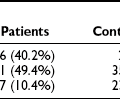Pancreatic and Islet Transplantation
Both type 1 and type 2 diabetes are major and growing worldwide health problems, largely due to their well-known vascular, eye, kidney, and neural complications. It is now accepted that these devastating problems are linked to hyperglycemia,1,2 which strongly implies that normalization of glucose levels with proper treatment early in the course of the disease will prevent these complications. The cost of these complications in personal and financial terms is enormous. Despite impressive improvements in treatment, including self-monitoring of glucose, advances in insulin therapy, new medications, and higher standards of care, too many people with diabetes continue to develop disabling complications. Transplantation of pancreatic tissue, as either the intact pancreas or isolated islets, is a treatment option for some individuals with diabetes. A successful pancreas or islet transplant offers normal or near-normal blood glucose control with decreased risk of hypoglycemia, as compared to intensive insulin therapy. Intact pancreas transplantation offers many recipients sustained, durable glycemic control but at the risk of surgical, postoperative, and immunosuppression-related complications. Islet transplantation is attractive as a less invasive alternative, but currently it does not achieve the same level of tight glucose control or durability as intact pancreas transplantation and has its own potential complications.
Pancreas Transplantation
The first clinical vascularized whole pancreas transplant was a duct-ligated segmental graft with a simultaneous kidney transplant performed by Drs. William Kelly and Richard Lillehei in 1966 at the University of Minnesota.3 Unfortunately, the first two pancreas recipients died within 6 months of transplantation. Over the next 15 years, the procedure was performed in relatively few centers and with an initial low success rate, but as surgical techniques improved and new immunosuppression approaches became available, by the mid-1980s the procedure became more widespread. As reported by the International Pancreas Transplant Registry (IPTR, www.iptr.umn.edu), over 23,000 pancreas transplants were performed through 2004, with more than 17,000 being done in the United States and approximately 6000 elsewhere.4 The increased numbers are explained by both improved outcomes and better coverage by insurance.
Indications and Types of Pancreas Transplants
Pancreas transplantation is most often performed in combination with kidney transplantation for patients who have renal failure secondary to diabetic nephropathy, either simultaneously (e.g., simultaneous pancreas and kidney [SPK]) or after kidney transplant (e.g., pancreas after kidney [PAK]). This is in agreement with the American Diabetes Association clinical practice recommendation that barring other contraindications, “pancreas transplants should be considered an acceptable therapeutic alternative to continued insulin therapy in diabetic patients with imminent or established end-stage renal disease who have had or plan to have a kidney transplant, since the successful addition of a pancreas does not jeopardize patient survival, may improve kidney survival, and will restore normal glycemia.”5 These individuals will already require immunosuppression as part of the medical regimen for the renal transplantation, and thus the primary additional risk is the operative procedure.
There has been considerable debate around pancreas transplant alone (PTA) in the absence of indications for kidney transplantation, with its risks of mortality, morbidity, and immunosuppression. Current recommendations are that PTA only be considered if the following criteria are met: (1) frequent, acute, and severe metabolic complications; (2) incapacitating clinical and emotional problems with exogenous insulin therapy; and (3) consistent failure of insulin-based management to prevent acute complications.5
Beyond the immunosuppressive risks, PTA has historically had a much higher graft failure rate than SPK. Much of the difference in graft survival rates between SPK and PTA is caused by a higher rate of graft loss due to rejection or thrombosis in the PTA group, but recent improvements have reduced these failure rates, increasing interest in PTA. Comparing 1987-1992 and 2004-2005, the 1-year pancreas graft survival rates improved from 76% to 86% for SPK and from 55% to 80% for PTA.6,7 During this same time, there have been improvements in insulin-based therapies for diabetes, including the introduction of more physiologic insulin regimens. Several conflicting analyses comparing patients on the waiting list to those undergoing pancreas transplantation have been published, with most of the divergence in the analyses attributable to differences in calculation of waiting list–associated mortality.8–10 Currently, SPK has significant impact on reduction of mortality, whereas a solitary pancreas transplant (PAK or PTA), when compared with forgoing the transplant, appears not to have a significant beneficial effect on survival. However, the benefits of improved quality of life, including reversing hypoglycemic unawareness and a modest amelioration of complications, could tilt patients towards this surgical option. The very poor survival rates of patients on the SPK waiting list may push patients to find living kidney donors, which will not only expand the pool of available kidneys but also facilitate their having a PAK procedure. In 2006, there were 1386 pancreas transplants reported to the U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients (OPTN/SRTR), with 67% (n = 924) SPK, 21% (n = 293) PAK, and 12% (n = 169) PTA recipients.7
Surgical Techniques
The evolution of surgical approaches to pancreas transplants has been previously detailed by Sutherland et al.11 Various early approaches to drainage of exocrine secretion included duct ligation, duct obliteration, enteric drainage, and bladder drainage. One of the major advances that led to improved results and thus wider acceptance of pancreas transplantation was the use of a duodenum-to-bladder drainage as pioneered by Sollinger’s group,12 which reduced the rate of transplant exocrine secretion leaks and allowed monitoring for evidence of early graft rejection by measurement of amylase activity in the urine. Unfortunately, loss of the bicarbonate-rich pancreatic secretions into the bladder as seen with this technique is associated with a number of problems: metabolic acidosis, dehydration, cystitis, urethritis, reflux pancreatitis, stricture formation, and bladder stones. Patients with bladder drainage of the pancreas may require daily oral bicarbonate and fluid replacement to offset the associated acidosis and fluid losses. By 3 years posttransplant, approximately 15% of such patients require reversal of bladder drainage, with redirection of secretions into the small bowel.6,13 By the late 1990s, improvements in immunosuppression and experience with direct percutaneous needle biopsy had reduced the need for urinary amylase monitoring to identify rejection, and many groups moved to enteric drainage, first with a Roux-en-Y technique, and now more commonly a side-to-side anastomosis of the donor duodenum and the recipient ileum (Fig. 24-1). In the United States, of the pancreas transplants performed in 2002-2003, enteric drainage was employed in 82% of SPK, 72% of PAK, and 57% of PTA procedures.4
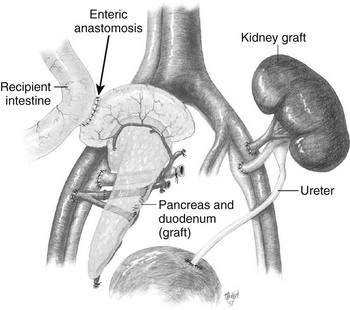
FIGURE 24-1 Combined pancreas and kidney transplant. Digestive juices of the pancreas are drained into the intestine via an enteric anastomosis between the donor duodenum and the recipient ileum. Venous outflow can be either into the portal vein or to the peripheral circulation via the iliac vein as shown. (Drawing courtesy Dr. David Sutherland.)
Venous Drainage
The majority of the venous drainage of the native pancreas is via the pancreaticoduodenal veins into the portal vein. Classically, venous drainage from a transplanted pancreas has been directed into the systemic circulation, but some centers are now employing the superior mesenteric vein to allow drainage into the portal vein, which is considered more physiologic but also more technically demanding.14,15 It has been suggested that presentation of pancreas alloantigens to the liver via portal drainage may provide an immunologic benefit,16 but this remains controversial and has not been confirmed in several prospective studies. An analysis of United Network for Organ Sharing (UNOS) registry data suggests that any immunologic advantage is dwarfed by current immunosuppression regimens.17 The portal vein route does result in lower peripheral circulating insulin levels.18,19 The continued use of portal drainage, despite being more technically demanding, highlights concerns that hyperinsulinemia may accelerate atherosclerosis. However, more atherogenic lipid profiles have not been identified with systemic drainage, and any potential benefit may be relatively small compared to the lipid changes associated with current immunosuppression regimens. Anecdotally, systemic drainage has also been linked to allograft-associated hypoglycemia, but these cases are rare and have not been well studied.20 Systemic venous drainage remains the more commonly performed procedure, and of enteric-drained grafts performed in the United States in the period 2002-2003, it was used in 80% of SPK, 79% of PAK, and 72% of PTA.4
Antirejection Therapy
In 2006 in the United States, the most common maintenance immunosuppression for pancreas transplantation was tacrolimus and mycophenolate mofetil (MMF), followed by tacrolimus and sirolimus, although other agents such as cyclosporine and azathioprine are used in varying combinations at some centers. Although the majority of initial regimens still include corticosteroids, since 2000 there has been a movement towards “steroid-free” or early steroid withdrawal immunosuppression protocols in an attempt to reduce the adverse effects of corticosteroids. Induction immunosuppression is routinely employed, with the majority of recipients being given T-cell-depleting antibody treatment, particularly with polyclonal rabbit antithymocyte globulin (rATG—produced from rabbits immunized with human thymus thymoglobulin) or monoclonal alemtuzumab (Campath-1H). The nondepleting anti-CD25 antibodies basiliximab (Simulect) and daclizumab (Zenapax) are used less frequently. When compared to basiliximab induction, alemtuzumab has been associated with lower rates of acute rejection but higher risk of cytomegalovirus disease.21 Although rATG has been available for over 15 years, there has been a recent increase in interest after its use was recognized to expand antigen-specific CD4+CD25+Foxp3+ regulatory T cells, while the anti-CD25 antibodies or alemtuzumab do not.22
In pancreas transplant recipients, immunosuppression is often needed not only to control allograft rejection but also the autoimmunity of type 1 diabetes. In the setting of inadequate immunosuppression, autoimmunity may lead to rapid destruction of the insulin-producing cells over a matter of weeks, without rejection of the exocrine pancreas or other transplanted organs.23,24
Many adverse effects are seen with these immunosuppression agents, but only a few will be mentioned here. In general, higher doses of immunosuppressives are required for pancreas transplants than for kidney transplants alone, which is of concern, since there are increased risks of infection and malignancy with higher doses or duration of these medications.25 Post–renal transplant data indicate that the risk of skin cancer for a patient on immunosuppression varies by country and is cumulative; at 10 and 20 years, respectively, it goes from 10% to 40% in the Netherlands and 45% to 70% in Australia.26 As with any transplant requiring immunosuppression, posttransplant diabetes mellitus (PTDM) may develop after pancreas transplantation.27,28 In a series of 144 transplants followed for 39 months by the Mayo Clinic, 19% developed diabetes despite functioning grafts.29 Contributing variables included high pre-transplant BMI, high pre-transplant insulin requirements, and episodes of acute rejection.
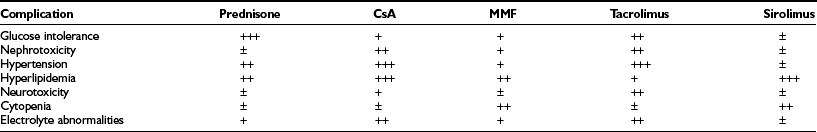
Glucocorticoids may cause insulin resistance and also have direct inhibitory effects upon β-cell function.30,31 The impairment of glucose-dependent insulin secretion by tacrolimus is well documented,32 and worrisome negative effects of sirolimus, including decreased insulin release and deleterious effects on islet viability and proliferation, have also been identified.33–35 There are data suggesting that MMF can directly inhibit islet neogenesis as well.36 A final concern is the nephrotoxicity of calcineurin inhibitors.37 In spite of the proven beneficial effects of a pancreas transplant on a transplanted kidney,38,39 presumably due to euglycemia, drug-induced nephrotoxicity can be severe. Side effects seen with standard maintenance immunosuppression include nephrotoxicity, hypertension, hyperlipidemia, microvascular disease, glucose intolerance, gastrointestinal problems, weight gain, electrolyte abnormalities, skin changes, alopecia, and hirsutism (Table 24-1).
Judging efficacy of immunosuppression regimens is more difficult with pancreas transplants than for other solid organ transplants such as liver or kidney, where functional tests are readily available. For SKP transplants, rejection of the pancreas may be followed using kidney function as a surrogate marker. Detection of problems with the pancreas prior to permanent end-organ damage is more difficult for PAK and PTA procedures. For patients with bladder drainage, amylase output has been thought to be useful by some, although current approaches are more likely to employ serum amylase and lipase levels and pancreas biopsies.40,41
Transplant Outcomes and Impact
The complex surgical procedures required for pancreas transplantation may be accompanied by significant mortality and morbidity, with patients potentially having long hospitalizations and readmissions for such problems as vascular thrombosis, intraabdominal infection, hemorrhage, graft pancreatitis, anastomotic leaks, and a variety of related problems.41,42 These risks, as well as those associated with lifelong immunosuppression, are weighed against the potential benefits of insulin independence and normoglycemia.
In an analysis of 937 transplants done at the University of Minnesota from 1994 to 2003, 313 grafts failed.43 Of these, 123 (39%) were technical failures, 119 (38%) were from rejection, 63 (20%) were from death with functioning grafts, and 4 (1%) were from primary nonfunction. Of the 123 technical failures, 52% were from thrombosis, 19% from infection, 20% from pancreatitis, 6.5% from leaks, and 2.4% from bladder drainage problems. There are many varied explanations for these outcomes, but an important variable appears to be the condition of the donated pancreas. For the follow-up period ending 2005, 1-year, 3-year, 5-year, and 10-year unadjusted pancreas graft survival by transplant type is shown in Fig. 24-2.7 Despite recent improvements in short-term graft function after solitary pancreas transplant, long-term graft function remains superior for SPK. Graft function at 5 years was 78% for SPK, 58% for PAK, and 51% for PTA.7 Graft function may be maintained for 10 or more years, greater than 20 years in some individuals, with fasting glucose and response to a meal or glucose challenge similar to nondiabetic individuals.44
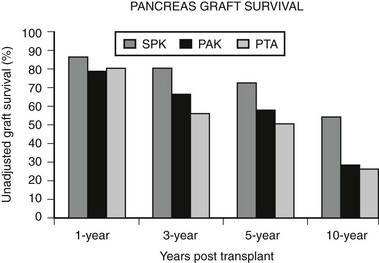
FIGURE 24-2 Pancreas graft survival among pancreas transplant recipients by type of transplant. (Adapted from 2007 OPTN/SRTR Annual Report, Table 1.13.7)
Survival
For patients awaiting SPK, the survival rates are much lower than for patients with normal kidney function waiting for a solitary pancreas,10 no doubt owing to the dangerous combination of end-stage renal failure and hyperglycemia. It is clear that transplantation of a kidney alone improves survival in patients with diabetes and renal failure,45 and survival is better for living-donor kidney recipients than those receiving cadaver kidneys.46 However, even with the benefits of living-donor kidneys, SPK transplant recipients have better long-term survival as determined by analysis of UNOS data,46 but the difference is modest.
Retinopathy
Many studies have examined the impact of pancreas transplantation, often in combination with kidney transplantation, on preexisting retinopathy. Many of these studies have included mainly patients with longstanding, advanced retinopathy and showed little impact of transplantation on retinopathy.47–49 However, in patients with less advanced retinopathy, in particular nonproliferative retinopathy, stability50,51 or regression52,53 and improvement in macular edema51,54 has been noted after transplantation. In a recent study of individuals with GFR > 50 mL/min who underwent PTA, nonproliferative retinopathy improved (in 50%) or remained stable (in 50%) after transplant. In individuals with preexisting proliferative retinopathy, there was deterioration in some but to a lesser degree than in matched controls—14% and 57%, respectively.52
Nephropathy
The benefits of pancreas transplantation to the kidney are more apparent. Biopsy-proven diabetic nephropathy of transplanted kidneys is slowed by subsequent pancreas transplantation or even prevented by simultaneous transplant.38,55 Importantly, a longitudinal study found improvement of native kidneys after pancreas transplantation. Clinical improvement in urinary albumin was noted at 5 and 10 years posttransplant. On biopsy, no benefit was seen at 5 years, but biopsies at 10 years showed an impressive reversion of histology towards normal.39 This finding is complemented by recent findings of reduced albumin excretion after PTA.56
Neuropathy
The course of diabetes neuropathy benefits from pancreas transplantation, as demonstrated by modest improvements in nerve conduction velocity, parameters of autonomic neuropathy, and symptoms.57–60 Gastric emptying can also improve,61 but it is not clear how much can be attributed to glycemic control versus improved renal function.
Vascular Disease
The course of macrovascular disease after pancreas transplantation has not been extensively studied, but it is clear that risk factors are reduced.62–64 Moreover, reports suggest that carotid intima media thickness can diminish,64 and the progression of coronary atherosclerosis can be slowed and even regress.65 These latter findings still need to be confirmed with larger studies.
Quality of Life
The most obvious benefit of pancreas transplants is that patients feel their quality of life is improved, particularly with freedom from insulin injections, hypoglycemic episodes, and food restrictions. It must be remembered, however, that quality of life is a difficult parameter to evaluate.66,67 For example, it has been difficult to show that patients’ lives are improved using such standard criteria as whether they are more active or have better performance at work, and it has been difficult to show striking improvement using a variety of study methods.68 Nonetheless, patients seem happy to be free of their diabetes, which is of undeniable importance.
Costs and Organ Availability
The costs of pancreas transplantation have been extensively analyzed by Stratta.69 For a typical SKP transplant in the United States, the cost is between $80,000 and $120,000, which includes hospital costs, professional fees, and organ acquisition charges. Complications requiring additional surgery and hospitalization can lead to much higher costs. In addition, the annual cost of related medications is about $15,000, and posttransplant monitoring is an additional $10,000 per year. It is important to consider this in the context of the pancreas transplant being added to the cost of a kidney transplant, which has a more pronounced impact upon health. The cost of dialysis is about $50,000 per year, while the cost of a kidney transplant from a cadaver donor is about $40,000 and from a living donor about $90,000.
Since SPK is the more commonly performed transplant type, pancreas transplants are closely linked to kidney availability. Unfortunately, there is a serious shortage of kidneys, which is particularly important for individuals with diabetes and end-stage renal disease, because once on dialysis, almost half are dead within 4 years. To further extend the donor pool by increasing living-donor renal transplants, there has been an effort to perform living-donor renal transplants at the same time as a cadaver pancreas becomes available (e.g., simultaneous cadaver-donor pancreas, living-donor kidney) or before a pancreas as a conventional PAK. Beyond encouraging living donation for kidneys, some centers have started to use extended donor criteria, including donation after cardiac death and wider acceptable age range for donation. In 2006, the number of pancreata recovered for transplantation compared to 1997 increased by 53%.7 Short-term outcomes with these extended donor criteria organs in SPK transplants have been comparable to those seen with conventional donors.70 Unfortunately, waiting-list time to transplantation has risen as well; for example, for SPK, median time to transplant went from 380 days in 1997 to 451 days in 2005.7 To further ease the shortage of organs, the U.S. Health Resources and Services Administration (HRSA) has recently launched a program to increase the numbers of available kidneys and pancreases.71
Islet Transplantation
The hypothesis that replacement of the endocrine pancreas by transplanting fragments of pancreas is not novel, with the first attempts to do so predating the discovery of insulin.72 The history of modern islet transplantation begins some 40 years ago when Paul Lacey proposed transplantation of the isolated islets of Langerhans.73 For a review of this fascinating history see Ref. 74. Progress has been slow and incremental. During the 1980s, reports suggested the feasibility of autologous islet transplantation in patients who underwent pancreatectomy for relief of chronic pain from pancreatitis.75 Despite improvements in islet isolation and purification, allotransplantation in recipients with preexisting type 1 diabetes proved significantly more difficult. Of 267 islet allografts performed between 1990 and 1998 in patients with type 1 diabetes, more than half completely failed within the first 2 months, with only 35% showing evidence of continuing function (C-peptide over 0.5 ng/mL), and 8% insulin independence beyond 1 year.76 More recently, insulin-independence rates of up to approximately 60% at 1 year posttransplant have been achieved, but only a minority (about 10%) maintain insulin independence at 5 years posttransplant.77
Islet Isolation and Transplantation
Islets are either cultured for a short period of time (usually less than 48 hours) or immediately prepared for infusion. In allotransplantation, recipient-donor matching is made according to ABO compatibility, but strict human leukocyte antigen (HLA) matching is not currently done. Most commonly, the islets are infused into the hepatic portal vein via a percutaneous transhepatic ultrasound and angiography guided approach, but some centers perform small laparotomies with general anesthesia. The transplanted islets become wedged in the terminal branches of the portal vein and engraft (Fig. 24-3).
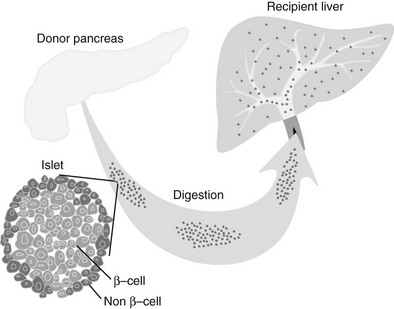
FIGURE 24-3 Islet transplantation in humans is usually done starting with a cadaver pancreas that is digested with a collagenase/protease mixture. The isolated islets are then introduced into the portal vein either by transhepatic angiography or via laparoscopy. The islets then are carried downstream and wedge in the hepatic sinusoids, whereupon they are vascularized by vessels from the recipient.
Recent Progress
In 2000, Shapiro and colleagues78 reported 100% insulin independence in seven individuals with type 1 diabetes after islet allotransplantation from multiple donors under what has become known as the Edmonton protocol. This result provided impetus to the islet transplant field worldwide. Existing centers accelerated their efforts, and other institutions embarked on establishing new islet programs. In 2001, the National Center for Research Resources of the National Institutes of Health established 10 islet resource centers in the United States with the mission of providing human islets for both clinical and research use. In addition, the Immune Tolerance Network of the National Institutes of Health, with help from the Juvenile Diabetes Research Foundation, began a trial at 9 centers to perform transplants in 40 subjects to try to reproduce the Edmonton results. As with the Edmonton trial, this multicenter trial recruited patients without kidney transplants who had life-threatening hypoglycemia episodes, severe glycemic instability, or advancing complications. The Immune Tolerance Network has confirmed that the Edmonton results can be reproduced in other centers, but better success rates occurred in more experienced centers than in the others.79 As reported by the Collaborative Islet Transplant Registry (CITR) in their 2008 Fifth Annual Report,80 between 1999 and 2007, 31 centers in North America performed 649 islet infusions in 325 recipients, while more than 200 patients received transplants in other parts of the world.
With increasing numbers of transplants, a number of advances have been reported. The Miami and Baylor groups reported successful transplants between cities, with pancreases flown from Houston to Miami and isolated islets flown back to Houston for implantation.81 This study and work from other groups shows that islets do not need to be transplanted immediately after isolation, as was done in the Edmonton trial, but can be placed in culture and transplanted 1 to 2 days later. Success in obtaining insulin independence from single donor transplants has also become more common.82,83 Building upon the Edmonton islets-alone approach, transplantation of islets after a kidney transplant has been explored and is now favored by a number of centers, as these patients will already require immunosuppression.84,85
In 2005, the Edmonton group reported 5-year follow-up data. Of the 44 Edmonton subjects achieving insulin independence, over half were back on insulin within 2 years, and at 5 years, only 10% were insulin independent.77 This general lack of durability has been confirmed in the 2008 CITR report. Of the 279 islet-alone recipients, as analyzed from the last infusion, 54% were insulin independent 6 months later, but only 22% were at year 3. The number on exogenous insulin with persistent graft function was about 25%, and the group with no detectable C-peptide was 34% at year 3 (the remaining 19% represents missing data). Over 60% of the recipients had hemoglobin A1c values below 7.0% at 6 months, and these values did not fall much over the 3 years, in part no doubt due to residual C-peptide secretion and treatment with exogenous insulin (Fig. 24-4).
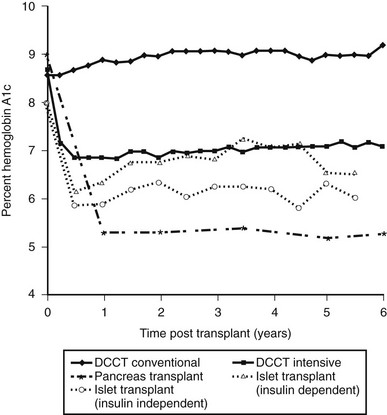
FIGURE 24-4 Comparison of hemoglobin A1c levels after successful pancreas or islet transplantation to levels obtained during the Diabetes Control and Complications Trial (DCCT). (Data was obtained from the DCCT Research Group,1 Robertson et al,219 and Ryan et al.87)
The 2008 CITR report found a variety of variables that influenced insulin independence. Low recipient insulin requirements, low BMI, and better control led to better outcomes, as did shorter cold ischemia of the donated pancreas and the number of IEQs infused per body weight. There was also benefit for longer duration of diabetes and older age of the recipient. Islets from donors positive for CMV given to CMV-negative recipients led to poorer results. Donor O blood type also was correlated with success.80
Risks and Benefits
Several adverse side effects of islet transplants were highlighted by a report on six recipients from the National Institutes of Health islet transplantation program. This study employed the Edmonton approach of transplanting patients who were receiving immunosuppression for the first time.86 Several of the patients found the side effects of the immunosuppression to be so problematic that the drugs had to be discontinued, in one case due to sirolimus-induced pneumonitis.
Immunosuppression
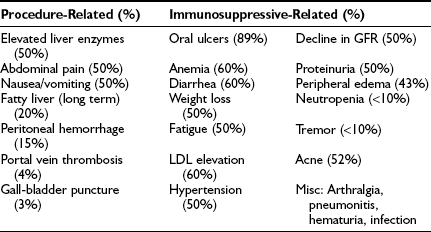
The side effects of the immunosuppression under the Edmonton protocol are similar to those seen after other transplants and have been described in a number of clinical reports, as summarized in Table 24-2.77,87,88 The major side effects of sirolimus have been mouth ulcers, diarrhea, malaise, hypertension, and hyperlipidemia. Especially worrisome is the nephrotoxicity that can be found with calcineurin inhibitors—in particular, the tacrolimus that is part of the Edmonton protocol. As with other transplants, there is concern about immunosuppression leading to opportunistic infections and malignancy.25 The complications found in these early studies predicted similar events in other trials. Indeed, this is now well documented in the 2008 CITR report, in which out of 325 transplanted subjects, there were 440 severe adverse events, most related to immunosuppression.
Islet Infusions
Portal pressure is monitored during the islet infusion, which usually is a packed-cell volume of 2 to 6 mL; in the Edmonton series, pressures on average rose from 12 to 17 mm Hg, with there being a correlation with increasing packed-cell volume.89 Furthermore, when compared with the first transplant, pressure increases are higher with the second and third transplants. While this has not been associated with any complications, it does indicate that islet infusions lead to changes in the portal vasculature. Immediately following the islet infusions, transaminase levels often increase to levels two to five times normal, peaking at about 7 days and then returning to normal within 2 months.90 More modest increases in alkaline phosphatase are also found.
Hepatic Steatosis
Magnetic resonance imaging (MRI) evidence of hepatic steatosis has been reported by several groups.91,92 Of the 30 patients studied with MRI in Edmonton, 20% had patchy steatosis, which in one case disappeared following graft failure. Histologic findings concur with these imaging studies.92 This steatosis is likely due to high concentrations of insulin released locally by clumps of islet tissue, promoting localized triglyceride storage in hepatocytes. The possibility that this could lead to deleterious scarring has not been excluded, but liver function is being carefully followed in the patients, and no problems have been reported.
Glycemic Control and Hypoglycemia
The major benefit of islet transplantation has been improvement in glycemia. Protection from severe hypoglycemic events is striking; a prevalence of about 85% pre-transplant has been reduced to less than 10% throughout the first year after the last infusion. It was expected that there would be little hypoglycemia when subjects are insulin independent, but even when insulin treatment has been required, recipients enjoyed relative freedom from severe insulin reactions. Moreover, hypoglycemia awareness as assessed by Clarke hypoglycemic score was restored in both insulin-independent and insulin-requiring patients. In these patients, glycemic thresholds for activation of counterregulatory hormones were restored to normal even though the glucagon and epinephrine responses were not fully normalized.93 The glucagon secretion in response to hypoglycemia from islet grafts in the liver was very modest,94 which contrasts with the essentially normal responses found in recipients of whole pancreases.95
Unfortunately, even when recipients become insulin independent, they typically are not truly normoglycemic but instead can be classified as having impaired glucose tolerance, and some even meet the criteria for diabetes. This is in contrast with pancreas transplant recipients, who usually have normal glucose tolerance. As glucose levels deteriorate, some have reasoned that insulin sensitizers should be beneficial, yet anecdotal reports suggest that the use of metformin and thiazolidinediones have been of limited value. There has been some anxiety about using sulfonylureas because of their putative potential to cause β-cell apoptosis, but this risk in clinical situations is far from proven. There has been limited experience with exendin-4 treatment, with mixed results.96,97 Even after there is a return to insulin use, for patients accustomed to the marked glycemic fluctuations of brittle diabetes, this improved control with a far less demanding regimen is a welcome benefit, which often justifies continuation of immunosuppression until graft function is lost and unstable diabetes returns.
Relatively small studies from Milan, Miami, Edmonton, and a few other groups indicate that successful islet transplantation may have a protective effect on long-term diabetic complications,98,99 but larger well-controlled studies are still lacking. Several surrogate markers for vascular disease, including intima media thickness and endothelial function, have been shown to improve,84,100 and there are indications of some beneficial effects on other diabetic complications. Considering quality of life, most patients, even many of those still needing insulin, are very thankful to be free of hypoglycemia unawareness and severe insulin reactions.101
Costs and Organ Availability
As with other transplants, the costs of islet transplantation are considerable. The startup costs include developing a pathogen-free space, purchasing equipment, and assembling an isolation team of five or more individuals, which typically costs over 2 million U.S. dollars. Costs for the actual transplants vary depending on healthcare costs.102 A recent example of costs for a patient in the United States was $188,000, which covered two isolations ($26,000), two transplants ($30,000), hospitalization ($14,000), medication for 1 year ($61,000), and monitoring for 1 year ($37,000), along with other costs (R.A. Dickey, oral communication, 2003).
There has been much discussion about the availability and allocation of pancreases that can provide islets for clinical trials and for research. In 2005, OPTN developed a pancreas allocation program to direct pancreases from donors older than 50 and/or a BMI of over 30 to islet transplantation.103 Pancreases from obese donors often provide high yields of excellent islets. While there has been the impression that islet yields from older donors are smaller and do less well, the pooled data in the 2008 CITR report failed to substantiate this concern. There continues to be a need to increase the number of high-quality pancreases available for islet and whole pancreas programs.103 Although the problem is not acute in 2008, the projected new clinical trails over the next few years may encounter shortages. There has been some preliminary success with the use of islets from pancreases of non-heart-beating donors,104 and there will be continued exploration of ways to expand the donor pool. The cost of pancreases for islet transplantation have remained problematic, since it is not uncommon for two or more isolations to be performed before a suitable islet preparation is obtained. This financial burden under current CMS accounting practices for the acquisition costs of organ procurement has been constraining, as laid out in a recent White Paper.105
Frontiers in Transplantation
Immune Modulation and Tolerance
One of the dreams of transplantation is to induce tolerance, which means that treatment given only at the time of transplantation will somehow trick the recipient’s immune system into accepting transplanted foreign tissue as its own. In contrast to solid-organ transplants, the procedural risks of islet transplantation are considered modest, and graft failure is not acutely life threatening. The ability of islets to survive in culture also makes it possible to more readily manipulate the graft prior to transplant. As such, many investigators consider islet transplantation a good system in which to study tolerogenic strategies. Many different approaches currently being studied could lead to full or operational tolerance (Table 24-3).
Table 24-3
Approaches to Tolerance Induction
Recipient Treatment Strategies
Chimerism (thymic irradiation)
Graft Treatment Strategies
Central tolerance refers to the process by which autoreactive T-cell clones are deleted as they migrate through the thymus. It has long been hoped that a similar process could be used to eliminate alloreactive T-cell clones. Several donor-specific tolerance strategies have been developed that mimic this central tolerance by leading to near-total and permanent elimination of donor-specific T-cell clones in the recipient. One such strategy is to directly introduce antigen into the thymus to aid in thymic selection. Posselt and colleagues demonstrated that injection of pancreatic islets directly into the thymus after treatment with anti-lymphocyte serum led to donor-specific unresponsiveness and long-term survival of islet grafts in a rodent model.106 To date, there has only been one report describing an attempt to apply this technique in a clinical setting with computed tomography (CT)-guided fine-needle intrathymic inoculation and simultaneous portal vein islet infusion and immunosuppression with an azathioprine, cyclosporine–based regimen. Although the patient never became insulin independent, there was evidence of continued graft function at 14 months.107 This approach has interesting immunologic implications, but the need for intrathymic injection, the temporal limitations of transplantation, and age-related atrophy of the thymus has made its clinical application difficult at best.
Another tactic is to use strategies that rely on the creation of mixed donor and recipient multilineage hematopoietic chimeras. This technique takes advantage of the ability of hematopoietic cells to home to areas of T-cell selection such as the thymus and to become dendritic cells capable of mediating selection. Thus, in chimeric hosts, donor-specific T cells are destroyed in peripheral immune tissues, and new thymic emigrants are depleted of donor-reactive T cells in the thymus.108 Using this approach, a small number of patients with refractory hematologic malignancies and renal failure have demonstrated tolerance after myeloablation followed by bone marrow transplantation and kidney transplant from the same donor.109,110 In islet transplantation, the induction of tolerance through mixed chimerism may have the added benefit of preventing autoimmune recurrence. Experiments by Megan Sykes’s group using nonmyeloablative conditioning in nonobese diabetic mice have demonstrated reversal of autoimmunity and acceptance of islet grafts.111 An ongoing trial at the University of Miami is using a steroid-free immunosuppressive regimen combined with infusion of CD34-enriched donor bone marrow cells.
While some immunosuppressive drugs interfere with the survival, expansion, and/or function of Tregs, sirolimus treatment has been shown to expand Tregs in murine and human models. It is therefore often used as an immunosuppressive agent in combination with immune modulators. Most of these immune modulators have been developed in other clinical settings and then applied in islet transplant trials. For example, the serine protease α1-antitrypsin (AAT) reduces inflammation but is permissive of interleukin 2 (IL-2) activity. In a murine islet allotransplant model, treatment with AAT monotherapy for 14 days allowed graft function for up to 120 days without additional treatment and the development of antigen-specific T-regulatory cells.112 Since AAT is already available for the treatment of α1-antitrypsin deficiency and is considered relatively safe, it is expected to be rapidly translated to human clinical islet transplant trials. Investigation of regimens consisting of thymoglobulin and the anti-TNF agent etanercept or the anti-CD3 monoclonal antibody hOKT3γ1(Ala-Ala) as induction agents in clinical islet transplants at the University of Minnesota are also underway.
The group of Judy Thomas at the University of Alabama has developed a primate anti-CD3-specific diphtheria-based immunotoxin. This compound is believed to deplete naive and memory T cells in blood and lymphatic compartments. 15-Deoxyspergualin is given concurrently to block proinflammatory cytokine production and maturation of dendritic cells by inhibiting nuclear translocation of nuclear factor κB (NF-κB).113 Using this technique, they have reported operational tolerance in approximately 85% of animals with normal β-cell function approximately 2 years after transplantation.114
In the two-signal model of T-cell activation, the costimulatory signal is essential for T-cell activation and effector function. Costimulatory blockade prevents clonal expansion and can achieve peripheral tolerance in several animal models. A number of costimulatory modifying agents, including anti-CD154 and CTLA4-Ig, are being actively investigated. Studies by Kenyon and colleagues have shown anti-CD154 treatment to be effective in a primate islet transplant model.115 Kirk and colleagues found that although administering anti-CD154 or CTLA4-Ig could significantly prolong renal allograft survival, treatment with both agents was much more efficacious. In a primate model, they saw improved graft survival and evidence of durable tolerance with subsequent nonresponsiveness to skin allografts from the original donors.116 Unfortunately, while anti-CD154/CTLA4-Ig combination therapy may produce some degree of transplantation tolerance in many animal models, this regimen does not enable long-term graft survival in mice with overt autoimmune diabetes.117 Given these findings, it is unlikely that simple deletion or costimulatory blockade alone will clinically prevent islet rejection in individuals already with underlying autoimmune diabetes. Despite these and other encouraging results in nonhuman primates, the future of anti-CD154 therapy is uncertain. CD154 is also expressed on platelets, and early clinical trials for rheumatoid arthritis with a humanized anti-CD154 blocking antibody (Hu5C8) were halted in the setting of thromboembolic complications.
On the other hand, CTLA4-Ig is progressing towards clinical trials. LEA29Y (belatacept), a mutant CTLA4-Ig molecule with increased binding activity, has been evaluated by Adams and colleagues in an allogeneic islet primate model. Their data suggest that this agent has promise as a component in a minimal immunosuppression regimen but not as a sole therapy.118 A clinical trial using sirolimus- and belatacept-based immunosuppression is underway.
Zheng et al have developed a strategy that avoids nonspecific inhibition, blockade of T-cell receptor signaling, or costimulation altogether and instead is designed to selectively eliminate activated cytopathic donor-reactive T cells while sparing immunoregulatory networks.119 Their approach takes advantage of activation-induced cell death (AICD), a method by which the body rapidly eliminates effector cells after antigen-dependent clonal expansion. AICD and passive cell death are both routine downstream consequences of T-cell activation and help maintain peripheral tolerance. By using sirolimus, which blocks the proliferation but not the apoptotic effects of IL-2, early clonal expansion can be limited without affecting subsequent apoptotic clearance via AICD. When sirolimus is given in combination with agonist IL-2 immunoglobulin fusion protein and antagonist IL-15 related cytolytic immunoglobulin fusion protein, this cocktail induces durable transplantation tolerance in mice with prior autoimmune diabetes.119 While the IL-2 promotes apoptosis of proliferating effector T cells, it is also a survival factor for immunoregulatory CD4+CD25+T cells. The mutant IL-15 operates by blocking normal IL-15-triggered antiapoptotic signals. Through these mechanisms, this treatment biases the immune response to one of Tregulatory cells over pathogenic T cells. Preliminary islet transplant experiments in nonhuman primates seem promising, with durable tolerance and no evidence of opportunistic infections (M. Koulmanda, X.X. Zheng, and T.B. Strom, unpublished observations).120
Immunobarrier Technologies
Semipermeable membranes that create an immunobarrier can protect transplanted islet tissue from immune destruction.121 These membranes have openings large enough for glucose, oxygen, and nutrients to reach the encapsulated islets and for insulin to be released to enter the bloodstream, yet the holes are small enough so that white blood cells and larger molecules cannot penetrate the membrane to reach the islet cells. Recently it has been found that merely maintaining a distance between lymphocytes and islet cells may be enough to prevent autoimmune and allorejection destruction,122 whereas protection of xenotransplants seems to require more restrictive membranes to limit passage of smaller molecules.
Macroencapsulation employs devices like hollow fibers or parallel flat sheets sealed at the edges, in which many islets are contained within a single device.123,124 Large gel beads or even slabs made of either agarose or alginate can also be used for macroencapsulation. One of the major advantages of such an approach is that the devices could be implanted in a variety of locations and yet still be retrieved or reloaded. However, it has been difficult to achieve a practical packing density, which means that too much surface area would be required to support the encapsulated islet cells. Moreover, there are still questions about whether insulin will be released quickly enough to adequately control blood glucose levels, particularly from large gel beads.
Microencapsulation is an approach in which single or small numbers of islets are contained within a membrane. The most commonly used method employs alginate obtained from seaweed that can form a gel after exposure to calcium or barium (Fig. 24-5).125 Thus, islets can be captured in a small gel bead (usually less than 1 mm in diameter) which can be coated with a material such as poly-L-lysine that can provide permselectivity. Because poly-L-lysine can generate an inflammatory tissue reaction, an outer layer of alginate is usually added to make the capsules more biocompatible, although simple barium alginate microcapsules without a poly-L-lysine coating can successfully protect against autoimmunity, allorejection, and even xenoreactivity in mice.122,126 Agarose has also been employed for microcapsules,127 as has alginate mixed with other polymers such as cellulose sulfate.128 Another similar approach that is being explored is to use a polyethylene glycol with photopolymerization to form a coating.129

FIGURE 24-5 Alginate microcapsules containing porcine neonatal pancreatic cell clusters. The alginate gel creates a semipermeable membrane that protects islet cells or aggregates of cells from immune destruction. A common approach is the use of small beads of alginate covered by poly-L-lysine, but in some situations the poly-L-lysine is not used. These microcapsules are usually between 500 and 1000 µm in diameter. The membrane will prevent penetration by cells and limit the entrance of antibodies but must be permeable enough to allow passage of glucose, nutrients, and oxygen into the islets and insulin out to diffuse into small vessels. (Photograph courtesy Dr. Abdulkadir Omer.)
Much of the optimism about immunobarriers has been based on successes in rodents, and until recently it is has been difficult to show efficacy in large-animal models or humans. However, allotransplanted islets coated with conformal PEG have succeeded when placed subcutaneously into nonhuman primates (David Scharp, Novocell Inc., oral communication). Canine islets microencapsulated in alginate/cellulose-sulfate have reversed diabetes in dogs.128 Also encouraging is a trial of human islets in alginate microcapsules placed in the peritoneal cavity of type 1 diabetics, which showed C-peptide production for several months.130 Work to find better biomaterials and strategies continues.
Sources of Insulin-Producing Tissue
At present the only source of islets for transplantation into humans is cadaver pancreases, which are in very short supply. In the United States, it would be a major challenge to obtain 3000 usable cadaver pancreases per year, yet the incidence of type 1 diabetes is about 30,000 cases per year, and more than 10 times as many people develop type 2 diabetes. Although there have been some successes with single donor transplants, usually two or more pancreases are required to provide insulin independence. There has been discussion of using human fetal tissue, but in spite of much effort,131 transplants to date have shown no benefit, and attention has turned elsewhere. A number of potential alternative sources for insulin-producing cells are listed in Table 24-4.
Table 24-4
Potential Sources of Insulin-Producing Cells
Human Sources
Live donors—could be a source of precursor cells
Expansion of existing human β-cells in vitro or in vivo
Precursor cells—embryonic or adult stem/precursor cells
Transdifferentiation of liver, intestinal, pancreatic acinar or other cells
Xeno Sources
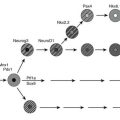
β-Cell Replication and Neogenesis
It is now appreciated that new β cells are generated throughout adult life, both from replication of preexisting β cells and through the formation of new β cells from precursor cells.132 β Cell hypertrophy in response to increased demand can also contribute to increased β-cell mass.133 In rodents, there is impressive capacity for expansion of β-cell mass through replication, as has been best shown in genetic models of insulin resistance.134 Although it has been contended that replication was the only path to new β cells in mice,135 it is now clear that new islet formation from precursor cells (neogenesis) occurs in adult life and is quantitatively important.136–138 Evidence is accumulating that pancreatic duct cells are the multipotent progenitor cells responsible for neogenesis.138,139 Nonetheless, in mice the dominant path to β-cell expansion is replication, but in humans the situation is unclear. β Cell turnover is very slow, and the relative contributions of replication and neogenesis are not known.140
Even though the replication rate of β cells in human pancreas is very low, the mechanisms of replication are being intensively studied in the hope that some pharmaceutical or genetic intervention could be clinically useful. It has also been proposed that β cells might be able to dedifferentiate through some kind of epithelial mesenchymal transition, be expanded in vitro, and then redifferentiated to form β cells that can then be transplanted. Results from these efforts have been inconsistent but provocative.141 The possibility that pancreatic duct cells can be exploited to generate more β cells continues to be of great interest. One possibility is that duct cells might be expanded in vitro and then stimulated to form β cells that could be used for transplantation.142,143 Another possibility is that duct cells might be stimulated to form islets in vivo, such that new β cells might be produced in either the native pancreas or in a graft site from duct cells contained within impure human islet preparations. Other candidate precursor cells have also been found in the pancreas. Two groups have found single cells from mouse pancreas that can form aggregates which can be differentiated to express β cell markers and even some insulin.144,145 In addition, very small embryonic-like (VSEL) stem cells have been found in pancreas, which also express islet markers.146 These pancreatic non-duct cells have developmental similarity to islets, but it remains to be seen if they can become fully differentiated to islet cells or are quantitatively sufficient.
There has been considerable interest in the possibility that circulating bone marrow stem cells, monocytes, or spleen cells could transdifferentiate to become β cells, but if this occurs at all it is a rare event.147 Yet bone marrow cells may turn out to be useful, as suggested by the provocative finding of multipotent adult precursor cells derived from long-term cultures of bone marrow stroma, which can generate ectodermal, endodermal, and mesodermal progeny.148 Other potentially interesting cells are amniotic fluid–derived stem cells, which express stem-cell markers and are impressively multipotent.149
Embryonic and iPS Cells
The expected potential of embryonic stem cells led to great optimism around the year 2000, but early excitement became more subdued as the difficulty of the challenge became apparent. However, a group from Novocell Inc. made impressive advances with human embryonic stem cells by using a complicated step-wise protocol that drove stem cells to definitive endoderm, then to pancreatic progenitor cells, and then to an islet cell phenotype.150 Although they were not able to obtain fully differentiated glucose-responsive β cells in vitro, when the pancreatic precursor cells were transplanted into immunocompromised mice, full differentiation to β cells was found, and diabetes in the mice could be cured. The excitement about this important accomplishment is tempered by the finding that teratomas were found in some of the transplanted mice. Finding ways to avoid teratoma formation so that transplants will be safe will be a major research priority. Another major advance with potential benefit for diabetes is a method for producing pluripotent stem cells from fully differentiated cells. The group of Yamanaka found that the introduction of four transcription factors into mouse fibroblasts could reprogram the cells to an undifferentiated state similar to ESC.151 These cells, which are called induced pluripotent stem (iPS) cells may be able to make pancreatic islet cells and will not face the same political hurdles as human embryonic stem cells. Compared with somatic cell nuclear transfer technology, this may be a much more convenient way for a transplant recipient to obtain genetically identical islet cells.
Genetic Engineering
Through advances in molecular and cell biology, it is now theoretically possible to manipulate the differentiation of cells with genetic engineering so they can be used for transplantation. One approach is to try to create a β-cell equivalent by using molecular techniques to stimulate or inhibit gene expression. This is very challenging because the genetic phenotype of β cells is so complex. On the other hand, as more is learned about the master switches that control the differentiation of cells, more promising results may emerge. Nonetheless, transplanted β cells will almost certainly need to have a near fully intact phenotype to provide the exquisite insulin secretory capabilities of normal β cells required to cope with meals, fasting, and exercise.152 A more attractive approach may be to transform β cells with the SV40 T antigen or some other oncogene, expand these cells in culture, and then turn off the oncogene genetically with resultant redifferentiation. Some success with this approach has been reported.153 Questions arise as to whether the non–β cells of the islet or some equivalent should be included in the transplanted cell aggregates. Non–β cells may not be required, as suggested by experiments in which relatively pure β-cell populations, prepared by flow cytometry, functioned very well when transplanted into diabetic rodents.154
Transdifferentiation
Transdifferentiation of acinar cells appears to be a particularly promising way to make new β cells. The injection of a combination of adenoviruses expressing three transcription factors—PDX-1, MafA, and Ngn-3—into pancreas has been found to generate what appear to be new, fully differentiated β cells of acinar cell origin in mice.155 This simple approach could have clinical applicability. Another possible avenue is to use hepatocytes, which are attractive because of their endodermal derivation. Several groups have made progress in creating insulin-producing cells by introducing genes expressing PDX-1, NeuroD, and betacellulin.156–159 There has also been a suggestion that hepatic oval cells can differentiate into insulin-producing cells.160
Xenotransplantation
Much of the focus in xenotransplantation has been on using porcine islets for clinical transplantation. Pigs have had particular appeal because pig insulin has been used in the past for treatment of diabetes, pigs have glucose levels similar to humans, and people seem to be comfortable about the prospect of using this source. Unfortunately, it continues to be difficult to generate high-quality islets from adult pigs.161 Much work has also been done to use either fetal tissue or neonatal xeno-islet tissue, which are attractive sources because of their growth potential.162–164 One of the problems is that the cells are immature, which means they can take weeks or months to normalize glucose levels in transplant recipients. Another potential problem is that porcine tissue contains porcine endogenous retroviruses (PERV) that can be transferred to human cells in tissue culture.165 Concerns about the infectious risk potentially posed by PERVs led to a reduction in research support in this area. Subsequently, guidelines have been developed aimed at reducing the infectious risk of xenotransplantation, and lines of pigs without infectious PERV have been identified. The failure to see direct transmission in several animal models has also been encouraging.
Rejection of xenografts is a complex and aggressive process. There is an early attack called hyperacute rejection mediated by antibodies and complement that can lead to destruction of transplanted organ within minutes and then a later cellular response. These preformed IgM antibodies recognize a glycoprotein called the Gal-a-Gal epitope (Gal epitope) that is strongly expressed on the surface of endothelial cells. This is a particular problem for organ transplants because the attack on endothelial cells produces ischemia that leads to rapid death of the transplanted tissue. Cell transplants may not be as vulnerable to hyperacute rejection, because adult pig islet cells have little Gal epitope.166 There has been great progress not only in generating transgenic pigs that do not express the Gal epitope but also in molecular interventions that could inhibit complement activation, thrombosis, and inflammation; these may be useful as a source for transplanted tissue.167 Others have turned to encapsulation to mask the xenoproteins.
A New Zealand study employing intraperitoneal transplants of alginate-encapsulated porcine islets was aborted in 1996 because of concern over PERVs. Of interest, capsules containing viable islet cells were retrieved from a patient in that study 9.5 years after the transplant.168 Further clinical study of intraperitoneal alginate-encapsulated porcine islet cells has been recently reinitiated. Co-transplantation of porcine neonatal islets and Sertoli cells have been attempted by a group in Mexico, but even with many transplants being performed, ethical concerns have been raised, and there has been no convincing evidence of any glycemic improvement.169,170
Despite some failures and obvious challenges, there is some optimism for the hope that transplanted porcine islet cells may become a viable treatment for diabetes; several groups have recently achieved impressive results. The Larsen group transplanted porcine neonatal pancreatic cell clusters into diabetic rhesus monkeys and could normalize glucose levels for a mean of over 140 days.171 Short-term immunosuppression employed antibodies to the IL-2 receptor and CD154, and maintenance immunosuppression used sirolimus and anti-CTLA4Ig. The Hering group had similar success with transplants of adult porcine islets into diabetic cynomolgus monkeys.172 Their induction immunosuppression was antibodies against the IL-2 receptor, and maintenance was with anti-CD154, FTY720 (or tacrolimus), everolimus, and leflunomide.
Enhancing Islet Engraftment, Survival, and Function
β-Cell Mass and Islet Cell Function
Questions about the β-cell mass and function of islet grafts in the liver are of fundamental importance to the field. Recipients receiving islets from one to three pancreases usually receive a total of over 10,000 islet equivalents per kg, which means they are given β-cell mass roughly equivalent to that present in a normal pancreas. Yet the usual outcome of impaired glucose tolerance indicates that the grafts are not nearly as efficacious as a normal pancreas. A study that is in general agreement with others found that in insulin-independent subjects, first-phase C-peptide responses to intravenous glucose were absent, acute responses to arginine were 32% of normal, and responses to a mixed-meal tolerance test were 36% of normal.173 It is of interest that subjects with type 2 diabetes who presumably had a β-cell mass about 50% of normal have insulin responses to arginine that are near normal.174 This might suggest that the β-cell mass of the transplants is much less than 50% of normal, but it is entirely possible that secretion of insulin per unit of β-cell mass is reduced because of the islets being in a foreign, unfavorable environment. At present, estimates of β-cell mass in a liver site are merely guesses. While one might conclude that islet function in the liver is impaired, it must be noted that in nonhuman primates (NHP) with islet transplants, insulin responses to stimulation are closer to normal.115 A possible clue to the difference may be that NHP islet preparations are usually much purer and healthier than those obtained from human pancreases.
Vascularization of Islet Grafts
During transplantation, there is some obligatory loss of cells due to local hypoxia occurring in nonvascularized large islets and clumps of islet tissue lodged in the terminal branches of the portal vein, as has been demonstrated in islets transplanted under the kidney capsule.175 Isolated islets removed from their normal blood supply face a fundamental problem with oxygen delivery after transplantation, because they are not fully vascularized for 10 to 14 days, as determined by studies in experimental animals.176 Although a few donor endothelial cells may take part in the process, most of the new vessels are supplied by the recipient.177 Eventually, the islet arterial supply comes mainly from the hepatic artery.178 There are many studies showing that hyperglycemia during this critical period has an adverse influence on the outcome of islet transplants,179 with one mechanism appearing to be slowing of angiogenesis.180 Hyperglycemia may also increase oxygen consumption in β cells, which would further reduce oxygen tension in their local environment. These findings provide a rationale for treating patients aggressively with insulin during the first 10 days posttransplant.
The vasculature of transplanted islets is known to be very different than islets in the pancreas, which is likely to have an impact upon function. In their normal location in the pancreas, islets have a highly specialized vasculature with a fenestrated endothelium; arterioles break into capillaries within the β cells’ core and then exit through the islet mantle that contains glucagon-secreting α cells.181 Because of this blood flow pattern, it is thought that insulin secreted from upstream has an important suppressive influence on downstream α cells, but that β cells see little if any downstream glucagon.182 This relationship between the different types of islet cells is not reestablished in islet grafts. However, transplants of dispersed reaggregated rat islet cells greatly enriched in β cells by flow cytometry were both effective and durable when transplanted without non–β cells.154
Importantly, the vasculature of islet grafts is considerably less dense than in islets in the pancreas.183 This probably explains why transplanted islets have much lower oxygen tension (about 5 mm Hg) than islets in the pancreas (about 40 mm Hg).184 Because of the likelihood that this poor vascularization has an adverse effect upon insulin secretion, efforts are underway to stimulate angiogenesis in transplanted islets with a variety of techniques.185 This is very challenging; normal islet vasculature is formed during development, so recreating truly normal vasculature in a graft may not be possible. Autonomic innervation also contributes to normal islet function. The efficacy of neural control in transplanted islets is unknown, but some reinnervation takes place over time.186
β-Cell Turnover in Islet Grafts
The regenerative potential of transplanted islets remains poorly defined. The lack of durability of islet grafts indicates that whatever capacity might be present is overwhelmed, presumably by immunosuppression and continuing immune killing. The possibility that pancreatic duct cells are the precursor cells responsible for islet neogenesis143 may be important, because infused islet preparations are far less pure than those from other species. The composition of 24 transplantable islet preparations from the Joslin Diabetes Center was 32% β cells, 12% non-β islet cells, 23% duct cells, and 27% acinar cells (S. Bonner-Weir, unpublished). The dynamics of β-cell turnover in human pancreas is a subject of intense interest. Current thinking is that β-cell replication is extremely low,140 and it has not been possible to measure neogenesis or the rate of apoptosis. Nonetheless, some slow rate of turnover must exist. It is possible, however, that transplanted islets behave differently, as suggested by experiments in which human islets transplanted into mice were found to have substantial rates of replication.187
Inhibiting Inflammation
A nonspecific inflammatory response accompanied by localized clotting188 may be seen immediately after islet transplant, enhancing the immune processes of rejection and autoimmunity and activating the innate immune system.189 This has been termed the instant blood-mediated inflammatory reaction (IBMIR), in which tissue factor and macrophage chemoattractant protein have been implicated.188,190 It has been found that expression of these factors is increased after the isolation of human islets but falls as islets are cultured and can be further suppressed by nicotinamide.190 Coating islets with heparin also seems to hold some promise.191
Role of HLA-Matching
The benefits of immunologic matching of donor and recipient have not been carefully studied, with most transplants having been matched only to blood type and not for histocompatibility antigens. In a recent study of islet transplants in Belgium, it was found that failure to achieve insulin independence with was associated with T-cell autoreactivity but not with the presence of autoantibodies or cellular alloreactivity.192 Supporting this, in a mouse model, HLA matching has been shown to accelerate autoimmune destruction of islet allografts.193 Similarly, recurrence of autoimmune disease and selective loss of islets has been reported in non-immunosuppressed or minimally immunosuppressed recipients of HLA-matched pancreas transplants, most notably with recipients of pancreas from an identical twin.194 Others have suggested that a rise in autoantibodies may be associated with decline of graft function.195 On the other hand, concerns are now being raised about HLA sensitization after islet transplantation. In the Edmonton series,196 11 of 69 (16%) were broadly sensitized with panel reactive antibody, which is problematic for future transplants. In individuals who discontinued immunosuppression, 10 of 14 (71%) were broadly sensitized.
Making Stronger β Cells for Transplantation
An ambitious goal is to create stronger β cells that will withstand the ischemia of early transplantation, early inflammatory assaults, toxic effects or immunosuppressive agents, and the challenge of immune attack. Individual strengthening measures might provide protection against multiple affronts. Genes can be transduced into β cells with a variety of techniques, as have been reviewed,197 and there are a variety of other ways in which islets can be treated either before or after transplantation, as is summarized in Table 24-5.
Table 24-5
Strategies to Improve β-Cell Function and Survival
1. Removal of surface antigens: Class I MHC antigens.
2. Engineer islets to secrete protective peptides: CTLA4-Ig, TGF-β, IL-1 receptor antagonist protein (IRAP), IL-10.
3. Genetic addition of antiapoptotic genes: A20, bcl-2, IκB repressor, MyD88, FLIP.
4. Treat with inhibitors of apoptosis: Z-DEVD-FMK (an inhibitor of caspace-3).
5. Enhance antioxidant protection: Overexpress manganese superoxide dismutase, catalase or heme oxygenase-1. Carbon monoxide treatment. Inhibit c-Jun NH2-terminal kinase.
6. Strengthening actions of growth factors: Hepatocyte growth factor (HGF) glucagon-like peptide 1 (GLP-1), enhance PI-3 kinase and Akt signaling.
The concept behind this approach is to remove cell-surface antigens that elicit immune recognition. An example of this is class I major histocompatibility complex (MHC) antigens, whose expression can be lowered by viral infection. Studies have shown that this can make β-cells resistant to both allorejection and autoimmunity.198
A variety of peptides can modulate the destructive actions of invading immune cells. For example, costimulation blockade with locally produced CTLA4-Ig can delay allograft rejection.199 Viral transduction of islets with transforming growth factor (TGF) β cells can also lead to improved islet graft survival.200 To protect β cells against the presumed toxic effects of IL-1, islets engineered to secrete the IL-1 receptor antagonist protein have also been found to have beneficial effects that may be helpful during transplantation.201 Another approach has employed the use of IL-10 or IL-4, which can push immune responses to a T-helper 2 phenotype. Islets transduced to express these cytokines are more resistant to rejection.202,203 There has also been interest in transplanting islets with Sertoli cells, which provide immunoprotection probably through production of TGF-β and as yet unknown mechanisms,204 but clinical applicability is not promising because of the large number of Sertoli cells that would likely need to be transplanted.
As information increases about cell death pathways in β cells,205 an increasing number of opportunities for islet protection have emerged. Transduction of the antiapoptotic gene bcl-2 can provide protection of islets against cytokines and improve performance with transplantation.206 Another potential target for protecting β cells might be NF-κB, but this has turned out to be very complex because NF-κB induced both proapoptotic and antiapoptotic mediators. Protection of β cells during transplantation has been found with viral overexpression of A20, which blocks NF-κB activation but has other effects.207 Other death pathways have been exploited, including the use of the dominant-negative mutant of the IL-1 receptor interacting protein MyD88208 and FLICE inhibitory protein (FLIP), which can inhibit caspase-3.209 Another way to reduce apoptosis is through inhibition of caspases. The potential has been shown with overexpression of X-linked inhibitor of apoptosis (XIAP)210 and the caspase inhibitor EP1013 (zVD-FMK),211 which is also effective and is a drug that could be used in a clinical situation. Yet another target is the proapoptotic pathway c-Jun NH2-terminal kinase (JNK), which is accessible to a variety of inhibitors that can improve islet transplant outcomes.212
β-Cell defenses against oxidant injury appear to be relatively weak,213 so bolstering these mechanisms is an attractive option. Enzymes that have received a lot of attention include manganese superoxide dismutase, catalase, and glutathione peroxidase; results of work in this area have been reviewed197 and are very contradictory. Another mechanism that provides protection against oxidative injury is the heme oxygenase system, which can be induced through a variety of maneuvers to provide improvements in graft survival.214
Growth factor signaling is being shown to be very important for β-cell function and survival. Glucagon-like peptide 1 (GLP-1) has antiapoptotic effects on β cells that appear to be dependent on cyclic adenosine monophosphate and IRS-2, with the later steps of this signaling probably being exerted through Akt.215 GLP-1 agonists have been shown to have beneficial effects on experimental islet transplantation216 and are now being evaluated clinically. Hepatocyte growth factor overexpression through both transgenesis and adenoviral transduction has been found to improve β-cell resistance to apoptosis and improve survival of transplanted islets.217 When the growth factor IGF-1 is overexpressed in transgenic mice with autoimmune diabetes,218 there is evidence of regeneration and protection from insulitis, making this another approach that could help islet transplantation.
References
1. The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986.
2. Group UKPDS. Intensive blood-glucose control with sulfonylureas of insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352:837–853.
3. Kelly, WD, Lillehei, RC, Merkel, FK, et al. Allotransplantation of the pancreas and duodenum along with the kidney in diabetic nephropathy. Surgery. 1967;61:827–837.
4. Gruessner, AC, Sutherland, DE. Pancreas transplant outcomes for United States (US) and non-US cases as reported to the United Network for Organ Sharing (UNOS) and the International Pancreas Transplant Registry (IPTR) as of June 2004. Clin Transplant. 2005;19:433–455.
5. Robertson, RP, Davis, C, Larsen, J, et al. Pancreas and islet transplantation in type 1 diabetes. Diabetes Care. 2006;29:935.
6. Sutherland, DE, Gruessner, RW, Dunn, DL, et al. Lessons learned from more than 1,000 pancreas transplants at a single institution. Ann Surg. 2001;233:463–501.
7. Health Resources and Services Administration HSB, Division of Transplantation 2007 Annual Report of the U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients: Transplant Data 1997–2006. (Rockville, MD, 2007) at < http://www.ustransplant.org/annual_reports/current/default.htm.
8. Gruessner, RW, Sutherland, DE, Gruessner, AC. Survival after pancreas transplantation. JAMA. 2005;293:675.
9. Gruessner, RW, Sutherland, DE, Gruessner, AC. Mortality assessment for pancreas transplants. Am J Transplant. 2004;4:2018–2026.
10. Venstrom, JM, McBride, MA, Rother, KI, et al. Survival after pancreas transplantation in patients with diabetes and preserved kidney function. JAMA. 2003;290:2817–2823.
11. Sutherland, DER, Groth, CG, Hakim, N, et al. History of pancreas transplantation. Oxford: Oxford University Press, 2002;1–13.
12. Sollinger, HW, Cook, K, Kamps, D, et al. Clinical and experimental experience with pancreaticocystostomy for exocrine pancreatic drainage in pancreas transplantation. Transplant Proc. 1984;16:749–751.
13. Larsen, JL. Pancreas transplantation: indications and consequences. Endocr Rev. 2004;25:919–946.
14. Stratta, RJ, Shokouh-Amiri, MH, Egidi, MF, et al. Long-term experience with simultaneous kidney-pancreas transplantation with portal-enteric drainage and tacrolimus/mycophenolate mofetil-based immunosuppression. Clin Transplant. 2003;17(Suppl 9):69–77.
15. Cattral, MS, Bigam, DL, Heming, AW, et al. Portal venous and enteric exocrine drainage versus systemic venous and bladder exocrine drainage of pancreas grafts: clinical outcome of 40 consecutive transplant recipients. Ann Surg. 2000;232:688–695.
16. Philosophe, B, Farney, AC, Schweitzer, EJ, et al. Superiority of portal venous drainage over systemic venous drainage in pancreas transplantation: a retrospective study. Ann Surg. 2001;234:689–696.
17. Troppmann, C, Gjertson, DW, Cecka, JM, et al. Impact of portal venous pancreas graft drainage on kidney graft outcome in simultaneous pancreas-kidney recipients reported to UNOS. Am J Transplant. 2004;4:544–553.
18. Katz, HH, Nguyen, TT, Velosa, JA, et al. Effects of systemic delivery of insulin on plasma lipids and lipoprotein concentrations in pancreas transplant recipients. Mayo Clin Proc. 1994;69:231–236.
19. Carpentier, A, Patterson, BW, Uffelman, KD, et al. The effect of systemic versus portal insulin delivery in pancreas transplantation on insulin action and VLDL metabolism. Diabetes. 2001;50:1402–1413.
20. Shen, J, Gaglia, J. Hypoglycemia following pancreas transplantation. Curr Diab Rep. 2008;8:317–323.
21. Magliocca, JF, Odorico, JS, Pirsch, JD, et al. A comparison of alemtuzumab with basiliximab induction in simultaneous pancreas-kidney transplantation. Am J Transplant. 2008;8:1702–1710.
22. Lopez, M, Clarkson, MR, Albin, M, et al. A novel mechanism of action for anti-thymocyte globulin: induction of CD4+CD25+Foxp3+ regulatory T cells. J Am Soc Nephrol. 2006;17:2844–2853.
23. Sutherland, DE, Goetz, FC, Sibley, RK. Recurrence of disease in pancreas transplants. Diabetes. 1989;38(Suppl 1):85–87.
24. Simone, EA, Wegmann, DR, Eisenbarth, GS. Immunologic “vaccination” for the prevention of autoimmune diabetes (type 1A). Diabetes Care. 1999;22(Suppl. 2):B7–B15.
25. London, NJ, Farmery, SM, Will, EJ, et al. Risk of neoplasia in renal transplant patients. Lancet. 1995;346:403–406.
26. Dreno, B. Skin cancers after transplantation. Nephrol Dial Transplant. 2003;18:1052–1058.
27. Jindal, RM, Revanur, VK, Jardine, AG, et al. Immunosuppression and diabetogenicity. Oxford: Oxford University Press, 2002;229–246.
28. First, MR, Gerber, DA, Hariharan, S, et al. Posttransplant diabetes mellitus in kidney allograft recipients: incidence, risk factors, and management. Transplantation. 2002;73:379–386.
29. Dean, PG, Kudva, YC, Larson, TS, et al. Posttransplant diabetes mellitus after pancreas transplantation. Am J Transplant. 2008;8:175–182.
30. Delaunay, F, Khan, A, Cintra, A, et al. Pancreatic beta cells are important targets for the diabetogenic effects of glucocorticoids. J Clin Invest. 1997;100:2094–2098.
31. Gremlich, S, Roduit, R, Thorens, B. Dexamethasone induces posttranslational degradation of GLUT2 and inhibition of insulin secretion in isolated pancreatic β cells. J Biol Chem. 1997;272:3216–3222.
32. Uchizono, Y, Iwase, M, Nakamura, U, et al. Tacrolimus impairment of insulin secretion in isolated rat islets occurs at multiple distal sites in stimulus-secretion coupling. Endocrinology. 2004;145:2264–2272.
33. Bell, E, Cao, X, Moibi, JA, et al. Rapamycin has a deleterious effect on MIN-6 cells and rat and human islets. Diabetes. 2003;52:2731–2739.
34. McDaniel, ML, Marshall, CA, Pappan, KL, et al. Metabolic and autocrine regulation of the mammalian target of rapamycin by pancreatic beta cells. Diabetes. 2002;51:2877–2885.
35. Zahr, E, Molano, RD, Pileggi, A, et al. Rapamycin impairs beta-cell proliferation in vivo. Transplant Proc. 2008;40:436–437.
36. Gao, R, Ustinov, J, Korsgren, O, et al. Effects of immunosuppressive drugs on in vitro neogenesis of human islets: mycophenolate mofetil inhibits the proliferation of ductal cells. Am J Transplant. 2007;7:1021–1026.
37. Nankivell, BJ, Borrows, RJ, Fung, CL, et al. The natural history of chronic allograft nephropathy. N Engl J Med. 2003;349:2326–2333.
38. Wilczek, HE, Jaremko, G, Tyden, G, et al. Evolution of diabetic nephropathy in kidney grafts. Evidence that a simultaneously transplanted pancreas exerts a protective effect. Transplantation. 1995;59:51–57.
39. Fioretto, P, Steffes, MW, Sutherland, DER, et al. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med. 1998;339:69–118.
40. Gaber, LW. Pancreas allograft biopsies in the management of pancreas transplant recipients: histopathologic review and clinical correlations. Arch Pathol Lab Med. 2007;131:1192–1199.
41. Lerner, SM. Kidney and pancreas transplantation in type 1 diabetes mellitus. Mt Sinai J Med. 2008;75:372–384.
42. Lipshutz, GS, Wilkinson, AH. Pancreas-kidney and pancreas transplantation for the treatment of diabetes mellitus. Endocrinol Metab Clin North Am. 2007;36:1015–1038.
43. Humar, A, Ramcharan, T, Kandaswamy, R, et al. Technical failures after pancreas transplants: why grafts fail and the risk factors—a multivariate analysis. Transplantation. 2004;78:1188–1192.
44. Robertson, RP. Consequences on beta-cell function and reserve after long-term pancreas transplantation. Diabetes. 2004;53:633–644.
45. Wolfe, RA, Ashby, VB, Milford, EL, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med. 1999;341:1725–1730.
46. Reddy, KS, Stablein, D, Taranto, S, et al. Long-term survival following simultaneous kidney-pancreas transplantation versus kidney transplantation alone in patients with type 1 diabetes mellitus and renal failure. Am J Kidney Dis. 2003;41:464–470.
47. Petersen, MR, Vine, AK. Progression of diabetic retinopathy after pancreas transplantation. The University of Michigan Pancreas Transplant Evaluation Committee. Ophthalmology. 1990;97:496–500.
48. Scheider, A, Meyer-Schwickerath, E, Nusser, J, et al. Diabetic retinopathy and pancreas transplantation: a 3-year follow-up. Diabetologia. 1991;34(Suppl 1):S95–S99.
49. Ramsay, RC, Goetz, FC, Sutherland, DE, et al. Progression of diabetic retinopathy after pancreas transplantation for insulin-dependent diabetes mellitus. N Engl J Med. 1988;318:208–214.
50. Pearce, IA, Ilango, B, Sells, RA, et al. Stabilisation of diabetic retinopathy following simultaneous pancreas and kidney transplant. Br J Ophthalmol. 2000;84:736–740.
51. Ulbig, M, Kampik, A, Thurau, S, et al. Long-term follow-up of diabetic retinopathy for up to 71 months after combined renal and pancreatic transplantation. Graefes Arch Clin Exp Ophthalmol. 1991;229:242–245.
52. Giannarelli, R, Coppelli, A, Sartini, MS, et al. Pancreas transplant alone has beneficial effects on retinopathy in type 1 diabetic patients. Diabetologia. 2006;49:2977–2982.
53. Konigsrainer, A, Miller, K, Steurer, W, et al. Does pancreas transplantation influence the course of diabetic retinopathy? Diabetologia. 1991;34(Suppl 1):S86–S88.
54. Friberg, TR, Tzakis, AG, Carroll, PB, et al. Visual improvement after long-term success of pancreatic transplantation. Am J Ophthalmol. 1990;110:564–565.
55. Bilous, RW, Mauer, SM, Sutherland, DE, et al. The effects of pancreas transplantation on the glomerular structure of renal allografts in patients with insulin-dependent diabetes. N Engl J Med. 1989;321:80–85.
56. Coppelli, A, Giannarelli, R, Vistoli, F, et al. The beneficial effects of pancreas transplant alone on diabetic nephropathy. Diabetes Care. 2005;28:1366–1370.
57. Solders, G, Tyden, G, Persson, A, et al. Improvement of nerve conduction in diabetic neuropathy. A follow-up study 4 yr after combined pancreatic and renal transplantation. Diabetes. 1992;41:946–951.
58. Muller-Felber, W, Landgraf, R, Scheuer, R, et al. Diabetic neuropathy 3 years after successful pancreas and kidney transplantation. Diabetes. 1993;42:1482–1486.
59. Navarro, X, Sutherland, DE, Kennedy, WR. Long-term effects of pancreatic transplantation on diabetic neuropathy. Ann Neurol. 1997;42:727–736.
60. Allen, RD, Al-Harbi, IS, Morris, JG, et al. Diabetic neuropathy after pancreas transplantation: determinants of recovery. Transplantation. 1997;63:830–838.
61. Hathaway, DK, Abell, T, Cardoso, S, et al. Improvement in autonomic and gastric function following pancreas-kidney versus kidney-alone transplantation and the correlation with quality of life. Transplantation. 1994;57:816–822.
62. Biesenbach, G, Margreiter, R, Konigsrainer, A, et al. Comparison of progression of macrovascular diseases after kidney or pancreas and kidney transplantation in diabetic patients with end-stage renal disease. Diabetologia. 2000;43:231–234.
63. Fiorina, P, La Rocca, E, Venturini, M, et al. Effects of kidney-pancreas transplantation on atherosclerotic risk factors and endothelial function in patients with uremia and type 1 diabetes. Diabetes. 2001;50:496–501.
64. Larsen, JL, Ratanasuwan, T, Burkman, T, et al. Carotid intima media thickness decreases after pancreas transplantation. Transplantation. 2002;73:936–940.
65. Jukema, JW, Smets, YF, van der Pijl, JW, et al. Impact of simultaneous pancreas and kidney transplantation on progression of coronary atherosclerosis in patients with end-stage renal failure due to type 1 diabetes. Diabetes Care. 2002;25:906–911.
66. Holohan, TV. Simultaneous pancreas-kidney and sequential pancreas-after-kidney transplantation. Health Technol Assess Rep. 1995;4:1–53.
67. Robertson, RP, Holohan, TV, Genuth, S. Therapeutic controversy: pancreas transplantation for type I diabetes. J Clin Endocrinol Metab. 1998;83:1868–1874.
68. Sureshkumar, KK, Patel, BM, Markatos, A, et al. Quality of life after organ transplantation in type 1 diabetics with end-stage renal disease. Clin Transplant. 2006;20:19–25.
69. Stratta, RJ. The economics of pancreas transplantation. Graft. 2000;3:19–23.
70. Singh, RP, Rogers, J, Farney, AC, et al. Outcomes of extended donors in pancreatic transplantation with portal-enteric drainage. Transplant Proc. 2008;40:502–505.
71. Leichtman, AB, Cohen, D, Keith, D, et al. Kidney and pancreas transplantation in the United States, 1997–2006: the HRSA Breakthrough Collaboratives and the 58 DSA Challenge. Am J Transplant. 2008;8:946–957.
72. Williams, PW. Notes on diabetes treated with extract and by grafts of sheep’s pancreas. Br Med J. 1894:1303–1304.
73. Lacy, PE, Kostianovsky, M. Method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes. 1967;16:35–39.
74. Gaglia, JL, Shapiro, AM, Weir, GC. Islet transplantation: progress and challenge. Arch Med Res. 2005;36:273–280.
75. Najarian, JS, Sutherland, DE, Baumgartner, D, et al. Total or near total pancreatectomy and islet autotransplantation for treatment of chronic pancreatitis. Ann Surg. 1980;192:526–542.
76. Hering, BJ, Ricordi, C. Islet transplantation for patients with type 1 diabetes. Graft. 1999;2:12–27.
77. Ryan, EA, Paty, BW, Senior, PA, et al. Five-year follow-up after clinical islet transplantation. Diabetes. 2005;54:2060–2069.
78. Shapiro, AM, Lakey, JR, Ryan, EA, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med. 2000;27:230–238.
79. Shapiro, AM, Ricordi, C, Hering, BJ, et al. International trial of the Edmonton protocol for islet transplantation. N Engl J Med. 2006;355:1318–1330.
80. Alejandro, R, Barton, FB, Hering, BJ, et al. 2008 Update from the Collaborative Islet Transplant Registry. Transplantation. 2008;86:1783–1788.
81. Goss, JA, Schock, AP, Brunicardi, FC, et al. Achievement of insulin independence in three consecutive type-1 diabetic patients via pancreatic islet transplantation using islets isolated at a remote islet isolation center. Transplantation. 2002;74:1761–1766.
82. Markmann, JF, Deng, S, Huang, X, et al. Insulin independence following isolated islet transplantation and single islet infusions. Ann Surg. 2003;237:741–749.
83. Hering, BJ, Kandaswamy, R, Harmon, JV, et al. Transplantation of cultured islets from two-layer preserved pancreases in type 1 diabetes with anti-CD3 antibody. Am J Transplant. 2004;4:390–401.
84. Fiorina, P, Folli, F, Bertuzzi, F, et al. Long-term beneficial effect of islet transplantation on diabetic macro-/microangiopathy in type 1 diabetic kidney-transplanted patients. Diabetes Care. 2003;26:1129–1136.
85. Kessler, L, Bucher, P, Milliat-Guittard, L, et al. Influence of islet transportation on pancreatic islet allotransplantation in type 1 diabetic patients within the Swiss-French GRAGIL network. Transplantation. 2004;77:1301–1304.
86. Hirshberg, B, Rother, KI, Digon, BJ, 3rd., et al. Benefits and risks of solitary islet transplantation for type 1 diabetes using steroid-sparing immunosuppression: the National Institutes of Health experience. Diabetes Care. 2003;26:3288–3295.
87. Ryan, EA, Lakey, JR, Paty, BW, et al. Successful islet transplantation: continued insulin reserve provides long-term glycemic control. Diabetes. 2002;51:2148–2157.
88. Paty, BW. Islet transplantation. In Endotext.org at < http://diabetesmanagerpbwikicom/Islet-Transplantation-#ClinicalandMetabolicOutcomes, 2009.
89. Casey, JJ, Lakey, JR, Ryan, EA, et al. Portal venous pressure changes after sequential clinical islet transplantation. Transplantation. 2002;74:913–915.
90. Rafael, E, Ryan, EA, Paty, BW, et al. Changes in liver enzymes after clinical islet transplantation. Transplantation. 2003;76:1280–1284.
91. Maffi, P, Angeli, E, Bertuzzi, F, et al. Minimal focal steatosis of liver after islet transplantation in humans: a long-term study. Cell Transplant. 2005;14:727–733.
92. Bhargava, R, Senior, PA, Ackerman, TE, et al. Prevalence of hepatic steatosis after islet transplantation and its relation to graft function. Diabetes. 2004;53:1311–1317.
93. Leitao, CB, Tharavanij, T, Cure, P, et al. Restoration of hypoglycemia awareness after islet transplantation. Diabetes Care. 2008;31:2113–2115.
94. Rickels, MR, Schutta, MH, Mueller, R, et al. Islet cell hormonal responses to hypoglycemia after human islet transplantation for type 1 diabetes. Diabetes. 2005;54:3205–3211.
95. Diem, P, Redmon, JB, Abid, M, et al. Glucagon, catecholamine and pancreatic polypeptide secretion in type I diabetic recipients of pancreas allografts. J Clin Invest. 1990;86:2008–2013.
96. Froud, T, Faradji, RN, Pileggi, A, et al. The use of exenatide in islet transplant recipients with chronic allograft dysfunction: safety, efficacy, and metabolic effects. Transplantation. 2008;86:36–45.
97. Ghofaili, KA, Fung, M, Ao, Z, et al. Effect of exenatide on beta cell function after islet transplantation in type 1 diabetes. Transplantation. 2007;83:24–28.
98. Warnock, GL, Thompson, DM, Meloche, RM, et al. A multi-year analysis of islet transplantation compared with intensive medical therapy on progression of complications in type 1 diabetes. Transplantation. 2008;86:1762–1766.
99. Fiorina, P, Shapiro, AM, Ricordi, C, et al. The clinical impact of islet transplantation. Am J Transplant. 2008;8:1990–1997.
100. Fiorina, P, Folli, F, Maffi, P, et al. Islet transplantation improves vascular diabetic complications in patients with diabetes who underwent kidney transplantation: a comparison between kidney-pancreas and kidney-alone transplantation. Transplantation. 2003;75:1296–1301.
101. Tharavanij, T, Betancourt, A, Messinger, S, et al. Improved long-term health-related quality of life after islet transplantation. Transplantation. 2008;86:1161–1167.
102. Guignard, AP, Oberholzer, J, Benhamou, PY, et al. Cost analysis of human islet transplantation for the treatment of type 1 diabetes in the Swiss-French Consortium GRAGIL. Diabetes Care. 2004;27:895–900.
103. Andreoni, KA, Brayman, KL, Guidinger, MK, et al. Kidney and pancreas transplantation in the United States, 1996–2005. Am J Transplant. 2007;7:1359–1375.
104. Markmann, JF, Deng, S, Desai, NM, et al. The use of non-heart-beating donors for isolated pancreatic islet transplantation. Transplantation. 2003;75:1423–1429.
105. Markmann, JF, Kaufman, DB, Ricordi, C, et al. Financial issues constraining the use of pancreata recovered for islet transplantation: a white paper. Am J Transplant. 2008;8:1588–1592.
106. Posselt, AM, Barker, CF, Tomaszewski, JE, et al. Induction of donor-specific unresponsiveness by intrathymic islet transplantation. Science. 1990;249:1293–1295.
107. Arias-Diaz, J, Vara, E, Balibrea, JL, et al. CT-guided fine-needle approach for intrathymic islet transplantation in a diabetic patient. Pancreas. 1996;12:100–102.
108. Sykes, M. Mixed chimerism and transplant tolerance. Immunity. 2001;14:417–424.
109. Sayegh, MH, Fine, NA, Smith, JL, et al. Immunologic tolerance to renal allografts after bone marrow transplants from the same donors. Ann Intern Med. 1991;114:954–955.
110. Cosimi, AB, Sachs, DH. Mixed chimerism and transplantation tolerance. Transplantation. 2004;77:943–946.
111. Nikolic, B, Takeuchi, Y, Leykin, I, et al. Mixed hematopoietic chimerism allows cure of autoimmune diabetes through allogeneic tolerance and reversal of autoimmunity. Diabetes. 2004;53:376–383.
112. Lewis, EC, Mizrahi, M, Toledano, M, et al. Alpha1-antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci U S A. 2008;105:16236–16241.
113. Contreras, JL, Wang, PX, Eckhoff, DE, et al. Peritransplant tolerance induction with anti-CD3-immunotoxin: a matter of proinflammatory cytokine control. Transplantation. 1998;65:1159–1169.
114. Contreras, JL, Jenkins, S, Eckhoff, DE, et al. Stable alpha- and beta-islet cell function after tolerance induction to pancreatic islet allografts in diabetic primates. Am J Transplant. 2003;3:128–138.
115. Kenyon, NS, Fernandez, LA, Lehmann, R, et al. Long-term survival and function of intrahepatic islet allografts in baboons treated with humanized andti-CD154. Diabetes. 1999;48:1473–1481.
116. Kirk, AD, Harlan, DM, Armstrong, NN, et al. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci U S A. 1997;94:8789–8794.
117. Demirci, G, Strom, TB, Li, XC. Islet allograft rejection in nonobese diabetic mice involves the common gamma-chain and CD28/CD154-dependent and -independent mechanisms. J Immunol. 2003;171:3878–3885.
118. Adams, AB, Shirasugi, N, Durham, MM, et al. Calcineurin inhibitor-free CD28 blockade-based protocol protects allogeneic islets in nonhuman primates. Diabetes. 2002;51:265–270.
119. Zheng, XX, Sanchez-Fueyo, A, Sho, M, et al. Favorably tipping the balance between cytopathic and regulatory T cells to create transplantation tolerance. Immunity. 2003;19:503–514.
120. Ricordi, C, Strom, TB. Clinical islet transplantation: advances and immunological challenges. Nat Rev Immunol. 2004;4:259–268.
121. Colton, CK. Implantable biohybrid artificial organs. Cell Transplant. 1995;4:415–436.
122. Duvivier-Kali, VF, Omer, A, Parent, RJ, et al. Complete protection of islets against allorejection and autoimmunity by a simple barium-alginate membrane. Diabetes. 2001;50:1698–1705.
123. Suzuki, K, Bonner-Weir, S, Hollister-Lock, J, et al. Number and volume of islets transplanted in immunobarrier devices. Cell Transplant. 1998;7:47–52.
124. Lacy, PE, Hegre, OD, Gerasimidi-Vazeou, A, et al. Maintenance of normoglycemia in diabetic mice by subcutaneous xenografts of encapsulated islets. Science. 1991;254:1782–1784.
125. Lim, F, Sun, AM. Microencapsulated islets as bioartificial endocrine pancreas. Science. 1980;210:908–910.
126. Omer, A, Duvivier-Kali, VF, Trivedi, N, et al. Survival and maturation of microencapsulated porcine neonatal pancreatic cell clusters transplanted into immunocompetent diabetic mice. Diabetes. 2003;52:69–75.
127. Kobayashi, T, Aomatsu, Y, Iwata, H, et al. Survival of microencapsulated islets at 400 days posttransplantation in the omental pouch of NOD mice. Cell Transplant. 2006;15:359–365.
128. Wang, T, Adcock, J, Kuhtreiber, W, et al. Successful allotransplantation of encapsulated islets in pancreatectomized canines for diabetic management without the use of immunosuppression. Transplantation. 2008;85:331–337.
129. Cruise, GM, Hegre, OD, Lamberti, FV, et al. In vitro and in vivo performance of porcine islets encapsulated in interfacially photopolymerized poly(ethylene glycol) diacrylate membranes. Cell Transplant. 1999;8:293–306.
130. Calafiore, R, Basta, G, Luca, G, et al. Microencapsulated pancreatic islet allografts into nonimmunosuppressed patients with type 1 diabetes: first two cases. Diabetes Care. 2006;29:137–138.
131. Beattie, GM, Otonkoski, T, Lopez, AD, et al. Functional β-cell mass after transplantation of human fetal pancreatic cells. Diabetes. 1997;46:244–248.
132. Bonner-Weir, S, Weir, GC. New sources of pancreatic beta-cells. Nat Biotechnol. 2005;23:857–861.
133. Jonas, JC, Sharma, A, Hasenkamp, W, et al. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J Biol Chem. 1999;274:14112–14121.
134. Bruning, JC, Winnay, J, Bonner-Weir, S, et al. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell. 1999;88:561–572.
135. Dor, Y, Brown, J, Martinez, OI, et al. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46.
136. Bonner-Weir, S. Two pathways of beta cell growth in the regenerating rat pancreas: implications for islet transplantation. Diab Nutr Metab. 1992;5(Suppl 1):21–25.
137. Xu, X, D’Hoker, J, Stange, G, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132:197–207.
138. Inada, A, Nienaber, C, Katsuta, H, et al. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci U S A. 2008;105:19915–19919.
139. Yatoh, S, Dodge, R, Akashi, T, et al. Differentiation of affinity-purified human pancreatic duct cells to beta-cells. Diabetes. 2007;56:1802–1809.
140. Butler, AE, Janson, J, Bonner-Weir, S, et al. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110.
141. Ouziel-Yahalom, L, Zalzman, M, Anker-Kitai, L, et al. Expansion and redifferentiation of adult human pancreatic islet cells. Biochem Biophys Res Commun. 2006;341:291–298.
142. Gao, R, Ustinov, J, Korsgren, O, et al. Maturation of in vitro-generated human islets after transplantation in nude mice. Mol Cell Endocrinol. 2007;264:28–34.
143. Bonner-Weir, S, Taneja, M, Weir, GC, et al. In vitro cultivation of human islets from expanded ductal tissue. Proc Natl Acad Sci U S A. 2000;97:7999–8004.
144. Seaberg, RM, Smukler, SR, Kieffer, TJ, et al. Clonal identification of multipotent precursors from adult mouse pancreas that generate neural and pancreatic lineages. Nat Biotechnol. 2004;22:1115–1124.
145. Suzuki, A, Nakauchi, H, Taniguchi, H. Prospective isolation of multipotent pancreatic progenitors using flow-cytometric cell sorting. Diabetes. 2004;53:2143–2152.
146. Ratajczak, MZ, Machalinski, B, Wojakowski, W, et al. A hypothesis for an embryonic origin of pluripotent Oct-4(+) stem cells in adult bone marrow and other tissues. Leukemia. 2007;21:860–867.
147. Lechner, A, Yang, YG, Blacken, RA, et al. No evidence for significant transdifferentiation of bone marrow into pancreatic beta-cells in vivo. Diabetes. 2004;53:616–623.
148. Jiang, Y, Jahagirdar, BN, Reinhardt, RL, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418:41–49.
149. De Coppi, P, Bartsch, G, Jr., Siddiqui, MM, et al. Isolation of amniotic stem cell lines with potential for therapy. Nat Biotechnol. 2007;25:100–106.
150. Kroon, E, Martinson, LA, Kadoya, K, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol. 2008;26:443–452.
151. Nakagawa, M, Koyanagi, M, Tanabe, K, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106.
152. Weir, GC. Can we make surrogate beta-cells better than the original? Semin Cell Dev Biol. 2004;15:347–357.
153. Narushima, M, Kobayashi, N, Okitsu, T, et al. A human beta-cell line for transplantation therapy to control type 1 diabetes. Nat Biotechnol. 2005;23:1274–1282.
154. King, AJ, Fernandes, JR, Hollister-Lock, J, et al. Normal relationship of beta- and non-beta-cells not needed for successful islet transplantation. Diabetes. 2007;56:2312–2318.
155. Zhou, Q, Brown, J, Kanarek, A, et al. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–632.
156. Zalzman, M, Gupta, S, Giri, RK, et al. Reversal of hyperglycemia in mice by using human expandable insulin-producing cells differentiated from fetal liver progenitor cells. Proc Natl Acad Sci U S A. 2003;100:7253–7258.
157. Kojima, H, Fujimiya, M, Matsumura, K, et al. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat Med. 2003;9:596–603.
158. Li, WC, Horb, ME, Tosh, D, et al. In vitro transdifferentiation of hepatoma cells into functional pancreatic cells. Mech Dev. 2005;122:835–847.
159. Ber, I, Shternhall, K, Perl, S, et al. Functional, persistent, and extended liver to pancreas transdifferentiation. J Biol Chem. 2003;278:31950–31957.
160. Yang, L, Li, S, Hatch, H, et al. In vitro trans-differentiation of adult hepatic stem cells into pancreatic endocrine hormone-producing cells. Proc Natl Acad Sci U S A. 2002;99:8078–8083.
161. Davalli, AM, Ogawa, Y, Scaglia, L, et al. Function, mass, and replication of porcine and rat islets transplanted into diabetic nude mice. Diabetes. 1995;44:104–111.
162. Mandel, TE, Koulmanda, M, Kovarik, J, et al. Transplantation of organ cultured fetal pig pancreata in non-obese diabetic (NOD) mice and primates (Macaca fascicularis). Xenotransplantation. 1996;2:128–132.
163. Yoon, KH, Quickel, RR, Tatarkiewicz, K, et al. Differentiation and expansion of beta cell mass in porcine neonatal pancreatic cell clusters transplanted into nude mice. Cell Transplant. 1999;8:673–689.
164. Korbutt, GS, Elliott, JF, Ao, Z, et al. Large scale isolation, growth, and function of porcine neonatal islet cells. J Clin Invest. 1996;97:2119–2129.
165. van der Lean, LJ, Lockey, C, Griffeth, BC, et al. Infection by porcine endogenous retrovirus after islet xenotransplantation in SCID mice. Nature. 2000;407:90–94.
166. McKenzie, IFC, Koulmanda, M, Sandrin, MS, et al. Expression of gal(1,3)gal by porcine islet cells and its relevance to xenotransplantation. Xenotransplantation. 1996;2:139–142.
167. d’Apice, AJ, Cowan, PJ. Gene-modified pigs. Xenotransplantation. 2008;15:87–90.
168. Elliott, RB, Escobar, L, Tan, PL, et al. Live encapsulated porcine islets from a type 1 diabetic patient 9.5 yr after xenotransplantation. Xenotransplantation. 2007;14:157–161.
169. Valdes-Gonzalez, RA, Dorantes, LM, Garibay, GN, et al. Xenotransplantation of porcine neonatal islets of Langerhans and Sertoli cells: a 4-year study. Eur J Endocrinol. 2005;153:419–427.
170. Sykes, M, Cozzi, E, d’Apice, A, et al. Clinical trial of islet xenotransplantation in Mexico. Xenotransplantation. 2006;13:371–372.
171. Cardona, K, Korbutt, GS, Milas, Z, et al. Long-term survival of neonatal porcine islets in nonhuman primates by targeting costimulation pathways. Nat Med. 2006;12:304–306.
172. Hering, BJ, Wijkstrom, M, Graham, ML, et al. Prolonged diabetes reversal after intraportal xenotransplantation of wild-type porcine islets in immunosuppressed nonhuman primates. Nat Med. 2006;12:301–303.
173. Rickels, MR, Schutta, MH, Markmann, JF, et al. β-Cell function following human islet transplantation for type 1 diabetes. Diabetes. 2005;54:100–106.
174. Palmer, JP, Benson, JW, Walter, RM, et al. Arginine-stimulated acute phase of insulin and glucagon secretion in diabetic subjects. J Clin Invest. 1976;58:565–570.
175. Davalli, AM, Scaglia, L, Zangen, DH, et al. Vulnerability of islets in the immediate posttransplantation period. Diabetes. 1996;45:1161–1167.
176. Menger, MD, Vajkoczy, P, Beger, C, et al. Orientation of microvascular blood flow in pancreatic islet isografts. J Clin Invest. 1994;93:2280–2285.
177. Brissova, M, Fowler, M, Wiebe, P, et al. Intra-islet endothelial cells contribute to revascularization of transplanted pancreatic islets. Diabetes. 2004;53:1318–1325.
178. Jansson, L, Carlsson, PO. Graft vascular function after transplantation of pancreatic islets. Diabetologia. 2002;45:749–763.
179. Juang, JH, Bonner-Weir, S, Wu, YJ, et al. Beneficial influence of glycemic control upon the growth and function of transplanted islets. Diabetes. 1994;43:1334–1339.
180. Vasir, B, Reitz, P, Xu, G, et al. Effects of diabetes and hypoxia on gene markers of angiogenesis (HGF, cMET, uPA and uPAR, TGF-alpha, TGF-beta, bFGF and Vimentin) in cultured and transplanted rat islets. Diabetologia. 2000;43:763–772.
181. Bonner-Weir, S, Orci, L. New perspectives on the microvasculature of the islets of Langerhans in the rat. Diabetes. 1982;31:883–939.
182. Stagner, JI, Samols, E. The vascular order of islet cellular perfusion in the human pancreas. Diabetes. 1992;41:93–97.
183. Carlsson, PO, Palm, F, Mattsson, G. Low revascularization of experimentally transplanted human pancreatic islets. J Clin Endocrinol Metab. 2002;87:5418–5423.
184. Carlsson, PO, Palm, F, Andersson, A, et al. Markedly decreased oxygen tension in transplanted rat pancreatic islets irrespective of the implantation site. Diabetes. 2001;50:489–495.
185. Brissova, M, Powers, AC. Revascularization of transplanted islets: can it be improved? Diabetes. 2008;57:2269–2271.
186. Korsgren, O, Andersson, A, Jansson, L, et al. Reinnervation of syngeneic mouse pancreatic islets transplanted into renal subcapsular space. Diabetes. 1992;41:130–135.
187. Tyrberg, B, Ustinov, J, Otonkoski, T, et al. Stimulated endocrine cell proliferation and differentiation in transplanted human pancreatic islets: effects of the ob gene and compensatory growth of the implantation organ. Diabetes. 2001;50:301–307.
188. Bennet, W, Sundberg, B, Groth, CG, et al. Incompatibility between human blood and isolated islets of Langerhans: a finding with implications for clinical intraportal islet transplantation? Diabetes. 1999;48:1907–1914.
189. Medzhitov, R, Janeway, JC. Innate immunity. N Engl J Med. 2000;343:338–343.
190. Moberg, L, Olsson, A, Berne, C, et al. Nicotinamide inhibits tissue factor expression in isolated human pancreatic islets: implications for clinical islet transplantation. Transplantation. 2003;76:1285–1288.
191. Cabric, S, Sanchez, J, Lundgren, T, et al. Islet surface heparinization prevents the instant blood-mediated inflammatory reaction in islet transplantation. Diabetes. 2007;56:2008–2015.
192. Huurman, VA, Hilbrands, R, Pinkse, GG, et al. Cellular islet autoimmunity associates with clinical outcome of islet cell transplantation. PLoS ONE. 2008;3:e2435.
193. Makhlouf, L, Kishimoto, K, Smith, RN, et al. The role of autoimmunity in islet allograft destruction: major histocompatibility complex class II matching is necessary for autoimmune destruction of allogeneic islet transplants after T-cell costimulatory blockade. Diabetes. 2002;51:3202–3210.
194. Sutherland, DER, Goetz, FC, Sibley, RK. Recurrence of disease in pancreas transplants. Diabetes. 1989;38:85–87.
195. Bosi, E, Braghi, S, Maffi, P, et al. Autoantibody response to islet transplantation in type 1 diabetes. Diabetes. 2001;50:2464–2471.
196. Campbell, PM, Senior, PA, Salam, A, et al. High risk of sensitization after failed islet transplantation. Am J Transplant. 2007;7:2311–2317.
197. McCabe, C, Samali, A, O’Brien, T. Cytoprotection of beta cells: rational gene transfer strategies. Diabetes Metab Res Rev. 2006;22:241–252.
198. Efrat, S, Fejer, G, Brownlee, M, et al. Prolonged survival of pancreatic islet allografts mediated by adenovirus immunoregulatory transgenes. Proc Natl Acad Sci U S A. 1995;92:6947–6951.
199. Feng, S, Quickel, RR, Hollister-Lock, J, et al. Prolonged xenograft survival of islets infected with small doses of adenovirus expressing CTLA4Ig. Transplantation. 1999;67:1607–1613.
200. Suarez-Pinzon, WL, Marcoux, Y, Ghahary, A, et al. Gene transfection and expression of transforming growth factor-beta1 in nonobese diabetic mouse islets protects beta-cells in syngeneic islet grafts from autoimmune destruction. Cell Transplant. 2002;11:519–528.
201. Giannoukakis, N, Rudert, WA, Ghivizzani, SC, et al. Adenoviral gene transfer of the interleukin 1 receptor antagonist protein to human islets prevents IL-1-beta-induced beta-cell impairment and activation of islet cell apoptosis in vitro. Diabetes. 1999;48:1730–1736.
202. Gallichan, WS, Kafri, T, Krahl, T, et al. Lentivirus-mediated transduction of islet grafts with interleukin 4 results in sustained gene expression and protection from insulitis. Hum Gene Ther. 1998;18:2717–2726.
203. Carter, JD, Ellett, JD, Chen, M, et al. Viral IL-10-mediated immune regulation in pancreatic islet transplantation. Mol Ther. 2005;12:360–368.
204. Suarez-Pinzon, W, Korbutt, GS, Power, R, et al. Testicular Sertoli cells protect islet beta cells from autoimmune destruction in NOD mice by a transforming growth factor beta 1–dependent mechanism. Diabetes. 2000;49:1810–1818.
205. Cnop, M, Welsh, N, Jonas, JC, et al. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(Suppl 2):S97–107.
206. Contreras, JL, Bilbao, G, Smyth, CA, et al. Cytoprotection of pancreatic islets before and soon after transplantation by gene transfer of the anti-apoptotic Bcl-2 gene. Transplantation. 2001;71:1015–1023.
207. Grey, ST, Longo, C, Shukri, T, et al. Genetic engineering of a suboptimal islet graft with A20 preserves beta cell mass and function. J Immunol. 2003;170:6250–6256.
208. Dupraz, P, Cottet, S, Hamburger, F, et al. Dominant negative MyD88 proteins inhibit interleukin-1β/interferon-γ-mediated induction of nuclear factor κB-dependent nitrite production and apoptosis in β cells. J Biol Chem. 2000;275:37672–37678.
209. Maedler, K, Fontana, A, Ris, F, et al. FLIP switches Fas-mediated glucose signaling in human pancreatic beta cells from apoptosis to cell replication. Proc Natl Acad Sci U S A. 2002;99:8236–8241.
210. Emamaullee, JA, Rajotte, RV, Liston, P, et al. XIAP overexpression in human islets prevents early posttransplant apoptosis and reduces the islet mass needed to treat diabetes. Diabetes. 2005;54:2541–2548.
211. Emamaullee, JA, Davis, J, Pawlick, R, et al. The caspase selective inhibitor EP1013 augments human islet graft function and longevity in marginal mass islet transplantation in mice. Diabetes. 2008;57:1556–1566.
212. Noguchi, H, Nakai, Y, Ueda, M, et al. Activation of c-Jun NH2-terminal kinase (JNK) pathway during islet transplantation and prevention of islet graft loss by intraportal injection of JNK inhibitor. Diabetologia. 2007;50:612–619.
213. Lenzen, S, Drinkgern, J, Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20:463–466.
214. Lee, SS, Gao, W, Mazzola, S, et al. Heme oxygenase-1, carbon monoxide, and bilirubin induce tolerance in recipients toward islet allografts by modulating T regulatory cells. FASEB J. 2007;21:3450–3457.
215. Li, Y, Hansotia, T, Yusta, B, et al. Glucagon-like peptide-1 receptor signaling modulates beta-cell apoptosis. J Biol Chem. 2003;278:471–478.
216. King, A, Lock, J, Xu, G, et al. Islet transplantation outcomes in mice are better with fresh islets and exendin-4 treatment. Diabetologia. 2005;48:2074–2079.
217. Fiaschi-Taesch, NM, Berman, DM, Sicari, BM, et al. Hepatocyte growth factor (HGF) enhances engraftment and function of non-human primate islets. Diabetes. 2008.
218. Casellas, A, Salavert, A, Agudo, J, et al. Expression of IGF-1 in pancreatic islets prevents lymphocytic infiltration and protects mice from type 1 diabetes. Diabetes. 2006;55:3246–3255.
219. Robertson, RP, Sutherland, DE, Kendall, DM, et al. Metabolic characterization of long-term successful pancreas transplants in type 1 diabetes. J Investig Med. 1996;44:549–555.