CHAPTER 37 Malignant Hyperthermia
Deaths during anesthesia associated with high fever and tachycardia were described repeatedly in the first half of the 20th century (Moschcowitz, 1916; Burford, 1940). These deaths were sometimes ascribed to heat stroke and dehydration, because at that time intravenous hydration and air conditioning were not available. In 1915 and 1919, two deaths in one family occurred during chloroform anesthesia; in these cases, severe muscle spasm was observed before cardiac arrest. In 1987 a young child from this same family died during anesthesia for dental surgery. Subsequent in vitro contracture testing demonstrated susceptibility to malignant hyperthermia (MH) in several relatives in the family (Harrison and Isaacs, 1992). Even though MH is often suspected in cases such as these, in the 21st century it can still take years to complete MH diagnostic testing for a family.
Denborough was the first to investigate MH as a clinical entity (Denborough and Lovell, 1960; Denborough et al., 1962). Dr. James Villiers anesthetized a young Australian man for repair of a broken leg; the man was afraid to undergo general anesthesia because many of his relatives had died during ether anesthesia. Although during halothane anesthesia the patient became febrile, tachycardic, cyanotic, and hypotensive, he was aggressively cooled in the recovery room and he survived. Villiers asked his medical colleagues to investigate this patient and family. They found a pattern of anesthetic deaths consistent with an autosomal dominant trait. In 2009, despite many investigations, the genetic change associated with abnormal muscle calcium homeostasis in this family was still not identified (Ball, 2007).
An episode of MH is recognized by documentation of an increasing metabolic rate after exposure to a triggering agent such as a potent inhalation anesthetic. Rhabdomyolysis may also be observed. Susceptibility to MH is rarely associated with clinically evident hypermetabolism in the absence of drugs used during anesthesia (Gronert et al., 1980; Tobin et al., 2001). Halothane and succinylcholine, two drugs formerly used often in pediatric anesthesia, are two of the most potent triggers of MH. A review of 25 years of halothane anesthetics in one pediatric hospital found the incidence of acute MH episodes to range from 1:20,000 to 1:40,000 anesthesia procedures (Warner et al., 1984). Because choices of anesthetic and neuromuscular blocker influence the development of this potentially lethal syndrome, MH is a disease of particular concern to the pediatric anesthesiologist who chooses to administer inhalation anesthetics or succinylcholine. Indeed, in 2008 administration of succinylcholine during potent inhalation anesthesia precipitated cardiac arrest and death from MH (North American Malignant Hyperthermia Registry, 2008). The purpose of this chapter is to review the clinical manifestations, management, and underlying pathophysiology of MH.
Muscle physiology
Normal Muscle Physiology
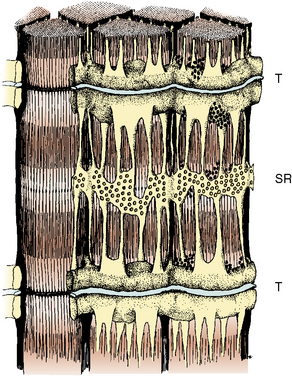
The propagation of an action potential down a motor nerve fiber produces depolarization of that fiber and release of acetylcholine (ACh) at the neuromuscular junction. When two molecules of ACh interact with the ACh receptor, the channel opens, allowing sodium to enter the cell and potassium to flow out. This initiates a wave of depolarization along the muscle cell membrane or sarcolemma. The propagated depolarization wave spreads internally via transverse tubules that abut onto the sarcoplasmic reticulum (SR) (Fig. 37-1). The SR is an internal cellular structure and a major storage site for calcium ions. The wall of the transverse tubule contains the dihydropyridine receptor, a voltage-dependent calcium channel that interacts with the calcium-sensitive ryanodine receptor (RYR), the footplate between the transverse tubule and the SR. Within a voltage window, calcium is released from the SR (Melzer and Dietze, 2001) into the sarcoplasm through RYR type 1 (RYR1). Increased calcium in the sarcoplasm also induces further calcium release from the SR.
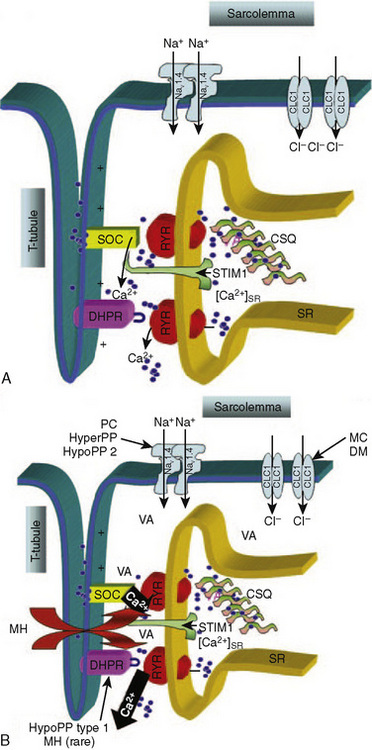
Furthermore, when calcium has been depleted from the SR, a process known as store-operated calcium entry (SOCE) allows calcium entry into the sarcoplasm from the extracellular milieu (Fig. 37-2) (Zhao et al., 2006). Excitation-coupled calcium entry (ECCE) refers to the process by which extracellular calcium enters the sarcoplasm through the plasma membrane after depolarization (Cherednichenko et al., 2004; Lyfenko and Dirksen, 2008). The RYR has influence on both SOCE and ECCE.
Type 1 RYRs are found in all skeletal muscle, smooth muscle, neurons, and B lymphocytes. Type 2 RYRs are found in cardiac muscle, brain, and some hematopoietic cells. Type 3 RYRs are found in skeletal and smooth muscle and to a lesser extent in the brain. The function of RYR1 is modified by other proteins and by the redox state of the cell (Fig. 37-3) (Aracena-Parks et al., 2006; Goonasekera et al., 2007; Durham et al., 2008; Cornea et al., 2009; Protasi et al., 2009).
Pathophysiology of Malignant Hyperthermia
In classic MH, mutation of the RYR1 gene produces enhanced activation of RYR1 by intraluminal calcium and reduced threshold for spontaneous calcium release during store calcium overload (store-overload–induced calcium release) (Jiang et al., 2008). In pigs, a specific single-point mutation in the RYR1 exists in all major breeds that are susceptible to malignant hyperthermia (Fuji et al., 1991). This point mutation is seen in a minority of human families who are susceptible to MH; however RYR1 on chromosome 19q13.1 has been linked to MH susceptibility in the majority of families studied. Of the four other loci proposed as linked to MH susceptibility, only the CACNA1S on chromosome 1q13.1, coding for the α1 subunit of the dihydropyridine receptor, has been linked to MH in more than two human families (Carpenter et al., 2009). The heterogeneous nature of human MH may be the result of a number of different mutations in RYR1 or to abnormalities in other proteins such as the dihydropyridine receptor (Gallant and Lentz, 1992). In humans who have MH susceptibility, RYR1 compound heterozygotes have been observed in several countries. This supports the claim that the incidence of a MH-causative genotype could be as great as 1 in 3000 people (Monnier et al., 2002; Ibarra et al., 2006).
The metabolic dysfunction of MH is caused by increased intracellular ionized calcium (Mickelson and Louis, 1996). A direct result of increased intracellular calcium is an increased need for intracellular ATP to drive pumps that transfer calcium into the SR, across the sarcolemma into the extracellular fluid, or into mitochondria. Increased demands for ATP lead to the clinically detectable manifestations of MH (e.g., an increase in metabolism). The elevation of intracellular calcium observed in MH muscle is decreased by dantrolene (Lopez et al., 1985; Mickelson and Louis, 1996; Cherednichenko et al., 2008).
Motor nerve activity and neuromuscular transmission are normal in patients who have MH susceptibility (Gronert, 1980). However, susceptible porcine muscle has a lower mechanical threshold than does normal muscle, and compared with normal muscle, less depolarization is required to initiate contraction (Moulds and Denborough, 1974; Okumura et al., 1979; Gronert, 1980). Calcium release from the SR occurs at more negative potentials in the presence of RYR1 MH mutations, and calcium-induced calcium release from the SR is abnormal in subjects susceptible to MH. The abnormal RYR1 is resistant to the inhibitory effects of both calcium and magnesium (Mickelson and Louis, 1996).
During an episode of MH in pigs, lactate release from muscle increases before mixed venous oxygen tension decreases (Gronert and Theye, 1976a). This sequence is consistent with intracellular ATP depletion. Magnetic resonance imaging (MRI) techniques showed an increased ratio of inorganic phosphate to phosphocreatine in vivo in human muscle susceptible to MH. This ratio is an indicator of the energy state of the muscle (Chance et al., 1985a, 1985b). An increased ratio suggests either impaired synthesis of ATP or increased breakdown of ATP.
Mechanism of Action of Dantrolene
Since dantrolene was introduced in the 1970s, its use has remarkably improved the treatment and survival of patients with MH. It has been claimed that dantrolene inhibits the release of calcium from the SR by limiting the activation of the calcium-dependent RYR1, but more recently it was shown that dantrolene blocks ECCE across the plasma membrane (Fruen et al., 1997; Cherednichenko et al., 2008). Dantrolene does not act at the neuromuscular junction and has no effect on the passive or active electrical properties of the surface and tubular membranes of skeletal muscle fibers. Patients who have been given dantrolene have normal neuromuscular transmission and depressed force of muscle contraction. The protective effects of dantrolene against MH require significant depression of the force of contraction (Flewellen et al., 1983). Dantrolene can produce clinically significant weakness and decreased metabolism in normal muscle as well as in MH-susceptible muscle (Flewellen et al., 1983). It can reduce elevated creatine kinase (CK) in diseased muscle, independent of the presence of RYR1 mutations (Bertorini et al., 1991; Kamper and Rodemann, 1992).
The dose-response relationship of dantrolene in children has not been reported. Available data suggest that the half-life of dantrolene in children is somewhat shorter than that in adults: 7.3 to 9.8 hours and 12.1 hours, respectively (Fig. 37-4) (Lietman et al., 1974; Flewellen et al., 1983; Lerman et al., 1989). In adults, a cumulative dose of 2.2 to 2.5 mg/kg of dantrolene administered intravenously over 125 minutes produced a steady plasma concentration of dantrolene for longer than 5 hours (Flewellen et al., 1983). Orally administered dantrolene, a total of 5 mg/kg in 3 or 4 divided doses administered every 6 hours to adults with MH susceptibility, has also been shown to produce protective plasma concentrations of dantrolene for at least 6 hours after induction of anesthesia (Allen et al., 1988). In children, intravenous administration of 2.4 mg/kg of dantrolene infused over 10 minutes produced stable blood levels of about 3.5 mcg/mL for 4 hours, after which a slow decline in plasma concentration occurred (Lerman et al., 1989). Hence, it may be reasonable to repeat 1 mg/kg of dantrolene every 5 to 7 hours for prophylaxis. Alternatively, pharmacokinetic modeling with parameters derived in adults recommends a continuous infusion of dantrolene (Podranski et al., 2005). It is likely that when plasma concentrations of dantrolene are sufficient to inhibit an episode of MH, the patient experiences weakness and possibly disequilibrium.
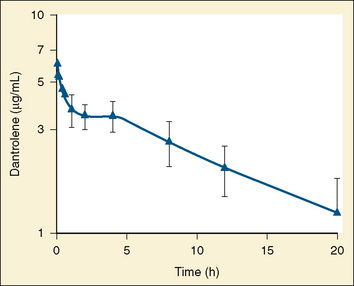
FIGURE 37-4 Plasma decay of dantrolene in children.
(From Lerman J et al: Pharmacokinetics of intravenous dantrolene in children, Anesthesiology 70:625, 1989.)
A dynamometer could be used to assess grip strength objectively, but this requires the patient’s cooperation. In a study of adults, the dose of dantrolene that produced maximal depression of grip strength and evoked force of thumb contraction had no significant effect on vital capacity (Flewellen et al., 1983). Similar studies have not been performed in children. Clinical experience suggests that less than 2 mg/kg of dantrolene administered intravenously to a child preoperatively can be associated with significant hypotonia in the postoperative period (Brandom and Carroll, unpublished observations). Weakness induced by dantrolene could compromise the ability to swallow and even necessitate artificial protection of the airway and mechanical ventilation, although this has never been reported in the literature (Flewellen et al., 1983). Intravenous dantrolene should be administered in settings where support of airway and ventilation can be easily provided.
Clinical presentation
The initial clinical signs of an impending episode of MH are nonspecific. The most commonly reported initial signs are hypercarbia, sinus tachycardia, and masseter spasm or jaw rigidity (Larach et al., 2010). Arrhythmias and tachypnea may be present (Box 37-1). Increased or rapidly increasing temperature is often an early sign of MH, and metabolic acidosis is often not observed before treatment of MH (Larach et al., 2010).
Box 37-1 Steps in Identifying and Treating Malignant Hyperthermia
Clinical events during MH
Rigidity of the extremities and the entire body are signs of fulminant MH. When MH is fulminant, there is severe metabolic acidosis (base deficit often greater than 8 mEq/L), respiratory acidosis (arterial carbon dioxide tension [Paco2] of greater than 60 mm Hg), tachycardia with arrhythmias, a rapid increase in body temperature to 39.5° C or greater, hyperkalemia, myoglobinuria, and often, but not always, a marked increase in serum CK (Gronert, 1980; Newmark et al., 2007). However, because of modern comprehensive monitoring and increased awareness of MH, fulminant MH is rarely observed. The clinical diagnosis of MH is often considered before metabolism and temperature reach these extremes (Karan et al., 1994). The patient’s medical history and clinical course usually, but not always, help to differentiate fulminant MH from other metabolic crises such as porphyria, thyroid storm, untreated pheochromocytoma, and Ecstasy exposure (Allen and Rosenberg, 1990). Some other conditions that produce signs similar to MH are sepsis, allergic drug reactions, intracranial trauma, and hypoxic encephalitis.
Ideally, MH should be recognized and treated before it becomes fulminant. Treatment is more likely to be successful the earlier it is started, but because the signs of MH are not highly specific it is difficult for anesthesia practitioners to distinguish between early MH and many other complications. If there are only mild symptoms or signs suggestive of MH (e.g., moderate increases in heart rate, blood pressure, and temperature along with a slight metabolic or respiratory acidosis), yet evidence is later obtained that the patient is indeed susceptible to MH, this constellation of clinical findings could be called abortive MH. In such cases, masseter spasm may or may not occur (Ellis et al., 1990). There may be a moderate increase in CK and serum myoglobin. When considering the implications of these tests, note that myoglobin appears in the plasma within minutes of muscle injury by succinylcholine. However, CK continues to increase for 8 to 20 hours after a transient injury, even in patients who are not susceptible to MH (Florence et al., 1985). The incidence of abortive MH is as high as 1:4200 anesthesias when succinylcholine is used in combination with potent inhalation anesthetics (Ording, 1985). However, in the first decade of the 21st century, the incidence of MH cases identified by hospital discharge diagnosis codes was estimated to be about 1:100,000 anesthesias (Brady et al., 2009).
The diagnosis of MH may be mistakenly applied to patients suffering hyperkalemic cardiac arrest or sudden exacerbation of chronic rhabdomyolysis, which has occurred in pediatric patients after the administration of succinylcholine (Kovarik and Morray, 1995; Larach et al., 1997; Piotrowski and Fendler, 2007). In this condition, the sudden severe increase in plasma potassium can be fatal, but metabolic abnormalities are secondary to cardiac failure and not increased metabolism in skeletal muscle, as in MH (Delphin et al., 1987; Rosenberg and Gronert, 1992; Tang et al., 1992; Larach et al., 2001).
In a group of 48 children, 17 of whom later proved to be susceptible to MH by muscle biopsy, two or more adverse signs or abnormal laboratory findings were present in all patients with positive in vitro contracture tests (Larach et al., 1987). However, similar adverse events occurred in 83% of the children who had negative muscle-biopsy findings. Generalized muscle rigidity was the single factor significantly associated with positive biopsy findings for MH. However, generalized muscle rigidity is not an absolute predictor of MH susceptibility. Three of 24 patients, who were referred for muscle-contracture testing to evaluate MH susceptibility and had negative contracture test results, had experienced generalized muscle rigidity during induction of anesthesia. Signs consistent with abortive MH, such as tachycardia, premature ventricular contractions, elevated end-tidal carbon dioxide, and increase in tension of the masseter muscle may be observed in the normal pediatric patient who is given halothane or sevoflurane and succinylcholine (Van der Spek et al., 1987; 1988; Lanier et al., 1990). Larach and others (1987) could not identify the patient with MH susceptibility on the basis of these signs (Hackl et al., 1990). Therefore, when MH is suspected clinically, after treating the patient arrangements should be made for confirmatory testing (see related sections in this chapter).
If events during the induction of anesthesia require explanation beyond light anesthesia, transient rigidity and tachycardia during inhalation of sevoflurane, or hypoventilation, further investigation to rule out the diagnosis of MH must be undertaken immediately, before surgery begins. The anesthesiologist must document the presence of increased metabolic rate, rather than the decreased metabolic rate that usually follows induction of anesthesia. Venous or arterial blood should be obtained for measurement of partial pressure of carbon dioxide [Pco2], lactate, potassium, and CK. Mixed venous blood is most likely to show significant alterations in Pco2, but it may not be readily available (Gronert and Theye, 1976a, 1976b). During anesthesia there is increased arterial-to-venous shunting through the skin. Despite this fact, blood from a large peripheral vein, femoral or antecubital, may demonstrate increasing Pco2 and worsening base deficit before these changes are found in arterial blood. Increasing end-tidal carbon dioxide concentrations, particularly with increased minute ventilation, supports the presumptive diagnosis of MH. Evidence of muscle injury, such as the presence of myoglobin in the serum and urine and elevated CK and other enzymes in the blood, may not be observed if MH is treated very quickly. With anesthesia techniques common in the 21st century, MH may not be recognized until the second or third hour of anesthesia. MH may first be recognized in the postoperative recovery room. Fatal MH can occur without exposure to anesthetics (Brown et al., 2000). Once a hypermetabolic state is recognized, appropriate actions must be taken without delay, as described later.
Masseter spasm
Masseter spasm (also termed masseter muscle rigidity or trismus) is a marked increase in tension of the masseter that prevents opening of the mouth when succinylcholine has produced neuromuscular blockade. Masseter spasm may be an early sign of MH. However, succinylcholine can produce increased tension in normal muscle at the same time that it produces block of neuromuscular transmission (van der Spek et al., 1987, 1988). Many anesthesiologists believe that only if the jaw cannot be forced open should this phenomenon be called masseter spasm.
Hannallah and Kaplan (1994) distinguish between masseter muscle rigidity and trismus. In masseter muscle rigidity, the mouth cannot be fully opened even with firm pressure on the incisors, but the mouth can be opened far enough to permit intubation of the trachea. In trismus, the mouth cannot be opened enough to permit intubation of the trachea. Using these definitions, Hannallah and Kaplan (1994) noted rigidity in 0.2% of 500 children anesthetized with halothane and administered succinylcholine, but none of these 500 patients experienced trismus. It may be that the several-fold greater incidence of masseter spasm noted in the 1980s included cases of incomplete jaw relaxation (i.e., the mouth opens fully with firm manual separation of the teeth), which was observed in 4.4% of these 500 pediatric patients. The 22 patients with incomplete jaw relaxation in this study continued to receive halothane anesthesia with no apparent complications. Masseter spasm has been touted as a specific early warning sign of MH. Undoubtedly, deaths from MH have occurred during anesthetic procedures in which masseter spasm was observed and in cases in which it was not observed. It may be that when masseter spasm is accompanied by rigidity of the entire body, MH is likely to occur. However, transient increase in jaw stiffness, or resting tension of jaw muscles, is a normal response to succinylcholine (van der Spek et al., 1987, 1988; Plumley et al., 1990). Increase in masseter muscle tension occurs in normal mammals after the administration of succinylcholine with prior administration of epinephrine (Pryn and van der Spek, 1990). There is a greater increase in jaw tension after administration of succinylcholine during halothane anesthesia than in the presence of barbiturates. Jaw tension is also increased in animals that are febrile as opposed to those that are normothermic (Storella et al., 1993). Temporomandibular joint abnormalities may confuse the diagnosis of masseter spasm by interfering with jaw opening.
Although masseter spasm usually occurs after anesthesia induction with halothane and the administration of succinylcholine, it may occur with other anesthetic agents (Larach et al., 1987; Marohn and Nagia, 1992; Takamatsu et al., 1996). Masseter spasm occurs despite abolition of evoked muscle function in the extremities. Tachycardia or other nonspecific arrhythmias may accompany masseter spasm. MH may follow masseter spasm immediately; in the continued presence of anesthetic trigger agents, however, a period of 10 or more minutes often intervenes between masseter spasm and the clinical presentation of MH when the clinical presentation of MH is defined as arterial Pco2 greater than or equal to 50 mm Hg, pH less than 7.25, and base deficit more negative than 8 mEq/L (O’Flynn et al., 1994).
Anesthesia with halothane has been continued after isolated masseter spasm with no increased metabolism or cardiovascular instability. Littleford and others (1991) reported on 57 such children, of whom 33% experienced transient arrhythmias intraoperatively. Most of these children also had some degree of hypercarbia or metabolic acidosis. CK levels measured 18 to 24 hours postoperatively were elevated in all but one of these children, and CK levels greater than 20,000 units/L were observed in many. However, there were 11 children who experienced generalized rigidity in combination with masseter muscle spasm. Anesthesia was aborted for four of these children and continued without inhalation agents in three of the children. None of these children developed fulminant MH in the perioperative period. The remaining four patients who developed generalized rigidity received dantrolene. Kaplan and Rushing (1992) documented a case of masseter spasm in which clinical abnormalities prompted administration of dantrolene, and postoperative CK was 40,000 IU. Nine years later, this healthy adolescent had completed extensive evaluation for neuromuscular disorders, including in vitro testing for MH with the caffeine-halothane contracture test (CHCT). The patient and family remained without signs, symptoms, or diagnosis of any myopathy. If this patient had been labeled as susceptible to MH, it would have been a misdiagnosis. Total body rigidity accompanying masseter muscle rigidity does not absolutely guarantee that the patient has MH (Larach et al., 1987). Anesthetic depth may have been misjudged; however, if rigidity does not abate when deep neuromuscular block was produced with nondepolarizing blockers, muscle must be abnormal. Either fulminant MH is occurring or the patient may have occult myotonia (Neuman and Kopman, 1993).
Inhalation anesthesia without succinylcholine was associated with fewer episodes of both fulminant (2 vs. 8) and abortive (17 vs. 110) MH than was succinylcholine with potent inhalation anesthetics in a Danish population (Ording, 1985). Thus, avoiding succinylcholine administration to pediatric patients anesthetized with inhalation anesthetics not only avoids the diagnostic uncertainties associated with masseter spasm but also produces fewer episodes of MH and other adverse events (Delphin et al., 1987; Rosenberg and Gronert, 1992). Pediatric anesthesiologists may choose to administer succinylcholine only when definite, strong indications for this drug have been identified.
Management of Masseter Spasm
There is no agreement among experienced clinicians concerning the preferred management of patients with incomplete relaxation of the masseter after the administration of succinylcholine (Kaplan et al., 1993). If jaw stiffness is mild, so that the mouth can be opened with increased effort, there is no rigidity in the rest of the body, and cardiovascular function is stable, anesthesia may be continued with careful documentation of capnography and core temperature. Fluid deficits should be replaced completely so that urine output is greater than 3 mL/kg per hour. Urine should be obtained in the early postoperative period to check for the presence of myoglobin. Blood should be obtained for measurement of electrolytes and CK. It is not necessary to terminate anesthetic administration unless signs of increasing metabolic rate occur. If jaw stiffness is so great that the mouth cannot be opened, there are several reasons to terminate elective anesthetic administration, not the least of which may be the need to clear the upper airway. If trismus and extensor rigidity are present, biochemical signs of MH should be sought and treatment of MH initiated. Venous blood should be obtained for gas analysis and measurement of electrolytes, myoglobin, and CK. If rigidity persists in the presence of neuromuscular blockade either the patient has MH or a form of myotonia.
After an episode of masseter spasm, myalgia and occasionally weakness may be present for several days or longer. In patients who were not susceptible to MH and undergoing ophthalmic surgery with halothane anesthesia who received succinylcholine intraoperatively, an increase in CK level was noted 24 hours after surgery. The highest postanesthetic CK level in these otherwise healthy patients was 40 times that of normal (Inness and Stromme, 1973). Myoglobin appears quickly in the plasma after halothane anesthesia and succinylcholine administration, even in children who had no masseter spasm (Plotz and Braun, 1982). If radioimmunoassay is used to measure serum myoglobin concentrations, increases in myoglobin can be measured within the first hour after succinylcholine administration in normal children anesthetized with isoflurane or halothane.
Postoperative renal failure has occurred in patients who had myoglobinuria after administration of succinylcholine during anesthesia. In any situation in which injury to muscle may occur, it is important to document that myoglobinuria is not present. If increased muscle stiffness is noted after administration of succinylcholine or the if child complains of muscle pain postoperatively, urine should be obtained. If there is no blood in the urine as assessed by orthotolidine (Hematest), then there is no myoglobin present (Bosch et al., 2009). If the response to blood is positive on the dipstick, urine should be examined for the presence of red blood cells, and free hemoglobin and myoglobin should be measured. If myoglobin is present, the patient should remain in the hospital. The patient should be observed for signs of MH, evaluated for the presence of occult muscle disease, and well hydrated. Alkaline urine may reduce the risk of renal tubular injury from myoglobin (Elsayed and Reilly, 2010).
Treatment of an acute episode of malignant hyperthermia
When the diagnosis of MH is strongly suspected, the most important step to take is to administer dantrolene. The other steps in management are to discontinue the triggering anesthetic agents immediately, increase minute ventilation several-fold with 100% oxygen at the highest possible flow rate, check core temperature, and alert the surgeon that the procedure must be concluded promptly. Other anesthesiologists, paramedical personnel, or both should be called in at once for assistance. If core temperature is higher than 39° C, cooling measures should be applied until temperature is below 38° C (see www.mhaus.org).
Dantrolene must be diluted with sterile, preservative-free, distilled water that should be stored in large quantities with the drug (Table 37-1). It is important to store sterile water in clearly labeled containers of a different size from those used for routine intravenous solutions and to keep a mixing system nearby. New formulations of dantrolene may go into solution faster than did dantrolene manufactured before 2008. The initial intravenous dantrolene dose should be 2.5 mg/kg, although more than 10 mg/kg may be needed to control the episode (Larach et al., 2010). Repeated dosing should be guided by clinical and laboratory signs. A flow sheet including minute ventilation, end-tidal carbon dioxide concentration, heart rate and rhythm, arterial blood pressure, central venous pressure, core temperature, and urine output, along with arterial and venous blood gas tensions, serum electrolytes, and glucose and total fluid intake, provides a useful guide for continued therapeutic interventions. Dantrolene must be administered until respiratory and metabolic acidosis, tachycardia and stiffness have resolved. The usual upper limit of 10 mg/kg may be exceeded as necessary. The most common side effects of this drug are muscle weakness and phlebitis.
TABLE 37-1 Drugs and Dosages Used to Treat an Acute Episode of Malignant Hyperthermia
| Dantrolene* | 2.5-10 mg/kg or more (Sterile water must be available to dilute dantrolene.) |
| Sodium bicarbonate | 2 mmol/kg as needed |
| Iced normal saline solution | As needed (10-12 L for 50-kg patient) |
| Mannitol | 300 mg/kg (Note that there is 150 mg of mannitol per milligram of dantrolene in the vial.) |
| Furosemide | 0.5-1 mg/kg |
| Insulin (regular) | 10 units regular insulin in 50 mL of 50% dextrose titrated to produce normokalemia |
| Lidocaine | 1 mg/kg |
* Dantrolene administration should be repeated until physical and chemical signs have returned to normal. When this degree of physiologic stability has been obtained, dantrolene (1 mg/kg or more) should be repeated approximately every 6 hours until all signs of MH, including muscle stiffness, have abated and creatine kinase has decreased consistently. If the patient is metabolically stable, vital signs are normal, and muscle weakness is present, the interval between dantrolene doses can be increased to 8 hours.
The anesthesia machine need not always be switched to a standby unit that has been kept free of inhalation anesthetics. However, because of both the increased internal gas volume of and the increased anesthetic solubility in modern anesthesia work stations, more time may be required to eliminate traces of anesthetic. For example, over 100 minutes of 10 L/min fresh gas flow (10 L/min FGF) was needed to reduce residual inhalation anesthetic to 5 ppm in the Drager Fabius anesthesia machine, whereas after 20 minutes residual sevoflurane was less than 5 ppm in the Drager Narcomed GS machine (Gunter et al., 2008; Whitty et al., 2009). Isoflurane washout can require more than 45 minutes of 10 L/min FGF in the Drager Primus machine, more than 50 minutes in the Datex-Ohmeda machine, and more than 75 minutes in the Aestiva machine (Crawford et al., 2007; Birgenheier et al., 2009). If the carbon dioxide absorber and circuit tubing are not changed, 30 minutes of 10 L/min FGF is needed to reach 2 ppm isoflurane in the Datex-Ohmeda work station (Schonell et al., 2003). In summary, replacement of all exposed elements, including the ventilator diaphragm and breathing system with autoclaved or new parts, provides the fastest preparation of the anesthetic work station without the addition of charcoal filters. Continuation of 10 L/min FGF for the duration of the anesthesia has been recommended to avoid increasing concentrations of agent (Crawford et al., 2007; Whitty et al., 2009). If MH occurs after the first hour of exposure to an inhalation anesthetic, there is a substantial volume of the anesthetic trigger agent in the patient’s body. It is likely that placement of a charcoal filter on the inspiratory limb of the anesthesia circuit and administration of 10 L/min FGF could greatly speed elimination of potent anesthetic from the breathing circuit (Gunter et al., 2008; Birgenheier et al., 2009.)
Procedures to cool the body should be instituted quickly. The goal is to reduce muscle metabolism and avoid exposure to a critical core temperature of greater than 40° C (Bouchama and Knochel, 2002). Furthermore, a temperature that is higher than normal may accelerate an MH episode in humans, as has been observed in animals (Chelu et al., 2006; Dainese et al., 2009). Drapes should be removed, heated humidifiers and hot air blowers should be turned off, and water mattresses should be turned to cooling temperatures. Room temperature normal saline solution should be given intravenously to maintain normal central venous pressure. The stomach can be irrigated with iced saline solution through an orogastric tube. Open body cavities can also be lavaged with iced saline solution, and ice packs can be placed in the groin and axillae where large vessels come close to the skin surface.
Hyperkalemia results when cell membranes are disrupted and when acidosis is severe. This is recognized on the electrocardiogram as increased T-wave amplitude in the early stages and later by widening QRS complexes, intraventricular conduction delays and blocks, and finally no organized rhythm at all. Intravenous calcium is appropriate emergency treatment of the hyperkalemia associated with MH (Gronert et al., 1986). Glucose and insulin (10 units of regular insulin in 50 mL of 50% glucose titrated to effect) can be administered to lower serum potassium temporarily. β-agonists can also be useful to move potassium intracellularly.
Large losses of intravascular volume should be anticipated; evaporative loss of fluid may be great, and edema formation may occur in muscles and in other tissues during fulminant MH. Intravenous fluids should be given to maintain normal cardiac filling pressures, as evidenced by adequate perfusion pressure, urine output, and capillary refill. Alkaline osmotic diuresis is induced by mannitol in the current formulation of dantrolene (150 mg of mannitol/mg of dantrolene in Dantrium). This protects renal tubule function in the presence of myoglobinuria and promotes acute intravascular volume loss. The management of the acute MH episode is summarized in Box 37-2.
Box 37-2 Management of the Acute Malignant Hyperthermia Episode
Follow treatment guidelines prepared by MHAUS (see www.mhaus.org).
Because calcium channel blockers might interfere with excitation-contraction coupling and conserve energy reserves, it is reasonable to ask what effects such drugs might have on MH-susceptible muscle (Lynch et al., 1986). Not all calcium channel-blocking drugs have the same effects in subjects with MH susceptibility. Diltiazem inhibits halothane-induced contracture in MH-susceptible pig muscle, thus confirming a single similar observation in human muscle (Illias et al., 1985). Verapamil, however, is not a therapeutic agent in porcine MH (Gallant et al., 1985). Furthermore, verapamil and dantrolene can interact to produce severe hyperkalemia and myocardial depression (Lynch et al., 1986; Rubin and Zablocki, 1987). Nifedipine administration has been associated with the development of MH in a child with underlying neuromuscular disease (Cook and Henderson-Tilton, 1985). At this time, it seems prudent to administer calcium channel-blocking drugs to patients with a history of MH or neuromuscular disease only with caution. Calcium channel blockers are not recommended in the management of acute MH. If dantrolene must be administered to a patient who is also receiving calcium channel-blocking drugs, invasive hemodynamic monitoring and frequent measurement of serum potassium levels have been recommended (Lynch et al., 1986; Rubin and Zablocki, 1987).
Postanesthesia Considerations
A patient who has been successfully treated for MH in the operating room requires intensive care to continue treatment and to monitor for late manifestations of the disorder. Continuation of treatment is necessary, because recrudescence of MH can occur after an episode that has apparently been successfully treated. This usually happens in the first few hours after the initial event. Patients with a muscular body type, temperature increase, or longer interval between induction of anesthesia and beginning of the MH reaction had increased risk of recrudescence (Burkman et al., 2007). As much as 12 mg/kg of dantrolene has been required to treat recurrences over 12 hours (Pollock et al., 1992). Continuous monitoring of vital signs and minute ventilation, and frequent measurement of venous lactate, blood gases, and electrolytes should detect metabolic changes. Dantrolene should be administered intravenously as necessary, not only until no evidence of metabolic acidosis remains, but also until serum myoglobin levels decrease toward normal or there is no myoglobinuria. The half-life of myoglobin in the blood is normally 1 to 3 hours. In contrast, CK peaks 24 to 36 hours after injury and usually decreases about 40% per day thereafter (Salluzzo, 1992).
After an acute episode, the patient with MH may die of a recrudescence, disseminated intravascular coagulopathy, or other nonspecific systemic injury. Disseminated intravascular coagulopathy is a common finding in fatal MH (Larach et al., 2008). The administration of dantrolene should be continued to stop the disruption of muscle, the presumed underlying cause of disseminated intravascular coagulopathy. Supportive intensive care should be given as indicated, and coagulation function must be carefully monitored during and after an episode of MH.
Late manifestations of an episode of MH range from mild muscle pain to multiorgan system failure. Cerebral edema may occur. Fulminant cases of MH may have permanent neurologic sequelae (e.g., coma or paralysis) for no apparent reason. Even satisfactory care during anesthesia may not prevent these neurologic complications (Gronert, 1980). Rehabilitation can take months after an episode of fulminant MH.
Pulmonary edema may occur because of marked shifts in intravascular volume, myocardial dysfunction, and the effects of heat shock. The presence of pulmonary edema requires more careful assessment of the circulatory status to improve cardiac filling pressures and inotropic state. Areas of myocardium may have abnormal conduction, decreased contractility, or both. It is important to maintain adequate renal perfusion, because massive myoglobinuria produced by fulminant MH can cause acute renal failure. Sufficient muscle damage to produce myoglobinuria and acute renal failure can occur in the absence of pigmenturia or dramatic elevation of CK (Grossman et al., 1974). If myoglobin (or hemoglobin) is present, urine gives a positive reaction with orthotolidine (Hematest). In the presence of myoglobinuria, normal saline solution should be given to force a diuresis of at least 3 mL/kg per hour; if urine output is less than this, then mannitol (1 mL of 25% solution) and bicarbonate (1 mEq) in 5% dextrose in water (D5W; 8 mL) should be given at twice the maintenance fluid rate. Rapidly increasing serum creatinine signals the onset of renal failure.
All cases of MH, anesthetic-related episodes of increased metabolism or rhabdomyolysis, isolated masseter spasm, and anesthetics administered to patients who have undergone a CHCT should be reported to the Malignant Hyperthermia Registry, so that the epidemiologic study of MH may have as broad a scope and as complete a collection of data as possible. Report forms may be obtained by telephone (888-274-7899) or printed from www.mhreg.org.
Prophylactic management
Indications for Muscle Biopsy and Genetic Evaluation of Type 1 Ryanodine Receptor
Indications may vary depending on the particular goals being addressed. Individuals, both patients and physicians, concerned with improving the diagnostic tests for MH may urge all patients with any symptoms consistent with MH and those with a clear history of fulminant MH to undergo muscle biopsy and CHCT. Others prefer to advise that this invasive test be performed only when the predictive value of the test is likely to be helpful for patient management. The CHCT is currently the only method that can rule out MH susceptibility. The Clinical Grading Scale (CGS) was developed using the Delphi process to provide clinical definition of a case that is almost certain to be MH for the purpose of defining the diagnostic criteria of the CHCT (Larach et al., 1994). Box 37-1 contains many of the elements of the CGS. The five processes evaluated by the CGS (rigidity, muscle breakdown, respiratory acidosis, temperature increase, and cardiac involvement) each contribute a limited number of points to the score. Scores are further divided into categories of likelihood of the patient having MH. Category 6 is almost certain MH; category 5 indicates that MH is very likely; category 4 means that MH is somewhat greater than likely; category 3 indicates that MH is somewhat less than likely; category 2 implies that MH is unlikely; and in category 1, the chance of a patient having MH is almost none. If the total score of the CGS is 50 or more, the case is category 6, almost certain to be MH. If the score is between 35 and 49 the case is category 5, very likely to be MH. The CGS must be applied with medical judgment to estimate the probability that a given anesthesia event was MH. It is important to document the adequacy of minute ventilation when interpreting carbon dioxide tensions. Increased minute ventilation may be the only sign of MH. This produces a category 3 score on the CGS, indicating that MH is somewhat less than likely and might suggest that further testing is not warranted. However, if the anesthesiologist feels that carbon dioxide production was increased and this was reversed by elimination of potent anesthetic, the patient should be referred for testing of MH susceptibility. On the other hand, a child who has sepsis with cerebral palsy, or one of many other medical conditions, may have a high score on the CGS. Referral of a patient whose illness is explained by other processes for evaluation of MH susceptibility would be a costly waste of effort.
A new benefit of undergoing CHCT is that patients with contracture tests indicating MH susceptibility are candidates for genetic study. Initial studies of the RYR1 gene in these subjects with MH susceptibility were limited to the three hot spots in RYR1. However, as a larger proportion of the RYR1 gene is studied, the percentage of patients who have MH susceptibility with RYR1 mutations or variants of unknown significance (VUS) has increased to 50% or more (Robinson et al., 2003; Sambuughin et al., 2005; Ibarra et al., 2006; Levano et al., 2009). The process of evaluating MH susceptibility in relatives can begin with genetic evaluation in search of the familial mutation (Urwyler et al., 2001; Girard et al., 2004) after an RYR1 mutation has been identified in the proband. Because fewer than 50% of all people who undergo contracture testing may be diagnosed as susceptible to MH by this test, if evaluation of MH status begins with screen of RYR1, the yield of positive diagnoses will be low (Girard et al., 2004).
A panel of 17 RYR1 exons, encompassing all the acknowledged MH mutations, is available as a clinically useful genetic test of MH susceptibility (Sei et al., 2004) (see www.mhaus.org for the addresses of the diagnostic laboratories). It may detect a RYR1 abnormality in about 30% of North American patients who would be diagnosed as susceptible to MH by CHCT. With this test, genetic counseling is available at the University of Pittsburgh, Center for Medial Genetics (800-54-8155). It is expected that genetic testing of MH susceptibility will be less sensitive but also less invasive and less costly than muscle biopsy and CHCT, because the genetics of MH susceptibility is not completely known (Nelson et al., 2004). Therefore, clinical genetic testing is of most value for determining affected family members in a family in which an RYR1 mutation has been found in an individual who experienced severe MH or had a positive CHCT. The incidence of compound heterozygotes is relatively high when the entire RYR1 gene is examined. Therefore, if an individual does not have the familial RYR1 variant, contracture testing must be performed to prove that the patient is not susceptible to MH.
Individuals who have had positive muscle contracture tests after experiencing MH episodes should undergo genetic screening and document the result in the MH Registry (www.mhreg.org; 888-74-7899). If no abnormality is found in the RYR1 gene in such patients, these are important subjects for experimental study of MH genetics.
The sensitivity and specificity of the CHCT have been determined; therefore, general statements about the predictive value of this test can be made (Larach et al., 1992; Larach, 1993; Ording et al., 1997; Allen et al., 1998). The clinician needs to know the predictive value of the test because the clinician can obtain the results of the diagnostic test. Before applying the test, the clinician cannot be sure whether or not the patient really has the disease.
The positive predictive value (PPV) of a test is the probability that an individual whose test result indicates that the disease is present does indeed have the disease of interest. The negative predictive value (NPV) of a test is the probability that an individual whose test result indicates that disease is absent does not have the disease in question. By definition, the predictive value depends on the probability that the individual has the disease in question before the test results are obtained, as well as the sensitivity and specificity of the test (Rosner, 1990). The sensitivity of a diagnostic test is the probability that a test result will indicate the disease is present, when in fact the individual tested has the disease in question. Specificity is the probability that a test result will indicate disease is not present, when in fact the individual tested does not have the disease in question. The definition of a positive result of the CHCT was set to make this test sensitive, so that fewer than 1% of the people who would be at risk of MH events would fail to be identified by the CHCT. Therefore, the CHCT could not also be highly specific. To apply Bayes Theorem and estimate the PPV or NPV, given the sensitivity and specificity of the CHCT, a clinician also has to make a decision about the probability that the patient is MH susceptible before the diagnostic results are known (Rosner, 1990). This probability may be thought of as the prior probability of the individual having the disease of interest or, if a population rather than an individual is considered, the incidence of the disease in the population (Box 37-3).
Box 37-3 Interpretation of a Test
Rather than speculate on what a group of clinical findings suggests about the probability of MH susceptibility in an individual, it is possible to use the test characteristics (sensitivity and specificity) to calculate the predictive value of the CHCT over a wide range of prior probabilities (Fig. 37-5, A and B). For example, if a patient with a history of isolated masseter spasm is thought to have less than a 25% chance of having MH, then if the sensitivity of the CHCT is 99% and the specificity is 85%, the positive predictive value of the CHCT in that individual is less than 69% and the negative predictive value is greater than 99%. In other words, the chance of that individual having a false-positive result of CHCT is at least 30%, and the chance of a false-negative result is less than 1%. In contrast, if a patient with a strong family history of MH has many of the clinical signs of MH, it might be judged that the probability of that individual having MH, before obtaining the result of the CHCT, would be 80%. The positive and negative predictive values of the CHCT would be 96% and 99%, respectively, for such a patient. These statements are oversimplifications in that these calculations assume that the disease being tested for has similar manifestations in all affected individuals. Certainly this is not the case for MH. Nevertheless, appreciation of the concept of the predictive value of a test is important when questions arise regarding the meaning of clinical events and test results. These concepts have been used to argue that the index case should be the first person to undergo contracture testing, followed by first-degree relatives (Loke and MacLennan, 1998a; Larach and MacLennan, 1999). The same order of testing is appropriate for genetic analysis.
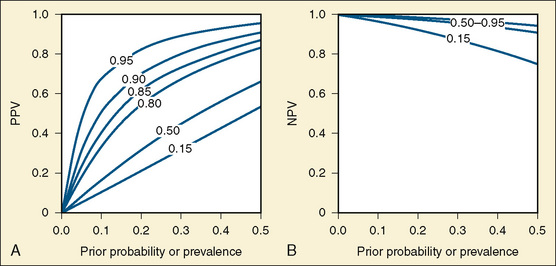
FIGURE 37-5 A, The y-axis is the PPV of the CHCT. This figure illustrates the fact that when the sensitivity is 95% and the specificity is 85%, the positive predictive value is less than 50% when the prior probability is less than 15%. B, The y-axis is the NPV of the CHCT. NPV is less altered by specificity than PPV when prior probability is less than 50%. Thus, the NPVs coincide for specificities from 85% to 95%. These two images illustrate the relationship between the sensitivity and specificity of a test, the prior probability of the disease, and the predictive value of that test (see definitions in Box 37-3). The probabilities on the graphs are shown as decimals between 0 and 1. In both figures, the x-axis is prior probability of MH susceptibility in the individual under consideration. This may be thought of as the probability that the individual under examination is susceptible to MH before the results of the CHCT are considered. This probability ranges from 0 to 0.5, or 50%, in these figures. In both figures, the sensitivity of the test is 0.95, or 95%. In both figures, the specificity of the CHCT varies between 0.95 (95%) and 0.15 (15%), as labeled on the dotted lines. The heavy line is the predictive value of the CHCT when sensitivity is 95% and specificity is 85%.
Currently, muscle biopsy and contracture testing of viable muscle is the only method that can confirm the diagnosis of not MH susceptible. The procedure can be viewed at www.mhaus.org. For satisfactory in vitro testing, 1 g of muscle must be removed from the thigh. A child weighing less than 20 kg may be too small to undergo muscle biopsy for CHCT. In general, children younger than 10 years of age are too young to undergo CHCT. Diagnostic standards were developed in adults.
Parents of an affected child may wish to have a muscle biopsy and contracture test performed (Figs. 37-6 and 37-7 and www.mhaus.org). The relatives of the parent whose findings are negative (assuming autosomal dominant inheritance) can then be reassured, without further testing, that they have no increased risk of MH. Ideally, siblings and first cousins on the affected side should be informed and offered diagnostic testing. Financial and geographic considerations often discourage families in these endeavors. However, the costs are less for genetic testing than for muscle contracture testing. Clinical genetic testing does not require travel to the location of the testing lab, as is necessary for CHCT. At the very least, relatives of an patient with MH susceptibility should be informed about the presence of MH susceptibility in their family and its implications.
A valuable self-help resource is the Malignant Hyperthermia Association of the United States (MHAUS; PO Box 1069, 11 East State St., Sherburne, NY 13460; fax 1-607-674-7910). This organization offers information, expert consultation and referral, and many useful resources for families and health care providers. The MHAUS newsletter contains up-to-date information and reviews of the recent professional literature on topics related to MH. MHAUS maintains a 24-hour, professionally staffed telephone line to provide information on diagnosis, treatment, and referral of patients with MH (1-800-644-9737). See www.mhaus.org for addresses of the clinical laboratories at which genetic testing and muscle contracture testing are performed.
Care of Patients with a History of Malignant Hyperthermia
CK levels, at rest, are of no predictive value in the general population (Britt et al., 1976; Paasuke and Brownell, 1986). If a relative of a patient known to be susceptible to MH has an elevated CK level, that individual has an increased likelihood of susceptibility; however, this will not hold true for patients with other problems producing CK elevations (e.g., Duchenne’s muscular dystrophy [DMD]). In some populations, more than 25% of the patients with elevated CK levels were not susceptible to MH on in vitro testing (Ellis et al., 1975).
Preoperative Care of Patients Who Are Susceptible to Malignant Hyperthermia
When it is clear that the suspected episode was MH, and especially when contracture testing has already been done and shows a typical MH pattern, several decisions must be made. If it is at all possible, regional or local anesthesia should be chosen. Although some theoretic objections to amide-type local anesthetics exist, data in animals and humans have not shown any local anesthetic to trigger MH. Review of anesthetics during muscle biopsy suggests that nerve blocks with small volumes of amide local anesthetics do not provoke an episode of MH (Berkowitz and Rosenberg, 1985). Regional anesthesia with lidocaine, bupivacaine, ropivacaine, or another local anesthetic is an acceptable choice for the patient who is susceptible to MH.
Preoperative prophylactic dantrolene is not indicated. Because many muscle biopsies and other operations without preoperative administration of dantrolene or postoperative complications have been performed in patients with positive CHCT, it is considered acceptable to withhold preoperative dantrolene (Ording et al., 1991). These patients must be closely monitored during anesthesia with nontriggering agents (Table 37-2). If there are no signs of increased metabolism or rhabdomyolysis, and if no dantrolene was administered, then patients with MH susceptibility may be safely discharged on the day of surgery, even as soon as 2.5 hours after leaving the operating room (Yentis et al., 1992; Pollock et al., 2004, 2006).
TABLE 37-2 Malignant Hyperthermia and Drugs Used During Anesthesia
| Drugs Likely to Trigger MH | Drugs that Do Not Trigger MH |
| Potent inhalation anesthetics: | Narcotics: |
| Sevoflurane | Benzodiazepines |
| Desflurane | Barbiturates |
| Isoflurane | Propofol |
| Halothane | Ketamine |
| Enflurane | Nondepolarizing (competitive) blockers |
| Ether | Anticholinesterases and anticholinergics |
| Depolarizing neuromuscular blocker succinylcholine | Local anesthetics |
| Nonsteroidal antiinflammatory drugs | |
| Calcium |
Perioperative Care of Patients Who Are Suspected of Being Susceptible to Malignant Hyperthermia
An anesthesiologist may encounter patients who have had an anesthetic course or who have a family history that suggests susceptibility to MH but who have not had that possibility evaluated by means of muscle biopsy and in vitro contracture testing. A reasonable approach to providing anesthesia for such patients is to administer a nontriggering anesthetic and monitor the patient carefully for signs of MH (when this is consistent with safe anesthesia in that patient). It may not be possible to diagnose the underlying muscle disorder, but it is practical to watch for the processes that may hurt the patient postoperatively if an occult myopathy is present; these processes are hyperkalemic cardiac arrest, MH in the postanesthesia care unit (PACU), hyperthermia later resulting in multiorgan system failure, and myoglobinuric renal failure. Electrocardiogram can be monitored in the PACU in children. If there are no signs of MH in the first hour postoperatively, MH is unlikely to occur later (Litman et al., 2008). Rectal acetaminophen could block the increase in thermoregulatory setpoint that may produce postoperative temperature elevation and appropriate fluid replacement can promote dissipation of heat during recovery from anesthesia. Urine should be examined before discharge if a diagnosis of MH or other occult muscle disease is suspected. Dipstick of the urine with no blood demonstrates that there is no myoglobin in the urine; if there is no myoglobin in the urine, the patient has no increased risk of renal injury.
Anesthesia techniques
Physiologic responses to stress may play a part in the initiation of an episode of MH in humans, as well as in pigs (Gronert and Theye, 1976a, 1976b; Gronert et al., 1980). Anesthesia for the patient who is susceptible to MH should be designed to be as stress free as possible, so that any tachycardia and arrhythmias that may occur are more likely to be associated with impending MH than with the stress of anesthetic, induction, or surgery.
There are many nontriggering techniques for general anesthesia (see Table 37-2). Local anesthetic cream can produce topical analgesia of the skin, which may facilitate intravenous catheter placement. Preoperative medication with midazolam, 0.3 to 0.7 mg/kg orally or 0.2 to 0.3 mg/kg nasally, often produces sedation adequate to facilitate the placement of an intravenous catheter in a child. In the opinion of some anesthesiologists, preoperative sedation is an important part of the anesthetic management for patients who are susceptible to MH. After placement of an intravenous catheter, anesthesia may be induced with barbiturates or propofol. narcotics, and α2-agonists (e.g., dexmedetomidine), as indicated by the planned surgery and other characteristics of the patient (Raff and Harrison, 1989; Harrison, 1991). Intramuscular administration of general anesthetics may be an alternative. Prior administration of benzodiazepine can reduce chest and truncal rigidity commonly observed after the administration of a synthetic narcotic. A nondepolarizing neuromuscular blocking agent may be administered if necessary. It is helpful to use a peripheral nerve stimulator when a neuromuscular blocker is administered so that the dose of drug can be titrated to the desired effect. Similarly, an anticholinesterase should be administered as indicated by the results of peripheral nerve stimulation.
There are rare reports of changes compatible with MH occurring after the use of such “safe” general anesthetic drugs (Fitzgibbons, 1980; Pollock et al., 1992; Shukry et al., 2006). For susceptible patients, there is no absolutely safe general anesthetic technique—merely drugs that are less likely to trigger MH. It is advisable to avoid drugs that may affect temperature regulation and sympathetic tone to such an extent that it might be difficult to detect early signs of insidious MH. Furthermore, serotonergic agonists and some psychotropic drugs (e.g., Ecstasy) have produced MH episodes in susceptible pigs (Wappler et al., 1997; Fiege et al., 2003). Large doses of phenothiazines and anticholinergics are not drugs of choice for the patient who is susceptible to MH. Atropine is administered only when there is significant risk of bradycardia. However, some anesthesiologists have found ketamine to be a useful anesthetic in patients who are susceptible to MH.
It is helpful to keep drugs and supplies to treat MH in a portable container, such as a carry-all or rolling cart that is immediately accessible in the operating room and recovery room and can easily be transported to other areas of the hospital (see Table 37-1). The necessary supplies include at least 5 to 10 mg/kg of dantrolene and liter quantities of sterile, preservative-free, distilled water in which to dissolve the dantrolene. Ice or cold packs should be available, and large volumes of normal saline solution should be available in a nearby refrigerator.
Anesthetic Management for Muscle Biopsy for Contracture Testing
The preceding recommendations for the anesthetic care of patients with MH susceptibility apply to the patient undergoing diagnostic muscle biopsy. Biopsy for CHCT can be done only in one of the four specialized centers in the United States that support the performance of this in vitro test. The list of currently active centers may be obtained from www.mhaus.org.
The child scheduled for a biopsy usually weighs more than 20 kg, and some biopsy center directors will test only children who are at least 10 years old. The patient may be mature enough to have regional anesthesia before biopsy; in a cooperative child the use of lateral femoral-cutaneous nerve block and intravenous sedation is often successful (Berkowitz and Rosenberg, 1985). If the child cannot cooperate, general anesthesia may be induced and maintained with various agents, including benzodiazepines, narcotics, propofol or barbiturates, dexmedetomidine, nondepolarizing relaxants, or nitrous oxide.
Postoperative Care of Patients Who Are Susceptible to Malignant Hyperthermia
If anesthesia has proceeded uneventfully and prophylactic dantrolene has not been administered, the patient should be transported to the recovery room for the usual monitoring of vital signs, electrocardiogram, mental status, and urine output and color. A dipstick should be used to check urine for blood. The reaction of heme with orthotolidine occurs if myoglobin, free hemoglobin, or red blood cells are present (Bosch et al., 2009). If no sign of MH appears, the patient may be transferred to the floor after an ordinary length of stay in the recovery room. After observing such a patient for 2.5 hours after the end of anesthesia without any sign of MH, the patient may be discharged from the hospital. In this way, it is possible for a child who is susceptible to MH to be treated successfully as an outpatient (Yentis et al., 1992; Pollock et al., 2004, 2006).
Disorders associated with malignant hyperthermia
A number of disorders have been thought to be related to the MH syndrome because their presence appears to increase the risk of MH during anesthesia, but solid evidence is often lacking (Table 37-3) (Davis and Brandom, 2009). A complete understanding of these syndromes could contribute to understanding the pathophysiology of MH. Although not all myopathic patients are susceptible to MH, the presence of a known or suspected myopathy should alert the anesthesiologist that there may be increased potential for MH susceptibility or anesthesia complications that may mimic MH (Heytens et al., 1992).
TABLE 37-3 Association of Various Diseases with Malignant Hyperthermia
| Disease | Observed Anesthetic Complications |
| Duchenne’s muscular dystrophy (DMD) | Rarely, chronic rhabdomyolysis increases and may result in hyperkalemic cardiac arrest. |
| Becker muscular dystrophy | As in DMD, and growth of new muscle increases likelihood of hyperkalemia after succinylcholine. |
| Noonan syndrome | Not related to MH |
| Osteogenesis imperfecta | Increase in temperature is common but not related to MH. |
| King-Denborough syndrome | Susceptible to MH |
| Carnitine palmitoyltransferase II deficiency | MH susceptibility plausible but unproven |
| Myophosphorylase B deficiency (McArdle disease) | Rhabdomyolysis is common with any stress; CHCT may be diagnostic of susceptibility to MH, but few anesthesia experiences have been recorded. |
| Myoadenylate deaminase deficiency | As in McArdle disease |
| Brodie disease | Not susceptible to MH |
| Asymptomatic hyperkalemia | Causes of this syndrome may be many. Patients are likely to have hyperkalemia after succinylcholine and may have normal CHCT. Alternatively, an individual with increased CK may be a patient with central core disease (CCD) who is asymptomatic. MH is likely in CCD. |
| Myotonia congenita | No increased risk of MH over that in the general population |
| Paramyotonia congenita | No increased risk of MH over that in the general population |
| Potassium-aggravated myotonia | No increased risk of MH over that in the general population |
| Myotonia fluctuans | No increased risk of MH over that in the general population |
| Myotonia permanens | No increased risk of MH over that in the general population |
| Acetazolamide-responsive myotonia | No increased risk of MH over that in the general population |
| Hyperkalemic periodic paralysis ± myotonia | No increased risk of MH over that in the general population |
| Myotonic dystrophy type I (Steinert disease) | No increased risk of MH over that in the general population |
| Myotonic dystrophy type II | No increased risk of MH over that in the general population, but there is greatly increased risk of adverse respiratory events postoperatively, as well as heart block. See detailed texts on this disease. |
| Hypokalemic periodic paralysis | Unclear, but there may be greater risk of MH than in the general population. There is potential for dangerous stiffness after succinylcholine, but metabolism may not increase as it does in MH. |
| Central core myopathy (CCD) | Susceptible to MH |
| Multiminicore disease with RYR1 mutation | Susceptible to MH |
| Multiminicore disease without RYR1 mutation | Maybe susceptible to MH |
| Nemaline rod myopathy without RYR1 mutation | Maybe susceptible to MH |
| Nemaline rod myopathy with RYR1 mutation | Susceptible to MH |
From Davis PJ, Brandom BW: The association of malignant hyperthermia and unusual disease: when you are hot you are hot or maybe not, Anesth Analg 109:1001, 2009.
Many patients with DMD have received potent inhalation anesthetics without occurrence of MH, but dystrophic muscle is fragile and can have RYR1 dysfunction (Peluso and Bianchini, 1992; Bellinger et al., 2009). Even mild exercise in patients with DMD results in a marked egress of sarcoplasmic components into the plasma, most notably myoglobin, CK, and potassium (Florence et al., 1985). These patients can have rhabdomyolysis during anesthesia with potent inhalation anesthetics even without the administration of succinylcholine (Rubiano et al., 1987). It is not surprising that anesthetic complications with many of the qualities of MH occur in patients with abnormal dystrophin (Kleopa et al., 2000) (see Chapter 32, Systemic Disorders). Nevertheless, there has been at least one patient who had dystrophinopathy and a negative contracture test, which ruled out MH susceptibility in that patient (Gronert et al., 1992).
Less common than dystrophinopathy, enzyme deficiencies and mitochondrial disease in muscle may also produce rhabdomyolysis, which can be easily exacerbated. Nonspecific signs of MH may occur during anesthesia (Hogan and Vladutiu, 2009). If the patient has a history of recurrent rhabdomyolysis, blood-based genetic testing of carnitine palmitoyltransferase II (CTP2), myophosphorylase (PYGM; deficiency of PYGM is McArdle disease), and myoadenylate deaminase (AMPD1) may determine the cause. When these blood tests are not diagnostic, muscle biopsy may be useful for diagnosis of these enzyme deficiencies as well as deficiencies in the glycolytic pathway and mitochondrial diseases.
Several extensive family studies of central core disease (CCD) have documented morphologically abnormal muscle—i.e., central cores of oddly aligned fibers—in some patients with MH (Shy and Magee, 1956; Byrne et al., 1982; Jeong et al., 2008). CCD is the first myopathy for which a definite link to MH was shown. Multiminicore disease is a different histologic diagnosis that has also been associated with susceptibility to MH and may be related to CCD (Ferreiro et al., 2002; Jeong et al., 2008). Several mutations in RYR1 cause both CCD and MH susceptibility; therefore, it is advisable to treat a patient with CCD as susceptible to MH (Loke and MacLennan, 1998b; Tilgen et al., 2001; Robinson et al., 2006). The risk of MH in CCD, multiminicore disease, and nemaline rod myopathy was recently reviewed by Klinger and colleagues (2009).
Another familial myopathy, the King-Denborough syndrome, is associated with MH susceptibility (King and Denborough, 1973). Affected individuals have proximal muscle weakness, postural imbalances, cryptorchidism, webbed neck, pectus deformities, delayed development, and elevated levels of CK (Jurkat-Rott et al., 2000). Variants in the RYR1 gene have been found in some patients with King-Denborough syndrome.
In the past, other disorders in which patients appeared to have MH susceptibility included myotonia congenita, osteogenesis imperfecta, Schwartz-Jampel syndrome (dwarfism, craniofacial and skeletal abnormalities, blepharophimosis, and muscle stiffness), and possibly arthrogryposis (Fowler et al., 1974; Rampton et al., 1984; Baines et al., 1986). These syndromes may be associated with many of the symptoms of MH, but these symptoms are not specific for MH. Many patients with these and other muscular disorders have received inhalation anesthetics without complications. For example, a pyloromyotomy was performed in an infant with paramyotonia congenita during sevoflurane anesthesia (Ay et al., 2004). Sometimes evaluation of suspect cases has found CHCT to be negative (Hopkins et al., 1991). A recent review of myotonias concluded that with the exception of hypokalemic periodic paralysis (hypoKPP), the risk of MH in myotonic patients is not increased (Parness et al., 2009). For theoretic reasons, although there are no published case reports of MH-like adverse events in patients with hypoKPP, risk of MH may be increased in this condition. Definitive diagnosis of myotonias and other channelopathies (including MH) should be pursued, because specific treatment may be available (Lehmann-Horn et al., 2008).
The neuroleptic malignant syndrome (NMS) is a potentially fatal disorder recognized by psychiatrists that may clinically resemble MH (Cohen et al., 1985; Guzé and Baxter, 1985; Mann et al., 2003). It occurs in 1:200 patients taking neuroleptic drugs that produce dopaminergic blockade. A 24-hour consultant service is available through the Neuroleptic Malignant Syndrome Information Service (NMSIS; www.nmsis.org; 888-667-8367). Most patients with NMS are young men with schizophrenia or mania treated with the potent piperazine phenothiazines or haloperidol, but more than 25 drugs have been implicated (Heiman-Patterson, 1993). NMS may also occur when the administration of antiparkinsonian drugs is stopped. The manifestations of NMS are altered mental status, hypermetabolism with fever, tachycardia, muscle rigidity, and myoglobinuria. NMS has all of the clinical features of MH, including acute renal failure and multiorgan failure, but it progresses over hours to days rather than minutes.
The inciting events of NMS differ from those of MH. Blockade of dopamine receptors can produce hyperthermia and rigidity, and bromoscriptine, (dopamine agonist) as well as dantrolene and benzodiazepines have been used to successfully treat this syndrome (Caroff, 1980; Granati et al., 1983; Heiman-Patterson, 1993). The results of CHCT in patients with a history of NMS have been inconsistent (Caroff et al., 1987; Adnet et al., 1989). Drugs that are known triggers of MH have been well tolerated in patients who have had NMS. Repeated exposure of pigs with MH susceptibility to a serotonin-2 receptor agonist can also induce typical MH symptoms without causing the same syndrome in normal animals (Gerbershagen et al., 2003). Mann et al. (2003) demonstrated that in humans the serotonin syndrome (tremor, diaphoresis, shivering, and myoclonus in the presence of serotonergic medication) could be elicited easily in individuals susceptible to MH. Dantrolene can delay serotonin-induced contractures (Wappler et al., 1997).
Contracture test
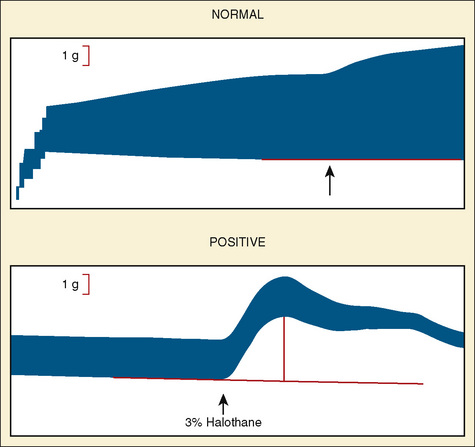
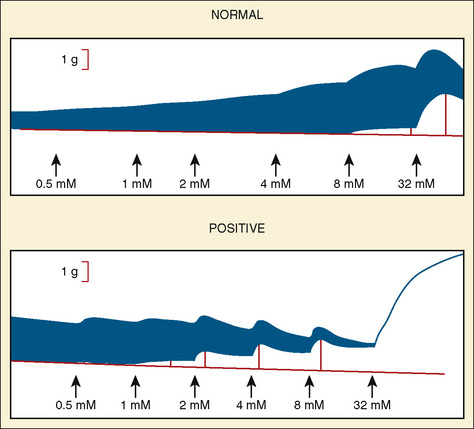
The CHCT is the best laboratory test available to investigate susceptibility to MH. The methods for performance of the test have been standardized in North America and slightly differently in Europe (Larach et al., 1989; Ording and Bendixen, 1992). Both tests provide consistent normal population values for comparison with diagnostic biopsies. In both North America and Europe, the CHCT is a concentration-response curve to caffeine alone and halothane alone. Unlike usual concentration-response phenomena, differences between persons susceptible to MH and those who are not do not appear as altered (median effective dose) ED50 or as a change in slope, but as a change in threshold. Patients must travel to the MH diagnostic center where this bioassay can be performed. The muscle strips to be tested must respond with active twitch to electrical stimulation (i.e., they must be viable). The test should be completed within 5 hours of the excision of muscle from the patient’s thigh.
The North American protocol includes exposure of muscle strips to 0.5, 1, 2, 4, 8, and 32 mmol/L caffeine. A positive CHCT is often defined as an increase in tension of 0.2 g in the presence of 2 mmol/L caffeine or less. However, a cutoff of 0.3 g or 0.4 g may be preferred to increase the specificity of the test (Larach, 1989). A positive halothane contracture test is the development of more than 0.2 g to 0.7 g tension (depending on the controls in that laboratory) in the presence of 3% halothane (Larach et al., 1989). In the United States, if one muscle strip produces a positive reaction in either caffeine or halothane, the patient is said to be susceptible to MH.
Other laboratory tests have been evaluated for their diagnostic usefulness in MH; these include calcium uptake into frozen muscle, skinned fiber testing, platelet nucleotide depletion measurement, and the measurement of abnormal proteins in MH muscle. None of these is generally accepted, because none has been shown to reproduce the results of the in vitro muscle contracture tests (Lee et al., 1985; Britt and Scott, 1986; Whistler et al., 1986; Nagarjan et al., 1987). Several of the proposed tests produced inconsistent results (Ording et al., 1990; Quinlan et al., 1990).
MH is a disorder of muscle that is usually subclinical until the muscle is stressed. Tests discern MH susceptibility only if they impose a stress on the intact tissue or organism or detect a genetic difference that has been demonstrated to be causative (Urwyler et al., 2003). MH muscle testing is generally designed to avoid false-negative diagnoses; hence, there may be false-positive results of the CHCT. Findings suggest that as with all tests, there are some rare false-negative contracture test results (Larach, 1993). However, overall MH contracture testing appears accurate, and patients with negative findings on CHCT can receive safe anesthetics with drugs that could trigger MH (Ording et al., 1991).
Failure to detect an RYR1 mutation does not imply that the patient is not susceptible to MH. If a patient undergoes genetic evaluation before contracture testing and no MH-causative mutation is found, the patient must undergo contracture testing in order to support the diagnosis of not susceptible to MH. All patients should be monitored for signs of MH during anesthesia. Furthermore, MH can occur without the use of triggering drugs such as succinylcholine and potent inhalation anesthetics (Fitzgibbons, 1980; Pollock et al., 1992; Shukry et al., 2006).
Anesthesiologists’ responsibility to other physicians
Physicians who are aware of the potential seriousness of MH may ask what the implications of the diagnosis of MH susceptibility are for the patient’s daily life. MH susceptibility has been associated with fourfold increases in plasma catecholamines with graded exercise and rapid exhaustion after intense exercise; however, individuals susceptible to MS have performed farm labor in the hot sun without precipitating an MH attack (Wappler et al., 2000; Rueffert et al., 2004). Early studies of metabolic responses during noncompetitive, low-intensity, steady-state exercise found no difference between control and patients with MH susceptibility (Green et al., 1987). Slower recovery of muscle pH and phosphocreatine/inorganic phosphate ratios have been observed in patients with MH susceptibility (Allsop et al., 1991; Olgin et al., 1991). Patients who are susceptible to MH should be encouraged to refrain from strenuous exercise if they experience cramps or fever under such circumstances (Davis et al., 2002). Some people with MH susceptibility are unable to sustain exercise in the heat as well as they do in cool environments, and some patients with exertional heat stroke are actually susceptible to MH (Bendahan et al., 2001; Tobin et al., 2001; Wappler et al., 2001). Dantrolene may be therapeutic when these symptoms occur (Gronert, 1980).
For questions and answers on topics in this chapter, go to “Chapter Questions” at www.expertconsult.com.
Adnet P.J., Krivosic-Horber R.M., Adamantidis M.M., et al. The association between neuroleptic malignant syndrome and malignant hyperthermia. Acta Anaesth Scand. 1989;33:676.
Allen G.C., Cattran C.B., Peterson R.G., et al. Plasma levels of dantrolene following oral administration in malignant hyperthermia-susceptible patients. Anesthesiology. 1988;69:900.
Allen G.C., Rosenberg H. Phaeochromocytoma presenting as acute malignant hyperthermia: a diagnostic challenge. Can J Anaesth. 1990;37:593.
Allen G.C., Larach M.G., Kunselman A.R., et al. The sensitivity and specificity of the caffeine-halothane contracture test: a report from the North American Malignant Hyperthermia Registry. Anesthesiology. 1998;88:579.
Allsop P., Jorfeldt L., Rutberg H., et al. Delayed recovery of muscle pH after short duration, high intensity exercise in malignant hyperthermia susceptible subjects. Br J Anaesth. 1991;66:541.
Aracena-Parks P., Goonasekera S.A., Gilman C.P., et al. Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J Biol Chem. 2006;281:40354.
Ay B., Gercek A., Dogan V.I., et al. Pyloromyotomy in a patient with paramyotonia congenital. Anesth Analg. 2004;98:68.
Baines D.B., Douglas I.D., Overton J.H., et al. Anaesthesia for patients with arthrogryposis multiplex congenita: what is the risk of malignant hyperthermia? Anaesth Intensive Care. 1986;14:370.
Ball C. Unraveling the mystery of malignant hyperthermia. Anaesth Int Care. 2007;35(Suppl):26.
Bellinger A.M., Reiken S., Carlson C., et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15:325.
Bendahan D., Kozak-Ribbens G., Confort-Gouny S., et al. A noninvasive investigation of muscle energetics supports similarities between exertional heat stroke and malignant hyperthermia. Anesth Analg. 2001;93:683.
Berkowitz A., Rosenberg H. Femoral block with mepivacaine for muscle biopsy in malignant hyperthermia patients. Anesthesiology. 1985;62:651.
Bertorini T.E., Palmieri G.M., Griffin J., et al. Effect of dantrolene in Duchenne muscular dystrophy. Muscle and Nerve. 1991;14:503.
Birgenheier N.M., Westenskow D.R., Orr J.A. Using activated charcoal filters to prepare newer anesthesia machines for MH-susceptible patients. ASA. 2009. abstract # A302,
Bosch X., Poch E., Grau J.M., et al. Rhabdomyolysis and acute kidney injury. N Engl J Med. 2009;361:62.
Bouchama A., Knochel J.P. Heat stroke. N Engl J Med. 2002;346:1978.
Brady J.E., Sun L.S., Rosenberg H., et al. Prevalence of malignant hyperthermia due to anesthesia in New York State. Anesth Analg. 2009;109:1162.
Britt B.A., Endrenyi L., Peters P.L., et al. Screening of malignant hyperthermia susceptible families by creatine phosphokinase measurement and other clinical investigations. Can Anaesth Soc J. 1976;23:263.
Britt B.A., Scott E.A. Failure of the platelet-halothane nucleotide depletion test as a diagnostic or screening test for malignant hyperthermia. Anesth Analg. 1986;65:171.
Brown R.L., Pollock A.N., Couchman K.G., et al. A novel ryanodine receptor mutation and genotype-phenotype correlation in a large malignant hyperthermia New Zealand Maori pedigree. Hum Mol Genet. 2000;9:1515.
Burford G.E. Hyperthermia following anesthesia: consideration of control of body temperature during anesthesia. Anesthesiology. 1940;1:208.
Burkman J.M., Posner K.L., Domino K.B., et al. Analysis of the clinical variables associated with recrudescence after malignant hyperthermia reactions. Anesthesiology. 2007;106:901.
Byrne E., Blumbergs P.C., Hallpike J.F., et al. Central core disease: study of a family with five affected generations. J Neurol Sci. 1982;53:77.
Caroff S.N. The neuroleptic malignant syndrome. J Clin Psychiatry. 1980;41:79.
Caroff S.N., Rosenberg H., Fletcher J.E., et al. Malignant hyperthermia susceptibility in neuroleptic malignant syndrome. Anesthesiology. 1987;67:20.
Carpenter D., Ringose C., Leo V., et al. The role of CACNA1S in predisposition to malignant hyperthermia. BMC Med Genet. 2009;10:104.
Chance B., Clark B.J., Nioka S., et al. Phosphorus nuclear magnetic resonance spectroscopy in vivo. Circulation. 1985;72:103.
Chance B., Leigh J.S., Clark B.J., et al. Control of oxidative metabolism and oxygen delivery in human skeletal muscle: a steady-state analysis of the work/energy cost transfer function. Proc Natl Acad Sci U S A. 1985;82:8384.
Chelu M.G., Goonasekera S.A., Durham W.J., et al. Heat- and anesthesia-induced malignant hyperthermia in an RyR1 knock-in mouse. FASEB. 2006;20:329.
Cherednichenko G., Hurne A.M., Fessenden J.D., et al. Conformational activation of Ca 2+ entry by depolarization of skeletal myotubes. Proc Natl Acad Sci U S A. 2004;101:15793.
Cherednichenko G., Ward C.W., Feng W., et al. Enhanced excitation-coupled calcium entry in myotubes expressing malignant hyperthermia mutation R163C is attenuated by dantrolene. Mol Pharmacol. 2008;73:1203.
Cohen B.M., Baldessamini R.J., Pope H.G., et al. Neuroleptic malignant syndrome. N Engl J Med. 1985;313:1293.
Cook J.D., Henderson-Tilton A.C. Malignant hyperthermic reaction to nifedipine in a child with Schwartz-Jampel syndrome. Ann Neurol. 1985;18:402.
Cornea R.L., Nitu F., Gruber S., et al. FRET-based mapping of calmodulin bound to the RyR1 Ca 2+ release channel. Proc Natl Acad Sci U S A. 2009;106:6128.
Crawford M.W., Prinzhausen H., Petroz G.C., et al. Accelerating the washout of inhalation anesthetics from the Drager Primus anesthetic workstation: effect of exchangeable internal components. Anesthesiology. 2007;106:289.
Dainese M., Quarta M., Lyfenko A.D., et al. Anesthetic- and heat-induced sudden death in calsequestrin-1-knockout mice. FASEB. 2009;23:1710.
Davis M., Brown R., Dickson A., et al. Malignant hyperthermia associated with exercise-induced rhabdomyolysis or congenital abnormalities and a novel RYR1 mutation in New Zealand and Australian pedigrees. Br J Anaesth. 2002;88:508.
Davis P.J., Brandom B.W. The association of malignant hyperthermia and unusual disease: when you’re hot you’re hot, or maybe not. Anesth Analg. 2009;109:1001.
Delphin E., Jackson D., Rothstein P., et al. Use of succinylcholine during elective pediatric anesthesia should be re-evaluated. Anesth Analg. 1987;66:1190.
Denborough M.A., Lovell R.R.H. Anaesthetic deaths in a family. Lancet. 1960;2:45.
Denborough M.A., Forster F.A., Lovell R.R.H., et al. Anaesthetic deaths in a family. Br J Anaesth. 1962;34:395.
Durham W.J., Aracena-Parks P., Long C., et al. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:33.
Ellis F.R., Clarke M.C., Modgill M., et al. Evaluation of creatine phosphokinase in screening patients for malignant hyperthermia. Br Med J. 1975;3:511.
Ellis F.R., Halsall P.J., Christian A.S., et al. Clinical presentation of suspected malignant hyperthermia during anesthesia in 402 probands. Anaesthesia. 1990;45:838.
Elsayed E., Reilly R.F. Rhabdomyolysis: a review, with emphasis on the pediatric population. Pediatr Nephrol. 2010;25:7.
Ferreiro A., Monnier N., Romero N.B., et al. A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann Neurol. 2002;51:750.
Fiege M., Wappler M., Weisshorn R., et al. Induction of malignant hyperthermia in susceptible swine by 3,4-methylenedioxymethamphetamine (“ecstasy”). Anesthesiology. 2003;99:1132.
Fitzgibbons D.C. Malignant hyperthermia following preoperative oral administration of dantrolene. Anesthesiology. 1980;54:73.
Flewellen E.H., Nelson T.E., Jones W.P., et al. Dantrolene dose response in awake man: implications for management of malignant hyperthermia. Anesthesiology. 1983;59:275.
Florence J.M., Fox P.T., Planer J., et al. Activity, creatine kinase, and myoglobin in Duchenne muscular dystrophy: a clue to etiology? Neurology. 1985;35:758.
Fowler W.M., Fayzer R.B., Taylor R.G., et al. The Schwartz-Jampel syndrome: its clinical, physiological and histologic expressions. J Neurol Sci. 1974;22:127.
Fruen B.R., Mickelson J.R., Louis C.F., et al. Dantrolene inhibition of sarcoplasmic reticulum Ca2+ release by direct and specific action at skeletal muscle ryanodine receptors. J Biol Chem. 1997;272:26965.
Fuji J., Otsu K., Zorzato F., et al. Identification of a point mutation in porcine ryanodine receptor associated with malignant hyperthermia. Science. 1991;253:448.
Gallant E.M., Foldes F.F., Rempel W.E., et al. Verapamil is not a therapeutic adjunct to dantrolene in porcine MH. Anesth Analg. 1985;64:601.
Gallant E.M., Lentz L.R. Excitation-contraction coupling in pigs heterozygous for malignant hyperthermia. Am J Physiol. 1992;262:C422.
Gerbershagen M.U., Wappler F., Fiege M., et al. Effects of a 5HT2 receptor agonist on anesthetized pigs susceptible to malignant hyperthermia. Br J Anaesth. 2003;91:281.
Girard T., Treves S., Voronkov E., et al. Molecular genetic testing for malignant hyperthermia susceptibility. Anesthesiology. 2004;100:1076.
Goonasekera S.A., Beard N.A., Groom L., et al. Triadin binding to the C-terminal luminal loop of the ryanodine receptor is important for skeletal muscle excitation contraction coupling. J Gen Physiol. 2007;130:365.
Granati J.E., Stern B.J., Ringel A., et al. Neuroleptic malignant syndrome: successful treatment with dantrolene and bromocriptine. Ann Neurol. 1983;14:89.
Green J.H., Ellis F.R., Halsall P.J., et al. Thermoregulation, plasma catecholamine and metabolite levels during submaximal work in individuals susceptible to malignant hyperthermia. Acta Anaesthesiol Scand. 1987;31:122.
Gronert G.A., Theye R.A. Halothane-induced porcine malignant hyperthermia: metabolic and hemodynamic changes. Anesthesiology. 1976;44:36.
Gronert G.A., Theye R.A. Suxamethonium-induced porcine malignant hyperthermia. Br J Anaesth. 1976;48:513.
Gronert G.A. Malignant hyperthermia. Anesthesiology. 1980;53:395.
Gronert G.A., Thompson R.L., Onofrio B.M., et al. Human malignant hyperthermia: awake episodes and correction by dantrolene. Anesth Analg. 1980;59:377.
Gronert G.A., Ahern C.P., Milde J.H., et al. Effect of CO2, calcium, digoxin, and potassium on cardiac and skeletal muscle metabolism in malignant hyperthermia susceptible swine. Anesthesiology. 1986;64:24.
Gronert G.A., Fowler W., Cardinet G.H., et al. Absence of malignant hyperthermia contractures in Becker-Duchenne dystrophy at age 2. Muscle Nerve. 1992;15:52.
Grossman R.A., Hamilton R.W., Morse B.M., et al. Nontraumatic rhabdomyolysis and acute renal failure. N Engl J Med. 1974;291:807.
Gunter J.B., Ball J., Than-Win S., et al. Preparation of the Drager Fabius anesthesia machine for the malignant-hyperthermia susceptible patient. Anesth Analg. 2008;107:61936-61945.
Guzé B.H., Baxter L.R. Neuroleptic malignant syndrome. N Engl J Med. 1985;313:163.
Hackl W., Mauritz W., Schemper M., et al. Prediction of malignant hyperthermia susceptibility: statistical evaluation of clinical signs. Br J Anaesth. 1990;64:425.
Hannallah R.S., Kaplan R.F. Jaw relaxation after a halothane/succinylcholine sequence in children. Anesthesiology. 1994;81:99.
Harrison G.G. Propofol in malignant hyperthermia. Lancet. 1991;337:503.
Harrison G.G., Isaacs H. Malignant hyperthermia: an historical vignette. Anaesthesia. 1992;47:54.
Heiman-Patterson T.D. Neuroleptic malignant syndrome and malignant hyperthermia. Med Clin North Am. 1993;77:477.
Heytens L., Martin J.J., Van de Kleft E., et al. In vitro contracture tests in patients with various neuromuscular diseases. Br J Anaesth. 1992;68:72.
Hogan K.J., Vladutiu G.D. Malignant hyperthermia-like syndrome and carnitine palmitoyltransferase II deficiency with heterozygous R503C mutation. Anesth Analg. 2009;109:1070.
Hopkins P.M., Ellis F.R., Halsall P.J., et al. Hypermetabolism in arthrogryposis multiplex congenital. Anaesthesia. 1991;46:374.
Ibarra M.C.A., Wu S., Murayama K., et al. Malignant hyperthermia in Japan: mutation screening of the entire ryanodine receptor type 1 gene coding region by direct sequencing. Anesthesiology. 2006;104:1146.
Illias W.K., Williams C.H., Fulfer R.T., et al. Diltiazem inhibits halothane-induced contractions in malignant hyperthermia-susceptible muscles in vitro. Br J Anaesth. 1985;57:994.
Inness R.K.R., Stromme J.H. Rise in serum creatine phosphokinase associated with agents used in Anaesthesia. Br J Anaesth. 1973;45:185.
Jeong S.K., Kim D.C., Cho Y.G., et al. A double mutation of the ryanodine receptor type 1 gene in a malignant hyperthermia family with multiminicore myopathy. J Clin Neurol. 2008;4:123.
Jiang D., Chen W., Xiao J., et al. Reduced threshold for luminal Ca2+ activation of RyR1 underlies a causal mechanism of porcine malignant hyperthermia. J Biol Chem. 2008;283:20813.
Jurkat-Rott K., McCarthy T., Lehmann-Horn F., et al. Genetics and pathogenesis of malignant hyperthermia. Muscle Nerve. 2000;23:4.
Kamper A., Rodemann P. Alterations of protein degradation and 2-D protein pattern in muscle cells of MDX and DMD origen. Biochem Biophys Res Commun. 1992;189:1484.
Kaplan R.F., Rushing E. Isolated masseter muscle spasm and increased creatine kinase without malignant hyperthermia susceptibility or other myopathies. Anesthesiology. 1992;77:820.
Kaplan R.F., Iaizzo P., Van der Spek A.F.L., et al. Management of incomplete jaw relaxation after succinylcholine-responses from MH hotline consultants (abstract). Anesthesiology. 1993;79:A919.
Karan S.M., Crowl F., Muldoon S.M., et al. Malignant hyperthermia masked by capnographic monitoring. Anesth Analg. 1994;78:590.
Kleopa K.A., Rosenberg H., Heiman-Patterson T., et al. Malignant hyperthermia-like episode in Becker muscular dystrophy. Anesthesiology. 2000;93:1535.
King J.O., Denborough M.A. Anesthetic-induced malignant hyperthermia in children. J Pediatr. 1973;83:37.
Klinger W., Rueffert H., Lehmann-Horn F., et al. Core myopathies and risk of malignant hyperthermia. Anesth Analg. 2009;109:1167.
Kovarik W.D., Morray J.P. Hyperkalemic cardiac arrest after succinylcholine administration in a child with purpura fulminans. Anesthesiology. 1995;83:211.
Lanier W.L., Iaizzo P.A., Milde J.H., et al. The effects of intravenous succinylcholine on cerebral function and muscle afferent activity following complete ischemia in halothane-anesthetized dogs. Anesthesiology. 1990;73:465.
Larach M.G., Rosenberg H., Larach D.R., et al. Prediction of malignant hyperthermia susceptibility by clinical signs. Anesthesiology. 1987;66:547.
Larach M.G., North American Malignant Hyperthermia Group. standardization of the caffeine halothane muscle contracture test. Anesth Analg. 1989;69:511.
Larach M.G., Landis J.R., Bunn J.S., et al. The North American Malignant Hyperthermia Registry: prediction of malignant hyperthermia susceptibility in low-risk subjects. Anesthesiology. 1992;76:16.
Larach M.G. Should we use muscle biopsy to diagnose malignant hyperthermia susceptibility? Anesthesiology. 1993;79:1.
Larach M.G., Localio A.R., Allen G.C., et al. A clinical grading scale to predict malignant hyperthermia susceptibility. Anesthesiology. 1994;80:771.
Larach M.G., Rosenberg H., Gronert G.A., et al. Hyperkalemic cardiac arrest during anesthesia in infants and children with occult myopathies. Clin Pediatr. 1997;36:9.
Larach M.G., MacLennan D.H. How carefully can we phenotype patients suspected of malignant hyperthermia susceptibility? Anesthesiology. 1999;90:645.
Larach M.G., Rosenberg H., Gronert G.A., et al. Did anesthetics trigger cardiac arrests in patients with occult myopathy? Anesthesiology. 2001;94:933.
Larach M.G., Brandom B.W., Allen G.A., et al. Cardiac arrests and deaths associated with malignant hyperthermia in North America from 1987 to 2006. Anesthesiology. 2008;108:603.
Larach M.G., Gronert G.A., Allen G.C., et al. Clinical presentation, treatment and complications of malignant hyperthermia in North America from 1987–2006. Anesth Analg. 2010. (in press)
Lee M.B., Adragna M.G., Edwards L., et al. Use of a platelet nucleotide assay as a possible diagnostic test for malignant hyperthermia. Anesthesiology. 1985;63:311.
Lehmann-Horn F., Jurkat-Rott K., Rudel R., et al. Diagnostics and therapy of muscle channelopathies: guidelines of the Ulm Muscle Centre. Acta Myol. 2008;27:98.
Lerman J., McLeod M.E., Strong H.A., et al. Pharmacokinetics of intravenous dantrolene in children. Anesthesiology. 1989;70:625.
Levano S., Vukcevic M., Singer M., et al. Increasing the number of diagnostic mutations in malignant hyperthermia. Hum Mutat. 2009;30:590.
Lietman P.S., Haslam R.H.A., Walcher J.R., et al. Pharmacology of dantrolene sodium in children. Arch Phys Med Rehabil. 1974;55:388.
Litman R.S., Flood C.D., Kaplan R.F., et al. Postoperative malignant hyperthermia. Anesthesiology. 2008;109:825.
Littleford J.A., Patel L.R., Bose D., et al. Masseter muscle spasm in children: implications of continuing the triggering anesthetic. Anesth Analg. 1991;72:151.
Loke J., MacLennan D.H. Bayesian modeling of muscle biopsy contracture testing for malignant hyperthermia susceptibility. Anesthesiology. 1998;88:589.
Loke J., MacLennan D.H. Malignant hyperthermia and central core disease: disorders of Ca2+ release channels. Am J Med. 1998;104:470.
Lopez J.R., Alamo L., Caputo C., et al. Intracellular ionized calcium concentrations in muscles from humans with malignant hyperthermia. Muscle Nerve. 1985;8:355.
Lyfenko A.D., Dirksen R.T. Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J Physiol. 2008;586:4815.
Lynch C., Durbin C.G., Fisher N.A., et al. Effects of dantrolene and verapamil on atrioventricular conduction and cardiovascular performance in dogs. Anesth Analg. 1986;65:252.
Mann S.C., Caroff S.N., Keck P.E., et al. Neuroleptic malignant syndrome and related conditions, ed 2. Washington, DC: American Psychiatric Association Publishing, Inc, 2003.
Marohn M.L., Nagia A.H. Masseter muscle rigidity after rapid-sequence induction of anesthesia. Anesthesiology. 1992;77:205.
Melzer W., Dietze B. Malignant hyperthermia and excitation-contraction coupling. Acta Physiol Scand. 2001;171:367.
Mickelson J.R., Louis C.F. Malignant hyperthermia: excitation-contraction coupling, Ca2+ release channel, and cell Ca2+ regulation defects. Physiol Rev. 1996;76:537.
Monnier N., Krivosic-Horber R., Payen J.F., et al. Presence of two different genetic traits in malignant hyperthermia families: implications for genetic analysis, diagnosis, and incidence of malignant hyperthermia susceptibility. Anesthesiology. 2002;97:1067.
Moschcowitz A.V. Postoperative heat stroke. Surg Gynecol Obstet. 1916;28:443.
Moulds R.F.W., Denborough M.A. Biochemical basis of malignant hyperpyrexia. Br Med J. 1974;2:241.
Nagarjan K., Fishbein W.N., Muldoon S.M., et al. Calcium uptake in frozen muscle biopsy sections compared with other predictors of malignant hyperthermia susceptibility. Anesthesiology. 1987;66:680.
Nelson T.E., Rosenberg H., Muldoon S., et al. Genetic testing for malignant hyperthermia in North America. Anesthesiology. 2004;100:212.
Neuman G.G., Kopman A.F. Dyskalemic periodic paralysis and myotonia. Anesth Analg. 1993;76:426.
Newmark J., Voelkel M., Brandom B.W., et al. Delayed onset of malignant hyperthermia without creatine kinase elevation in a geriatric, ryanodine receptor type 1 gene compound heterozygous patient. Anesthesiology. 2007;107:350.
North American Malignant Hyperthermia Registry. 2008.
O’Flynn R., Shutack J., Rosenberg H., et al. Masseter muscle rigidity and malignant hyperthermia susceptibility in pediatric patients: an update on management and diagnosis. Anesthesiology. 1994;80:1228.
Okumura F., Cracker B.D., Denborough M.A., et al. Identification of susceptibility to malignant hyperthermia in swine. Br J Anaesth. 1979;51:171.
Olgin J., Rosenberg H., Gregory A., et al. A blinded comparison of non-invasive, in vivo phosphorus nuclear magnetic resonance spectroscopy and the in vitro halothane/caffeine contracture test in the evaluation of malignant hyperthermia susceptibility. Anesthesiology. 1991;72:36.
Ording H. Incidence of malignant hyperthermia in Denmark. Anesth Analg. 1985;64:700.
Ording H., Foder B., Scharff O., et al. Cytosolic free calcium concentrations in lymphocytes from malignant hyperthermia susceptible patients. Br J Anaesth. 1990;64:341.
Ording H., Hedegran A.M., Skovgaard L.T., et al. Evaluation of 119 anaesthetics received after investigation for susceptibility to malignant hyperthermia. Acta Anaesthesiol Scand. 1991;35:711.
Ording H., Bendixen D. Sources of variability in halothane and caffeine contracture tests for susceptibility to malignant hyperthermia. Eur J Anaesthesiol. 1992;9:367.
Ording H., Brancadoro V., Cozzolino S., et al. In vitro contracture test for diagnosis of malignant hyperthermia following the protocol of the European MH Group: results of testing patients surviving fulminant MH and unrelated low-risk subjects. Acta Anaesthesiol Scand. 1997;41:955.
Paasuke R.T., Brownell A.K.W. Serum creatine kinase level as a screening test for susceptibility to malignant hyperthermia. JAMA. 1986;255:769.
Parness J., Bandschapp O., Girard T., et al. The myotonias and susceptibility to malignant hyperthermia. Anesth Analg. 2009;109:1054.
Peluso A., Bianchini A. Malignant hyperthermia susceptibility in patients with Duchenne’s muscular dystrophy. Can J Anaesth. 1992;39:1117.
Piotrowski A.J., Fendler W.M. Hyperkalemia and cardiac arrest following succinylcholine administration in a 16-yr-old boy with acute nonlymphoblastic leukemia and sepsis. Pediatr Crit Care Med. 2007;8:183.
Plotz J., Braun J. Failure of “self-taming” doses of succinylcholine to inhibit increases in post-operative serum creatine kinase activity in children. Anesthesiology. 1982;56:207.
Plumley M.H., Bevan J.C., Saddler J.M., et al. Dose-related effects of succinylcholine on the adductor pollicis and masseter muscles in children. Can J Anaesth. 1990;37:15.
Podranski T., Bouillon T., Schumacher P.M., et al. Compartmental pharmacokinetics of dantrolene in adults: do malignant hyperthermia association dosing guidelines work? Anesth Analg. 2005;101:1695.
Pollock N., Hodges M., Sendall J., et al. Prolonged malignant hyperthermia in the absence of triggering agents. Anaesth Intensive Care. 1992;20:520.
Pollock N., Langton E., Stowell K., et al. Safe duration of postoperative monitoring for malignant hyperthermia susceptible patients. Anaesth Intensive Care. 2004;32:502.
Pollock N., Langton E., McDonnell N., et al. Malignant hyperthermia and day stay surgery. Anaesth Intensive Care. 2006;34:40.
Protasi F., Paolini C., Dainese M., et al. Calsequestrin-1: a new candidate gene for malignant hyperthermia and exertional/environmental heat stroke. J Physiol. 2009;587:3095.
Pryn S.J., van der Spek A.F. Comparative pharmacology of succinylcholine on jaw, eye, and tibialis muscle (abstract). Anesthesiology. 1990;73:A859.
Quinlan J.G., Wedel D.J., Iaizzo P.A., et al. Multiple-pulse stimulation and dantrolene in malignant hyperthermia. Muscle Nerve. 1990;13:904.
Raff M., Harrison G.G. The screening of propofol in MHS swine. Anesth Analg. 1989;68:750.
Rampton A.J., Kelly D.A., Shanahan E.C., et al. Occurrence of malignant hyperpyrexia in a patient with osteogenesis imperfecta. Br J Anaesth. 1984;56:1443.
Robinson R.L., Anetseder M.J., Brancadoro V., et al. Recent advances in the diagnosis of malignant hyperthermia susceptibility: how confident can we be of genetic testing? Eur J Hum Genet. 2003;11:342.
Robinson R.L., Carpenter D., Shaw M.-A., et al. Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat. 2006;27:977.
Rosenberg H., Gronert G.A. Intractable cardiac arrest in children given succinylcholine. Anesthesiology. 1992;77:1054.
Rosner B. Fundamentals of biostatistics, ed 3. Boston: PWS Kent Publishing Co, 1990.
Rubiano R., Chang J.L., Carroll J., et al. Acute rhabdomyolysis following halothane anesthesia without succinylcholine. Anesthesiology. 1987;67:856.
Rubin A.S., Zablocki A.D. Hyperkalemia, verapamil, and dantrolene. Anesthesiology. 1987;66:246.
Rueffert H., Olthoff D., Deutrich C., et al. Spontaneous occurrence of the disposition to malignant hyperthermia. Anesthesiology. 2004;100:731-733.
Salluzzo R.F.. Rhabdomyolysis. Rosen P., Barkin R.M., editors. Emergency medicine: concepts and clinical practice. vol. 3. St. Louis: Mosby-Year Book; 1992:2237.
Sambuughin N., Holley H., Muldoon S., et al. Screening of the entire ryanodine receptor type 1 coding region for sequence variants associated with malignant hyperthermia susceptibility in the North American population. Anesthesiology. 2005;102:515.
Schonell L.H., Sims C., Bulsara M., et al. Preparing a new generation of anaesthetic machine for patients susceptible to malignant hyperthermia. Anaesth Intensive Care. 2003;31:58.
Sei Y., Sambuughin N., Muldoon S., et al. Malignant hyperthermia genetic testing in North America Working Group Meeting. Anesthesiology. 2004;100:464.
Shukry M., Guruli Z.V., Ramadhyani U., et al. Suspected malignant hyperthermia in a child with laminin alpha 2 (merosin) deficiency in the absence of a triggering agent. Pediatr Anesth. 2006;16:462.
Shy G.M., Magee K.R. A new congenital non-progressive myopathy. Brain. 1956;79:610.
Storella R.J., Keykhah M.M., Rosenberg H., et al. Halothane and temperature interact to increase succinylcholine-induced jaw contracture in the rat. Anesthesiology. 1993;79:1261.
Takamatsu F., Taoda M., Uchihashi Y., et al. Rhabdomyolysis induced by succinylcholine chloride and sevoflurane in an elderly man. Masui. 1996;45:1406.
Tang T.T., Oechler H.W., Siker D., et al. Anesthesia-induced rhabdomyolysis in infants with unsuspected Duchenne dystrophy. Acta Paediatr. 1992;81:716.
Tilgen N., Zorzato F., Halliger-Keller B., et al. Identification of four novel mutations in the C-terminal membrane spanning region of the ryanodine receptor 1: association with central core disease and alteration of calcium homeostasis. Hum Mol Genet. 2001;10:2879.
Tobin J.R., Jason D.R., Challa V.R., et al. Malignant hyperthermia and apparent heat stroke. JAMA. 2001;286:168.
Urwyler A., Duefel T., McCarthy T., European Malignant Hyperthermia Group. guidelines for molecular genetic detection of susceptibility to malignant hyperthermia. Br J Anaesth. 2001;86:283.
Urwyler A., Halsall P.J., Mueller C., et al. Ryanodine receptor gene (RYR1) mutations for diagnosing susceptibility to malignant hyperthermia. Acta Anaesth Scand. 2003;47:492.
van der Spek A.F.L., Fang W.B., Ashton-Miller J.A., et al. The effects of succinylcholine on mouth opening. Anesthesiology. 1987;67:459.
van der Spek A.F.L., Fang W.B., Ashton-Miller J.A., et al. Increased masticatory muscle stiffness during limb muscle flaccidity associated with succinylcholine administration. Anesthesiology. 1988;69:11.
Wappler F., Scholz J., von Richthofen V., et al. Attenuation of serotonin-induced contractures in skeletal muscle from malignant hyperthermia-susceptible patients with dantrolene. Acta Anaesth Scand. 1997;41:1312.
Wappler F., Fiege M., Antz M., et al. Hemodynamic and metabolic alterations in response to graded exercise in a patient susceptible to malignant hyperthermia. Anesthesiology. 2000;92:268.
Wappler F., Fiege M., Steinfath M., et al. Evidence for susceptibility to malignant hyperthermia in patients with exercise-induced rhabdomyolysis. Anesthesiology. 2001;94:95.
Warner L.O., Beach T.P., Garvin J.P., et al. Halothane and children: the first quarter century. Anesth Analg. 1984;63:838.
Whistler T., Isaacs H., Badenhorst M., et al. No abnormal low molecular weight proteins identified in human malignant hyperthermic muscle. Anesthesiology. 1986;64:795.
Whitty R.J., Wong G.K., Petroz G.C., et al. Preparation of the Drager Fabius GS ™ workstation for malignant hyperthermia-susceptible patients. Can J Anesth. 2009;56:497.
Yentis S.M., Levine M.F., Hartley E.J., et al. Should all children with suspected or confirmed malignant hyperthermia susceptibility be admitted after surgery? A 10-year review. Anesth Analg. 1992;75:345.
Zhao X., Weisleder N., Han X., et al. Azumolene inhibits a component of store-operated calcium entry coupled to the skeletal muscle ryanodine receptor. J Biol Chem. 2006;281:33477.