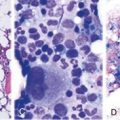
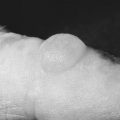
Chapter 5 Paroxysmal Nocturnal Hemoglobinuria
Paroxysmal Nocturnal Hemoglobinuria
Published on 04/03/2015 by admin
Filed under Hematology, Oncology and Palliative Medicine
Last modified 22/04/2025
Print this pagePublished on 04/03/2015 by admin
Filed under Hematology, Oncology and Palliative Medicine
Last modified 22/04/2025
Print this page