CHAPTER 15 Pain Management
“We must all die. But that I can save (a person) from days of torture, that is what I feel as my great and ever new privilege. Pain is a more terrible lord of mankind than even death itself” (Albert Schweitzer). The treatment and alleviation of pain is a basic human right that exists regardless of age (Yaster et al., 1997; Schechter et al., 2003). The old “wisdom” that young children neither respond to, nor remember, painful experiences to the same degree that adults do is simply untrue (Taddio and Katz, 2005). Many, if not all, of the nerve pathways essential for the transmission and perception of pain are present and functioning by 24 weeks’ gestation (Lee et al., 2005; Lowery et al., 2007). Furthermore, recent research in newborn animals has revealed that the failure to provide analgesia for pain results in “rewiring” the nerve pathways responsible for pain transmission in the dorsal horn of the spinal cord and results in increased pain perception with future painful insults (Fitzgerald and Beggs, 2001; Pattinson and Fitzgerald, 2004). This confirms human newborn research in which the failure to provide anesthesia or analgesia for newborn circumcision resulted not only in short-term physiologic perturbations but also in longer term behavioral changes, particularly during immunization (Taddio et al., 1995; Maxwell et al., 1987; Taddio and Katz, 2005; Anand et al., 2006).
Providing effective analgesia to infants, preverbal children, adolescents, and the mentally and physically disabled poses unique challenges to those who practice pediatric medicine and surgery. In the past, several studies documented that physicians, nurses, and parents underestimate the amount of pain experienced by children and that they overestimate the risks inherent in the drugs used in the treatment of pain (Schechter et al., 1986; Finley et al., 1996; McGrath and Finley, 1996). This is not at all surprising; the guiding principle of medical practice is to do no harm, primum non nocere. Physicians are taught throughout their training that opioids, the analgesics most commonly prescribed in moderate to severe pain, cause respiratory depression, cardiovascular collapse, depressed levels of consciousness, constipation, nausea, vomiting, and, with repeated use, tolerance and addiction. Less potent analgesics, such as nonsteroidal antiinflammatory drugs (NSAIDs), can also cause problems such as bleeding, liver dysfunction, coagulopathies, and impaired wound and bone healing. Thus, physicians at times prescribe insufficiently potent analgesics, recommend inadequate doses, or use pharmacologically irrational dosing regimens because of their overriding concern that children may be harmed by the use of these drugs. The resulting conundrum often results in inadequate treatment for pain and for painful procedures. On the other hand, the adverse effects of pain and the failure to treat it are rarely discussed. In addition to its impact on neurodevelopment, it is known that unrelieved pain interferes with sleep, leads to fatigue and a sense of helplessness, enhances the stress and inflammatory response, and may result in increased morbidity or mortality (Anand et al., 1987).
Nurses may be wary of physicians’ orders (and patients’ requests) as well. The most common prescription order for potent analgesics is “to give as needed” (pro re nata, or PRN). Thus, the patient must know or remember to ask for pain medication, or the nurse must identify when a patient is in pain. These requirements may not always be met by children in pain. Children younger than 3 years of age may be unable to adequately verbalize when or where they hurt. Alternatively, they may be afraid to report their pain. Many children withdraw or deny their pain if pain relief involves yet another terrifying and painful experience—the intramuscular injection or “shot.” Finally, several studies have documented the inability of nurses, physicians, and parents to correctly identify and treat pain, even in postoperative pediatric patients (McGrath and Finley, 1996; Romsing et al., 1996; Fortier et al., 2009).
Societal fears of opioid addiction and lack of advocacy are also causal factors in the under treatment of pediatric pain. Unlike adult patients, pain management in children is often dependent on the ability of parents to recognize and assess pain and on their decision to treat or not treat it (Romsing and Walther-Larsen, 1996; Sutters and Miaskowski, 1997). Even in hospitalized patients, most of the pain that children experience is managed by the patient’s parents (Greenberg et al., 1999; Krane, 2008). Parental misconceptions concerning pain assessment and pain management may therefore result in inadequate pain treatment (Romsing and Walther-Larsen, 1996; Fortier et al., 2009). This is particularly true in patients who are too young or too developmentally handicapped to report their pain themselves. Parents may fail to report pain, either because they are unable to assess it or they are afraid of the consequences of pain therapy. False beliefs about addiction and the proper use of acetaminophen and other analgesics resulted in the failure to provide analgesia to children (Forward et al., 1996; Fortier et al., 2009). In another study, the belief that pain was useful or that repeated doses of analgesics lead to medication not working well resulted in the failure of the parents to provide or ask for prescribed analgesics to treat their children’s pain (Finley et al., 1996). Parental education is therefore essential if children are to be adequately treated for pain. Unfortunately, the ability to educate parents properly about this issue is often limited by insufficient resources, time, and personnel (Greenberg et al., 1999).
Fortunately, the past 25 years have seen an increase in research and interest in pediatric pain management and in the development of pediatric pain services, primarily under the direction of pediatric anesthesiologists (Shapiro et al., 1991; Nelson et al., 2009). Pediatric-pain service teams provide pain management for acute, postoperative, terminal, neuropathic, and chronic pain. This chapter reviews the recent advances in opioid and local anesthetic pharmacology, as well as the various modalities that are useful in the treatment of acute childhood pain.
Pain assessment
The International Association for the Study of Pain (IASP) defines pain as “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage” (Merskey et al., 1979). Pain is a subjective experience; operationally it can be defined as “what the patient says hurts” and exists “when the patient says it does hurt.” Infants, preverbal children, and children between the ages of 2 and 7 (Piaget’s Preoperational Thought stage) may be unable to describe their pain or their subjective experiences. This has led many to conclude incorrectly that these children don’t experience pain in the same way as adults. Clearly, children do not have to know (or be able to express) the meaning of an experience in order to have the experience (Anand and Craig, 1996). On the other hand, because pain is essentially a subjective experience, focusing on the child’s perspective of pain is an indispensable facet of pediatric pain management and an essential element in the specialized study of childhood pain. Indeed, pain assessment and management are interdependent and one is essentially useless without the other. The goal of pain assessment is to provide accurate data about the location and intensity of pain, as well as the effectiveness of measures used to alleviate or abolish it.
Multiple validated instruments currently exist to measure and assess pain in children of all ages (von Baeyer and Spagrud, 2007; Crellin et al., 2007; Franck et al., 2000). The sensitivity and specificity of these instruments have been widely debated and have resulted in a plethora of studies to validate their reliability and validity. The most commonly used instruments that measure the quality and intensity of pain are “self-report measures.” In older children and adults, the most commonly used self-report instruments are visual analogue scales (VASs) and numerical rating scales (0 = no pain; 10 = worst pain). However, pain intensity or severity can also be measured in children as young as 3 years of age by using pictures or word descriptors to describe pain. Two common examples include the Oucher Scale (developed by Dr. Judy Beyer), a two-part scale with a vertical numerical scale (0–100) on one side and six photographs of a young child on the other, or the Six-Face Pain Scale, first developed by Dr. Donna Wong and later modified by Bieri et al. (Fig. 15-1) (Beyer and Wells, 1989; Wong and Baker, 1988; Beyer et al., 1990). Because of its simplicity, the Six-Face Pain Scale-Revised is commonly used (Hicks et al., 2001; Bieri et al., 1990). Alternatively, color, word-graphic rating scales, and poker chips have been used to assess the intensity of pain in children. One obvious limitation of all of these self-report measures is their inability to be used in cognitively impaired children or in intubated, sedated, and paralyzed patients.
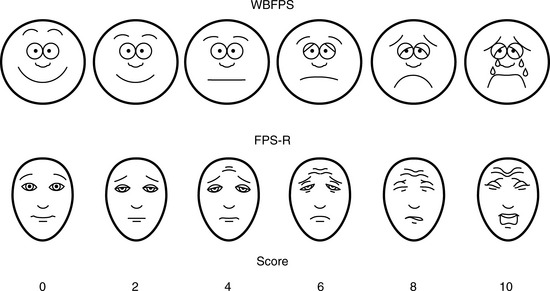
FIGURE 15-1 Six-Face Pain Scale (Top). Original scale developed by Wong et al. Lower, Modified scale by Bieri et al.
(From Bieri D et al.: The Faces Pain Scale for the self-assessment of the severity of pain experienced by children: development, initial validation, and preliminary investigation for ratio scale properties, Pain 41:139, 1990; Beyer JE et al.: Discordance between self-report and behavioral pain measures in children aged 3-7 years after surgery, J Pain Symptom Manage 5:350, 1990.)
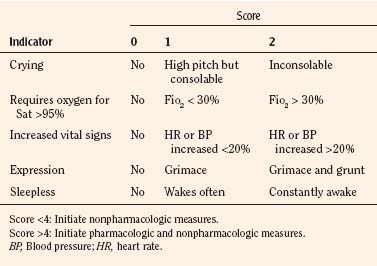
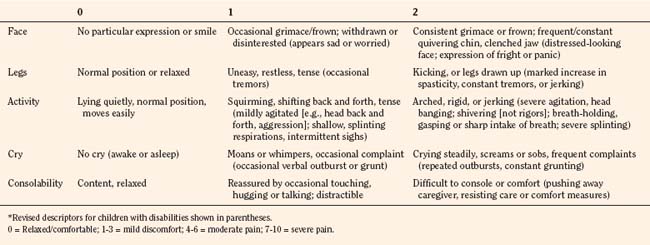
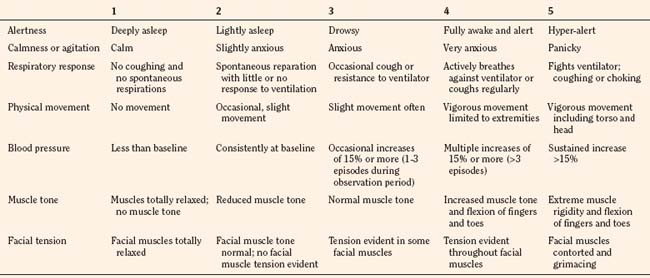
In infants, newborns, and the cognitively impaired, pain has been assessed by measuring physiologic responses to nociceptive stimuli, such as blood pressure and heart rate changes (observational pain scales, OPSs), or by measuring levels of adrenal stress hormones (Krechel and Bildner, 1995). Alternatively, behavioral approaches have used facial expression, body movements, and the intensity and quality of crying as indices of response to nociceptive stimuli. The most appropriate are the Crying, Requires oxygen, Increased vital signs, Expression, and Sleepless (CRIES) score for newborns and the revised Face, Legs, Activity, Cry, and Consolability (FLACC) pain tool for children who have difficulty verbalizing pain (Tables 15-1 and 15-2) (Grunau et al., 1990; Hadjistavropoulos et al., 1994; Voepel-Lewis et al., 2002). Another commonly used pain and sedation tool that uses both behaviors and physiologic parameters is the COMFORT scale, which relies on the measurement of five behavioral variables (alertness, facial tension, muscle tone, agitation, and movement) and three physiologic variables (heart rate, respiration, and blood pressure) (Table 15-3) (Ambuel et al., 1992; Bear and Ward-Smith, 2006). Each is assigned a score ranging from 1 to 5, to give a total score ranging from 8 (deep sedation) to 40 (alert and agitated). A modified COMFORT scale that eliminates physiologic parameters has also been developed (Ista et al., 2005). It is also important to define accurately the location of pain. This is readily accomplished by using either dolls or action figures or by using drawings of body outlines, both front and back. Finally, in the research laboratory, sophisticated new tools such as functional magnetic resonance imaging (MRI) are being used to objectively assess and map pain and its pathways through the central nervous system (CNS) (Tracey and Mantyh, 2007).
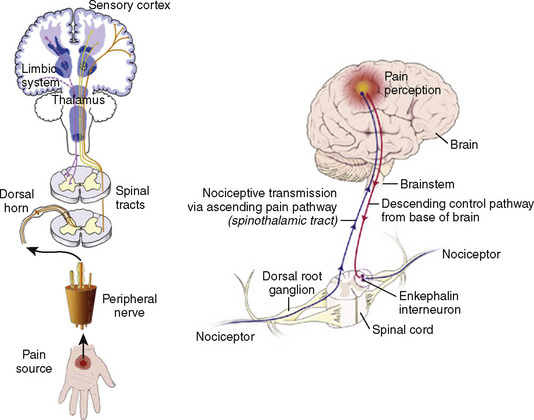
Neurophysiology of pain
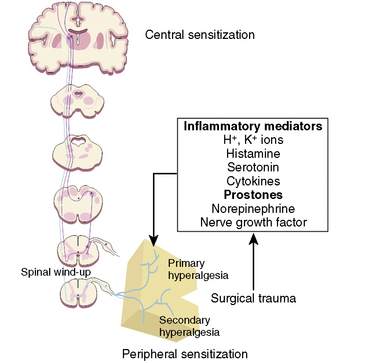
Many if not all of the nerve pathways essential for the transmission, perception, and modulation of pain are present and functioning by 24 weeks of gestation (Fig. 15-2) (Lowry et al., 2007; Lee et al., 2005). Although neural transmission in peripheral nerves is slower in neonates because myelination is incomplete at birth, the major nociceptive neurons in neonates as well as in adults are either unmyelinated C fibers or thinly myelinated Aδ fibers. After an acute injury such as surgical or accidental trauma, inflammatory mediators are released; they lower the pain threshold at the site of injury (primary hyperalgesia) and in the surrounding uninjured tissue (secondary hyperalgesia). These inflammatory mediators, which include hydrogen and potassium ions, histamine, leukotrienes, prostaglandins, cytokines, serotonin (5-HT), bradykinins, and nerve-growth factors make a “sensitizing soup,” which together with repeated stimuli of the nociceptive fibers cause decreased excitatory thresholds and result in peripheral sensitization. They are also targets of therapeutic intervention (Fig. 15-3). Secondary effects of peripheral sensitization include hyperalgesia, the increased response to a noxious stimulus and allodynia, whereby non-nociceptive fibers transmit noxious stimuli resulting in the sensation of pain from non-noxious stimuli.
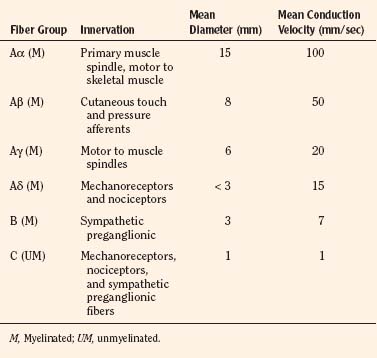
Sensory afferent neurons have a unipolar cell body located in the dorsal root ganglion and are classified by fiber size into three major groups (A, B, C) (Table 15-4). Group A is further subclassified into four subgroups. Sensory fibers that respond to noxious stimulation include small caliber myelinated (Aδ) or fine unmyelinated C fibers. These fibers originate as free nerve endings that can be characterized by their response to specific stimuli such as pressure, heat, and chemical irritants and arise from epidermal and internal receptive fields, including the periosteum, joints, and viscera. The Aδ nociceptors transmit “first pain,” which is well localized, sharp, and lasts as only as long as the original stimulus. The C-fiber, polymodal nociceptors display a slow conduction velocity and respond to mechanothermal and chemical stimuli. This “second pain” is diffuse, persistent, burning, slow to be perceived, and lasts well beyond the termination of the stimulus.
As the primary afferent neurons enter the spinal cord they segregate and occupy a lateral position in the dorsal horn (Fig. 15-4). The Aδ fibers terminate in laminae I, II (substantia gelatinosa), V (nucleus proprius), and X (central canal). The C fibers terminate in laminae I, II, and V, and some enter the dorsal horn through the ventral root. These afferent neurons release one or more excitatory amino acids (e.g., glutamate and aspartate) or peptide neurotransmitters (e.g., substance P, neurokinin A, calcitonin gene-related peptide [CGRP], cholecystokinin, and somatostatin). Second-order neurons that receive these chemical signals integrate the afferent input with facilitatory and inhibitory influences of interneurons and descending neuronal projections. It is this convergence within the dorsal horn that is responsible for much of the processing, amplification, and modulation of pain. Furthermore, the ability to simultaneously process noxious and innocuous stimuli underlies the gate-control theory of pain described by Melzack and Wall (Melzack and Wall, 1965; DeLeo, 2006).

Nociceptive activity in the spinal cord and the ascending spinothalamic, spinoreticular, and spinomesencephalic tracts carry messages to supraspinal centers (e.g., periaqueductal gray, locus coeruleus, hypothalamus, thalamus, and cerebral cortex) where they are modulated and integrated with autonomic, homeostatic, and arousal processes. This modulation, particularly by the endogenous opioids, γ-aminobutyric acid (GABA), and norepinephrine (NE), can either facilitate pain transmission or inhibit it. Modulating pain at peripheral, spinal, and supraspinal sites helps achieve better pain management than targeting only one site and is the underlying principle of treating pain in a multimodal fashion (Fig. 15-3).
Although pain pathways are present at birth, they are often immature (Lee et al., 2005; Lowery et al., 2007). As a result, there are considerable differences in how an infant responds to injury compared with the way an adult does. Within the developing nervous system, inhibitory mechanisms in the dorsal horn of the spinal cord are immature, and inhibition of nociceptive input in the dorsal horn of the spinal cord is less than in the adult. Furthermore, dorsal horn neurons in the newborn have wider receptive fields and lower excitatory thresholds than those in older children (Torsney and Fitzgerald, 2003; Bremner and Fitzgerald, 2008; Fitzgerald and Walker, 2009). Thus, compared with adults, young infants have exaggerated reflex responses to pain. Furthermore, recent research in newborn animals has revealed that the failure to provide analgesia for pain results in “rewiring” the nerve pathways responsible for pain transmission in the dorsal horn of the spinal cord and results in increased pain perception for future painful insults (Fitzgerald and Beggs, 2001; Pattinson and Fitzgerald, 2004). This confirms human newborn research in which the failure to provide anesthesia or analgesia for newborn circumcision resulted not only in short-term physiologic perturbations but also in longer term behavioral changes, particularly during immunization (Maxwell et al., 1987; Taddio et al., 1995; Taddio and Katz, 2005).
Preemptive Analgesia, Preventive Analgesia, and Multimodal Analgesia
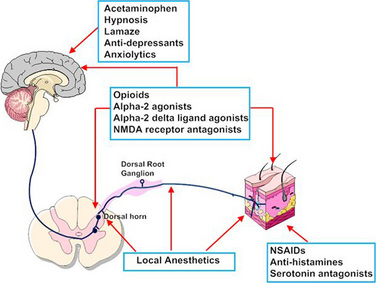
The possibility that pain after surgery might be preemptively prevented or ameliorated by the use of opioids or local anesthetics given preoperatively has been a concept under review (Katz and McCartney, 2002; Moiniche et al., 2002; Oong et al., 2005). Over the past few years the concept of preemptive analgesia has expanded and evolved to include the reduction of nociceptive inputs during and after surgery. This expanded conceptual framework, which includes preoperative, intraoperative, and postoperative analgesia, targets multiple sites along the pain pathway and is referred to as preventive or multimodal analgesia (Ballantyne, 2001; Katz and McCartney, 2002). Indeed, acute pediatric (and adult) pain management is increasingly characterized by a multimodal or “balanced” approach in which smaller doses of opioid and nonopioid analgesics, such NSAIDs, local anesthetics, NMDA antagonists, and α2-adrenergic agonists, are combined to maximize pain control and minimize drug-induced adverse side effects (Fig. 15-5) (DeLeo, 2006). Additionally, a multimodal approach also uses nonpharmacologic complementary and alternative medicine therapies. These alternative medical therapies include distraction, guided imagery, hypnosis, relaxation techniques, biofeedback, transcutaneous nerve stimulation, and acupuncture (Rusy and Weisman, 2000). Taking this approach, activation of peripheral nociceptors can be attenuated with the use of NSAIDs, antihistamines, 5-HT antagonists, and local anesthetics (Fig. 15-5). Within the dorsal horn, nociceptive transmission and processing can be further affected by the administration of local anesthetics, neuraxial opioids, α2-adrenergic agonists (e.g., clonidine and dexmedetomidine), and NMDA receptor antagonists (e.g., ketamine and methadone). Within the CNS, pain can be ameliorated by systemic opioids, α2-agonists, anticonvulsants (e.g., gabapentin and pregabalin), pharmacologic therapies (e.g., benzodiazepines, α2-agonists), and nonpharmacologic therapies (e.g., hypnosis, Lamaze, and acupuncture) that reduce anxiety and induce rest and sleep.
Pharmacologic management of pain: the conundrum of “off-label” drug use
Unfortunately, very few studies have evaluated the pharmacokinetic and pharmacodynamic properties of drugs in children (Conroy and Peden, 2001; Katz and Kelly, 1993). Most pharmacokinetic studies are performed using healthy adult volunteers, adult patients who are only minimally ill, or adult patients in a stable phase of a chronic disease. These data are then extrapolated to infants, children, and adolescents, and the medications are prescribed “off-label.” So little pharmacokinetic and pharmacodynamic testing has been performed in children that they are often considered “therapeutic orphans” (Blumer, 1999). In addition, drug formulations designed for adults are often manipulated and altered by practitioners for use in children (e.g., tablets are dissolved to make a liquid formulation, or suppositories are cut in half). The U.S. Congress has enacted the Best Pharmaceuticals for Children Act, the Pediatric Research Equity Act, and the Food and Drug Administration (FDA) Amendments Act to promote standards and requirements for the use and labeling of pediatric drugs (BPCA, 2002; PREA, 2003; FDAAA, 2007).
Pharmacologic management of pain
Nonopioid Analgesics (or Weaker Analgesics with Antipyretic Activity)
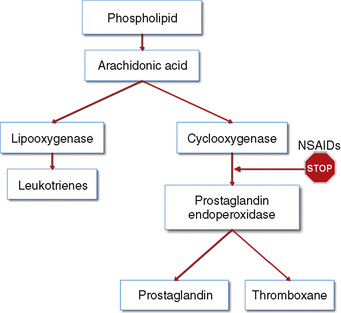
The weaker or milder analgesics with antipyretic activity, of which acetaminophen (paracetamol), salicylate (aspirin), ibuprofen, naproxen, ketoprofen, and diclofenac are common examples, comprise a heterogenous group of NSAIDs and nonopioid analgesics (Table 15-5) (Agency for Health Care Policy and Research, 1992; Yaster, 1997; Tobias, 2000b; Kokki, 2003). They produce their analgesic, antiinflammatory, antiplatelet, and antipyretic effects primarily by blocking peripheral and central prostaglandin and thromboxane production by inhibiting cyclooxygenase (COX) types 1, 2, and 3 (Fig. 15-6). These metabolites of cyclooxygenase sensitize peripheral nerve endings and vasodilate the blood vessels causing pain, erythema, and inflammation.
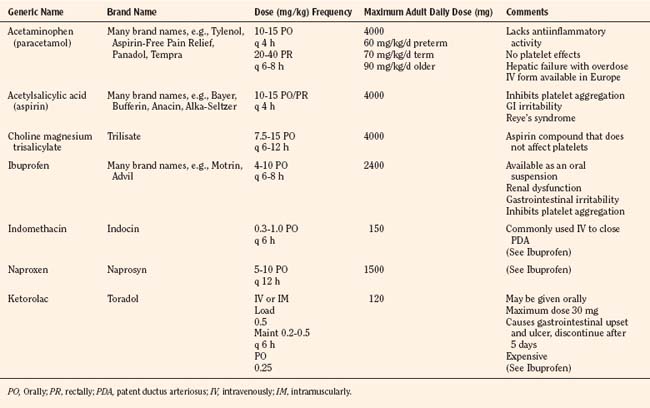
These analgesic agents are administered enterally via the oral or, on occasion, the rectal route and are particularly useful for inflammatory, bony, or rheumatic pain. Parenterally administered agents, such as ketorolac and acetaminophen, are available for use in children in whom the oral or rectal routes of administration are not possible (Murat et al., 2005). Unfortunately, regardless of dose, the nonopioid analgesics are limited by a “ceiling effect” above which pain cannot be relieved by these drugs alone. Because of this, these weaker analgesics are often administered in oral combination forms with opioids such as codeine, oxycodone, or hydrocodone.
Only a few trials have compared the efficacy of these drugs in head to head competition, and in general these studies have shown that there are no major differences in their analgesic effects when appropriate doses of each drug are used. The commonly used NSAIDs, such as ketorolac, diclofenac, ibuprofen, and ketoprofen, have reversible antiplatelet adhesion and aggregation effects that are attributable to the inhibition of thromboxane synthesis (Niemi et al., 1997; Munsterhjelm et al., 2006). As a result, bleeding times are usually slightly increased, but in most instances they remain within normal limits in children with normal coagulation systems. Nevertheless this side effect is of such great concern, particularly in surgical procedures in which even a small amount of bleeding can be catastrophic (e.g., tonsillectomy and neurosurgery), that few clinicians prescribe them even though the evidence supporting increased bleeding is equivocal at best (Moiniche et al., 2003; Cardwell et al., 2005). Finally, many orthopedic surgeons are also concerned about the negative influence of all NSAIDs, both selective and nonselective COX inhibitors, on bone growth and healing (Simon et al., 2002; Einhorn, 2003; Dahners and Mullis, 2004). Thus, most pediatric orthopedic surgeons have recommended that these drugs not be used in their patients in the postoperative period.
The discovery of at least three COX isoenzymes (COX-1, COX-2, and COX-3) has enhanced our knowledge of NSAIDs (Cashman, 1996; Vane and Botting, 1998). The COX isoenzymes share structural and enzymatic similarities, but they are specifically regulated at the molecular level and may be distinguished by their functions. Protective prostaglandins, which preserve the integrity of the stomach lining and maintain normal renal function in a compromised kidney, are synthesized by COX-1 (Vane and Botting, 1998; Moiniche et al., 2003; Levesque et al., 2005). The COX-2 isoenzyme is inducible by proinflammatory cytokines and growth factors, implying a role for COX-2 in both inflammation and control of cell growth. In addition to the induction of COX-2 in inflammatory lesions, it is expressed constitutively in the brain and spinal cord, where it may be involved in nerve transmission, particularly for pain and fever. Prostaglandins made by COX-2 are also important in ovulation and in the birth process (Vane and Botting, 1998; Moiniche et al., 2003; Levesque et al., 2005). The discovery of COX-2 has made possible the design of drugs that reduce inflammation without removing the protective prostaglandins in the stomach and kidneys made by COX-1. In fact, developing a more specific COX-2 inhibitor was a “holy grail” of drug research, because this class of drug was postulated to have all of the desired antiinflammatory and analgesic properties with none of the gastrointestinal and antiplatelet side effects. Unfortunately, the controversy regarding the potential adverse cardiovascular risks of prolonged use of COX-2 inhibitors has dampened much of the enthusiasm for these drugs and has led to the removal of rofecoxib from the market by its manufacturer (Johnsen et al., 2005; Levesque et al., 2005).
Aspirin
Aspirin, one of the oldest and most effective nonopioid analgesics, has been largely abandoned in pediatric practice because of its possible role in Reye’s syndrome, its effects on platelet function, and its gastric irritant properties. Despite these problems, a “sister” compound, choline-magnesium trisalicylate is still prescribed, particularly in the management of postoperative pain and in the child with cancer. Choline-magnesium trisalicylate is a unique aspirin-like compound that does not bind to platelets and therefore has minimal, if any, effects on platelet function (Yaster, 1997). As a result, it can be prescribed to patients with low platelet counts (cancer patients), dysfunctional platelets (uremia), and in the postoperative period. It is a convenient drug to give to children because it is available in both a liquid and tablet form and is administered either twice a day or every 6 hours. However, the association of salicylates with Reye’s syndrome limits its use, even though the risk of developing this syndrome postoperatively is extremely unlikely.
Acetaminophen
The most commonly used nonopioid analgesic in pediatric practice remains acetaminophen, although its analgesic effectiveness in the neonate is unclear (Shah et al., 1998; Anderson, 2008). Unlike aspirin and other NSAIDs, acetaminophen produces analgesia centrally as a COX-3 inhibitor and via activation of descending serotonergic pathways (Graham and Scott, 2005; Anderson, 2008). It is also thought to produce analgesia as a cannabinoid agonist and by antagonizing NMDA and substance P in the spinal cord (Bertolini et al., 2006). Acetaminophen is an antipyretic analgesic with minimal, if any, antiinflammatory and antiplatelet activity and takes about 30 minutes to provide effective analgesia. When administered orally in standard doses, 10 to 15 mg/kg acetaminophen is extremely safe, effective, and has few serious side effects. When administered rectally, higher doses of 25 to 40 mg/kg are required (Rusy et al., 1995; Birmingham et al., 1997). Because of its known association with fulminant hepatic necrosis, the daily maximum acetaminophen dosages, regardless of formulation or route of delivery, in the preterm infant, full-term infant, and older child are 60, 80, and 90 mg/kg respectively (Table 15-5). Thus, when administering acetaminophen rectally it should be given every 8 hours rather than every 4 hours. Finally, an intravenous formulation of acetaminophen is now available in Europe and can be used in patients in whom the enteral route is unavailable. This formulation has been associated with better analgesia than oral acetaminophen in clinical trials in adult patients and is equally effective and less painful than the prodrug formulation of the drug in children (Murat et al., 2005).
Opioids
Overview
Over the past 30 years, multiple opioid receptors and subtypes have been identified and classified. There are 3 primary opioid receptor types, designated mu (µ) (for morphine), kappa (κ), and delta (δ). These receptors are primarily located in the brain and spinal cord, but they also exist peripherally on peripheral nerve cells, immune cells, and other cells (e.g., oocytes) (Sabbe and Yaksh, 1990; Snyder and Pasternak, 2003; Stein and Rosow, 2004). The μ-receptor is further subdivided into several subtypes such as the μ1 (supraspinal analgesia), μ2 (respiratory depression, inhibition of gastrointestinal motility), and μ3 (antiinflammation, leukocytes), which affects the pharmacologic profiles of different opioids (Pasternak, 2001a, 2001b, 2005; Bonnet et al., 2008). Both endogenous and exogenous agonists and antagonists bind to various opioid receptors.
Opioid receptors, which are found anchored to the plasma membrane both presynaptically and postsynaptically, decrease the release of excitatory neurotransmitters from terminals carrying nociceptive stimuli. These receptors belong to the steroid superfamily of G protein–coupled receptors. Their protein structure contains seven transmembrane regions with extracellular loops that confer subtype specificity and intracellular loops that mediate subreceptor phenomena (Stein and Rosow, 2004). These receptors are coupled to guanine nucleotide (GTP)-binding regulatory proteins (G proteins) and regulate transmembrane signaling by regulating adenylate cyclase (and therefore cyclic adenosine monophosphate [cAMP]), various ion channels (K+, Ca,++, Na+) and transport proteins, neuronal nitric oxide synthetase, and phospholipase C and A2 (Fig. 15-7) (Standifer and Pasternak, 1997; Maxwell et al., 2005; Pasternak, 2005). Signal transduction from opioid receptors occurs via bonding to inhibitory G proteins (Gi and Go). Analgesic effects are mediated by decreased neuronal excitability from an inwardly rectifying K+ current, which hyperpolarizes the neuronal membrane, decreases cAMP production, increases nitric oxide synthesis, and increases the production of 12-lipoxygenase metabolites. Indeed, synergism between opioids and NSAIDs occurs as a result of the greater availability of arachidonic acid for metabolism by the 12-lipooxygenase pathway, after blockade of prostaglandin production by NSAIDs (Vaughan et al., 1997). Some of the unwanted side effects of opioids, such as pruritus, may be the result of opioid binding to stimulatory G proteins (Gs) and may be antagonized by low dose infusions of naloxone (Fig. 15-7) (Crain and Shen, 1998, 1996; Maxwell et al., 2005).
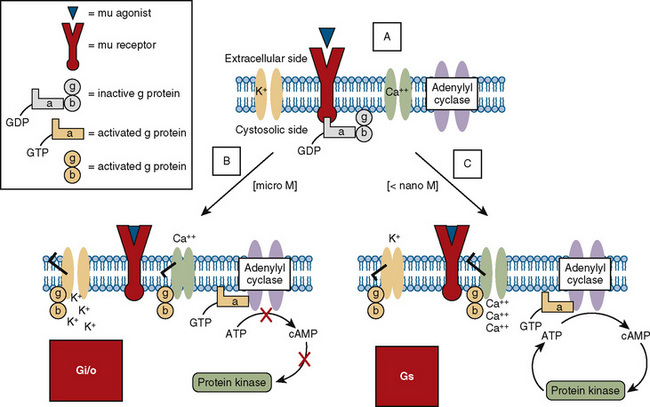
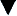 ) (e.g., morphine), it physically associates with the α subunit, causing the latter to release GDP and bind GTP. The GTP binding causes the dissociation of the α subunit from the β-γ subunit and from the receptor. Free α and β-γ subunits are functionally active and directly regulate a number of effector proteins, such as ion channels, adenylyl cyclase, and phospholipase C. B, Classically, the opioid receptor is thought to be a Gi/o coupled receptor. Adenylyl cyclase is inhibited, the potassium channel is open, and the calcium channel is closed. C, At picomolar or nanomolar concentrations, opioid receptors are coupled to Gs proteins. Adenylyl cyclase is activated, the calcium channel is open, and the potassium channel is closed.
) (e.g., morphine), it physically associates with the α subunit, causing the latter to release GDP and bind GTP. The GTP binding causes the dissociation of the α subunit from the β-γ subunit and from the receptor. Free α and β-γ subunits are functionally active and directly regulate a number of effector proteins, such as ion channels, adenylyl cyclase, and phospholipase C. B, Classically, the opioid receptor is thought to be a Gi/o coupled receptor. Adenylyl cyclase is inhibited, the potassium channel is open, and the calcium channel is closed. C, At picomolar or nanomolar concentrations, opioid receptors are coupled to Gs proteins. Adenylyl cyclase is activated, the calcium channel is open, and the potassium channel is closed.Pharmacokinetics
For opioids to effectively relieve or prevent most pain, the agonist must reach the receptor in the CNS. There are essentially two ways that this occurs, either via the bloodstream (after intravenous, intramuscular, oral, nasal, transdermal, or mucosal administration) or by direct application into the cerebrospinal fluid (intrathecal or epidural). Agonists administered via the bloodstream must cross the blood-brain barrier, a lipid membrane interface between the endothelial cells of the brain vasculature and the extracellular fluid of the brain, to reach the receptor. Normally, highly lipid-soluble agonists, such as fentanyl, rapidly diffuse across the blood-brain barrier, whereas agonists with limited lipid solubility, such as morphine, have limited brain uptake. This rule, however, does not hold true for patients of all ages. The blood-brain barrier may be immature at birth and is known to be more permeable in neonates to morphine. Indeed, Kupferberg and Way (1963) demonstrated in a classic paper that morphine concentrations were two to four times greater in the brains of younger rats than older rats despite equal blood concentrations. Spinal administration, either intrathecally (subarachnoid) or epidurally, bypasses the blood and directly places an agonist into the cerebrospinal fluid, which bathes the receptor sites in the spinal cord (substantia gelatinosa) and brain. This “back door” to the receptor significantly reduces the amount of agonist needed to relieve pain and to induce opioid side effects such as pruritus, urinary retention, and respiratory depression (Cousins and Mather, 1984; Sabbe and Yaksh, 1990). After spinal administration, opioids are absorbed by the epidural veins and redistributed to the systemic circulation, where they are metabolized and excreted. Hydrophilic agents such as morphine cross the dura more slowly than more lipid-soluble agents such as fentanyl or meperidine. This physical chemical property is responsible for the more prolonged duration of action of spinal morphine and its very slow onset of action after epidural administration (Sabbe and Yaksh, 1990).
Biotransformation
Effects of Age and Disease
Morphine, meperidine, methadone, codeine, and fentanyl are biotransformed in the liver before excretion by the kidneys. Many of these reactions are catalyzed in the liver by glucuronidation or microsomal mixed-function oxidases that require the cytochrome P450 system, nicotinamide adenine dinucleotide phosphate (NADPH), and oxygen. The cytochrome P450 system is immature at birth and does not reach adult levels of activity until the first month or two of life. The immaturity of this hepatic enzyme system may explain the prolonged clearance or elimination of some opioids in the first few days to weeks of life (Anderson and Lynn, 2009). On the other hand, the P450 system can be induced by various drugs (such as phenobarbital) and substrates, and once the infant is born, it matures regardless of gestational age. Thus, it is the age from birth and not the duration of gestation that determines how premature and full-term infants metabolize drugs.
Morphine is primarily glucuronidated into two forms: an inactive form, morphine-3-glucuronide, and an active form, morphine-6-glucuronide. Both glucuronides are excreted by the kidney. In patients with renal failure, morphine-6-glucuronide can accumulate and cause toxic side effects including respiratory depression (Murtagh et al., 2007; Lotsch, 2005). This is important to consider not only when prescribing morphine but when administering other opioids that are metabolized into morphine, such as codeine.
Distribution and Clearance
The pharmacokinetics of morphine have been extensively studied in adults, older children, and in premature and full-term newborns (Lynn et al., 1991, 1993). After an intravenous bolus, 30% of morphine is protein-bound in the adult vs. only 20% in the newborn. This increase in unbound (“free”) morphine allows a greater proportion of active drug to penetrate the brain. This may in part explain the observation of Way et al. (1965) of increased brain levels of morphine in the newborn and its more profound respiratory depressant effects. The elimination half-life of morphine in adults and older children is 3 to 4 hours and is consistent with its duration of analgesic action. The half-life of elimination (t½β) is more than twice as long in newborns younger than 1 week of age than in older children and adults and is even longer in premature infants (Lynn and Slattery, 1987). Clearance is similarly decreased in the newborn compared with the older child and adult. Thus, infants younger than 1 month of age attain higher serum levels that decline more slowly than older children and adults. This may also account for the increased respiratory depression associated with morphine in this age group. Interestingly, the t½β and clearance of morphine in children older than 2 months of age are similar to adult values; thus, the hesitancy in prescribing and administering morphine in children younger than 1 year of age may not always be warranted. On the other hand, the use of any opioids in children who were born prematurely (fewer than 37 weeks’ gestation) and are less than 52 to 60 weeks’ postconceptional age or who were born at term and are younger than 2 months of age must be restricted to a monitored setting.
Based on its relatively short half-life (3 to 4 hours), one would expect older children and adults to require morphine supplementation every 2 to 3 hours when being treated for pain, particularly if the morphine is administered intravenously. This has led to the use of continuous infusion regimens of morphine and patient-controlled analgesia (see related section) or, alternatively, administration of longer-acting agonists such as methadone. When administered by continuous infusion, the duration of action of opioids that are rapidly redistributed (such as fentanyl, sufentanil, and alfentanil) and are thought to be short-acting may be longer than would be predicted simply by their pharmacokinetics (see Chapter 7, Pharmacology of Pediatric Anesthesia). An exception to this is remifentanil, a μ-opioid receptor agonist with unique pharmacokinetic properties (Burkle et al., 1996).
The pharmacokinetics of remifentanil are characterized by a small volume of distribution, rapid clearances, and low interpatient variability, compared with other intravenous anesthetic and analgesic agents. The drug has a rapid onset of action (the half-time for equilibration between blood and the effect compartment is 1.3 minutes) and a short context-sensitive half-life (3 to 5 minutes) (Bailey, 2002; Welzing and Roth, 2006). This latter property is attributable to hydrolytic metabolism of the compound by nonspecific tissue and plasma esterases. Virtually all (99.8%) of an administered remifentanil dose is eliminated during the half-life of redistribution t½α (0.9 minutes) and the t½β (6.3 minutes). The pharmacokinetics of remifentanil suggest that within 10 minutes of starting an infusion, remifentanil blood levels will have nearly reached steady state. Thus, changing the infusion rate of remifentanil produces rapid changes in drug effect. The rapid metabolism of remifentanil and its small volume of distribution mean that remifentanil does not accumulate. Discontinuing the drug rapidly terminates its effects and has significant intraoperative implications. When remifentanil is administered intraoperatively as part of a balanced or primary opioid general anesthetic, some patients have reportedly awakened in severe pain. This can be because of inadequate loading of a longer-acting opioid, as well as “opioid-induced hyperalgesia,” a paradoxical process by which opioid administration, even for short periods of time, increases the sensitivity to pain and worsens pain when the opioid is discontinued (Crawford et al., 1992; Chu et al., 2008). Finally, remifentanil may be a reasonable alternative to inhaled general anesthetics in newborn infants undergoing surgery, because it only briefly interferes with the control of breathing and because this effect is terminated shortly after discontinuing the drug (Davis et al., 2001; Galinkin et al., 2001).
Commonly Used Oral Opioids
Codeine, oxycodone (the opioid in Tylox and Percocet), and hydrocodone (the opioid in Vicodin and Lortab) are opioids that are commonly used to treat pain in children and adults, and are quite useful when making the transition from parenteral to enteral analgesia (Table 15-6). Methadone and sustained release formulations of morphine, oxycodone, oxymorphone, and hydromorphone are commonly used to treat chronic medical pain (e.g., cancer) and in postoperative surgical or trauma patients with long recuperative times (e.g., pectus excavatum or posterior spine surgery). Codeine, oxycodone, and hydrocodone are most commonly administered in the oral form, often in combination with acetaminophen. Although the combination drugs are convenient, and acetaminophen potentiates the analgesia produced by codeine—allowing the practitioner to use less opioid to achieve satisfactory analgesia—the combination of drugs significantly increases the risk of acetaminophen toxicity. Acetaminophen toxicity may result from a single toxic dose, from repeated ingestion of large doses of acetaminophen (e.g., in adults 7.5 to 10 g/day for 1 to 2 days, in children 60 to 420 mg/kg per day for 1 to 42 days), or from chronic ingestion. However, hepatotoxicity can also occur inadvertently when patients with poorly controlled pain increase the number of combination tablets they need to control their pain or when they are receiving more than one source of acetaminophen (Heubi et al., 1998). The latter occurs because so many prescription and over-the-counter drug products contain acetaminophen (e.g., cold remedies). Because of this risk, the preferred method is to prescribe opioids and acetaminophen (or ibuprofen) separately.
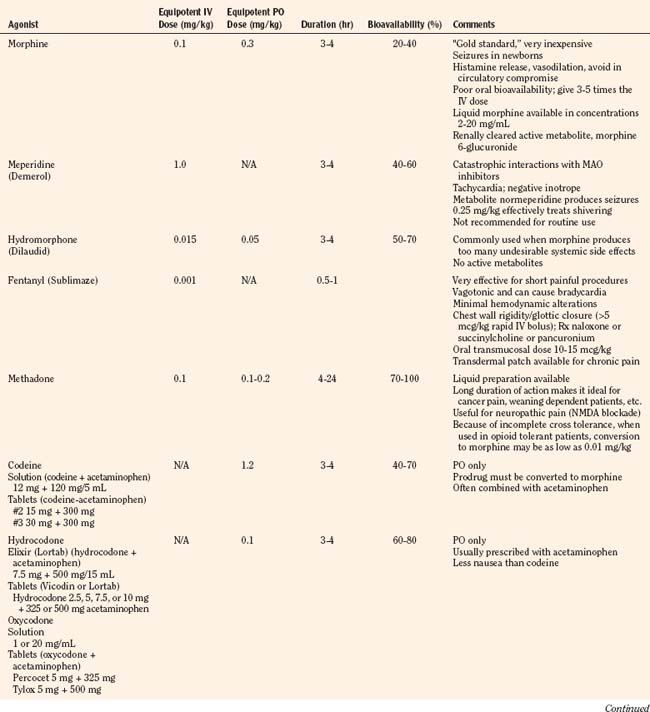
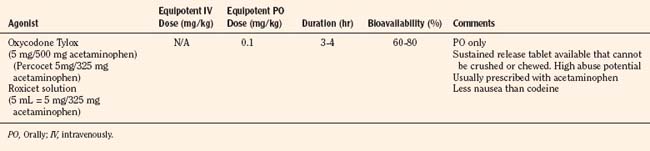
In equipotent doses, oral analgesics have similar effects and side effects including analgesia, sedation, cough suppression, pruritus, nausea, vomiting, constipation and respiratory depression (Table 15-6). Yet the responses of patients to individual opioids can vary markedly, even among these μ-opioid agonists (Pasternak, 2001a, 2005). Understanding this variability greatly enhances the ability to treat patients appropriately. Codeine is a case in point; although readily available, it is very nauseating, and many patients claim they are allergic to it because it so commonly induces vomiting. On the other hand, some differences may be more based on folklore rather than reality. Many physicians falsely believe that meperidine has less of an effect on the sphincter of Oddi than other opioids and therefore prescribe it for patients with gallbladder disease. In fact, meperidine offers no advantage over other opioids and has a serious disadvantage, namely catastrophic interactions with monoamine oxidase (MAO) inhibitors.
Patient-Controlled Analgesia
Historically, pain medications have been administered on a demand or PRN basis (Krane, 2008). When drugs are given PRN, the patient or caregiver must recognize that pain exists, summon a nurse and await the preparation and administration of the analgesic. Even in the best of circumstances, there is a delay between the patient’s request and the provider’s response (Krane, 2008). Around-the-clock administration of analgesics at intervals based on population pharmacokinetics (e.g., every 4 hours) is not always effective because there are enormous individual variations in pain perception and opioid metabolism. Knowledge of opioid pharmacokinetics suggests that intravenous boluses of intermediate-acting opioids such as morphine may be needed as often as every 1 to 2 hours in order to avoid marked fluctuations in plasma drug levels, but generally they are ordered no more frequently than every 4 hours. One way to achieve this goal is via continuous intravenous opioid infusions. This approach provides steady analgesic levels and has been used with great safety and efficacy in children; however, because neither the perception nor the intensity of pain is constant, continuous opioid infusions do not adequately treat pain in all patients (Lynn et al., 2000). For example, a postoperative patient may be very comfortable resting in bed and may require little adjustment in opioid dosing. This same patient may experience excruciating pain when coughing, voiding, or getting out of bed. Receiving the same dose of opioid in both instances may result in either oversedation or under treatment. Thus, rational pain management requires some form of titration to effect whenever any opioid is administered. In order to give patients some measure of control over their pain therapy, analgesia on demand or patient-controlled analgesia (PCA) devices were developed (Berde et al., 1991; Yaster et al., 1997). These are microprocessor driven pumps that the patient controls to self-administer intermittent, predetermined, small doses of opioid whenever a need for more pain relief is felt. The opioid, usually morphine, hydromorphone, or fentanyl is administered either intravenously or subcutaneously (Table 15-7). The dosage of opioid, number of demand doses (“boluses”) per hour, and the time interval between boluses (the “lock-out period”) are programmed into the equipment by the pain-service physician to allow maximum patient flexibility and sense of control with minimal risk of overdosage. Generally, because older patients know that if they have severe pain they can obtain relief immediately, many prefer dosing regimens that result in mild to moderate pain in exchange for fewer side effects such as nausea or pruritus. Morphine is the most commonly prescribed opioid. Typically it is prescribed at a bolus dose of 20 mcg/kg, at a rate of up to 5 boluses/hour, and with a lock-out interval between each bolus of 6 to 8 minutes (Table 15-7). Variations include larger boluses (30 to 50 mcg/kg) and shorter time intervals (5 minutes).
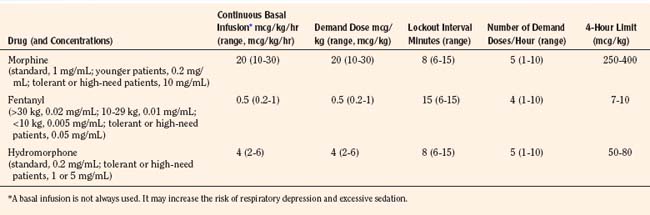
The PCA pump computer stores within its memory how many boluses the patient has received, as well as how many attempts the patient has made at receiving boluses. This allows the provider to evaluate how well the patient understands the use of the pump and provides information to program the pump more efficiently. In addition, most PCA units allow low, continuous “background” infusions (e.g., morphine, 20 to 30 mcg/kg per hour, hydromorphone 3 to 4 mcg/kg per hour or fentanyl 0.5 mcg/kg per hour) in addition to self-administered boluses. Continuous background infusions can facilitate more restful sleep by preventing the patient from awakening in pain (Doyle et al., 1993). Although in adults a background infusion increases the potential for overdosage without significantly improving analgesia, this has not been the experience in pediatrics and in adult cancer patients (Fleming and Coombs, 1992). In these patients, a continuous infusion improves analgesia and makes the life easier for the health care provider, because with better analgesia there are fewer phone calls to rewrite orders or to change therapy (Monitto et al., 1998; Yildiz et al., 2003; Nelson et al., 2009). Contraindications to the use of PCA include inability to push the bolus button (because of weakness or arm restraints), inability to understand how to use the machine, and a patient’s desire not to assume responsibility for personal care. Difficulties with PCA include increased costs; patient age limitations; and the need for physician, nursing, pharmacy protocols, education; and storage arrangements that must be instituted before its implementation. These policies, procedures, and protocols are essential for the safe use of opioids regardless of the method of administration. Essential features include age-appropriate parameters for monitoring respiratory status (e.g., respiratory rate and oxygen saturation), patient alertness, and pain assessment, as well as weight-based dosing. Additionally, because accurate prescribing requires a correct weight, proper conversion of pounds to kilograms, and the choice of an appropriate medication preparation and concentration, the computerization of analgesic medication prescribing is an important patient safety strategy (Wrona et al., 2007; Lee et al., 2008). Figure 15-8 is an example of an electronic, computerized PCA provider order set used in the Children’s Center of the Johns Hopkins Hospital that incorporates many of these features.
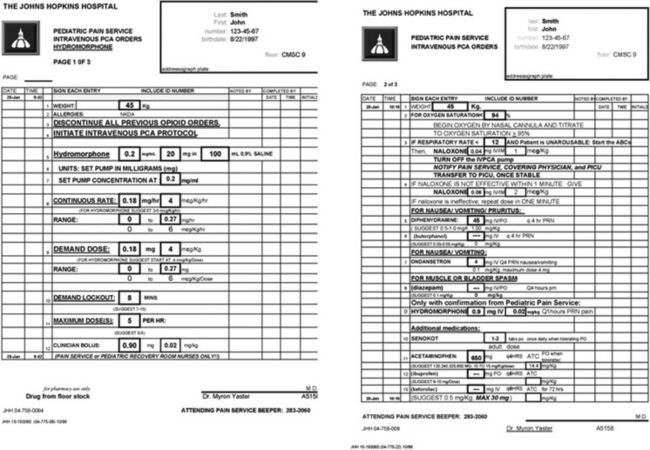
Parent- and Nurse-Controlled Analgesia (Surrogate PCA or PCA by Proxy)
Independent use of PCA requires a patient with sufficient understanding, manual dexterity, and strength to initiate a demand dose. Thus, it was initially limited to adolescents and teenagers, but over time the lower age limit of patients has fallen. In general, many have found that any child able to play a video game can successfully operate a PCA pump independently. However, in very young children or children with developmental or physical handicaps, a similar but alternate mode of therapy is parent- or nurse-controlled analgesia (PNCA), sometimes referred to as PCA by proxy or surrogate PCA (Monitto et al., 2000). When using this technique, the child receives a basal opioid infusion when PCA bolus doses are initiated by a designated surrogate, generally a parent or nurse, when they perceive that the child appears to be in pain (Monitto et al., 2000; Nelson et al., 2009).
Allowing parents or nurses to initiate a PCA bolus is controversial. In 2004, the Joint Commission on Accreditation of Healthcare Organizations (JCAHO) issued a sentinel event alert warning that serious adverse events can result when surrogates become involved in administering analgesia by proxy (Joint Commission on Accreditation of Healthcare Organizations, 2004). Of note, the JCAHO based this warning on a series of adverse events in adults who for the most part developed complications as a result of the preemptive use of the PCA bolus by spouses, children, or nurses when the patient was asleep. Although this alert was not meant to address cases in which caregivers were authorized to administer PCA boluses, it nevertheless raised serious concerns in the pediatric pain management community and changed the practice at some hospitals. Published studies have reported that up to 1% to 3% of patients receiving intravenous PCA by proxy may receive naloxone to treat cardiopulmonary complications (Monitto et al., 2000; Anghelescu et al., 2005; Voepel-Lewis et al., 2008). Interestingly, in a study comparing pediatric intravenous PCA and intravenous PCA by proxy, complication rates were similar between the two modes of therapy, but the use of naloxone was higher in the younger patients. However, it was unclear whether providers had a lower threshold for treating these children with naloxone given their younger age and an associated increased incidence of comorbidities (Voepel-Lewis et al., 2008). Because respiratory depression does occur, safe institution of PCA by proxy as well as PCA requires close patient monitoring, established nursing protocols, and an understanding by surrogates of the appropriate use of the bolus dose.
Transdermal and Transmucosal Fentanyl
Because fentanyl is extremely lipophilic, it can be readily absorbed across any biological membrane, including the skin. Thus, fentanyl can be administered painlessly by nonintravenous routes, including transmucosal (nose and mouth) and transdermal routes. The transmucosal route of fentanyl administration is extremely effective for acute pain relief. When given intranasally (2 mcg/kg), it produces rapid analgesia that is equivalent to intravenously administered fentanyl (Galinkin et al., 2000). For transoral absorption, fentanyl is also available in a candy matrix (Actiq) attached to a plastic applicator device that looks like a lollipop. As the patient sucks on the lollipop, fentanyl is absorbed across the buccal mucosa and is rapidly absorbed (10 to 20 minutes) into the systemic circulation (Goldstein-Dresner et al., 1991; Streisand et al., 1989, 1991; Ashburn et al., 1993; Schechter et al., 1995). If excessive sedation occurs, the fentanyl is removed from the patient’s mouth by the applicator. Transmucosal absorption is more efficient than ordinary oral-gastric intestinal administration because it bypasses the efficient first-pass hepatic metabolism of fentanyl that occurs after enteral absorption into the portal circulation. The candy matrix of fentanyl has been approved by the FDA for use in children for premedication before surgery and for procedure-related pain (e.g., lumbar puncture or bone marrow aspiration) (Dsida et al., 1998). It is also useful in the treatment of cancer pain and as a supplement to transdermal fentanyl (Portenoy et al., 1999). When administered transmucosally, a fentanyl dose of 10 to 15 mcg/kg is effective within 20 minutes and lasts approximately 2 hours. Because approximately 25% to 33% of the given dose is absorbed, blood levels equivalent to 3 to 5 mcg/kg intravenous fentanyl are achieved with this dose. The major side effect of treatment is nausea and vomiting, which occurs in approximately 20% to 33% of patients who receive it (Epstein et al., 1996). Finally, a new, rapidly dissolving, effervescent fentanyl buccal tablet, Fentora, has become available for breakthrough pain in patients who are already receiving opioids for persistent pain and who are tolerant to opioid therapy (Messina et al., 2008; Weinstein et al., 2009). These tablets come in various doses (100, 200, 400, 600, and 800 mcg). In adults, fentanyl buccal tablets have an absolute bioavailability of 65%; approximately 50% of the total administered dose is absorbed transmucosally, and the remaining half is swallowed and undergoes slow absorption from the gastrointestinal tract. There is little published pediatric experience with this drug.
The transdermal route is commonly used to administer many drugs, including scopolamine, clonidine, and nitroglycerin. Many factors, including body site, skin temperature, skin damage, ethnicity, and age affect the absorption of transdermally administered drugs. The transdermal fentanyl patch has revolutionized adult and pediatric cancer pain management (Zernikow et al., 2007; Finkel et al., 2005). Placed in a selective semipermeable membrane patch, which is attached to the skin by a contact adhesive, a reservoir of fentanyl provides slow, steady-state absorption of drug across the skin. As fentanyl is painlessly absorbed across the skin, a substantial amount is stored in the upper skin layers, which then act as a secondary reservoir. The presence of this “skin depot” has several implications:
Indeed, the amount of fentanyl remaining within the system and skin depot after removal of the patch is substantial. At the end of a 24-hour period, a fentanyl patch releasing drug at the rate of 100 mcg/hr, 1.07 ± 0.43 mg fentanyl (approximately 30% of the total delivered dose from the patch) remains in the skin depot. Thus, even after removal of the patch, fentanyl continues to be absorbed from the subcutaneous fat reservoir for almost 24 hours (Grond et al., 2000).
Because of its long onset time, inability to rapidly adjust drug delivery, and long t½β, transdermal fentanyl is contraindicated for acute pain management. In fact, the use of this drug delivery system to treat acute pain has resulted in the death of an otherwise healthy patient. Transdermal fentanyl is appropriate only for patients who have developed opioid tolerance and for those with chronic pain (e.g., cancer) (Zernikow et al., 2007; Finkel et al., 2005). Even when transdermal fentanyl is appropriate, the vehicle imposes its own constraints; the patch with the lowest dose delivers 12.5 mcg of fentanyl per hour; others deliver 25, 50, 75, and 100 mcg of fentanyl per hour. The patches cannot be physically cut in smaller pieces to deliver less fentanyl.
Methadone
Primarily thought of as a drug to treat or wean patients who are addicted to or dependent on opioid, methadone is increasingly being used in the management of acute and chronic intractable pain. Unique among the opioids, methadone exists as a racemic mixture of two active isomers; one isomer binds as an agonist to the μ-receptor and the other as an antagonist at the NMDA receptor. It is this latter property that makes methadone unique among opioids. The NMDA system is involved in wind-up and the maintenance of chronic pain as well as opioid-induced hyperalgesia and the development of tolerance (Ebert et al., 1998; Gagnon and Bruera, 1999; Gorman et al., 1997). Thus, blockade of NMDA receptors can act to acutely enhance opioid-induced antinociception, impair the development of tolerance, and prevent the development of chronic pain (Raffa, 1996).
In addition, methadone is noted for its slow t½β, very long duration of effective analgesia, high oral bioavailability, and inactive metabolites. The t½β of methadone averages 19 hours, and clearance averages 5.4 mL/kg per minute in children 1 to 18 years of age (Berde et al., 1991). Methadone has the longest t½β of any of the commonly available opiates and can provide up to 12 to 36 hours of analgesia after a single intravenous or oral dose (Gourlay et al., 1986, 1982, 1984; Berde 1989; Shannon and Berde, 1989). Pharmacokinetically, children are indistinguishable from young adults. Because a single dose of methadone can achieve and sustain a high drug-plasma level, it is a convenient way to provide prolonged analgesia without requiring an intramuscular injection or a continuous infusion. Berde et al. (1989) recommend methadone as a “poor man’s PCA” and suggest loading patients with an initial dose of intravenous methadone (0.1 to 0.2 mg/kg) and then titrating in 0.05-mg/kg increments every 10 to 15 minutes until analgesia is achieved. Supplemental methadone can be administered in 0.05- to 0.1-mg/kg increments administered by slow intravenous infusion every 4 to 12 hours as needed. Shannon and Berde (1989) have also reported the use of small incremental doses administered by sliding scale. “Small increments of methadone are administered intravenously over 20 minutes every 4 hours via a ‘sliding’ scale on a ‘reverse prn’ (the nurse asks the patient) basis: 0.07 to 0.08 mg/kg for severe pain; 0.05 to 0.06 mg/kg for moderate pain; 0.03 mg/kg for little or no pain, if the patient is alert; and no drug if the patient has little pain and is somnolent” (Berde 1989; Shannon and Berde, 1989).
In addition, both methadone and sustained relief morphine can be used to wean patients who have become physically dependent on opioids after prolonged analgesic therapy (Yaster et al., 1996; Suresh and Anand, 1998; Tobias, 2000a). Finally, because methadone is extremely well absorbed from the gastrointestinal tract and has a bioavailability of 80% to 90%, it is extremely easy to convert intravenous dosing regimens to oral ones. Recently, however, the conversion dose of morphine to methadone has been challenged. Traditionally, it has been thought that the ratio of morphine to methadone was approximately 1:1; it now appears that when tolerance develops and morphine doses are “high,” it is closer to 1:0.25 or even 1:0.1 (Lawlor et al., 1998; Ripamonti et al., 1998a, 1998b; Gagnon and Bruera, 1999). Tolerance to morphine and other opioids such as fentanyl is a significant problem in patients being treated for pain chronically or acutely in the intensive care unit setting. When this occurs, substituting methadone for morphine or fentanyl at the lower doses discussed in the previous section can rapidly reestablish analgesia even though all of these opioids work at the same μ-opioid receptor. This occurs because of “incomplete cross tolerance,” which may be because methadone’s antagonist actions at the NMDA receptor or because of multiple μ-receptor subtypes (Gorman et al., 1997; Ebert et al., 1998; Trujillo and Akil, 1991; Pasternak, 2001a).
Among opioid analgesics, methadone is unique as a potent blocker of the delayed rectifier potassium ion channel. This results in QT prolongation, can produce torsade de pointes ventricular tachycardia in susceptible individuals, and may explain the sudden death associated with its use (Andrews et al., 2009). The effects of methadone on the QT interval may be enhanced by hypokalemia, drugs that increase the QT interval such as erythromycin and ondansetron, or by CYP 3A4 inhibitors such as fluoxetine, fluconazole, valproate, and clarithromycin (Ehret et al., 2006). An updated list of medications causing torsade de pointes ventricular tachycardia can be found at www.azcert.org. Indeed, this is such a serious consequence of therapy that some have recommended that all patients treated with methadone have routine screening ECGs before or during their treatment.
Tramadol
Tramadol, a synthetic 4-phenylpiperidine analogue of codeine, is a centrally acting synthetic analgesic that has been used for 30 years in Europe and was approved by the FDA for adult use in the United States in 1995 (Raffa 1996; Minto and Power, 1997). It is a racemic mixture of two enantiomers: + tramadol and –tramadol (Berde, 1989; Glare and Lickiss, 1992). The +enantiomer has a moderate affinity for the µ-opioid receptor that is greater than that of the −enantiomer (Raffa, 1993). In addition, the +enantiomer inhibits 5-HT uptake and the –enantiomer blocks the reuptake of NE, complementary properties that result in a synergistic antinociceptive interaction between the two enantiomers. Tramadol may also produce analgesia as an α2-agonist (Desmeules et al., 1996). A metabolite (O-desmethyltramadol) binds to opioid receptors with a greater affinity than the parent compound and could contribute to tramadol’s analgesic effects as well. However, in most animal tests and human clinical trials, the analgesic effect of tramadol is only partially blocked by the opioid antagonist naloxone, suggesting an important nonopioid mechanism. Thus, tramadol provides analgesia synergistically by opioid (direct binding to the μ-opioid receptor by the parent compound and its metabolite) and nonopioid mechanisms (an increase in central neuronal synaptic levels of 5-HT and NE).Finally, animal and human studies have suggested that tramadol may have a selective spinal and local anesthetic action on peripheral nerves. Tramadol has been shown to provide effective, long-lasting analgesia after extradural administration in both adults and children and prolongs the duration of action of local anesthetics when used for brachial plexus and epidural blockade (Kapral et al., 1999; Prosser et al., 1997).
Tramadol’s intravenous analgesic effect has been reported to be 10 to 15 times less than that of morphine and is roughly equianalgesic with NSAIDs (Raffa, 1996; Naguib et al., 1998). Unlike NSAIDs and opioid mixed agonist/antagonists, the therapeutic use of tramadol has not been associated with the clinically important side effects such as respiratory depression, constipation, or sedation. In addition, analgesic tolerance has not been a serious problem during repeated administration, and neither psychological dependence nor euphoric effects are observed in long-term clinical trials. Thus, tramadol may offer significant advantages in the management of pain in children by virtue of its dual mechanism of action, its lack of a ceiling effect, and its minimal respiratory depression.
Tramadol may be administered orally, rectally, intravenously, or epidurally (Gunes et al., 2004; Bozkurt, 2005). Oral and intravenous tramadol is administered in doses of 1 to 2 mg/kg; the higher dose provides a longer duration of action without increasing side effects (Finkel et al., 2002; Rose, 2003; Bozkurt, 2005).
Complications of opioid therapy
Regardless of the method of administration, all opioids commonly produce unwanted side effects such as pruritus, nausea and vomiting, constipation, urinary retention, cognitive impairment, tolerance, and dependence (Yaster et al., 2003). Many patients suffer needlessly from pain in order to avoid these debilitating induced side effects (Watcha and White, 1992). Additionally, physicians are often reluctant to prescribe opioids because of these side effects and because of their fear of other, less common but more serious side effects such as respiratory depression. Several clinical and laboratory studies have demonstrated that low-dose naloxone infusions (0.25 to 1 mcg/kg per hour) can treat or prevent opioid-induced side effects without affecting the quality of analgesia or opioid requirements in adults, children, and adolescents (Maxwell et al., 2005; Gan et al., 1997).
Opioid-Induced Bowel Dysfunction
Opioid-induced bowel dysfunction (OBD), often described as constipation, is a constellation of symptoms that includes delayed gastric emptying, slow bowel motility, incomplete evacuation, bloating, abdominal distention, and gastric reflux. OBD occurs whether opioids are administered acutely or chronically, is found in 90% of patients treated with opioids, and is a significant problem in 50% to 60% of adult patients with advanced cancer (Glare and Lickiss, 1992; Fallon and Hanks, 1999). OBD is not really a side effect of opioid therapy; rather the effects of opioids on gastric emptying, peristalsis, and bowel motility are intrinsic opioid actions. Indeed, opium has been used in the treatment of dysentery for thousands of years. However, unlike the analgesic effects of opioids, the gastrointestinal ones are not accompanied by the development of tolerance.
Therefore, patients treated with opioids, regardless of drug, route, or method of delivery, should be considered for a prophylactic bowel regimen of stool softeners and bulking agents (e.g., senna and lubiprostone) as soon as the patient can eat or drink. Indeed, there may be value in starting these agents preoperatively. Alternatively, several new peripherally-acting opioid antagonists, methylnaltrexone and alvimopan, have recently been approved by the FDA and may be of great utility (Moss and Rosow, 2008). These medications offer promise in the treatment of OBD in patients with late-stage advanced illness (methylnaltrexone), as well as postoperative ileus in adult patients (alvimopan). However, neither of these medications has been studied or approved for use in children. If these conservative measures fail, then stimulant laxatives (e.g., polyethylene glycol 3550) and enemas should be added to this regimen.
Opioid-Induced Pruritus
Opioid-induced pruritus (OIP) is one of the most common adverse side effects associated with opioid use. The incidence of OIP varies between 20% and 100% when opioids are administered neuroaxially (intrathecally or epidurally) and between 20% to 60% when administered intravenously (Ganesh and Maxwell, 2007). Surprisingly, the pathophysiology of clinical itch remains unclear. Over 300 years ago, the German physician Samuel Hafenreffer described itch as an “unpleasant sensation that elicits the desire to scratch,” and this definition is still valid today. Much of the current research on itch focuses on identifying the neuronal mechanisms and the mediators responsible for itch in dermatological and systemic disease. OIP is primarily mediated by binding of μ-agonists with central μ-opioid receptors in the brain and spinal cord and can be blocked with centrally acting μ-receptor antagonists (Ko and Naughton, 2000). Binding of the dopamine D2 receptor and the release of prostaglandin E1 and E2 have also been implicated in the development of OIP. Blockade of the D2 receptor by antagonists such as droperidol or by inhibiting prostaglandin production with NSAIDs significantly reduces OIP (Horta et al., 1996). Whereas histamine is a potent pruritic agent, opioid-induced histamine release from mast cells plays almost no part in OIP.
A wide variety of drugs with different mechanisms of action have been used to treat or prevent OIP. Antihistamines, such as diphenhydramine or hydroxyzine are the most common and perhaps least effective drugs used to treat established OIP. They primarily interrupt the itch-scratch cycle by providing needed sleep but are not really effective at reducing the severity of itch. A more effective approach is to prophylactically administer a low-dose intravenous infusion of the opioid antagonist naloxone (0.25 to 1 mcg/kg per hour) or the partial agonist/antagonist nalbuphine (50 mcg/kg, maximum 5 mg/dose) (Kendrick et al., 1996; Maxwell and Yaster, 2003; Nakatsuka et al., 2006). Other strategies involve rotating opioids, reducing opioid dose, or switching to the oral route of administration.
Opioid-Induced Nausea and Vomiting
A wide variety of drugs with different mechanism of actions have been used to treat or prevent opioid-induced nausea and vomiting (Watcha and White, 1992). The most commonly used antiemetics are antihistamines (diphenhydramine), phenothiazines (prochlorperazine and promethazine), butyrophenones (haloperidol and droperidol), benzamides (metoclopramide), 5-HT3-receptor antagonists (ondansetron, dolasetron, granisetron), and dexamethasone. A low-dose intravenous naloxone infusion (1 mcg/kg per hour) has also been shown to be effective (Maxwell and Yaster, 2003). Many of the agents listed above work synergistically. Although it does not make sense to combine two drugs that act via the same mechanism (e.g., two antihistamines such as diphenhydramine and hydroxyzine), combinations of drugs that act via different mechanisms (e.g., ondansetron, a selective inhibitor of 5-HT3 receptors and the steroid dexamethasone) can be effective (McKenzie et al., 1994). Finally, if these measures fail, opioid dose reduction, with or without the addition of NSAIDs, or opioid rotation may be effective.
Opioid-Induced Sedation
The sedating effects of opioids in opioid-naive patients are well known and can be thought of as either a desired effect or an adverse side effect. Although tolerance to sedation and drowsiness often develop, in some patients sedation and mental clouding are so debilitating that it leads to a reduced quality of life. Treatment is often dose reduction, opioid rotation, or the use of psychostimulants. Methylphenidate is the most commonly used stimulant to improve drowsiness, but others include dextroamphetamine, caffeine, and modafinil (Rozans et al., 2002; Prommer, 2006).
Tolerance, Dependence, and Withdrawal
Tolerance is the development of a need to increase the dose of an opioid (or benzodiazepine) agonist to achieve the same analgesic (or sedative) effect previously achieved with a lower dose (Nutt, 1996; Wise, 1996). Whereas tolerance to the sedative and analgesic effects of opioids usually develops after 5 to 21 days of morphine administration, tolerance to the constipating effects of opioids rarely occur. Additionally, cross-tolerance develops between all μ-opioid receptor agonists. However, because this cross tolerance is rarely complete, opioid rotation, that is, changing from one opioid (morphine, fentanyl, or hydromorphone) to another (usually methadone) can be helpful in preventing a continuous escalation in analgesic dosing. When it is necessary to switch, careful consideration must be given to the choice of opioid, dose, and expected degree of cross-tolerance. For example, when switching from high-dose morphine to methadone in patients who are tolerant of opioid, the equianalgesic dose is decreased by a factor of four- to fivefold. Even with this reduction, the calculated dose of methadone may be so high that it warrants a stepwise conversion while the patient remains in a high-surveillance care unit.
Physical dependence, sometimes referred to as neuroadaptation, is caused by repeated administration of an opioid that necessitates the continued administration of the drug to prevent the development of a withdrawal or abstinence syndrome characteristic for that particular drug (O’Brien, 1996). Physical dependence usually occurs after 2 to 3 weeks of morphine administration, but when high doses of opioid are administered it may occur after only a few days of therapy. Very young infants treated with high-dose fentanyl infusions after surgical repair of congenital heart disease and those who require extracorporeal membrane oxygenation (ECMO) have been identified to be at particular risk of developing dependence and withdrawal on discontinuation of therapy (Arnold et al., 1991, 1990; Kauffman 1991; Lane et al., 1991).
Physical dependence must be differentiated from addiction (O’Brien, 1996). Addiction is a term used to connote a severe degree of drug abuse and dependence that is an extreme of behavior in which drug use pervades the total life activity of the user and of the range of circumstances in which drug use controls the user’s behavior. Patients who are addicted to opioids often spend large amounts of time acquiring or using the drug, abandon social or occupational activities because of drug use, and continue to use the drug despite adverse psychological or physical effects. In a sense, addiction is a subset of physical dependence. Anyone who is addicted to an opioid is physically dependent; however, not everyone who is physically dependent is addicted. Patients appropriately treated with opioid agonists for pain can become tolerant and physically dependent. They rarely become psychologically dependent or addicted (Porter and Jick, 1980).
When physical dependence has been established, sudden discontinuation of an opioid or benzodiazepine agonist produces a withdrawal syndrome within 24 hours of drug cessation. Symptoms reach their peak within 72 hours and include abdominal cramping, vomiting, diarrhea, tachycardia, hypertension, diaphoresis, restlessness, insomnia, movement disorders, reversible neurologic abnormalities, and seizures (O’Brien, 1996; Anand and Arnold, 1994; Katz et al., 1994; Nestler 1994, 1996; Norton 1988).
Clinical and experimental data suggest that the duration of opioid receptor occupancy is an important factor in the development of tolerance and dependence. Thus, continuous infusions may produce tolerance more rapidly than intermittent therapy (Katz et al., 1994; Anand and Arnold, 1994). This is particularly true for highly lipid-soluble opioids such as fentanyl. Tolerance and dependence predictably develops after only 5 to 10 days (2.5 mg/kg total fentanyl dose) of continuous fentanyl infusions (Arnold et al., 1991, 1990; Anand and Arnold, 1994; Katz et al., 1994). Nevertheless, prolonged therapy in excess of 10 days, even by intermittent bolus administration, should be expected to produce opioid dependence. As a result, tolerance and physical dependence to both opioid and benzodiazepines is a common phenomenon in the intensive care unit (Suresh and Anand, 1998; Anand and Arnold, 1994; O’Brien, 1996; Yaster et al., 1996; Koob and Nestler, 1997; Tobias, 2000a).
Withdrawal Scales and Weaning Strategies
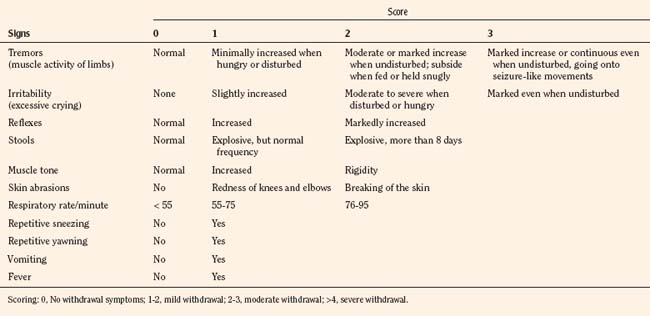
Withdrawal and Abstinence Scales for Infants and Children
The most widely used tool to assess neonatal abstinence is the Finnegan scale (Table 15-8). This tool is also among the most commonly used withdrawal scales in older infants and children, even though it has never been validated for this use. The Finnegan scale is a complicated assessment measure that uses a weighted scoring of 31 items that requires training and when used clinically, some assessment of interrater reliability. As a result, it may be too complicated for routine use. An alternative, the Lipsitz scale, offers the advantage of being a relatively simple numerical system, with a reported 77% sensitivity using a value greater than 4 as an indication of significant signs of withdrawal (Table 15-9) (Lipsitz, 1975). The Lipsitz scale and the need for a scoring system to guide therapy was recommended by the American Academy of Pediatrics (AAP) in a 1998 consensus statement (AAP Committee on Drugs, 1998).
| Sign/Symptoms | Score |
| Cry | |
| Excessive | 2 |
| Continuous | 3 |
| Sleep (# hours after feeding) | |
| <1 Hour | 3 |
| <2 Hours | 2 |
| <3 Hours | 1 |
| Moro Reflex | |
| Hyperactive | 2 |
| Markedly hyperactive | 3 |
| Tremors | |
| Mild | 1 |
| Moderate-severe | 2 |
| Moderate-severe when undisturbed | 3 |
| Increased tone | 2 |
| Frequent yawning | 2 |
| Sneezing | 1 |
| Nasal congestion | 1 |
| Nasal flaring | 2 |
| Respiratory Rate | |
| >60 Breaths per minute | 1 |
| >60 Breaths per minute with retractions | 2 |
| Excoriation | 1 |
| Seizures | 5 |
| Sweating | 1 |
| Fever | |
| 100-101oF | 1 |
| >101oF | |
| Mottling | 1 |
| Excessive sucking | 1 |
| Poor feeding | 2 |
| Regurgitation | 2 |
| Projectile vomiting | 3 |
| Stooling | |
| Loose | 2 |
| Watery | 3 |
Scoring: 0-7, Mild symptoms of withdrawal; 8-11, moderate withdrawal; 12-15, severe withdrawal.
When pharmacologic treatment is needed to treat withdrawal, the AAP recommends dilute tincture of opium for neonatal opiate withdrawal. For sedative-hypnotic withdrawal, phenobarbital is the agent of choice. Agthe and others (2009) demonstrated that when tincture of opium is supplemented with oral clonidine (1 mcg/kg) every 4 hours, the duration of pharmacotherapy for neonatal abstinence syndrome is dramatically reduced. However, despite clear, evidence-based recommendations from the AAP, the management of the newborn with psychomotor behavior consistent with withdrawal varies widely. In a recently published survey of neonatal withdrawal treatment, Sarkar and Donn (2006) found inconsistent policies, scale utilization, and treatment regimens between institutions and individual physicians. These results reflect similar findings of earlier studies and reemphasize the disparity between the published evidence and recommendations supporting the use of withdrawal scoring and current clinical practice for neonatal withdrawal treatment.
Franck et al. (2004) investigated the use of an adapted neonatal assessment tool to older children. This 21-item checklist was initially used for opioid weaning and modified for the evaluation of opioid and benzodiazepine withdrawal symptoms (Franck et al., 2004). Their small study demonstrated good interrater reliability and content validity of the tool, as well as applicability to a wide range of ages (6 to 28 months). Thus, this withdrawal scale, named the Opioid and Benzodiazepine Withdrawal Score (OBWS), has a wide range of applicability. Adult withdrawal assessment tools often involve personal reporting of symptoms by the patient. Recently, a clinician-administered tool (the Clinical Opiate Withdrawal Scale [COWS]) was developed to provide a simplified 11-question score to assess withdrawal symptoms and to review its applicability in iatrogenic and abuse-related opioid withdrawal scenarios (Wesson and Ling, 2003). Although simplistic and promising, its applicability to other agents (such as benzodiazepines) and to the pediatric age group is unknown.
Weaning Strategies in Infants and Children
As previously discussed, tolerance and physical dependence develop as a result of the drug, dose, method of delivery, and duration of therapy. When the risk of withdrawal is high, weaning patients should be slow (Yaster et al., 1997, 1996). Unfortunately, abrupt withdrawal of opioids and sedatives to facilitate extubation and transfer of patients out of the PICU is commonplace. If sedative or opioid use has been of short duration (i.e., less than 72 hours), acute discontinuation is reasonable. If a patient has required infusions or repeated administration of an agent for more than 5 days, then an agent-specific weaning strategy should be employed.
One approach to the weaning process is to convert all of the patient’s analgesic and sedative medications to intermittent parenteral therapy whenever possible. All forms of the drugs being used therapeutically must be counted in this conversion, including PRN medications. Furthermore, because it is quite common for patients to receive multiple opioids and sedatives, all of the opioids should be converted to morphine equivalents and the benzodiazepines to diazepam equivalents (Yaster et al., 1996). How to proceed with weaning beyond this first step is not always clear, and there are no published evidence-based studies.
The α2-adrenergic agents help prevent or mitigate the occurrence of drug withdrawal regardless of the drug causing addiction or dependence. Agthe and others (2009) have reported the use of clonidine in treating infants born to drug-addicted mothers as well as in patients who have become opioid and sedative dependent as a result of pain or sedation therapy. If tolerated hemodynamically, coadministered oral or transdermal clonidine (6 to 12 mcg/kg per day) can be used to ameliorate the signs and symptoms of withdrawal during the weaning process. In this instance, the clonidine is subsequently weaned. In addition, the use of dexmedetomidine has been reported to prevent withdrawal symptoms in patients dependent on opioids and sedatives (Finkel and Elrefai, 2004; Multz, 2003).
Analgesic adjuvants
Adjuvant pain medications are drugs with a primary function that is not to treat pain, but that may have analgesic properties in specific circumstances. Many of the drugs that are discussed in the following section were initially used to treat neuropathic and chronic pain but are now increasingly being used to treat acute pain as a part of a multimodal therapeutic regimen (Fig. 15-3).
Antidepressants
Because 5-HT and NE mediate descending inhibition of ascending pain pathways in the brain and spinal cord (Figs. 15-2 and 15-3), 5-HT and NE reuptake inhibitor antidepressant medications may have efficacy in relieving pain (Saarto and Wiffen, 2007). Antidepressants that enhance NE action are more effective analgesics than those that predominantly enhance 5-HT action, such as with many of the newer antidepressants (Saarto and Wiffen, 2007). Older antidepressants, particularly the tricyclic antidepressants (TCAs) such as amitriptyline, doxepin, and nortriptyline have been the most thoroughly studied and are thought to cause analgesia by NE and 5-HT reuptake inhibition (Wiffen et al., 2005). They also have other pharmacologic properties that may contribute to analgesia, such as reducing sympathetic activity, NMDA-receptor antagonism, anticholinergic activity, and sodium-channel blockade. Although generally administered orally, Collins et al. (1995) report using intravenous amitriptyline in eight children who could not tolerate oral medications. Newer non-TCAs seem to be less efficacious analgesics. Ironically, this may be in part because of their “cleaner” pharmacodynamic profiles. Of the newer antidepressants, duloxetine, a dual inhibitor of 5-HT and NE, has been shown to be effective in several randomized controlled trials in adult patients with fibromyalgia and other chronic pain conditions even in the absence of major depressive disorders (Arnold et al., 2009).
Antiepileptic Agents
Like the TCAs, antiepileptic adjuvant analgesics suffer an unfortunate name. Most families (and physicians who are unaware of their analgesic properties) question the use of an antiepileptic drug in a child who does not have seizures. Conceptually, these agents work by preventing “peripheral seizures” in the form of pathologic peripheral nerve discharge (Tanelian and Brose, 1991; Kingery, 1997; Rizzo, 1997). Carbamazepine is the most widely studied antiepileptic in the management of neuropathic pain, particularly in the treatment of lancinating neuropathic pain (such as pain caused by nerve-root compression or injury to a discrete peripheral nerve). Antiepileptic drugs can also help painful “glove and stocking” neuropathic conditions, disagreeable paresthesias, and intense sensitivity to innocuous stimuli (as seen in some human immunodeficiency virus [HIV] neuropathies and chemotherapeutic nerve injuries). Especially in oncology patients, carbamazepine’s propensity for drug-drug interactions and risk of blood dyscrasias are of concern. As a result, these drugs have largely been replaced by gabapentin and pregabalin, an interesting class of weak anticonvulsant drugs that bind at voltage-gated–calcium-channel α2-δ (Cav-α2-δ) proteins (Taylor, 2009). Interestingly, despite their names, these drugs are not GABAergic and produce analgesia by reducing the presynaptic release of pain-inducing neurotransmitters such as glutamate, NE, substance P, and CGRP in the spinal cord and CNS.
Gabapentin and pregabalin have been most widely studied and used for the treatment of chronic pain conditions such as postherpetic neuralgia, diabetic neuropathy, complex regional pain syndromes, malignant pain, HIV-related neuropathy, and headaches. Increasingly, they are being used in the perioperative period as a component of multimodal pain therapy (see Fig. 15-3) (Joshi, 2005; Ho et al., 2006; Kong and Irwin, 2007; White, 2008). Adult studies have demonstrated their effectiveness (1200 mg gabapentin, 300 mg pregabalin orally) at enhancing postoperative analgesia and preoperative anxiolysis, preventing chronic postsurgical pain, attenuating the hemodynamic responses to laryngoscopy and intubation, and reducing postoperative delirium (Ho et al., 2006). The main side effect of both drugs is somnolence.
Alpha2-Adrenergic Agonists: Clonidine, Tizanidine, and Dexmedetomidine
NE is involved in the control of pain by modulating pain-related responses through various pathways (Fig. 15-3). α2-Adrenergic agonists, such as clonidine, tizanidine, and dexmedetomidine, have well-established analgesic and sedative profiles and wide application in perioperative multimodal pain management. Clonidine is the prototype and most widely studied of this class of drugs. It can be administered via the epidural, oral, and transdermal routes. Clonidine is traditionally used as an antihypertensive and to minimize the symptoms of opioid withdrawal (Agthe, 2009). However, when administered orally, intravenously, or transdermally, clonidine may reduce opioid requirements and improve analgesia. Similarly, the addition of clonidine to local anesthetic solutions for neuraxial or peripheral nerve blocks may enhance and prolong analgesia. However, the analgesic benefits of clonidine remain controversial. Finally, clonidine can be a useful antineuropathic agent, especially in children who cannot tolerate oral medications or who have coexisting problems like steroid-induced hypertension (Kingery, 1997). Clonidine is empirically started at 1 to 2 mcg/kg per dose, every 8 hours, and increased incrementally over days to doses up to 4 mcg/kg per dose. Alternatively, a transdermal patch can be applied in order to administer 6 to 12 mcg/kg per day. Clonidine use is limited by its side effects, which include bradycardia, hypotension, and excessive sedation (Joshi, 2005; White, 2008).
N-Methyl-D-Aspartate Receptor Antagonists
NMDA receptor antagonists, such as ketamine and methadone, are important modulators of chronic pain and have been shown in some studies to be useful in preventive analgesia by reducing acute postoperative pain, analgesic consumption, or both when they are added to more conventional means of providing analgesia, such as opioids and NSAIDs, in the perioperative period (Fig. 15-3) (McCartney et al., 2004). NMDA receptor antagonists may reduce pain by two nonmutually exclusive mechanisms: a reduction in central hypersensitivity and a reduction of opioid tolerance. Nevertheless, the effectiveness of NMDA receptor antagonists in preventive analgesia has been equivocal at best (McCartney et al., 2004; Pogatzki-Zahn and Zahn, 2006). Ketamine is well known as a dissociative general anesthetic and may be an effective adjuvant in pain management when used in low doses (0.05 to 0.2 mg/kg per hour) (Tsui et al., 2007; Sveticic et al., 2008).
Regional anesthesia and analgesia
Overview
Since the late twentieth century, the use of local anesthetics and regional anesthetic techniques in pediatric practice has increased dramatically. Unlike most drugs used in medical practice, local anesthetics must be physically deposited at their sites of action by direct application and require patient cooperation and the use of specialized needles. Because of this, for decades children were considered poor candidates for regional anesthetic techniques. However, once it was recognized that regional anesthesia could be used as an adjunct and not a replacement for general anesthesia, its use increased dramatically. Regional anesthesia offers the anesthesiologist and pain specialist many benefits. It modifies the neuroendocrine stress response, provides profound postoperative pain relief, insures a more rapid recovery, and may shorten hospital stay with fewer opioid-induced side effects. Furthermore, because catheters placed in the epidural, pleural, femoral, sciatic, brachial plexus, and other spaces can be used for days or months, local anesthetics are increasingly being used not only for postoperative pain relief, but also for medical, neuropathic, and terminal pain (Dalens, 1989; Yaster and Maxwell, 1989; Giaufre et al., 1996; Golianu et al., 2000; Ross et al., 2000; Capdevila et al., 2003; Dadure et al., 2003). Peripheral nerve blocks provide significant pain relief after many common pediatric procedures. Techniques range from simple infiltration of local anesthetics to neuraxial blocks like spinal and epidural analgesia. To be used safely, a working knowledge of the differences in how local anesthetics are metabolized in infants and children is necessary (Table 15-10) (Dalens, 1989, 1995; Yaster et al., 1993).
Effects of Age on Metabolism of Local Anesthetics
All local anesthetics in current use are either amino amides or amino esters and achieve their intended effect by blocking gated sodium channels. The ester local anesthetics are metabolized by plasma cholinesterase. Neonates and infants up to 6 months of age have less than half of the adult levels of this plasma enzyme. Theoretically, clearance may be reduced and the effects of ester local anesthetics prolonged. In reality this is never the case. Amides, on the other hand, are metabolized in the liver and bound by plasma proteins. Neonates and young infants (younger than 3 months of age) have reduced liver-blood flow and immature metabolic degradation pathways. Thus, larger fractions of local anesthetics are not metabolized and remain active in the plasma compared with adults. More local anesthetic is excreted in the urine unchanged. Furthermore, neonates and infants may be at increased risk for the toxic effects of amide local anesthetics because of lower levels of albumin and α1-acid glycoproteins, which are proteins essential for drug binding (Lerman et al., 1989). This decreased binding leads to increased concentrations of free drug and potential toxicity, particularly with bupivacaine. On the other hand, the larger volume of distribution at steady state seen in the neonate for these (and other) drugs may confer some clinical protection by lowering plasma drug levels (see Chapter 7, Pharmacology of Pediatric Anesthesia).
Local Anesthetic Toxicity
Cardiovascular and CNS toxicity after local anesthetic administration in children is rare (Berde, 1992; McCloskey et al., 1992 ). Local anesthetic toxicity can be limited by careful attention to dose, route of administration, fractionating the dose, and rapidity of absorption of local anesthetic into the systemic circulation (Berde, 2004). Cardiovascular toxicity caused by bupivacaine is the most feared complication of local anesthetic administration, whether it is administered acutely (intermittent dosing) or continuously, because it presents as ventricular dysrhythmias that may be refractory to treatment. Neonates may be at increased risk for bupivacaine toxicity for reasons discussed earlier. Because of this, it is increasingly being replaced with either ropivacaine or levobupivacaine, which may have a greater therapeutic index and margin of safety (Dony et al., 2000; Groban et al., 2001). Patients who develop “lethal” cardiovascular collapse may be rescued with a 20% lipid solution bolus, 1 to 2 mL/kg or 150 mL for adults (Dalgleish and Katawaroo, 2005; Weinberg et al., 2006). This dose may be repeated while resuscitation continues. Once circulation is reestablished, a continuous lipid infusion of 0.5 mL/kg per minute is initiated, and the patient is transferred to an intensive care unit for further monitoring (Box 15-1). Other therapies include prolonged resuscitation efforts, extracorporeal membrane oxygenation, or another temporary circulatory-assistance device.
Topical Local Anesthetics EMLA and ELA-Max
There are several methods for providing topical anesthesia to minimize procedural pain (e.g., venipuncture, lumbar puncture, chest-tube insertion). These include injection of local anesthetic at the procedure site, application of topical anesthetic creams and ointments, iontophoresis, and laser-assisted delivery of anesthetics. An eutectic mixture of local anesthetics (EMLA) in the form of cream is a topical emulsion composed of 2.5% prilocaine and 2.5% lidocaine and produces complete anesthesia of intact skin after application. Unfortunately, for best effect, EMLA cream must be applied and covered with an occlusive dressing for 60 minutes before performing a procedure. This limits its use in the emergency room or office to situations in which the site can be prepared well in advance of anticipated use. Furthermore, if the procedure is a venipuncture, multiple sites must be prepared, in case the initial attempt is unsuccessful (a common problem in pediatric practice in general and more so when EMLA is applied, because it causes cutaneous blanching). Finally, as stated previously, the prilocaine component of EMLA can cause methemoglobinemia. Unfortunately, this has limited the use of EMLA in the newborn. Nevertheless, a single dose is safe and has been shown to be effective in the management of newborn circumcision (Taddio et al., 1997; Lehr and Taddio, 2007). Alternatives that do not contain prilocaine are readily available. Lidocane 4% (ELA-Max) is as effective as EMLA and requires only 30 minutes to become effective (Koh et al., 2004).
Interestingly, the effectiveness of topical local anesthetics at reducing pain is dependent on who makes the assessment. Soliman et al. (1988) studied the efficacy of EMLA cream compared with injected lidocaine at reducing the pain associated with venipuncture. Both an observer and a physician performing the procedure judged pain relief to be virtually complete in both groups. However, the children involved in the study were not so sanguine and were equally dissatisfied with both methods, particularly if the needle used for venipuncture was visible to them. Thus, despite the fact that two observers felt that the child was pain free, the child’s cooperation with venipuncture did not improve. Therefore, it is not clear whether the delay that is involved in the use of EMLA (60-minute wait for effect) is always justified. On the other hand, topical local anesthetics may be more effective in children accustomed to numerous medical procedures (e.g., oncology patients) or for procedures in which the child cannot see the needle, such as lumbar puncture or bone marrow aspiration (although there is little evidence to support the effectiveness of EMLA even in these situations).
Local Infiltration
Infiltration of wound edges with local anesthetics (field block) or by directly instilling local anesthetic into a wound (splash) effectively provides intraoperative and postoperative analgesia for many minor (e.g., inguinal herniorrhaphy, laceration repair, or tonsillectomy) and some major surgical procedures (e.g., craniotomy) (Wong et al., 1995; Casey et al., 1990). Many studies have demonstrated the effectiveness of tetracaine-adrenaline [epinephrine]-cocaine (TAC), lidocaine-epinephrine-tetracaine (LET) and bupivacaine-norepinephrine (BN) in the management of lacerations in children (Schilling et al., 1995; Ernst et al., 1996). Unfortunately, cocaine, a key ingredient in making the TAC drug combination effective, is toxic. Indeed, toxicity has been reported even when TAC has been applied appropriately and according to recommended guidelines.
The most commonly used local anesthetics for local infiltration are lidocaine, mepivacaine, bupivacaine, ropivacaine, and levobupivacaine. As mentioned previously, local anesthetic toxicity is primarily related to how rapidly and how much local anesthetic is absorbed (or deposited) in the blood. Toxicity can be limited by careful attention to dose, route of administration, and by limiting the rate of rise of local anesthetic into the systemic circulation (Table 15-11). No more than 2 to 2.5 mg/kg of bupivacaine or 5 to 7 mg/kg of lidocaine should be used. Dilute solutions of the local anesthetics can be used to provide adequate spread of the anesthetic solution without exceeding the maximum dose. Epinephrine can also be added to the solution in vascular areas to slow the uptake of the anesthetic and to prolong its action. However, in order to avoid ischemic injury, epinephrine must never be used in procedures involving end-arteries, such as the penis or distal extremities. Finally, other adjuvants, particularly clonidine, can be added to the local anesthetic solution to improve the quality and duration of neural blockade (Cucchiaro and Ganesh, 2007). When performing nerve blocks, the pain of local anesthetic administration can be minimized by using small-gauge needles (25 to 30) and warm, buffered anesthetic solutions, and by injecting slowly. Adding bicarbonate to local anesthetic solutions shortens the onset time (faster block) and reduces the pain of injection (Christoph et al., 1988; Orlinsky et al., 1992). This is best accomplished by adding 1 mL (1 mEq) of 8.4% sodium bicarbonate to 9 mL lidocaine or by adding 1 mL (1 mEq) of 8.4% sodium bicarbonate to 29 mL bupivacaine (Yaster et al., 1993, 1994b).
Neural Blockade
The anatomy of peripheral nerves and the techniques of nerve blockade are basically the same for children and adults. The use of nerve stimulators, ultrasonography, and a description of the most common nerve blocks used in pediatric practice are discussed in Chapter 16, Regional Anesthesia. In the next sections the administration of analgesics through these catheters is discussed.
Continuous Epidural Analgesia
Continuous or intermittent epidural analgesia uses local anesthetics administered either alone or in combination with opioids, α2-adrenergic agonists (clonidine) or NMDA receptor antagonists (ketamine) in order to block nociceptive impulses from entering the CNS and provide profound analgesia without systemic sedation (Dalens and Hasnaoui, 1989; Yaster and Maxwell, 1989; Llewellyn and Moriarty, 2007). Epidural analgesia has become the most commonly performed regional anesthetic technique for the intraoperative and postoperative management of patients with urologic, orthopedic, and general surgical procedures below the T4 dermatomal level in children. It has been used to provide continuous sympathetic blockade in children with vascular insufficiency secondary to intense vasoconstriction (e.g., purpura fulminans), in patients with cancer unresponsive to parenteral and enteral opioids, and in the management of patients who have sickle cell trait with vasoocclusive crisis (Yaster et al., 1994a). How long an indwelling caudal or lumbar epidural catheter can be left in place without risking local or systemic infection is unknown; however, serious systemic infections after short-term (3 to 5 days) continuous lumbar and caudal epidural analgesia are extremely rare (Strafford et al., 1995; Kost-Byerly et al., 1998).
Epidural catheters can be inserted at the caudal, lumbar, or thoracic level. The closer the tip of the catheter lies to the dermatome to be blocked, the smaller the amount of drug required to produce neural blockade. Because local anesthetic toxicity is directly related to the total amount of drug infused, catheter placement plays a very important role in the overall safety of this technique. Epidural placement via the caudal and lumbar approach is most common, although even thoracic placement is advocated by some (Bosenberg et al., 1998). Of note, because the epidural space of young children is filled with loosely packed fat and blood vessels (compared with adults), it is possible to thread a caudally (or lumbar) placed catheter as far as the thorax. Bosenberg et al. (1998) first reported the use of the caudal approach for thoracic placement of an epidural catheter in children younger than 2 years of age. Gunter and Eng (1992) extended this observation to older children as well. In children under 5 years of age, caudal insertion and threading 8 to 10 cm of catheter is the preferred epidural technique for most surgery below T4. The key to success is to use short (5-cm), 18-gauge needles, through which 19- to 20-gauge, styletted catheters are inserted. The large-bore catheter offers many advantages over smaller bore (21- to 24-gauge) catheters that were initially used in pediatric epidural analgesia. These large-bore catheters allow for less resistance to flow, less likelihood of occlusion (kinking), and less back-leakage at the site of insertion.
In North America, the most commonly used local anesthetics in continuous epidural blockade are bupivacaine and ropivacaine. Bupivacaine and ropivacaine are administered in concentrations ranging from 0.625 mg/mL (1/16th% solution) to as high as 2.5 mg/mL (0.25% solution). Concentrations above 1.25 mg/mL (1/8% solution) are rarely required for postoperative or medical analgesia and significantly increase the risks of toxicity and unwanted side effects (e.g., sensory, motor, autonomic dysfunction, urinary retention, and inability to walk). Berde, in the editorial accompanying McCloskey’s report of bupivacaine toxicity in children, recommended that bupivacaine infusions be kept below 0.4 mg/kg per hour in children and 0.2 mg/kg per hour in neonates (Berde, 1992; McCloskey et al., 1992). Although this has not been formally studied, these recommended doses have become dosing guidelines. The most commonly used (and easiest) epidural concentration of bupivacaine or ropivacaine is 0.1% (1 mg/mL). Because in concentrations of 1 mg/mL, bupivacaine and ropivacaine do not always produce reliable analgesia, opioids such as fentanyl (2 to 2.5 mcg/mL), hydromorphone (10 mcg/mL), or morphine (20 to 30 mcg/mL) are almost always added to this dilute epidural solution. Which opioid to use is based on the site of the surgical procedure. For surgical procedures performed above the umbilicus (e.g., Nissen fundoplication or thoracotomy), many practitioners prefer hydromorphone or morphine, because they are less lipophilic than fentanyl and may have better rostral spread. For pain below the umbilicus, the initial starting infusion is 0.2 mL/kg per hour (0.2 mg/kg per hour bupivacaine or ropivacaine; 0.4 to 0.5 mcg/kg per hour fentanyl, or 2 mcg/kg per hour hydromorphone, or morphine 6 mcg/kg per hour); for pain above the umbilicus, the initial starting epidural infusion is 0.3 mL/kg per hour (0.3 mg/kg per hour bupivacaine or ropivacaine; 0.6 to 0.75 mcg/kg per hour fentanyl or 3 mcg/kg per hour hydromorphone, or morphine 9 mcg/kg per hour). Generally, infusion rates do not exceed 14 to 16 mL/hour.
If bupivacaine or ropivacaine is used in older children for epidural PCA, the basal infusion is generally administered at a rate that provides 0.2 mg/kg per hour of local anesthetic, while half of the basal rate is given as a bolus (Birmingham et al., 2003). A maximum of two boluses are allowed per hour with a lockout period of 15 minutes. This approach provides a maximum of 0.4 mg/kg per hour bupivacaine or ropivacaine.
Continuous Peripheral Nerve Blockade
Local anesthetics can be infused in continuous peripheral nerve blockade using either disposable elastomeric pumps or standard electronic PCA pumps. The most commonly used solution consists of 0.125% bupivacaine, 0.15% ropivacaine, or 0.1% ropivacaine at rates of 0.1 to 0.15 mL/kg per hour (maximum rate of 12 mL/h). The choice of local anesthetic is based on the anesthesiologist’s preference and the need to prevent a motor blockade. Disposable elastomeric pumps have a rate selection of 2 to 14 mL/h (even numbers only) with a reservoir that can be filled with up to 400 mL of local anesthetic solution. They are not refillable but may be replaced to provide multiple days of peripheral neural blockade. In general, these catheters/pumps result in an opioid sparing effect as well as a low incidence of self-limited complications (Ganesh et al., 2007).
Chronic pain
Complex Regional Pain Syndromes
CRPS as a term was first proposed by Stanton-Hicks to describe a varied and dynamic presentation of symptoms including intense, almost incapacitating, regional pain and sensory, neurovascular, motor, and pseudomotor abnormalities (Stanton-Hicks et al., 1995; Stanton-Hicks, 2003; Berde and Lebel, 2005). Although the precise pathophysiology remains unclear, multiple abnormalities have been described in the peripheral nervous system and the CNS. Sympathetic nervous system involvement has been found in many but not all patients, as demonstrated by relief of pain by sympathetic blockade of the affected extremity or an abnormal hemodynamic response to tilt-table testing (Meier et al., 2006). The diagnosis of the syndrome continues to be based primarily on the patient’s history and physical examination. Presentations without identifiable trauma to nervous system structures are classified as CRPS type I, or reflex sympathetic dystrophy (RSD) in older taxonomy. In cases in which a definable peripheral nerve lesion exists, the diagnosis is CRPS type II, or causalgia. The signs and symptoms for CRPS types I and II are clinically indistinguishable.
CRPS type I has been described in children as young as 5 years of age, and type II has been observed in children as young as 3 years old (Tan et al., 2008). Although uncommon, CRPS is not rare and is increasingly being recognized and diagnosed (Sherry et al., 1999; Lee et al., 2002). Unlike in adults, in children and adolescents CRPS is predominately a disease of females (female/male ratio 5:1) and most commonly affects a lower extremity. Furthermore, if a preceding injury is described, it is usually minor. In addition, psychosocial factors are thought to play a greater role than in adults. Finally, noninvasive, cognitive, behavioral, and physical therapies are more effective in the pediatric population than in adults (Sherry et al., 1999; Lee et al., 2002; Stanton-Hicks, 2003). However, in a minority of patients, persistent pain and a high degree of disability exist despite multimodal therapy including interventional therapy (Wilder et al., 1992).
Diagnosis
The diagnosis of CRPS is based primarily on the patient’s history and physical examination rather than on laboratory testing. Examination or history often show that the affected child has sensory abnormalities such as intense, burning pain; allodynia; and hyperalgesia in the distal aspects of a single extremity. Even in CRPS type II, pain does not follow any sensory dermatomes but is present in a glove or stocking distribution. Whereas children may have all the symptoms generally associated with the syndrome, in adults there are several distinct differences: the temperature of the affected limb is often cooler in relation to the symmetric extremity, edema is less common, and atrophic changes are rare except for decreases in skeletal muscle mass (Tan et al., 2008). Just as in adults, spread of symptoms to other parts of the body, either in a continuous or in a discontinuous fashion, has been observed.
The most common laboratory evaluations in CRPS involve nuclear medicine, quantitative sensory testing, and most recently, functional MRI (Intenzo et al., 2005; Lebel et al., 2008). Although a patient’s nuclear medicine report often states that findings after bone scintigraphy are, or are not, consistent with CRPS, no particular pattern of hypofixation or hyperfixation on scintigraphic studies has been found to be diagnostic. Patients with a history and clinical presentation consistent with CRPS may have a decreased, increased, or normal uptake of tracer on a bone scan. Scintigraphy is more sensitive than plain x-rays and can be useful in supporting or confirming the diagnosis by helping to exclude other diagnoses such as arthritis, benign or malignant bony lesions, or metabolic bone diseases (Intenzo et al., 2005). Hypoperfusion corresponding to osteopenia on a plain radiograph may be used as a cautionary sign for conducting physical therapy sessions to avoid pathologic fractures.
Nerve-conduction studies (NCS) and electromyography (EMG) should be reserved for the occasional patient. These are painful tests and may not be well tolerated in highly sensitive patients. NCS and EMG are indicated when initially diffuse symptoms become more localized as therapy progresses, or when pain follows a distinct dermatomal pattern, because this may be indicative of a peripheral nerve entrapment. Quantitative sensory testing (QST) is primarily a research tool. Sethna et al. (2007) performed standardized neurologic examinations and QST in a group of 42 pediatric patients and found that cold allodynia was the most common abnormality. More than half of their patients also had mechanical, dynamic, and static allodynia and allodynia to punctuate temporal summation. The authors suggested that these abnormal hyperexcitable sensory patterns may be consistent with a central sensitization pattern.
Finally, the importance of psychological disturbances both in the diagnosis and management of CRPS have been hotly debated. At one point, CRPS was thought to be an entirely psychosomatic disorder. Although this view is no longer tenable, the importance of psychological dysfunction and multiple stressors in affected children is clear and an important target of therapeutic intervention (Sherry et al., 1999; Tan et al., 2008). Indeed, Sherry et al. (1999) emphasize the importance of individual and family psychological issues in the perpetuation of this condition.
Treatment
There are currently no therapeutic modalities that predictably lead to resolution of CRPS. All treatment is directed at aggressive mobilization of the affected limb early in the process to prevent disability, refractoriness, and treatment failure and must include the family as well as the patient (Lee et al., 2002). Unquestionably, the key to therapy is physical reconditioning and mobilization of the affected limb by exercise, ambulation, and range of motion exercises (Lee et al., 2002). With reconditioning, affect, endurance, and pain improve. Medication may help alleviate pain, improve sleep, and facilitate physical therapy. Many drug classes, including opioids, NSAIDs, TCAs, and anticonvulsants are prescribed, but none has been subjected to the rigors of randomized controlled trials in the treatment of these patients. Finally, cognitive behavioral therapy is as important as physical therapy and medication, because it promotes positive coping skills and eliminates reinforcement of maladaptive behaviors.
Occasionally more aggressive intervention therapy, such as sympathetic nerve blocks, are used when pain is not well controlled by these more conservative measures, when allodynia is so severe that physical therapy is impossible, or when there is no improvement or even further loss of function despite adequate therapy. Sympathetic nerve blocks are not required for diagnosis and no association between the use of blocks and long-term outcome has been made. There are multiple case series and reports on the use of continuous infusions of local anesthetic with or without adjuvants such as opioids, clonidine, or ziconitide administered via peripheral nerve, lumbar sympathetic, epidural, and intrathecal catheters in these disorders (Dadure et al., 2005; Farid et al., 2007; Meier et al., 2009). Most infusions were limited to a few days, although some catheters remained in place for several months. Meier et al. (2009) demonstrated variable effectiveness of lumbar sympathetic blockade in lower extremity CRPS in the first randomized controlled crossover trial ever conducted in adolescent patients with this disease. However, no association between the response to sympathetic nerve blockade and long-term outcome has been shown. On the other hand, Sherry et al. (1999) believe that nerve blocks and medication are unnecessary and may be counterproductive because they reinforce the patient taking a passive, rather than active, role in recovery.
Finally, the use of neuromodulation (spinal-cord stimulators) in the management of pediatric CRPS is unclear. Spinal-cord stimulators are increasingly being used in adults with CRPS whose conservative treatment has failed, who are psychologically stable, and who can achieve functional status sufficient to participate in exercise after the procedure (Nelson and Stacey, 2006). However, their effectiveness is unclear. Initial favorable reports in adults were followed by less encouraging functional results in the same population 2 to 5 years later (Kemler et al., 2000, 2004, 2008). A case series of seven adolescent girls reported that at least some of the patients who received spinal-cord stimulators entered remission and no longer required the device at a long-term follow-up session, suggesting this modality has potential therapeutic use in pediatric CRPS (Olsson et al., 2008). However, much larger randomized controlled trials are required before this therapy can be recommended in young patients.
Functional Gastrointestinal Disorder
Functional gastrointestinal disorder (FGID) encompasses a group of conditions characterized by chronic or recurrent symptoms that are not explained by biochemical, anatomic, or structural abnormalities (Saps and Di, 2009; Yacob and Di, 2009). Normal gastrointestinal functions include transport, digestion, and absorption of nutrients, and removal of waste products. The gut is also an important immune barrier. Patients with FGIDs experience a constellation of symptoms consistent with abnormalities in these gastrointestinal functions. These include dysmotility, secretory dysfunction, malabsorption, diarrhea or constipation, and allergic enteritis. However, significant weight loss, growth failure, unexplained fever, pain far from the umbilicus, bloody diarrhea, and repeated emesis are rarely associated with FGIDs. Presence of these signs or symptoms should result in a search for an organic process, such as a tumor, mechanical obstruction, infection, or inflammatory bowel disease.
Treatment
Therapy for FGIDs is largely supportive and has to address contributing psychosocial factors. There are no magic pills, and there is limited evidence to justify the use of drugs or herbal preparations outside of clinical trials (AAP, 2005; Huertas-Ceballos et al., 2008, 2009; Saps and Di, 2009; Yacob and Di, 2009). Pharmacologic therapy is focused on the control of symptoms with prokinetic, antispasmotic, secretory, and coating agents. Anxiolytics and antidepressants are used because of their ability to modulate pain transmission and perception, as well as their potential to address psychological comorbidities. However, if using drugs as a therapeutic trial, clinicians should be aware that these are fluctuating conditions and any response may reflect the natural history of the condition or a placebo effect rather than drug efficacy (Huertas-Ceballos et al., 2008). Indeed, this was confirmed in a large multicenter, randomized, placebo-controlled study of amitriptyline in 83 children with functional abdominal pain that found no statistical difference between amitriptyline and placebo therapy (Saps et al., 2009).
Chronic Abdominal Wall Pain
Although chronic abdominal pain arising from a visceral source is the most common type of abdominal pain, it must be differentiated from pain originating in the abdominal wall. Abdominal wall pain is localized, not diffuse, and on physical examination is exquisitely tender with abdominal wall tensing (Carnett’s sign). The abdominal wall is innervated by the seventh to twelfth intercostal nerves. Entrapment of these nerves at the level of the anterior cutaneous branch, as well as in the rectus sheath, has been described in children (Skinner and Lauder, 2007). Successful treatment with rectus sheath blocks has been described and should be made easier with ultrasound guidance (Willschke et al., 2006; Skinner and Lauder, 2007).
Headache
Headaches are a universal feature of the human experience. Studies of Swedish schoolchildren have indicated that 40% of children experience a headache by age 7, 75% experience a headache by age 15, and migraine (one of the most common causes of headache in childhood) occurs in 1% of children by age 7 and 5% of children by age 15 (Bille, 1962). Headaches have a significant impact on the lives of children by causing school absences, poor school performance, and decreased extracurricular activities. Although the majority of patients have benign causes of headaches that can usually be diagnosed by a careful history and physical examination, radiologic evaluation using computed tomography (CT), MRI, or both may be necessary in select cases. Proper diagnosis, treatment, and close monitoring of these patients are extremely important to ensure that serious etiologies are not overlooked.
Diagnosis
Standardized criteria have been developed to diagnose headache and divides headache into primary and secondary etiologies (Headache Classification Committee of the International Headache Society, 1988). Primary headaches are those that are directly attributed to a neurologic basis and include migraine, tension-type headaches, cluster headaches, and trigeminal autonomic cephalalgias. Secondary headaches are headaches directly attributed to another medical condition, such as headaches associated with space-occupying lesions, inflammation, sinusitis, and abnormalities of intracranial pressure (both high and low pressure) such as pseudotumor cerebri, Arnold-Chiari malformation, or hydrocephalus. Headaches that are associated with focal neurologic signs or symptoms, or that progressively worsen in severity or frequency are suggestive of intracranial pathology, and require neuroimaging by MRI or CT scan as appropriate and subsequent focused therapy. On physical examination, the clinician should look carefully for changes in consciousness, attention, language or memory, cranial nerve asymmetry or papilledema, nuchal rigidity, abnormal tone, gait ataxia, or any new neurologic abnormality.
Migraines and tension type headaches are the most common primary headaches of childhood. Approximately 10% of school-aged children suffer from migraine (Diamond et al., 2007; Silberstein et al., 2007). Migraine duration is generally between 1 to 48 hours. Migraine quality can be unilateral or bilateral, pulsating, moderate to severe in intensity, and aggravated by routine activity. The headache may be accompanied by nausea and vomiting or by photophobia and phonophobia (Headache Classification Committee of the International Headache Society, 1988). About 14% to 30% of children with migraine also experience a migraine aura, indicating focal cortical or brainstem dysfunction. Typical auras include spots, colors, images distortions, and visual scotoma. Interestingly, in adult patients a high prevalence of right-to-left shunting has been described in patients with migraine, especially migraine with aura, and symptoms have improved in some patients after closure of a patent foramen ovale (Nahas et al., 2009).
Migraine variants are headaches that are accompanied or manifested by transient neurologic symptoms. For example, hemiplegic migraine is characterized by the abrupt onset of hemiparesis that is usually followed by a headache. Basilar artery migraine is characterized by dizziness, weakness, ataxia, and severe occipital headache, and opthalmoplegic migraine is associated with orbital or periorbital pain and third, fourth, or sixth cranial nerve involvement. Cyclic vomiting and recurrent abdominal pain, in the absence of primary gastrointestinal disease, are also considered migraine variants. Status migraine is defined as a severely painful, continuous unremitting headache of more than 72 hours’ duration (Olesen and Lipton, 2004).
Therapy
Mechanisms of migraine prevention are not completely understood. In general, therapies have been focused on the three major theories proposed to explain migraine pathophysiology. The vascular theory attributes migraine pain to vasodilation. The second hypothesis focuses on cortical spreading depression, a neuronal depolarization wave followed by a suppression of bioelectric activity. The third theory postulates that migraine is related to the release of inflammatory neuropeptides from the trigeminal system, which subsequently dilate meningeal blood vessels (Galletti et al., 2009). Interestingly, mutations in voltage-gated calcium and sodium channels have been described in some patients with familial hemiplegic migraine, but whether similar pathologies occur in more standard cases of migraine is unknown (Pietrobon, 2005, 2007).
Prophylactic therapies include β-blockers (e.g., nadolol, propanolol), antidepressants (e.g., amitriptylline), cyproheptadine (periactin—an antihistaminergic, antiserotonergic drug), anticonvulsants (valproic acid, topiramate, gabapentin), and calcium channel blockers (flunarizine, verapamil). Emerging treatments in adults include angiotensin converting enzyme inhibitors, angiotensin II type 1 receptor blockers, and botulinum toxin. These classes of drugs can target multiple cortical and subcortical structures and also modulate peripheral neurogenic inflammation (Galletti et al., 2009).
β-Blockade with propranolol (1 to 3 mg/kg per day divided in 2 or 3 doses), calcium channel blockade with flurarizine, and topiramate treatment have been shown to be effective in ameliorating pediatric migraine (Victor and Ryan, 2003; Cruz et al., 2009). β-Blockade may decrease the frequency and intensity of migraines by increasing arterial tone and hampering vasodilation, or by reducing sympathetic tone. In addition, β-blockade reduces firing of some central noradrenergic neurons, and it may also interact with the serotonergic system, which plays an important role in migraine. Calcium channel blockers such as verapamil (4 to 8 mg/kg per day in three divided doses) may prevent migraine headaches by impairing the activation of neurogenic inflammation, a calcium-dependent process, or by increasing pain thresholds. Anticonvulsants can limit neuronal hyperexcitability through effects on voltage-gated sodium channels. In addition, antidepressants, anticonvulsants, and calcium channel blockers can also influence the serotonergic and dopaminergic systems, which play an important role in migraine pathophysiology.
Once a headache develops, the goals of therapy are to abort an attack and suppress pain, nausea, and vomiting. Often inducing or promoting sleep in a dark and quiet room is helpful in diminishing symptoms. Relaxation techniques, biofeedback, behavioral techniques, and acupuncture may also be helpful. Pharmacologic treatments to interrupt or abort the headache include a number of different classes of drugs. Mild analgesics such as acetaminophen and NSAIDs are often effective in children (Hamalainen et al., 1997a; Hamalainen, 2006). Sumatriptan and other members of the triptan family are approved by the FDA for the treatment of adult migraine. Although commonly used in children, none has FDA approval for this indication. These medications are 5-HT1 receptor agonists. Sumatriptan is available as an oral form (25 mg in adolescents), an injectable form (subcutaneous, 3 to 6 mg), and a nasal spray (5 to 20 mg). Nasal sumatriptan has been shown to be effective in pediatric migraine, whereas unlike in adults, the oral form of the drug has not (Hamalainen et al., 1997; Ueberall and Wenzel, 1999; Ahonen et al., 2004). Adverse effects associated with the use of triptans include tingling, dizziness, warm sensations, chest pain, and cardiac arrhythmias. Isometheptene and ergotamines have also been reported to be effective, especially when administered at the onset of the aura or start of the headache (Hamalainen, 2006). However, Hamalainen et al. (1997b) did not see an improvement in headaches after oral dihydroergotamine as compared with placebo in children with therapy-resistant migraines. In those children whose conservative therapy fails and who come to the hospital with status migraine, treatment with multiple doses of intravenous dihydroergotamine has been shown to be effective, although there is a significant incidence of adverse side effects, especially anxiety, nausea, and vomiting (Kabbouche et al., 2009). It should be noted that all abortive medications carry the risk of causing a headache from overuse of secondary medication, defined as a headache on more than 15 days of a month with overuse of acute treatment drugs for more than 3 months and headache that has developed or worsened during the period of medication overuse (Silberstein et al., 2005). When medication overuse headache occurs, a need for multisystem treatment approach may be beneficial (Pakalnis et al., 2007).
Sickle Cell Disease
Sickle cell disease (SCD) is an autosomal-recessive chronic hemolytic anemia characterized by the production of abnormal hemoglobin. A point mutation in the sickle cell β-globin gene results in the substitution of the hydrophobic amino acid valine for a glutamic acid at position 6 in the globin chain. When hemoglobin S tetramers become deoxygenated, valine is able to interact with other hydrophobic residues on neighboring globin chains to form insoluble globin polymers. Whereas polymerization rapidly reverts to normal once hemoglobin is reoxygenated, cells can become irreversibly sickled as a result of oxidative damage to the cell membrane after repeated cycles of sickling and unsickling. Sickle cells display abnormal adhesion to endothelial cells and initiate microvascular occlusion (Vijay et al., 1998; Kaul et al., 1989). Resultant hypoxia causes further sickling, tissue infarction, and the release of inflammatory cytokines. Repeated vasoocclusive crises (VOC) predispose to multiorgan dysfunction and shorten survival (Platt et al., 1991, 1994). In addition, patients with SCD are susceptible to bacterial infections caused by splenic autoinfarction as well as abnormal cell-mediated immunity. Other consequences of SCD include gallstone disease as a result of chronic hemolysis, priapisim, and stroke. Acute chest syndrome, caused by infection or embolic phenomena, is the leading cause of death (Vichinsky, 1991) (see Chapter 36, Systemic Disorders).
VOC are often characterized by excruciating and at times incapacitating pain. It is the most common and debilitating problem encountered by patients with SCD. Many factors have been associated with the onset of acute pain crises, including cold, dehydration, alcohol intake, stress, and intercurrent infection. However, over one half of episodes have no identifiable precipitant. Acute sickle cell pain is primarily the result of tissue ischemia and occlusion of the microcirculation, whereas acute bone pain appears to be the result of avascular necrosis of the bone marrow (Shapiro, 1989; Stinson and Naser, 2003). Painful episodes can begin as early as 6 months of age. Younger children often suffer from finger, toe, and limb pain, whereas in adolescents back and abdominal pain may be the most prominent symptoms. Acute painful episodes include a prodromal period followed by an infarctive phase. Subsequently, a postinfarctive phase characterized by signs of inflammation and persistent severe pain develops and is then followed by a resolving phase during which the pain gradually remits.
Patients coming to the hospital with sickle cell pain have commonly failed home therapy or may be unable to tolerate oral analgesics because of nausea and vomiting. Sickle cell crisis pain can be excruciating and is the leading cause of emergency room visits and hospital admissions in these patients. When studied, average VAS scores of 9.5 + 0.63 out of 10 have been reported (Ballas, 1997). In that setting, the use of parenteral opioids is common. Once pain has been appropriately assessed, medication is titrated to provide pain relief (Benjamin et al., 1999). Initial doses of opioid are based on a history of what has provided adequate analgesia in the past. Although historically, meperidine was the most commonly prescribed opioid for treatment of SCD pain, it is no longer a first line treatment of acute pain because of the CNS toxicity of its metabolite, normeperidine (Latta et al., 2002). In general, titration schemes involve administration of a loading dose of opioid (e.g., morphine 0.1 to 0.2 mg/kg or hydromorphone 0.01 to 0.04 mg/kg) followed by additional smaller bolus doses (generally one fourth or one half of the loading dose) or initiation of PCA (National Heart, Lung, and Blood Institute, 2002). A prospective controlled trial of morphine PCA showed that PCA was as effective as intermittent nurse-administered intravenous doses of morphine, with 80% of patients describing the PCA analgesic regimen as good to excellent (Shapiro et al., 1993). Once pain has stabilized and the patient can tolerate oral intake, analgesics can be transitioned to an equianalgesic sustained-release oral opioid, in conjunction with rescue analgesia, to treat breakthrough pain.
Unfortunately, even with aggressive, proactive pain management, pain in SCD is often difficult to treat, resulting in therapeutic failure and frustration for both patients and their health care providers. Patients with acute sickle cell crisis pain often report higher pain scores than postoperative patients until they enter the resolving phase of their crisis. They often report 10 out of 10 on a pain scale, even when they appear to their physicians and nurses to be comfortable. This disconnect between subjective and objective assessments can result in a therapeutic dilemma in which patients request more and more opioids and their physicians prescribe less and less. This lack of trust is in part caused by race and a fear of producing drug addiction (Geller and O’Connor, 2008). SCD is a disease primarily of African Americans and it is clear from other studies that African Americans are less likely than Caucasian and Hispanic Americans to be prescribed opioids for even common conditions in the emergency department, such as migraine headaches and fractures (Tamayo-Sarver et al., 2004, 2003).
Although patients with SCD do develop tolerance, as well as physical dependence and at times hyperalgesia, to opioids if they are administered for prolonged periods of time, they do not develop drug addiction at a higher rate than patients with other conditions (Waldrop and Mandry, 1995). Nevertheless, the failure to provide adequate analgesia often results in pseudoaddiction (Elander et al., 2003). Pseudoaddiction arises when a patient’s pain is inadequately managed, and the response to this undertreatment is used as evidence for the diagnosis of drug addiction. Pseudoaddiction is postulated to progress through three phases. The cycle begins with “as-needed” dosing of inadequate analgesics for the treatment of continuous or recurrent pain. Initially, the patient merely requests more pain medication. When these requests are overlooked or ignored, the patient then tries to convince the physician of pain by moaning, grimacing, or crying. The physician interprets this behavior as aberrant and again refuses the requested dose escalation. Finally, the crisis phase occurs when the patient increases the level of bizarre, drug-seeking behavior. The cycle continues, with the patient persistently trying to acquire the drug and the physician consistently refusing to treat the pain, resulting in a lack of trust between the two parties and ultimately in the patient being viewed as a drug addict (Weissman and Haddox, 1989).
Opioids, even at high doses, often have limited efficacy in ameliorating sickle cell pain and often produce unwanted side effects such as sedation, constipation, nausea, and vomiting. Thus, other treatments have been used in place of or in conjunction with opioids in an attempt to diminish these side effects. Whereas single doses of an NSAID do not appear to significantly impact on opioid use, multidose parenteral NSAID infusions do seem to result in a significant opioid-sparing effect. Thus, ketorolac is often added to parenteral opioids in an attempt to improve analgesia and diminish opioid consumption. However, because of its impact on renal function, it is recommended that it not be given for more than 5 days in a given month (Feldman et al., 1997). Studies of parenteral corticosteroids also suggest that pain and length of hospital stay can be shortened by their use without producing short-term adverse effects (Dunlop and Bennett, 2006). However, concerns regarding increases in recurrent pain episodes, as well as the side effects of chronic steroids, have limited their clinical use in this setting (Couillard et al., 2007). Although not routinely employed, epidural analgesia with local anesthetic and opioid has also been shown to be effective in treating sickle cell pain and improving respiratory status in patients whose conventional therapy has failed (Yaster et al., 1994a). However, this modality is only effective if pain is localized to areas of the body that can be effectively blocked by epidural analgesia (e.g., chest, abdomen, and lower extremities). In addition, the impact of repeated epidural placements for the treatment of pain in patients with repeated pain crises is unknown. Finally, self-hypnosis, biofeedback, relaxation, and acupuncture have all been reported to be effective in reducing pain in some patients (Zeltzer et al., 1979; Sodipo, 1993).
In addition to recurrent, acute pain, patients with SCD are also at risk of developing chronic pain that can be physically and psychologically debilitating. Causes of chronic pain include arthritis, arthropathy, avascular necrosis, skin ulcers, and vertebral body collapse (National Heart, Lung, and Blood Institute, 2002). Although there are no studies addressing the management of chronic pain in patients with sickle cell disease, sustained-release opioids, such as MS-Contin or methadone, are at times prescribed to provide consistent analgesia (Dunlop and Bennett, 2006). Adjuvant drugs can be prescribed as well.
Ultimately, the best method of sickle cell pain control may involve therapies that decrease the likelihood of VOC by preventing sickling. Hydroxyurea was approved by the FDA for treatment of adults with sickle cell anemia in 1998, and in 2002 the National Heart, Lung, and Blood Institute issued recommendations for its use in children with SCD (National Heart, Lung, and Blood Institute, 2002; Strouse et al., 2008). Hydroxyurea acts in part by inducing the induction of fetal hemoglobin production, which inhibits hemoglobin polymerization and sickling. It also decreases hemolysis and reduces the expression of cell-adhesion molecules that contribute to vasoocclusion (Benkerrou et al., 2002). Studies in children have shown an increase in fetal hemoglobin concentration, a decline in the yearly rate of hospitalizations, and a reduction in pain crises, but long-term effects of hydroxyurea treatment in SCD are still unknown (Strouse et al., 2008).
Palliative care
Despite dramatic advances in the diagnosis and treatment of many pediatric diseases, death during childhood remains a persistent reality, and caring for children during their final days remains a compelling clinical responsibility for pediatric health care providers (Kang et al., 2007). The past decade has seen an enormous shift in the attitudes, beliefs, and practices of pediatric palliative medicine. Palliative care is increasingly being recommended for a variety of pediatric illnesses, including those for which a cure remains possible or even likely (Field and Behrman, 2009; Goldman et al., 2009). The most obvious are those life-threatening diseases for which curative treatment is possible but might fail, such as cancer. Less intuitive, are conditions with long periods of treatment devoted to prolonging life, but without cure, such as Duchenne’s muscular dystrophy or children with severe neurologic disabilities (Wusthoff et al., 2007).
The goal of palliative care is to achieve the best quality of life possible for patients and their families. Control of pain, other physical symptoms, and psychological, social, and spiritual problems are vital components of this care. Pediatric palliative care focuses on three prominent aspects of care (Kang et al., 2007; Field and Behrman, 2009; Goldman et al., 2009). First, and arguably most important, is communication. Understanding how disease processes and a person’s innate abilities and liabilities affect that communication is essential to building a foundation of collaboration and a sense of teamwork with the patient and family. Second, psychosocial aspects of pediatric palliative care are important to the present and future well-being of the child, the family, and the practitioner. Acknowledging and facilitating a family’s spiritual needs and involvement in religious traditions that are comforting to them often helps provide meaning to this distressing experience (McSherry et al., 2007). For many families and health care providers the incorporation of religion and spirituality into the medical care of their child and patient can be an integral part of providing comprehensive high-quality palliative care (McSherry et al., 2007). Focusing on these and other issues that affect the children’s, families’, and practitioners’ physical and mental health must be central when caring for dying children. Finally, the aspect that is most easily translated into clinical practice is caring for the specific physical needs of the child being treated. In the ensuing discussion, strategies are provided for managing common symptoms, including pain, dyspnea, agitation, gastrointestinal complaints, and seizures (Lin et al., 2009).
Pain Management
When prescribing opioids for palliative care (or chronic pain) it is essential for the prescriber to ascertain if the patient’s local pharmacy has the drug and if the pharmacy will dispense it with subsequent refills. Furthermore, to avoid licensing authority investigation and error, prescribers must use best-prescribing practice (i.e., the prescriptions must be dated, signed, and clearly state that the opioid is being prescribed for a legitimate purpose [palliative care or chronic pain]) (Lee et al., 2008). Unfortunately, even when complying with these regulations, many pharmacies do not fill prescriptions for oral or parenteral methadone or sustained-release oxycodone. Furthermore, individuals who write these prescriptions are at risk of punitive investigations by their licensing authorities and the Drug Enforcement Agency (Jung and Reidenberg, 2006). Indeed, a practice with a large percentage of its patients who are patients with chronic pain and treated with opioids may be a trigger for an investigation.
Gastrointestinal Symptoms
The management of gastrointestinal symptoms, such as nausea, vomiting, constipation, diarrhea, anorexia (loss of appetite), and cachexia (involuntary weight loss and wasting) is fundamental in palliative care (Stanton-Hicks et al., 1995; American Academy of Pediatrics Subcommittee on Chronic Abdominal Pain, 2005). Although opioids are a common cause of these complaints, other causes must be suspected and ruled out. These include increased intracranial pressure, other medications (e.g., chemotherapeutics and antibiotics), metabolic abnormalities (e.g., uremia or hepatic failure), intestinal obstruction (e.g., gastric outlet or bowel), and mucositis. The management of opioid-induced nausea, vomiting, and constipation has been discussed previously, and the treatment strategies are the same in patients receiving palliative care.
Weight loss and malnutrition are often unavoidable symptoms experienced by patients at the end of life and are commonly associated with poor clinical outcome and increased morbidity and mortality (Stanton-Hicks et al., 1995; American Academy of Pediatrics Subcommittee on Chronic Abdominal Pain, 2005). Furthermore, eating and food can hold important meaning for patients and families; thus, anorexia and cachexia can have a negative impact on quality of life far in excess of their clinical impact. Mealtime commonly holds cultural, emotional, and religious significance, and the inability to enjoy food can affect the entire family. Families may believe that providing nutrition may stave off or reverse wasting; however, for children with certain malignant tumors or advanced disease, weight loss may be largely irreversible. Even so, several drugs have shown some benefit in palliating the cachexia associated with advanced cancer and disease. Appetite stimulants such as progestational drugs, corticosteroids, and cannabinoids can offer some improvement in appetite and weight gain, albeit primarily in adipose and not skeletal muscle tissue (Stanton-Hicks, 2003; Tan et al., 2008). In addition, megestrol acetate and medroxyprogesterone, synthetic progestins, tend to increase a sense of well-being in addition to stimulating appetite (Stanton-Hicks, 2003; Tan et al., 2008).
Neurologic Symptoms
Many children who require end-of-life care may experience seizures, agitation, and somnolence or loss of consciousness as a result of a primary neurologic illness, an overwhelming systemic illness (chronic or acute), a metabolic derangement (e.g., hyponatremia, hypernatremia, or hypoglycemia), or disease progression (e.g., cerebral metastases) (Sherry et al., 1999). Although it is beyond the scope of this chapter to discuss the etiologies and management of these problems in great detail, a general overview of management is provided.
Seizures are paroxysmal discharges of neurons in the brain resulting in alteration of function or behavior. They are an indication of CNS irritability or disease and typically cause significant stress and distress to patients and families. Parents who have witnessed a child seizing commonly state that they believed their child was dying and that they never want to see their child have another seizure. Patients are more likely to express the fear of future mental handicap and embarrassment about losing control of consciousness, bladder, and/or bowel function in front of friends and family. Many seizures are provoked by fever, infection, and electrolyte abnormalities (e.g., hypoglycemia, hyponatremia, hypocalcemia, hypomagnesemia, or hypooxia). If no precipitating cause is found and the events witnessed are real seizures (some conditions, such as syncope, cardiac arrhythmias, and migraines, mimic seizures), the next step in management is to determine the type of seizure that is occurring, because most antiepileptic drugs are prescribed based on seizure type (Sherry et al., 1999).
The most common types of seizures are generalized and partial-onset seizures. Generalized seizures have no apparent focal onset, whereas partial-onset seizures have a focal onset and may remain focal or secondarily generalize. Table 15-12 lists the most common anticonvulsants and their routes of administration. Like opioids, which anticonvulsant to use depends in part on the anticonvulsant’s formulation and the route of administration. For children who have recurrent seizures, physicians and caregivers must also plan for the possibility of status epilepticus. Status epilepticus is a single continuous seizure or repetitive seizures lasting more than 30 minutes without recovery of consciousness. Enabling caregivers to initiate treatment of prolonged or repetitive seizures can improve outcome, decrease anxiety, and prevent visits to an emergency department. Several abortive therapies are available. When intravenous access is not available, the most widely used anticonvulsant is rectal diazepam gel (0.2 to 0.5 mg/kg), although intranasal and buccal midazolam are effective alternatives (Farid and Heiner, 2007; Dadure et al., 2009; Rochette et al., 2009). The risk for respiratory compromise is lower with rectal diazepam than with intravenous formulations of the same medication, because absorption is slower and peak concentrations remain lower. When intravenous access is available, status epilepticus is best treated with lorazepam (0.05 to 0.1 mg/kg, at a rate of 2 mg/min) or with a slow infusion of fosphenytoin (20 mg/kg administered at a rate of 3 mg/min).
Agitation is an unpleasant state of increased arousal. It may present as loud or angry speech, crying, increased muscle tension, increased autonomic arousal (such as diaphoresis and tachycardia), or irritable affect. It can and often does evolve into delirium with sleep disturbance, confusion, and impaired attention. There are many treatable causes of agitation including pain, dyspnea, muscle spasm, bowel dysmotility, and bladder distention. In addition, acute withdrawal from several drugs, including opioids, benzodiazepines, corticosteroids, and some anticonvulsants can also cause agitation. The initial management of agitation is intuitive and nonpharmacologic. Familiar objects from home, a gentle touch, or a soothing voice are always the first steps. If these fail, and they often do, pharmacologic intervention may be warranted with either benzodiazepines, antipsychotics, or barbiturates (Truog et al., 1992; Sherry et al., 1999; Intenzo et al., 2005).
Dyspnea
Dyspnea, or a feeling of breathlessness, occurs when the respiratory system is unable to meet the body’s need for oxygen uptake or carbon dioxide elimination. It commonly occurs in terminal illness either because of an increased oxygen demand (e.g., sepsis or organ system failure) or an inability to excrete carbon dioxide (e.g., muscle fatigue, pneumonia, interstitial lung disease, cystic fibrosis, or neoplasm) (Meier et al., 2009; Lin et al., 2009). There are several ways that dyspnea can be treated, namely positive pressure mechanical ventilation, noninvasive promotion of gas exchange (continuous positive airway pressure [CPAP] or supplemental oxygen), or pharmacologic measures to decrease or suppress the sensation of dyspnea. Positive pressure ventilation and CPAP may improve the physiologic disturbances causing dyspnea, but because they cannot reverse the underlying process, they are generally inappropriate in end-of-life situations. However, supplemental oxygen is not. Although supplemental oxygen can often ameliorate the symptoms of hypoxemia and dyspnea, oxygen may also reverse the hypoxic drive to breathe and precipitate apnea.
Opioids are the drugs of choice in the treatment of dyspnea (McGrath et al., 2008; Meier et al., 2009; Saps and Di, 2009). All opioids raise the apneic threshold and shift the carbon dioxide response curve to the right; thus, there is no particular opioid that is superior to another for dyspnea. In a patient already receiving opioids, an increase of 25% in baseline dose effectively treats dyspnea. The use of nebulized morphine is controversial but has its advocates(McGrath et al., 2008; Meier et al., 2009; Saps and Di, 2009). It is hypothesized that the receptors in the lungs involved in the sensation of dyspnea are J-type stretch receptors and that the output from these receptors is attenuated by topically applied inhaled opioids (Sethna et al., 2005). Finally, dyspnea can cause anxiety, which can further worsen the sense of dyspnea. When this occurs, benzodiazepines are useful therapeutic adjuncts.