[level-membership-for-neurology-category]
Chapter 84 Paraneoplastic Syndromes Affecting the Nervous System
Introduction
Paraneoplastic neurologic syndromes (PNS) may be the presenting symptom of cancer [Tuzun and Dalmau, 2007]. The disorders may also occur in patients with known cancer and can be mistaken for treatment effects, opportunistic infections, or the results of other causes. Neurologic complications occur in approximately 20 percent of patients treated for cancer. Direct effects of the primary tumor and/or metastases are the most common cause, while treatment-related complications are secondary causes.
The focus of this chapter is PNS, a constellation of rare but well-described syndromes associated with cancer. The chapter will provide a brief overview of the history of PNS, describe the most common syndromes, and discuss the diagnosis, treatment, and management of several syndromes that specifically affect pediatric patients. For ease of reference, the clinically relevant aspects of PNS are summarized in Table 84-1.
Table 84-1 Paraneoplastic Syndromes and Associated Tumors and Antibodies
| Clinical Syndrome | Associated Tumors | Associated Antibodies* |
|---|---|---|
| Limbic encephalitis | SCLC, testicular cancer, thymoma, teratoma, Hodgkin’s disease, non-SCLC | Anti-Hu(ANNA-1), anti-Yo(PCA-1), anti-Ri(ANNA-2), ANNA-3, anti-Ma1, anti-Ma2(Ta), anti-amphiphysin, anti-CRMP5s anti CV2, PCA-2, anti-CRMP3,4, anti-NR2B,NR2A, antineuropil antibodies GABAB, AMPAR (anti GABAB receptor; anti-AMPA receptor) |
| Cerebellar degeneration | Breast cancer, ovarian cancer, SCLC, Hodgkin’s disease | Anti-Yo(PCA-1), anti-Hu(ANNA-1), anti-Tr, anti-Ri(ANNA-2), anti-mGluR1, anti-VGCC, anti-Ma1, anti-RMP5(CV2), anti-Zic4 |
| Opsoclonus-myoclonus | Neuroblastoma, SCLC, breast cancer | Anti-Ri(ANNA-2), anti-Yo(PCA-1), anti-Hu(ANNA-1), anti-Ma1, anti-Ma2(Ta), anti-amphiphysin, anti-CRMP5(CV2) |
| Stiff person syndrome | Breast cancer, SCLC, Hodgkin’s disease | Anti-amphiphysin, anti-GAD, anti-Ri(ANNA-2), anti-gephyrin |
| Retinopathies | SCLC, melanoma, breast cancer | Anti-recoverin, anti-enolase, anti-TULP1, anti-PTB-like protein, anti-photoreceptor cell-specific nuclear receptor, anti-CRMP5(CV2) |
| Motor neuron disease | Lymphoproliferative disorders, SCLC, breast cancer, ovarian cancer | Anti-Hu(ANNA-1), anti-Yo(PCA-1), anti-MAG, anti-SGPS, anti-gangliosides GM1, GM2, GD1a, and GD1b |
| Peripheral neuropathy | SCLC, thymoma, lymphoproliferative disorders | Anti-Hu(ANNA-1), anti-CRMP5(CV2), anti-MAG, anti-SGPS, anti-gangliosides GM1, GM2, GD1a, and GD1b |
| Neuromyotonia | Thymoma, Hodgkin’s disease, SCLC | Anti-VGKC, anti-Hu(ANNA-1) |
| Lambert–Eaton syndrome | SCLC | Anti-P/Q VGCC |
| Myasthenia gravis | Thymoma | Anti-AChR, anti-titin, anti-ryanodine |
| Inflammatory myopathies | Non-Hodgkin’s lymphoma, ovarian cancer, lung cancer, gastric cancer, pancreatic cancer, bladder cancer | Anti-Jo-1, anti-JO, anti-Mi-2, anti-p155 |
AMPAR, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; ANNA, antineuronal nuclear antibody; CRMP, collapsin response mediator protein; GABAB, gamma-aminobutyric acid type B receptors; GAD, glutamic acid decarboxylase; MAG, myelin-associated glycoprotein; NR, N-methyl-d-aspartate (NMDA) receptor; PCA, Purkinje cell autoantibody; PTB, polypyrimidine tract binding; SCLC, small-cell lung cancer; SGPS, sulfated glucuronic acid paragloboside; TULP1, tubby-like protein 1; VGCC, voltage-gated calcium channels; VGKC, voltage-gated potassium channel.
* Well-characterized onconeural antibodies are shown in bold.
(Adapted with permission from Toothaker TB, Rubin M. Paraneoplastic neurological syndromes: a review. Neurologist Jan 2009;15[1]:21–33.)
History of Paraneoplastic Syndromes
The 19th-century French physician, Armand Trousseau, is credited with describing the first paraneoplastic syndrome in 1865, when he made the association between cancer and thromboses [Trousseau, 1865]. In 1948, Derek Denny-Brown published a paper describing two patients with sensory neuronopathy and lung cancer [Denny-Brown, 1948]. Henson and Urich described the first case of encephalomyelitis and carcinoma in 1965 [Henson et al., 1965]. The term “paraneoplastic syndrome” was first used in the 1956 by Cabanne and colleagues, who reported on three patients with paraneoplastic polyneuritis in the setting of cancer [Cabanne et al., 1956]. Graus and colleagues identified the first antibody associated with PNS in 1985 [Graus et al., 1985]. In the past two decades, 16 identifiable syndromes and at least 10 paraneoplastic antibodies have been described. Neuroblastoma-associated opsoclonus-myoclonus-ataxia syndrome was the first PNS identified in children in 1968 by Solomon and Chutorian (see below). More recently, anti-N-methyl-d-aspartate (NMDA) antibodies have been associated with an encephalomyelitis that may be paraneoplastic in adults but is only rarely associated with an identifiable neoplasm in pediatric patients.
Definition
PNS are defined as illnesses resulting from the indirect effects of cancer. There are five groups of neurologic paraneoplastic disorders: vascular, infectious, metabolic, treatment-related, and remote effects of cancer on the nervous system [DeAngelis and Posner, 2009]. This chapter will focus on the remote effects of cancer on the nervous system. PNS affect less than 0.01 percent of cancer patients. Exceptions include Lambert–Eaton myasthenic syndrome (LEMS), which affects about 3 percent of patients with small-cell lung cancer (SCLC) [Payne et al., 2010]; and myasthenia gravis, which is associated with thymoma in about 5 percent of cases [Wirtz et al., 2003].
PNS may cause injury anywhere along the neuraxis. Paraneoplastic and/or limbic encephalitis (LE) can affect the supratentorial compartment of the brain, while the cerebellum is involved in paraneoplastic cerebellar degeneration and opsoclonus-myoclonus-ataxia syndrome. The visual pathway is affected in cancer-associated retinopathy, while the spinal cord can be injured in subacute motor neuronopathy. Sensory neuronopathy involves the sensory nerves and, in LEMS, the neuromuscular junction is the target. Finally, inflammatory myopathies, including dermatomyositis, polymyositis, and inclusion body myositis, are PNS that affect the muscle. Dermatomyositis and polymyositis may be PNS, but no more than 10 percent of patients with these neuromuscular disorders have cancer; immunosuppressive treatment is often effective [Darnell and Posner, 2006].
The PNS are divided into two major categories: classical syndromes that strongly suggest an associated malignancy, and nonclassical ones that are only occasionally associated with malignancy. In 2004, Graus and colleagues published official diagnostic criteria developed by a panel of neurologists [Graus et al., 2004]. They defined classic syndromes and nonclassic syndromes (Box 84-1). Classic syndromes include encephalomyelitis, cerebellar degeneration and sensory neuronopathy. In addition, Graus and colleagues distinguished between definite PNS and possible ones (Box 84-2) [Graus et al., 2004]. The neurologic symptoms of paraneoplastic syndromes typically present before the tumor is diagnosed. The tumors are often small and difficult to detect, and may spontaneously regress. Patients often survive the tumor but with persistent neurologic effects. The hypothesis is that patients develop neurologic symptoms because the tumor expresses onconeural antigens that cause a cellular and/or humoral response; this then cross-reacts with specific antigens in varied regions of the nervous system.
















Box 84-2 Diagnostic Criteria for Paraneoplastic Neurologic Syndromes
Definite PNS
Lambert–Eaton Myasthenic Syndrome
Several neurologic paraneoplastic disorders may affect the neuromuscular junction: LEMS, myasthenia gravis, and neuromyotonia. LEMS is considered one of the classical neurologic PNS and is a disorder of presynaptic neuromuscular transmission; this is in contradistinction to myasthenia gravis, which is a postsynaptic disorder. Initially, patients complain of weakness and fatigue, followed by sleepiness, autonomic dysfunction, muscle weakness, and diminished deep tendon reflexes. Electromyography is helpful in distinguishing between LEMS and myasthenia gravis; patients with LEMS have an incremental response to repetitive muscle stimulation, while those with myasthenia gravis have a decremental response. The Tensilon test may also be helpful in distinguishing LEMS from myasthenia gravis. As edrophonium chloride blocks acetylcholinesterase, patients with myasthenia gravis will have a positive response to the Tensilon test, while LEMS patients have antibodies against the presynaptic receptor and thus do not respond to Tensilon. Between 40 and 60 percent of patients with LEMS harbor a malignancy, and, as in the other PNS, neurologic symptoms often precede tumor diagnosis. The most common malignancy associated with LEMS is SCLC. Recently, Payne and colleagues examined 63 patients with the diagnosis of SCLC and found that 5 (8 percent) had high titers of antibodies to voltage-gated calcium channels present in the serum, with 2 (3 percent) patients exhibiting clinical and electrophysiologic evidence of LEMS [Payne et al., 2010]. In pediatric patients, there have been several reports of neuroblastoma-associated LEMS [Bosdure et al., 2006; de Buys Roessingh et al., 2009]. In adults, both myasthenia gravis (thymoma) and LEMS (SCLC) may be the presenting complaint in a patient with an underlying cancer. Both illnesses respond to treatment [Darnell and Posner, 2006].
Paraneoplastic Cerebellar Degeneration
Paraneoplastic cerebellar degeneration (PCD) was actually the first paraneoplastic syndrome to be described and, as such, is the best described and most easily recognized. In 1919, Brouwer et al., described a patient with rapid onset of a cerebellar syndrome [Brouwer, 1919]. At autopsy, the patient had Purkinje cell loss in the cerebellum and a pelvic tumor, most likely ovarian carcinoma. In 1938, Brouwer went on to postulate the association between PCD and cancer [Brouwer, 1938]. PCD may occur in isolation but may be associated with other PNS, including LE and LEMS.
PCD is most commonly associated with SCLC, gynecologic cancers, and Hodgkin’s lymphoma. Symptoms often begin explosively, with dizziness, nausea, and vomiting. Patients may develop diplopia and, subsequently, both truncal and appendicular ataxia, dysarthria, and dysphagia. Cerebrospinal fluid may have mild pleocytosis and magnetic resonance imaging (MRI) may reveal edema of the cerebellum. Autopsy usually shows massive Purkinje cell destruction. Patients with SCLC and PCD may also develop LEMS (see below) due to the presence of antibodies to voltage-gated calcium channels, which may cause both syndromes. Several paraneoplastic antibodies have been identified in patients with PCD. The target of these antibodies is typically the Purkinje cell layer in the cerebellum. The anti-Yo antibody is the most frequently detected antibody in patients with PCD and ovarian or breast cancer. Patients with gynecologic cancers who develop PCD invariably harbor anti-Yo antibodies, which recognize a major protein, cDR2 (cerebellar degeneration-related protein), expressed in the cytoplasm of Purkinje cells. All patients with PCD-associated breast and ovarian cancer express cDR2 antigens on their tumor cells. Approximately 60 percent of patients with ovarian cancer who do not have PCD express cDR2 on their tumor cells. Anti-Yo antibody titers are usually higher in cerebrospinal fluid than serum. Cerebrospinal fluid may show mild pleocytosis with elevated protein. About 50 percent of patients with SCLC and PCD do not harbor classic onconeural antibodies. Recently, Sabater and colleagues described the Zic zinc finger protein-associated syndrome, in which MRI is initially normal but, as the disease progresses, cerebellar atrophy may become more prominent [Sabater et al., 2008].
Retinopathies
Paraneoplastic visual disorders are rare. Amongst these, cancer-associated retinopathy (CAR) and melanoma-associated retinopathy (MAR) are the most common syndromes [Adamus, 2009]. CAR was first reported in 1976 by Sawyer and colleagues, who evaluated three women with visual loss before or after the diagnosis of cancer was made [Sawyer et al., 1976]. In 50 percent of patients, CAR precedes the diagnosis of cancer, typically SCLC and gynecologic cancers.
Patients complain of painless visual loss, which may begin with night blindness. On physical examination, the retina may initially appear normal, but later is mottled with optic disc pallor and retinal artery narrowing. CAR affects rods and cones and, histologically, shows loss of both inner and outer layers with occasional inflammation. Vitreous and cerebrospinal fluid may contain inflammatory cells. Several antibodies have been identified, but most commonly, anti-recoverin is found. Recoverin is a member of a family of calcium-binding proteins involved in the transduction of light by photoreceptors. Recoverin has been found to be expressed in the lung tumor of a CAR patient, but not in similar tumors obtained from individuals without the associated retinopathy [Polans et al., 1995].
Subacute Sensory Neuronopathy
PNS can affect peripheral nerves, neuromuscular junction, or muscle. In one series, 9 percent of patients referred to a center for evaluation of a peripheral neuropathy had cancer as the underlying cause [Antoine et al., 1999]. The neuropathy may be axonal or demyelinating, or, in the case of a pure sensory neuropathy, a dorsal root ganglionopathy. In patients older than 50 years, particularly smokers and those with acutely developing, purely sensory neuropathy, a PNS is likely.
In 1948, Denny-Brown described two patients with a rapidly developing and debilitating sensory neuronopathy. Pathologic evaluation revealed inflammatory infiltrates within the dorsal root ganglia and degeneration of the posterior columns of the spinal cord [Denny-Brown, 1948]. In 1985, Graus and colleagues identified an antibody called anti-Hu, associated with patients who have SCLC and subacute sensory neuronopathy (SSN). Anti-Hu patients typically have sensory loss and paresthesias, which may affect the extremities initially, but may also include the neck and trunk and can occasionally be asymmetric. All sensory modalities are involved, but proprioceptive loss can severely impair gait and use of the hands. Anti-Hu antibody is a 35–40 kDa protein, which immunoreacts with the nuclei of neurons. The antigen is expressed in the nuclei of most neurons in the central nervous system, including those in the dorsal root ganglia, explaining why the anti-Hu neurologic paraneoplastic syndrome may cause encephalomyelitis (PEMS) and/or SSN in the same patient. In addition, most SCLCs express the antigen. In 1992, Dalmau and colleagues identified 71 patients with PEMS/SSN in association with the presence of anti-Hu antibodies [Dalmau et al., 1992]. Seventy-eight percent had SCLC. In 9 patients, no tumor was detected. In the others, tumors included prostate cancer (2), adenocarcinoma of the lung (1), chondromyxosarcoma (1), and, interestingly, neuroblastoma (1).
Opsoclonus-Myoclonus-Ataxia Syndrome
Case 1
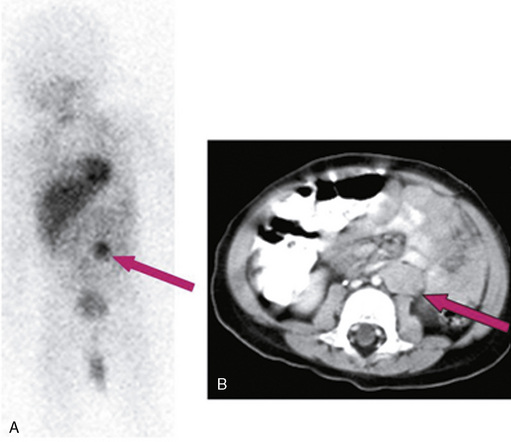
MS is a previously healthy, 18-month-old girl who developed sudden onset of ataxia, chaotic rapid eye movements, and irritability. MRI of the brain was normal and meta-iodo-benzyl-guanidine (MIBG) testing revealed uptake in the left adrenal gland (Figure 84-1). Thin-cut, contrast-enhanced computed tomography (CT) of the chest/abdomen/pelvis confirmed a homogeneously enhancing mass in the left adrenal gland. Tumor was resected and found to be consistent with favorable histology; it was a non-MYCN amplified neuroblastoma. The patient did not require further therapy for neuroblastoma; however, for treatment of refractory OMA syndrome, she received monthly intravenous gammaglobulin for 2 years, intramuscular adrenocorticotropic hormone (ACTH) for 14 months, and a 4-week course of rituximab. Motor symptoms of OMA completely resolved after 24 months of therapy. The patient is now 6 years old and “mainstreamed” for education; gross motor symptoms have resolved and she receives weekly occupational and speech therapy. Her neuroblastoma has been in remission since surgery.
History
Orzechowski first coined the term in 1913, based on the Greek term “opso” meaning eye and “clonus” meaning confused: “the ocular globes are in a state of continuous agitation, being shaken and increasingly displaced by very rapid unequal movements, which generally take place in the horizontal plane.” In 1927, he noted the association between opsoclonus and myoclonus [Orzechowski, 1927]. Kinsbourne described a syndrome of myoclonic encephalopathy in 1962 [Kinsbourne, 1962], and finally, in 1968, Solomon and Chutorian described the association between neuroblastoma and OMA [Solomon and Chutorian, 1968].
Incidence
The incidence of OMA is estimated at 1 in 10 million, with the syndrome occurring in 2–3 percent of all neuroblastoma patients. Up to 50 percent of children with OMA harbor a neuroblastoma; some researchers speculate that the incidence of neuroblastoma in children with OMA is actually higher, but a cellular and/or antibody-mediated response destroys the tumor before detection. OMA in children has been reported in certain infections, including those caused by Epstein–Barr virus, Streptococcus spp., and Mycoplasma pneumoniae [Huber et al., 2010]. OMA is paraneoplastic in 20 percent of adults. Typically, patients have breast or gynecologic cancers.
Antibodies
The most common antibody associated with OMA in adults is the anti-Ri antibody, which binds to Nova, an RNA binding protein important in regulating inhibitory neurotransmitters in the brainstem. Despite multiple attempts to isolate a specific antibody in pediatric patients with OMA, none has been identified to date. Approximately 75 percent of neuroblastomas express anti-Hu antigen, but only 5–15 percent of serum samples contain the anti-Hu antibody. Classically, OMA precedes the diagnosis of neuroblastoma. Recently, Kurian and colleagues described a 23-year-old woman with OMA syndrome plus other neurologic abnormalities (facial paresis, frontal lobe behavior) [Kurian et al., 2010]. Extensive evaluation for malignancy was negative but the patient did have anti-NMDA receptor antibodies present in the cerebrospinal fluid.
Evaluation and Treatment
MRI of the brain is typically normal at onset of OMA, although it may reveal cerebellar atrophy at a later time. Lumbar puncture early in the course of the illness may contain normal to slightly elevated protein, with no cells or predominantly lymphocytes. Children with OMA should have an MIBG scan in an effort to identify the neuroblastoma. Thin-cut CT through the area of increased uptake will help identify the tumor. Patients usually have lower-stage/grade neuroblastoma, with favorable histology and unamplified MYCN [Rudnick et al., 2001]. Treatment of the OMA begins with surgical resection of the tumor, and is typically followed by immunomodulation. For 2 days perioperatively, intravenous gammaglobulin is administered at a dose of 1 g/kg, as surgery may transiently increase symptoms. There are numerous protocols for immune suppression thereafter but, clearly, immunomodulation is the next treatment of choice. In 1997, Russo and colleagues suggested that children with OMA-associated neuroblastoma had better neurologic outcome if they received cyclophosphamide chemotherapy as part of the treatment for neuroblastoma [Russo et al., 1997]. Pranzatelli and colleagues at the National Pediatric Myoclonus Center have advocated the use of ACTH as follows: within 1 week of surgery, patients are started on ACTH 75 IU/m2/day intramuscularly twice daily. The ACTH is tapered quickly in the first month and then more slowly over the next 11 months. If OMA recurs, the previous dose of ACTH is reinstituted. Recently, high-dose pulse dexamethasone has been successfully used with lower cost of treatment and fewer long-term side effects than ACTH [Ertle et al., 2008]. Pranzatelli and colleagues identified B-cell expansions in the cerebrospinal fluid of many of these patients and found that a short course of rituximab (375 mg/m2/week for 4 consecutive weeks) improves symptoms [Pranzatelli et al., 2006]. If a prominent T-cell population is noted in the cerebrospinal fluid of refractory patients, then cyclophosphamide is recommended at a dose of 750 mg/m2/day monthly for 3–6 months. Patients with OMA should not receive immunizations, as any stimulus to the immune system may exacerbate the symptoms.
Prognosis
Patients with neuroblastoma-associated OMA typically have better overall survival than patients without OMA [Rudnick et al., 2001]. Their neurologic and cognitive outcomes, however, are generally not as good. In a recent publication and subsequent follow-up abstract by Catsman-Berrevoets, 9 children with OMA (4 with neuroblastoma) were followed for a median of 11 years. Despite severe motor deficits initially, patients actually recovered these functions quite well; in 7 out of 9 patients, however, severe cognitive and behavioral impairments persisted [Catsman-Berrevoets et al., 2009a, b].
Pathophysiology
As yet, the pathophysiology of OMA is poorly understood. In 2003, Helmchen and colleagues performed functional MRI in two adults with OMA syndrome and demonstrated disinhibition of the fastigial nucleus in the cerebellum [Helmchen et al., 2003]. As mentioned above, unlike adults, children with OMA do not have a specific antibody present in the cerebrospinal fluid and serum. The initial suggestion that the syndrome is humorally mediated is being challenged as more evidence supports a cellular etiology. Though the neuroblasts of OMA-associated neuroblastomas appear histologically identical to those tumors in patients without OMA, the tumors themselves are quite distinctive. Patients with OMA-associated neuroblastoma have large lymphocytic infiltrates present in the tumor specimens. In a study by Cooper and colleagues, the pathologist could identify which patients had OMA simply by looking at the tumors in 92 percent of cases [Cooper et al., 2001].
Paraneoplastic Encephalitides
The paraneoplastic encephalitides include LE and, more recently, anti-NMDA receptor encephalitis.
Limbic Encephalitis
Paraneoplastic LE was first described in 1968 by Corsellis and colleagues, who published reports on a clinicopathologic series of patients with carcinoma and neurologic symptoms consisting of seizures and memory loss [Corsellis et al., 1968]. Since then, investigators have further characterized paraneoplastic antibodies associated with LE.
In LE, the presentation is generally abrupt (days to weeks), with onset of personality/behavior changes, seizures and alteration of consciousness. Acute and persistent memory loss with behavioral changes may be the first presenting symptom of malignancies such as SCLC [Tuzun and Dalmau, 2007]. When these symptoms occur in a previously well person, LE, either paraneoplastic or non-paraneoplastic (autoimmune), must be considered. Transient memory loss, as in transient global amnesia, is a more common problem, but not a specific complication of cancer.
In any adult patient with acute memory loss, even when MRI does not reveal the characteristic medial temporal lobe lesions, one should consider assaying for paraneoplastic antibodies. If no paraneoplastic antibodies are found and polymerase chain reaction rules out herpes simplex encephalitis, cancer must be sought, usually by body positron emission tomography (PET) imaging [McKeon et al., 2010].
History
In 1997, Nokura and colleagues reported a case of a 19-year-old woman with psychosis and confusion, who was found to have an ovarian teratoma [Nokura et al., 1997]. Once the teratoma was removed, the patient made a partial recovery. At that time, no clear paraneoplastic antibody was identified. Other similar cases were subsequently described, but the first antibody was not identified until 2005, when Vitaliani and colleagues reported four young women with ovarian teratoma and acute behavioral and psychiatric changes, neurologic deterioration, seizures, orofacial dyskinesias, and central hypoventilation [Vitaliani et al., 2005]. Cerebrospinal fluid and serum revealed a novel antibody against EFA6A, a protein expressed in the cytoplasm of hippocampal neurons. In 2007, Dalmau and colleagues further characterized these antibodies as acting against the NR1 subunit of the NR1/NR2 heteromers of the NMDA receptor, also expressed on the tumors [Dalmau et al., 2007]. In 2008, the same group analyzed 100 cases of patients with anti-NMDA receptor encephalitis [Dalmau et al., 2008]. Ninety-one percent of patients were women; 59 percent had tumors (primarily ovarian teratomas), and 1 man had SCLC. Twenty-two were 18 years old or less, and 55 percent of these patients had an underlying tumor. Severe neurologic deficits, including poor responsiveness, catatonia, abnormal movements, and central hypoventilation, were present in 93 patients. Ninety-two percent of patients had abnormal EEGs; 95 percent had abnormal cerebrospinal fluid profiles, and 55 percent had FLAIR or T2 signal abnormalities on MRI. Despite severe neurologic deficits, 75 percent of patients made a complete recovery, while 25 percent had severe deficits or died. In 2009, Florance and colleagues reported 81 patients with NMDA receptor encephalitis from several institutions [Florance et al., 2009]. Thirty-two patients were 18 years or younger. Seventy-seven percent of pediatric patients had seizures, 84 percent had movement disorders, and 60 percent had personality or behavioral changes. Teratomas were identified in 25 percent of patients, all of whom were adults. As mentioned above, Kurian et al. reported a 23-year-old woman with OMA syndrome plus features of LE, who was diagnosed with NMDA receptor encephalitis [Kurian et al., 2010]. A tumor was not identified in this patient and, although she made a significant recovery with immunotherapy, she had persistent frontal lobe behavioral abnormalities 5 months after diagnosis. In 2009, at the European Paediatric Neurologic Society meeting, multiple groups reported individual children with, and pediatric patient case series of, NMDA receptor encephalitis, none of whom had evidence of malignancy; most of them had a favorable neurologic outcome [Gowda et al., 2009; Poloni et al., 2009].
Other Encephalitides
Recently, two new paraneoplastic encephalitides have been described in adults: gamma-aminobutyric acid type B receptors (anti-GABAB) and anti-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis [Lancaster et al., 2010; Lai et al., 2009].
Recent findings indicate that certain subgroups of LE, such as those caused by autoantibodies against the GABAB1 subunit of the GABAB receptor, are potentially treatable. Neurologic symptoms in GABAB LE include early or prominent seizures; MRI and EEG findings are consistent with predominant limbic dysfunction. In some patients, it is associated with SCLC and other autoantibodies [Lancaster et al., 2010]. Another recently described subgroup of LE caused by antibodies against neuronal cell-surface antigens were found to be against the glutamate receptor 1 (GluR1) and GluR2 subunits of the AMPA receptor [Lai et al., 2009].
Cancer is identified in less than 70 percent of patients with AMPA receptor LE [Lai et al., 2009], 50 percent of whom have antibodies against the GABAB receptor [Lancaster et al., 2010], and 20 percent of whom have antibodies against voltage-gated potassium channels [Vincent et al., 2004].
Antibodies against the GABAB receptor are reported as frequently being associated with those against glutamic acid decarboxylase 65 (GAD65) [Lancaster et al., 2010], as already reported for antibodies against the AMPA receptor [Lai et al., 2009; Honnorat, 2010]. The neurological presentation and the outcome of patients with LE and antibodies against neuronal cell-surface antigens, such as AMPA receptor or GABAB, differ from those of paraneoplastic LE, in which antibodies are directed against intracytoplasmic targets. In addition to symptoms from LE, patients with antibodies directed against intracytoplasmic targets also present with other neurological symptoms. For example, sensory neuronopathy or encephalomyelitis is frequently present in LE with anti-Hu antibodies [Honnorat et al., 2009].
More than 95 percent of patients with antibodies against intracytoplasmic targets have cancer, and the outcome is poor with limited response to treatment [Dalmau et al., 2004; Honnorat et al., 2009]. By contrast, association with cancer seems to be less frequent in patients with antibodies against neuronal cell-surface antigens.
In the 15 described patients with GABAB receptor LE, 7 had tumors, 5 of which were SCLC, and 7 patients had non-neuronal autoantibodies. Although 9 of 10 patients who received treatment showed neurological improvement, none of the 4 who were not similarly treated improved. Similarly, AMPA receptor LE seems to respond to treatment but has a tendency to relapse [Polans et al., 1995].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Adamus G. Autoantibody targets and their cancer relationship in the pathogenicity of paraneoplastic retinopathy. Autoimmun Rev. 2009;8(5):410-414.
Antoine J.C., Mosnier J.F., Absi L., et al. Carcinoma associated paraneoplastic peripheral neuropathies in patients with and without anti-onconeural antibodies. J Neurol Neurosurg Psychiatry. 1999;67(1):7-14.
Bosdure E., Attarian S., Mancini J., et al. Lambert-Eaton myastenic syndrome revealing neuroblastoma in 2 children. Arch Pediatr. 2006;13(8):1121-1124.
Brouwer B. Beitrag zur kenntnis der chronischen diffusen kleinhirnerkrankungen. Mendels neurologisches Zentralblblatt. 1919;38:674-682.
Brouwer B. Les affections parenchymateuses du cervelet et leur signification du point de vue de l’anatomie et de la physiologie de cet organ. J Belge Neurol Pscychiat. 1938;38:691-757.
Cabanne F., Fayolle J., Guichard A., et al. Polyneuritis in cancer patients and neoplastic polyneuritis; three case reports. Lyon Med. 1956;88(41):309-329.
Catsman-Berrevoets C.E., Aarsen F.K., van Hemsbergen M.L., et al. Improvement of neurological status and quality of life in children with opsoclonus myoclonus syndrome at long-term follow-up. Pediatr Blood Cancer. 2009;53(6):1048-1053.
Catsman-Berrevoets C.A.F., van Hemsbergen M.L.C., Van Noesel M.M., et al. Persistent improvement of neurologic status and quality of life in children with opsoclonus-myoclonus syndrome at long term. European Pediatric Neurology Society. 2009:S100. United Kingdom
Cooper R., Khakoo Y., Matthay K.K., et al. Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: histopathologic features-a report from the Children’s Cancer Group. Med Pediatr Oncol. 2001;36(6):623-629.
Corsellis J.A., Goldberg G.J., Norton A.R. “Limbic encephalitis” and its association with carcinoma. Brain. 1968;91(3):481-496.
Dalmau J., Gleichman A.J., Hughes E.G., et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091-1098.
Dalmau J., Graus F., Rosenblum M.K., et al. Anti-Hu–associated paraneoplastic encephalomyelitis/sensory neuronopathy. A clinical study of 71 patients. Medicine (Baltimore). 1992;71(2):59-72.
Dalmau J., Graus F., Villarejo A., et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain. 2004;127(Pt 8):1831-1844.
Dalmau J., Tüzün E., Wu H.-Y, et al. Paraneoplastic anti-N-methyl-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36.
Darnell R.B., Posner J.B. Paraneoplastic syndromes affecting the nervous system. Semin Oncol. 2006;33(3):270-298.
DeAngelis L.M., Posner J.B. Neurologic complications of cancer, ed 2. Oxford University Press; 2009.
de Buys Roessingh A.S., Loriot M.-H., Wiesenauer C., et al. Lambert-Eaton myasthenic syndrome revealing an abdominal neuroblastoma. J Pediatr Surg. 2009;44(8):e5-e7.
Denny-Brown D. Primary sensory neuropathy with muscular changes associated with carcinoma. J Neurol Neurosurg Psychiatry. 1948;11(2):73-87.
Ertle F., Behnisch W., Mulla N.A.A., et al. Treatment of neuroblastoma-related opsoclonus-myoclonus-ataxia syndrome with high-dose dexamethasone pulses. Pediatr Blood Cancer. 2008;50(3):683-687.
Florance N.R., Davis R.L., Lam C., et al. Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol. 2009;66(1):11-18.
Gowda V.M.B., Pike M.G., Hewertson J., et al. Two children with NMDAr +ve limbic encephalitis. United Kingdom: European Pediatric Neurology Society; 2009:S102.
Graus F., Cordon-Cardo C., Posner J.B. Neuronal antinuclear antibody in sensory neuronopathy from lung cancer. Neurology. 1985;35(4):538-543.
Graus F., Delattre J.Y., Antoine J.C., et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75(8):1135-1140.
Helmchen C., Rambold H., Sprenger A., et al. Cerebellar activation in opsoclonus: an fMRI study. Neurology. 2003;61(3):412-415.
Henson R., Hoffman H., Urich H. Encephalomyelitis with carcinoma. Brain. 1965;88(3):449-464.
Honnorat J.. Autoimmune limbic encephalitis: an expanding concept. 2010.http://www.mdconsult.com/das/article/body/263531790-853/jorg=journal&source=&sp=22810799&sid=0/N/730278/1.html?issn=1474-4422-r09703333007.
Honnorat J., Cartalat-Carel S., Ricard D., et al. Onco-neural antibodies and tumour type determine survival and neurological symptoms in paraneoplastic neurological syndromes with Hu or CV2/CRMP5 antibodies. J Neurol Neurosurg Psychiatry. 2009;80(4):412-416.
Huber B.M., Strozzi S., Steinlin M., et al. Mycoplasma pneumoniae associated opsoclonus-myoclonus syndrome in three cases. Eur J Pediatr. 2010;169(4):441-445.
Kinsbourne M. Myoclonic encephalopathy of infants. J Neurol Neurosurg Psychiatry. 1962;25:271-276.
Kurian M., Lalive P.H., Dalmau J.O., et al. Opsoclonus-myoclonus syndrome in anti-N-methyl-D-aspartate receptor encephalitis. Arch Neurol. 2010;67(1):118-121.
Lai M., Hughes E.G., Peng X., et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65(4):424-434.
Lancaster E., Lai M., Peng X., et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 2010;9(1):67-76.
McKeon A., Apiwattanakul M., Lachance D.H., et al. Positron Emission Tomography- Computed Tomography in Paraneoplastic Neurologic Disorders: Systematic Analysis and Review. Arch Neurol. 2010. Jan 11
Nokura K., Yamamoto H., Okawara Y., et al. Reversible limbic encephalitis caused by ovarian teratoma. Acta Neurol Scand. 1997;95(6):367-373.
Orzechowski K. De l’ataxie dysmetrique des yeux: Remarques sur l’ataxie des yeux dite myoclonique (opsoclonie, opsochorie). J Psychol Neurol. 1927;35:1-18.
Payne M., Bradbury P., Lang B., et al. Prospective study into the incidence of Lambert Eaton myasthenic syndrome in small cell lung cancer. J Thorac Oncol. 2010;5(1):34-38.
Polans A.S., Witkowska D., Haley T.L., et al. Recoverin, a photoreceptor-specific calcium-binding protein, is expressed by the tumor of a patient with cancer-associated retinopathy. Proc Natl Acad Sci USA. 1995;92(20):9176-9180.
Poloni C.K.C., King M., Deonna T., et al. Severe childhood encephalopathy with dyskinesia and cognitive disturbances: evidence for andi-NMDA receptor encephalopathy. United Kingdom: European Pediatric Neurology Society; 2009:S99.
Pranzatelli M.R., Tate E.D., Travelstead A.L., et al. Rituximab (anti-CD20) adjunctive therapy for opsoclonus-myoclonus syndrome. J Pediatr Hematol Oncol. 2006;28(9):585-593.
Rudnick E., Khakoo Y., Antunes N.L., et al. Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: clinical outcome and antineuronal antibodies-a report from the Children’s Cancer Group Study. Med Pediatr Oncol. 2001;36(6):612-622.
Russo C., Cohn S.L., Petruzzi M.J., et al. Long-term neurologic outcome in children with opsoclonus-myoclonus associated with neuroblastoma: a report from the Pediatric Oncology Group. Med Pediatr Oncol. 1997;28(4):284-288.
Sabater L., Bataller L., Suarez-Calvet M., et al. ZIC antibodies in paraneoplastic cerebellar degeneration and small cell lung cancer. J Neuroimmunol. 2008;201–202:163-165.
Sawyer R.A., Selhorst J.B., Zimmerman L.E., et al. Blindness caused by photoreceptor degeneration as a remote effect of cancer. Am J Ophthalmol. 1976;81(5):606-613.
Solomon G.E., Chutorian A.M. Opsoclonus and occult neuroblastoma. N Engl J Med. 1968;279(9):475-477.
Trousseau A. Phlegmasia alba dolens. Clin Med Hotel Dieu de Paris. 1865;3:94.
Tuzun E., Dalmau J. Limbic encephalitis and variants: classification, diagnosis and treatment. Neurologist. 2007;13(5):261-271.
Vincent A., Buckley C., Schott J.M., et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004;127(Pt 3):701-712.
Vitaliani R., Mason W., Ances B., et al. Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol. 2005;58(4):594-604.
Wirtz P.W., Nijnuis M.G., Sotodeh M., et al. The epidemiology of myasthenia gravis, Lambert-Eaton myasthenic syndrome and their associated tumours in the northern part of the province of South Holland. J Neurol. 2003;250(6):698-701.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 84 Paraneoplastic Syndromes Affecting the Nervous System
Introduction
Paraneoplastic neurologic syndromes (PNS) may be the presenting symptom of cancer [Tuzun and Dalmau, 2007]. The disorders may also occur in patients with known cancer and can be mistaken for treatment effects, opportunistic infections, or the results of other causes. Neurologic complications occur in approximately 20 percent of patients treated for cancer. Direct effects of the primary tumor and/or metastases are the most common cause, while treatment-related complications are secondary causes.
The focus of this chapter is PNS, a constellation of rare but well-described syndromes associated with cancer. The chapter will provide a brief overview of the history of PNS, describe the most common syndromes, and discuss the diagnosis, treatment, and management of several syndromes that specifically affect pediatric patients. For ease of reference, the clinically relevant aspects of PNS are summarized in Table 84-1.
Table 84-1 Paraneoplastic Syndromes and Associated Tumors and Antibodies
| Clinical Syndrome | Associated Tumors | Associated Antibodies* |
|---|---|---|
| Limbic encephalitis | SCLC, testicular cancer, thymoma, teratoma, Hodgkin’s disease, non-SCLC | Anti-Hu(ANNA-1), anti-Yo(PCA-1), anti-Ri(ANNA-2), ANNA-3, anti-Ma1, anti-Ma2(Ta), anti-amphiphysin, anti-CRMP5s anti CV2, PCA-2, anti-CRMP3,4, anti-NR2B,NR2A, antineuropil antibodies GABAB, AMPAR (anti GABAB receptor; anti-AMPA receptor) |
| Cerebellar degeneration | Breast cancer, ovarian cancer, SCLC, Hodgkin’s disease | Anti-Yo(PCA-1), anti-Hu(ANNA-1), anti-Tr, anti-Ri(ANNA-2), anti-mGluR1, anti-VGCC, anti-Ma1, anti-RMP5(CV2), anti-Zic4 |
| Opsoclonus-myoclonus | Neuroblastoma, SCLC, breast cancer | Anti-Ri(ANNA-2), anti-Yo(PCA-1), anti-Hu(ANNA-1), anti-Ma1, anti-Ma2(Ta), anti-amphiphysin, anti-CRMP5(CV2) |
| Stiff person syndrome | Breast cancer, SCLC, Hodgkin’s disease | Anti-amphiphysin, anti-GAD, anti-Ri(ANNA-2), anti-gephyrin |
| Retinopathies | SCLC, melanoma, breast cancer | Anti-recoverin, anti-enolase, anti-TULP1, anti-PTB-like protein, anti-photoreceptor cell-specific nuclear receptor, anti-CRMP5(CV2) |
| Motor neuron disease | Lymphoproliferative disorders, SCLC, breast cancer, ovarian cancer | Anti-Hu(ANNA-1), anti-Yo(PCA-1), anti-MAG, anti-SGPS, anti-gangliosides GM1, GM2, GD1a, and GD1b |
| Peripheral neuropathy | SCLC, thymoma, lymphoproliferative disorders | Anti-Hu(ANNA-1), anti-CRMP5(CV2), anti-MAG, anti-SGPS, anti-gangliosides GM1, GM2, GD1a, and GD1b |
| Neuromyotonia | Thymoma, Hodgkin’s disease, SCLC | Anti-VGKC, anti-Hu(ANNA-1) |
| Lambert–Eaton syndrome | SCLC | Anti-P/Q VGCC |
| Myasthenia gravis | Thymoma | Anti-AChR, anti-titin, anti-ryanodine |
| Inflammatory myopathies | Non-Hodgkin’s lymphoma, ovarian cancer, lung cancer, gastric cancer, pancreatic cancer, bladder cancer | Anti-Jo-1, anti-JO, anti-Mi-2, anti-p155 |
AMPAR, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; ANNA, antineuronal nuclear antibody; CRMP, collapsin response mediator protein; GABAB, gamma-aminobutyric acid type B receptors; GAD, glutamic acid decarboxylase; MAG, myelin-associated glycoprotein; NR, N-methyl-d-aspartate (NMDA) receptor; PCA, Purkinje cell autoantibody; PTB, polypyrimidine tract binding; SCLC, small-cell lung cancer; SGPS, sulfated glucuronic acid paragloboside; TULP1, tubby-like protein 1; VGCC, voltage-gated calcium channels; VGKC, voltage-gated potassium channel.
* Well-characterized onconeural antibodies are shown in bold.
(Adapted with permission from Toothaker TB, Rubin M. Paraneoplastic neurological syndromes: a review. Neurologist Jan 2009;15[1]:21–33.)
History of Paraneoplastic Syndromes
The 19th-century French physician, Armand Trousseau, is credited with describing the first paraneoplastic syndrome in 1865, when he made the association between cancer and thromboses [Trousseau, 1865]. In 1948, Derek Denny-Brown published a paper describing two patients with sensory neuronopathy and lung cancer [Denny-Brown, 1948]. Henson and Urich described the first case of encephalomyelitis and carcinoma in 1965 [Henson et al., 1965]. The term “paraneoplastic syndrome” was first used in the 1956 by Cabanne and colleagues, who reported on three patients with paraneoplastic polyneuritis in the setting of cancer [Cabanne et al., 1956]. Graus and colleagues identified the first antibody associated with PNS in 1985 [Graus et al., 1985]. In the past two decades, 16 identifiable syndromes and at least 10 paraneoplastic antibodies have been described. Neuroblastoma-associated opsoclonus-myoclonus-ataxia syndrome was the first PNS identified in children in 1968 by Solomon and Chutorian (see below). More recently, anti-N-methyl-d-aspartate (NMDA) antibodies have been associated with an encephalomyelitis that may be paraneoplastic in adults but is only rarely associated with an identifiable neoplasm in pediatric patients.
Definition
PNS are defined as illnesses resulting from the indirect effects of cancer. There are five groups of neurologic paraneoplastic disorders: vascular, infectious, metabolic, treatment-related, and remote effects of cancer on the nervous system [DeAngelis and Posner, 2009]. This chapter will focus on the remote effects of cancer on the nervous system. PNS affect less than 0.01 percent of cancer patients. Exceptions include Lambert–Eaton myasthenic syndrome (LEMS), which affects about 3 percent of patients with small-cell lung cancer (SCLC) [Payne et al., 2010]; and myasthenia gravis, which is associated with thymoma in about 5 percent of cases [Wirtz et al., 2003].
PNS may cause injury anywhere along the neuraxis. Paraneoplastic and/or limbic encephalitis (LE) can affect the supratentorial compartment of the brain, while the cerebellum is involved in paraneoplastic cerebellar degeneration and opsoclonus-myoclonus-ataxia syndrome. The visual pathway is affected in cancer-associated retinopathy, while the spinal cord can be injured in subacute motor neuronopathy. Sensory neuronopathy involves the sensory nerves and, in LEMS, the neuromuscular junction is the target. Finally, inflammatory myopathies, including dermatomyositis, polymyositis, and inclusion body myositis, are PNS that affect the muscle. Dermatomyositis and polymyositis may be PNS, but no more than 10 percent of patients with these neuromuscular disorders have cancer; immunosuppressive treatment is often effective [Darnell and Posner, 2006].
The PNS are divided into two major categories: classical syndromes that strongly suggest an associated malignancy, and nonclassical ones that are only occasionally associated with malignancy. In 2004, Graus and colleagues published official diagnostic criteria developed by a panel of neurologists [Graus et al., 2004]. They defined classic syndromes and nonclassic syndromes (Box 84-1). Classic syndromes include encephalomyelitis, cerebellar degeneration and sensory neuronopathy. In addition, Graus and colleagues distinguished between definite PNS and possible ones (Box 84-2) [Graus et al., 2004]. The neurologic symptoms of paraneoplastic syndromes typically present before the tumor is diagnosed. The tumors are often small and difficult to detect, and may spontaneously regress. Patients often survive the tumor but with persistent neurologic effects. The hypothesis is that patients develop neurologic symptoms because the tumor expresses onconeural antigens that cause a cellular and/or humoral response; this then cross-reacts with specific antigens in varied regions of the nervous system.
Box 84-2 Diagnostic Criteria for Paraneoplastic Neurologic Syndromes
Definite PNS
Lambert–Eaton Myasthenic Syndrome
Several neurologic paraneoplastic disorders may affect the neuromuscular junction: LEMS, myasthenia gravis, and neuromyotonia. LEMS is considered one of the classical neurologic PNS and is a disorder of presynaptic neuromuscular transmission; this is in contradistinction to myasthenia gravis, which is a postsynaptic disorder. Initially, patients complain of weakness and fatigue, followed by sleepiness, autonomic dysfunction, muscle weakness, and diminished deep tendon reflexes. Electromyography is helpful in distinguishing between LEMS and myasthenia gravis; patients with LEMS have an incremental response to repetitive muscle stimulation, while those with myasthenia gravis have a decremental response. The Tensilon test may also be helpful in distinguishing LEMS from myasthenia gravis. As edrophonium chloride blocks acetylcholinesterase, patients with myasthenia gravis will have a positive response to the Tensilon test, while LEMS patients have antibodies against the presynaptic receptor and thus do not respond to Tensilon. Between 40 and 60 percent of patients with LEMS harbor a malignancy, and, as in the other PNS, neurologic symptoms often precede tumor diagnosis. The most common malignancy associated with LEMS is SCLC. Recently, Payne and colleagues examined 63 patients with the diagnosis of SCLC and found that 5 (8 percent) had high titers of antibodies to voltage-gated calcium channels present in the serum, with 2 (3 percent) patients exhibiting clinical and electrophysiologic evidence of LEMS [Payne et al., 2010]. In pediatric patients, there have been several reports of neuroblastoma-associated LEMS [Bosdure et al., 2006; de Buys Roessingh et al., 2009]. In adults, both myasthenia gravis (thymoma) and LEMS (SCLC) may be the presenting complaint in a patient with an underlying cancer. Both illnesses respond to treatment [Darnell and Posner, 2006].
Paraneoplastic Cerebellar Degeneration
Paraneoplastic cerebellar degeneration (PCD) was actually the first paraneoplastic syndrome to be described and, as such, is the best described and most easily recognized. In 1919, Brouwer et al., described a patient with rapid onset of a cerebellar syndrome [Brouwer, 1919]. At autopsy, the patient had Purkinje cell loss in the cerebellum and a pelvic tumor, most likely ovarian carcinoma. In 1938, Brouwer went on to postulate the association between PCD and cancer [Brouwer, 1938]. PCD may occur in isolation but may be associated with other PNS, including LE and LEMS.
[/not-level-membership-for-neurology-category]
