[level-membership-for-neurology-category]
12 Pain Pathophysiology and Management
Neuropathic Pain Syndromes
Pathophysiology
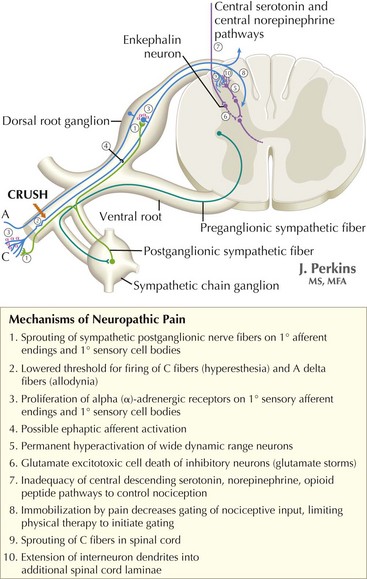
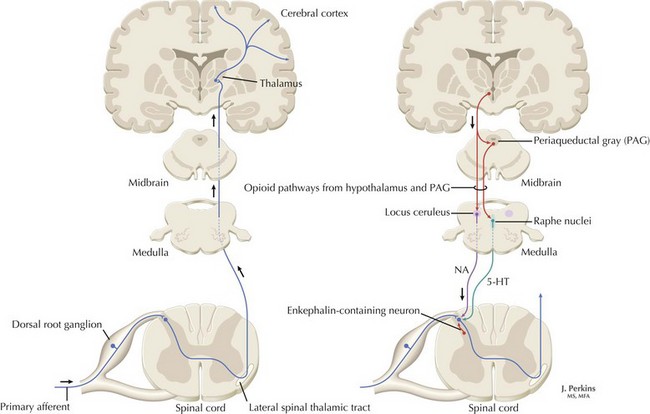
The understanding of the relationship between the clinical features of neuropathic pain and underlying molecular mechanisms in humans remains in its infancy. Following nerve injury, neuronal remodeling occurs, with microscopic structural changes in the neuronal membrane and individual membrane bound ion channels. Animal models suggest that neuronal remodeling alters the membrane electrical properties, resulting in a state of hyperexcitability wherein thresholds are lowered, action potentials are propagated more easily, and the duration of nerve impulses is prolonged. These aberrant action potentials are reproduced at multiple anatomic levels: from the primary sensory neuron, to the sensory ganglia, and neurons within the dorsal horn of the spinal cord (Fig. 12-1). Such a pattern of aberrant nerve discharges may account for the positive symptoms of neuropathic pain. The cognizant perception of painful symptoms is based on the neural pathways commencing in the periphery at the primary sensory nerve ending, transmitted through the dorsal root ganglia, the spinal cord, up to the thalamus and finally the somatosensory parietal cerebral cortex (Fig. 12-2). During the passage of nerve potentials along this pathway, a number of opportunities are available at the various synapses for modulation of the impulses per se and thus the eventual perception of the original stimulus.
Diagnosis and Clinical Manifestations
In addition to the history and physical examination, ancillary studies may aid in diagnosis. These are very important for confirming or excluding the presence of underlying etiologies for neuropathic pain. Magnetic resonance imaging is used to assess the integrity of central neuroanatomic structures involved in pain signaling pathways. These include the spinal cord, brainstem, thalamus, and cortices (see Fig. 12-1). Peripheral sources for the pain can sometimes be defined by electromyography and nerve conduction studies. The latter particularly assess the function of large myelinated nerve fibers. Nonneurologic tests such as oral glucose tolerance, Tzanck prep (a rapid test previously performed to diagnose infections caused by herpes viruses) and enzyme-linked immunosorbent assay. This can be used to confirm or exclude the presence of a number of underlying diseases that may lead to sequelae of neuropathic pain. Psychiatric evaluation is also useful in evaluating possible somatization disorders in patients with multisystem complaints that may date back into early developmental stages.
Treatment
First-Line Prescription Agents
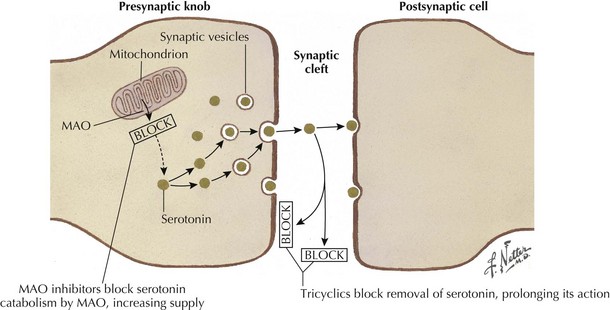
Tricyclic antidepressants historically are thought to modulate pain signaling by affecting both serotonergic and noradrenergic reuptake in descending inhibitory supraspinal pathways (Fig. 12-3). Desipramine and nortriptyline are preferred for neuropathic pain because of their favorable side-effect profiles relative to amitriptyline. Although their anticholinergic side effects demand careful consideration when these are used in patients with comorbidities, TCAs will relieve pain in PHN, postmastectomy pain syndrome, and painful diabetic neuropathy (PDN) and nondiabetic peripheral neuropathy. These agents should be tried alone initially, and then may be used in combination to pursue symptomatic improvement if necessary, provided that the clinician carefully monitors their interactions and side effects.
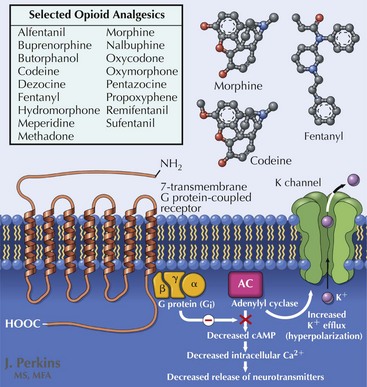
Opioid analgesia (Fig. 12-4) is considered by many experts to be a first-line agent as well, although considerable debate surrounds its role in management of neuropathic pain because of issues of tolerability, long-term efficacy, and the risks of misuse and abuse. Multiple trials support the use of these various pharmacologic modalities in chronic conditions such as PDN and PHN with evidence of superior efficacy and reduced side-effect burden when prescribed in combination with a calcium channel modulator. In clinical trials, opioid side effects, including nausea, sedation, and constipation, contributed to significant patient dropout, despite reported reductions in pain. Tramadol exerts its analgesic effects through both opioid and descending inhibitory pathways; it has been shown to be effective in reducing pain associated with nerve injury and improving quality of life.
A Ten-Step Process for opioid therapy utilized over the long term has been suggested by the International Society of Interventional Pain Physicians (Box 12-1). This provides the clinician with responsible guidelines for maintaining careful control of a potentially very useful therapeutic modality.
Box 12-1
Ten-Step Process for Long-Term Opioid Therapy in Chronic Pain
Adapted from Trescot AM, Boswell MV, Atluri SL, et al: Opioid guidelines in the management of chronic non-cancer pain. Pain Physician 9:1-39, 2006.
Painful Diabetic Neuropathy
Overview
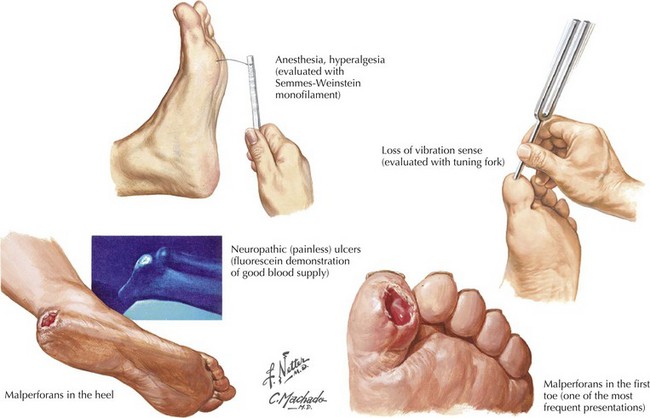
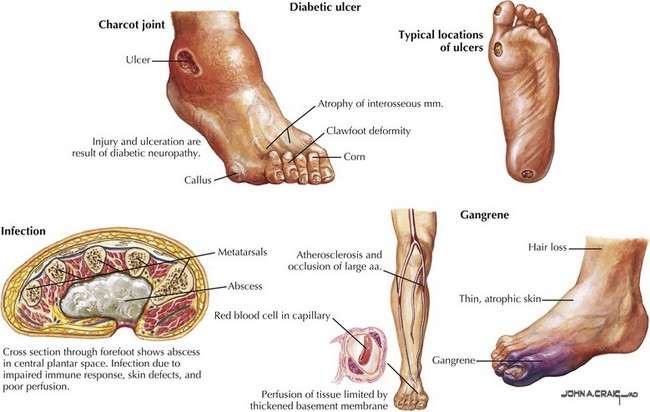
This is an increasingly common complication of diabetes mellitus, developing in up to 66% of insulin-dependent (type 1) and 59% of non–insulin-dependent (type 2) diabetics. Of these patients, studies suggest that up to 20% will experience symptoms of pain, loss of sensation, numbness, and tingling of at least 3 months’ duration, meeting the criteria for symptomatic peripheral neuropathy. Diabetic polyneuropathies also occur in other divisions of the nervous system and may present as autonomic neuropathies involving the gastrointestinal, cardiovascular, or genitourinary system, or as amyotrophic neuropathies with focal motor involvement especially in the thigh. Long-term complications of diabetic neuropathy include foot ulcers, which occur in 15% of diabetic patients and account for 85% of amputations in the same group (Fig. 12-5). PDN is a serious condition responsible for debilitating pain as well as serious long-term sequelae (Fig. 12-6), with early diagnosis and management crucial in preventing long-term complications and reductions in quality of life.
Complex Regional Pain
Clinical Vignette
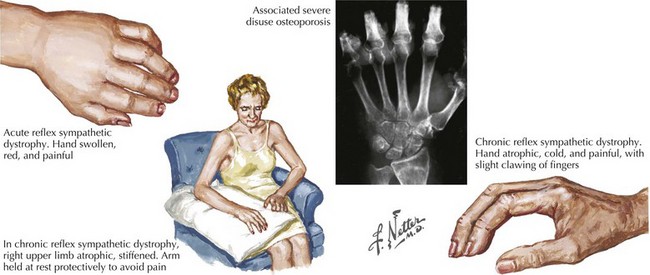
Seven weeks postoperatively, the patient presents back to the orthopedic surgeon complaining of swelling, redness, and stiffness of her right hand. She also notes that her hand has become more sensitive to painful stimuli and that she experiences significant discomfort when her hand contacts virtually anything. On exam, the hand is warm, red, and swollen, with hyperhydrosis noted on the palmar surface (Fig. 12-7).
Clinical Features and Diagnosis
Additionally, sympathetic dysfunction is present, mediating pain symptoms as well as autonomic instability (see Fig. 12-7). Patients may initially experience a warm, anhydrotic, edematous extremity with progression to a cold, hyperhidrotic limb and resolving edema. Trophic changes in the affected limb also occur, with hypertrophic or atrophic hair and nail growth and degeneration of the skin and subcutaneous fat. Connective tissues may become adherent to each other, leading to contractures and loss of function in the affected extremity. Rapid bone loss also develops, resulting in demineralization and osteoporosis. Finally, weakness, tremor, and other motor anomalies may occur in the affected extremity, with profound loss of function.
Eisenberg E, McNicol ED, Carr DB. Efficacy and safety of opioid agonists in the treatment of neuropathic pain of nonmalignant origin: systematic review and meta-analysis of randomized controlled trials. JAMA. 2005 Jun 22;293(24):3043-3052.
Miller A. Neuropathic Pain. In: American Academy of Neurology. Continuum. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:15. No 5
O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med. 2009 Oct;122(Suppl. 10):S22-S32.
Treede RD, Jensen TS, Campbell JN, et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. 2008 Apr 29;70(18):1630-1635.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
12 Pain Pathophysiology and Management
Neuropathic Pain Syndromes
Pathophysiology
The understanding of the relationship between the clinical features of neuropathic pain and underlying molecular mechanisms in humans remains in its infancy. Following nerve injury, neuronal remodeling occurs, with microscopic structural changes in the neuronal membrane and individual membrane bound ion channels. Animal models suggest that neuronal remodeling alters the membrane electrical properties, resulting in a state of hyperexcitability wherein thresholds are lowered, action potentials are propagated more easily, and the duration of nerve impulses is prolonged. These aberrant action potentials are reproduced at multiple anatomic levels: from the primary sensory neuron, to the sensory ganglia, and neurons within the dorsal horn of the spinal cord (Fig. 12-1). Such a pattern of aberrant nerve discharges may account for the positive symptoms of neuropathic pain. The cognizant perception of painful symptoms is based on the neural pathways commencing in the periphery at the primary sensory nerve ending, transmitted through the dorsal root ganglia, the spinal cord, up to the thalamus and finally the somatosensory parietal cerebral cortex (Fig. 12-2). During the passage of nerve potentials along this pathway, a number of opportunities are available at the various synapses for modulation of the impulses per se and thus the eventual perception of the original stimulus.
Diagnosis and Clinical Manifestations
In addition to the history and physical examination, ancillary studies may aid in diagnosis. These are very important for confirming or excluding the presence of underlying etiologies for neuropathic pain. Magnetic resonance imaging is used to assess the integrity of central neuroanatomic structures involved in pain signaling pathways. These include the spinal cord, brainstem, thalamus, and cortices (see Fig. 12-1). Peripheral sources for the pain can sometimes be defined by electromyography and nerve conduction studies. The latter particularly assess the function of large myelinated nerve fibers. Nonneurologic tests such as oral glucose tolerance, Tzanck prep (a rapid test previously performed to diagnose infections caused by herpes viruses) and enzyme-linked immunosorbent assay. This can be used to confirm or exclude the presence of a number of underlying diseases that may lead to sequelae of neuropathic pain. Psychiatric evaluation is also useful in evaluating possible somatization disorders in patients with multisystem complaints that may date back into early developmental stages.
Treatment
First-Line Prescription Agents
Tricyclic antidepressants historically are thought to modulate pain signaling by affecting both serotonergic and noradrenergic reuptake in descending inhibitory supraspinal pathways (Fig. 12-3). Desipramine and nortriptyline are preferred for neuropathic pain because of their favorable side-effect profiles relative to amitriptyline. Although their anticholinergic side effects demand careful consideration when these are used in patients with comorbidities, TCAs will relieve pain in PHN, postmastectomy pain syndrome, and painful diabetic neuropathy (PDN) and nondiabetic peripheral neuropathy. These agents should be tried alone initially, and then may be used in combination to pursue symptomatic improvement if necessary, provided that the clinician carefully monitors their interactions and side effects.
[/not-level-membership-for-neurology-category]
