Parkinsonism and akinetic–rigid disorders
PARKINSON’S DISEASE (PD)
 PARKINSON’S DISEASE
PARKINSON’S DISEASE


 CAUSES OF PARKINSONISM
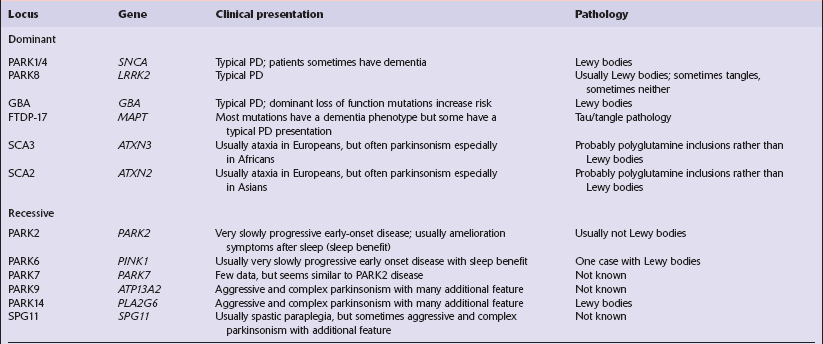
CAUSES OF PARKINSONISM Parkinson’s disease (PD) (Table 28.1). In autopsy studies, 20–30% of patients diagnosed clinically as having Parkinson’s disease have been found to have an alternative cause for their parkinsonism.
Parkinson’s disease (PD) (Table 28.1). In autopsy studies, 20–30% of patients diagnosed clinically as having Parkinson’s disease have been found to have an alternative cause for their parkinsonism.
Table 28.1
Parkinson’s disease and parkinsonism: genetic, clinical and pathologic correlation
Hardy (2010); Hardy et al. (2009).















LEWY BODIES
The pathologic hallmark of Parkinson’s disease is the presence of neuronal inclusions called Lewy bodies. There are two main types, termed ‘classical’ and ‘cortical’ (Table 28.2), which are found in different locations. The presence of Lewy bodies defines several conditions, termed Lewy body disorders (see Tables 28.3 and 28.4).

Table 28.3
Distribution of Lewy bodies in different disorders and their clinical-pathologic correlation
| Disorder | Main site of Lewy body pathology | Clinical correlate |
| Parkinson’s disease (PD) | Substantia nigra | Akinetic–rigid syndrome |
| Parkinson’s disease with dementia (PDD) | Substantia nigra, cerebral cortex | Dementia occurs ≥1 year after a clinical diagnosis of PD |
| Dementia with Lewy bodies (DLB) | Cerebral cortex, substantia nigra | Dementia with akinetic–rigid syndrome. Dementia occurs within a year of onset of parkinsonian features |
| Autonomic failure | Sympathetic neurons in spinal cord | Autonomic failure |
| Lewy body dysphagia | Dorsal vagal nucleus | Dysphagia |
Table 28.4
Anatomical regions susceptible to Lewy pathology, according to two major classification schemes
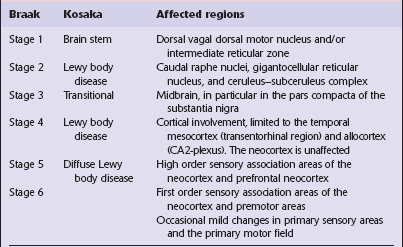
Braak et al. (2003); Kosaka et al. (1988).
MACROSCOPIC APPEARANCES
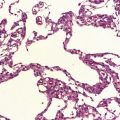
Sections through the midbrain and pons reveal loss of pigment from the substantia nigra and locus ceruleus (Fig. 28.1 – note that pallor of the substantia nigra is normal in childhood and adolescence, the slate-gray color being acquired during early adulthood). The globus pallidus, putamen, and caudate nucleus appear normal.
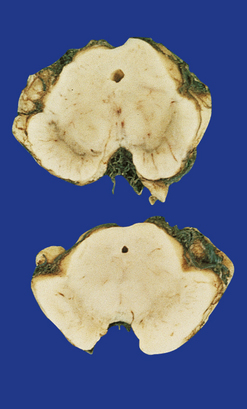
28.1 Substantia nigra in Parkinson’s disease.
Sections through midbrain of normal control (top) and patient with PD (bottom), showing the abnormal pallor of the substantia nigra in PD.
 LEWY BODIES
LEWY BODIESThe three major pathologic forms of α-synuclein-containing inclusions are:
 Neuronal Lewy bodies and Lewy neurites (Figs 28.4, 28.5).
Neuronal Lewy bodies and Lewy neurites (Figs 28.4, 28.5).
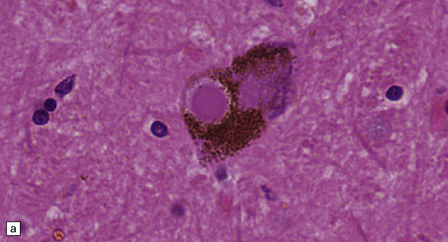
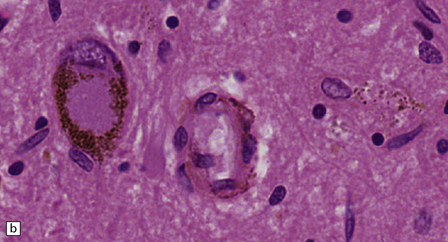
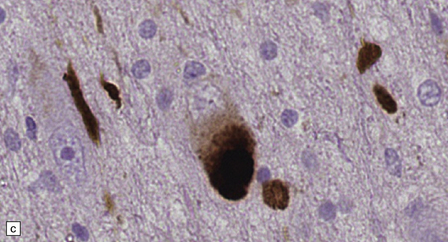
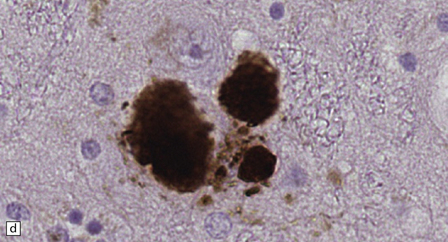
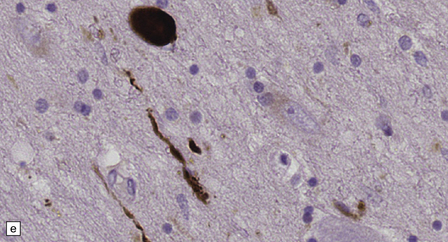
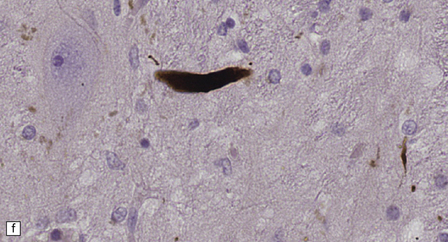
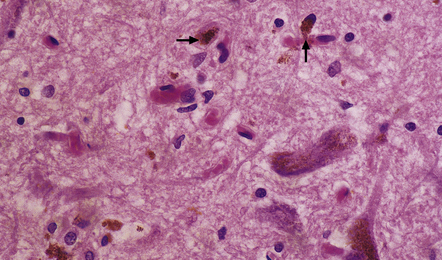
28.4 Lewy bodies in the substantia nigra.
(a) Classical Lewy body with a hyaline eosinophilic core and a pale halo. The core may comprise concentric lamellae of differing staining intensity. (b) A pale body: a round zone of finely granular or homogeneous, weakly eosinophilic material displacing the neuromelanin. In contrast to Lewy bodies, pale bodies do not have a halo. (c) Classical Lewy body with an intensely labeled core and lighter, granular staining in the halo. The nucleus of the neuron is visible above the Lewy body. Several Lewy neurites are also labeled. (d) Occasionally, aggregated or multiple Lewy bodies are present in the cytoplasm. (e) Beaded Lewy neurites. A classical Lewy body is also present, towards the top of the image. (f) Lewy body away from the perikaryon.
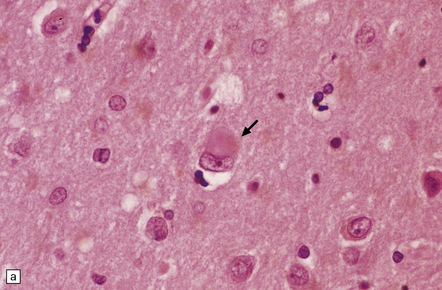
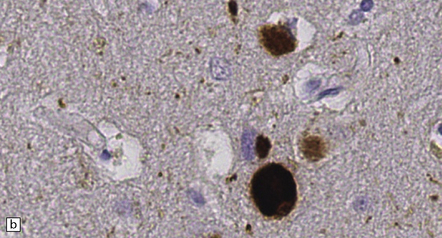
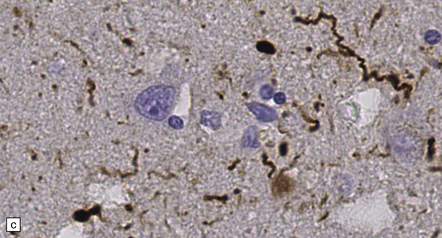
28.5 Cortical Lewy bodies.
These are homogeneous eosinophilic structures that have an ill-defined edge and usually lack an obvious halo (arrow). (a) Typically, a cortical Lewy body appears as a round eosinophilic inclusion which pushes the nucleus to one side of the cell. The nuclei of affected neurons usually appear somewhat vesicular with a prominent nucleolus. (b) α-synuclein immunostaining: cortical Lewy body with an intensely labeled core. Note too, the dot-like immunopositivity in the neuropil, often present, particularly in patients with many neocortical Lewy bodies. (c) α-synuclein immunostaining of cortical Lewy neurites.


Functional proteins that make up the structure of the Lewy body:
 α-synuclein (seen in all Lewy bodies). Mutations in the gene for α-synuclein, SNCA, cause some familial cases of PD.
α-synuclein (seen in all Lewy bodies). Mutations in the gene for α-synuclein, SNCA, cause some familial cases of PD.
 Neurofilament proteins, which form the cytoskeleton of the inclusion.
Neurofilament proteins, which form the cytoskeleton of the inclusion.


Incorporated proteins, probably in the process of being degraded:



Immunohistochemical staining is a sensitive way of detecting cortical Lewy bodies. Antibodies to α-synuclein are most sensitive but antibodies to p62 or ubiquitin are also useful. Pale bodies occur in neurons of the substantia nigra and locus ceruleus (Fig. 28.4b) and have a similar immunohistochemical profile to that of Lewy bodies. Although not always associated with Lewy bodies, the latter should be carefully sought if pale bodies are present. Pale bodies may represent precursors of Lewy bodies.
MICROSCOPIC APPEARANCES
The substantia nigra and other pigmented brain stem nuclei show:

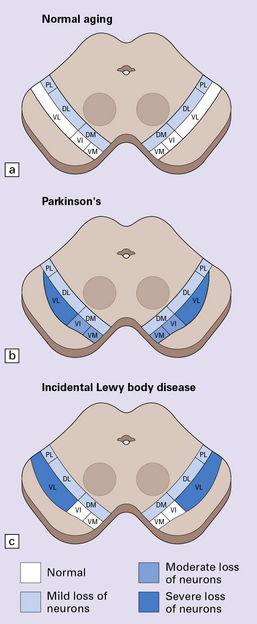
28.2 Cell loss from the substantia nigra is not random, but occurs in a region-specific manner.
The pars compacta of the substantia nigra can be divided into ventral and dorsal tiers, which project to different brain areas, and each tier can be further subdivided into regions (medial to lateral). (a) In normal aging, the estimated rate of cell loss from the dorsal tier of the substantia nigra is 7% per decade, leading to 40–50% cell loss by 65 years of age. (b) In PD, cell loss is greatest in the ventrolateral tier (VL). Typically, 70–90% have been lost by the time a patient dies. The ventromedial tier (VM) is next most affected. Cell loss from the dorsal tier is not significantly different from that in normal aging. (c) It has been suggested that symptoms of PD occur only after 50% of ventral tier neurons have been lost. This is preceded by a subclinical phase that can be regarded as incidental Lewy body disease, in which there is less pronounced cell loss, largely confined to the ventrolateral tier (VL). The age-specific prevalence of Lewy bodies rises from 3.8% to 12.8% between the 6th and 9th decades.
 accumulation of neuromelanin in macrophages (Figs 28.6, 28.7)
accumulation of neuromelanin in macrophages (Figs 28.6, 28.7)
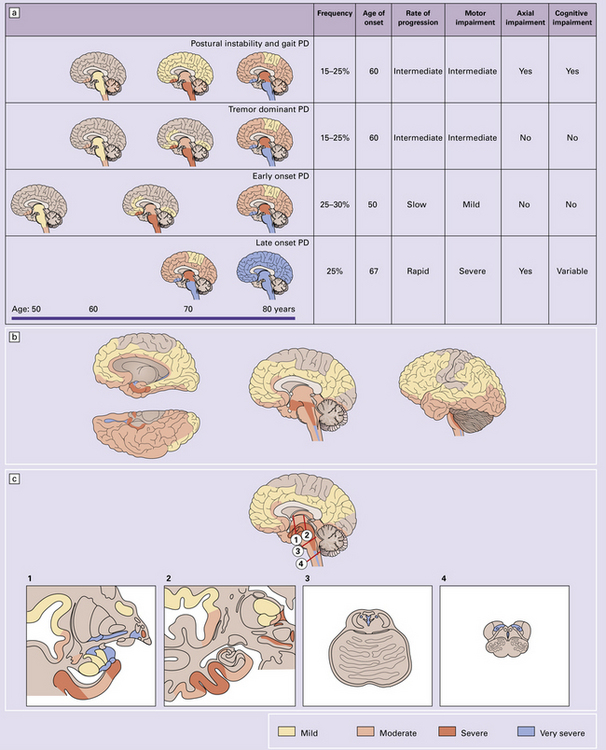
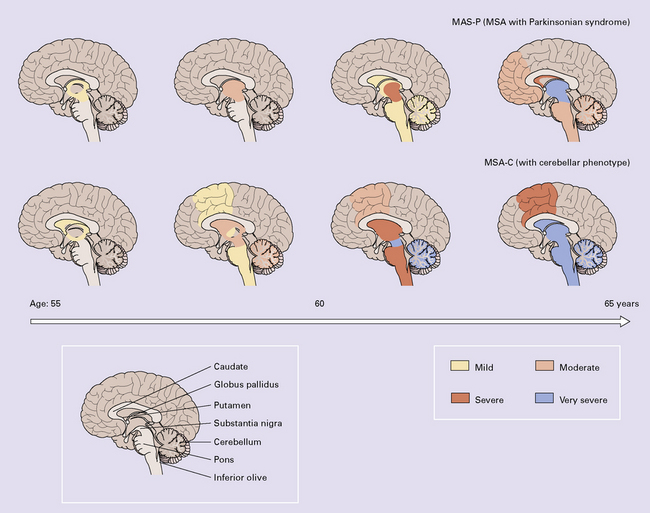
28.3 α-Synuclein pathology in Parkinson’s disease. (a)
Frequency and clinical features of the four major types phenotypes of PD. Brain schematics show increasing severity with age (blue bar). (b,c) Anatomic distribution of α-synuclein pathology. (Adapted from Halliday GM et al. 2011. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol 122(2):187–204 and Braak H et al. 2003. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211.)
28.6 Phagocytosis of neuromelanin.
Neuromelanin from neurons that have degenerated is taken up into macrophages (arrows).
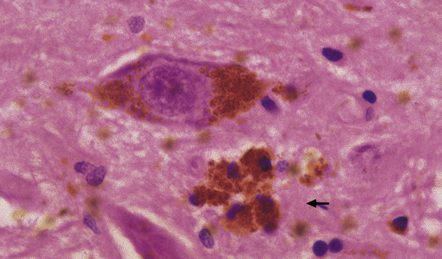
28.7 Phagocytosis of neuromelanin.
A cluster of pigment-laden macrophages (arrow) marks the site of degeneration of a nigral neuron.

 Lewy bodies and pale bodies (see Fig. 28.5) in some remaining neurons
Lewy bodies and pale bodies (see Fig. 28.5) in some remaining neurons

A distinctive form of neuritic degeneration (Lewy neurites), demonstrable by immunostaining for α-synuclein or ubiquitin, but not by silver impregnation, occurs in Lewy body diseases, including PD (see Figs 28.4, 28.5). The Lewy neurites may be detected in the substantia nigra, CA2/3 region of the hippocampus, dorsal motor nucleus of the vagus, nucleus basalis of Meynert, and amygdala.
DRUG AND TOXIN-RELATED PARKINSONISM
Other causes of drug- and toxin-related parkinsonism are described in Chapter 25.
PROGRESSIVE SUPRANUCLEAR PALSY (PSP) (STEELE–RICHARDSON–OLSZEWSKI SYNDROME)
The cause of PSP is not known, but the disease is strongly associated with the H1 haplotype of MAPT, the tau gene (this haplotype is also associated with CBD, see below). In the brain of patients with PSP, as in CBD (and also argyrophilic grain disease, see Chapter 31), four-repeat tau predominates, i.e. tau that is synthesized from transcripts that include exon 10 and therefore encode four microtubule-binding domains rather than three. Approximately 1–8% of patients diagnosed clinically as having PD have PSP.
MACROSCOPIC APPEARANCES
There is loss of pigment from the substantia nigra and locus ceruleus (Fig. 28.8) and, occasionally, atrophy of the midbrain, pontine tegmentum, and globus pallidus.
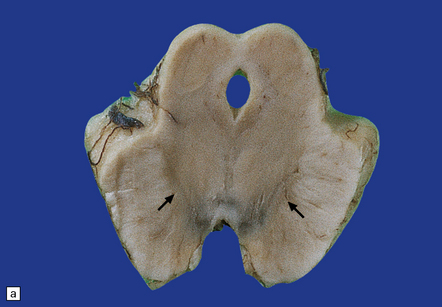
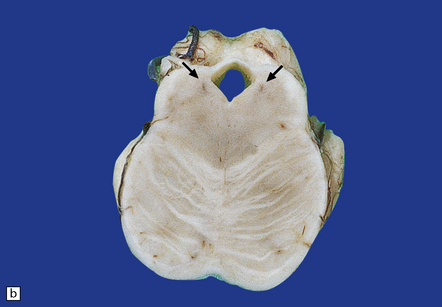
28.8 PSP.
(a) Atrophy of the midbrain and pallor of the substantia nigra (arrows). (b) Pallor of the locus ceruleus (arrows).
 PROGRESSIVE SUPRANUCLEAR PALSY
PROGRESSIVE SUPRANUCLEAR PALSY



MICROSCOPIC APPEARANCES
Certain abnormalities are common to several regions of the CNS (Fig. 28.9):
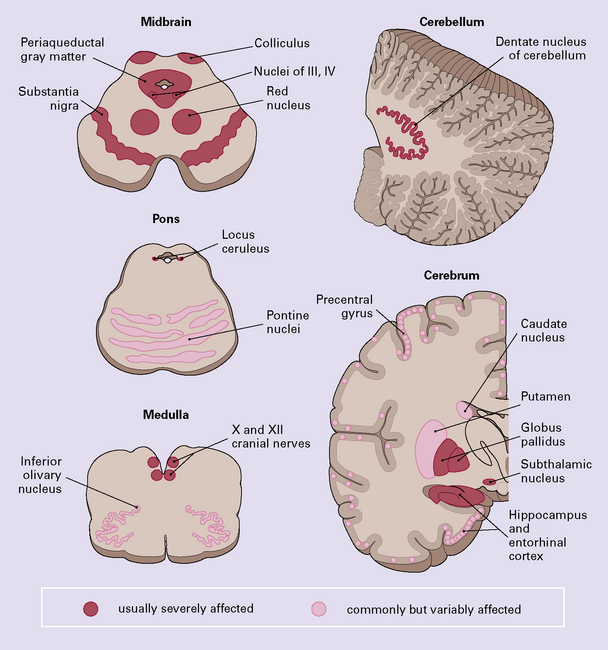
28.9 Pathologic changes in PSP.
The areas affected in PSP can be divided into those that are consistently and severely affected, and those that are less consistently affected.
 Neuronal accumulation of abnormal tau protein (Fig. 28.10), either diffusely distributed and detectable only immunohistochemically, or aggregated into neurofibrillary tangles, many of which are also demonstrable by silver impregnation. The tangles stain poorly for ubiquitin.
Neuronal accumulation of abnormal tau protein (Fig. 28.10), either diffusely distributed and detectable only immunohistochemically, or aggregated into neurofibrillary tangles, many of which are also demonstrable by silver impregnation. The tangles stain poorly for ubiquitin.
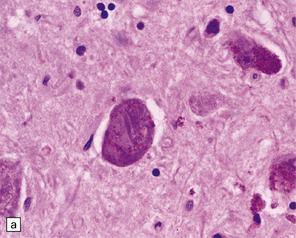
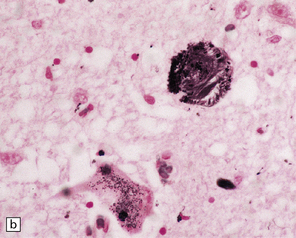
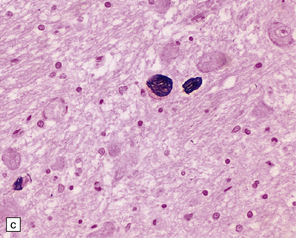
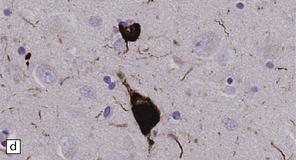
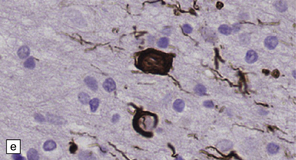
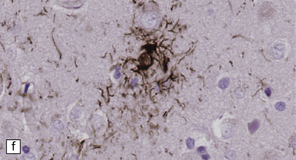
28.10 Types of tau-positive inclusion in PSP as demonstrated by labeling with the AT8 antibody.
(a) Typical basophilic, rounded or globose tangle in the substantia nigra. (b) Gallyas silver impregnation is a sensitive method of detecting the tangles, as in this section of substantia nigra. (c) Tangles in the pontine nuclei. (d) Cortical neurons containing tau-positive tangles. Scattered neurites are also labeled. (e) Oligodendroglial tau inclusions (coiled bodies and interfascicular threads) in the subcortical white matter. (f) Tufted astrocytes in the cortex. This form of astrocytic tau is specific for PSP and is distinct from the astrocytic plaques of CBD.
 Glial accumulation of tau protein. Tufted astrocytes seen in gray matter are especially characteristic of PSP (Fig. 28.10f).
Glial accumulation of tau protein. Tufted astrocytes seen in gray matter are especially characteristic of PSP (Fig. 28.10f).

The findings vary in different regions of the CNS:
 In the cerebral cortex there are commonly neuronal tangles, tau-immunoreactive glia, and neuropil threads, particularly in the precentral gyrus, entorhinal cortex, and hippocampus. An occasional neuron in the cerebral cortex and basal ganglia may appear swollen and achromasic. An abundance of swollen neurons suggests corticobasal degeneration (CBD).
In the cerebral cortex there are commonly neuronal tangles, tau-immunoreactive glia, and neuropil threads, particularly in the precentral gyrus, entorhinal cortex, and hippocampus. An occasional neuron in the cerebral cortex and basal ganglia may appear swollen and achromasic. An abundance of swollen neurons suggests corticobasal degeneration (CBD).





Criteria for pathologic diagnosis of PSP have been proposed (Table 28.5).
Table 28.5

 DIFFERENTIAL DIAGNOSIS OF PROGRESSIVE SUPRANUCLEAR PALSY
DIFFERENTIAL DIAGNOSIS OF PROGRESSIVE SUPRANUCLEAR PALSY
 Supranuclear palsy may be associated with several other neurologic diseases, including vascular disease, Lewy body disease, corticobasal degeneration (CBD), and Creutzfeldt–Jakob disease.
Supranuclear palsy may be associated with several other neurologic diseases, including vascular disease, Lewy body disease, corticobasal degeneration (CBD), and Creutzfeldt–Jakob disease.


• Postencephalitic parkinsonism: the distribution of tangles is similar to that in PSP. The clinical picture and history of encephalitis are important distinguishing features. Rarely, parkinsonian patients who have no history of encephalitis are found to have tangles restricted to the substantia nigra and locus ceruleus (tangle-only parkinsonism).
• CBD: the inclusions are confined to the superficial part of the cerebral cortex and the locus ceruleus and substantia nigra, the medial part of which is relatively spared. Finding many swollen neurons in the cerebral cortex also favors a diagnosis of CBD.
• Alzheimer’s disease: tangles may occur in the basal nuclei and brain stem, but are also present in a characteristic distribution in the cerebral cortex where they are accompanied by neuritic plaques (see Chapter 32).
POSTENCEPHALITIC PARKINSONISM
 DIFFERENTIAL DIAGNOSIS OF POSTENCEPHALITIC PARKINSONISM
DIFFERENTIAL DIAGNOSIS OF POSTENCEPHALITIC PARKINSONISM
 There is considerable pathologic overlap between postencephalitic parkinsonism, PSP, and Guamanian parkinsonism, and they may be indistinguishable on histologic grounds alone. Rarely, tangles confined to the substantia nigra and locus ceruleus occur in parkinsonian patients with no history of encephalitis (tangle-only parkinsonism).
There is considerable pathologic overlap between postencephalitic parkinsonism, PSP, and Guamanian parkinsonism, and they may be indistinguishable on histologic grounds alone. Rarely, tangles confined to the substantia nigra and locus ceruleus occur in parkinsonian patients with no history of encephalitis (tangle-only parkinsonism).

 POSTENCEPHALITIC PARKINSONISM
POSTENCEPHALITIC PARKINSONISM

MACROSCOPIC AND MICROSCOPIC APPEARANCES
Histologically, the substantia nigra is gliotic and severely depleted of neurons (Fig. 28.11), the locus ceruleus moderately so. A mononuclear inflammatory infiltrate is typically present, but this tends to be very sparse in long-surviving patients. Neurofibrillary tangles composed of abnormal tau protein and biochemically and ultrastructurally identical to those in Alzheimer’s disease (see Chapter 31), are present in the remaining neurons in the substantia nigra, and also in the locus ceruleus, hippocampus, and the entorhinal, temporal, frontal, parietal, and insular cortex. There are tau-immunoreactive glial cells in the affected regions.
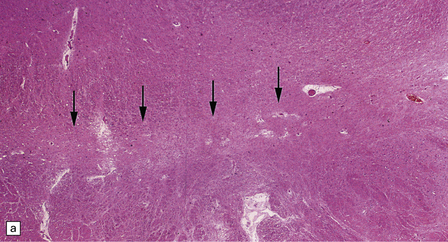
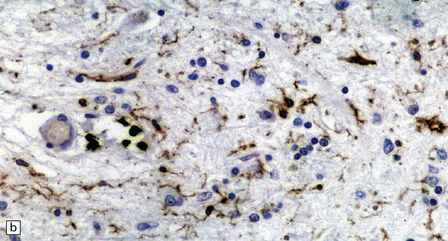
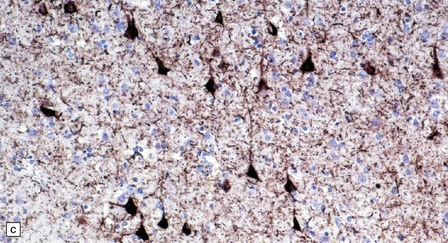
28.11 Postencephalitic parkinsonism.
(a) The substantia nigra (arrows) is severely depleted of neurons. (b) A higher-magnification view of the substantia nigra, showing sparse perivascular lymphocytes, and scattered lymphocytes and microglia within the parenchyma. (c) The medial temporal cortex contains numerous tangle-bearing neurons and neuropil threads that can be immunostained for tau with monoclonal antibody AT8.
MULTIPLE SYSTEM ATROPHY (MSA)



 MULTIPLE SYSTEM ATROPHY
MULTIPLE SYSTEM ATROPHY



 CLINICOPATHOLOGIC CORRELATIONS IN MSA
CLINICOPATHOLOGIC CORRELATIONS IN MSA
 Nigrostriatal degeneration causes parkinsonism that may be responsive to levodopa in the early stages.
Nigrostriatal degeneration causes parkinsonism that may be responsive to levodopa in the early stages.
 Degeneration of pontine nuclei and Purkinje cells results in progressive cerebellar ataxia.
Degeneration of pontine nuclei and Purkinje cells results in progressive cerebellar ataxia.

 Atrophy of the posterior cricoarytenoid (abductor) muscles causes laryngeal stridor (Fig. 28.12).
Atrophy of the posterior cricoarytenoid (abductor) muscles causes laryngeal stridor (Fig. 28.12).
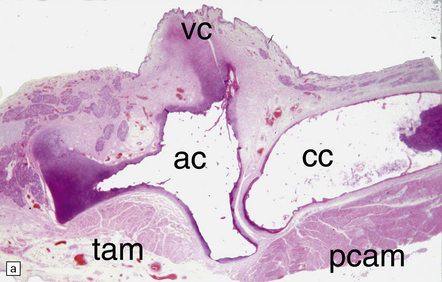
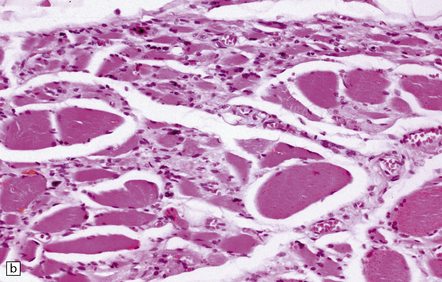
28.12 Neurogenic atrophy of posterior cricoarytenoid muscle in MSA.
(a) Parasagittal section through larynx from a patient with MSA who was found dead unexpectedly. The section includes the arytenoid and cricoid cartilages (ac and cc), posterior attachment of the vocal cord (vc), transverse arytenoid muscle (tam) and posterior cricoarytenoid muscle (pcam). (b) A higher magnification view of the posterior cricoarytenoid muscle shows it to be fibrotic and to contain sheets of severely atrophic fibers with only scattered fibers of normal or near-normal caliber. Other laryngeal muscles were less severely affected. Laryngeal obstruction due to weakened abduction of the vocal cords is a common cause of sudden death in patients with MSA. (Courtesy of Dr Simon Rose, Royal United Hospital, Bath, UK.)
 Loss of neurons from Onufrowicz nucleus leads to sphincter disturbances.
Loss of neurons from Onufrowicz nucleus leads to sphincter disturbances.
 Frontal lobe dysfunction and dementia are related to involvement of the cerebral cortex.
Frontal lobe dysfunction and dementia are related to involvement of the cerebral cortex.
 CRITERIA FOR POSSIBLE MSA
CRITERIA FOR POSSIBLE MSAA sporadic, progressive, adult–onset (>30 years) disease characterized by:
1. Parkinsonism (bradykinesia with rigidity, tremor, or postural instability) or
2. A cerebellar syndrome (gait ataxia with cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction) and
3. At least one feature suggesting autonomic dysfunction (otherwise unexplained urinary urgency, frequency or incomplete bladder emptying, erectile dysfunction in males, or significant orthostatic blood pressure decline that does not meet the level required in probable MSA) and


Possible MSA-P (with predominant parkinsonism)
 Rapidly progressive parkinsonism.
Rapidly progressive parkinsonism.

 Postural instability within 3 years of motor onset.
Postural instability within 3 years of motor onset.
 Gait ataxia, cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction.
Gait ataxia, cerebellar dysarthria, limb ataxia, or cerebellar oculomotor dysfunction.
 Dysphagia within 5 years of motor onset.
Dysphagia within 5 years of motor onset.
 Atrophy on MRI of putamen, middle cerebellar peduncle, pons, or cerebellum.
Atrophy on MRI of putamen, middle cerebellar peduncle, pons, or cerebellum.
 Hypometabolism on FDG-PET in putamen, brain stem, or cerebellum.
Hypometabolism on FDG-PET in putamen, brain stem, or cerebellum.




Gilman et al. (2008).
MACROSCOPIC APPEARANCES
Depending on the systems involved, there may be:
 atrophy of the cerebellum, middle cerebellar peduncles, and pons (Fig. 28.13)
atrophy of the cerebellum, middle cerebellar peduncles, and pons (Fig. 28.13)
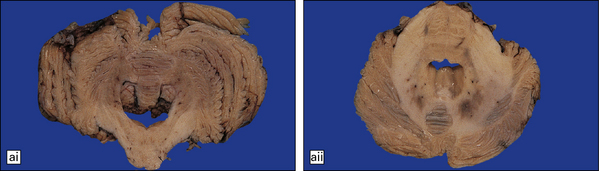
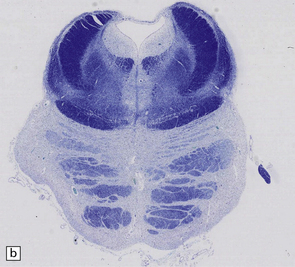
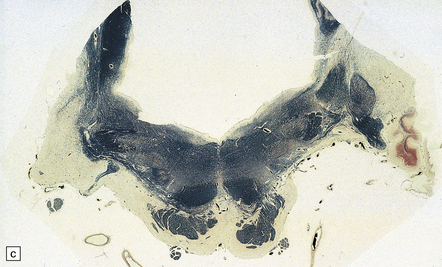
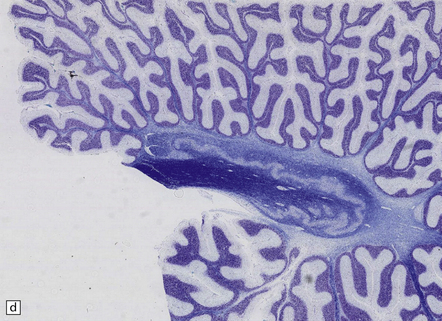
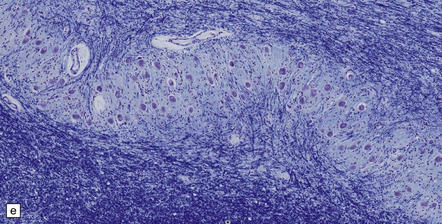
28.13 MSA-OPCA pattern.
Horizontal sections through the pons and medulla of a patient with OPCA pattern of MSA (ai) and a normal control (aii). In OPCA there may be marked atrophy of the cerebellum, middle cerebellar peduncles, and base of the pons, the last two features giving the pons a ‘beaked’ appearance. (b) Marked atrophy of the base of the pons with loss of transverse pontine fibers. The superior cerebellar peduncles are preserved. (c) Loss of pontocerebellar fibers, but preservation of the pontine tegmentum and superior cerebellar peduncles. (d) Atrophy of the olivary nucleus sin MSA. At higher magnification (e) the neuronal loss and a gliosis become visible.
 pallor of the substantia nigra and locus ceruleus (Fig. 28.14)
pallor of the substantia nigra and locus ceruleus (Fig. 28.14)
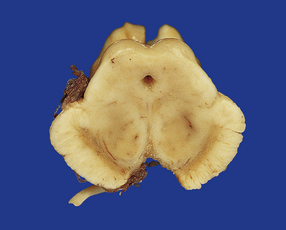
28.14 Striatonigral-type MSA.
Pallor of the substantia nigra in MSA, reflecting the loss of pigmented neurons. The loss of nigral neurons contributes in part to the extrapyramidal features of MSA.
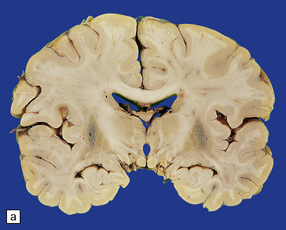
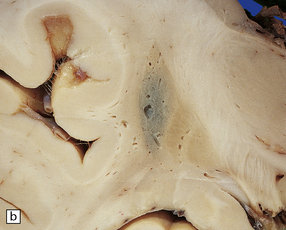
 atrophy and gray-brown discoloration of the putamen (Fig. 28.15).
atrophy and gray-brown discoloration of the putamen (Fig. 28.15).
MICROSCOPIC APPEARANCES
Glial cytoplasmic inclusions (Fig. 28.16)
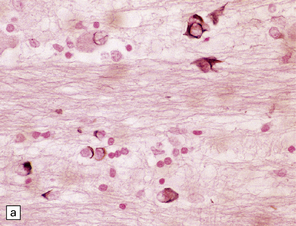
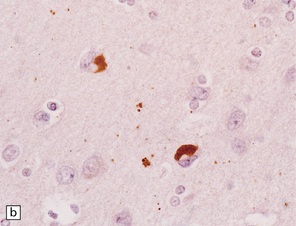
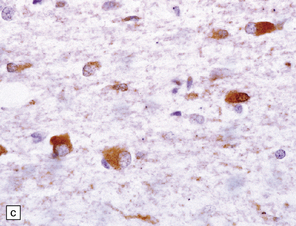
28.16 Glial cytoplasmic inclusions in MSA.
(a) Flame- or sickle-shaped glial cytoplasmic inclusions. (b) The glial cytoplasmic inclusions are immunoreactive for α-synuclein. (c) Glial cytoplasmic inclusions immunoreactive for ubiquitin.







Neuronal cytoplasmic inclusions
 Rounded filamentous structures, predominantly seen in the pontine nuclei, putamen, subthalamic nucleus, amygdala, hippocampus, dentate fascia, substantia nigra, inferior olivary nucleus, and the brain stem reticular formation.
Rounded filamentous structures, predominantly seen in the pontine nuclei, putamen, subthalamic nucleus, amygdala, hippocampus, dentate fascia, substantia nigra, inferior olivary nucleus, and the brain stem reticular formation.
 Demonstrable by silver impregnation (Gallyas is particularly sensitive).
Demonstrable by silver impregnation (Gallyas is particularly sensitive).
 Immunoreactive for α-synuclein and ubiquitin.
Immunoreactive for α-synuclein and ubiquitin.

 Present in large numbers in the basis pontis and putamen only.
Present in large numbers in the basis pontis and putamen only.
Nuclear inclusions
 Sparse in most cases, these inclusions are seen predominantly in the basis pontis and putamen.
Sparse in most cases, these inclusions are seen predominantly in the basis pontis and putamen.
 Demonstrable in neuronal and glial nuclei by Gallyas silver impregnation as well as α-synuclein immunohistochemistry (Fig. 28.17).
Demonstrable in neuronal and glial nuclei by Gallyas silver impregnation as well as α-synuclein immunohistochemistry (Fig. 28.17).
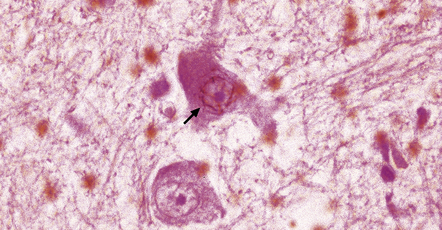
 Evident as a web of fine fibrils beneath the nuclear membrane in neurons (Fig. 28.18) and as small rods in glia.
Evident as a web of fine fibrils beneath the nuclear membrane in neurons (Fig. 28.18) and as small rods in glia.
Neuropil threads
 Numerous only in the basis pontis and putamen.
Numerous only in the basis pontis and putamen.
 Immunoreactive for α-synuclein and ubiquitin, but not tau-protein.
Immunoreactive for α-synuclein and ubiquitin, but not tau-protein.
 DIFFERENTIAL DIAGNOSIS OF MSA
DIFFERENTIAL DIAGNOSIS OF MSA MSA enters the differential diagnosis of parkinsonism, cerebellar ataxia, and autonomic failure.
MSA enters the differential diagnosis of parkinsonism, cerebellar ataxia, and autonomic failure.
 An OPCA pattern of atrophy is often found in inherited or sporadic spinocerebellar ataxias.
An OPCA pattern of atrophy is often found in inherited or sporadic spinocerebellar ataxias.
 Shy–Drager syndrome can be caused by Lewy body pathology.
Shy–Drager syndrome can be caused by Lewy body pathology.
 Multiple system degeneration may occur in association with the inclusions of motor neuron disease.
Multiple system degeneration may occur in association with the inclusions of motor neuron disease.
CORTICOBASAL DEGENERATION (CBD)
CBD is one of the tauopathies. Other terms for the same disorder include: corticodentatonigral degeneration with neuronal achromasia, cortical–basal ganglionic degeneration, and corticonigral degeneration. Along with other tau disorders it is now recognized that some cases are associated with mutations in the tau gene. However, most are sporadic, although as in PSP there is a strong genetic association with the H1 tau gene haplotype. In the brain in corticobasal degeneration, four-repeat tau predominates, i.e. tau synthesized from transcripts that contain exon 10 and therefore include four microtubule-binding domains rather than three. This is as occurs in progressive supranuclear palsy and also argyrophilic grain disease (see Chapter 31).
MACROSCOPIC APPEARANCES
 CORTICOBASAL DEGENERATION
CORTICOBASAL DEGENERATION Average age of onset is approximately 60 years.
Average age of onset is approximately 60 years.
 Three phases of illness can usually be distinguished:
Three phases of illness can usually be distinguished:
• Early phase (years 1–3): asymmetric clumsiness, stiffness, or jerking of an arm or leg
• Middle phase (years 3–5): dystonic rigidity and akinesia of the limbs, ‘alien limb’ phenomenon, lower limb apraxia, pyramidal deficits, and cortical sensory disturbances
• Late phase (years 5–8): cognitive dysfunction and frontotemporal neurobehavioral disorder.


 DIFFERENTIAL DIAGNOSIS OF CBD
DIFFERENTIAL DIAGNOSIS OF CBDProminent swollen cortical neurons are a feature of several neurodegenerative diseases.
 Large numbers of prominent swollen cortical neurons can be seen in Pick’s disease with Pick bodies, in CBD, and occasional cases of Creutzfeldt–Jakob disease.
Large numbers of prominent swollen cortical neurons can be seen in Pick’s disease with Pick bodies, in CBD, and occasional cases of Creutzfeldt–Jakob disease.
 Small numbers of prominent swollen cortical neurons may be seen in dementia with Lewy bodies, Alzheimer’s disease, and FTLD-MND dementia (see Chapter 31).
Small numbers of prominent swollen cortical neurons may be seen in dementia with Lewy bodies, Alzheimer’s disease, and FTLD-MND dementia (see Chapter 31).
 Very occasional swollen cortical neurons are seen in PSP and Nasu–Hakola disease (membranous lipodystrophy, see Chapter 33).
Very occasional swollen cortical neurons are seen in PSP and Nasu–Hakola disease (membranous lipodystrophy, see Chapter 33).
There is considerable overlap in the distribution of basal and cortical tau-related pathology between CBD, PSP and disease caused by mutation in the tau gene (FTDP-17, see Chapter 31). The presence of large numbers of swollen cortical neurons, sparing of the cerebellar dentate nucleus, and a paucity of tangles in the pons and medulla favor a diagnosis of CBD rather than PSP. Mutational analysis of the tau gene is appropriate in familial cases.
MICROSCOPIC APPEARANCES
In affected cortical areas
 Neuronal loss and astrogliosis, leading to thinning of the cortical ribbon (Fig. 28.19).
Neuronal loss and astrogliosis, leading to thinning of the cortical ribbon (Fig. 28.19).
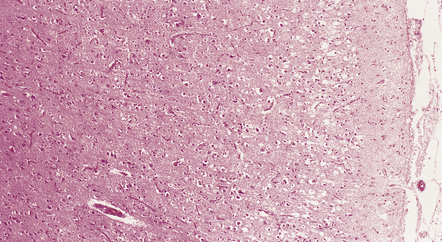
28.19 CBD showing cortical microvacuolation.
The microvacuolation is initially restricted to the superficial cortical layers. Later it affects all layers and is associated with marked astrocytic gliosis.
 Swollen or ballooned neurons (BN) are scattered in third, fifth, and sixth cortical layers (Fig. 28.20). BN are eosinophilic or amphophilic and are often vacuolated. They may be weakly argyrophilic, and show loss of Nissl substance (best demonstrated by staining with cresyl violet). BN occasionally contain granulovacuolar bodies.
Swollen or ballooned neurons (BN) are scattered in third, fifth, and sixth cortical layers (Fig. 28.20). BN are eosinophilic or amphophilic and are often vacuolated. They may be weakly argyrophilic, and show loss of Nissl substance (best demonstrated by staining with cresyl violet). BN occasionally contain granulovacuolar bodies.
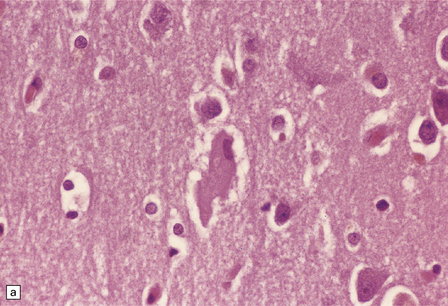
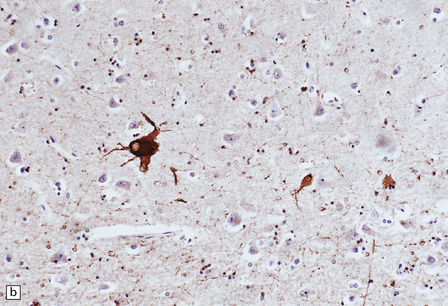
28.20 Swollen cortical neurons (also termed achromasic or ballooned neurons) are a distinctive finding in CBD.
(a) They are easily detected in hematoxylin and eosin-stained sections and are most frequent in layers III, IV, and VI, in the peri-Rolandic, posterior frontal, and parietal cortices. In advanced cases such swollen neurons may be seen in the insular and frontal cortex and the tip of the temporal lobe. (b) Cortex stained to show αB-crystallin. The swollen cells strongly express phosphorylated neurofilament proteins and αB-crystallin. Here immunostaining of αB-crystallin emphasizes the distortion of the axons and dendrites of the swollen neurons.
 BN are immunoreactive for phosphorylated neurofilament proteins, αB-crystallin, heat shock protein 27, and sometimes ubiquitin. They do not contain Alzheimer-type tangles but often show diffuse tau positivity, particularly at the periphery of the cytoplasm (Fig. 28.21).
BN are immunoreactive for phosphorylated neurofilament proteins, αB-crystallin, heat shock protein 27, and sometimes ubiquitin. They do not contain Alzheimer-type tangles but often show diffuse tau positivity, particularly at the periphery of the cytoplasm (Fig. 28.21).
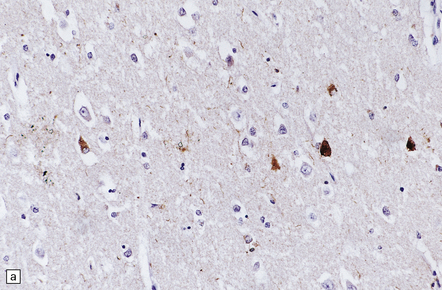
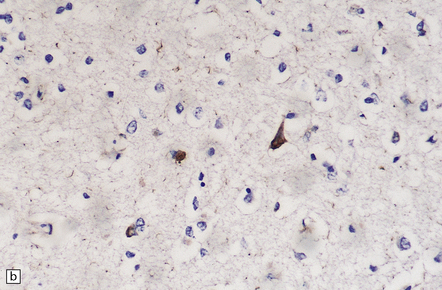
28.21 CBD cortical inclusion bodies.
(a,b) These are sections from superficial cortex immunostained to show tau-proteins. The tau-immunoreactive globular or angular inclusions (which are not ubiquitinated) are present in small numbers of neurons in the superficial cortex. The inclusions occur in residual neurons in areas of superficial microvacuolation, but may not be detectable in severely gliotic cortex.



Patterns of tau immunoreactivity
 Diffuse or granular cytoplasmic immunoreactivity typical of so-called ‘pre-tangles’ (this is the pattern in most of the tau-positive neurons).
Diffuse or granular cytoplasmic immunoreactivity typical of so-called ‘pre-tangles’ (this is the pattern in most of the tau-positive neurons).
 Densely-packed small tau-positive inclusions reminiscent of Pick bodies or small NFT.
Densely-packed small tau-positive inclusions reminiscent of Pick bodies or small NFT.
 Dispersed or skein-like cytoplasmic filamentous staining.
Dispersed or skein-like cytoplasmic filamentous staining.

 Many fine, tau-immunopositive cell processes within the neuropil (it is incorrect to use the term neuropil threads in the context of CBD, as the processes are astrocytic) (Fig. 28.22).
Many fine, tau-immunopositive cell processes within the neuropil (it is incorrect to use the term neuropil threads in the context of CBD, as the processes are astrocytic) (Fig. 28.22).
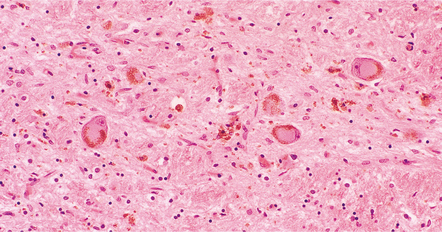
28.22 Nigral neuronal loss and astrocytic gliosis in CBD.
Argyrophilic filamentous inclusions termed ‘corticobasal inclusions’ are present in residual nigral neurons. In H&E-stained sections the inclusions are moderately to weakly basophilic and displace the neuromelanin. Similar inclusions may be seen in the locus ceruleus. The inclusions are immunoreactive for tau-protein. Ultrastructurally, some consist of paired helical filaments, while others are composed of straight tubules and resemble the tangles of PSP.
 Tau-positive argyrophilic inclusions in oligodendroglia (termed oligodendroglial microtubular masses, or coiled bodies); these tau-positive oligodendroglial inclusion differ from the flame-shaped α-synuclein-positive oligodendroglial inclusions in MSA (Fig. 28.23).
Tau-positive argyrophilic inclusions in oligodendroglia (termed oligodendroglial microtubular masses, or coiled bodies); these tau-positive oligodendroglial inclusion differ from the flame-shaped α-synuclein-positive oligodendroglial inclusions in MSA (Fig. 28.23).
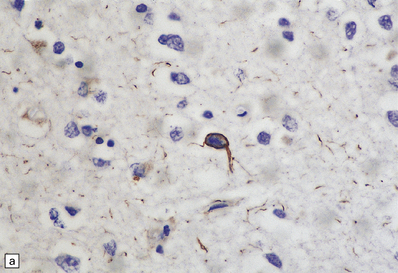
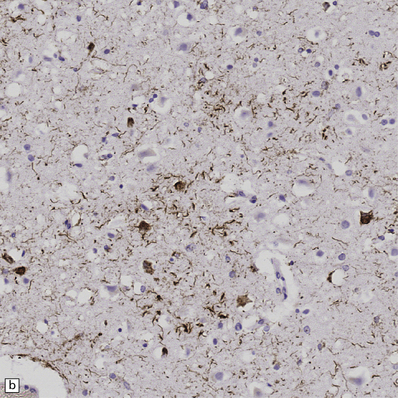
28.23 Tau-immunoreactive glial inclusions in CBD.
(a) Glial inclusions which are immunoreactive for tau protein are present in large numbers in CBD. Most appear as coils and grains as shown here. These can also be detected by Gallyas silver impregnation. (b) Some glial tau-reactive inclusions resemble those seen in MSA but are present in low density and do not stain with α-synuclein.
 RECOMMENDED TISSUE AREAS FOR TISSUE SAMPLING IN CBD
RECOMMENDED TISSUE AREAS FOR TISSUE SAMPLING IN CBD














ARTERIOSCLEROTIC PSEUDOPARKINSONISM
 ARTERIOSCLEROTIC PSEUDOPARKINSONISM
ARTERIOSCLEROTIC PSEUDOPARKINSONISM


MACROSCOPIC AND MICROSCOPIC APPEARANCES
Pathologically, there are usually lacunar infarcts in the basal ganglia (Fig. 28.24) or ischemic lesions in the white matter of the frontal lobes (Fig. 28.25). Infarction of the substantia nigra is a very rare cause of parkinsonism.
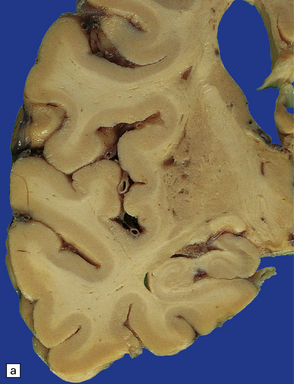
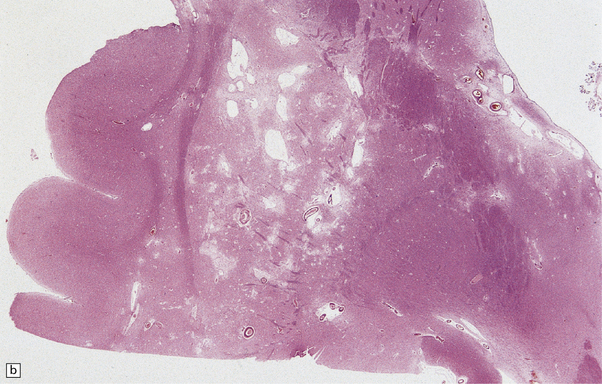
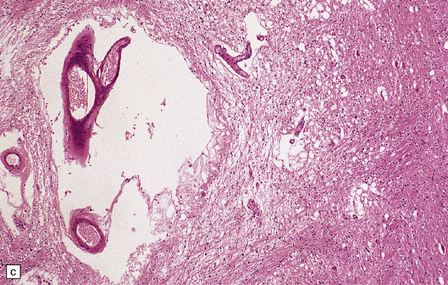
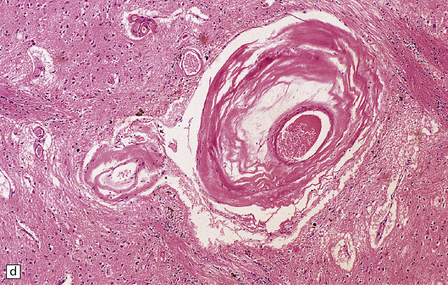
28.24 Arteriosclerotic pseudoparkinsonism.
(a) Multiple lacunar infarcts in the putamen. (b) Histology of basal ganglia shows dilated perivascular spaces with rarefaction of parenchyma. (c) Astrocytic gliosis is present around the dilated perivascular spaces. (d) Vessels in the basal ganglia are markedly arteriosclerotic.
GUAM PARKINSONISM-DEMENTIA
 GUAM PARKINSONISM-DEMENTIA
GUAM PARKINSONISM-DEMENTIA


MACROSCOPIC AND MICROSCOPIC APPEARANCES
The brain usually shows generalized atrophy, with pallor of the substantia nigra and locus ceruleus.
Histologically (Fig. 28.26), neuronal loss and astrocytic gliosis are associated with neurofibrillary tangles in:
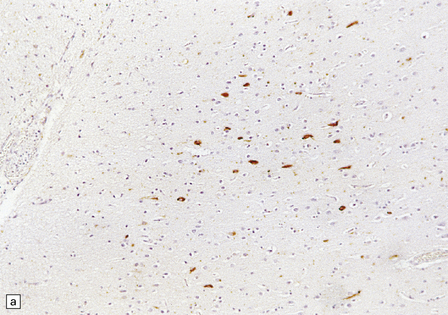

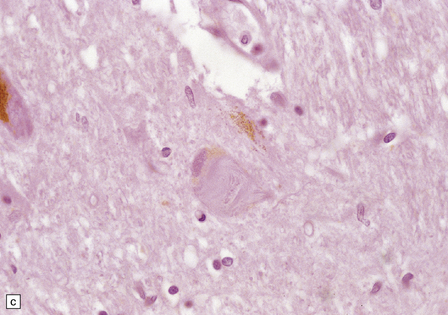
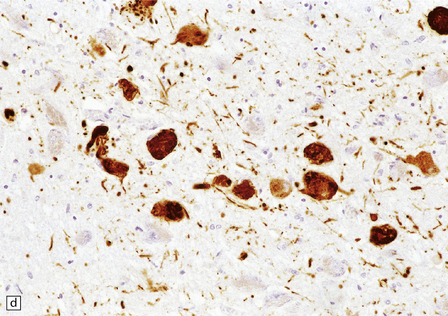
28.26 Guam parkinsonism–dementia.
(a) Tau-immunoreactive neurofibrillary tangles in the superficial laminae of the neocortex. (b) Silver impregnation of two adjacent gyri reveals numerous tangles in one and relatively few in the other. (c) Tangle in a remaining neuron in the substantia nigra. (d) Numerous tau-immunoreactive tangles and neurites in the oculomotor nucleus.








Aggregates of α-synuclein may be present in the amygdala.
 DISEASES CHARACTERIZED BY ABNORMAL STIFFNESS
DISEASES CHARACTERIZED BY ABNORMAL STIFFNESS
 An autoimmune disease that presents in the 4th and 5th decades and results from reduced GABAergic inhibition in the spinal cord with a resulting increase in the activity of spinal flexor reflex pathways.
An autoimmune disease that presents in the 4th and 5th decades and results from reduced GABAergic inhibition in the spinal cord with a resulting increase in the activity of spinal flexor reflex pathways.



The stiff man-plus syndrome generally presents in one of three patterns:
 Progressive encephalomyelitis with rigidity, in which there is severe muscle stiffness and axial rigidity with a progressive subacute course and development of multiple cranial nerve signs leading to death in about 3 years.
Progressive encephalomyelitis with rigidity, in which there is severe muscle stiffness and axial rigidity with a progressive subacute course and development of multiple cranial nerve signs leading to death in about 3 years.

 ‘Stiff limb syndrome’ clinically dominated by stiffness and spasm in one or more limbs.
‘Stiff limb syndrome’ clinically dominated by stiffness and spasm in one or more limbs.
Histology shows neuronal loss and lymphocytic infiltration in the brain stem and spinal cord.
NECROPSY IN CASES OF AKINETIC–RIGID SYNDROME
 The brain should be fixed intact for neuropathologic examination.
The brain should be fixed intact for neuropathologic examination.







 In akinetic–rigid disease with cognitive decline, the brain should be examined as for dementia (see Chapter 32), with particular emphasis on dementia with cortical Lewy bodies, Alzheimer’s disease, and CBD.
In akinetic–rigid disease with cognitive decline, the brain should be examined as for dementia (see Chapter 32), with particular emphasis on dementia with cortical Lewy bodies, Alzheimer’s disease, and CBD.
REFERENCES
Braak, H., Del Tredici, K., Rüb, U., et al. Del Tredici, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211.
Dickson, D.W., Braak, H., Duda, J.E., et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 2009;8(12):1150–1157.
Goker-Alpan, O., Schiffmann, R., LaMarca, M.E., et al. Parkinsonism among Gaucher disease carriers. J Med Genet.. 2004;41(12):937–940.
Halliday, G.M., Holton, J.L., Revesz, T., et al. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 2011;122(2):187–204.
Hardy, J. Genetic analysis of pathways to Parkinson disease. Neuron. 2010;68(2):201–206.
Hardy, J., Lewis, P., Revesz, T., et al. The genetics of Parkinson’s syndromes: a critical review. Curr Opin Genet Dev. 2009;19(3):254–265.
Houlden, H., Baker, M., Morris, H.R., et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology. 2001;56(12):1702–1706.
Jellinger, K.A. Neuropathological spectrum of synucleinopathies. Mov Disord. 2003;18(Suppl 6):S2–12.
Kosaka, K., Tsuchiya, K., Yoshimura, M. Lewy body disease with and without dementia: a clinicopathological study of 35 cases. Clin Neuropathol. 1988;7:299–305.
Lees, A.J., Hardy, J., Revesz, T. Parkinson’s disease. Lancet. 2009;373:2055–2066.
Polymeropoulos, M.H. Genetics of Parkinson’s disease. Ann N Y Acad Sci. 2000;920:28–32.
Spillantini, M.G., Schmidt, M.L., Lee, V.M., et al. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840.
Yoshino, H., Tomiyama, H., Tachibana, N., et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology. 2010;75(15):1356–1361.
Progressive supranuclear palsy
Houlden, H., Baker, M., Morris, H.R., et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology. 2001;56(12):1702–1706.
Komori, T. Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. Brain Pathol. 1999;9(4):663–679.
Liu, W.K., Le, T.V., Adamson, J., et al. Relationship of the extended tau haplotype to tau biochemistry and neuropathology in progressive supranuclear palsy. Ann Neurol. 2001;50(4):494–502.
Morris, H.R., Gibb, G., Katzenschlager, R., et al. Pathological, clinical and genetic heterogeneity in progressive supranuclear palsy. Brain. 2002;125(Pt 5):969–975.
Wakabayashi, K., Takahashi, H. Pathological heterogeneity in progressive supranuclear palsy and corticobasal degeneration. Neuropathology. 2004;24(1):79–86.
Geddes, J.F., Hughes, A.J., Lees, A.J., et al. Pathological overlap in cases of parkinsonism associated with neurofibrillary tangles. A study of recent cases of postencephalitic parkinsonism and comparison with progressive supranuclear palsy and Guamanian parkinsonism-dementia complex. Brain. 1993;116(Part 1):281–302.
Gilman, S., Wenning, G.K., Low, P.A., et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670–676.
Halliday, G.M., Holton, J.L., Revesz, T., et al. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 2011;122(2):187–204.
Jellinger, K.A. Neuropathological spectrum of synucleinopathies. Mov Disord. 2003;18(Suppl 6):S2–12.
Trojanowski, J.Q., Revesz, T. Neuropathology Working Group on MSA. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol. 2007;33(6):615–620.
Ahmed, Z., Doherty, K.M., Silveira-Moriyama, L., et al. Globular glial tauopathies (GGT) presenting with motor neuron disease or frontotemporal dementia: an emerging group of 4-repeat tauopathies. Acta Neuropathol. 2011;122(4):415–428.
Dickson, D.W., Bergeron, C., Chin, S.S., et al. Office of Rare Diseases’ neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61(11):935–946.
Forman, M.S., Schmidt, M.L., Kasturi, S., et al. Tau and alpha-synuclein pathology in amygdala of Parkinsonism-dementia complex patients of Guam. Am J Pathol. 2002;160(5):1725–1731.
Oyanagi, K., Makifuchi, T., Ohtoh, T., et al. Amyotrophic lateral sclerosis of Guam: the nature of the neuropathological findings. Acta Neuropathol. 1994;88(5):405–412.
Oyanagi, K., Tsuchiya, K., Yamazaki, M., et al. Substantia nigra in progressive supranuclear palsy, corticobasal degeneration, and parkinsonism-dementia complex of Guam: specific pathological features. J Neuropathol Exp Neurol. 2001;60(4):393–402.
Plato, C.C., Galasko, D., Garruto, R.M., et al. ALS and PDC of Guam: forty-year follow-up. Neurology. 2002;58(5):765–773.