Parenteral drug delivery
Robert Lowe
Chapter contents
Reasons for choosing parenteral administration
Routes of parenteral administration
Intravenous injections and infusions
Intra-arterial and intracardiac injections
Category-specific requirements
Absorption from injection sites
Key points
• Parenteral preparations are administered to a patient by injection.
• All parenteral products must be sterile.
• Parenteral preparations intended for multiple use must contain an antimicrobial preservative.
Introduction
In medicine and pharmacy, enteral administration is the term used to describe drug administration via the gastrointestinal tract. The majority of medicines are administered orally via this route in the form of tablets, capsules or liquids. The enteral route also encompasses rectal administration utilizing dosage forms such as suppositories, enemas or rectal ointment. In contrast to this, the term parenteral administration literally means any method of drug administration which does not utilize the gastrointestinal tract, such as by inhalation or application to the skin. In practice however, parenteral administration is commonly taken to mean drug administration by injection.
In this chapter, we will explore why the parenteral route of administration may be chosen by the clinician or the manufacturer of a medicine. The routes available for parenteral administration and the tissues, organs and anatomical spaces that can be accessed by injection are outlined. The various forms or types of parenteral product commonly manufactured are described and the pharmacopoeial standards for injectable products are discussed. The ingredients of formulated injectable products with regard to vehicles or solvents, excipients and preservatives are described along with physiological considerations, such as the pH and tonicity of the product prior to administration. Finally the containers, closures and primary packaging commonly used for parenteral products are discussed.
Reasons for choosing parenteral administration
The vast majority of patients would prefer to receive their medication as an oral tablet or liquid to swallow, or as a cream, ointment or transdermal patch to apply to the skin rather than receive treatment via injection, which can be painful or stressful (indeed some patients suffer from needle phobia). From a manufacturer’s point of view it is often simpler and much cheaper to prepare medicines such as tablets or liquids, particularly given the less stringent requirements for manufacturing premises for these non-sterile products, compared to the costs associated with manufacturing sterile medicines, such as injections, in highly specialized, controlled environments. There are, however, a number of clinical advantages associated with parenteral administration.
Many medicines are administered parenterally simply because the drug molecule itself would be rapidly broken down in the gastrointestinal tract and would thus become inactivated before it could be absorbed into the circulatory system. Good examples of this are aminoglycoside antibiotics, such as gentamicin. The injectable route may be chosen to provide a highly localized effect. This is particularly true when the injection route accesses a particular anatomical area or organ system. Examples of this include the injection of drugs, such as steroids, into joint spaces (intra-articular injection), intra-ocular injections to treat eye diseases or intrathecal injections where medicines are administered into the spinal column to deliver drugs into the cerebrospinal fluid, that otherwise might not accumulate sufficiently in this tissue to achieve the desired effect.
Intravenous injection delivers the drug directly into the circulatory system, where it is then rapidly distributed around the body. This is important clinically as the drug will rapidly produce an effect, whereas peak blood levels may not be achieved for one to two hours after a drug is administered orally. This rapid onset of action for an intravenously administered drug may be critical in emergency situations. Conversely, by choosing to administer a drug by intramuscular injection, the release of the medicine from the injection site into the circulation can be delayed and prolonged. Indeed, as will be seen later, by manipulation of the formulation of intramuscular injections it is possible to provide prolonged drug release allowing doses to be required at only once-monthly intervals. Finally, the intravenous route of injection is routinely used to administer medication to the unconscious patient who is unable to swallow. This route is also employed in conscious or unconscious patients if the gastrointestinal tract is not working. In this scenario, not only are medicines, but fluids for hydration and electrolyte replacement, plus all the nutrients, vitamins and trace elements normally obtained from a healthy diet supplied by parenteral nutrition provided intravenously.
Routes of parenteral administration
As noted above medicines are injected by many different routes and the choice of route is governed by the purpose of the treatment and the volume of medicine to be administered.
Intravenous injections and infusions
Intravenous (IV or i.v.) injections and infusions are administered into an easily accessible prominent vein near the surface of the skin, typically on the back of the hand or in the internal flexure of the elbow. The volumes administered can range from 1 mL for an intravenous injection, up to several litres for an intravenous infusion. Medicines administered by intravenous injection (or intravenous bolus dose) will rapidly increase the concentration of the drug in the plasma and produce a rapid effect. If the medicine is first added into a large volume of fluid (500 mL to 1 L infusion bag) and then administered by intravenous infusion at a slow and controlled rate, often utilising a pump, the drug will enter the circulation at a much slower and controlled rate. By altering the infusion rate it is possible for the clinician to titrate the dose against the effect required, e.g. controlling blood pressure, by manipulating the infusion rate of, for example, an inotropic drug such as dobutamine.
Drug solutions at high or low pH or highly concentrated hypertonic solutions (see below) will damage the cells lining the vein and cause localized pain and inflammation (thrombophlebitis). In order to avoid this problem a central line may be inserted. This is a long, indwelling catheter inserted into a vein in the neck or forearm with the end of the catheter sited in the superior vena cava close to the right atrium of the heart (see Fig. 36.1). Medicines administered intravenously, via a central line, become rapidly diluted in a large volume of blood and do not cause local irritation to the blood vessel. It is worth noting that injections which are formulated either as water in oil emulsions or suspensions must not be administered by the intravenous route. This is because the suspended drug particles can physically block blood capillaries and the oil phase of a water-in-oil injection could cause a fat embolism, again blocking blood vessels.
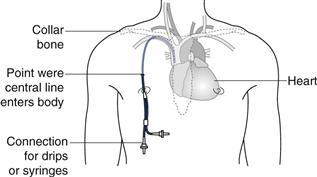
Fig. 36.1 Central-line placement.
Intra-arterial and intracardiac injections
The large majority of drugs that are administered parenterally are given intravenously. As noted above, this delivers the drug directly into the blood stream to provide a rapid and predictable clinical effect. However, it is not the only way that medicines can be administered into the vascular system.
Intra-arterial administration is essentially the same as intravenous administration except that the drug is administered into an artery rather than a vein. Arteries are not as readily accessible as veins and this technique is much more invasive, and carries a greater risk than simple intravenous administration. For this reason it is seldom used. Intra-arterial administration is sometimes used when intravenous access cannot easily be established, such as in very premature infants, due to the very small size of their veins in relation to the catheter tubes used to maintain vascular access. Intra-arterial administration has also been used in the treatment of some cancers (such as liver cancer) where the anti-cancer medicines are injected into an artery upstream of the tumour site to ensure the maximum amount of drug reaches the tumour before distribution elsewhere around the body. However, the benefits of this method of administration do not appear to outweigh the risks to any significant degree.
Intracardiac injections are used to administer a drug (a common example being an aqueous solution of adrenaline) directly into either cardiac muscle or into a ventricle of the heart. This is undertaken only in life threatening emergencies to produce a rapid, local effect in the heart during a heart attack or in circulatory collapse.
Intradermal injections
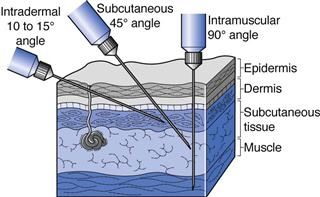
Intradermal (ID or i.d.) injections are given into the skin between the epidermal and dermal layers (Fig. 36.2). Volumes of up to 0.2 mL can be given by this route and absorption from the intradermal injection site is slow. This route is used for immunological diagnostic tests, such as allergy tests, or the injection of tuberculin protein to determine immunity against tuberculosis. Some vaccines such as BCG (tuberculosis) are administered by intradermal injection.
Subcutaneous injections
Subcutaneous (SC, s.c. also called hypodermic) injections are administered into the loose connective and adipose tissues immediately beneath the dermal skin layer (Fig. 36.2). Typical injection sites are the abdomen, upper arms and upper legs. Volumes of up to 1 mL can be administered comfortably, and aqueous solutions or suspensions of drugs are administered by this route. As this tissue is highly vascular, drugs administered by the subcutaneous route are fairly rapidly and predictably absorbed from this site. A common example of a drug administered by subcutaneous injection is insulin.
Intramuscular injections
Intramuscular (IM or i.m.) injections are preferably administered into the tissue of a relaxed muscle (Fig. 36.2). The muscle sites commonly used for intramuscular injection are the buttock, thigh or shoulder muscles. Aqueous or oily solutions or suspensions can be administered in volumes of up to 4 mL. In adults, the gluteal, or buttock, muscle will be used for larger volume injections, whereas in children the thigh muscle is usually larger and thus preferred. Drugs administered by the intramuscular route are more slowly absorbed from the injection site into the systemic circulation compared to those administered by the subcutaneous route.
Intraspinal injections
Intraspinal injections are given between the vertebrae of the spine into the area of the spinal column (Fig. 36.3). Only drugs in aqueous solution are administered by this route. Intrathecal (IT or i.t.) injections are administered into the cerebrospinal fluid (CSF) in the subarachnoid space between the arachnoid mater and the pia mater, the two innermost protective membranes surrounding the spinal cord. This route can be used for spinal anaesthesia, and in this case the specific gravity of the injection can be manipulated to localize the site of action and thus the area of the body anaesthetized. Intrathecal injections are also given to introduce drug substances into the CSF that would otherwise not diffuse across the blood brain barrier. Typically this could be antibiotics to treat meningitis or anticancer agents such as methotrexate or cytarabine. Volumes up to 10 mL can be administered by intrathecal injection.
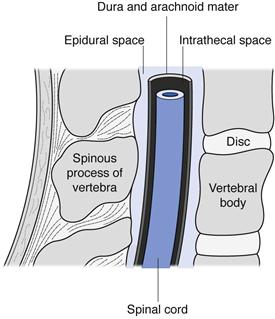
Fig. 36.3 Spinal anatomy.
Intracisternal injections are given between the atlas and axis vertebrae into the cisterna magna. This route is also used for antibiotic administration into the CSF, or to withdraw CSF for diagnostic purposes.
Epidural injections or infusions are given into the peridural space between the dura mater (the outermost protective membrane covering the spinal cord) and the vertebrae. This route is commonly used for spinal anaesthesia, for example during childbirth.
Intra-articular injections
Intra-articular (IA or i.a.) injections are given into the synovial fluid of joint cavities such as the knee. Aqueous solutions or suspensions can be administered by this route. This route of injection produces a local effect, and typically anti-inflammatory drugs are administered to treat arthritic conditions or sports injuries.
Ophthalmic injections
Ophthalmic injections are administered either around or into the eye; in the latter case these are referred to as intraocular injections (Chapter 41). Subconjunctival injections usually of 1 mL or less are administered under the conjunctiva or into the skin surrounding the eye (inside the eyelid for instance). Intraocular injections can be further classified as intracameral injections into the anterior chamber of the eye (in front of the lens), or intravitreal injections into the vitreous chamber (behind the lens). Intracameral injections can be from 0.1 mL to 1 mL in volume depending on whether the drug is left in the eye or administered during surgery on the open eye. This route has been used to administer antibiotics or local anaesthetics during eye surgery (e.g. cataract surgery). Intravitreal injections are used to administer a number of different drugs used to treat various ocular diseases. Because of the danger caused by raising intraocular pressure which can damage the retina, a maximum volume of only 0.1 mL can be administered by the intravitreal route. Ophthalmic drug delivery by injection is discussed further in Chapter 41.
Pharmacopoeial requirements
General requirements
Almost all pharmacopoeias lay down the requirements for parenteral products in a series of general monographs, with which all parenterally administered medicines are expected to comply. Most pharmacopoeias are very similar in their requirements, although details do differ and these need to be checked closely. Several different categories of parenteral preparation are described and further requirements specific to a given medicinal form are specified. This is discussed below.
Sterility
All parenteral preparations are sterile preparations intended for injection, infusion or implantation into the body. The requirement for sterility is vital as the method of administration of these products bypasses the body’s natural defence systems and barriers (such as the skin or gastrointestinal system), and introduces the medicine directly into the bloodstream or other body tissues. The methods of sterilization of parenteral medications are discussed in Chapters 16 and 17.
Excipients
Excipients may be added to parenteral preparations to serve a number of purposes. They can be added to make the preparation isotonic in relation to human blood, to adjust the pH, to increase the solubility of the drug substance, to increase the stability of the drug substance and increase the shelf-life of the product or to act as a preservative. The use of such excipients will be discussed more fully below. However it is worth noting here that the use of excipients should not adversely affect the action of the drug substance, or cause any side effects or toxicity at the concentrations used in a given formulation.
Containers
Containers for parenteral preparations should be made, wherever possible, from materials that are sufficiently transparent to allow the contents to be visually inspected for particles, prior to use. Containers can be made from glass or plastic. Whatever the type of container, it should be effectively sealed to prevent the enclosed medicine becoming contaminated with microorganisms or other contaminants during storage prior to use. Containers should therefore be airtight and also preferably tamper evident. The types of containers and closures will be discussed further below.
Endotoxins and pyrogens
As well as being sterile, parenteral preparations must be practically free from endotoxins and pyrogens. These substances are bacterial products that may be released from certain types of bacteria when they are alive, or after they die. They may therefore be present in sterile products as a by-product of the sterilization process which kills the bacteria during manufacture. When they are injected into a patient they can cause fever, and even shock if present in sufficient quantities. Therefore parenteral products must comply with the test for bacterial endotoxins or the test for pyrogens. For further information on endotoxins and pyrogens and on depyrogenation of containers equipment and raw materials see Chapter 16 and 17.
Particulates
The final general test with which certain parenteral products much comply is for particulate contamination. They must be free of visible particles and contain only very low numbers of sub-visible particles. This is of particular importance for medicines administered intravenously. Particles inadvertently injected with a medicine will travel through the venous system to the heart and from there to the lungs. In the lungs the vascular system narrows to a network of capillaries around each alveolus and any suspended particles may become entrapped at this point preventing blood from flowing, resulting in a pulmonary embolism.
Pharmacopoeias have standards for particulate matter in injections for intravenous use, for example the European Pharmacopeia (PhEur) has limits on the number of 10 and 25 µm particles per container of injectable product. The PhEur notes that these levels would not be appropriate for suspensions for injection. Suspensions for injection are meant to be administered by the intramuscular, intra-articular or subcutaneous routes of administration.
Obviously suspensions are not (supposed to be) injected intravenously for the reasons noted above, but when injected intramuscularly or subcutaneously or into a joint space, the suspended particles will dissolve slowly and provide a prolonged effect. This may be many hours in the case of subcutaneous insulin suspension, or perhaps many weeks for a steroid suspension injected into a joint. The required dissolution characteristics will to a great extent determine the size and nature of the solid drug particles (e.g. amorphous or crystalline).
Emulsions can be injected intravenously, but here the maximum droplet size will be linked to capillary diameter. The droplet size must be controlled and is usually less than 3 µm in diameter to prevent oil embolisms forming in the blood-stream. However there is evidence that certain oil droplets may deform to some extent, to pass through a capillary without occluding it, such that oil droplets slightly larger than the diameter of a capillary can be administered.
Category-specific requirements
Pharmacopoeias usually recognize several distinct categories of parenteral product. These are injections, infusions, concentrates for injection or infusion, powders for injection or infusion and gels for injection.
Injections
Injections can be sterile solutions, emulsions or suspensions. They are prepared by dissolving, emulsifying or suspending the drug substance (or substances), together with any required excipients, in water or non-aqueous liquid or a mixture of aqueous and non-aqueous vehicles. Solutions for injection are clear and free from visible particles. Emulsions for injection must not show any evidence of phase separation (creaming or cracking – see Chapter 27). Suspensions for injection may show sediment, but if they do this must be readily resuspended on shaking to give a suspension that is sufficiently stable to allow a uniform dose to be withdrawn from the container.
With regard to the resuspension of sedimented injections in practice, it is not usual for particle size to be tested in a quantitative manner on a routine basis prior to use. Indeed resuspended injections can be administered in patients’ homes or at GP clinics where particle size determination apparatus is not available. Thus, it is the responsibility of the original formulator/manufacturer of the suspension to provide the necessary data to convince the regulatory authorities that resuspension can be readily achieved by a patient, medical practitioner or nurse, by simple shaking prior to administration.
Aqueous injections that are designed for multiple dosing must contain an antimicrobial preservative, unless it can be shown that the preparation itself has sufficient antimicrobial properties to be self-preserving. Preservatives must not be used when the volume to be routinely administered in a single dose exceeds 15 mL. Preservatives must not be used if the product is intended to be injected intracisternally, epidurally or intrathecally (or any other route giving access to the cerebrospinal fluid), or if it is injected into the eye. Unpreserved injections should be presented in single-dose containers (ampoules or prefilled syringes) rather than vials. This is because vials allow more than one dose to be withdrawn and therefore may become contaminated with micro-organisms if used for multiple doses.
Infusions
Infusions are sterile aqueous solutions or emulsions with water as the continuous phase. They are usually made isotonic with respect to blood. They are large volume parenterals, typically ranging in volume from 100 mL to 1000 mL, but they may be larger. Infusions do not contain antimicrobial preservatives. Solutions for infusion are clear and free from visible particles. Emulsions do not show any sign of phase separation.
Concentrates for injection or infusions
Concentrates for injection or infusion are sterile solutions intended for injection or infusion only after dilution. They are diluted to a prescribed volume usually with an aqueous liquid such as saline (0.9% w/v sodium chloride) or water before administration. After dilution, they comply with the requirements for injections or infusions given above.
Powders for injection or infusion
Powders for injection or infusion are dry solid, sterile substances sealed in their final container. When dispensed, a volume of the prescribed sterile diluent (usually an aqueous liquid) is added and shaken with the powder. This should rapidly form either a clear, particle-free solution or a uniform suspension. After dissolution or suspension they comply with the requirements for injections or infusions.
Freeze-dried (lyophilized) products for parenteral use are considered to be powders for injection or infusion. Freeze-drying is often used for drug substances that are not stable in solution (e.g. degrade by hydrolysis). In this process, a solution of the drug is prepared, sterilized by filtration and filled into the final container (usually a vial). The solution is then freeze-dried by reducing the temperature and applying a vacuum, so that the water in the drug solution is removed by sublimation, leaving a sterile plug of the drug substance in the vial which is then closed and sealed. For further details of this process see Chapters 17 and 29.
Absorption from injection sites
Factors affecting absorption from the injection site
For a drug to exert its pharmacological effect (i.e. provide a clinical action) it must be able to reach its appropriate site of action. The movement of a drug from the site of administration (injection site) into the bloodstream is the process of drug absorption. From this it can be seen that there is no absorption process if the drug is injected intravenously into the bloodstream or into a similar fluid of distribution such as the cerebrospinal fluid (intrathecal and intracisternal injections), or directly into the site of action e.g. intra-articular or intraocular injections. In contrast, drugs that are injected intradermally, subcutaneously or intramuscularly must undergo absorption to reach the systemic circulation. This occurs by diffusion of the drug through the tissues surrounding the injection site followed by penetration through the walls of blood capillaries or the lymphatic system. Both the subcutaneous area and muscle tissue are richly supplied with blood capillaries. Lymph vessels are found extensively in subcutaneous tissue and in connective tissue sheaths around muscles, but are found only in small numbers within muscle tissue itself.
Subcutaneous and intramuscular injections may be either solutions or suspensions. Intradermal injections are usually solutions, but generally are only used for diagnostic purposes (e.g. allergy testing) and act locally at the site of administration. When aqueous solutions of drugs are administered by subcutaneous or intramuscular injection drug absorption is usually comparable to that seen with oral administration, and absorption is usually complete within 30 minutes, though the lipid solubility of the drug can play a role and delay the absorption of a drug injected into subcutaneous fatty tissue. The fairly rapid absorption of aqueous injections from subcutaneous or intramuscular injection sites depends on an unimpaired blood flow surrounding the injection site. Vasoconstrictors such as adrenaline may be incorporated into the formulation of other drugs to prolong their retention at the injection site. This is commonly used to prolong the action of local anaesthetic drugs at and around the injection site (e.g. during dental surgery). Large molecules such as proteins and peptides (e.g. insulin) and colloidal particles (e.g. injectable iron complexes) with molecular weights greater than 20,000 Da are absorbed by lymph vessels rather than the capillary network at subcutaneous or intramuscular injection sites. The speed of lymphatic flow and thus systemic uptake can be increased by exercising the injected muscle or massaging the injection site for subcutaneous injections.
Formulation factors
If a drug is formulated as a suspension, the suspended drug must first dissolve from its solid state before it can be absorbed from the injection site. This means that drug absorption from an injected suspension is much slower than from a solution injected into the same site. This slow and prolonged release from the site of action can be used to reduce the frequency of dosing that might otherwise be required if a drug was administered intravenously or orally. Drugs salts with low aqueous solubility may be specifically chosen for intramuscular injection to provide a prolonged effect. Benzathine penicillin injected intramuscularly as a suspension forms a depot from which the active penicillin is slowly released. Such a product is used in the treatment of early syphilis. Procaine penicillin is another depot forming salt of penicillin.
Several corticosteroids such as hydrocortisone acetate, prednisolone acetate and triamcinolone acetinide are formulated as suspensions for intramuscular injection. When given by this route, they release drug into the systemic circulation over a period of 2 days to 1 week depending on the dose given. Corticosteroid aqueous suspensions can also be given by intra-articular injection into joint spaces to produce a prolonged anti-inflammatory action over many weeks, usually to treat arthritic conditions. However, as the suspended drug substance can irritate the joint cartilage, a joint should be treated in this manner no more than three times a year.
The rate of release of a drug from a suspension is governed by the solubility of the drug in the tissue fluids and the surface area of the suspended drug particles. Differences in particle size and crystal structure have been used to alter the absorption rate of drugs from subcutaneous injection sites. Most notably this has been employed with insulin to give a range of insulin injections with different times for the onset and different durations of action. Subcutaneous injection of insulin causes few problems, but lipodystrophy may occur if injections are given repeatedly into the same area. Aside from causing subcutaneous indentations there is a reduction of vascularity in the affected area and therefore slower absorption of the insulin from the injection site. This can be avoided by moving the site of subsequent injections around the body.
Soluble insulin is the short-acting form of the drug, which is usually injected 15 to 30 minutes prior to eating. When injected subcutaneously, it has an onset of action of 30 to 60 minutes, a maximal effect between 2 and 4 hours after injection, and a duration of action up to 8 hours. Soluble insulin can also be injected intravenously in response to diabetic emergencies, such as diabetic ketoacidosis. When injected intravenously, soluble insulin is rapidly eliminated and its effect disappears within 30 minutes. In the treatment of the unconscious diabetic patient a slow intravenous infusion of soluble insulin may be more appropriate.
Intermediate and long-acting insulins have an onset of action approximately 1 to 2 hours after subcutaneous injection, a maximal effect 4 to 12 hours after injection and duration of 16 to 35 hours. They are administered once or twice daily in conjunction with short-acting insulin. Isophane insulin is an intermediate-acting insulin in the form of a suspension of soluble insulin complexed with protamine sulfate. The insulin protamine complex forms rod shaped crystals greater than 1 µm in length but rarely exceeding 60 µm. Insulin Zinc Suspension (Mixed) is a long-acting insulin and is a mixture of amorphous and regular crystals. The amorphous crystals dissolve more rapidly than the regular crystals. The ratio of the amorphous and crystalline insulin must be controlled. The British and European Pharmacopoeias, for example, direct that Insulin Zinc Suspension consists of 30% amorphous crystals of no uniform shape but not exceeding 2 µm in size, and 70% crystalline insulin consisting of rhombohedral crystals between 10 to 40 µm in size. Insulin Zinc Suspension is obtained by adding a suitable zinc salt such as zinc chloride to soluble insulin.
Oily intramuscular injections are solutions or suspensions of drug substances, often steroids, hormones or fat soluble vitamins in a suitable metabolizable oil, such as arachis or sesame oils, as the vehicle. This approach can be used to administer drugs that are insoluble in water, or water soluble drug substances can be chemically modified to produce an oil soluble compound specifically for administration in an oily injection. The use of undecanoate, enanthate or propionate esters to obtain the oil soluble form of a drug is commonplace. As oily injections are much more viscous than aqueous injections, the injected solution does not spread along the muscle fascias when injected intramuscularly. This means a depot is formed in the muscle tissue. The drug must partition from the oil into the aqueous tissue fluid before it can be absorbed, therefore release from oily intramuscular injections is very slow. Many antipsychotic medicines are given as oily intramuscular injections as they require dosing only every 2 to 4 weeks rather than daily oral dosing, thus compliance with treatment can be better managed.
Excipients
It is very unusual for an injectable medicine to be composed entirely of the drug substance and no other ingredient. The drug, unless presented as a dried powder for reconstitution before use, will be dissolved or suspended in a vehicle such as water or a saline solution, or a non-aqueous liquid. Other ingredients (excipients) may be present in the formulation to aid the dissolution or suspension of the drug in the vehicle. Other excipients may be incorporated to comply with pharmacopoeial requirements such as the incorporation of a preservative if the preparation is for multiple use rather than a single dose product. Excipients are often included in parenteral products to prevent, reduce or delay the degradation of the drug product over time and thus improve the product’s shelf-life. Finally, excipients are frequently added to adjust the pH and tonicity of the product to make it comparable to the physiological pH and tonicity of the tissue into which it is being injected (usually comparable to human plasma values). This is done to reduce pain and irritation that may otherwise be caused to blood vessels or tissues by the administration process.
Vehicles for injections
‘Water for injections’ is the most common vehicle used for parenteral products. Water for injections is a highly purified grade of water which is subject to pharmacopoeial standards with respect to production methods and purity. Water is of course well tolerated by the body and it is a solvent for a wide range of drug substances. For those drugs which are poorly soluble in water, water-miscible non-aqueous solvents such as ethanol, glycerol or propylene glycol may be added as co-solvents to improve the solubility of a drug substance.
Solubilizing agents may be added to an injection formulation to aid the dissolution of drugs with poor aqueous solubility. Polyoxyethylene castor oil derivatives will solubilize hydrophobic drugs into aqueous solutions for injections and are used, for instance, for formulations of paclitaxel, diazepam and cisplatin. Cyclodextrins are cyclic oligosaccharide molecules (see Fig. 24.1) with a bucket-like structure containing a hydrophobic central cavity, while the outer surface is hydrophilic. Cyclodextrins can form inclusion complexes with a variety of poorly soluble drug molecules. The ‘hydrophobic’ drugs are held within the cyclodextrin ‘bucket’ with the outer surface of the complex remaining hydrophilic. Both alpha and gamma-cyclodextrin can be used in parenteral products, but beta-cyclodextrin should not be used as it causes severe kidney damage.
Water-insoluble drugs may be administered parenterally by dissolving the drug in a suitable oil and forming an oil-in-water emulsion using a suitable emulsifying agent to stabilize the emulsion (see Chapter 27 for further details on emulsions). Droplet size must be controlled and is usually less than 3 µm in diameter to prevent oil embolisms forming in the blood-stream. Lecithin and various sorbitan fatty acid esters have been used as emulsifying agents in parenteral products. Finally, oils such as arachis oil or sesame oil may be chosen as a vehicle for intramuscular injections, for drug release over a prolonged period of time (depot injections).
Preservatives
Antimicrobial preservatives are added to injections which are designed for multiple use. Such products are usually packaged in glass vials or cartridges with a synthetic rubber septum that can be punctured on a number of occasions to withdraw a dose of the drug for administration (see the section below on containers). A preservative is included to inhibit the growth of any microorganisms that may be inadvertently introduced into the product during repeated use by the patient or healthcare professional.
Excipients such as ethanol, glycerol and propylene glycol which may be added to a formulation as co-solvents to aid drug dissolution also will provide an antimicrobial effect. Ethanol is effective at concentrations above 10% v/v, glycerol at 10–20% v/v and propylene glycol at 15–30% v/v. Some of the commonly used antimicrobial preservatives suitable for parenteral administration are given in Table 36.1. It should be noted that fatal toxic reactions in low birth-weight neonates have been linked to benzyl alcohol preserved injections. Thus, parenteral products preserved with benzyl alcohol should not be administered to neonates. Also, as noted above, preservatives should not be added to large volume parenterals (infusions), or products intended for intraspinal or intraocular injection. Preservation of pharmaceutical products, including injections is discussed in detail in Part 6 of this book.
Table 36.1
| Preservative | Typical concentration (% w/v) |
| Benzalkonium Chloride | 0.01 |
| Benzoic Acid | 0.17 |
| Benzyl Alcohol | 1–2 |
| Chlorobutanol | 0.1–0.5 |
| Chlorocresol | 0.1 |
| Cresol | 0.15–0.3 |
Antioxidants
If the drug substance to be injected is prone to degradation by oxidation a number of formulation processes and excipients can be used to reduce the rate of drug degradation in the product and thus improve the shelf-life or expiry date.
It is common practice to use pharmaceutical grade compressed nitrogen gas (filtered through a 0.2 µm pore size filter) during the manufacturing process. Nitrogen is bubbled through the solution containing the drug prior to filling into the final packaging. The nitrogen gas displaces any dissolved oxygen from the drug solution. This process is known as ‘sparging’. A nitrogen overlay may also be applied during the filling operation prior to sealing the final containers of the drug product. This will displace air from the headspace between the surface of the product and the top of the container (for example in a vial or ampoule) thereby removing oxygen.
An antioxidant may also be included in the formulation. Antioxidants are chemicals that have a lower oxidation potential than the drug substance and thus will react with any oxygen present in the product in preference to the drug. Vitamin C (ascorbic acid) and Vitamin E (alpha-tocopherol) can be used for this purpose, both in pharmaceutical products and in food. Alpha-tocopherol is highly lipophilic and can be used in oil-based parenteral products usually in the range of 0.001–0.05% v/v. Ascorbic acid is used in aqueous parenteral products at a concentration of 0.01–0.1% w/v. Ascorbic acid can also be used to adjust the pH of the formulation (see below). Butylated hydroxyanisole (BHA) and butylated hydroxytoluene (BHT) are structurally similar antioxidants used in parenteral preparations either separately or in combination. For intramuscular injections they are usually used at a concentration of 0.03% w/v and for intravenous injections 0.0002–0.002% w/v is used. The most commonly used antioxidants are the sulphite salts. Sodium metabisulphite is used at concentrations between 0.01–0.1% w/v and also has some preservative properties. It is used as an antioxidant for acidic parenteral products. If the product is of neutral pH sodium bisulphite is used, whereas sodium sulphite is used as an antioxidant in alkali parenterals.
The activity of an antioxidant can be enhanced by the inclusion of an antioxidant synergist, also referred to as chelating or sequestering agents. Antioxidant synergists reduce oxidation by removing trace levels of metal ions from the product by forming chelates with them. Metal ions, particularly copper, iron and manganese are believed to catalyse oxidation reactions between oxygen and drug substances. Examples of chelating agents used in parenteral products include: citric acid at concentrations between 0.3–2.0% w/v and derivatives of edetic acid (ethylenediaminetetraacetic acid; EDTA) at concentrations between 0.0005–0.01% w/v. Citric acid can also be used to adjust the pH of formulations and edetate compounds possess preservative properties.
If the primary mechanism of drug degradation in the product over time is hydrolysis rather than oxidation, then the shelf-life of the product will be improved by removal of all water from the product. Injectable products which are presented as freeze-dried powders (see Chapter 29 for the freeze-drying process and Chapter 17 for the sterilization of these powders) are usually formulated in this manner to improve the stability of a readily hydrolyzed drug substance.
pH adjustment and buffers
The physiological pH of plasma and extracellular fluid is 7.4; therefore ideally all injectable products should be formulated to this pH value. However, there is likely to be an optimum pH value at which the drug substance is most stable. The solubility of the drug in the vehicle may also be dependent on pH. Therefore, the pH chosen for a parenteral product is likely to be a compromise between the requirements for stability, solubility and physiological compatibility. Injectable products should have a pH value between 3.0 and 9.0 prior to administration. pH values above or below this range are too corrosive and will cause tissue damage at the site of injection. The pH of a parenteral formulation can be adjusted using acidifying or alkalizing agents. Acidifying agents include hydrochloric, citric and sulphuric acids. Alkalizing agents include sodium bicarbonate, sodium citrate and sodium hydroxide.
Buffers are included in parenteral products to maintain the pH of the product at the desired optimum value. Changes in pH may arise due to interactions between an ingredient in the formulation and the container, or from changes in storage temperature. Buffer ingredients commonly used in parenteral products include citric acid, sodium citrate, sodium acetate, sodium lactate and mono- and dibasic sodium phosphate.
Tonicity adjusting agents
An aqueous solution of sodium chloride at a concentration of 0.9% w/v or 9 g per L has a measured osmolarity of 286 mmol per L and is isotonic (meaning has the same osmotic pressure (see Chapter 3) with human plasma, which has an osmolality of between 280–295 mmol per kg.
Hypotonic solutions have a lower osmotic pressure than plasma. If mixed with blood they would cause the blood cells to swell and burst as water would be driven into the cells by osmosis. Hypertonic solutions have a higher osmotic pressure than plasma. If mixed with blood they would cause the blood cells to lose water by osmosis and shrink.
Hypotonic injection solutions are made isotonic by the addition of sodium chloride, dextrose or mannitol. Hypertonic injection solutions must be made isotonic by dilution prior to administration. Pharmacopoeias direct that intravenous infusions should be made isotonic with human plasma. Whilst not a pharmacopoeial requirement, it is considered desirable for subcutaneous, intradermal and intramuscular injections also to be isotonic. Intrathecal and intraocular injections should also be isotonic to avoid serious changes in osmotic pressure in the cerebrospinal fluid and the eye.
There are a number of methods available for calculating the amount of additional substance to be added to a hypotonic drug solution to render it isotonic, including freezing point depression, the use of sodium chloride equivalents, molar concentrations and calculations based on serum osmolarity. One method is demonstrated below:
Isotonicity calculation based on freezing point depression
The presence of solutes in water will increase osmolarity and depress the freezing point of water. These effects (colligative properties; Chapter 3) are dependent on the concentration of solute particles. Consequently, the freezing point of a solution can be used as a measure of its osmolarity. The freezing point of blood serum/plasma and tears is −0.52 °C. Therefore, an aqueous solution that freezes at −0.52 °C is isotonic. For high concentrations of electrolytes there may be a slight deviation in the direct relationship between concentration and freezing point depression, but in most cases the relationship holds true. Reference sources, such as the Pharmaceutical Codex give the freezing point depressions produced by a wide range of soluble materials.
The required amount of adjusting substance required to make a hypotonic solution isotonic is given by the equation:
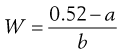 (36.1)
(36.1)
where W is the % w/v of adjusting substance in the final solution, a is the freezing point depression of unadjusted solution (i.e. freezing point depression of 1% solution × strength in % w/v) and b is the freezing point depression of water due to 1% w/v of adjusting substance, usually sodium chloride or glucose. A worked example is presented in Box 36.1.
Box 36.1 Use of freezing point depression to adjust solutions to isotonicity
You are requested to make a solution containing 0.28% w/v potassium chloride isotonic by the addition of anhydrous glucose. From the literature: the freezing point depression of a 1% solution of potassium chloride is 0.439 and for anhydrous glucose it is 0.101.
Using Equation 36.1:
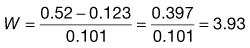
Thus, you require the addition of 3.93% w/v (i.e. 3.93 g per 100 mL) anhydrous glucose to make the potassium chloride solution isotonic with plasma.
Suspending agents
Drugs presented as suspensions for injection may require a suspending agent to ensure that the drug can be readily and uniformly resuspended prior to use. A water soluble cellulose derivative such as methylcellulose can be used in intramuscular and intra-articular injectable suspensions. Povidone has been used in the past for this purpose, but safety concerns about this compound when injected intramuscularly have led to it falling out of favour. A suitable non-ionic injectable surfactant such as a polysorbate may also be included in a suspension formulation to aid the uniform dispersion of the suspended drug substance.
Containers
As noted above (see pharmacopoeial requirements) the container or primary packaging of a parenteral product should ideally be transparent to allow the product to be examined prior to use. This is particularly important for injections supplied as powders for reconstitution, as the healthcare practitioner needs to be able to see that the drug substance has completely dissolved in the diluent prior to withdrawal of the dose. Large volume infusion fluids often have other drugs added to them, so again the clarity of the container is important to allow the proper mixing of the product to be assessed and to check that large particles (e.g. from the rubber closure) have not been inadvertently introduced.
Whatever type of container is used it must be tightly sealed to maintain the sterility of the injection prior to use and to prevent other contaminants entering the product which may lead to degradation of the drug substance (e.g. oxygen). The container should not interact with the drug product or other excipients it contains. It should also be robust enough to withstand the chosen sterilization process. In preference, parenteral products are manufactured and filled into the primary container which is then sealed and the product is then terminally sterilized inside the container, for example by using moist heat in an autoclave (see Chapters 16 and 17). If the drug substance cannot withstand this process then the containers will be sterilized first, the drug product sterilized by filtration and then filled under conditions of strict asepsis and the container sealed. This latter method of aseptic production obviously carries the risk of microbial contamination of the product during the filling and sealing process, hence the preference for sterilization of the drug product inside the sealed container after filling.
Ampoules
Small-volume parenteral products are often packaged in glass or plastic ampoules. The use of glass and plastics as packaging materials for pharmaceutical products is discussed in Chapter 47. Ampoules are used for single use, unpreserved products. Glass ampoules range in size typically from 1 mL up to 10 mL in volume, though larger sizes are available. The glass chosen is referred to as Type I or borosilicate glass which is less alkaline than the usual glass used for beverages and other purposes. Ampoules are supplied as open necked containers that are sealed by fusion of the narrow glass neck after filling (see Fig. 36.4). Usually the neck of the ampoule has a painted ceramic ring on it. Due to the baking process required to fuse the ceramic to the glass, this acts as a weak point at which the ampoule can be easily snapped open by hand. The main disadvantages of glass ampoules are the fragility of the container, the potential for deposition of glass particles into the drug product on opening and the potential for injury to the fingers of the person opening the ampoule. The problem of fragility is overcome by using robust secondary packaging. Glass particles can be removed from the product by drawing up the contents of the ampoule through a filter straw or quill into a syringe. The advantages of glass ampoules are low cost and (if Type I glass is used) very little interaction between the container and the product.

Fig. 36.4 Open and sealed glass ampoules.
Plastic ampoules are prepared using a highly automated blow-fill-seal product process. The filling machine is loaded with the drug solution to be filled and with plastic granules (polyethylene and/or polypropylene) which are then melted. The molten plastic is blown into the ampoule mould to form the body of the ampoule, the body of the ampoule is filled with product and then the lid of the ampoule is moulded onto the top of the ampoule to form a seal. All of the above happens as a single process which can take less than a second to complete (see Fig. 36.5). The sealed ampoule is opened by twisting off the lid and very few particles are generated to contaminate the product. Plastic ampoules are also much more robust than glass ampoules. Disadvantages are that this is a more costly process, and is only suitable for drug products formulated as simple solutions (for instance freeze-drying processes cannot be undertaken using plastic ampoules). A full comparison of glass and plastics as materials for pharmaceutical packaging is provided in Chapter 47.
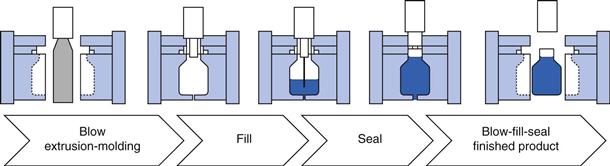
Fig. 36.5 Blow-fill-seal process.
Vials
Vials are containers usually made of Type I borosilicate glass with a re-usable synthetic rubber closure. Vials have advantages as containers as they permit multiple withdrawals and are made in sizes usually ranging from 5 mL to 100 mL. Vials are sealed with a bromobutyl or chlorobutyl synthetic rubber closure held in place by an aluminium seal crimped around the neck of the glass vial. The rubber closure (or septum) is usually protected by a plastic flip-off cap (Fig. 36.6). This acts purely as a dust cap and does not provide a covering that maintains the sterility of the septum prior to use.

To withdraw a dose from a vial, the cap is removed and the septum disinfected with a sterile alcohol wipe. A syringe and needle is used to puncture the rubber closure and remove the required amount of product. The rubber septum is self-sealing to a high degree and so more than one withdrawal can be made from a vial. However, only a limited number of punctures can be made through the rubber closure before it will lose its integrity as a seal. Products packaged in vials for multiple use will therefore incorporate a preservative to prevent any microorganisms accidentally introduced into the product during use from proliferating.
The glass is inert and does not interact with the drug and the use of synthetic rubber closures reduces the likelihood of the drug or other excipients reacting with or being adsorbed into the rubber on storage. Synthetic rubber is also latex-free, which is important as sensitization to latex is an increasing problem for healthcare workers. The main disadvantage is that puncturing the rubber closure can cause large rubber particles to be introduced into the drug product.
Infusion bags and bottles
Large volume parenteral products are packaged in glass bottles, collapsible plastic bags and semi-rigid plastic bottles, though the use of glass bottles for large volume parenterals is becoming much less commonplace. These products range in size from 100 mL up to 1000 mL, though larger sizes (e.g. 3000 mL) can be used, particularly for parenteral nutrition products.
Collapsible bag presentations are the most common form of container (see Fig. 36.7). They are manufactured from PVC or more increasingly polyolefin plastic. Collapsible bags usually have an additive port to allow other injectable drugs to be added to the infusion fluid. The main advantage of collapsible bags is that they collapse under atmospheric pressure as the contents are removed from them, therefore they do not require an air inlet system to equilibrate air pressure between the outside and inside of the container, as do rigid glass bottles. The main disadvantage of PVC bags is that drugs may become adsorbed onto the plastic (e.g. insulin) or react with the plastic (e.g. etoposide). Additionally, components can leach out of the plastic such as monomers and phthalate plasticizers which may be toxic in long-term exposure. Polyolefin is much less reactive and is now replacing PVC for this reason in infusion bags.

Fig. 36.7 Collapsible infusion bag.
Semi-rigid plastic containers are often made of polyethylene (see Fig. 36.8). These containers may have an additive port to allow other drugs to be added to them. As they do not fully collapse during use, air equilibration may be required. Large volume glass bottles are essentially the same as glass vials, but on a larger scale. All large volume parenteral products are meant for single-use only.

Fig. 36.8 Semi-rigid infusion container.
Nowadays, parenteral products may be packaged in syringes and thus be presented to the healthcare professional or patient in a ready to use format. This requires aseptic filling using specialist equipment. Drug can be administered from the syringe using an infusion device, for instance in patient controlled analgesia, postoperatively or as part of palliative care.
Bibliography
1. Ansel HC, Allen Jnr LV, Popovich NG. Pharmaceutical Dosage Forms and Drug Delivery Systems. 7th edn Baltimore: Lippincott Williams & Wilkins; 1999.
2012. British Pharmacopoeia. London: British Pharmacopoeia Commission Stationery Office; 2012.
3. Lund W, ed. Pharmaceutical Codex. 12th edn London: Pharmaceutical Press; 1994.
4. Rowe RC, Sheskey PJ, Cook WG, Fenton ME, eds. Handbook of Pharmaceutical Excipients. 7th edn London: Pharmaceutical Press; 2012.
5. Wade A. Pharmaceutical Handbook. 19th edn London: Pharmaceutical Press; 1980.
6. Winfield AJ, Richards RME. Pharmaceutical Practice. Edinburgh: Elsevier/Churchill Livingstone; 2004.