CHAPTER 46 Parasitic Infections
Parasitic diseases of the central nervous system (CNS) affect millions of people in the developing world, where most infections are linked to poverty and related conditions. In addition, increased tourism and immigration have turned some formerly geographically restricted parasitic infections into widespread conditions.1 Parasites are divided primarily into protozoa and helminths. Protozoa are simpler unicellular microorganisms, whereas helminths are multicellular organisms with functional structures and complex life cycles that usually involve two or more hosts. Helminth parasites include nematodes (roundworms), trematodes (flukes), and cestodes (tapeworms). Infection of the CNS by parasites causes pleomorphic and nonspecific clinical syndromes, including seizure disorders, subacute or chronic meningitis, acute or subacute encephalitis, space-occupying brain lesions, stroke, and myelopathy.2 In this chapter we review the most common parasitic infections of the CNS.
Protozoal Infections
Malaria
Plasmodium infections have a complex biologic cycle. Humans are infected when the sporozoite forms of the parasite are inoculated through the skin during a blood meal by a female Anopheles mosquito. Sporozoites are carried to the liver of the host, where they hide and mature into tissue schizonts that liberate many merozoites, or products of asexual division. Merozoites enter the bloodstream, parasitize red blood cells, mature into trophozoites, and again divide to produce schizonts, which will rupture and simultaneously release many merozoites into the circulation. These merozoites invade a new group of red cells and the cycle continues. A proportion of trophozoites transform into male or female gametocytes. The life cycle is completed when the mosquito ingests gametocytes in infected human red blood cells and they reproduce sexually in the mosquito to form sporozoites.3 Of the four species of malaria parasites that can infect humans, only Plasmodium falciparum causes cerebral malaria.
Clinical Manifestations
Cerebral malaria is a major cause of mortality in the world, mostly in Africa. The current definition of cerebral malaria requires all of the following: (1) unarousable coma, (2) evidence of acute infection with P. falciparum, and (3) no other identifiable cause of coma.3 Fever is the initial complaint. This is followed by progressive somnolence associated with seizures, extensor posturing, and disconjugate gaze. Retinal hemorrhages suggest a poor prognosis. Some patients, particularly children, have focal signs related to cerebral infarcts or hemorrhage. Hypoglycemia, pulmonary edema, renal failure, bleeding diathesis, and hepatic dysfunction may complicate the course of the disease.4 Up to 25% of patients die despite medical care. Permanent sequelae, more common in children, include mental retardation, epilepsy, blindness, and motor deficits.5
Diagnosis
P. falciparum may be seen by examining thin and thick blood smears with Giemsa stain; repeated examinations may be needed because the parasitemia is cyclic. Although cerebrospinal fluid (CSF) is usually normal, routine CSF examination is mandatory to exclude other causes of encephalopathy. Neuroimaging studies may show brain swelling or small hemorrhages in severe cases.6
Pathology
Brain edema and small ring hemorrhages in the subcortical white matter are found in almost 80% of fatal cases. The hemorrhaging is caused by extravasation of erythrocytes as a result of endothelial damage. The erythrocytes that form the ring hemorrhages are not parasitized, thus suggesting that the damage to blood vessels is related to the liberation of cytokines and vasoactive substances (humoral hypothesis). Capillaries and venules are plugged by clumped, parasitized erythrocytes, which causes brain damage because of obstruction of the cerebral microvasculature, reduced cerebral blood flow, increased concentrations of lactic acid, and ischemic hypoxia (mechanical hypothesis).7 The brains of patients who survive the acute phase of the disease have granulomatous lesions (Dürck’s nodules) at the site of the ring hemorrhages.
Treatment
Because of chloroquine-resistant strains of P. falciparum, quinine is the drug of choice for cerebral malaria. After an initial loading dose (20 mg/kg), the maintenance dose of quinine should be adjusted according to plasma concentrations to prevent accumulation. Quinidine may be used when quinine is not available. More recent clinical trials have shown that artemether, an artemisinin derivative, is equally as effective as but less toxic than quinine for the treatment of cerebral malaria.8 Artesunate, another artemisinin derivative, can reduce mortality by more than a third in comparison to quinine.9 Systemic complications must be recognized and treated. Symptomatic measures include anticonvulsants, sedatives, and osmotic diuretics. Corticosteroids are harmful to comatose patients with cerebral malaria.10
Toxoplasmosis
After the acquired immunodeficiency syndrome (AIDS) epidemic, toxoplasmosis has become a highly common parasitic disease of the CNS.11 Toxoplasma gondii is a protozoan acquired by the ingestion of contaminated cat feces or by eating undercooked meat. In AIDS patients, CNS toxoplasmosis most often results from reactivation of a dormant infection with T. gondii. CNS toxoplasmosis may also develop in immunocompetent hosts during acute infections, and fetuses may be involved as a result of placental transmission of tachyzoites from women who acquire the disease during pregnancy.
Clinical Manifestations
Neurological symptoms rarely develop in immunocompetent hosts, although an acute encephalitis with fever, irritability, seizures, and drowsiness progressing to coma occurs in some cases. Immunocompromised hosts are also susceptible to an acute encephalitic syndrome or, more frequently, a subacute disease characterized by focal signs associated with seizures and signs of intracranial hypertension.12,13 Apparently, this focal form predominates in AIDS patients who contracted latent infection before the depletion of CD4+ cells, whereas the diffuse encephalitic form, in which multiple parasite-containing microglial nodules are disseminated through the brain, is more common in those who became infected after they were immunosuppressed.14 Meningeal signs rarely occur in patients with cerebral toxoplasmosis because the pathologic lesions are usually confined to the brain parenchyma and do not disseminate through the subarachnoid space.14 In AIDS patients, the clinical picture is usually complicated because of concurrent infections.
Diagnosis
In normal hosts, a fourfold rise in serum antibody titer is a sensitive indicator of acute infection. The sustained persistence of specific IgM antibodies and high IgG titers in a significant proportion of individuals in the general population complicates the serologic interpretation for discrimination between latent infection and active infection, regardless of their human immunodeficiency virus serologic status.15–17 There is controversy regarding the positive predictive value of high IgG titers for presumed CNS toxoplasmosis in AIDS patients.18,19 The role of serology in AIDS patients is to establish at-risk status for active disease. Absence of antibodies in AIDS patients with CNS toxoplasmosis is rarely seen and should raise the possibility of an alternative diagnosis.15,20 Neuroimaging studies show ring-enhancing lesions surrounded by edema; the lesions are usually multiple and may be located in the subcortical white matter, the basal ganglia, or the brainstem.21 However, ring-enhancing lesions are not pathognomonic for cerebral toxoplasmosis because they may be observed in other diseases affecting AIDS patients, so definitive diagnosis requires histologic demonstration of the parasite. Empirical therapy followed by repeated neuroimaging studies at 3 weeks has been proposed as an alternative to biopsy in AIDS patients. Polymerase chain reaction (PCR) techniques have been introduced with promising results for the detection of T. gondii DNA from the CSF of patients with cerebral toxoplasmosis.22
Pathology
T. gondii may produce a focal or diffuse necrotizing encephalitis associated with perivascular inflammation. Cerebral abscesses may also occur and are most often located at the corticosubcortical junction, basal ganglia, and upper brainstem.23 They consist of a necrotic center and a periphery in which multiple tachyzoites and cysts are seen together with patchy areas of necrosis and perivascular cuffing of lymphocytes. Glial nodules composed of astrocytes and microglial cells are common in the surrounding brain tissue.
Treatment
The combination of pyrimethamine (100 to 200 mg the first day, followed by 50 to 75 mg/day for 6 weeks) and sulfadiazine (4 to 6 g/day for 6 weeks) is the therapy of choice for CNS toxoplasmosis.24 Clindamycin, clarithromycin, trimetrexate, piritrexim, and atovaquone are alternative drugs in patients in whom skin reactions to sulfadiazine develop.25 In AIDS patients, permanent maintenance therapy with pyrimethamine and sulfadiazine is usually advised to decrease the risk for relapse.
African Trypanosomiasis
Clinical Manifestations
T. brucei invades the CNS very shortly after inoculation and remains latent for a long time.26,27 Thereafter, the disease enters into a stage in which the symptoms—fever, hepatosplenomegaly, and cervical lymphadenopathy (Winterbottom’s sign)—suggest activation of the reticuloendothelial system. Somnolence, apathy, involuntary movements, and rigidity then appear. The neurological manifestations progress to dementia, stupor, coma, and death.
Diagnosis
T. brucei may be isolated from blood smears and from CSF, lymph node, and bone marrow aspirates. Repeated examinations and concentration techniques may be necessary. CSF examination may reveal lymphocytic pleocytosis, increased protein, and the typical Mott cells (plasma cells filled with eosinophilic inclusions of IgM). Demonstration of motile trypanosomes in CSF confirms CNS involvement. Chronic disease may be diagnosed by immune tests performed with serum or CSF. Neuroimaging findings are nonspecific and include diffuse changes in white matter, hyperintensity in the basal ganglia, and ventricular enlargement.28,29
Treatment
When CNS symptoms and signs of CSF inflammation have appeared, therapy requires the use of melarsoprol. This arsenic drug produces a severe reactive encephalopathy in about 10% of patients, half of whom die of it. The role of pretreatment with corticosteroids to prevent this reaction is unclear.30
American Trypanosomiasis
Triatomine bugs (“kissing bugs”), found mostly in the genus Triatoma, are the vector for T. cruzi. These insects infect humans by biting them to feed on their blood and defecating in the area. T. cruzi parasites in the insect’s feces are then exposed to the bite wound or facial mucosae (eyes, mouth), usually by the bitten person when scratching. More rarely, infection can be acquired through uncooked food contaminated with infected bug feces, from blood/organ donation, or congenitally from an infected mother. An estimated 10 to 12 million people are currently infected with T. cruzi in Latin America, approximately 30% of whom, or 3 to 4 million, have or will eventually have consequent life-threatening cardiac or gastrointestinal disease, or both.31–33 CNS disease is rare and occurs mostly in immunocompromised individuals.
Clinical Manifestations
Unilateral orbital edema (Romaña’s sign) is considered the typical sign of acute Chagas’ disease and reflects the site of a bite. The edema is associated with mild constitutional symptoms, although early invasion of the CNS by trypanosomes may cause a diffuse encephalopathy, particularly in infants and patients with AIDS.34 Chronic disease is not usually associated with primary neurological complications; however, cardioembolic brain infarcts develop in some patients as a result of chagasic dilated cardiomyopathy. In addition, immunocompromised patients can experience reactivation of chronic infections, which results in a rapidly fatal meningoencephalitic syndrome similar to that observed in acute infections.35
Pathology
The brains of patients with Chagas’ disease involving the brain show multiple areas of hemorrhagic necrosis, glial proliferation, and perivascular infiltrates of inflammatory cells.36
Treatment
Antitrypanosomal drug therapy with either nifurtimox or benznidazole can cure most, if not all, congenitally infected infants when treated early in life, as well as 60% or more of infected children. The earlier the treatment in the course of infection, the higher the probability of cure. Chronic Chagas’ disease has no specific treatment. Nevertheless, recent studies suggest that adults with clinical signs of early chronic Chagas’ disease may also benefit from specific drug therapy.37 However, antitrypanosomal drug therapy requires prolonged administration of one of the only two available drugs, both with significant side effects and elevated cost.
Free-Living Amebae
Free-living amebae of the genera Acanthamoeba, Balamuthia, and Naegleria may invade the CNS.38 Acanthamoeba spp. and Balamuthia mandrillaris are opportunistic pathogens that affect mainly immunocompromised patients, and they invade the CNS by the hematogenous route from a primary infection of the skin or the respiratory tract. In contrast, Naegleria fowleri infection occurs in normal hosts and is acquired during swimming in warm fresh water; the parasites enter through the nasal cavity and migrate through olfactory nerves to the CNS.
Clinical Manifestations
Acanthamoeba spp. and B. mandrillaris produce a subacute disease called granulomatous amebic encephalitis characterized by low-grade fever, focal signs, seizures, intracranial hypertension, and behavioral changes; the disease runs a progressive course over a 2- to 8-week period.38–40 Balamuthia infection is usually manifested initially by a centrofacial skin lesion, which allows the diagnosis. N. fowleri causes primary amebic meningoencephalitis, a fulminant disease carrying a grim prognosis that resembles acute bacterial meningitis.38,41
Diagnosis
Neuroimaging studies usually show multiple ring-enhancing lesions in patients infected with Acanthamoeba spp. and B. mandrillaris and diffuse edema in those infected with N. fowleri.42 Examination of fresh CSF may reveal mobile trophozoites in patients with N. fowleri encephalitis. In contrast, the diagnosis of infection with Acanthamoeba spp. and B. mandrillaris usually rests on the demonstration of parasites in biopsy specimens.
Pathology
CNS infection by Acanthamoeba spp. and B. mandrillaris results in the formation of hemorrhagic brain abscesses surrounded by a granulomatous inflammatory infiltrate. Invasion of arterial walls by trophozoites causes a necrotizing angiitis that may lead to cerebral infarcts.39,40 N. fowleri induces purulent meningitis associated with diffuse hemorrhagic necrosis of the brain parenchyma; involvement is more prominent in the frontal lobes and the olfactory bulbs, around the portal of entry of the microorganisms.41
Amebiasis by Entamoeba histolytica
The intestinal parasite E. histolytica normally causes dysenteric diarrhea or liver abscesses. It may invade the CNS from the colon or liver in patients with severe infections (usually in the setting of advanced systemic amebiasis) and produce a multifocal encephalopathy.43 Neuroimaging studies generally show multiple ring-enhancing lesions. The diagnosis of E. histolytica amebiasis is made by demonstration of parasites in biopsy specimens. Pathologic examination of tissue infected with E. histolytica shows multiple ill-defined brain abscesses formed by a central hemorrhagic area and a rim of necrotic tissue.43 Surgery and metronidazole are advised for E. histolytica brain abscesses.43
Helminthic Infections
Cysticercosis
Cysticercosis occurs when humans become intermediate hosts of Taenia solium by ingesting its eggs from contaminated food or through contact with the feces of T. solium carriers. After ingestion, the eggs mature into oncospheres, which are then carried into the tissues of the host, where cysticerci develop. Invasion of the parasites into the CNS causes neurocysticercosis (NCC).44
Clinical Manifestations
Epilepsy is the most common manifestation of NCC,45 but a variety of focal neurological signs have also been described in patients with NCC. These manifestations usually follow a subacute course, thus making it difficult to differentiate NCC from neoplasia or other infections of the CNS. Hydrocephalus, mass effect, and cysticercotic encephalitis are the most common causes of intracranial hypertension in patients with NCC.46,47 As a result of intense inflammation around cysticerci and diffuse cerebral edema, cysticercotic encephalitis is a particularly severe form of NCC characterized by headache, vomiting, generalized seizures, decreased visual acuity, and clouding of consciousness.47 Manifestations of spinal cysticercosis include motor and sensory deficits, which vary according to the level of the lesion.48
Diagnosis
Accurate diagnosis of NCC is possible with proper interpretation of clinical data together with neuroimaging findings and results of immunologic tests.49 Neuroimaging studies provide objective evidence about the location of lesions and the degree of the host inflammatory response against the parasites. The most typical findings are cystic lesions showing the scolex and parenchymal brain calcifications.50 Other neuroimaging findings in NCC, such as ring-enhancing lesions, abnormal enhancement of the leptomeninges, hydrocephalus, and cerebral infarctions, are nonspecific because many other conditions cause similar changes on neuroimaging studies.51 CSF may be normal in patients with parenchymal NCC. In the subarachnoid and ventricular forms of the disease, CSF analysis demonstrates a lymphocytic pleocytosis, an increased protein concentration, and a normal glucose concentration. The most accurate serologic test is immunoblotting.52 However, false-positive results occur in patients who have cysticerci outside the CNS or antibodies arising from exposure only, and false-negative results are common in patients with a single cyst.53
Pathology
Within the CNS, cysticerci may be located in the brain parenchyma, subarachnoid space, ventricular system, or spinal cord. Parenchymal brain cysts usually lodge in the cerebral cortex or the basal ganglia. Subarachnoid cysts are most commonly located in the sylvian fissure or in the cisterns at the base of the brain. Ventricular cysticerci may be attached to the choroid plexus or may be floating free in the ventricular cavities. Spinal cysticerci may be found in both the cord parenchyma and the subarachnoid space.54 Cysticerci may remain viable within the CNS for years and elicit few inflammatory changes in the surrounding tissue. In other cases, cysticerci enter the CNS and prompt a complex immune attack from the host that results in a process of degeneration ending with death of the parasite. The inflammation around cysticerci induces changes in cerebral tissues, including edema, gliosis, thickening of the leptomeninges, entrapment of the cranial nerves, angiitis, hydrocephalus, and ependymitis. Calcified cysticerci have been seen as inert lesions that do not cause further neuropathologic changes in the CNS. However, there is growing evidence that periodic remodeling of calcifications may be associated with the release of cysticercal antigens into the brain, which in turn may induce recurrent inflammatory reactions and relapsing symptoms.55
Treatment
Therapeutic approaches to patients with NCC include the use of cysticidal agents, as well as anticonvulsants and other symptom-relieving drugs. Surgical procedures are needed in some cases.56 Praziquantel and albendazole have been used with success to treat NCC; these drugs destroy 60% to 80% of parenchymal brain cysticerci after a course of therapy. The most accepted regimens of cysticidal drugs are albendazole, 15 mg/kg per day for 1 week, and praziquantel, 50 mg/kg per day for 2 weeks.57,58 It seems that patients with giant subarachnoid cysts require higher doses of albendazole or longer courses of therapy.59 Although cysticidal drugs have changed the prognosis of most patients with NCC, the anecdotal nature of the first studies on these drugs generated criticism about their usefulness. Some authors claimed that cysticidal drugs do not modify the natural course of the disease. In a recent double-blind, placebo-controlled trial, albendazole was found to be safe and effective for the treatment of viable parenchymal brain cysticerci.60 In this study, treated patients had better seizure control than the placebo group did, as reflected by a 67% reduction in the number of seizures. In addition, the number of cystic lesions that resolved was significantly higher in patients receiving albendazole. A recent meta-analysis of randomized trials of cysticidal drug therapy evaluated the effect of cysticidal drugs on neuroimaging and clinical outcomes of NCC.61 According to this meta-analysis, cysticidal drug therapy results in better resolution of both colloidal and vesicular cysticerci, a lower risk for recurrence of seizures in patients with colloidal cysticerci, and a reduction in the rate of generalized seizures in patients with vesicular cysticerci.
Patients with cysticercotic encephalitis should not receive cysticidal drugs because they may exacerbate symptoms in this form of the disease. These patients must be managed with high doses of corticosteroids and osmotic diuretics.47 Cysticidal drugs must be used with caution in patients with giant subarachnoid cysticerci because the host inflammatory response to destruction of the parasites may occlude small leptomeningeal vessels surrounding the cyst. In such cases, concomitant corticosteroid therapy is mandatory.62 Patients with calcifications alone should not receive cysticidal drugs because these lesions represent dead parasites.
Hydrocephalus secondary to cysticercotic arachnoiditis requires placement of a ventricular shunt. The main complication of a ventricular shunt is the high incidence of shunt dysfunction. The high mortality rate (up to 50%) associated with the development of hydrocephalus from NCC is related to the number of surgical interventions for revision of the shunt.63 A shunt that functions at a constant flow rate and does not allow spinal CSF to enter the ventricular system has been developed to treat these patients.64 Inversion of CSF transit is the most important cause of shunt dysfunction because it allows parasitic debris to enter the ventricular system. The efficacy of this new shunt was compared with that of a conventional Pudenz-type shunt. After 1 year of follow-up, withdrawal of the shunt was necessary in 45% of patients with the Pudenz-type shunt but in only 30% of those with the new shunt.65
Although albendazole destroys many ventricular cysts, the inflammatory reaction may cause acute hydrocephalus if the cysts are located within the fourth ventricle or near the interventricular foramina of Monro. Ventricular cysts can be removed more safely by surgical excision or endoscopic aspiration.66 The surgeon must consider the possibility of cyst migration between the time of diagnosis and the surgical procedure, and such migration must be ruled out with a neuroimaging study before surgery to avoid unnecessary craniotomies.67 Shunt placement should follow or even precede the excision of ventricular cysts associated with ependymitis.
Echinococcosis (Hydatid Disease)
There are two main forms of echinococcosis: cystic hydatid disease (caused by Echinococcus granulosus) and alveolar hydatid disease (caused by Echinococcus multilocularis). In the cycle of hydatid disease, a herbivore usually harbors the larvae, mostly in the viscera, and a carnivore becomes infected with the adult tapeworm by eating raw viscera. In turn, the herbivore intermediate host acquires a larval infection by ingesting tapeworm eggs in pasture. Humans acquire the infection when they become intermediate hosts of these tapeworms by accidental ingestion of the eggs of Echinococcus spp.68 After entering the body, the eggs transform into cysts that grow in the liver, lungs, heart, and CNS. In the latter, cysts may also result from metastatic dissemination of a visceral cyst.
Cystic Hydatid Disease (Echinococcus granulosus)
Clinical Manifestations
Cystic hydatid disease results in seizures or increased intracranial pressure of subacute onset and has a progressive course, often in association with focal neurological deficits.69 Orbital involvement in patients with cystic hydatid disease is manifested as proptosis and ophthalmoplegia.70
Diagnosis
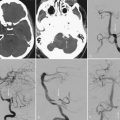
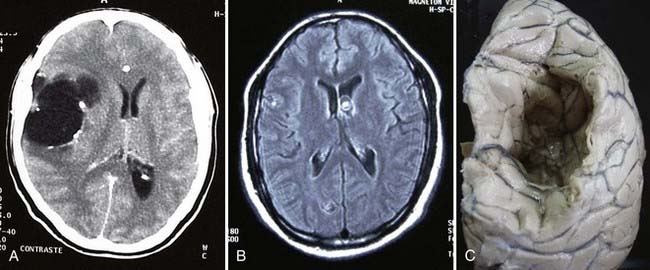
On neuroimaging studies, cystic hydatid disease is characterized by a large nonenhancing vesicle that is well demarcated from the surrounding brain parenchyma (Fig. 46-1). Some lesions may be calcified.71 Cystic lesions located in the subarachnoid space may be multiple and confluent. Intracranial or spinal epidural cysts have a biconvex shape or a multilocular appearance and may be associated with bone erosion. Immunologic diagnosis of cystic hydatid disease by enzyme-linked immunosorbent assay (ELISA) or enzyme-linked immunoelectrotransfer blot (EITB) is not accurate because of cross-reactions with other parasitic diseases. Moreover, these tests yield false-negative results in up to 50% of patients with intact brain cystic hydatid lesions.
Pathology
E. granulosus cysts are large, spherical, and well demarcated from surrounding tissue.72 Within the CNS, these cysts may be located in the brain parenchyma, ventricular system, subarachnoid space, epidural space, orbits, and both the epidural and subarachnoid spaces in the spinal canal; epidural cysts tend to be associated with vertebral bone erosion.73 Primary hydatid disease of the heart may be the source of an embolic cerebral infarction, usually in the territory of the middle cerebral artery.
Treatment
Current therapy for hydatid disease of the CNS is largely empirical, and experience is limited to anecdotal cases and uncontrolled studies. Surgical resection has been the classic approach to most hydatid cysts of the CNS.74 Antiparasitic drugs are usually given before surgical resection in the case of intraoperative rupture of cysts or postoperatively to treat recurrent hydatid disease.75 Therapy for hydatid disease of the CNS is differentiated into three categories: cystic hydatid disease of the brain, cystic hydatid disease of the spine, and alveolar hydatid disease.
Cystic Hydatid Disease of the Brain
Most hydatid cysts are removed with Dowling’s technique, which consists of hydrostatic expulsion of the entire cyst by irrigation of saline solution between the lesion and the surrounding nervous tissue.76 The aim of this technique is to remove the cyst without damaging its walls; however, accidental intraoperative rupture of the cyst occurs in 25% of cases.74 Such rupture is associated with spillage of the cyst’s contents (including the protoscolices), which may in turn cause an allergic reaction or recurrent hydatid disease.76 Other complications associated with the Dowling technique include subdural effusions and intracranial hemorrhages. To avoid these complications, some surgeons puncture the cyst, aspirate its contents, irrigate the cyst with a hypertonic saline solution, and then remove the shrunken cyst. This is a modification of the percutaneous aspiration, injection, and reaspiration (PAIR) technique that is the currently accepted approach for removal of most hydatid cysts of the liver. Albendazole at doses ranging from 10 to 15 mg/kg per day, given for several 1-month cycles with therapy-free intervals of 14 days between cycles, cured 28% of patients and improved the condition of 51% of the 72% of remaining patients. Albendazole may be used in patients who are not candidates for surgical resection of lesions, as prophylactic therapy for those at risk for accidental rupture of the cysts perioperatively, or to treat recurrent cystic hydatid disease after surgery.
Praziquantel is not effective against hydatid cysts; however, the drug has protoscolicidal activity at doses of 40 mg/kg once per week and may have a role in the prevention of secondary reactions related to accidental spillage of protoscolices during surgery. Two trials suggested that combined albendazole and praziquantel therapy may be more effective than albendazole alone for preoperative prophylactic treatment of hydatid cysts.77,78
Cystic Hydatid Disease of the Spine
The surgical approach to patients with spinal hydatid disease usually includes a combination of decompressive laminectomy, removal of cysts, excision of involved bone, and stabilization of the spine.79 Almost 50% of these lesions may rupture during surgery because of the narrow space in which the surgeon has to work. Moreover, involvement of adjacent bone and multiplicity of lesions make complete removal of spinal cysts difficult. Hydatid disease recurs after surgery in up to 40% of patients, and this complication is associated with neurological deterioration. The use of albendazole, in a regimen similar to that recommended for patients with intracranial cystic hydatid disease, is advised to reduce such complications.80
Alveolar Hydatid Disease (Echinococcus multilocularis)
Clinical Manifestations
The neurological manifestations progress more rapidly and are more severe with alveolar hydatid disease than with cystic hydatid disease. Alveolar hydatid disease is characterized by focal neurological deficits, seizures, and intracranial hypertension.81 Spinal cord involvement, associated with root pain and motor or sensory deficits below the level of the lesion, is more common in cystic hydatid disease than in alveolar hydatid disease, but it may be observed in both.
Diagnosis
On neuroimaging studies, alveolar hydatid disease is characterized by multiple lesions surrounded by edema, with ring-like enhancement mimicking other infectious or neoplastic diseases of the CNS.82 Alveolar hydatidosis of the spinal canal may be visualized by magnetic resonance imaging (MRI), although the findings are nonspecific; computed tomography (CT) is better than MRI for demonstrating lytic lesions in vertebral bodies. Immunologic diagnosis is better with alveolar echinococcosis than with cystic hydatid disease. ELISA using purified and recombinant antigens (Em2 and II/3-10 ELISA) is the test of choice.83
Pathology
E. multilocularis cysts are small, group in clusters, elicit a severe inflammatory reaction from the host, and tend to metastasize both locally and distantly. They are usually located within the brain parenchyma.81 Primary hydatid disease of the heart may be the source of an embolic cerebral infarction that is generally located in the territory of the middle cerebral artery.
Treatment
Alveolar hydatid disease is invasive, and total surgical removal usually requires resection of adjacent tissue. This approach may cause neurological deficits from cysts located in eloquent cerebral areas. Administration of albendazole should follow or even precede the surgical procedure, or it may be used as primary therapy in patients with inoperable alveolar hydatid disease.84 The drug is given in regimens similar to those described for cystic hydatid disease. With a combination of surgery and cysticidal therapy, 50% of lesions regress, 40% remain static, and 10% continue to grow.85
Paragonimiasis
Paragonimiasis is caused by flukes of the genus Paragonimus. Humans acquire the infection by ingesting metacercariae in undercooked crustaceans. Metacercariae liberate larvae, which cross the intestinal wall and migrate to the lungs, where they mature into adult worms. Erratic migration of worms along the jugular veins and carotid arteries or hematogenous dissemination of larvae results in CNS involvement.86
Clinical Manifestations
Most patients have acute meningitis associated or not with focal neurological signs as a result of cerebral infarction secondary to arteritis.86 Other patients with parenchymal brain granulomas have seizures, focal neurological deficits, and intracranial hypertension. Cerebral hemorrhages may occur along tracks of larval migration or as a result of the necrotizing vasculitis that occurs during granuloma formation.87 Spinal paragonimiasis is associated with radicular pain, weakness, and sensory disturbances.88
Diagnosis
In patients with meningitis, analysis of CSF reveals a mild eosinophilic pleocytosis with increased protein concentration and normal glucose levels. Neuroimaging abnormalities include cystic or ring-enhancing parenchymal lesions, hemorrhages, or multiple calcifications that are typically located in the occipital and temporal lobes, are closely related to each other, and have the appearance of “soap bubbles.”89,90 The diagnosis rests on the demonstration of specific antibodies in blood and CSF or by finding Paragonimus eggs in sputum or adult worms in tissue samples.86
Pathology
The CNS lesions associated with paragonimiasis include necrotic tracks in the brain parenchyma, cystic lesions, granulomatous reactions, diffuse arachnoiditis, and obstructive hydrocephalus.86 Parenchymal brain lesions predominate in the occipital and temporal lobes. Spinal lesions may be located in both the subdural and epidural spaces.91
Treatment
There is scant experience with the use of antiparasitic drugs in patients with cerebral paragonimiasis. Bithionol (40 mg/kg every other day for 1 month) improved the neurological status of 9 of 24 patients with different forms of cerebral involvement.92 This study was performed before the availability of CT, and the results are difficult to interpret. Praziquantel (75 mg/kg per day for 2 days) has also been used with success in a few patients.86 Corticosteroids are the primary form of therapy for patients with arachnoiditis to prevent further cranial nerve and blood vessel damage. Corticosteroids also reduce the edema surrounding active lesions and ameliorate the adverse effects related to destruction of parasites by praziquantel or bithionol. Antiepileptic drugs must be used in patients with cystic lesions and parenchymal brain calcifications who have seizures.
Surgical resection of intracranial lesions has a limited role in management of this disease.91 The large size and multiplicity of lesions make radical resection difficult without damaging the surrounding brain parenchyma. Patients with calcified “soap bubble” lesions should not undergo surgery because resection of these lesions is rarely associated with clinical improvement.93 Surgery is useful to remove cystic lesions located in the spinal subdural and epidural spaces and to decompress the spinal cord.91 A ventricular shunt for relief of hydrocephalus may be necessary in patients with arachnoiditis.
Schistosomiasis (Schistosoma mansoni, haematobium, and japonicum)
Schistosomiasis, or infection with the flukes S. mansoni, S. haematobium, or S. japonicum, affects approximately 200 million individuals, mostly in sub-Saharan Africa; 10% have severe liver or urinary disease and 100,000 die per year. Schistosomiasis is endemic in most of sub-Saharan Africa, Asia, and Latin America. Other less frequent species include Schistosoma mekongi and Schistosoma intercalatum.94
CNS infection, a rare complication of schistosomiasis, is caused by aberrant location of eggs transported by the circulatory system. Among the different species. S. japonicum is associated more often with this rare complication (2% to 5% of cases), which contributes to seizures and epilepsy. CNS symptoms include seizures, focal neurological deficits, mass effect, or diffuse encephalitis. Nodular, ring-enhancing intraparenchymal lesions are seen on CT or MRI. Occasionally, eggs in the medulla may cause transverse myelitis as a result of granulomatous lesions (S. mansoni). Treatment involves the administration of praziquantel or oxamniquine. Surgery may be required for refractory epilepsy.94,95
Toxocariasis (Toxocara canis and cati)
Toxocariasis is a cosmopolitan nematode infection caused by T. canis or T cati. Most infected humans are asymptomatic, as shown by multiple seroepidemiologic studies demonstrating antibody prevalence ranging from 20% to 55%.96 Human disease is mostly related to allergic, lung, or liver complications of larval migration. Larvae (measuring less than 0.5 mm) originate from the intestine and migrate through the circulatory system to the liver, lungs, and other locations (visceral larva migrans) or the eye (ocular larva migrans). Apparently, symptomatic CNS invasion is infrequent. It has, however, been associated with seizures, motor and sensory problems, meningitis, encephalitis, and other neurological syndromes.96–98 Treatment with anti-inflammatory or anthelmintic drugs may be considered for severe complications of the brain. Given the scarce number of proven cases, there are no controlled trials to define therapy. Benzimidazoles and diethylcarbamazine seem to be the more useful regimens. As with other brain parasites (e.g., cysticercosis), in the initial days of treatment an intense inflammatory response may occur along with a subsequent increase in symptoms after liberation of antigen from the injured larvae. There are also some reports of successful management with corticosteroids, with or without specific antilarval therapy. Surgery is rarely if ever required.
Carod-Artal FJ. Neurological complications of Schistosoma infection. Trans R Soc Trop Med Hyg. 2008;102:107-116.
Cobo F, Yarnoz C, Sesma B, et al. Albendazole plus praziquantel versus albendazole alone as a pre-operative treatment in intra-abdominal hydatidosis caused by Echinococcus granulosus. Trop Med Int Health. 1998;3:462-466.
Del Brutto OH, Roos KL, Coffey CS, et al. Meta-analysis: cysticidal drugs for neurocysticercosis: albendazole and praziquantel. Ann Intern Med. 2006;4:43-51.
Del Brutto OH, Santibañez R, Noboa CA, et al. Epilepsy due to neurocysticercosis: analysis of 203 patients. Neurology. 1992;42:389-392.
Derouin F, Leport C, Pueyo S, et al. Predictive value of Toxoplasma gondii antibody titres on the occurrence of toxoplasmic encephalitis in HIV infected patients. AIDS. 1996;10:1521-1527.
Dondorp A, Nosten F, Stepniewska K, et al. Artesunate versus quinine for treatment of severe falciparum malaria: a randomized trial. Lancet. 2005;366:717-725.
Duma RJ, Ferrell HW, Nelson EC, et al. Primary amebic meningoencephalitis. N Engl J Med. 1969;281:1315-1323.
Finsterer J, Auer H. Neurotoxocarosis. Rev Inst Med Trop Sao Paulo. 2007;49:279-287.
Garcia HH, Pretell EJ, Gilman RH, et al. A trial of antiparasitic treatment to reduce the rate of seizures due to cerebral cysticercosis. N Engl J Med. 2004;15:249-258.
Gottstein B, Jacquier P, Bresson-Hadni S, et al. Improved primary immunodiagnosis of alveolar echinococcosis in humans by an enzyme-linked immunosorbent assay using the Em2plus antigen. J Clin Microbiol. 1993;31:373-376.
Kennedy PG. Human African trypanosomiasis of the CNS: current issues and challenges. J Clin Invest. 2004;113:496-504.
Kirchhoff LV. Changing epidemiology and approaches to therapy for Chagas disease. Curr Infect Dis Rep. 2003;5:59-65.
Kusner DJ, King CH. Cerebral paragonimiasis. Semin Neurol. 1993;13:201-208.
Mohamed AE, Yasawy MI, Al Karawi MA. Combined albendazole and praziquantel versus albendazole alone in the treatment of hydatid disease. Hepatogastroenterology. 1998;45:1690-1694.
Phillips RE, Warrell DA. The pathophysiology of severe falciparum malaria. Parasitol Today. 1986;2:271-282.
Porter SB, Sande MA. Toxoplasmosis of the central nervous system in the acquired immunodeficiency syndrome. N Engl J Med. 1992;327:1643-1648.
Proaño JV, Madrazo I, Avelar F, et al. Medical treatment for neurocysticercosis characterized by giant subarachnoid cysts. N Engl J Med. 2001;345:879-885.
Raffi F, Aboulker J, Michelet C, et al. A prospective study of criteria for the diagnosis of toxoplasmic encephalitis in 186 AIDS patients. AIDS. 1997;11:177-184.
Viotti R, Vigliano C, Lococo B, et al. A. Long-term cardiac outcomes of treating chronic Chagas disease with benznidazole versus no treatment: a nonrandomized trial. Ann Intern Med. 2006;144:724-734.
Warrell DA, Looareesuwan S, Warrell MJ, et al. Dexamethasone proves deleterious in cerebral malaria. A double-blind trial in 100 comatose patients. N Engl J Med. 1982;306:313-319.
1 Nishimura K, Hung T. Current views on geographic distribution and modes of infection of neurohelminthic diseases. J Neurol Sci. 1997;145:5-14.
2 Del Brutto OH. Non-viral forms of encephalitis. In: Joynt RJ, Griggs RC, editors. Baker’s Clinical Neurology, vol 2. Philadelphia: Lippincott Williams & Wilkins; 1998:1-117.
3 Warrell DA. Cerebral malaria. In: Shakir RA, Newman PK, Poser CM, editors. Tropical Neurology. London: WB Saunders; 1996:213-245.
4 Román GC, Senanayake N. Neurological manifestations of malaria. Arq Neuropsiquiatr. 1992;50:3-9.
5 Waller D, Krishna S, Crawley J, et al. Clinical features and outcome of severe malaria in Gambian children. Clin Infect Dis. 1995;21:577-587.
6 Looareesuwan S, Wilairatana P, Krishna S, et al. Magnetic resonance imaging of the brain in patients with cerebral malaria. Clin Infect Dis. 1995;21:300-309.
7 Phillips RE, Warrell DA. The pathophysiology of severe falciparum malaria. Parasitol Today. 1986;2:271-282.
8 Tran TH, Day NP, Nguyen HP, et al. A controlled trial of artemether or quinine in Vietnamese adults with severe falciparum malaria. N Engl J Med. 1996;335:76-83.
9 Dondorp A, Nosten F, Stepniewska K, et al. Artesunate versus quinine for treatment of severe falciparum malaria: a randomized trial. Lancet. 2005;366:717-725.
10 Warrell DA, Looareesuwan S, Warrell MJ, et al. Dexamethasone proves deleterious in cerebral malaria. A double-blind trial in 100 comatose patients. N Engl J Med. 1982;306:313-319.
11 Price RW. Neurological complications of HIV infection. Lancet. 1996;348:445-452.
12 Porter SB, Sande MA. Toxoplasmosis of the central nervous system in the acquired immunodeficiency syndrome. N Engl J Med. 1992;327:1643-1648.
13 Wijdicks EF, Borleffs JC, Hoepelman AI, et al. Fatal disseminated hemorrhagic toxoplasmosis encephalitis as the initial manifestation of AIDS. Ann Neurol. 1991;29:683-686.
14 Gray F, Gherardi R, Wingate E, et al. Diffuse “encephalitic” cerebral toxoplasmosis in AIDS. Report of four cases. J Neurol. 1989;236:273-277.
15 Raffi F, Aboulker J, Michelet C, et al. A prospective study of criteria for the diagnosis of toxoplasmic encephalitis in 186 AIDS patients. AIDS. 1997;11:177-184.
16 Candolfi E, Partisani ML, De Mautort E, et al. Séropréevalence de la toxoplasmose chez 346 sujets infectés par le VIH dans l’Est de la France. Suivi sérologique des sujets non contaminés par Toxoplasma gondii. Presse Med. 1992;21:394-395.
17 Borges AS, Figueiredo JF. Detecção de imunoglobulinas IgG, IgM e IgA anti-Toxoplasma gondii no soro, líquor e saliva de pacientes com síndrome da imunodeficiência adquirida e neurotoxoplasmose. Arq Neuropsiquiatr. 2004;62:1033-1037.
18 Derouin F, Leport C, Pueyo S, et al. Predictive value of Toxoplasma gondii antibody titres on the occurrence of toxoplasmic encephalitis in HIV infected patients. AIDS. 1996;10:1521-1527.
19 Raffi F, Franck J, Pelloux H, et al. Specific anti-toxoplasmic IgG antibody immunoblot profiles in patients with AIDS-associated Toxoplasma encephalitis. Diagn Microbiol Infect Dis. 1999;34:51-56.
20 Luft BJ, Brooks RG, Conley FK, et al. Toxoplasmic encephalitis in patients with acquired immunodeficiency syndrome. JAMA. 1984;252:913-917.
21 Ciricillo SF, Rosenblum ML. Use of CT and MR imaging to distinguish intracranial lesions and to define the need for biopsy in AIDS patients. J Neurosurg. 1990;73:720-724.
22 Roberts TC, Storch GA. Multiplex PCR for diagnosis of AIDS-related central nervous system lymphoma and toxoplasmosis. J Clin Microbiol. 1997;35:268-269.
23 Marinz P, Bosler E, Luft BJ. Toxoplasmosis. In: Berger JR, Levy RM, editors. AIDS and the Nervous System. 2nd ed. Philadelphia: Lippincott-Raven; 1997:41-659.
24 Leport C, Raffi F, Matheron S, et al. Treatment of central nervous system toxoplasmosis with pyrimethamine/sulfadiazine combination in 35 patients with the acquired immuno-deficiency syndrome. Efficacy of long-term continuous therapy. Am J Med. 1988;84:94-100.
25 Araujo FG, Remington JS. Recent advances in the search for new drugs for treatment of toxoplasmosis. Int J Antimicrob Agents. 1992;1:153-164.
26 Pentreath VW. Trypanosomiasis and the nervous system. Pathology and immunology. Trans R Soc Trop Med Hyg. 1995;89:9-15.
27 Boa YF, Traore MA, Doua F, et al. Les différents tableaux cliniques actuels de la trypanosomiase humaine africaine á T. b. gambiense. Analyse de 300 dossiers du foyer de Daloa, Cote d’Ivoire. Bull Soc Pathol Exot Filiales. 1988;81:427-444.
28 Sabbah P, Brosset C, Imbert P, et al. Human African trypanosomiasis: MRI. Neuroradiology. 1997;39:708-710.
29 Gill DS, Chatha DS, del Carpio-O’Donovan R. MR Imaging findings in African trypanosomiasis. Am J Neuroradiol. 2003;24:1383-1385.
30 Kennedy PG. Human African trypanosomiasis of the CNS: current issues and challenges. J Clin Invest. 2004;113:496-504.
31 Dias JC, Silveira AC, Schofield CJ. The impact of Chagas disease control in Latin America: a review. Mem Inst Oswaldo Cruz. 2002;97:603-612.
32 Kirchhoff LV. American trypanosomiasis (Chagas’ disease). Gastroenterol Clin North Am. 1996;25:517-533.
33 Kirchhoff LV. Changing epidemiology and approaches to therapy for Chagas disease. Curr Infect Dis Rep. 2003;5:59-65.
34 Villanueva MS. Trypanosomiasis of the central nervous system. Semin Neurol. 1993;13:209-218.
35 Leiguarda R, Roncoroni A, Taratuto AL, et al. Acute CNS infection by Trypanosoma cruzi (Chagas’ disease) in immunosuppressed patients. Neurology. 1990;40:850-851.
36 Pittella JE. Central nervous system involvement in Chagas’ disease. An updating. Rev Inst Med Trop Sao Paolo. 1993;35:111-116.
37 Viotti R, Vigliano C, Lococo B, et al. A. Long-term cardiac outcomes of treating chronic Chagas disease with benznidazole versus no treatment: a nonrandomized trial. Ann Intern Med. 2006;144:724-734.
38 Campbell S. Amebic brain abscess and meningoencephalitis. Sem Neurol. 1993;13:153-160.
39 Griesemer DA, Barton LL, Reese CM, et al. Amebic meningoencephalitis caused by Balamuthia mandrillaris. Pediatr Neurol. 1994;10:249-254.
40 Ofori-Kwakye SK, Sidebottom DG, Herbert J, et al. Granulomatous brain tumor caused by Acanthamoeba. Case report. J Neurosurg. 1986;64:505-509.
41 Duma RJ, Ferrell HW, Nelson EC, et al. Primary amebic meningoencephalitis. N Engl J Med. 1969;281:1315-1323.
42 Schumacher DJ, Tien RD, Lane K. Neuroimaging findings in rare amebic infections of the central nervous system. Am J Neuroradiol. 1995;16(suppl 4):S930-S935.
43 Becker GL, Knep S, Lance KP, et al. Amebic abscess of the brain. Neurosurgery. 1980;6:192-194.
44 Del Brutto OH, Sotelo J, Román GC. Neurocysticercosis. A Clinical Handbook. Lisse, Netherlands: Swets & Zeitlinger; 1988.
45 Del Brutto OH, Santibañez R, Noboa CA, et al. Epilepsy due to neurocysticercosis: analysis of 203 patients. Neurology. 1992;42:389-392.
46 Estañol B, Kleriga E, Loyo M, et al. Mechanisms of hydrocephalus in cerebral cysticercosis: implications for therapy. Neurosurgery. 1983;13:119-123.
47 Rangel R, Torres B, Del Brutto O, et al. Cysticercotic encephalitis. A severe form in young females. Am J Trop Med Hyg. 1987;36:387-392.
48 Firemark HM. Spinal cysticercosis. Arch Neurol. 1978;35:250-251.
49 Del Brutto OH, Rajshekhar V, White ACJr, et al. Proposed diagnostic criteria for neurocysticercosis. Neurology. 2001;57:177-183.
50 Suss RA, Maravilla KR, Thompson J. MR imaging of intracranial cysticercosis: comparison with CT and anatomopathologic features. AJNR Am J Neuroradiol. 1986;7:235-242.
51 Dumas JL, Visy JM, Belin C, et al. Parenchymal neurocysticercosis: follow-up and staging by MRI. Neuroradiology. 1997;39:12-18.
52 Richards FJr, Schantz PM. Laboratory diagnosis of cysticercosis. Clin Lab Med. 1991;11:1011-1028.
53 Rajshekhar V, Oommen A. Serological studies using ELISA and EITB in patients with solitary cysticercus granuloma and seizures. Neurol Infect Epidemiol. 1997;2:177-180.
54 Pittella JE. Neurocysticercosis. Brain Pathol. 1997;7:681-693.
55 Nash TE, Pretell J, Garcia HH. Calcified cysticerci provoke perilesional edema and seizures. Clin Infect Dis. 2001;33:1649-1653.
56 Del Brutto OH, Sotelo J, Roman GC. Therapy for neurocysticercosis: a reappraisal. Clin Infect Dis. 1993;17:730-735.
57 Sotelo J, Escobedo F, Rodriguez-Carbajal J, et al. Therapy of parenchymal brain cysticercosis with praziquantel. N Engl J Med. 1984;301:1001-1007.
58 Garcia HH, Gilman RH, Horton J, et al. Albendazole therapy for neurocysticercosis: a prospective double-blind trial comparing 7 versus 14 days of treatment. Neurology. 1997;48:1421-1427.
59 Proaño JV, Madrazo I, Avelar F, et al. Medical treatment for neurocysticercosis characterized by giant subarachnoid cysts. N Engl J Med. 2001;345:879-885.
60 Garcia HH, Pretell EJ, Gilman RH, et al. A trial of antiparasitic treatment to reduce the rate of seizures due to cerebral cysticercosis. N Engl J Med. 2004;15:249-258.
61 Del Brutto OH, Roos KL, Coffey CS, et al. Meta-analysis: cysticidal drugs for neurocysticercosis: albendazole and praziquantel. Ann Intern Med. 2006;4:43-51.
62 Del Brutto OH, Sotelo J, Aguirre R, et al. Albendazole therapy for giant subarachnoid cysticerci. Arch Neurol. 1992;49:535-538.
63 Sotelo J, Marin C. Hydrocephalus secondary to cysticercotic arachnoiditis: a long-term follow-up review of 92 cases. J Neurosurg. 1987;66:686-689.
64 Sotelo J. A new ventriculoperitoneal shunt for treatment of hydrocephalus. Experimental results. BM Eur J Biomed Technol. 1993;15:257-262.
65 Sotelo J, Rubalcaba MA, Gomez-Llata S. A new shunt for hydrocephalus that relies on CSF production rather than on ventricular pressure: initial clinical experience. Surg Neurol. 1995;43:324-332.
66 Neal JH. An endoscopic approach to cysticercosis cysts of the posterior third ventricle. Neurosurgery. 1995;36:1040-1043.
67 Zee CS, Segall HD, Apuzzo MLJ, et al. Intraventricular cysticercal cysts: further neuroradiological observations and neurosurgical implications. Am J Neuroradiol. 1984;5:727-730.
68 Schantz PM. Echinococcosis. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical Infectious Diseases. Principles, Pathogens & Practice. Philadelphia: Churchill Livingstone; 1999:1005-1025.
69 Patrikar DM, Mitra KR, Bhutada VR. Cerebral hydatid disease. Autralas Radiol. 1993;37:226-227.
70 Ergün R, Okten AI, Yüksel M, et al. Orbital hydatid cysts: report of four cases. Neurosurg Rev. 1997;20:33-37.
71 Micheli F, Lehkuniec E, Giannaula R, et al. Calcified cerebral hydatid cyst. Eur Neurol. 1987;27:1-4.
72 Moro PL, Gonzalez AE, Gilman RH. Cystic hydatid disease. In: Strickland GT, editor. Hunter’s Tropical Medicine and Emerging Infectious Diseases. 8th ed. Philadelphia: WB Saunders; 2000:866-871.
73 Ciurea AV, Valisescu G, Nuteanu L, et al. Cerebral hydatid disease in children. Experience of 27 cases. Childs Nerv Syst. 1995;11:679-686.
74 Singounas EG, Leventis AS, Sakas DE, et al. Successful treatment of intracranial hydatid cysts with albendazole: case report and review of literature. Neurosurgery. 1992;31:571-574.
75 Arana-Iñiguez R, San Julián J. Hydatid cysts of the brain. J Neurosurg. 1955;12:323-335.
76 Sandhu P, Saggar K, Sodhi KS. Neurological picture. Multiple hydatid cysts of the brain after surgery. J Neurol Neurosurg Psychiatry. 2000;68:97.
77 Mohamed AE, Yasawy MI, Al Karawi MA. Combined albendazole and praziquantel versus albendazole alone in the treatment of hydatid disease. Hepatogastroenterology. 1998;45:1690-1694.
78 Cobo F, Yarnoz C, Sesma B, et al. Albendazole plus praziquantel versus albendazole alone as a pre-operative treatment in intra-abdominal hydatidosis caused by Echinococcus granulosus. Trop Med Int Health. 1998;3:462-466.
79 Pandey M, Chaudhari MP. Primary hydatid cyst of sacral spinal canal: case report. Neurosurgery. 1997;40:407-409.
80 Baykaner MK, Dogulu F, Ozturk G, et al. A viable residual spinal hydatid cyst cured with albendazole: case report. J Neurosurg. 2000;93(suppl 1):142-144.
81 Jacquet G, Godard J, Czorny A. Echinococcose alvéolaire cérébrale. A propos d’un cas opéré. Revue de la littérature. Neurochirurgie. 1992;38:362-367.
82 Weber M, Vespignani H, Jacquier P, et al. Manifestations neurologiques de l’échinococcose alvéolaire. Rev Neurol (Paris).. 1988;144:104-112.
83 Gottstein B, Jacquier P, Bresson-Hadni S, et al. Improved primary immunodiagnosis of alveolar echinococcosis in humans by an enzyme-linked immunosorbent assay using the Em2plus antigen. J Clin Microbiol. 1993;31:373-376.
84 Todorov T, Vutova K, Mechkov G, et al. Experience in the chemotherapy of severe, inoperable echinococcosis in man. Infection. 1992;20:19-24.
85 Gilman RH, Lee BH. Alveolar hydatid disease. In: Strickland GT, editor. Hunter’s Tropical Medicine and Emerging Infectious Diseases. 8th ed. Philadelphia: WB Saunders; 2000:872-875.
86 Kusner DJ, King CH. Cerebral paragonimiasis. Semin Neurol. 1993;13:201-208.
87 Brenes Madrigal R, Rodriguez-Ortiz B, Vargas Solano G, et al. Cerebral hemorrhagic lesions produced by Paragonimus mexicanus. Report of three cases in Costa Rica. Am J Trop Med Hyg. 1982;31:522-526.
88 Oh SJ. Spinal paragonimiasis. J Neurol Sci. 1968;6:125-140.
89 Yoshida M, Moritaka K, Kuga S, et al. CT findings of cerebral paragonimiasis in the chronic state. J Comput Assist Tomogr. 1982;6:195-196.
90 Cha SH, Chang KH, Cho SY, et al. Cerebral paragonimiasis in early active stage: CT and MR features. AJR Am J Roentgenol. 1994;162:141-145.
91 Hung T-P, Chen E-R. Paragonimiasis of the central nervous system. Neurol Infect Epidemiol. 1996;1:11-29.
92 Oh SJ. Bithionol treatment in cerebral paragonimiasis. Am J Trop Med Hyg. 1967;16:585-590.
93 Toyonaga S, Kurisaka M, Mori K, et al. Cerebral paragonimiasis. Report of five cases. Neurol Med Chir (Tokyo). 1992;32:157-162.
94 Carod-Artal FJ. Neurological complications of Schistosoma infection. Trans R Soc Trop Med Hyg. 2008;102:107-116.
95 Lei T, Shu K, Chen X, Li L. Surgical treatment of epilepsy with chronic cerebral granuloma caused by Schistosoma japonicum. Epilepsia. 2008;49:73-79.
96 Nicoletti A, Sofia V, Mantella A, et al. Epilepsy and toxocariasis: a case-control study in Italy. Epilepsia. 2008;49:594-599.
97 Hoffmeister B, Glaeser S, Flick H, et al. Cerebral toxocariasis after consumption of raw duck liver. Am J Trop Med Hyg. 2007;76:600-602.
98 Finsterer J, Auer H. Neurotoxocarosis. Rev Inst Med Trop Sao Paulo. 2007;49:279-287.