling in growth and rupture of human intracranial aneurysms. Stroke. 2008;39:886.
72 Chyatte D, Bruno G, Desai S, et al. Inflammation and intracranial aneurysms. Neurosurgery. 1999;45:1137.
73 Krischek B, Kasuya H, Tajima A, et al. Network-based gene expression analysis of intracranial aneurysm tissue reveals role of antigen presenting cells. Neuroscience. 2008;154:1398.
74 Aoki T, Kataoka H, Ishibashi R, et al. Cathepsin B, K, and S are expressed in cerebral aneurysms and promote the progression of cerebral aneurysms. Stroke. 2008;39:2603.
75 Aoki T, Kataoka H, Ishibashi R, et al. Simvastatin suppresses the progression of experimentally induced cerebral aneurysms in rats. Stroke. 2008;39:1276.
76 Aoki T, Kataoka H, Morimoto M, et al. Macrophage-derived matrix metalloproteinase-2 and -9 promote the progression of cerebral aneurysms in rats. Stroke. 2007;38:162.
77 Aoki T, Kataoka H, Moriwaki T, et al. Role of TIMP-1 and TIMP-2 in the progression of cerebral aneurysms. Stroke. 2007;38:2337.
78 Aoki T, Kataoka H, Shimamura M, et al. NF-κB is a key mediator of cerebral aneurysm formation. Circulation. 2007;116:2830.
79 Aoki T, Moriwaki T, Takagi Y, et al. The efficacy of apolipoprotein E deficiency in cerebral aneurysm formation. Int J Mol Med. 2008;21:453.
80 Fukuda S, Hashimoto N, Naritomi H, et al. Prevention of rat cerebral aneurysm formation by inhibition of nitric oxide synthase. Circulation. 2000;101:2532.
81 Jamous MA, Nagahiro S, Kitazato KT, et al. Endothelial injury and inflammatory response induced by hemodynamic changes preceding intracranial aneurysm formation: experimental study in rats. J Neurosurg. 2007;107:405.
82 Morimoto M, Miyamoto S, Mizoguchi A, et al. Mouse model of cerebral aneurysm: experimental induction by renal hypertension and local hemodynamic changes. Stroke. 2002;33:1911.
83 Moriwaki T, Takagi Y, Sadamasa N, et al. Impaired progression of cerebral aneurysms in interleukin-1beta–deficient mice. Stroke. 2006;37:900.
84 Sadamasa N, Nozaki K, Hashimoto N. Disruption of gene for inducible nitric oxide synthase reduces progression of cerebral aneurysms. Stroke. 2003;34:2980.
85 Sadamasa N, Nozaki K, Kita-Matsuo H, et al. Gene expression during the development of experimentally induced cerebral aneurysms. J Vasc Res. 2008;45:343.
86 Sibon I, Mercier N, Darret D, et al. Association between semicarbazide-sensitive amine oxidase, a regulator of the glucose transporter, and elastic lamellae thinning during experimental cerebral aneurysm development: laboratory investigation. J Neurosurg. 2008;108:558.
87 Boussel L, Rayz V, McCulloch C, et al. Aneurysm growth occurs at region of low wall shear stress: patient-specific correlation of hemodynamics and growth in a longitudinal study. Stroke. 2008;39:2997.
88 Yamaguchi R, Ujiie H, Haida S, et al. Velocity profile and wall shear stress of saccular aneurysms at the anterior communicating artery. Heart Vessels. 2008;23:60.
89 Jou LD, Lee DH, Morsi H, et al. Wall shear stress on ruptured and unruptured intracranial aneurysms at the internal carotid artery. AJNR Am J Neuroradiol. 2008;29:1761.
90 Hansson GK, Holm J, Jonasson L. Detection of activated T lymphocytes in the human atherosclerotic plaque. Am J Pathol. 1989;135:169.
91 Jonasson L, Holm J, Skalli O, et al. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986;6:131.
92 Stemme S, Faber B, Holm J, et al. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci U S A. 1995;92:3893.
93 Houtkamp MA, de Boer OJ, van der Loos CM, et al. Adventitial infiltrates associated with advanced atherosclerotic plaques: structural organization suggests generation of local humoral immune responses. J Pathol. 2001;193:263.
94 Kaartinen M, Penttila A, Kovanen PT. Accumulation of activated mast cells in the shoulder region of human coronary atheroma, the predilection site of atheromatous rupture. Circulation. 1994;90:1669.
95 Lappalainen H, Laine P, Pentikainen MO, et al. Mast cells in neovascularized human coronary plaques store and secrete basic fibroblast growth factor, a potent angiogenic mediator. Arterioscler Thromb Vasc Biol. 2004;24:1880.
96 Bobryshev YV, Lord RS. Co-accumulation of dendritic cells and natural killer T cells within rupture-prone regions in human atherosclerotic plaques. J Histochem Cytochem. 2005;53:781.
97 Yla-Herttuala S, Palinski W, Butler SW, et al. Rabbit and human atherosclerotic lesions contain IgG that recognizes epitopes of oxidized LDL. Arterioscler Thromb. 1994;14:32.
98 Meuwissen M, van der Wal AC, Niessen HW, et al. Colocalisation of intraplaque C reactive protein, complement, oxidised low density lipoprotein, and macrophages in stable and unstable angina and acute myocardial infarction. J Clin Pathol. 2006;59:196.
99 Oksjoki R, Jarva H, Kovanen PT, et al. Association between complement factor H and proteoglycans in early human coronary atherosclerotic lesions: implications for local regulation of complement activation. Arterioscler Thromb Vasc Biol. 2003;23:630.
100 Hansson GK, Libby P, Schonbeck U, et al. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. 2002;91:281.
101 Glass CK, Witztum JL. Atherosclerosis: the road ahead. Cell. 2001;104:503.
102 Bolger C, Phillips J, Gilligan S, et al. Elevated levels of lipoprotein (a) in association with cerebrovascular saccular aneurysmal disease. Neurosurgery. 1995;37:241.
103 Tateshima S, Tanishita K, Omura H, et al. Intra-aneurysmal hemodynamics in a large middle cerebral artery aneurysm with wall atherosclerosis. Surg Neurol. 2008;70:454. discussion 462
104 Santiago-Sim T, Mathew-Joseph S, Pannu H, et al. Sequencing of TGF-beta pathway genes in familial cases of intracranial aneurysm. Stroke. 2009;40:1604.
105 Santiago-Sim T, DePalma SR, Ju ISL, et al. Genomewide linkage in a large caucasian family maps a new locus for intercranial aneurysms to chromosome 13q. Stroke. 2009;40:S57.