Chapter 56 Pancreatic and periampullary tumors
Classification and pathologic features
Tumors of the Pancreas
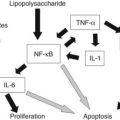
Pancreatic neoplasms are classified based on the degree to which they recapitulate one of the cellular components of the pancreas (Hruban et al, 2007; Klimstra & Adsay, 2009; Klimstra et al, 2007b). To appreciate the various tumor types that occur in the pancreas, basic functional and histomorphologic characteristics of the elements that constitute this gland, both neuroendocrine and exocrine, must be recognized. The neuroendocrine component consists of the islets of Langerhans, small nests of up to a few hundred cells, scattered throughout the pancreas, usually forming round, well-delineated units amid the exocrine elements. Islet cells synthesize hormones (i.e., insulin, glucagon, and somatostatin) and store them in neurosecretory granules; under appropriate stimuli, these hormones are released into the surrounding capillary network. Pancreatic neoplasms that recapitulate the islet cells were previously referred to as islet cell tumors but are now called pancreatic neuroendocrine tumors (PanNETs; see Chapter 61).
The ductal component of the pancreas is responsible for most of the neoplasms (see Chapter 58A, Chapter 58B ), but it is perhaps the least sophisticated. Ducts are responsible for transporting the acinar secretions to the duodenum. The ductal system starts with the centroacinar cells, and through intralobar and interlobar ducts, the enzymatic secretions are carried to the main pancreatic duct and eventually to the duodenum through the ampulla of Vater. The larger ducts produce and secrete mucins for lubrication and protection, to aid the passage of acinar products, and to create the proper milieu for the enzymes during transportation. However, intracellular mucin is not normally detectable in cells of the interlobular or smaller ducts. Recapitulating the normal ducts, ductal neoplasms are characterized by gland, duct, or cyst formation with or without papillary structures. Production of variable amounts of mucin is also typical and ranges from focal intracellular to abundant intracystic or stromal. As in any organ of the body, supportive tissues (mesenchymal elements) and neighboring organs may also give rise to tumors in the pancreas, although these are very rare. This section reviews the general characteristics of pancreatic neoplasms.
Invasive Ductal Adenocarcinoma
Invasive ductal adenocarcinoma is the most common neoplasm of the pancreas. Its clinical characteristics are discussed in detail elsewhere in this text (see Chapter 58A, Chapter 58B ). Most are solid, ill-defined masses; conversely, most solid, ill-defined lesions in the pancreas also prove to be ductal adenocarcinoma. Ductal adenocarcinomas have a remarkable tendency for rapid dissemination and insidious infiltration. Typically, it spreads in the abdomen in a multinodular fashion (intraabdominal carcinomatosis) or is already widely metastatic by the time the primary tumor grows to 5 to 6 cm in size. This feature is so characteristic that a larger solitary pancreatic mass is unlikely to be ductal adenocarcinoma. Interestingly, however, despite the high frequency of distant metastases, some ductal carcinomas cause death of the patient as a result of predominantly local extension; such cases appear to be molecularly distinctive.
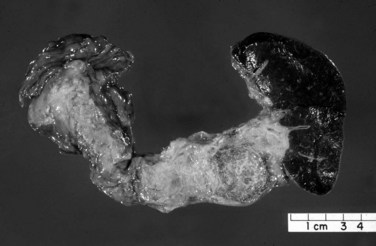
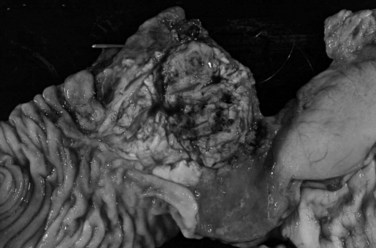
Macroscopically, ductal adenocarcinoma is a scirrhous (scar-forming) type of carcinoma; it is associated with abundant desmoplastic stroma, in which the neoplastic glands are widely scattered. This creates a challenge in the diagnosis of ductal adenocarcinoma, because often only a few cancer cells are present, if any, for evaluation in biopsy specimens. This is also a problem for cancer researchers, because most procured “tumor” specimens may in fact contain much more host tissue than carcinoma cells. Also, because ductal adenocarcinoma is typically very sclerotic, it can be difficult to distinguish from fibrosis of chronic pancreatitis, not only radiologically and intraoperatively but also for pathologists examining the gross specimen. The characteristics that distinguish ductal adenocarcinoma are its firm, gray, and gritty cut surface rather than the rubbery, milky white appearance of benign fibrotic lesions (Fig. 56.1).
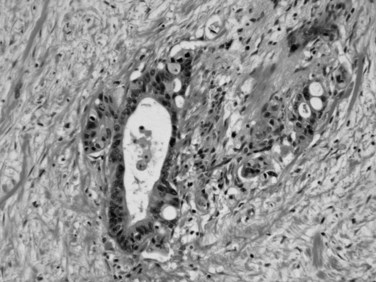
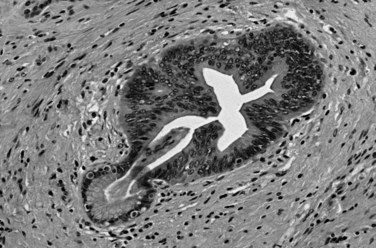
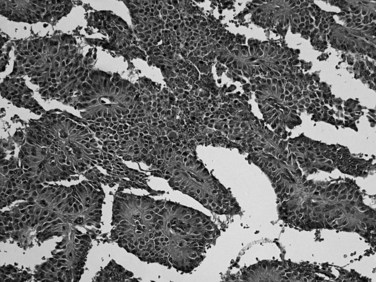
Microscopically, ductal adenocarcinoma is characterized by infiltrating tubular glands, often widely scattered, embedded in a desmoplastic stroma of variable cellularity (Fig. 56.2). The ducts are lined by cuboidal to columnar cells, with variable amounts of intracellular mucin and nuclear atypia (Klimstra & Adsay, 2009). The columnar cells may form cords or nests, or they may grow as individual cells. Perineural invasion is present in more than 60% of cases and is often identifiable in the retroperitoneal region or around the common bile duct, even away from the grossly evident tumor (Fig. 56.3). Neoadjuvant therapy seems to cause substantial alterations in the morphology of the tumor cells. Also, residual foci of previously treated ductal adenocarcinoma may be patchy and may require more careful examination. Recently, scoring systems have been devised in an attempt to evaluate the efficacy of chemotherapy; however, the relevance of these proposals requires further study.
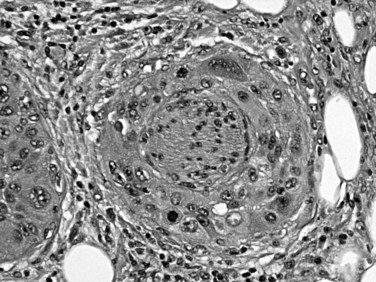
The difficulty in distinguishing ductal adenocarcinoma from chronic pancreatitis (see Chapter 55A, Chapter 55B ) also applies to the microscopic diagnosis and is regarded to be one of the most difficult distinctions in diagnostic pathology (Adsay et al, 2004a). Chronic pancreatitis may be associated with epithelial atypia, both architectural and cytologic, in pancreatic tissue; conversely, ductal adenocarcinoma is notorious for its deceptively bland appearance. Features favoring a malignant diagnosis include abnormal location of glands (adjacent to muscular arteries, within the duodenal muscularis, adjacent to adipocytes in the peripancreatic tissue, or around nerves), architectural abnormalities in the shape of the glands (cribriforming, angulation, or incomplete gland formation), and nuclear abnormalities (variation in the shape and size of nuclei among the cells within an individual gland). A fourfold variation in nuclear volume between adjacent cells, otherwise known as the 4-to-1 rule, is helpful to recognize carcinoma. Diagnostic difficulty also extends to the differential diagnosis of ductal adenocarcinoma in metastatic sites, because ductal adenocarcinoma often retains its well-differentiated appearance and mimics benign or low-grade neoplasms of these sites. Common pitfalls include misinterpretation of metastatic ductal adenocarcinoma in the ovary as a primary borderline ovarian cystadenoma (Young, 2007); in the lung, as bronchioloalveolar carcinoma; and in the liver, as bile duct adenoma. In the last instance, the converse—misinterpretation of bile duct adenoma as metastatic ductal adenocarcinoma—seems to be more common.
No uniformly applied pathologic grading system exists for ductal adenocarcinoma. Schemes advocated by Western and Asian experts show major philosophical differences in principle and results. The current American Joint Commission on Cancer–endorsed tumor-node-metastasis (TNM) grading system (Edge et al, 2010) is similar to the grading of other adenocarcinomas of the gastrointestinal (GI) tract: well differentiated means 95% or more composed of glandular structures; moderately differentiated, 50% to 95% glandular in pattern, the remainder occurring as solid nests or individual cells; and poorly differentiated, with more than 50% solid nest and individual cells. The World Health Organization (WHO) has adopted the complex grading scheme proposed by Klöppel and colleagues, which is difficult to employ and not widely used in daily practice (Fukushima, 2010). Intratumoral heterogeneity seems to be an important problem in the grading of ductal adenocarcinoma, and for this reason, a simpler, more practical, and more clinically relevant grading scheme that accounts for this heterogeneity by scoring the patterns of infiltration has been proposed (Adsay et al, 2005).
The pathologic evaluation of a pancreatectomy specimen is important both for staging and in determining the adequacy of resection (Lüttges et al, 1998; see Chapter 58A, Chapter 58B ). Extrapancreatic extension and involvement of neighboring organs are documented in the pathology report, and determination of tumor size is important. As stated earlier, it is often difficult to appreciate the boundaries of ductal adenocarcinomas; careful macroscopic examination and close correlation with the microscopic findings are necessary to establish the correct size of the tumor. Microscopic foci of carcinoma are often present within the pancreas distant from the main tumor (Bandyopadhyay et al, 2009). Metastasis to lymph nodes is considered one of the most important predictors of outcome in resected ductal adenocarcinomas. Generally, at least 12 lymph nodes should be identified in a simple pancreatoduodenectomy specimen (Adsay et al, 2009). Most of these lymph nodes are embedded in the surfaces of the pancreas or in the groove between the pancreas and duodenum.
Proper identification of margins and their adequate sampling also are important components in the pathologic evaluation of a pancreatoduodenectomy specimen (Ferrone et al, 2008). For pancreatic ductal adenocarcinoma, the most important margin seems to be the retroperitoneal margin, also termed the uncinate, superior mesenteric vein, or vascular margin (Staley et al, 1996; Lüttges et al, 1998). The retroperitoneal margin at the posteroinferior aspect of the head of the pancreas can be distinguished by its nodular, granular, and irregular appearance, with the vascular bed of the superior mesenteric vessels appearing as a groove to its left. Studies have shown that the retroperitoneal margin should be sampled thoroughly, because this margin is often microscopically positive for grossly undetectable disease. Common bile duct, distal pancreatic (pancreatic ductal), and GI mucosal margins also should be evaluated.
As expected, ductal adenocarcinoma shows immunohistochemical evidence of ductal differentiation. Mucin-related glycoproteins and oncoproteins, including carbohydrate antigen 19-9 (CA19-9), carcinoembryonic antigen (CEA), B72.3, DUPAN2, MUC1, and MUC5AC are commonly detected in ductal adenocarcinoma (Basturk et al, 2010b; Hruban et al, 2007; Klimstra, 1998). Expression of these and the differential labeling for cytokeratins (CK7 positive, CK20 variable) sometimes may help distinguish ductal adenocarcinoma from other carcinomas, although no profile of immunohistochemical markers is specific for ductal adenocarcinoma. Scattered cells with neuroendocrine differentiation may be present, and acinar enzymes typically are not expressed.
Substantial developments have occurred in understanding the molecular carcinogenesis of ductal adenocarcinoma (see Chapter 8A). Mutation in codon 12 of the KRAS oncogene is found in more than 90% of ductal adenocarcinoma and seems to be an early event (Hruban & Adsay, 2009). Mutation of P16 or methylation of the promoter also is common (>80%) and represents the pathogenetic link with the familial atypical multiple mole–melanoma syndrome (Hruban et al, 2001a). Overexpression of TP53 (Barton et al, 1991; Hameed et al, 1994) and loss of SMAD4/DPC4 are detected in about half of cases (Tascilar et al, 2001). The latter appears to have a modest degree of specificity for pancreatic ductal adenocarcinoma. Immunohistochemical demonstration of these abnormalities may be useful in the diagnosis of carcinoma (Fig. 56.4). BRCA2 and Peutz-Jeghers gene mutations have been implicated in about 5% of ductal adenocarcinoma cases (Hruban & Adsay, 2009). Fanconi anemia gene alterations also have been identified (van der Heijden et al, 2003). Abnormalities in mismatch repair proteins and microsatellite instability are uncommon, although pancreatic ductal adenocarcinomas can occur as one of the less common manifestations of Lynch syndrome.
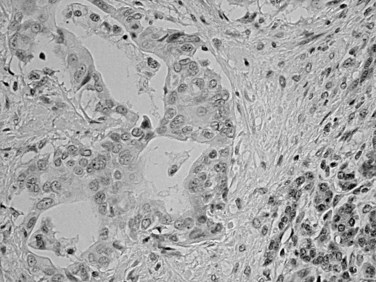
Pancreatic Intraepithelial Neoplasia (PanIN)
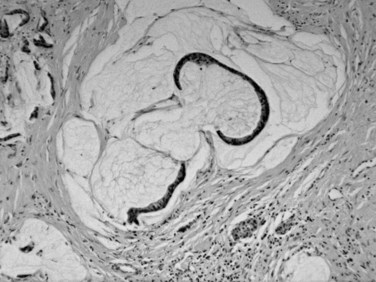
It has been speculated for decades that proliferative lesions in the ducts precede the development of invasive ductal carcinoma (Klöppel et al, 1980). These proliferations have been referred to by a plethora of terms, including hyperplasia and dysplasia, but these are now designated pancreatic intraepithelial neoplasia (Hruban et al, 2001b) in recognition of their neoplastic nature (Fig. 56.5). The spectrum of changes is classified in three grades (Hruban et al, 2001b). Replacement of the normal cuboidal, nonmucinous ductal epithelium with columnar cells that contain abundant apical mucin, but without architectural complexity (e.g., papilla formation) or cytologic atypia, is regarded as PanIN1A. Cytologically similar lesions that form papillae constitute PanIN1B. When a substantial pseudostratification of the cells and some degree of cytologic atypia are seen, the lesion is PanIN2. When irregular papillary architecture is present with tufting, cytologic atypia, necrosis, suprabasal mitoses, and loss of cell polarity, it is regarded as PanIN3.
A progressive accumulation of molecular alterations is reported from PanIN1 to invasive carcinoma (Hruban & Adsay, 2009). Some alterations, such as KRAS mutations, are early events; others, such as TP53 overexpression, occur at the more advanced end of this spectrum. Low-grade PanINs are very common incidental findings in the normal population (Andea et al, 2003); therefore they are generally believed to not to require any further clinical attention, if encountered in isolation or at resection margins. PanIN3, on the other hand, is seldom seen in the absence of an invasive carcinoma (Andea et al, 2003), and for this reason, if PanIN3 is encountered in a pancreas, the likelihood of pancreas cancer elsewhere in the gland is very high.
Other Invasive Carcinomas Related to Ductal Adenocarcinoma
Certain types of carcinomas are closely related to, and often seen in association with, ductal adenocarcinoma (Klimstra & Adsay, 2009). Undifferentiated carcinoma can be regarded as the least differentiated form of ductal adenocarcinoma, in which characteristic tubule formation is no longer evident or only focal. Such tumors are rare, and their demographics do not seem to differ from ordinary ductal adenocarcinoma, except that they may have even more aggressive behavior. Undifferentiated carcinomas include sarcomatoid (spindle cell) carcinoma, anaplastic giant cell carcinoma, and carcinosarcoma. Rarely, the sarcomatoid components of these tumors may show aberrant differentiation, including bone and cartilage formation.
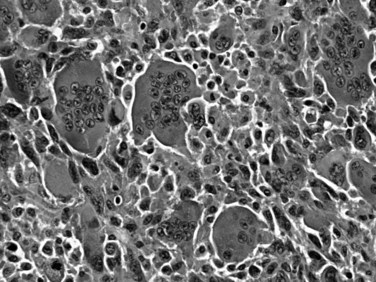
Also known as osteoclastic giant cell carcinoma, undifferentiated carcinoma with osteoclast-like giant cells is the name commonly used for a distinctive tumor type characterized by an abundance of such cells in the background of a sarcomatoid carcinoma (Fig. 56.6; Hoorens et al, 1998; Westra et al, 1998). Studies have shown that the osteoclastic giant cells are nonneoplastic histiocytic cells (Westra et al, 1998); the reasons for this chemotaxis are unknown. The true neoplastic cells in this tumor type are the sarcomatoid mononuclear cells. An adenocarcinoma component or, in some cases, high-grade PanIN or mucinous cystic neoplasm precursors may be present. Undifferentiated carcinomas with osteoclast-like giant cells often appear well demarcated and form a large solitary mass. If examined carefully, many such tumors appear to have substantial intraductal growth. These are clearly malignant neoplasms, most exhibiting an aggressive clinical course; however, some examples with minimal ductal adenocarcinoma components have a protracted clinical course.
Squamous differentiation is seen in some conventional ductal adenocarcinomas (i.e., adenosquamous carcinomas; Fig. 56.7; Kardon et al, 2001), but rare pure examples of squamous cell carcinoma without any glandular components also may be seen. They may have variable degrees of keratinization. Squamous cell carcinoma and adenosquamous carcinoma of this region are highly aggressive tumors (Kardon et al, 2001) with a prognosis that is even worse then that of ordinary ductal adenocarcinoma.
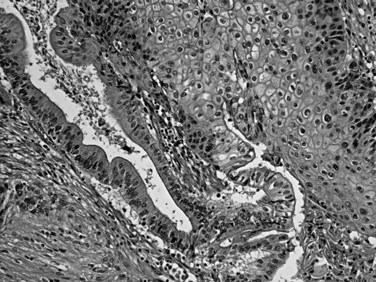
FIGURE 56.7 Adenosquamous carcinoma. Glandular differentiation and pavement-like squamous morphology are apparent.
Colloid carcinoma, pure mucinous or mucinous noncystic carcinoma (Adsay et al, 2001; Hruban et al, 2007; Seidel et al, 2002), is characterized by extensive stromal mucin deposition (Fig. 56.8). Those tumors composed almost exclusively of the colloid pattern—in which the mucin/epithelium ratio is very high, and most carcinoma cells are floating within the mucin (detached from the stroma)—have a different biology with an unusually protracted clinical course. Anecdotal evidence suggests that open biopsy of colloid carcinomas may contribute to dissemination, presumably owing to the adherent nature of the mucin. Colloid carcinomas also tend to be larger and better demarcated than ductal adenocarcinomas, and their molecular alterations seem to be somewhat unique. They usually are associated with intraductal papillary mucinous neoplasms (IPMNs) of the intestinal type (see Chapter 57) and also exhibit immunohistochemical evidence of intestinal differentiation (see later).
Medullary carcinomas, akin to those seen in the GI tract associated with microsatellite instability, also have been reported in the pancreas (Goggins et al, 1998; Wilentz et al, 2000). Syncytial nodules of large, poorly differentiated epithelioid cells with a pushing pattern of invasion characterize medullary carcinomas. In one study (Goggins et al, 1998), these tumors were found to have a more protracted clinical course, but further data are necessary to define the prognosis of these rare tumors.
Intraductal Neoplasms
Intraductal neoplasms constitute an increasingly encountered and important category of pancreatic tumors of ductal origin, characterized by intraductal polypoid/papillary nodules that are often associated with cystic dilatation of the ducts (Basturk et al, 2009). Intraductal neoplasms constitute a wide spectrum of morphologic variants, but they are all regarded to represent precursors to invasive carcinoma; that is, they may be associated with, or may progress to, invasive carcinoma. As preinvasive neoplasms, they are similar to PanINs; but in contrast to PanINs, which are incidentally detected microscopic forms of dysplasia, intraductal tumors form clinically detectable masses (defined as >1.0 cm), conceptually similar to adenomas of the GI tract. It is estimated that about 2% of invasive adenocarcinomas in the pancreas arise from these tumor types. The neoplasms that are included under this umbrella are intraductal papillary mucinous neoplasms and intraductal tubulopapillary neoplasms.
Intraductal Papillary Mucinous Neoplasms (See Chapter 57)
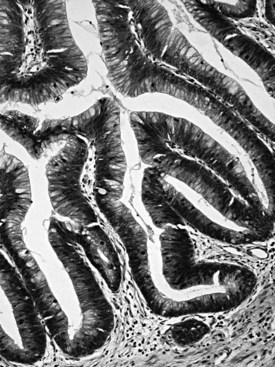
Intraductal papillary mucinous neoplasms (IPMNs) arise predominantly in the head of the gland and occur more commonly in elderly men (males ≥ females; mean age, 64 years; Basturk et al, 2009). The mucin produced by these tumors may exude from the ampulla of Vater, a finding that is virtually diagnostic of an IPMN. Radiographic findings of ductal dilatation with irregularities also are often diagnostic. A history of pancreatitis is noted in some patients. An adenoma-to-carcinoma progression is seen in IPMNs, which are correspondingly classified as having low-, intermediate- or high-grade dysplasia based on the greatest degree of atypia (Hruban et al, 2007; Adsay et al, 2010); invasive carcinomas that arise in IPMNs are recognized separately and are graded and staged like other ductal-type carcinomas. IPMNs that predominantly involve the secondary ducts (branch duct type; Tanaka et al, 2006; Schnelldorfer et al, 2008; Nagai et al, 2009; Koizumi et al, 2009; Tanno et al, 2008, 2010; Terris et al, 2000) tend to be small (<3 cm) and noncomplex, and they generally prove to have low- to intermediate-grade dysplasia, with simple, gastric foveolar-type epithelium by microscopic examination. IPMNs that mostly involve the major ducts (main duct type; Fig. 56.9) (Tanaka et al, 2006) more commonly harbor high-grade dysplasia or invasive carcinoma (Salvia et al, 2004).

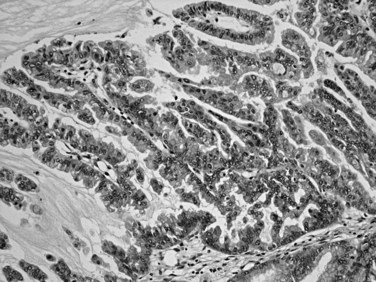
Distinctive pathologic subsets of IPMNs are emerging (Adsay et al, 2004b; Furukawa et al, 2005) with different histologic characteristics. In intestinal-type IPMNs, the papillary nodules are morphologically identical to colonic villous adenomas (Fig. 56.10), and the invasive carcinomas that develop in these tend to be of the relatively indolent colloid type (Adsay et al, 2001; Seidel et al, 2002). Intestinal-type IPMNs and colloid carcinomas typically express intestinal differentiation markers (MUC2 and CDX2) not found in ductal adenocarcinomas or in the nonintestinal subtypes of IPMNs discussed below, indicating that they represent a distinct “intestinal pathway” of carcinogenesis in the pancreas (Adsay et al, 2004b). Other IPMNs have more complex papillae that resemble papillary carcinomas of the biliary tree (Fig. 56.11). These pancreatobiliary-type IPMNs tend to be associated with tubular-type invasive carcinoma (conventional ductal adenocarcinoma) and appear to have more aggressive behavior (Sadakari et al, 2010). What is also known as intraductal oncocytic papillary neoplasm (Adsay et al, 1996) is now regarded as a distinct subtype of IPMN, characterized not only by the oncocytic nature of the cells but also by the complexity of the papillary nodules, which have an arborizing pattern. Oncocytic IPMNs are often large and very floridly proliferative; however, the carcinomas arising from them tend to be small, the incidence of metastasis is low, and the overall prognosis appears to be favorable. They also have different molecular changes from other IPMNs, with a much lower incidence of KRAS mutation and frequent expression of MUC6, suggesting a pyloropancreatic lineage distinct from the intestinal-type IPMNs (Basturk et al, 2010b).
IPMNs can be multifocal in up to 40% of patients. They seem not only to be precursors but also “markers” of invasive adenocarcinoma (i.e., in some cases, invasive carcinoma is detected elsewhere in the gland, away from the IPMN; Fujii et al, 1996; Yamaguchi et al, 2002). Invasive carcinoma of either the colloid or tubular type (Adsay et al, 2004b) is present in more than one third of resected IPMNs. Colloid-type invasion occurs almost exclusively in intestinal-type IPMNs. It is generally believed that IPMNs proven devoid of high-grade dysplasia or invasive carcinoma by thorough histologic examination are innocuous, curable neoplasms. Patients with high-grade dysplasia may on occasion experience recurrences and metastases (White et al, 2007), presumably the result of undetected foci of invasive carcinoma; invasive carcinoma has a malignant clinical course, albeit often significantly more protracted than that of ordinary ductal adenocarcinoma. It should be kept in mind that most patients with IPMNs are older, often with comorbid diseases—including another malignant neoplasm in one third of patients (Eguchi et al, 2006; Sugiyama & Atomi, 1999; Adsay et al, 2002a; Adsay, 2008). The prognosis and management of IPMNs are discussed in detail elsewhere in this book (see Chapter 58A, Chapter 58B ).
Intraductal Tubulopapillary Neoplasms
Intraductal tubulopapillary neoplasm (Adsay et al, 2004b; Tajiri et al, 2005; Yamaguchi et al, 2009; Klimstra et al, 2007c) is a recently recognized category of mass-forming (>1.0 cm) intraductal neoplasm that is fairly similar to an IPMN, from which it is distinguished microscopically by its mucin-poor nature and distinctive tubular architecture. First reported by Tajiri and colleagues (2005) under the heading of intraductal tubular adenocarcinoma, the entity is now being designated intraductal tubulopapillary neoplasm in the new WHO classification. It is a rare tumor seen at an average age of 53 years, and it presents with nonspecific symptoms. The clinical findings are often indistinguishable from those of IPMNs. Most cases have cystic ducts filled with tumor nodules. Intraductal tubulopapillary neoplasm occurs predominantly in the head of the pancreas but may involve any part. It is often large (mean, 7 cm; range, ≤15 cm).
It should be noted that a variety of pancreatic neoplasms can show prominent intraductal growth, including acinar cell carcinomas (Basturk et al, 2007; Ban et al, 2010), pancreatic neuroendocrine tumors, osteoclastic giant-cell carcinomas (Basturk et al, 2010c), and even metastatic tumors; thus, they fall into the differential diagnosis of these intraductal neoplasms.
Mucinous Cystic Neoplasms (See Chapter 57)
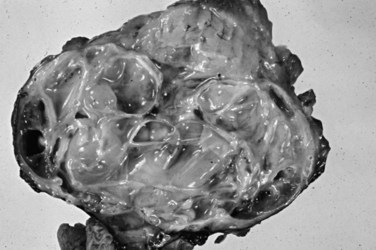
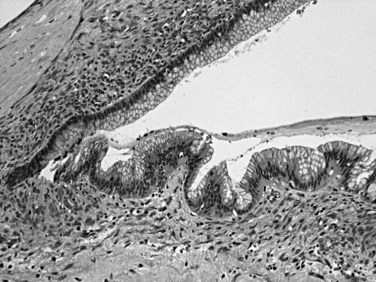
Mucinous cystic neoplasms (MCNs) are seen almost exclusively in perimenopausal women (>95% female; mean age, 48 years; Tanaka et al, 2006; Thompson et al, 1999; Zamboni et al, 1999). One of the characteristic features, which essentially has become a requirement for the diagnosis, is an ovarian-type subepithelial stroma that is not only histologically similar to the ovarian cortex but that also expresses progesterone and estrogen receptors by immunohistochemistry. This distinctive mesenchyme helps distinguish MCNs from similar neoplasms, especially IPMNs (see above). MCNs typically form a thick-walled multilocular cyst in the body or tail of the pancreas (Fig. 56.12). Some examples may mimic pseudocysts, although the surrounding pancreatic tissue typically does not show evidence of chronic pancreatitis. In most cases, cysts do not communicate with the ductal system. The cyst fluid is rich in mucin-related glycoproteins and oncoproteins, such as CEA (Pitman et al, 2010; Deshpande et al, 2006), which may help distinguish these tumors from other cystic lesions. The cysts are lined by mucinous epithelium, which may exhibit varying degrees of cytologic and architectural atypia (Fig. 56.13), graded as low-, intermediate-, or high-grade dysplasia based on the most atypical region. Invasive carcinoma typically occurs in larger and more complex cases that show florid papillary nodules in the cysts.
Invasive carcinomas are found in 10% to 15% of MCNs (Pitman et al, 2010; Zamboni et al, 1999; Reddy et al, 2004). The carcinoma may be limited to microscopic foci within the septa of the cysts, or it may invade out of the cyst into the peripancreatic tissues. Invasive carcinomas resemble ductal adenocarcinoma histologically, including the occasional occurrence of an uncommon histologic variant such as sarcomatoid or giant-cell carcinoma. Although some studies have questioned the utility of grading MCNs, it has become clear recently that if these tumors are examined thoroughly, and the presence of invasive carcinoma is ruled out by rigorous pathologic examination, patients typically are cured by complete excision (Zamboni et al, 1999; Sakorafas & Sarr, 2005; Hruban & Zamboni, 2009). Because all MCNs have the potential to harbor invasive carcinoma, most authors agree that these tumors should be completely resected.
Serous Cystic Tumors (See Chapter 57)
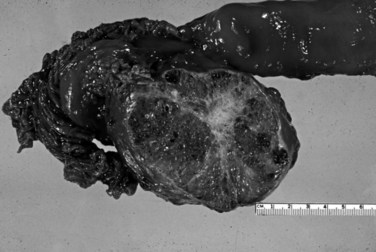
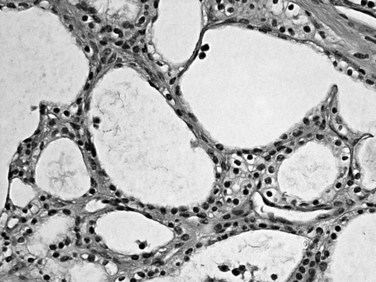
Serous cystadenomas (Bassi et al, 2003; Compton, 2000) form relatively large masses (mean size, 9 cm, some 25 cm) that tend to be well demarcated, predominantly in elderly women (mean age, 63 years; female/male ratio, 3 : 1). Typically, serous cystadenomas are composed of thousands of small cysts, each measuring only millimeters and creating the diagnostic macroscopic appearance of a spongelike configuration, hence the synonym microcystic adenoma (Fig. 56.14). Rare macrocystic and solid examples of serous adenomas also have been described. Microcystic serous cystadenomas often have a central stellate scar. The defining microscopic feature is a simple, nonmucinous, cuboidal epithelium that contains intracytoplasmic glycogen, resulting in characteristic clear cytoplasm (Fig. 56.15). Serous cystic tumors are one of the few ductal neoplasms of the pancreas that do not produce mucin, possibly reflecting a recapitulation of the centroacinar cells that are nonmucinous. Along the same lines, the cyst contents are devoid of the mucin-related glycoproteins and oncoproteins that typically are found in mucinous pancreatic tumors, a feature that may help in the preoperative diagnosis (Brugge et al, 2004). Microscopically, these lesions are similar to the cysts seen in von Hippel–Lindau (VHL) disease, and some serous cystadenomas do show VHL gene alterations. Serous cystadenomas often are reported to coexist or “collide” with other pancreatic neoplasms and with congenital pathologic conditions (Compton, 2000).
Malignant serous tumors (serous cystadenocarcinomas) (Matsumoto et al, 2005; Shintaku et al, 2005; Strobel et al, 2003) are exceedingly rare. Only the presence of recurrence, metastases, or angioinvasive growth separates these tumors from their benign counterparts (i.e., the cytologic appearance is entirely bland). For practical purposes, serous cystadenomas limited to the pancreas are regarded as uniformly benign.
Pancreatic Neuroendocrine Tumors (See Chapter 61)
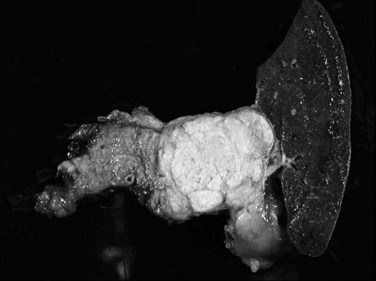
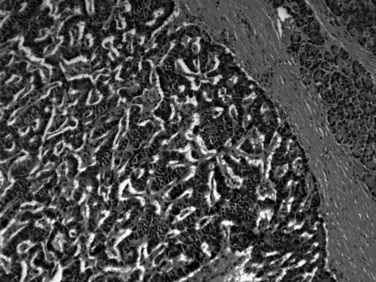
Most neuroendocrine-type tumors of the pancreas are well-differentiated, low- to intermediate-grade neuroendocrine tumors, previously referred to as islet cell tumors and now designated as pancreatic neuroendocrine tumors (PanNETs) by the World Health Organization (WHO) (Klimstra et al, 2010a; Solcia et al, 1991), with low-grade malignant behavior (Hochwald et al, 2002). Grossly, PanNETs are usually solid, circumscribed, and fleshy tumors, although multinodular and sclerotic examples occur (Fig. 56.16). Rarely, cystic degeneration may be seen (Adsay & Klimstra, 2000; Deshpande & Lauwers, 2007), with a central unilocular cyst lined by a cuff of viable tumor; this occurs more often in the setting of multiple endocrine neoplasia (MEN) syndrome type I. PanNETs recapitulate the morphologic features of islet cells by forming nested, gyriform, trabecular, or rarely acinar or glandular patterns (Fig. 56.17). The cells have characteristic neuroendocrine features, including round, monotonous nuclei with a coarsely stippled chromatin pattern and moderate amounts of cytoplasm (Fig. 56.18).
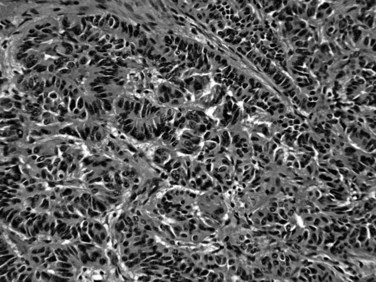
Almost half of PanNETs are clinically functional, and patients come to medical attention with signs or symptoms of inappropriate production and serologic activity of one or more hormones. These functional PanNETs are named based on the hormonal syndrome: insulinoma, glucagonoma, gastrinoma, somatostatinoma, and VIPoma (vasoactive intestinal peptide tumor) for example. The correlation of serum hormone levels and expression of the corresponding hormone in the tumor itself by immunohistochemistry is often imperfect. Nonfunctional PanNETs (Hochwald, et al, 2001; Klimstra & Adsay, 2009; La Rosa et al, 1996) constitute an ever-enlarging proportion of PanNETs because they are being detected more commonly as incidental findings on abdominal imaging studies. Most insulinomas follow a benign clinical course, likely because insulinomas typically are highly symptomatic, even when they are small, which leads to their early detection. Glucagonomas, on the other hand, tend to be large at diagnosis and have a more aggressive course. PanNETs associated with MEN type I or other syndromes tend to be multifocal and less aggressive (Anlauf et al, 2006, 2007; Klimstra & Adsay, 2009; Perigny et al, 2009). In addition to grossly evident and usually functional PanNETs, patients with MEN type I have numerous neuroendocrine microadenomas, defined as PanNETs (<0.5 cm). Most sporadically occurring functional and nonfunctional PanNETs are clinically low-grade malignancies. More than half of patients have recurrence or metastasis after resection, and many patients come to attention only after the development of metastatic disease. Nonetheless, there may be a relatively protracted clinical course even in patients with metastatic disease. It has been difficult, as in neuroendocrine tumors of other organs, to determine which PanNETs are more likely to metastasize and which metastatic cases are likely to progress most rapidly (Pelosi et al, 1996; White et al, 1994). Findings associated with more aggressive behavior include size greater than 3 cm, a functional PanNET other than insulinoma, extrapancreatic or vascular invasion, high mitotic activity, high proliferation index (based on immunohistochemical staining for Ki-67) (Jamali & Chetty, 2008; Rindi et al, 2007; Chatzipantelis et al, 2009), CK19 expression (La Rosa et al, 2007), and c-KIT expression (Zhang et al, 2009); however, some PanNETs lacking all of these features still may metastasize.
The 2004 WHO classification (Heitz et al, 2004) separated well-differentiated pancreatic neuroendocrine tumors, those lacking metastases or gross local invasion, from well-differentiated pancreatic neuroendocrine carcinomas, with metastases or gross local invasion; they further predicted “benign behavior” or “uncertain behavior” in the former group based on size less than 2 cm, mitotic rate below 2/10 high-power fields (HPFs), Ki-67 labeling index below 2%, and absence of vascular or perineural invasion. However, studies have demonstrated that the patients who fall in the uncertain behavior group still experience recurrence and metastasis in long-term follow-up in up to 40% of the cases (Schmitt et al, 2007); therefore it is clear that even this group represents a low-grade malignancy.
Another more recent proposal (Ferrone et al, 2007) divides PanNETs into low-grade and intermediate-grade groups based on the mitotic rate (2 mitoses per 50 HPFs) and necrosis. The two groups exhibit a highly significant difference in prognosis, although no subset specifically is regarded to be utterly benign. Recently, a multidisciplinary group of international experts have proposed a set of parameters to be included in pathology reports (Klimstra et al, 2010a). It was emphasized that PanNETs ought to be evaluated by the approach used for any other malignancy, and accordingly, the grade and stage should be reported separately (Klimstra et al, 2010b; Klöppel et al, 2010a). For grading, in 2010 the WHO adopted the system originally devised and tested by the European Neuroendocrine Tumor Society (ENETS), which grades PanNETs based on the mitotic count and Ki-67 labeling index. Grade 1 if the mitotic rate is less than 2/10 HPFs and the Ki-67 index is below 3%; grade 2 if the mitotic rate is 2 to 20/10 HPFs or the Ki-67 index is 3% to 20%; and grade 3 if either is above 20. For staging, the AJCC has adapted the TNM-based staging system used for adenocarcinomas, but the ENETS system is somewhat different (Klöppel et al, 2007).
On the other end of the spectrum of neuroendocrine neoplasia, very rarely, poorly differentiated neuroendocrine carcinoma, related to small cell carcinoma, may be encountered in the pancreas (Tranchida et al, 2000). Some are composed of large cells with more abundant cytoplasm (high-grade neuroendocrine carcinomas of a “large cell” type). According to WHO, high-grade neuroendocrine carcinomas are classified as grade 3 PanNETs, although the true nature of such tumors is difficult to characterize because of their rarity, and they appear to be a distinct tumor type with morphology and behavior usually very similar to that of high-grade neuroendocrine carcinomas of the lung rather than being advanced forms of well-differentiated PanNETs. In fact, especially when the small cell morphology is present, poorly differentiated neuroendocrine carcinomas must be distinguished from metastases to the pancreas from a pulmonary primary.
Acinar Neoplasms
Acinar cell carcinomas (Kitagami et al 2007; Klimstra, 2007; Wisnoski, 2008) form relatively large tumors (mean, 10 cm), usually in older men (mean age, 63 years), although some occur in children (Shorter et al, 2002). In a small percentage of cases (10%), patients experience a “lipase hypersecretion syndrome” (Klimstra & Adsay, 2001) characterized by subcutaneous fat necrosis, polyarthralgia, and peripheral eosinophilia. Serum α-fetoprotein (AFP) levels may be elevated in some cases (Cingolani et al, 2000). Half of acinar cell carcinomas have metastases at the time of diagnosis, usually in the liver and/or regional lymph nodes (Holen et al, 2002). Once believed to be almost as aggressive clinically as ductal adenocarcinomas with a 5-year survival of 10% (Holen et al, 2002), more recent studies place acinar cell carcinomas in a somewhat more indolent category (Klimstra et al, 1992) with some studies reporting a 5-year survival over 40% (Kitagami et al, 2007; Wisnoski et al, 2008). In contrast to ductal adenocarcinomas, acinar cell carcinomas are stroma-poor cellular tumors composed of sheets of cells and small glands (acini) composed of polygonal cells with prominent nucleoli (Labate et al, 1997) and cytoplasmic eosinophilia (Fig. 56.19), often with periodic acid-Schiff (PAS)-positive granules. Acinar enzymes, especially trypsin and chymotrypsin, are demonstrable by immunohistochemistry in essentially all cases (Klimstra et al, 1992). A subset of acinar cell carcinomas is characterized by prominent intraductal growth; such cases are reportedly associated with a more protracted clinical course (Basturk et al, 2007; Ban et al, 2010). Molecular genetic findings of acinar cell carcinomas markedly differ from those of ductal adenocarcinomas (Cao et al, 2005), with absence of the common alterations of ductal adenocarcinoma in genes such as KRAS, TP53, P16, or SMAD4.
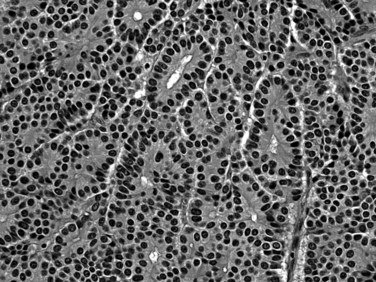
Immunohistochemistry discloses scattered neuroendocrine cells in 30% to 40% of acinar cell carcinomas. Some cases have a significant neuroendocrine component that may be evident microscopically (Klimstra et al, 1992). If the latter constitutes more than 25% of the tumor, it is classified as mixed acinar-neuroendocrine carcinoma (Fig. 56.20), a tumor that seems to be biologically similar to pure acinar cell carcinoma (Klimstra et al, 1994; Ohike et al, 2004). Similarly, acinar cell carcinomas with more than 25% ductal differentiation are classified as mixed acinar-ductal carcinoma (Stelow et al, 2010). Rarely, acinar cell carcinomas may show a grossly cystic pattern and are designated acinar cell cystadenocarcinoma (Cantrell et al, 1981; Stamm et al, 1987).
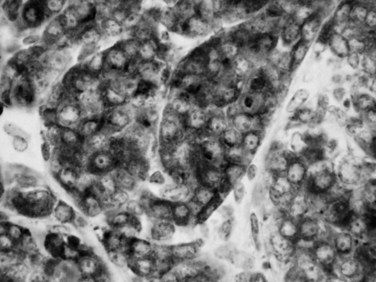
Acinar Cell Cystadenoma
Acinar cell cystadenoma, also referred to as acinar cystic transformation, is a rare entity (Albores-Saavedra, 2002; Zamboni et al, 2002). Acinar cell cystadenomas are usually small, incidental cysts lined by benign-appearing acinar cells, although some may form a mass that measures a few centimeters. Some examples have a patchy distribution that spans to a large segment of the organ. They are considered to be benign.
Pancreatoblastoma
Pancreatoblastoma is an extremely rare childhood tumor of the pancreas (mean age of 4 years, with a second small peak in the third decade) (Dhebri et al, 2004; Klimstra et al, 1995; Shorter et al, 2002). Pancreatoblastomas are usually large (7 to 18 cm). Some cases are associated with elevated serum AFP levels, and occasional cases are seen in association with Beckwith-Wiedemann syndrome (Drut & Jones, 1988; Kerr et al, 2002) or familial adenomatous polyposis (FAP) syndrome (Abraham et al, 2001). Pancreatoblastomas are malignant tumors with a 5-year survival of about 25%, although children diagnosed before the development of metastases have been cured. Pancreatoblastomas are predominantly acinar neoplasms. Microscopically, they have sheets of primitive-appearing epithelial cells and acinar formations. A characteristic and peculiar microscopic finding is the squamoid nest, which is a small morular arrangement of squamoid cells specific for this tumor type in the pancreas (Fig. 56.21). Typically, pancreatoblastomas exhibit all three lines of pancreatic differentiation—acinar, neuroendocrine, and ductal—with acinar elements being the most consistently present (Klimstra et al, 1995). Molecular genetic findings of pancreatoblastomas differ from the findings of ductal adenocarcinoma; they are associated with β-catenin pathway alterations (Cao et al, 2005). The squamoid nests (morules) are interesting in this regard because they are also often encountered in other cancer types in which this pathway is abnormal (Nakatani et al, 2004).
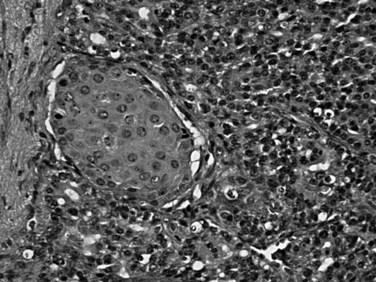
Solid Pseudopapillary Neoplasm
Solid pseudopapillary neoplasm is seen predominantly in young women (mean age, 25 years; >80% female) (Klimstra et al, 2000; Kosmahl et al, 2000; Papavramidis & Papavramidis, 2005; Tang et al, 2005). It is a peculiar neoplasm of unknown origin, and its obscure nature is reflected in the various descriptive names assigned to this tumor in the past, including papillary cystic tumor, solid and papillary tumor, solid and cystic tumor, and Frantz tumor (Klöppel et al 1981; Lieber et al, 1987; Pettinato et al, 1992; Stommer et al, 1991). It is an indolent malignant neoplasm for which complete resection is curative in most cases. In a small percentage (approximately 15%), metastases are noted, usually to liver or peritoneum and often at the time of diagnosis. Even patients with metastases have a protracted clinical course, and death as a result of this tumor is rare; however, a few highly aggressive solid pseudopapillary neoplasms have been reported that exhibit high-grade malignant transformation from a conventional solid pseudopapillary neoplasm (Tang et al, 2005).
Solid pseudopapillary neoplasms are relatively large tumors (mean, 8 to 10 cm). Although they are typically solid cellular neoplasms, cystic degeneration is common (Fig. 56.22). Degenerative changes combined with the preservation of the cells around the microvasculature create the pseudopapillary microscopic pattern (Fig. 56.23). The cells are round to oval and often show nuclear grooves. Intracytoplasmic hyaline globules and aggregates of foamy macrophages are characteristic findings, and calcifications may also be seen. Overall, the histologic picture may resemble closely that of PanNETs. The immunophenotype of this tumor is quite distinctive but fails to disclose the line of differentiation of the cells. The tumor typically expresses vimentin, progesterone receptors, CD10, and some of the neuroendocrine markers, CD56 and synaptophysin (Klimstra et al, 2000; Kosmahl et al, 2000; Ladanyi et al, 1987). Chromogranin, the most specific neuroendocrine marker, is negative, which is important for the differential diagnosis with PanNETs, and pancreatic enzymes are not expressed. β-Catenin and cyclin D1 expression have been found in these tumors, suggesting an alteration in the WNT signaling pathway. E-cadherin and n-cadherin expression are also abnormal (Abraham et al, 2002; Tiemann et al, 2007).
Miscellaneous Cystic Pancreatic Lesions
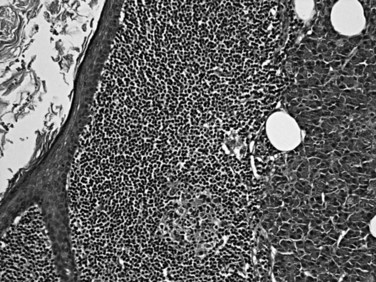
Other uncommon cystic lesions occur in the pancreas in addition to pseudocysts, intraductal cyst-forming neoplasms, mucinous cystic neoplasms, and serous cystic tumors (Adsay et al, 2000; Kosmahl et al, 2004). Lymphoepithelial cysts (Adsay et al, 2002b) are usually peripancreatic rather than intrapancreatic, and they occur predominantly in men (mean age, 52 years; male/female ratio, 3 : 1). In contrast to their salivary gland counterparts, pancreatic lymphoepithelial cysts do not show associations with autoimmune syndromes, human immunodeficiency virus (HIV), or lymphoma. Lymphoepithelial cysts may occur in any part of the pancreas and may be unilocular or multilocular. They are characterized by variably keratinized, squamous-lined cysts immediately surrounded by a rim of lymphoid tissue, some with lymphoid follicles and a capsule (Fig. 56.24). The cyst contents may extrude into the cyst wall and cause an inflammatory reaction, including granulomas. Dermoid cysts are similar to lymphoepithelial cysts but lack the lymphoid tissue and show skin adnexal elements, including sebaceous glands. Lymphangiomas (Paal et al, 1998) are seen in young women (mean age, 29 years; male/female ratio, 1 : 3) and form endothelial-lined cysts surrounded by a rim of lymphoid tissue. Congenital cysts and intestinal duplications also may form cystic lesions in the vicinity of the pancreas and periampullary region. These may have a variable lining, including respiratory type, intestinal, squamous, or transitional.
Pseudotumors
As discussed in the section on ductal adenocarcinoma, benign chronic inflammatory and fibrosing conditions of the pancreas may be difficult to distinguish from carcinomas both clinically and pathologically. Chronic pancreatitis of any cause—including alcohol, obstruction, and even causes with granulomatous inflammation—may lead to segmental fibrosis and a tumorous mass (see Chapter 55A, Chapter 55B ; Adsay et al, 2004c; Klöppel & Maillet, 1991). However, certain subtypes of chronic pancreatitis are especially prone to form pseudotumors and mimic carcinomas, as discussed below (Adsay et al, 2004c; Klöppel, 2007; Levenick et al, 2009).
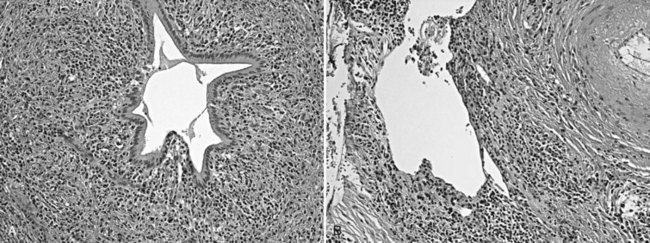
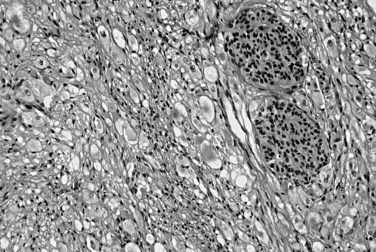
Autoimmune pancreatitis (AIP), also called lymphoplasmacytic sclerosing pancreatitis (Zamboni et al, 2004), is often misdiagnosed as carcinoma preoperatively (see Chapter 55A, Chapter 55B ). AIP typically is seen in patients in their 30s to 50s, and high serum immunoglobulin G4 (IgG4) levels are helpful in the preoperative diagnosis (Dhall et al, 2010; Morselli-Labate & Pezzilli, 2009; Sanchez-Castanon et al, 2009; Tabata et al, 2009). It may be associated with other autoimmune disorders, such as Sjögren syndrome, and with components of the multifocal fibrosclerosis syndrome: retroperitoneal and mediastinal fibrosis, Riedel thyroiditis, inflammatory pseudotumor of the orbit, and sclerosing cholangitis. Microscopically, dense periductal lymphoplasmacytic infiltrates, interstitial fibrosis with abundant myofibroblasts arranged in a storiform pattern, and obliterative venulitis are characteristic (Fig. 56.25).
Recently, a variant of AIP has been recognized (Klöppel et al, 2010b) and termed variably as type 2 autoimmune pancreatitis or pancreatitis with granulocytic epithelial lesions (GELs). This type is more commonly seen in patients with ulcerative colitis and does not seem to be associated with IgG4-producing plasma cells but rather shows neutrophilic destruction of the duct epithelium (Klöppel et al, 2010b).
Paraduodenal pancreatitis (Adsay & Zamboni, 2004; Coban et al, 2009) is the name recently proposed for a distinct but poorly recognized subset of chronic pancreatitis that creates a clinical picture often indistinguishable from pancreatic or periampullary carcinoma. This entity is also known as groove pancreatitis (Badia Bartolome et al, 2009; Castell-Monsalve et al, 2008; Ishigami et al, 2009; Levenick et al, 2009; Triantopoulou et al, 2009) or cystic dystrophy of heterotopic pancreas (Casetti et al, 2009; de Madaria et al, 2009). Affected patients are predominantly men in their 50s with a history of alcohol abuse. Endoscopically, in the second portion of the duodenum, proximal to the ampulla, mucosal nodularities are present that microscopically reveal inflamed mucosa, Brunner gland hyperplasia, or myoid spindle cell proliferation in the submucosa that can also extend to the mucosa. These pseudotumors are typically centered around the minor papilla or accessory duct and thus invade into the region between the common bile duct, duodenum, and pancreas (i.e., the “groove” region) (Badia Bartolome et al, 2009; Castell-Monsalve et al, 2008; Ishigami et al, 2009; Levenick et al, 2009; Triantopoulou et al, 2009). The process often exhibits a dense myofibroblastic proliferation within which lobules of pancreatic tissue are scattered, and cystic ducts contain inspissated enzymatic secretions. In some cases, cystic change in the duodenal wall can be prominent (cystic dystrophy of the duodenum) and may become large (paraduodenal wall cyst) (Casetti et al, 2009; de Madaria et al, 2009), mimicking pseudocysts, congenital cysts, or intestinal duplication. Some of the cysts are lined by granulation tissue without any epithelium.
Some developmental abnormalities also may lead to pseudotumors. A solid and cystic hamartoma and a cellular hamartoma (Pauser et al, 2005a, 2005b) have been reported to present as pancreatic tumors in adults. Lipomatous pseudohypertrophy (Altinel et al, 2009) of the pancreas also may lead to a mass that could be mistaken for carcinoma. Adenomyomatous hyperplasia (Handra-Luca et al, 2003) of the ampulla—a common finding in the general population, reported in 40% of autopsies—has been implicated as a cause of biliary obstruction that mimics a periampullary carcinoma.
Mesenchymal Tumors
Primary mesenchymal tumors of the pancreas are rare (Khanani et al, 2003; Lüttges et al, 1997), but mesenchymal tumors from neighboring sites may secondarily involve the pancreas. In particular, GI stromal tumors and retroperitoneal sarcomas may appear to be centered in the pancreas. A variety of benign mesenchymal tumors, including fibromatosis (desmoid tumor), solitary fibrous tumor (Lüttges et al, 1999b), and schwannoma (Paal et al, 1998), have been reported in the pancreas. Primary sarcomas include primitive neuroectodermal tumor (Movahedi-Lankarani et al, 2002), synovial sarcoma, desmoplastic small round-cell tumor, leiomyosarcoma, and malignant fibrous histiocytoma, all of which are largely documented in single case reports.
Secondary Tumors
A widely metastatic malignant neoplasm commonly may involve the pancreas; however, most of these are clinically undetected lesions identified only at autopsy (see Chapter 60; Adsay et al, 2004d). Autopsy studies have shown that most secondary tumors involving the pancreas are of pulmonary origin, followed by GI carcinomas; however, a few metastatic tumors are prone to involve the pancreas in the absence of other metastatic foci, mimicking a primary carcinoma. Lymphomas and renal cell carcinomas (Adsay et al, 2004d; Temellini et al, 1989) seem to be the most common tumor types responsible for such cases. Renal cell carcinomas in particular are known to form polypoid ampullary nodules or even to grow within the ducts (Klimstra & Adsay, 2009).
Tumors of the Ampulla of Vater (See Chapter 59)
Although the ampulla of Vater is anatomically a small structure, it and the surrounding periampullary duodenum give rise to a surprising number and variety of neoplasms that often present with dramatic symptoms because of the strategic location of the ampulla at the confluence of the pancreatic and biliary ducts. The ampulla itself includes several kinds of epithelia: the duodenal mucosa, covering the papilla; the pancreatic ductal epithelium and that of the distal common bile duct; and the epithelium lining the common channel—the short, if not hypothetical, union of the two ducts within the duodenal wall. The epithelia lining the ducts and the common channel are histologically similar pancreatobiliary-type epithelium, whereas the papilla is covered by intestinal-type epithelium (Klimstra et al, 2007a). Different cell types can be observed intimately admixed in the transitional region. Neoplasms arising in the ampulla may have predominantly intestinal or pancreatobiliary features, or patterns may be mixed. Most ampullary neoplasms are adenocarcinomas (Klimstra & Adsay, 2009), although some neuroendocrine tumors also affect this region. Benign neoplasms also arise in the ampulla, and techniques for local treatment, such as endoscopic polypectomy or transduodenal ampullectomy (Roggin et al, 2005) have introduced new challenges into the pathologic analysis of ampullary tumors (Klimstra & Adsay, 2009). This section reviews the pathologic features of these ampullary neoplasms.
Adenoma and Noninvasive Papillary Carcinoma
These tumors can be regarded in two categories: 1) those that arise from the duodenal surface of the ampulla, which are nearly identical to colonic-type adenomas and may occur in association with FAP syndrome (Albores-Saavedra et al, 2000; Alexander et al, 1989; Domizio et al, 1990; Klimstra & Adsay, 2009; Noda et al, 1992; Odze et al, 1994); and 2) those that arise within the ampulla (Fig. 56.26), which are not only of an intestinal type, similar to colonic-type adenomas, but are also of a pancreatobiliary type, referred as noninvasive papillary carcinomas in the WHO classification (Klimstra et al, 2010c). For the latter group, those occurring within the ampulla, the designation of intraampullary papillary tubular neoplasm (IAPN), akin to pancreatic IPMNs and intraductal tubulopapillary neoplasms, also has been recently proposed (Ohike et al, 2010a).
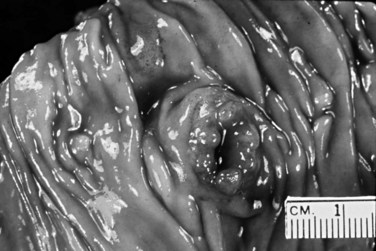
Adenomas of the Duodenal Surface of the Papilla of Vater
Adenomas of the duodenal surface of the ampulla are morphologically and genetically similar to adenomas of the large bowel. Although in most clinical studies, invasive carcinomas outnumber pure adenomas, residual adenomas are commonly found associated with invasive carcinomas. Most ampullary adenocarcinomas, especially those with an intestinal phenotype, are thought to arise from adenoma precursors. Conversely, many adenomas of the ampulla contain foci of invasive carcinoma, requiring a thorough histologic evaluation (Perzin & Bridge, 1981).
A significant percentage of adenomas of the small intestine involve the ampulla or the surrounding duodenal mucosa (Perzin & Bridge, 1981). They may occur sporadically, or they may be associated with FAP. The ampulla and periampullary duodenum are the most common sites of extracolonic adenomas in patients with FAP (Alexander et al, 1989; Domizio et al, 1990; Klimstra & Adsay, 2009; Noda et al, 1992; Odze et al, 1994). Sporadic ampullary adenomas are generally solitary, whereas those arising in patients with FAP usually are accompanied by numerous adenomas in the periampullary duodenum, sometimes forming a carpet of polyps. Ampullary adenomas in patients with FAP usually are detected at a younger age (mean, 41 years) compared with sporadic ampullary adenomas (mean, 62 years) (Albores-Saavedra et al, 2000). Symptomatic patients are generally seen initially with biliary obstruction, although ampullary and duodenal adenomas are increasingly detected in asymptomatic patients with FAP owing to endoscopic screening (Shemesh & Bat, 1985). The risk of an invasive carcinoma being found within an ampullary adenoma is proportional to the size of the adenoma, and it is greater for a given size compared to adenomas of the large bowel.
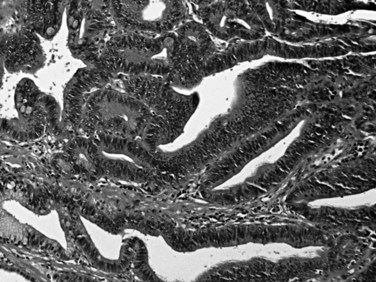
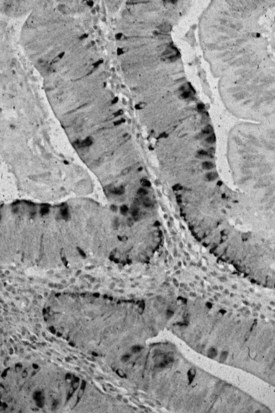
Microscopically, ampullary adenomas resemble their large bowel counterparts and may be tubular, villous, or mixed (tubulovillous) (Albores-Saavedra et al, 2000). The intestinal-type epithelium shows pseudostratification of elongated nuclei, and additional cytologic atypia and architectural complexity may be evident, depending on the degree of dysplasia (Fig. 56.27). Areas of high-grade dysplasia are characterized by complex, cribriform glands with considerable nuclear atypia and loss of epithelial polarity. Immunohistochemical stains often reveal abundant neuroendocrine cells and Paneth cells in ampullary adenomas (Fig. 56.28; Albores-Saavedra et al, 2000; Mora et al, 2004), and these two cell types may be prominent enough to be recognized in routinely stained material.
In some instances, ampullary adenomas may be treated effectively by endoscopic resection or transduodenal ampullectomy rather than pancreatoduodenectomy (see Chapters 27 and 62C; Bohra et al, 2002; Saurin et al, 2003). When this procedure is performed, it is essential for the pathologist to evaluate the lesion carefully to determine the completeness of resection and to search for subtle foci of invasive carcinoma. Sometimes this evaluation must be performed on frozen sections.
Intraampullary Papillary Tubular Neoplasms
Intestinal-type preinvasive neoplasms (adenomas), similar to those occurring on the duodenal surface, may also occur within the ampulla; however, in addition, noninvasive pancreatobiliary-type neoplasms (Klimstra et al, 2010c) may also be encountered in this region. Recently, the term intraampullary papillary tubular neoplasm (Ohike et al, 2010a) was proposed to unify all the tumoral intraepithelial neoplasms that arise predominantly within the ampulla—or the very terminal, intraampullary ends of the two ducts—under one category to avoid confusion and also to establish their kinship with IPMNs and intraductal tubulopapillary neoplasms of the pancreas. As in IPMNs, IAPNs occur predominantly in men at a mean age of 64 years. They form exophytic masses within the ampulla that lead to a protuberant ampulla with a widened orifice (see Fig. 56.26) through which a granular, polypoid tumor can be appreciated within the ampulla. Cut sections show friable, granular lesions with a mean size of 2.5 cm filling the ampullary lumen. Microscopically, half of the cases exhibit a mixture of papillary and tubular growth pattern, and in the remainder, one of the two patterns predominates. About three quarters of the cases have a more intestinal-type appearance, and the remainder have gastropancreatobiliary features; however, overlaps and hybrid types are common, presumably owing to the transitional nature of ampullary epithelium, which harbors different cell types. This is reflected also in the immunophenotype, which often shows discrepant and unexpected profiles using markers that are putatively lineage-specific. High-grade dysplasia is detected in the majority of the cases, and invasive carcinoma is found in more than 75%. Conversely, from the invasive carcinoma perspective, IAPNs are detected in the background of about 30% of invasive ampullary carcinomas. Invasive carcinoma often but not always follows the cell phenotype of the preinvasive component. Noninvasive carcinomas have a very good prognosis; however, they may occasionally develop recurrence in long-term follow-up. Invasive carcinomas arising from IAPNs have a malignant course but one that is significantly more protracted than that of invasive carcinomas unaccompanied by IAPNs (Ohike et al, 2010a).
Flat-Type Intraepithelial Neoplasia (Dysplasia and Carcinoma in Situ)
Much less common preinvasive neoplastic lesions of the ampulla include flat dysplasia (Albores-Saavedra et al, 2000), a lesion with epithelial dysplasia that does not form a grossly polypoid mass. Microscopically, a low papillary pattern may be apparent, because the epithelia of the papilla of Vater and the common channel normally have papillae. Otherwise, the dysplastic epithelium involves the native architecture of the ampulla.
Adenocarcinoma (See Chapter 59)
Similar to the adenomas and IAPNs from which many of them arise (Stolte & Pscherer, 1996), adenocarcinomas are more common in the ampulla than in other sites in the small bowel (Klimstra & Adsay, 2009). Also, they are prevalent in patients with FAP (Harned & Williams, 1982), generally presenting later than the large bowel carcinomas that these patients also have. The classic presentation of ampullary adenocarcinoma is considered to be biliary obstruction accompanied by a distended, palpable gallbladder (the Courvoisier sign); in truth, a palpable gallbladder is an uncommon finding. Accurate preoperative diagnosis depends on the extent of the invasive carcinoma, relative to any residual adenoma, and on the technique of endoscopic biopsy (Elek et al 2003; Rodriguez et al, 2002). The invasive components are commonly deeply situated within the adenoma, so superficial biopsy specimens may fail to reveal invasion. Especially for carcinomas located within the common channel, endoscopic papillotomy may be helpful to reach the diagnostic areas.
Ampullary adenocarcinomas are often relatively small when detected, presumably because of early symptoms. Nearly 20% are less than 1 cm, and more than 75% are less than 4 cm at diagnosis (Albores-Saavedra et al, 2000; Tajiri et al, 2009a). Because about a third of the cases arise in an adenoma or IAPN, the overall tumor size is often misleading. The gross configuration depends on the specific epithelia involved. Some ampullary carcinomas have the bulk of the lesion at the duodenal surface of the ampulla, forming a polypoid or ulcerated mass readily recognizable endoscopically, whereas other intraampullary carcinomas are covered by intact duodenal mucosa (Fig. 56.29). Larger tumors may replace the normal ampullary structures completely, making it difficult to determine precisely where the tumor arose. A subset of ampullary carcinomas forms schirrous, constrictive lesions in the wall of the distal tips of the ducts, in particular the common bile duct, without causing an overt exophytic mass in the ampulla or on the duodenal surface.
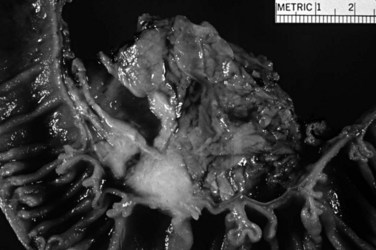
Microscopically, some ampullary adenocarcinomas resemble carcinomas of the large bowel and are referred to as intestinal type (Albores-Saavedra et al, 2000; Kimura et al, 1994). They are composed of individual, complex, cribriform glands with pseudostratified, elongated nuclei, commonly exhibiting extensive luminal necrosis (Fig. 56.30). Another subset of ampullary adenocarcinomas resembles ductal adenocarcinoma of the pancreas or common bile duct; these are referred to as a pancreatobiliary type, which show simple and relatively small glands with a single layer of cells with round nuclei and abundant stromal desmoplastic cells (Fig. 56.31). Most commonly, intestinal-type ampullary adenocarcinomas arise in association with a preinvasive neoplasm, although many pancreatobiliary-type adenocarcinomas appear to arise without a recognizable preinvasive lesion (Albores-Saavedra et al, 2000). Recent sudies have shown different biologic behavior for intestinal versus pancreatobiliary adenocarcinomas; however, it is also clear that a substantial portion of the cases has either hybrid or unclassifiable patterns, making this distinction highly subjective (Balci et al, 2010). Small, infiltrative clusters at the advancing edge of the tumor, so-called tumor budding, is seen quite commonly in ampullary adenocarcinomas when examined carefully and has been found to be an indicator of aggressive behavior (Ohike et al, 2010b). Perineural invasion is less common than it is in pancreatic cancer and may also be an indicator of adverse behavior (Lowe et al, 2009).
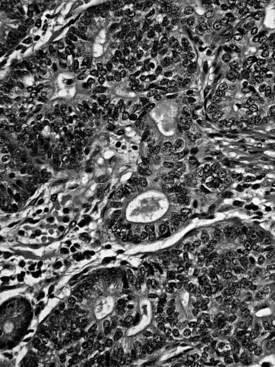
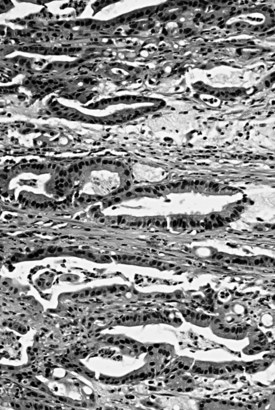
Ampullary adenocarcinomas typically invade into the muscularis of the sphincter of Oddi, extend through the duodenal submucosa and muscularis propria, and invade the pancreatic parenchyma or peripancreatic soft tissues. However, it is often difficult to determine the precise extent of the tumor at the three-dimensional level because of the anatomic complexity of this region. This may be partly responsible for the better survival recorded for stage pT2 cases compared with pT1 cases in the data from the National Cancer Database of American College of Surgeons (Edge et al, 2010). Furthermore, even relatively small invasive carcinomas can extend to the peripancreatic soft tissues, which creates problems in staging such cases (Tajiri et al, 2009a). Lymph node metastases may be found even in superficially invasive carcinomas and are detected in about 40% of patients, far less than the 75% associated with pancreatic ductal adenocarcinomas.
Numerous histologic variants of ampullary adenocarcinoma have been described, including adenosquamous carcinoma, clear cell carcinoma, mucinous adenocarcinoma, signet ring cell carcinoma, and micropapillary carcinoma, often associated with tumor infiltrating neutrophils (Albores-Saavedra et al, 2000; Hara et al, 2002; Khayyata et al, 2005; Ueno et al, 2002). Although the morphologic features of each of these subtypes are distinctive, specific clinical or genetic differences from the more common histologic types have yet to be described. Unusual carcinoma types, such as acinar, medullary, and osteoclastic giant cell, can also occur in the ampulla (see the discussion on other invasive carcinomas related to ductal adenocarcinoma).
The molecular genetic changes involved in the development of ampullary adenocarinomas roughly parallel those that occur in colorectal carcinomas: mutations in the KRAS oncogene are found in approximately 40% and include mutations in codon 12 and codon 13 (Scarpa et al, 1993). More than half of cases have mutations in TP53 (Albores-Saavedra et al, 2000; Howe et al, 1997; Kubota et al, 2003; Matsubayashi et al, 1999; Takashima et al, 2000; Younes et al, 1995), and loss of SMAD4 has been shown (McCarthy et al, 2003). However, abnormalities of DNA mismatch repair genes are uncommon in ampullary carcinomas (Park et al, 2003).
Ampullary carcinomas have a relatively favorable survival compared with ductal carcinomas of the pancreas; patients with localized disease have a 5-year survival of 50%, and patients with lymph node metastases have a 5-year survival of 28% (Albores-Saavedra et al, 2000; Howe et al, 1998; Tajiri et al, 2009b). Although some authors have questioned whether this outcome reflects a different biology for ampullary carcinoma, as opposed to simple detection at an earlier stage because of earlier onset of symptoms, even ampullary carcinoma patients with positive lymph nodes have a better survival than any group of pancreatic carcinoma patients (Howe et al, 1998).
High-Grade Neuroendocrine Carcinoma
Although high-grade neuroendocrine carcinomas of the ampulla are rare, they are more common in this location than in the pancreas (see Chapter 61). The clinical findings are the same as those of ampullary adenocarcinomas; hormonal symptoms are uncommon (Nassar et al, 2005). Many high-grade neuroendocrine carcinomas arise in association with an adenoma, and some contain mixed components of adenocarcinoma. Two histologic types occur, including small cell carcinoma (Zamboni et al, 1990) and large cell neuroendocrine carcinoma (Albores-Saavedra et al, 2000; Nassar et al, 2005). Small cell carcinomas have fusiform small cells with minimal cytoplasm, finely granular chromatin, and nuclear molding (Fig. 56.32). Large cell neuroendocrine carcinomas have moderate amounts of cytoplasm and round nuclei with prominent nucleoli. Both are distinguished from well-differentiated neuroendocrine tumors by their high mitotic rate (>20 per 10 HPFs) and abundant necrosis. Immunohistochemical evidence of neuroendocrine differentiation is generally identifiable and is required for the diagnosis of the large cell type, but staining for chromogranin and synaptophysin is less widespread than in carcinoid tumors (Fig. 56.33). Some of the molecular genetic abnormalities are different from those of ampullary adenocarcinomas, including frequent loss of retinoblastoma gene product expression (Nassar et al, 2005). High-grade neuroendocrine carcinomas are highly aggressive. Most patients are seen initially with lymph node involvement and suffer early recurrence and death from the tumor.
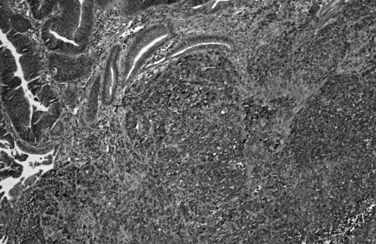
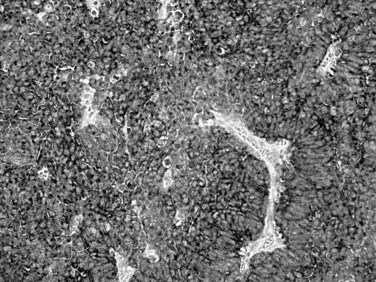
Well-Differentiated Neuroendocrine Neoplasms
Carcinoid Tumors
A family of well-differentiated neuroendocrine tumors occurs in and around the ampulla of Vater. Most of these have also been designated carcinoid tumors, although neuroendocrine tumor (NET) is now the preferred term (Burke et al, 1989; Hartel et al, 2005; Hatzitheoklitos et al, 1994; Niido et al, 2005). Ampullary NETs are distinguished from high-grade neuroendocrine carcinomas by their strikingly organoid growth pattern, with nests and ribbons of cells; their low mitotic rate (<20 mitoses per 10 HPFs); and their relative paucity of necrosis. They predominantly involve the submucosa and muscularis of the duodenum and ampulla, although extension into the mucosa is generally present, and endoscopic mucosal biopsy specimens usually yield diagnostic tissue (Bornstein-Quevedo & Gamboa-Dominguez, 2001). Most periampullary NETs are relatively small, ranging from a few millimeters to several centimeters. Tumors that directly involve the ampulla of Vater manifest early with biliary obstruction, whereas tumors situated in the surrounding duodenum are often asymptomatic unless associated with a neuroendocrine paraneoplastic syndrome. Periampullary NETs are associated with several hereditary genetic syndromes, including neurofibromatosis type 1 and MEN type I (Burke et al, 1989). Some NETs arising in these patients have distinctive clinical or morphologic features, representing either gastrinomas or so-called glandular duodenal carcinoid tumors. Most periampullary NETs are not associated with a clinical syndrome or distinctive morphology, however, and they only rarely give rise to the carcinoid syndrome, generally after the development of hepatic metastases. These NETs, not otherwise specified, resemble NETs of the stomach and upper GI tract, with a predominately solid, nested architecture; cytologic uniformity; coarsely granular chromatin pattern; and moderate amounts of amphophilic cytoplasm (Fig. 56.34; Makhlouf et al, 1999). By immunohistochemistry, the tumors strongly express the general neuroendocrine markers chromogranin and synaptophysin, and most express keratin as well (Albores-Saavedra et al, 2000; Burke et al, 1989). The low proliferative rate correlates with a low rate of immunolabeling for Ki-67, defined to be less than 20% and usually less than 5%. Small NETs (<1 cm) limited to the mucosa and submucosa generally lack lymph node metastases, and some may be treated effectively by local excision (Pyun et al, 2004); however, the lesion often proves to be more advanced than originally appreciated, and even small tumors may on occasion have metastases, thus these tumors can require pancreatoduodenectomy. As the size increases, the risk for nodal metastases also increases. Patients with nodal or hepatic metastases are difficult to cure, but the disease has a protracted clinical course, and survival for many years in the presence of liver metastases is not unusual.
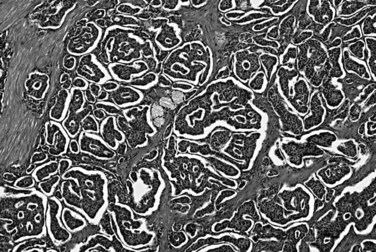
Gastrinomas of the periampullary region are NETs associated with hypersecretion of gastrin, resulting in peptic ulceration (Zollinger-Ellison syndrome). Periampullary gastrinomas are particularly prevalent in patients with MEN type I, and they outnumber pancreatic gastrinomas as the source of Zollinger-Ellison syndrome (Donow et al, 1991). In contrast to the pancreatic primaries, periampullary gastrinomas are often small (<1 cm), and regional lymph node metastases from these tumors may be larger than the primaries; and in some cases, the primary tumor is difficult to identify (Anlauf et al, 2008). For this reason, some authors advocate pancreatoduodenectomy for patients with gastrinoma syndrome with an unidentified primary to remove the most plausible culprit region, the “gastrinoma triangle.” Processing of such specimens in the pathology laboratory may require special protocols (Anlauf et al, 2008). Histologically, gastrinomas are indistinguishable from nonfunctioning NETs, but gastrin production is usually demonstrable by immunohistochemistry (Fig. 56.35). Although regional lymph node metastases are commonly present, periampullary gastrinomas have a better long-term prognosis than pancreatic gastrinomas, which are usually considerably larger at the time of diagnosis.
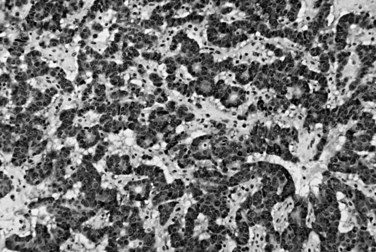
Glandular duodenal carcinoid tumors, also known as ampullary somatostatinomas or psammomatous somatostatinomas, are well-differentiated NETs with an exquisitely glandular growth pattern (Thirabanjasak et al, 2008). These tumors are highly characteristic of the periampullary region and are closely associated with neurofibromatosis type 1, although sporadic examples occur (Burke et al, 1990). Many of these tumors produce somatostatin (Dayal et al, 1983), and they almost never cause the somatostatinoma syndrome, in contrast to pancreatic somatostatinomas. In addition to their gland-forming architecture, glandular duodenal carcinoid tumors often exhibit psammoma bodies (Fig. 56.36; Albores-Saavedra et al, 2000; Dayal et al, 1983). By immunohistochemistry, expression of general neuroendocrine markers, such as chromogranin and synaptophysin, is consistent, and somatostatin is usually detectable. Although the nuclear features are those of a well-differentiated NET, the glandular architecture of glandular duodenal carcinoid tumors may cause confusion for adenocarcinoma. Also, abundant, lightly eosinophilic cytoplasm causes resemblance of the neoplastic glands to submucosal Brunner glands of the duodenum on endoscopic biopsy specimens. Glandular duodenal carcinoid tumors are relatively aggressive in their spread and are frequently associated with lymph node and hepatic metastases; however, limited data in the literature indicate that they may still be indolent neoplasms like other ampullary-region NETs (Thirabanjasak et al, 2008).
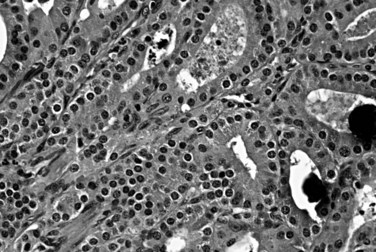
Gangliocytic Paraganglioma
A pathologically distinctive tumor that is essentially limited to the ampullary region, gangliocytic paraganglioma contains a variety of different cell types that have defied a logical histogenetic explanation. Most patients are adults who present with minimal symptoms, unless the tumor is close enough to the ampulla to cause biliary obstruction. Some cases are associated with neurofibromatosis type 1 (Burke & Helwig, 1989; Castoldi et al, 2001), but the association is less strong than for glandular duodenal carcinoid tumors. Gangliocytic paragangliomas are situated in the submucosa, similar to NETs, and some are polypoid or pedunculated and usually measure 1 to 3 cm. Microscopically, three different cellular components vary in proportion from case to case (Fig. 56.37; Albores-Saavedra et al, 2000; Burke & Helwig, 1989). Nests of epithelioid neuroendocrine cells are usually predominant. Individual ganglionic cells have abundant cytoplasm and large, round nuclei with prominent nucleoli. Finally, a spindle cell component represents Schwann cells. The neuroendocrine cells stain for chromogranin and synaptophysin and variably express keratin (Collina et al, 1991); this last finding establishes a relationship with NETs and distinguishes them from true paragangliomas. The ganglion cells express synaptophysin and neurofilaments, and the schwannian component stains for S100 protein. Most gangliocytic paragangliomas also produce pancreatic polypeptide (Altavilla et al, 2001). The origin of these tumors has been debated, because they contain endodermally derived (NET-like elements) and neural crest–derived (schwannian) components (Perrone, 1986). Most gangliocytic paragangliomas are benign, but rare metastases have been reported.
Abraham SC, et al. Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis–associated pancreatoblastomas: frequent alterations in the APC/beta-catenin pathway and chromosome 11p. Am J Pathol. 2001;159(5):1619-1627.
Abraham SC, et al. Solid-pseudopapillary tumors of the pancreas are genetically distinct from pancreatic ductal adenocarcinomas and almost always harbor beta-catenin mutations. Am J Pathol. 2002;160(4):1361-1369.
Adsay NV. Cystic neoplasia of the pancreas: pathology and biology. J Gastrointest Surg. 2008;12(3):401-404.
Adsay NV, Klimstra DS. Cystic forms of typically solid pancreatic tumors. Semin Diagn Pathol. 2000;17(1):81-88.
Adsay NV, Klimstra DS, Compton CC. Cystic lesions of the pancreas: introduction. Semin Diagn Pathol. 2000;17(1):1-6.
Adsay NV, Longnecker DS, Klimstra DS. Pancreatic tumors with cystic dilatation of the ducts: intraductal papillary mucinous neoplasms and intraductal oncocytic papillary neoplasms. Semin Diagn Pathol. 2000;17(1):16-30.
Adsay NV, Zamboni G. Paraduodenal pancreatitis: a clinico-pathologically distinct entity unifying “cystic dystrophy of heterotopic pancreas,” “para-duodenal wall cyst,” and “groove pancreatitis. Semin Diagn Pathol. 2004;21(4):247-254.
Adsay NV, et al. Intraductal oncocytic papillary neoplasms of the pancreas. Am J Surg Pathol. 1996;20(8):980-994.
Adsay NV, et al. Pseudotumoral pancreatitis: a clinicopathologic analysis of 33 patients with mass-forming pancreatitis with emphasis on the probable mechanisms (abstract). Mod Pathol. 2000;13(1):179A.
Adsay NV, et al. Colloid (mucinous noncystic) carcinoma of the pancreas. Am J Surg Pathol. 2001;25(1):26-42.
Adsay NV, et al. Intraductal papillary-mucinous neoplasms of the pancreas: an analysis of in situ and invasive carcinomas in 28 patients. Cancer. 2002;94(1):62-77.
Adsay NV, et al. Lymphoepithelial cysts of the pancreas: a report of 12 cases and a review of the literature. Mod Pathol. 2002;15(5):492-501.
Adsay NV, et al. Chronic pancreatitis or pancreatic ductal adenocarcinoma? Semin Diagn Pathol. 2004;21(4):268-276.
Adsay NV, et al. Pathologically and biologically distinct types of epithelium in intraductal papillary mucinous neoplasms: delineation of an “intestinal” pathway of carcinogenesis in the pancreas. Am J Surg Pathol. 2004;28(7):839-848.
Adsay NV, et al. Pancreatic pseudotumors: non-neoplastic solid lesions of the pancreas that clinically mimic pancreas cancer. Semin Diagn Pathol. 2004;21(4):260-267.
Adsay NV, et al. Secondary tumors of the pancreas: an analysis of a surgical and autopsy database and review of the literature. Virchows Arch. 2004;444(6):527-535.
Adsay NV, et al. A proposal for a new and more practical grading scheme for pancreatic ductal adenocarcinoma. Am J Surg Pathol. 2005;29(6):724-733.
Adsay NV, et al. The number of lymph nodes identified in a simple pancreatoduodenectomy specimen: comparison of conventional vs orange-peeling approach in pathologic assessment. Mod Pathol. 2009;22(1):107-112.
Adsay NV, et al. Tumours of the pancreas. Intraductal neoplasms of the pancreas. In: Bosman, FT, et al. WHO Classification of Tumours of the Digestive System. Lyon, France: International Agency for Research on Cancer, 2010.
Albores-Saavedra J. Acinar cystadenoma of the pancreas: a previously undescribed tumor. Ann Diagn Pathol. 2002;6(2):113-115.
Albores-Saavedra J, Henson DE, Klimstra DS. Tumors of the gallbladder, extrahepatic bile ducts, and ampulla of vater, 3rd series, fascicle 27. Atlas of Tumor Pathology. Washington, DC: Armed Forces Institute of Pathology. 2000.
Alexander JR, et al. High prevalence of adenomatous polyps of the duodenal papilla in familial adenomatous polyposis. Dig Dis Sci. 1989;34(2):167-170.
Altavilla G, Chiarelli S, Fassina A. Duodenal periampullary gangliocytic paraganglioma: report of two cases with immunohistochemical and ultrastructural study. Ultrastruct Pathol. 2001;25(2):137-145.
Altinel D, et al. Lipomatous pseudohypertrophy of the pancreas: a clinicopathologically distinct entity. Pancreas. 2009;39(3):392-397.
Andea A, Sarkar F, Adsay VN. Clinicopathological correlates of pancreatic intraepithelial neoplasia: a comparative analysis of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma. Mod Pathol. 2003;16(10):996-1006.
Anlauf M, Perren A, Klöppel G. Endocrine precursor lesions and microadenomas of the duodenum and pancreas with and without MEN1: criteria, molecular concepts and clinical significance. Pathobiology. 2007;74(5):279-284.
Anlauf M, et al. Microadenomatosis of the endocrine pancreas in patients with and without the multiple endocrine neoplasia type 1 syndrome. Am J Surg Pathol. 2006;30(5):560-574.
Anlauf M, et al. Primary lymph node gastrinoma or occult duodenal microgastrinoma with lymph node metastases in a MEN1 patient: the need for a systematic search for the primary tumor. Am J Surg Pathol. 2008;32(7):1101-1105.
Badia Bartolome C, et al. Groove pancreatitis and its differential diagnosis with pancreatic adenocarcinoma. Gastroenterol Hepatol. 2009;32(1):22-28.
Balci S, et al. Applicability and prognostic relevance of ampullary carcinoma histologic typing as pancreatobiliary versus intestinal. Mod Pathol. 2010;23(1):350A.
Ban D, et al. Pancreatic ducts as an important route of tumor extension for acinar cell carcinoma of the pancreas. Am J Surg Pathol. 2010;34(7):1025-1036.
Bandyopadhyay S, et al. Isolated solitary ducts (naked ducts) in adipose tissue: a specific but underappreciated finding of pancreatic adenocarcinoma and one of the potential reasons of understaging and high recurrence rate. Am J Surg Pathol. 2009;33(3):425-429.
Barton CM, et al. Abnormalities of the p53 tumour suppressor gene in human pancreatic cancer. Br J Cancer. 1991;64(6):1076-1082.
Bassi C, et al. Management of 100 consecutive cases of pancreatic serous cystadenoma: wait for symptoms and see at imaging or vice versa? World J Surg. 2003;27(3):319-323.
Basturk O, Coban I, Adsay NV. Pancreatic cysts: pathologic classification, differential diagnosis, and clinical implications. Arch Pathol Lab Med. 2009;133(3):423-438.
Basturk O, Farris AB, Adsay N. Immunohistology of pancreas, gallbladder, extrahepatic bile ducts, ampulla and liver. In: Dabbs, D, editor. Diagnostic Immunohistochemistry. Philadelphia: Elsevier, 2010.
Basturk O, et al. Intraductal and papillary variants of acinar cell carcinomas: a new addition to the challenging differential diagnosis of intraductal neoplasms. Am J Surg Pathol. 2007;31(3):363-370.
Basturk O, et al. Preferential expression of MUC6 in oncocytic and pancreatobiliary types of intraductal papillary neoplasms highlights a pyloropancreatic pathway, distinct from the intestinal pathway, in pancreatic carcinogenesis. Am J Surg Pathol. 2010;34(3):364-370.
Basturk O, et al. Pancreatobiliary osteoclastic–giant cell carcinomas (OGCCs) are often characterized by intraductal growth and polypoid pattern similar to sarcomatoid carcinomas (SCs) of other organs. Mod Pathol. 2010;23(1):350A.
Bohra AK, McKie L, Diamond T. Transduodenal excision of ampullary tumours. Ulster Med J. 2002;71(2):121-127.
Bornstein-Quevedo L, Gamboa-Dominguez A. Carcinoid tumors of the duodenum and ampulla of vater: a clinicomorphologic, immunohistochemical, and cell kinetic comparison. Hum Pathol. 2001;32(11):1252-1256.
Brugge WR, et al. Diagnosis of pancreatic cystic neoplasms: a report of the cooperative pancreatic cyst study. Gastroenterology. 2004;126(5):1330-1336.
Burke AP, Helwig EB. Gangliocytic paraganglioma. Am J Clin Pathol. 1989;92(1):1-9.
Burke AP, et al. Carcinoids of the duodenum: a histologic and immunohistochemical study of 65 tumors. Am J Surg Pathol. 1989;13(10):828-837.
Burke AP, et al. Somatostatin-producing duodenal carcinoids in patients with von Recklinghausen’s neurofibromatosis: a predilection for black patients. Cancer. 1990;65(7):1591-1595.
Cantrell BB, et al. Acinar cell cystadenocarcinoma of human pancreas. Cancer. 1981;47(2):410-416.
Cao D, et al. Expression of novel markers of pancreatic ductal adenocarcinoma in pancreatic nonductal neoplasms: additional evidence of different genetic pathways. Mod Pathol. 2005;18(6):752-761.
Casetti L, et al. “Paraduodenal” pancreatitis: results of surgery on 58 consecutives patients from a single institution. World J Surg. 2009;33(12):2664-2669.
Castell-Monsalve FJ, Sousa-Martin JM, Carranza-Carranza A. Groove pancreatitis: MRI and pathologic findings. Abdom Imaging. 2008;33(3):342-348.
Castoldi L, et al. Neurofibromatosis-1 and ampullary gangliocytic paraganglioma causing biliary and pancreatic obstruction. Int J Gastrointest Cancer. 2001;29(2):93-98.
Chatzipantelis P, et al. The role of cytomorphology and proliferative activity in predicting biologic behavior of pancreatic neuroendocrine tumors: a study by endoscopic ultrasound-guided fine-needle aspiration cytology. Cancer Cytopathol. 2009;117(3):211-216.
Cingolani N, et al. Alpha-fetoprotein production by pancreatic tumors exhibiting acinar cell differentiation: study of five cases, one arising in a mediastinal teratoma. Hum Pathol. 2000;31(8):938-944.
Coban I, et al. Paraduodenal pancreatitis is one of the main causes of pseudotumor in the pancreas and periampullary region: features of a distinct entity becoming clearer. Mod Pathol. 2009;22(S1):309A.
Collina G, Maiorana A, Trentini GP. Duodenal gangliocytic paraganglioma: case report with immunohistochemical study on the expression of keratin polypeptides. Histopathology. 1991;19(5):476-478.
Compton CC. Serous cystic tumors of the pancreas. Semin Diagn Pathol. 2000;17(1):43-56.
Dayal Y, et al. Psammomatous somatostatinomas of the duodenum. Am J Surg Pathol. 1983;7(7):653-665.
de Madaria E, et al. Cystic dystrophy of the duodenal wall and groove pancreatitis. Gastroenterol Hepatol. 2009;32(9):662-663.
Deshpande V, Lauwers GY. Cystic pancreatic endocrine tumor: a variant commonly confused with cystic adenocarcinoma. Cancer. 2007;111(1):47-53.
Deshpande V, et al. Autoimmune pancreatitis: a systemic immune complex mediated disease. Am J Surg Pathol. 2006;30(12):1537-1545.
Dhall D, et al. Use of immunohistochemistry for IgG4 in the distinction of autoimmune pancreatitis from peritumoral pancreatitis. Hum Pathol. 2010;41(5):643-652.
Dhebri AR, et al. Diagnosis, treatment and outcome of pancreatoblastoma. Pancreatology. 2004;4(5):441-451. discussion 452-443
Domizio P, et al. Upper gastrointestinal pathology in familial adenomatous polyposis: results from a prospective study of 102 patients. J Clin Pathol. 1990;43(9):738-743.
Donow C, et al. Surgical pathology of gastrinoma: site, size, multicentricity, association with multiple endocrine neoplasia type 1, and malignancy. Cancer. 1991;68(6):1329-1334.
Drut R, Jones MC. Congenital pancreatoblastoma in Beckwith–Wiedemann syndrome: an emerging association. Pediatr Pathol. 1988;8(3):331-339.
Edge SB, et al. AJCC Cancer Staging Manual, 7th ed. New York: Springer; 2010.
Eguchi H, et al. Patients with pancreatic intraductal papillary mucinous neoplasms are at high risk of colorectal cancer development. Surgery. 2006;139(6):749-754.
Elek G, et al. Histological evaluation of preoperative biopsies from ampulla vateri. Pathol Oncol Res. 2003;9(1):32-41.
Ferrone CR, et al. Determining prognosis in patients with pancreatic endocrine neoplasms: can the WHO classification system be simplified? J Clin Oncol. 2007;25(35):5609-5615.
Ferrone CR, et al. Pancreatic adenocarcinoma: the actual 5-year survivors. J Gastrointest Surg. 2008;12(4):701-706.
Fujii T, et al. Clinicopathological study of mucin-producing tumors of the pancreas: multi-centric development of carcinoma through aytpical hyperplasia. J Jpn Pancreatol Soc. 1996;11:344-352.
Fukushima N, et al. Ductal adenocarcinoma variants and mixed neoplasms of the pancreas. In: Bosman, FT, et al. WHO Classification of Tumours of the Digestive System. Lyon, France: International Agency for Research on Cancer, 2010.
Furukawa T, et al. Classification of types of intraductal papillary-mucinous neoplasm of the pancreas: a consensus study. Virchows Arch. 2005;447(5):794-799.
Goggins M, et al. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology: poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am J Pathol. 1998;152(6):1501-1507.
Hameed M, et al. Expression of p53 nucleophosphoprotein in in situ pancreatic ductal adenocarcinoma: an immunohistochemical study of 100 cases. Lab Invest. 1994;70:132A.
Handra-Luca A, et al. Adenomyoma and adenomyomatous hyperplasia of the Vaterian system: clinical, pathological, and new immunohistochemical features of 13 cases. Mod Pathol. 2003;16(6):530-536.
Hara T, et al. Signet-ring–cell carcinoma of the ampulla of Vater: a case report. Hepatogastroenterology. 2002;49(44):561-563.
Harned RK, Williams SM. Familial polyposis coli and periampullary malignancy. Dis Colon Rectum. 1982;25(3):227-229.
Hartel M, et al. Carcinoid of the ampulla of Vater. J Gastroenterol Hepatol. 2005;20(5):676-681.
Hatzitheoklitos E, et al. Carcinoid of the ampulla of Vater: clinical characteristics and morphologic features. Cancer. 1994;73(6):1580-1588.
Heitz PU, et al. Tumors of the endocrine pancreas. In: DeLellis, RA, et al. World Health Organization Classification of Tumors: Pathology and Genetics, Tumors of Endocrine Organs. Lyon: IARC Press; 2004:177-208.
Hochwald S, Conlon K, Brenann M. Nonfunctioning pancreatic islet cell tumors. In: Doherty, G, editor. Surgical Endocrinology. Philadelphia. Lippincott: Williams & Wilkins; 2001:361-373.
Hochwald SN, et al. Prognostic factors in pancreatic endocrine neoplasms: an analysis of 136 cases with a proposal for low-grade and intermediate-grade groups. J Clin Oncol. 2002;20(11):2633-2642.
Holen KD, et al. Clinical characteristics and outcomes from an institutional series of acinar cell carcinoma of the pancreas and related tumors. J Clin Oncol. 2002;20(24):4673-4678.
Hoorens A, et al. Undifferentiated carcinoma of the pancreas: analysis of intermediate filament profile and Ki-ras mutations provides evidence of a ductal origin. J Pathol. 1998;185(1):53-60.
Howe JR, et al. K-ras mutation in adenomas and carcinomas of the ampulla of vater. Clin Cancer Res. 1997;3(1):129-133.
Howe JR, et al. Factors predictive of survival in ampullary carcinoma. Ann Surg. 1998;228(1):87-94.
Hruban RH, Adsay NV. Molecular classification of neoplasms of the pancreas. Hum Pathol. 2009;40(5):612-623.
Hruban RH, Pitman MB, Klimstra DS. Tumors of the Pancreas. Armed Forces Institute of Pathology. Atlas of Tumor Pathology, 4th series, fascicle 6. Washington, DC: American Registry of Pathology. 2007.
Hruban RH, Zamboni G. Pancreatic cancer: special issue—insights and controversies in pancreatic pathology. Arch Pathol Lab Med. 2009;133(3):347-349.
Hruban RH, et al. Molecular pathology of pancreatic cancer. Cancer J. 2001;7(4):251-258.
Hruban RH, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25(5):579-586.
Ishigami K, et al. Differential diagnosis of groove pancreatic carcinomas vs. groove pancreatitis: usefulness of the portal venous phase. Eur J Radiol. 2009;74(3):e95-e100.
Iwama T, et al. Indications for local excision of ampullary lesions associated with familial adenomatous polyposis. J Am Coll Surg. 1994;179(4):462-464.
Jamali M, Chetty R. Predicting prognosis in gastroentero-pancreatic neuroendocrine tumors: an overview and the value of Ki-67 immunostaining. Endocr Pathol. 2008;19(4):282-288.
Kardon DE, et al. Adenosquamous carcinoma of the pancreas: a clinicopathologic series of 25 cases. Mod Pathol. 2001;14(5):443-451.
Kerr NJ, et al. Beckwith–Wiedemann syndrome, pancreatoblastoma, and the Wnt signaling pathway. Am J Pathol. 2002;160(4):1541-1542. author reply 1542
Khanani F, et al. Mesenchymal lesions involving the pancreas (abstract). Mod Pathol. 2003;16(1):279A.
Khayyata S, Basturk O, Adsay NV. Invasive micropapillary carcinomas of the ampullo-pancreatobiliary region and their association with tumor-infiltrating neutrophils. Mod Pathol. 2005;18(11):1504-1511.
Kimura W, et al. Different clinicopathologic findings in two histologic types of carcinoma of papilla of Vater. Jpn J Cancer Res. 1994;85(2):161-166.
Kitagami H, et al. Acinar cell carcinoma of the pancreas: clinical analysis of 115 patients from Pancreatic Cancer Registry of Japan Pancreas Society. Pancreas. 2007;35(1):42-46.
Klimstra DS. Cell lineage in pancreatic neoplasms. In: Sarkar, F, Dugan, MC. Pancreatic Cancer: Advances in Molecular Pathology, Diagnosis, and Clinical Management. Natick, MA, Biotechniques Books, 1998.
Klimstra DS. Nonductal neoplasms of the pancreas. Mod Pathol. 2007;20(Suppl 1):S94-S112.
Klimstra DS, Adsay NV. Acinar cell carcinoma of the pancreas: a case associated with lipase hypersecretion syndrome. Pathol Case Rev. 2001;6:121-126.
Klimstra DS, Adsay NV. Tumors of the pancreas and ampulla of Vater. In: Odze, RD, Goldblum, JR. Surgical Pathology of the GI Tract, Liver, Biliary Tract, and Pancreas. Philadelphia: Saunders; 2009:909-960.
Klimstra DS, Hruban RH, Pitman MB. Gallbladder and extrahepatic biliary system. In: Mills, SE, editor. Histology for Pathologists. Philadelphia. Lippincott: Williams & Wilkins; 2007:705-722.
Klimstra DS, Hruban RH, Pitman MB. Pancreas. In: Mills, SE, editor. Histology for Pathologists. Philadelphia. Lippincott: Williams & Wilkins; 2007:721-760.
Klimstra DS, Rosai J, Heffess CS. Mixed acinar-endocrine carcinomas of the pancreas. Am J Surg Pathol. 1994;18(8):765-778.
Klimstra DS, Wenig BM, Heffess CS. Solid-pseudopapillary tumor of the pancreas: a typically cystic tumor of low malignant potential. Semin Diagn Pathol. 2000;17(1):66-81.
Klimstra DS, et al. Acinar cell carcinoma of the pancreas: a clinicopathologic study of 28 cases. Am J Surg Pathol. 1992;16(9):815-837.
Klimstra DS, et al. Pancreatoblastoma: a clinicopathologic study and review of the literature. Am J Surg Pathol. 1995;19(12):1371-1389.
Klimstra DS, et al. Intraductal tubular carcinoma of the pancreas: clinicopathologic and immunohistochemical analysis of 18 cases. Mod Pathol. 2007;20(2):285A.
Klimstra DS, et al. Pathology reporting of neuroendocrine tumors: application of the Delphic consensus process to the development of a minimum pathology data set. Am J Surg Pathol. 2010;34(3):300-313.
Klimstra DS, et al. The pathologic classification of neuroendocrine tumors: a review of nomenclature, grading, and staging systems. Pancreas. 2010;39(6):707-712.
Klimstra DS, et al. Tumors of the ampulla: adenomas and other preinvasive neoplastic lesions. In: Bosman, FT, et al. WHO Classification of Tumours of the Digestive System. Lyon, France: International Agency for Research on Cancer, 2010.
Klöppel G. Chronic pancreatitis, pseudotumors and other tumor-like lesions. Mod Pathol. 2007;20(Suppl 1):S113-S131.
Klöppel G, Maillet B. Chronic pancreatitis: evolution of the disease. Hepatogastroenterology. 1991;38(5):408-412.
Klöppel G, et al. Intraductal proliferation in the pancreas and its relationship to human and experimental carcinogenesis. Virchows Arch A Pathol Anat Histol. 1980;387(2):221-233.
Klöppel G, et al. Solid and cystic acinar cell tumour of the pancreas: a tumour in young women with favourable prognosis. Virchows Arch Pathol Anat Histol. 1981;392(2):171-183.
Klöppel G, et al. Site-specific biology and pathology of gastroenteropancreatic neuroendocrine tumors. Virchows Arch. 2007;451(Suppl 1):S9-27.
Klöppel G, et al. The ENETS and AJCC/UICC TNM classifications of the neuroendocrine tumors of the gastrointestinal tract and the pancreas: a statement. Virchows Arch. 2010;456(6):595-597.
Klöppel G, et al. Autoimmune pancreatitis: the clinicopathological characteristics of the subtype with granulocytic epithelial lesions. J Gastroenterol. 2010;45(8):787-793.
Koizumi K, et al. Synchronous double invasive ductal carcinomas of the pancreas with multifocal branch duct intraductal papillary mucinous neoplasms of the pancreas. Nippon Shokakibyo Gakkai Zasshi. 2009;106(1):98-105.
Kosmahl M, et al. Solid-pseudopapillary tumor of the pancreas: its origin revisited. Virchows Arch. 2000;436(5):473-480.
Kosmahl M, et al. Cystic neoplasms of the pancreas and tumor-like lesions with cystic features: a review of 418 cases and a classification proposal. Virchows Arch. 2004;445(2):168-178.
Kubota K, et al. Usefulness of endoscopic biopsy using immunostaining of p53 and Ki-67 in tumors of the ampulla of Vater. Pathol Int. 2003;53(6):361-370.
Labate AM, Klimstra DL, Zakowski MF. Comparative cytologic features of pancreatic acinar cell carcinoma and islet cell tumor. Diagn Cytopathol. 1997;16(2):112-116.
Ladanyi M, et al. Estrogen and progesterone receptor determination in the papillary cystic neoplasm of the pancreas (with immunohistochemical and ultrastructural observations). Cancer. 1987;60(7):1604-1611.
La Rosa S, et al. Prognostic criteria in nonfunctioning pancreatic endocrine tumours. Virchows Arch. 1996;429(6):323-333.
La Rosa S, et al. Prognostic and biological significance of cytokeratin 19 in pancreatic endocrine tumours. Histopathology. 2007;50(5):597-606.
Levenick JM, et al. A comprehensive, case-based review of groove pancreatitis. Pancreas. 2009;38(6):e169-e175.
Lieber MR, et al. Solid and papillary epithelial neoplasm of the pancreas: an ultrastructural and immunocytochemical study of six cases. Am J Surg Pathol. 1987;11(2):85-93.
Lowe MC, et al. Important prognostic factors in adenocarcinoma of the ampulla of Vater. Am Surg. 2009;75(9):754-760. discussion 761
Lüttges J, Zamboni G, Klöppel G. Recommendation for the examination of pancreaticoduodenectomy specimens removed from patients with carcinoma of the exocrine pancreas: a proposal for a standardized pathological staging of pancreaticoduodenectomy specimens including a checklist. Dig Surg. 1999;16(4):291-296.
Lüttges J, et al. Malignant non-epithelial tumors of the pancreas. Pathologe. 1997;18(3):233-237.
Lüttges J, et al. The retroperitoneal resection margin and vessel involvement are important factors determining survival after pancreaticoduodenectomy for ductal adenocarcinoma of the head of the pancreas. Virchows Arch. 1998;433(3):237-242.
Lüttges J, et al. Solitary fibrous tumour of the pancreas: a new member of the small group of mesenchymal pancreatic tumours. Virchows Arch. 1999;435(1):37-42.
Makhlouf HR, Burke AP, Sobin LH. Carcinoid tumors of the ampulla of Vater: a comparison with duodenal carcinoid tumors. Cancer. 1999;85(6):1241-1249.
Matsubayashi H, et al. Differences in mucus and K-ras mutation in relation to phenotypes of tumors of the papilla of vater. Cancer. 1999;86(4):596-607.
Matsumoto T, et al. Malignant serous cystic neoplasm of the pancreas: report of a case and review of the literature. J Clin Gastroenterol. 2005;39(3):253-256.
McCarthy DM, et al. Role of the DPC4 tumor suppressor gene in adenocarcinoma of the ampulla of Vater: analysis of 140 cases. Mod Pathol. 2003;16(3):272-278.
Mora JI, et al. Paneth cell carcinoma of the ampulla of Vater. Arch Pathol Lab Med. 2004;128(8):908-910.
Morselli-Labate AM, Pezzilli R. Usefulness of serum IgG4 in the diagnosis and follow up of autoimmune pancreatitis: a systematic literature review and meta-analysis. J Gastroenterol Hepatol. 2009;24(1):15-36.
Movahedi-Lankarani S, et al. Primitive neuroectodermal tumors of the pancreas: a report of seven cases of a rare neoplasm. Am J Surg Pathol. 2002;26(8):1040-1047.
Nagai K, et al. Single-institution validation of the international consensus guidelines for treatment of branch duct intraductal papillary mucinous neoplasms of the pancreas. J Hepatobiliary Pancreat Surg. 2009;16(3):353-358.
Nakatani Y, et al. Biotin-rich, optically clear nuclei express estrogen receptor-beta: tumors with morules may develop under the influence of estrogen and aberrant beta-catenin expression. Hum Pathol. 2004;35(7):869-874.
Nassar H, Albores-Saavedra J, Klimstra DS. High-grade neuroendocrine carcinoma of the ampulla of vater: a clinicopathologic and immunohistochemical analysis of 14 cases. Am J Surg Pathol. 2005;29(5):588-594.
Niido T, et al. Carcinoid of major duodenal papilla. Gastrointest Endosc. 2005;61(1):106-107.
Noda Y, et al. Histologic follow-up of ampullary adenomas in patients with familial adenomatosis coli. Cancer. 1992;70(7):1847-1856.
Odze R, et al. Duodenal adenomas in familial adenomatous polyposis: relation of cell differentiation and mucin histochemical features to growth pattern. Mod Pathol. 1994;7(3):376-384.
Ohike N, Kosmahl M, Klöppel G. Mixed acinar-endocrine carcinoma of the pancreas: a clinicopathological study and comparison with acinar-cell carcinoma. Virchows Arch. 2004;445(3):231-235.
Ohike N, et al. Intraampullary papillary-tubular neoplasm (IAPN): characterization of tumoral intraepithelial neoplasia occuring within the ampulla: a clinicopathologic analysis of 82 cases. Am J Surg Pathol. 2010;34:1731-1748.
Ohike N, et al. Tumor budding as a strong prognostic indicator in invasive ampullary adenocarcinomas. Am J Surg Pathol. 2010;34:1417-1424.
Paal E, Thompson LD, Heffess CS. A clinicopathologic and immunohistochemical study of ten pancreatic lymphangiomas and a review of the literature. Cancer. 1998;82(11):2150-2158.
Papavramidis T, Papavramidis S. Solid pseudopapillary tumors of the pancreas: review of 718 patients reported in English literature. J Am Coll Surg. 2005;200(6):965-972.
Park S, et al. Lack of microsatellite instability in neoplasms of ampulla of Vater. Pathol Int. 2003;53(10):667-670.
Pauser U, et al. Cellular hamartoma resembling gastrointestinal stromal tumor: a solid tumor of the pancreas expressing c-kit (CD117). Mod Pathol. 2005;18(9):1211-1216.
Pauser U, et al. Pancreatic solid and cystic hamartoma in adults: characterization of a new tumorous lesion. Am J Surg Pathol. 2005;29(6):797-800.
Pelosi G, et al. Endocrine tumors of the pancreas: Ki-67 immunoreactivity on paraffin sections is an independent predictor for malignancy: a comparative study with proliferating-cell nuclear antigen and progesterone receptor protein immunostaining, mitotic index, and other clinicopathologic variables. Hum Pathol. 1996;27(11):1124-1134.
Perigny M, et al. Pancreatic endocrine microadenomatosis in patients with von Hippel–Lindau disease: characterization by VHL/HIF pathway proteins expression. Am J Surg Pathol. 2009;33(5):739-748.
Perrone T. Duodenal gangliocytic paraganglioma and carcinoid. Am J Surg Pathol. 1986;10(2):147-149.
Perzin KH, Bridge MF. Adenomas of the small intestine: a clinicopathologic review of 51 cases and a study of their relationship to carcinoma. Cancer. 1981;48(3):799-819.
Pettinato G, et al. Papillary cystic tumor of the pancreas: a clinicopathologic study of 20 cases with cytologic, immunohistochemical, ultrastructural, and flow cytometric observations, and a review of the literature (published erratum appears in Am J Clin Pathol 1993 99(6):764; see comments). Am J Clin Pathol. 1992;98(5):478-488.
Pitman MB, et al. Pancreatic cysts: preoperative diagnosis and clinical management. Cancer Cytopathol. 2010;118(1):1-13.
Pyun DK, et al. A carcinoid tumor of the ampulla of Vater treated by endoscopic snare papillectomy. Korean J Intern Med. 2004;19(4):257-260.
Reddy RP, et al. Pancreatic mucinous cystic neoplasm defined by ovarian stroma: demographics, clinical features, and prevalence of cancer. Clin Gastroenterol Hepatol. 2004;2(11):1026-1031.
Rindi G, et al. TNM staging of midgut and hindgut (neuro) endocrine tumors: a consensus proposal including a grading system. Virchows Arch. 2007;451(4):757-762.
Rodriguez C, et al. How accurate is preoperative diagnosis by endoscopic biopsies in ampullary tumours? Rev Esp Enferm Dig. 2002;94(10):585-592.
Roggin KK, et al. Limitations of ampullectomy in the treatment of nonfamilial ampullary neoplasms. Ann Surg Oncol. 2005;12(12):971-980.
Sadakari Y, et al. Invasive carcinoma derived from the nonintestinal type intraductal papillary mucinous neoplasm of the pancreas has a poorer prognosis than that derived from the intestinal type. Surgery. 2010;147(6):812-817.
Sakorafas GH, Sarr MG. Cystic neoplasms of the pancreas: what a clinician should know. Cancer Treat Rev. 2005;31(7):507-535.
Salvia R, et al. Main-duct intraductal papillary mucinous neoplasms of the pancreas: clinical predictors of malignancy and long-term survival following resection. Ann Surg. 2004;239(5):678-685. discussion 685-677
Sanchez-Castanon M, de Las Heras-Castano G, Lopez-Hoyos M. Autoimmune pancreatitis: an underdiagnosed autoimmune disease with clinical, imaging and serological features. Autoimmun Rev. 2009;9(4):237-240.
Saurin JC, et al. Long-term follow-up of patients with endoscopic treatment of sporadic adenomas of the papilla of vater. Endoscopy. 2003;35(5):402-406.
Scarpa A, et al. Neoplasia of the ampulla of Vater: Ki-ras and p53 mutations. Am J Pathol. 1993;142(4):1163-1172.
Schmitt AM, et al. WHO 2004 criteria and CK19 are reliable prognostic markers in pancreatic endocrine tumors. Am J Surg Pathol. 2007;31(11):1677-1682.
Schnelldorfer T, et al. Experience with 208 resections for intraductal papillary mucinous neoplasm of the pancreas. Arch Surg. 2008;143(7):639-646. discussion 646
Seidel G, et al. Almost all infiltrating colloid carcinomas of the pancreas and periampullary region arise from in situ papillary neoplasms: a study of 39 cases. Am J Surg Pathol. 2002;26(1):56-63.
Shemesh E, Bat L. A prospective evaluation of the upper gastrointestinal tract and periampullary region in patients with Gardner syndrome. Am J Gastroenterol. 1985;80(11):825-827.
Shintaku M, Arimoto A, Sakita N. Serous cystadenocarcinoma of the pancreas. Pathol Int. 2005;55(7):436-439.
Shorter NA, et al. Malignant pancreatic tumors in childhood and adolescence: the Memorial Sloan–Kettering experience, 1967 to present. J Pediatr Surg. 2002;37(6):887-892.
Solcia E, Sessa F, Rindi G. Pancreatic endocrine tumors: general concepts, nonfunctoning tumors and tumors with uncommon function. In: Dayal, Y, editor. Endocrine Pathology of the Gut and Pancreas. Boca Raton: CRC Press; 1991:105-131.
Staley CA, et al. The need for standardized pathologic staging of pancreaticoduodenectomy specimens. Pancreas. 1996;12(4):373-380.
Stamm B, Burger H, Hollinger A. Acinar cell cystadenocarcinoma of the pancreas. Cancer. 1987;60(10):2542-2547.
Stelow EB, et al. Pancreatic acinar cell carcinomas with prominent ductal differentiation: mixed acinar ductal carcinoma and mixed acinar endocrine ductal carcinoma. Am J Surg Pathol. 2010;34(4):510-518.
Stolte M, Pscherer C. Adenoma-carcinoma sequence in the papilla of Vater. Scand J Gastroenterol. 1996;31(4):376-382.
Stommer P, et al. Solid and cystic pancreatic tumors: clinical, histochemical, and electron microscopic features in ten cases. Cancer. 1991;67(6):1635-1641.
Strobel O, et al. Risk of malignancy in serous cystic neoplasms of the pancreas. Digestion. 2003;68(1):24-33.
Sugiyama M, Atomi Y. Extrapancreatic neoplasms occur with unusual frequency in patients with intraductal papillary mucinous tumors of the pancreas. Am J Gastroenterol. 1999;94(2):470-473.
Tabata T, et al. Serum IgG4 concentrations and IgG4-related sclerosing disease. Clin Chim Acta. 2009;408(1-2):25-28.
Tajiri T, et al. Intraductal tubular neoplasms of the pancreas: histogenesis and differentiation. Pancreas. 2005;30(2):115-121.
Tajiri T, et al. Proposal for a more applicable and clinically relevant staging evaluation of ampullary carcinomas. Mod Pathol. 2009;22(1):149A.
Tajiri T, et al. Prognostic differences between ampullary carcinomas and pancreatic ductal adenocarcinomas: the importance of size of invasive component. Mod Pathol. 2009;22(1):323A.
Takashima M, et al. Carcinoma of the ampulla of Vater associated with or without adenoma: a clinicopathologic analysis of 198 cases with reference to p53 and Ki-67 immunohistochemical expressions. Mod Pathol. 2000;13(12):1300-1307.
Tanaka M, et al. International consensus guidelines for management of intraductal papillary mucinous neoplasms and mucinous cystic neoplasms of the pancreas. Pancreatology. 2006;6(1-2):17-32.
Tang LH, et al. Clinically aggressive solid pseudopapillary tumors of the pancreas: a report of two cases with components of undifferentiated carcinoma and a comparative clinicopathologic analysis of 34 conventional cases. Am J Surg Pathol. 2005;29(4):512-519.
Tanno S, et al. Natural history of branch duct intraductal papillary-mucinous neoplasms of the pancreas without mural nodules: long-term follow-up results. Gut. 2008;57(3):339-343.
Tanno S, et al. Incidence of synchronous and metachronous pancreatic carcinoma in 168 patients with branch duct intraductal papillary mucinous neoplasm. Pancreatology. 2010;10(2-3):173-178.
Tascilar M, et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2001;7(12):4115-4121.
Temellini F, et al. Pancreatic metastasis 25 years after nephrectomy for renal cancer. Tumori. 1989;75(5):503-504.
Terris B, et al. Intraductal papillary mucinous tumors of the pancreas confined to secondary ducts show less aggressive pathologic features as compared with those involving the main pancreatic duct. Am J Surg Pathol. 2000;24(10):1372-1377.
Thirabanjasak D, et al. Glandular carcinoids of the ampulla with psammoma bodies (so-called ampullary somatostatinomas): analysis of 12 cases of an under-recognized entity. Mod Pathol. 2008;21(1):137A.
Thompson LDR, et al. Mucinous cystic neoplasm (mucinous cystadenocarcinoma of low malignant potential) of the pancreas: a clinicopathologic study of 130 cases. Am J Surg Pathol. 1999;23(1):1-16.
Tiemann K, et al. Solid pseudopapillary neoplasms of the pancreas show an interruption of the Wnt-signaling pathway and express gene products of 11q. Mod Pathol. 2007;20(9):955-960.
Tranchida P, et al. Poorly differentiated neuroendocrine (small cell) carcinomas of the pancreas: an analysis of 8 examples of a debated entity (abstract). Mod Pathol. 2000;13(1):190A.
Triantopoulou C, et al. Groove pancreatitis: a diagnostic challenge. Eur Radiol. 2009;19(7):1736-1743.
Ueno N, et al. Adenosquamous cell carcinoma arising from the papilla major. Oncol Rep. 2002;9(2):317-320.
van der Heijden MS, et al. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003;63(10):2585-2588.
Westra WH, et al. K-ras oncogene mutations in osteoclast-like giant cell tumors of the pancreas and liver: genetic evidence to support origin from the duct epithelium. Am J Surg Pathol. 1998;22(10):1247-1254.
White R, et al. Fate of the remnant pancreas after resection of noninvasive intraductal papillary mucinous neoplasm. J Am Coll Surg. 2007;204(5):987-993. discussion 993-985
White TJ, et al. Is there a prognostic difference between functional and nonfunctional islet cell tumors? Am J Surg. 1994;168(6):627-629. discussion 629-630
Wilentz RE, et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: a newly described and characterized entity. Am J Pathol. 2000;156(5):1641-1651.
Wisnoski NC, et al. 672 patients with acinar cell carcinoma of the pancreas: a population-based comparison to pancreatic adenocarcinoma. Surgery. 2008;144(2):141-148.
Yamaguchi K, et al. Intraductal papillary-mucinous tumor of the pancreas concomitant with ductal carcinoma of the pancreas. Pancreatology. 2002;2(5):484-490.
Yamaguchi H, et al. Intraductal tubulopapillary neoplasms of the pancreas distinct from pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2009;33(8):1164-1172.
Younes M, et al. p53 protein accumulation in tumors of the ampulla of Vater. Cancer. 1995;76(7):1150-1154.
Young RH. From Krukenberg to today: the ever present problems posed by metastatic tumors in the ovary. Part II. Adv Anat Pathol. 2007;14(3):149-177.
Zamboni G, et al. Small-cell neuroendocrine carcinoma of the ampullary region: a clinicopathologic, immunohistochemical, and ultrastructural study of three cases. Am J Surg Pathol. 1990;14(8):703-713.
Zamboni G, et al. Mucinous cystic tumors of the pancreas: clinicopathological features, prognosis, and relationship to other mucinous cystic tumors. Am J Surg Pathol. 1999;23(4):410-422.
Zamboni G, et al. Acinar cell cystadenoma of the pancreas: a new entity? Am J Surg Pathol. 2002;26(6):698-704.
Zamboni G, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch. 2004;445(6):552-563.
Zhang L, et al. KIT is an independent prognostic marker for pancreatic endocrine tumors: a finding derived from analysis of islet cell differentiation markers. Am J Surg Pathol. 2009;33(10):1562-1569.