[level-membership-for-dermatology-category]
Panniculitis
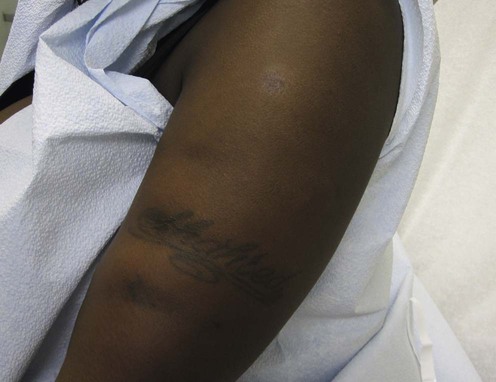










Lupus panniculitis
First-line therapies
 Antimalarials
Antimalarials Systemic or intralesional corticosteroids
Systemic or intralesional corticosteroids Topical corticosteroids under occlusion
Topical corticosteroids under occlusion Second antimalarial agent
Second antimalarial agentThird-line therapies
 IVIG
IVIG Rituximab
Rituximab Gold
Gold Bismuth
Bismuth Thalidomide
Thalidomide Cyclosporine
Cyclosporine Dapsone
DapsoneNodular vasculitis
First-line therapies
 Potassium iodide
Potassium iodide Treat underlying tuberculosis
Treat underlying tuberculosis NSAIDs
NSAIDs Antimalarials
Antimalarials Colchicine
Colchicine Gold
Gold Mycophenolate mofetil
Mycophenolate mofetil
Pancreatic panniculitis
First-line therapies
 Treat underlying pancreatic condition
Treat underlying pancreatic condition Resection of metastases of pancreatic cancer
Resection of metastases of pancreatic cancer Octreotide
OctreotideCytophagic histiocytic panniculitis
First-line therapies
 Treat underlying T-cell lymphoma (chemotherapy)
Treat underlying T-cell lymphoma (chemotherapy) Prednisone
Prednisone Cyclosporine
CyclosporineSecond-line therapies
 Tacrolimus
Tacrolimus Bone marrow transplantation
Bone marrow transplantation Potassium iodide
Potassium iodide Dapsone
Dapsone Cyclophosphamide
Cyclophosphamide Irradiation
Irradiation Anakinra
Anakinraα1-Antitrypsin Deficiency Panniculitis
First-line therapies
 Doxycycline
Doxycycline Dapsone
Dapsone α1-Antitrypsin concentrate
α1-Antitrypsin concentrate Liver transplant
Liver transplantSecond-line therapies
 Potassium iodide
Potassium iodide Plasmapheresis
Plasmapheresis Cyclophosphamide
Cyclophosphamide Colchicine
Colchicine Prednisone
Prednisone[/level-membership-for-dermatology-category][not-level-membership-for-dermatology-category]
Panniculitis
