CHAPTER 22 Anesthesia for Neurosurgery
Developmental considerations
Intracranial Pressure
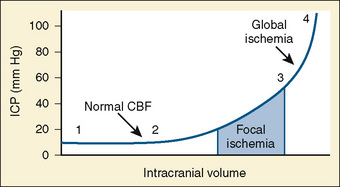
Normal intracranial pressure (ICP) is relatively low in premature infants and slightly higher in full-term infants (2 to 6 mm Hg) and children and adults (0 to 15 mm Hg). Intracranial compliance is defined as change in intracranial pressure relative to the intracranial volume (Fig. 22-1). At normal intracranial volumes (point 1 in Fig. 22-1), ICP is low but compliance is high and remains so despite small increases in volume. As intracranial volume acutely rises, the ability to compensate is rapidly overwhelmed. This occurs even when the ICP is still within normal limits, but the compliance is low (point 2 in Fig. 22-1). When ICP is already high, a threshold is reached, after which further volume expansion leads to rapid ICP elevation (point 3 in Fig. 22-1). Point 4 (see Fig. 22-1) reflects maximal intracranial volume and high ICP. Clinically, intracranial compliance can be assessed by a variety of devices that measure intracranial pressure (Wiegand and Richards, 2007).
The neonatal cranial vault is unique in that it is in a state of flux. Open fontanelles and cranial sutures lead to a compliant intracranial space (Fig. 22-2). The mass effect of a slow-growing tumor or insidious hemorrhage is often masked by a compensatory increase in intracranial volume accompanied by head growth. When ICP increases occur slowly, open fontanelles and cranial sutures separate and enlarge the intracranial space, and thus the mass effect of a slow-growing tumor or insidious intracranial bleeding is often shrouded by a compensatory increase in intracranial volume (widening of the fontanelles and cranial sutures). However, acute increases in cranial volume caused by massive hemorrhage or obstructed cerebrospinal fluid (CSF) flow cannot be attenuated by expansion of the cranial vault and frequently result in life-threatening intracranial hypertension in infants (Shapiro et al., 1980). Once the fontanelles and sutures have closed, children have a smaller cranial volume and lower intracranial compliance than adults. The posterior fontanelle is usually closed by 2 months of age. The anterior fontanelle closes after the posterior fontanelle. By 3 months of age, the anterior fontanelle is closed in 1% of infants. By 1 year it is closed in about 40%, and by 2 years it is closed in 96%. Contributory factors include a higher ratio of brain water content, less CSF volume, and a higher ratio of brain content to intracranial capacity (Arieff et al., 1992). Thus, children are at higher risk of herniation than adults.
Cerebral Blood Flow
The cerebral circulation is tightly regulated by a number of homeostatic mechanisms. The major influences of the cerebral circulation are metabolism, partial pressure of arterial carbon dioxide (Paco2), partial pressure of oxygen in arterial blood (Pao2), blood viscosity, and cerebral autoregulation. Coupling of flow and metabolism is the most significant regulator of the cerebral circulation and is preserved even during sleep (Madsen and Vorstrup, 1991; Lenzi et al., 2000) and during general anesthesia (Lam et al., 1995). Under normal conditions, cerebral blood flow (CBF) is tightly coupled to cerebral metabolism and cerebral metabolic rate of oxygen consumption (CMRo2) at global as well as regional levels. However, during periods of CNS activation, CBF increases more than CMRo2, resulting in a decrease in the cerebral oxygen extraction fraction (Fox and Raichle, 1986)
During development, CBF changes with age, mirroring changes in neural development. CBF increases during early childhood, peaking during early-to-mid childhood and plateauing at 7 to 8 years (Wintermark et al., 2004). The healthy brain receives about 15% of cardiac output, and normal adult CBF is approximately 50 mL/100 g per minute (Vavilala et al., 2002a). There are few data available from healthy children. Kennedy and Sokoloff (1957) found CBF to be much higher, in the order of 100 mL/100 g per minute, in conscious healthy children, and a study using arterial spin labeling found similar values in young children (Biagi et al., 2007), which then decreased and approached adult values during the teenage years. Even during low-dosage sevoflurane anesthesia, values for CBF in children are higher than those for adults, whose CBF ranges between 50 and 100 mL/100 g per minute (Settergren et al., 1980).
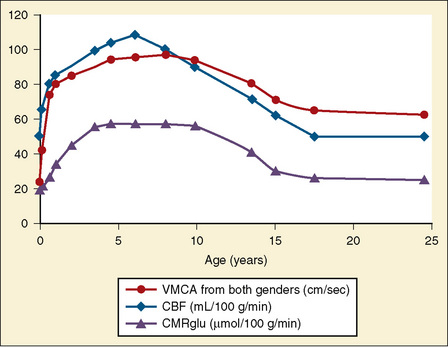
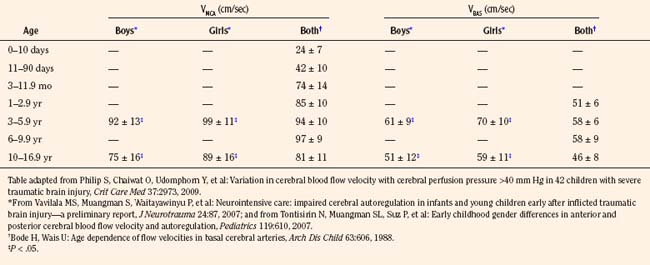
Transcranial Doppler (TCD) ultrasonography can be used to estimate CBF, assuming that changes in CBF are proportional to changes in cerebral blood flow velocity (CBFV), and that mean flow velocities are used to represent mean CBF. Systolic and diastolic flow velocity parameters are often used to examine resistance of the cerebral vessels and also brain death. Studies using TCD show that in healthy newborns, CBFV is approximately 24 cm/sec, thereafter increasing with age until 6 to 9 years (97 cm/sec) (Bode and Wais, 1988) (Fig. 22-3). Beyond 10 years of age, CBFV decreases, approximating adult values of about 50 cm/sec (Aaslid et al., 1984; Vavilala et al., 2002a). Sex differences have also been observed and may result from differences in hematocrit, hormones, vessel size, or cerebral metabolism (Kennedy and Sokoloff, 1957; Tontisirin et al., 2007). The mean CBFV estimates for middle cerebral artery (VMCA; anterior circulation) and basilar artery (VBAS; posterior circulation) by age and sex are listed in Figure 22-3 and Table 22-1 (Bode and Wais, 1988; Wintermark et al., 2004; Vavilala et al., 2005).
Cerebral Metabolic Rate
Global cerebral metabolic rate (CMR) for oxygen and glucose is higher in children than in adults (oxygen, 5.8 versus 3.5 mL/100 g brain tissue per minute, and glucose, 6.8 versus 5.5 mL/100 g brain tissue per minute, respectively) (Kennedy and Sokoloff, 1957). Like CMRo2, cerebral metabolic rate for glucose (CMRglu) is lower at birth (13 to 25 µmol/100 g per minute), increases during childhood, peaks by 3 to 4 years (49 to 65 µmol/100 g per minute), and remains high until 9 years of age. CMRglu decreases thereafter, approaching adult rates (19 to 33 µmol/100 g per minute) (Chugani et al., 1987); changes in CMRo2 and CRMglu mirror age-related changes in CBF (see Fig. 22-3).
Adenosine and nitric oxide (NO) are two purported mediators of flow–metabolism coupling and CBF increase with neuronal activation. Although the effect of adenosine on human CBF is controversial (Stange et al., 1997; Sollevi et al., 1987), up-regulation of NO production has been reported to increase CBF (Endres et al., 2004). Antagonists of both adenosine and NO attenuate the rise in CBF associated with neuronal activation, although neither mediator antagonist, alone or in combination, completely abolishes the CBF increase in response to neuronal activation (Gotoh et al., 2001). Therefore, other mediators, such as H+ ions, adenine nucleotides, potassium, prostaglandins, and vasoactive intestinal peptide, may also be involved in flow–metabolism coupling.
Hypothermia causes a reduction in CMRo2, thereby decreasing CBF via flow–metabolism coupling. CBF decreases approximately 5% to 7% per degree Celsius, and reduction of the brain temperature to 15° C will reduce CMRo2 to 10% of normothermic values (Vavilala et al., 2002). Hypothermia causes a reduction in both the basal metabolism required for maintenance of cellular integrity and the functional metabolism of the CNS.
Carbon Dioxide Vasoreactivity
Cerebral circulation is exquisitely sensitive to changes in Paco2 (Harper and Glass, 1965; Padayachee et al., 1986). Like that of CBF and metabolism, the vasoreactivity of CO2 may be higher in healthy children than in adults (13.8% and 10.3% change in mean CBFV per 1 mm Hg change in end-tidal CO2) during propofol anesthesia (Leon and Bissonnette, 1991; Rowney et al., 2002; Karsli et al., 2003) The cerebrovascular response to changing CO2 is attenuated at low blood pressure (Harper and Glass, 1965), increases in the presence of moderate hypoxia, decreases again during severe hypoxia (Quint et al., 1980), and may remain abnormally low after a hypoxic episode. Although there is an overall paucity of information regarding CO2 reactivity in healthy children and in pediatric disease states, older studies suggest that reactivity to CO2 is well developed even in healthy preterm infants (Pryds et al., 1990) and that CO2 reactivity in newborns correlates with the lowest pH and may reflect the severity of perinatal asphyxia (Baenziger et al., 1999).
There are many potential control mechanisms involved in reactivity to carbon dioxide (CO2R). Although neural regulation of CO2R may be important (Meyer et al., 1971; Bates et al., 1977; Rovere et al., 1977), the most prominent proposed mechanism is the biochemical/metabolic control of cerebral vasoreactivity. The brisk response of the cerebral vasculature to CO2 (the biochemical mechanism) is caused by the rapid diffusion of arterial CO2 across the blood-brain barrier (BBB) into the perivascular fluid and cerebral vascular smooth muscle. Changes in extracellular fluid pH by both CO2 and bicarbonate ions lead to cerebral vasodilation and increased CBF (Kontos et al., 1977). Although CO2 is a potent cerebral vasodilator, the role of arterial H+ ions is more controversial. Although pH is not known to generally affect the cerebral vasculature, because arterial H+ ions do not readily diffuse across the intact BBB (Harper and Bell, 1963), severe metabolic acidosis may cause vasodilation and increase CBF (Westerlind et al., 1994). Therefore, severe metabolic acidosis and alkalosis may affect cerebral vascular tone, as do respiratory acidosis and alkalosis.
During chronic hypercapnia maintained for 6 hours, an adaptive increase in the pH of the CSF occurs with a decrease in CBF (Warner et al., 1987). A decrease in pH is accompanied by an increase in CSF bicarbonate. Similarly, during chronic hypocapnia, CSF bicarbonate concentration decreases, CSF pH gradually decreases, and CBF increases (Muizelaar et al., 1988).
The mechanism of CO2R also appears to be regulated by local mediators rather than by chemoreceptors in the periphery. NO is partially responsible for CO2-mediated cerebral vasodilation. Schmetterer and coworkers (1997) demonstrated a significant reduction in mean VMCA to hypercapnia in healthy humans after administration of an NO synthase (NOS) inhibitor. However, NOS inhibitors do not completely ablate CO2R, and NO may be more important in regional than in global regulation of vasoreactivity. The cerebral cortex in primates was the only site in which NOS inhibitor attenuated the CBF response to increasing arterial CO2 concentration (McPherson et al., 1995). Another putative mediator of CO2R is prostaglandin (PG) E2. In contrast, indomethacin (a cyclooxygenase inhibitor) was shown to inhibit prostanoid synthesis and completely abolished CO2-induced cerebral vasodilation (Wagerle and Degiulio, 1994).
Hypoxic and Hyperoxic Control of CBF
Compared with Paco2, the influence of Pao2 on the cerebral circulation is of much less clinical significance. There are minimal changes in CBF with changes in Pao2 to greater than 50 mm Hg. Below that threshold of 50 mm Hg, CBF increases to maintain adequate cerebral oxygen delivery. Unlike CO2R, the equilibration of CBF is longer and takes approximately 6 minutes after the establishment of hypoxemia (Ellingsen et al., 1987).
Hypoxemia may induce cerebral vasodilation via anaerobic glycolysis and lactic acid production, causing decreased extracellular pH and subsequent vasodilation. However, pH changes during hypoxemia are only partially responsible for the increased CBF (Koehler and Traystman, 1982). Adenosine has been shown to be necessary for the vasodilatory response to hypoxemia (Morii et al., 1987; Brian et al., 1996; DiGeronimo et al., 1998). NO has also been implicated as a mediator, because NOS inhibitors attenuate the increase in CBF that occurs during hypoxemia (Hudetz et al., 1998). Opioids such as methionine enkephalin and leucine enkephalin have been observed to contribute to hypoxic pial artery dilation in the piglet. Vasopressin-induced CSF methionine enkephalin and leucine enkephalin release are attenuated in the presence of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) antagonists. These data show that both cAMP and cGMP contribute to vasopressin-induced pial artery dilation and the release of the opioids methionine enkephalin and leucine enkephalin (Rossberg and Armstead, 1997).
Although hypoxemia produces cerebrovasodilation, the influence of increases in Pao2 (hyperoxia) at normal atmospheric pressure is less well characterized and somewhat controversial (Armstead, 1998). Previous reports have documented both decrease in CBF (Kennedy et al., 1972) and no change in CBF (Busija et al., 1980) during hyperoxia. Animal studies demonstrate that hyperoxia elicits pial artery vasoconstriction during normocapnia, and that vasoconstrictor peptide endothelin-1 (ET-1) contributes to that vascular response (Armstead, 1999).
Effects of Viscosity on CBF
Viscosity of blood is primarily a function of hematocrit (Hct), and decrease in viscosity is usually secondary to hemodilution. During anemia, CBF increases as a result of improved rheology of the blood flow in the cerebral vessels and as a compensatory response to decreased oxygen delivery (Tomiyama et al., 1999). Although some data suggest that an Hct of less than 30% is associated with worse Glasgow Outcome Scale scores at discharge for adults with severe traumatic brain injury (TBI) (Carlson et al., 2006), neither the optimal duration for maintaining a specific hemoglobin (Hgb) level nor the relationship between target transfusion and neurologic outcome is fully known (Timmons, 2006). A recent study of critically ill children demonstrated that maintaining a Hgb of 7 g/dL rather than 9.5 g/dL can reduce requirements for blood transfusion, but none of these subjects had TBI (Lacroix et al., 2007).
Cerebral Autoregulation
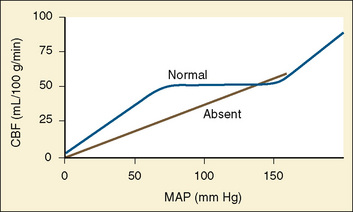
Cerebral autoregulation is a homeostatic process: arterioles dilate and constrict to maintain CBF nearly constant over a range of blood pressures. In healthy adults, changes in mean arterial pressure (MAP) between 60 and 160 mm Hg result in little or no change in CBF (Fig. 22-4) (Lassen, 1959; Paulson et al., 1990). This adaptive mechanism maintains constant (adequate) CBF by vasodilation or decreasing cerebrovascular resistance. Beyond these limits of autoregulation, hypotension may result in cerebral ischemia, and hypertension may cause cerebral hyperemia.
Healthy infants appear to autoregulate CBF as well as older children during low-dosage sevoflurane anesthesia, but the long-held assumption that the lower limit of autoregulation (LLA) is lower in young than in older children may not be valid (same LLA range for younger and older children, 46 to 76 mm Hg) (Vavilala et al., 2003). There are no data on the LLA in healthy neonates. Because blood pressure increases with age, young children may be at increased risk of cerebral ischemia as a result of lower blood pressure reserve (mean arterial pressure to LLA); however, neonates are especially vulnerable to cerebral ischemia and intraventricular hemorrhage because of this narrow autoregulatory range (Pryds et al., 2005) (Fig. 22-5). Sick premature neonates have CBF pressure-passivity, resulting in a linear correlation between CBF and systemic blood pressure (Boylan et al., 2000). Therefore, tight blood pressure control is essential in the management of neonates to minimize both cerebral ischemia during hypotension and intraventricular hemorrhage with increased blood pressure. Techniques such as deliberate hypotension in very young children or infants may not be desirable. Age-related increased latency may also occur in cerebral autoregulatory response in young children (Vavilala et al., 2002a), but no data exist in neonates. Animal data suggest that although CBF pressure autoregulation and reactivity to CO2 operate in the newborn rat, hypercapnia abolishes cerebral autoregulation (Pryds et al., 2005), that abolished autoregulation is associated with cerebral damage in asphyxiated infants, and that the combination of isoelectric electroencephalograms (EEGs) and cerebral hyperperfusion is an early indicator of very severe brain damage (Pryds et al., 1990).
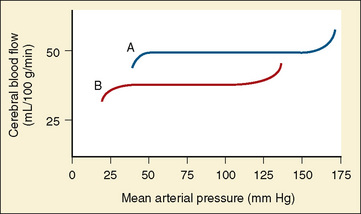
FIGURE 22-5 Autoregulation of cerebral circulation in neonates (curve B) and adults (curve A).
(Redrawn from Harris MM: Pediatric neuroanesthesia. In Berry FA, editor: Anesthetic management of difficult and routine pediatric patients, ed 2, New York, 1990, Churchill Livingstone, p 341. After data of Hernández MJ, Brennan RW, Vannucci RC, et al: Cerebral blood flow and oxygen consumption in the newborn dog, Am J Physiol 234:R209, 1978.)
Despite these clinical observations, the mechanisms of normal cerebral autoregulation in healthy children and adaptations in acute disease are not completely understood, and as occurs with changes in CBF, both anatomic and physiologic maturation might play a role in the development of a fully developed autoregulatory response. The mechanisms of cerebral autoregulation may involve a combination of myogenic, neurogenic, and metabolic processes. The metabolic mechanism stipulates that autoregulation is mediated by the release of a vasodilator substance that regulates the cerebrovascular resistance to maintain CBF constant. Although no specific substance fits all experimental observations, adenosine, a potent cerebral vasodilator, is formed from the breakdown of ATP when neuronal demand of oxygen exceeds supply (Winn et al., 1985). Adenosine can be found in increased concentration in cerebral tissue as systemic blood pressure falls toward the lower limit of autoregulation. In fact, brain adenosine concentration doubles within 5 seconds of decreasing blood pressure (Winn et al., 1980). Cortical activation via contralateral peripheral stimulation is also immediately followed by adenosine release and regional vasodilation (Ngai et al., 1998). It has been suggested that NO exerts an influence on basal and stimuli mediated cerebrovascular tone. The mechanism of NO-induced cerebral vasodilation probably involves cGMP and a decrease in intracellular calcium. It is unclear to what extent NO affects cerebral autoregulation in both healthy patients and in patients with TBI. Although earlier studies suggest that NO has no influence on cerebral autoregulation, Jones and coworkers (1999) described an increase in the lower limit of autoregulation with NOS inhibitors. Other transmitters or substances that have been proposed as mediators of autoregulation include protein kinase C (Jones et al., 1999), melatonin (Régrigny et al., 1999), prostacyclin, activated potassium channels, and intracellular second messengers (Faraci and Heistad, 1998).
The Neurogenic Mechanism
Perivascular innervation of the cerebral resistance vessels and the specific neurotransmitter contained in the perivascular nerve fibers may also modulate vascular response to change in blood pressure. However, the specific mechanisms by which the CNS exerts control on the cerebral vasculature are poorly understood. Although acetylcholine is the most abundant perivascular neurotransmitter, the list of neurotransmitters involved in this neural response includes norepinephrine, neuropeptide Y, cholecystokinin, acetylcholine, vasoactive intestinal peptide, and calcitonin gene-related peptide (Yoon et al., 1997). Experimentally sympathetic stimulation can shift the autoregulatory curve to the right, thus protecting the brain against severe elevation of MAP.
Effects of drugs on neurophysiology
The effects of the commonly used anesthetic agents on CBF, CMRo2, and CSF dynamics are discussed here and summarized in Table 22-2. The data are scant on these effects in infants and children. The responses in children are assumed to be the same as those in adults.
TABLE 22-2 Anesthetic Agents and Their Effects on Cerebral Hemodynamics and Neurophysiologic Monitors
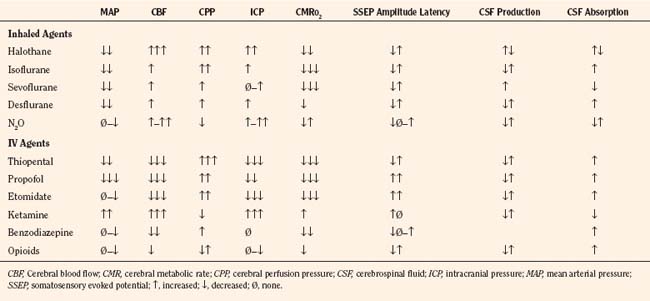
Intravenous anesthetic drugs have varied effects on cerebral hemodynamics. Thiopental and propofol maintain autoregulation and coupling and potently reduce CBF, cerebral blood volume (CBV), and CMRo2. Karsli and associates (2004) found that propofol maintained cerebrovascular reactivity to CO2 at levels greater than 30 mm Hg end-tidal CO2 in children. Furthermore, propofol, at high dosages, lowers CBFV and MAP values in children, which may be mediated by its cerebral vasoconstrictive properties (Karsli et al., 2002). Both thiopental and propofol have been touted as having neuroprotective properties, especially in preclinical studies, but their clinical efficacies in neurologically compromised pediatric patients have not been tested. Etomidate and ketamine are frequently used to induce anesthesia in hemodynamically compromised patients, as these drugs are less likely to cause hypotension than thiopental or propofol. However, CNS excitation and increased ICP have been associated with these drugs, respectively, and they may not be appropriate for many neurosurgical patients.
Intravenous Anesthetics
Propofol
Propofol is an intravenous (IV) induction agent with cerebrovascular properties similar to those of thiopental: both may depress systemic blood pressure but both potently decrease CMRo2, CBF, and ICP (Van Hemelrijck et al., 1990). Cerebral autoregulation and cerebral responsiveness to changes in arterial CO2 tension (Karsli et al., 2004) are well preserved during propofol anesthesia. Propofol is a good alternative to thiopental for induction of anesthesia for neurosurgery. In the presence of nitrous oxide and hypocapnia, propofol results in lower ICP, higher cerebral perfusion pressure (CPP), and less cerebral cortical edema than isoflurane or sevoflurane. In patients with or at risk for intracranial hypertension and decreased cerebral perfusion, maintenance of anesthesia with propofol is superior to inhaled halogenated anesthetics, at least until the dura has been opened.
Barbiturates
Barbiturates decrease CBF, CBV, and CMRo2 in a dosage-dependent manner (Pierce et al., 1962; Michenfelder, 1974; Albrecht et al., 1977) and therefore reduce ICP. Neither CBF nor cerebral metabolism is significantly altered by subanesthetic dosages of barbiturates. When the EEG becomes isoelectric, CBF and CMRo2 decrease to about 50% of normal, and additional doses of barbiturates have little further effect. Barbiturates may also be used to prevent increases in ICP that can occur with laryngoscopy and endotracheal intubation. Autoregulation and the cerebrovascular response to changes in Paco2 remain intact during barbiturate anesthesia. The rate of CSF formation and the resistance to reabsorption of CSF are not altered by barbiturates (Mann et al., 1979). In dosages that suppress the EEG, barbiturates reduce cerebral damage in animal and human models of focal cerebral ischemia (Smith et al., 1974; Nehls et al., 1987). In animals, barbiturates also reduce the extent of cerebral edema after a cortical freeze injury. This decrease in edema is in contrast to the response observed with the volatile anesthetics (Smith and Marque 1976).
Opioids
Opioids have little effect on CBF, CBV, or ICP unless respiration is depressed and Paco2 is increased (Miller et al., 1975; Misfeldt et al., 1976; Jobes, 1977; Moss et al., 1978).
Fentanyl
When combined with nitrous oxide, fentanyl decreases CBF by 47% and CMRo2 by 18% (Michenfelder, 1974). Autoregulation and CO2 responsiveness of the cerebral circulation are not altered. Fentanyl does not alter the rate of CSF formation, but it reduces the resistance to CSF reabsorption by 50% (Artru, 1984a, 1984c), the effect of which is to decrease CSF volume to a degree of unknown clinical significance. The neonatal cerebral circulation is also unaffected by fentanyl (Yaster et al., 1987). Sufentanil and alfentanil in high dosages (10 to 20 mcg/kg) reduce both CBF and CMRo2 by 25% to 30% (Stephan et al., 1991), whereas at exceedingly high dosages (10 to 200 mcg/kg), sufentanil may transiently increase CBF while reducing CMRo2 (Milde et al., 1990). However, at conventional dosages, sufentanil (Herrick et al., 1991; Weinstabl et al., 1991; Mayberg et al., 1993) does not appear to have adverse effects on the cerebral vasculature or on ICP in most patients. In a subset of patients with severe head injuries and very poor intracranial compliance, sufentanil may cause a small (e.g., <10 mm Hg) and transient increase in ICP that may be clinically significant in some settings (Sperry et al., 1992; Weinstabl et al., 1992; Albanese et al., 1993).
Remifentanil
Remifentanil is an ultra-short-acting to short-acting opioid that is rapidly metabolized by plasma and tissue esterases. The very short clinical duration of effect of remifentanil and its context-sensitive half-life, which is independent of the duration of infusion (Minto et al., 1997; Mertens et al., 2003), make it an appealing opioid for lengthy neurosurgical procedures after which rapid return of consciousness is desirable. As is the case with other opioids that have been studied, remifentanil does not increase CBF or ICP (Hoffman et al., 1993; Baker et al., 1997; Ostapkovich et al., 1998; Klimscha et al., 2003; Engelhard et al., 2004; Lagace et al., 2004). Return of consciousness is very rapid after remifentanil is discontinued, and the frequency of administration of naloxone to permit neurologic assessment is decreased (Guy et al., 1997). However, because remifentanil analgesia is very brief after its discontinuation, a long-acting opioid analgesic must be administered to prevent severe pain and rebound hypertension before or soon after remifentanil is discontinued (Gerlach et al., 2003; Cafiero et al., 2004).
Other Intravenous Anesthetics
Etomidate reduces ICP by decreasing CBF and CMRo2 by 34% and 45%, respectively. It, too, preserves the CO2 responsiveness of the cerebral circulation (Renou et al., 1978; Moss et al., 1979). A side effect of etomidate administration is myoclonus, which has been reported after prolonged continuous infusion of etomidate (Laughlin and Newberg, 1985). Lidocaine in clinical dosages decreases CBF and reduces the increase in ICP associated with endotracheal intubation (Sakabe et al., 1974; Donegan and Bedford, 1980). The benzodiazepines (diazepam, lorazepam, and midazolam) decrease CBF and CMRo2 by approximately 25% (Cotev and Shalit, 1975; Rockoff et al., 1980; Tateishi et al., 1981; Forster et al., 1982; Nugent et al., 1982; Nakahashi et al., 1991).
Dexmedetomidine
The neurophysiologic effects of dexmedetomidine (an α2-agonist) have not been extensively studied in humans. In animals, it has been noted to decrease CBF but was not associated with a proportional decrease in the cerebral metabolic rate (Zornow et al., 1990). In human studies, CBFV decreases after dexmedetomidine administration (Zornow et al., 1993) and CBF—both global and regional—decreases by 30% at clinically relevant concentrations of dexmedetomidine (Prielipp et al., 2002).
The effect of dexmedetomidine on ICP in patients with space-occupying lesions is unclear. In patients undergoing transsphenoidal hypophysectomy, dexmedetomidine had no effect on lumbar CSF pressure (Talke et al., 1997). Its effect on the amplitude and latency of cortical somatosensory evoked potential is minimal.
Ketamine
In contrast to the other IV anesthetic agents, ketamine is a potent cerebrovasodilator. It increases CBF by 60% with little change in CMRo2 (Dawson et al., 1971; Takeshita et al., 1972; Schwedler et al., 1982). The cerebrovascular response to administration of ketamine is thought to be the result of regional cerebral activation induced by the drug (Hougaard et al., 1974). Ketamine produces a marked increase in ICP, which can be reduced, but not prevented, by hyperventilation (Gardner et al., 1972; Takeshita et al., 1972; Wyte et al., 1972). The increase in CBF, and presumably in ICP, can be blocked by previous administration of thiopental (Dawson et al., 1971). Ketamine has been associated with sudden elevation of ICP and clinical deterioration when used in patients with hydrocephalus and other intracranial pathology (List et al., 1972; Lockhart and Jenkins, 1972; Wyte et al., 1972). Although ketamine is not currently used as a general anesthetic in patients with reduced intracranial compliance, a recent study in critically ill children reported a decrease in ICP following ketamine (Bar-Joseph et al., 2009), thereby challenging the notion that ketamine increases ICP.
Inhalational Anesthetics
Volatile anesthetic drugs are potent cerebrovasodilators and may produce uncoupling of CMRo2 and CBF, and different volatile anesthetics may have varying uncoupling profiles (Hansen et al., 1989). Uncoupling may lead to increased CBV and can exacerbate intracranial hypertension. Children have increased sensitivity to the vasodilatory effects of volatile drugs. TCD measurements reveal that the following volatile agents increase CBFV in descending order: halothane, desflurane, isoflurane, and sevoflurane. Autoregulation is also blunted in the presence of volatile drugs in a dosage-dependent fashion. However, this perturbation is mitigated by hypocapnia. Nitrous oxide has vasodilatory effects alone and in combination with other anesthetics. Because isoflurane and sevoflurane significantly decrease CMRo2 in children and maintain coupling, these two inhaled drugs are ideal anesthetics for the neurologically compromised patient. Sevoflurane does not significantly affect CBFV, and it preserves cerebral autoregulation in children anesthetized with up to a minimum alveolar concentration (MAC) of 1.5 (Fairgrieve et al., 2003; Wong et al., 2006).
Nitrous Oxide
Nitrous oxide is a weak cerebrovasodilator whose effects on CBF are offset by hyperventilation and barbiturate anesthesia (Algotsson et al., 1988). The variability of effects that nitrous oxide has on CBF and ICP in different reports results from differences in experimental species and background anesthesia. In many animals, nitrous oxide in subanesthetic dosages (60% to 70%) causes excitement and cerebral metabolic stimulation, with an accompanying increase in CBF (Theye and Michenfelder, 1968; Sakabe et al., 1978; Pelligrino et al., 1984; Drummond et al., 1987). Because nitrous oxide is not an adequate anesthetic in the absence of other inhalation or IV anesthetics, the modification of its cerebral effects by additional anesthetic drugs is particularly important. Nitrous oxide at 70% does not, for example, cause a change in CBF, but it does reduce CMRo2 by 15% to 20% during barbiturate and narcotic anesthesia (Sakabe et al., 1978). However, when nitrous oxide is added to a volatile anesthetic such as isoflurane (Cucchiara et al., 1974; Manohar and Parks, 1984) or halothane (Sakabe et al., 1976), both CBF and CMRo2 increase. When nitrous oxide is added to sevoflurane, cerebral hyperemia increases and autoregulation is impaired (Iacopino et al., 2003).
The cerebrovascular responses to changes in Paco2 and MAP are preserved during nitrous oxide anesthesia. ICP may increase in response to nitrous oxide in patients with intracranial mass lesions and reduced intracranial compliance (Henriksen and Jörgensen, 1973; Moss and McDowall, 1979; Iacopino et al., 2003). The increase in ICP with nitrous oxide, however, is readily reversible by diazepam and barbiturate anesthesia and simultaneously initiated hyperventilation (Phirman and Shapiro, 1977).
The use of nitrous oxide for pediatric neuroanesthesia remains controversial. Some anesthesiologists prefer to avoid it because of its ability to increase CMRo2 and reduce the cerebroprotective effects of barbiturates. Others are concerned because it readily diffuses into collections of intracranial air and may increase ICP in the presence of pneumocephalus (Artru, 1982; Skahen et al., 1986). Asymptomatic accumulation of intracranial air occurs commonly during craniotomies, especially those associated with posterior fossa surgery and drainage of CSF (Yates et al., 1994). Some anesthesiologists discontinue nitrous oxide before closure of the dura to reduce the incidence of tension pneumocephalus, and others administer nitrous oxide throughout the procedure without any obvious detrimental effects. Indeed, one randomized control trial comparing anesthetic techniques with and without nitrous oxide in patients undergoing sitting craniotomies showed no difference in the incidence or size of pneumocephalus between the three groups (Hernández-Palazón et al., 2003). It may be that nitrous oxide equilibrates with intracranial air before the dura is closed; if so, ICP would not increase during craniodural closure because air pockets would already contain nitrous oxide. In addition, the discontinuance of nitrous oxide would decrease ICP, as nitrous oxide diffused back into the bloodstream (Skahen et al., 1986). Maintenance with nitrous oxide until the end of the surgery may be advantageous because it permits rapid awakening and may reduce the intracranial gas volume and the likelihood of delayed tension pneumocephalus. Nitrous oxide is generally not contraindicated during sitting craniotomies, even though the volume of a venous air embolus (VAE) expands in the presence of nitrous oxide. In fact, this phenomenon actually increases the sensitivity of monitoring for VAE by capnography, while at the same time nitrous oxide neither increases the risk of VAE (Losasso et al., 1992) nor increases the hemodynamic consequences of VAE, provided that nitrous oxide is discontinued when VAE is first detected (Losasso et al., 1992).
Isoflurane
Isoflurane is frequently used for neuroanesthesia. Its popularity is based on the fact that it affects CBF less than does halothane at equivalent MAC dosages (Todd and Drummond, 1984; Drummond and Todd, 1985; Algotsson et al., 1988), the fact that 1 MAC isoflurane preserves cerebral autoregulation (McPherson and Traystman, 1988) and CO2 responsiveness (McPherson et al., 1989), and the belief that it may provide cerebral protection (Newberg and Michenfelder, 1983; Verhaegen et al., 1992). Cerebral autoregulation is less affected by isoflurane than by halothane (Todd and Drummond, 1984b). In addition, isoflurane does not change CSF production, and it reduces the resistance to reabsorption of CSF (Artru, 1984b). During hypocapnia, CBF is lower with 1.0 MAC isoflurane (with 75% nitrous oxide) than with nitrous oxide alone (Cucchiara et al., 1974; Drummond and Todd, 1985). In contrast, 1.0 MAC halothane (with 75% nitrous oxide) increases CBF. Despite their dissimilar effects on CBF, isoflurane and halothane increase ICP equally in an animal model of brain injury (Scheller et al., 1987). This is probably because isoflurane and halothane increase CBV to a similar degree (Artru 1984c; Archer et al., 1987). In patients with reduced intracranial compliance, isoflurane increases ICP. This increase can be attenuated by simultaneous initiation of hyperventilation (Adams et al., 1981).
Isoflurane may be safely used in patients with small supratentorial brain tumors (Madsen et al., 1987), but it may cause dangerous increases in ICP in patients with large intracranial mass lesions that are associated with a midline shift evident on a computed tomography (CT) scan (Grosslight et al., 1985). Like halothane, isoflurane should be avoided in patients with reduced intracranial compliance until the dura is open, if ICP is not being monitored. Isoflurane decreases CMRo2 by 30%, and it causes an isoelectric EEG at concentrations above 2.0 MAC (Newberg and Michenfelder, 1983). It is unique among the volatile agents in that it preserves normal cerebral energy states and aerobic metabolism at very low blood pressure (40 mm Hg), in contrast to the findings observed with hypotension induced by halothane, trimethaphan, or sodium nitroprusside (Newberg et al., 1984). In studies of mice exposed to 5% oxygen, isoflurane increased survival time and thus may have provided some degree of cerebral protection. In studies of incomplete global ischemia in isoflurane-anesthetized dogs, cerebral energy stores were increased, presumably through depression of cortical electrical activity and cerebral metabolism (Newberg and Michenfelder, 1983). Protective effects of isoflurane, however, were not observed in a primate model of regional cerebral ischemia (Nehls et al., 1987).
Sevoflurane
Sevoflurane is a fluorinated ether with a low blood-gas solubility. Studies in rabbits suggest that sevoflurane does not increase CBF at 0.5 to 1.0 MAC. Sevoflurane does cause increases in CBF and ICP and a decrease in cerebral oxygen consumption (Scheller et al., 1988) and CPP, similar to isoflurane (Petersen et al., 2003). Compared with isoflurane, sevoflurane allows more rapid emergence after lengthy neurosurgery, and thus more rapid neurologic assessment (Mönkhoff et al., 2001; Gauthier et al., 2002). Taken together, the available evidence suggests that sevoflurane is a more appropriate inhaled anesthetic than halothane during craniotomy, and that it is equivalent in its cerebrovascular effects to isoflurane, while allowing more rapid recovery after long anesthesia. As is the case with other halogenated agents, if ICP or intracranial compliance is compromised, sevoflurane should be withheld until the dura has been opened (Petersen et al., 2003).
Halothane
Halothane is a cerebral vasodilator that decreases cerebrovascular resistance and increases CBF in a dosage-dependent fashion (Wollman et al., 1964; Albrecht et al., 1977; Todd and Drummond, 1984; Brüssel et al., 1991). The increase in CBF is transient; CBF decreases to baseline levels after 150 minutes of halothane anesthesia (Albrecht et al., 1983). CBV, however, remains elevated by 11% to 12% over a 3-hour period of halothane administration (Artrub, 1983). Halothane reduces CMRo2 by 17% to 33% (Albrecht et al., 1977). The cerebral vasculature remains responsive to changes in arterial Paco2 (Alexander et al., 1964; Wollman et al., 1964; Drummond and Todd, 1985). Halothane in high concentrations (2.0 MAC) abolishes autoregulation of the cerebral circulation in response to changes in MAP in both adults (Miletich et al., 1976; Todd and Drummond, 1984) and infants (Messer et al., 1989). Halothane alters blood-brain barrier permeability, promoting the extravasation of plasma proteins into normal brain during periods of acute hypertension (Forster et al., 1978). Halothane reduces CSF formation by 30% in dogs and increases the resistance of reabsorption of CSF (Artru, 1983a, 1984b).
Because ICP is determined by CBV, CSF volume, and brain tissue volume, it is not surprising that ICP increases with halothane (Jennett et al., 1969; DiGiovanni et al., 1974). Peak increases are observed in 3 to 13 minutes, although the increase persists over 3 hours of halothane exposure (Artru, 1983b). The increase in ICP in patients with intracranial mass lesions can be attenuated, but not totally prevented, by establishing hyperventilation for 10 minutes before the introduction of halothane (Adams et al., 1981). If ICP is not being monitored, halothane should not be used in patients with reduced intracranial compliance until the dura is open and its effects on the brain can be seen.
Desflurane
Desflurane is an inhalation agent that is chemically similar to isoflurane. Its physicochemical properties are remarkable, with a blood-gas partition coefficient even lower than that of nitrous oxide, permitting rapid uptake and washout of the gas. The effects of desflurane on cerebral metabolism and hemodynamics are not as well studied as the effects of the other inhalation agents, but animal studies suggest that its effects are not unique in any way. Desflurane at clinical concentrations is a potent cerebral vasodilator, increasing ICP (Artru, 1994), increasing CBF by 50% at 1.5 MAC, and reducing autoregulation of CBF (Lutz and Milde, 1990). However, cerebrovascular responsiveness to hypocapnia is preserved during desflurane anesthesia in laboratory animals, protecting the animal from increases in ICP if hyperventilation occurs during the anesthesia (Lutz et al., 1991; Young, 1992). In patients with mass lesions, equivalent MAC dosages of desflurane and isoflurane are similar in terms of absolute CBF, the response to increasing dosages, and the preservation of CO2 reactivity (Ornstein et al., 1993; Fraga et al., 2003). Desflurane effects seen on the EEG are also similar to those of isoflurane. At increasing concentrations, the electroencephalographic frequency decreases and the amplitude increases. Burst suppression appears at about 1.24 MAC (Rampil et al., 1991).
Muscle Relaxants
Succinylcholine
Succinylcholine is very infrequently used in pediatric anesthesia because of its association with life-threatening hyperkalemia and cardiac arrest in children with undiagnosed myopathies. This has led to a black-box warning by the U.S. Food and Drug Administration, reserving its use in children for emergency intubation when securing the airway is necessary. Most often, the use of even high-dosage nondepolarizing neuromuscular blockers is appropriate for emergently securing the airway in children, but in some cases a very rapid and immediate pharmacologic paralysis is required, and succinylcholine remains the drug of choice in these circumstances. Life-threatening hyperkalemia has also been associated with administration of succinylcholine after many types of CNS disorders, including closed-head injury, even without motor deficits (Mazze et al., 1969; Thomas, 1969; Smith and Grenvik, 1970), cerebral hypoxia caused by near-drowning (Tong, 1987), subarachnoid hemorrhage (Iwatsuki et al., 1980), encephalitis (Cowgill et al., 1974), cerebrovascular accidents (Cooperman et al., 1970), and paraplegia (Cooperman, 1970; Tobey, 1970). The onset of the period of vulnerability is not well defined. It may begin as early as 24 to 48 hours after injury and may last up to 1 to 2 years after injury (Cooperman, 1970). Because the period of risk for succinylcholine-induced hyperkalemia after cerebral injury is undefined, succinylcholine should be avoided in these patients, except in the period immediately after injury. Succinylcholine can increase CBF and ICP in patients with reduced intracranial compliance (Cottrell et al., 1983; Minton et al., 1986; Stirt et al., 1987a; Thiagarajah et al., 1988), probably because of cerebral stimulation from succinylcholine-induced increases in afferent-muscle spindle activity (Lanier et al., 1986.). The increases in CBF and ICP can be blunted by deep general anesthesia or by previous paralyzing or “defasciculating” dosages of nondepolarizing muscle relaxants (Minton et al., 1986; Stirt et al., 1987a). In contrast, most nondepolarizing relaxants have little effect on CBV and ICP (Lanier et al., 1985; Minton et al., 1985; Stirt et al., 1987b; Rosa et al., 1991), unless associated with histamine release (d-tubocurarine, atracurium), which causes transient cerebrovasodilation and increased ICP (Tarkkanen et al., 1974). In dosages that do not release histamine, atracurium does not increase ICP, despite the accumulation of laudanosine, a major metabolic product of atracurium and a potential CNS arousal agent.
Nondepolarizing Muscle Relaxants
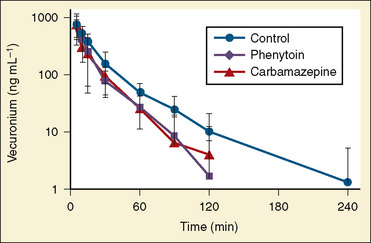
The presence of motor deficits or the administration of anticonvulsants may affect the dosage of nondepolarizing muscle relaxant necessary in neurosurgical patients. Hemiplegia from an upper motor neuron lesion (such as a stroke or a brain tumor) is associated with resistance to nondepolarizing relaxants on the paretic side (Graham, 1980; Moorthy and Hilgenberg, 1980; Shayevitz and Matteo, 1985). Excessive dosages of muscle relaxants may be given if the dosage is guided by a nerve stimulator monitoring a hemiplegic extremity. In contrast, an increased response to nondepolarizing muscle relaxants is observed in paretic-muscle lower motor neuron lesions (e.g., paraplegia and quadriplegia) (Brown et al., 1975). Acute administration of several anticonvulsants, including phenytoin, phenobarbital, trimethadione, and ethosuximide, enhances nondepolarizing neuromuscular blockade or delays its reversal (Gandhi et al., 1976; Spacek et al., 1999). Patients receiving chronic phenytoin or carbamazepine therapy are resistant to the effects of nondepolarizing relaxants, including pancuronium, (Roth and Ebrahim, 1987), metocurine (Ornstein et al., 1985), vecuronium (Ornstein et al., 1987; Alloul et al., 1996), and rocuronium (Spacek et al., 1999; Hernández-Palazón et al., 2001) but, interestingly, not mivacurium or atracurium (Ornstein et al., 1987; Spacek et al., 1996). The cause of phenytoin-induced resistance to nondepolarizing muscle relaxants and the reason for the lack of the same effect with mivacurium or atracurium are unclear. Finally, no data have yet been published describing the interactions of anticonvulsants (felbamate, gabapentin, levetiracetam, tiagabine, topiramate, sodium valproate, or valproic acid) with nondepolarizing neuromuscular blocking drugs (Fig. 22-6).
Vasodilators
The direct-acting vasodilators, including sodium nitroprusside, ATP, adenosine, nitroglycerin, diazoxide, and hydralazine, are cerebrovasodilators and may increase CBF and ICP (Stoyka and Schutz, 1975; Turner et al., 1977; Marsh et al., 1979; Cottrell et al., 1980; McDowall, 1985). The calcium channel blockers also raise CBF and ICP (Griffin et al., 1983; Mazzoni et al., 1985). These drugs should therefore be avoided in patients with reduced intracranial compliance, unless the dura is open or ICP is monitored. Indirect-acting antihypertensives, including trimethaphan (a ganglionic blocker), propranolol and esmolol (β-adrenergic blockers), and labetalol (a combined α- and β-blocker), do not increase CBF or ICP (Magness et al., 1973; Turner et al., 1977; McDowall, 1985). These agents are useful for the control of blood pressure in patients with elevated ICP. Trimethaphan may interfere with the neurologic examination by causing mydriasis, cycloplegia, or anisocoria. Sodium nitroprusside lowers the range of cerebral autoregulation. Brain-surface oxygen tension is greater (Maekawa et al., 1977) and metabolic disturbances in brain biochemistry (e.g., lactate, pyruvate, and phosphocreatine levels) are less during nitroprusside-induced hypotension than with trimethaphan- or hemorrhage-induced hypotension (Michenfelder and Theye, 1977). In addition, cortical blood flow and electrical activity are better preserved at lower MAP with sodium nitroprusside (Ishikawa and McDowall, 1981). Nitroprusside, however, induces more pronounced blood-brain barrier dysfunction than does trimethaphan (Ishikawa et al., 1983). Because CBF, brain oxygen tension, neuronal function, and brain metabolism are better maintained with sodium nitroprusside than with trimethaphan at a MAP of 50 mm Hg, sodium nitroprusside is the preferred agent for deliberate hypotension.
Central Nervous System Toxicity of Anesthetic Agents
Preclinical studies have demonstrated that prolonged exposure of fetal and neonatal laboratory animals to anesthetic drugs leads to accelerated neurodegeneration (Ikonomidou et al., 1999; Jevtovic-Todorovic et al., 2003; Slikker et al., 2007). The data generated in these reports have provoked a heated debate on its relevance to the practice of pediatric anesthesia (Anand and Soriano, 2004; Olney et al., 2004). There is no doubt that this phenomenon is valid in the experimental paradigms used in these laboratory investigations. The same model has also implicated dexamethasone and magnesium sulfate in accelerating neuroapoptosis in the development of rat brain (Noguchi et al., 2008; Dribben et al., 2009). However, a number of factors make the extrapolation of these findings to humans in the daily practice of clinical pediatric anesthesiology questionable. For example, these animal and in vitro studies have significant limitations in terms of agent dosage and duration of exposure (both absolute and in comparison with human studies) (Loepke and Soriano, 2008). Furthermore, no defined clinical markers or syndromes are associated with neonatal exposure to general anesthetics. Unlike general anesthetic agents, exposure to alcohol or anticonvulsant drugs during gestational development has clearly characterized clinical syndromes. Although histologic evidence of accelerated neurodegeneration has been verified by several investigational groups, discrepancies in neurocognitive outcomes exist (Loepke et al., 2009). Prospective clinical investigations are underway to verify these observations. Finally, the majority of neonatal and infant surgery is urgent or emergent, and anesthetic care is essential to proceed safely. It has been clearly demonstrated that inadequate anesthesia leads to poor postoperative outcomes in infants (Anand and Hickey, 1987; Anand and Soriano, 2004), and it is crucial to attenuate the stress response associated with surgical stimulation.
Several retrospective reports have positively linked exposure to general anesthesia before the age of 4 with an increased chance of developing learning deficits. Wilder and colleagues (2009) examined the extensive medical and school records of 5358 children in Rochester, Minnesota, and determined that there was an association between learning deficits in school and having more than two surgeries before age 4. A cross-sectional study done by Kalkman and colleagues (2009) in Utrecht, The Netherlands, surveyed a small number of parents of children who had had surgery at a young age and determined that there was a trend toward greater prevalence of learning deficits in children who had had early surgery. However, these reports need to be reconciled with a recent report demonstrating no causal relationship between anesthesia exposure and subsequent learning behavior in twin cohorts (Bartels et al., 2009). Clinicians should be aware of the rapid developments in the area of anesthetic-induced neurodegeneration and keep in mind the degree of necessity of infant surgical procedures and the more significant effects of the hypoxic and cardiovascular mechanisms of brain injury and death during anesthesia and surgery in these patients.
Preoperative evaluation and preparation
A proper and thorough evaluation and preparation of the pediatric patient for anesthesia and surgery are essential components of minimizing perioperative morbidity and mortality. Because of the urgent nature of many pediatric neurosurgical procedures, a thorough preoperative evaluation may be difficult. The perioperative management of pediatric patients undergoing neurosurgical procedures should be based on the developmental stage of the patient and is discussed in Chapter 8, Psychological Aspects, and Chapter 9, Preoperative Preparation. Given the systemic effects of general anesthesia and the physiologic stress of surgery, an organ system–based approach is optimal for anticipating potential physiologic derangements and coexisting disease states that may increase the risk of perioperative complications (Ferrari, 1999). Many potential perioperative problems can be preemptively addressed with such an approach. Because some children are either preverbal or do not fully understand their medical condition, their parents or primary caregivers should be interviewed carefully to obtain information regarding coexisting medical problems. A thorough review of the patient’s history can reveal conditions that may increase the risk of adverse reactions to anesthesia and perioperative morbidity and identify patients who need more extensive evaluation or whose medical condition needs to be optimized before surgery. There are also special perioperative concerns regarding children with neurologic abnormalities. However, certain aspects of this evaluation are of special relevance to neurosurgical patients. Specific concerns in these patients are listed in Table 22-3. Preoperative laboratory testing should be tailored to the proposed neurosurgical procedure.
TABLE 22-3 Common Perioperative Concerns for Infants and Children with Neurologic Lesions
| Condition | Anesthetic Implications |
| Denervation injuries | Hyperkalemia after succinylcholine Resistance to nondepolarizing muscle relaxants Abnormal response to nerve stimulation |
| Chronic anticonvulsant therapy | Hepatic and hematologic abnormalities Increased metabolism of anesthetic agents |
| Arteriovenous malformation | Potential congestive heart failure |
| Neuromuscular disease | Malignant hyperthermia Respiratory failure Sudden cardiac death |
| Chiari malformation | Apnea Aspiration pneumonia Stridor |
| Hypothalamic and pituitary lesions | Diabetes insipidus or syndrome of inappropriate antidiuretic hormone Hypothyroidism or hyperthyroidism Adrenal insufficiency or adrenal excess |
The chronicity and severity of the patient’s neurologic condition vary greatly and should dictate the perioperative management. Special attention should be given to symptoms of allergy to latex products, because anaphylaxis has been reported in a number of children who have undergone multiple previous operations, especially patients with meningomyeloceles (Holzman, 1997). Severe dehydration and electrolyte abnormalities can be the result of protracted vomiting from intracranial hypertension. Patients with diabetes insipidus can develop hypovolemia as a result of polyuria. During the perioperative period, steroids are frequently initiated to palliate cerebral swelling in patients with intracranial tumors. Therapeutic levels of anticonvulsants should be verified preoperatively and maintained perioperatively. Patients on long-term anticonvulsants may develop toxicity, especially if seizures are difficult to control. This reaction frequently manifests with abnormalities in either hematologic or hepatic function, or both. Patients on chronic anticonvulsant therapy may also require increased amounts of sedatives, nondepolarizing muscle relaxants, and narcotics because of enhanced metabolism of these drugs (Soriano et al., 2000b; Soriano et al., 2001; Soriano and Martyn, 2004).
Physical Examination
The physical examination should include a brief neurologic evaluation that includes consciousness level, motor and sensory function, normal and pathologic reflexes, integrity of the cranial nerves, and signs and symptoms of intracranial hypertension (Fig. 22-7 and Table 22-4). It is essential to document these factors in the preoperative examination to be able to gauge postoperative neurologic function. Lesions of the brainstem can manifest with cranial nerve dysfunction such as respiratory distress, impaired gag reflex and swallowing, and pulmonary aspiration. Evidence of muscle atrophy and weakness should be noted, particularly if the patient is hemparetic, hemiplegic, or bedridden, because up-regulation of acetylcholine receptors may precipitate sudden hyperkalemia after succinylcholine administration and induce resistance to nondepolarizing muscle relaxants in the affected limbs. Body weight should be accurately measured to guide the administration of drugs, fluids, and blood products. Physical signs of dehydration should be noted, especially in patients who have been chronically ill or have received osmotic or diuretic agents.
TABLE 22-4 Signs of Intracranial Hypertension in Infants and Children
| Infants | Children | Infants and Children |
| Irritability | Headache | Decreased consciousness |
| Full fontanelle | Diplopia | Cranial nerve (III and VI) palsies |
| Widely separated cranial sutures | Papilledema | Loss of upward gaze (setting-sun sign) |
| Cranial enlargement | Vomiting | Signs of herniation, Cushing’s triad, pupillary changes |
Radiologic and Laboratory Evaluation
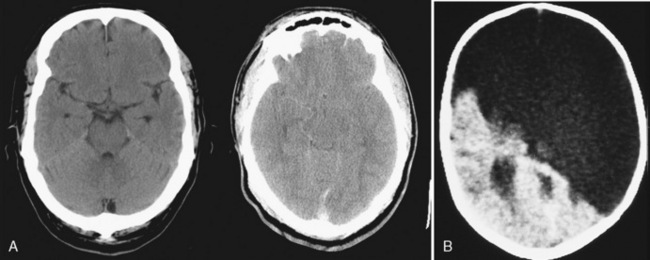
Most neurosurgical patients have a magnetic resonance imaging (MRI) or CT scan of the head as part of the preoperative assessment. These scans should be reviewed with the neurosurgeon to confirm the primary lesion and the presence of evolving neurologic conditions (hydrocephalus, compressed cisterns, midline shifts) (Fig. 22-8). Preoperative laboratory tests should be tailored to the proposed neurosurgical procedure. Given the risk of significant blood loss associated with surgery, the hematocrit, prothrombin time, and partial thromboplastin time should be obtained to uncover any insidious hematologic disorder. Patients with suprasellar pathology should have an endocrinology evaluation. Type- and cross-matched blood should be ordered before all craniotomies. Endocrinologists may be needed to help optimize the patient’s condition before surgery and to assist in postoperative management. Liver function tests and a hematologic profile should be obtained if not recently reviewed for children taking long-term anticonvulsants.
Premedication
Perioperative anxiety plays a significant role in the care of the pediatric neurosurgical patient. Anxiety issues are related to the cognitive development and age of the child (see Chapter 2, Behavioral Development). Preoperative sedatives given before the induction of anesthesia can ease the transition from the preoperative holding area to the operating room (Kain et al., 2004). Oral midazolam is particularly effective in relieving anxiety and producing amnesia. If an indwelling IV catheter is in place, midazolam can be slowly titrated to achieve sedation. Sedation is usually withheld from pediatric neurosurgical patients until they arrive in the preoperative area to avoid the problem of an oversedated, neurologically impaired patient with inadequate supervision; however, heavy premedication may be warranted to avoid agitation in patients with moyamoya disease or an intracranial aneurysm or arteriovenous malformation that has recently bled. Narcotics are best withheld preoperatively, as they may cause nausea or respiratory depression, especially in patients with increased ICP.
General principles of intraoperative management
Induction of Anesthesia
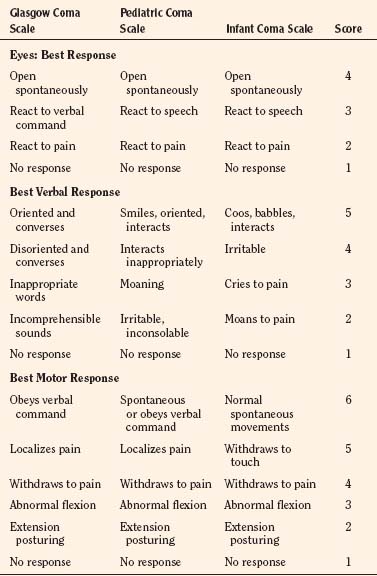
The patient’s preoperative status dictates the appropriate technique and drugs for induction of anesthesia. A modified Glasgow Coma Scale for infants and children is useful for assessing the mental status of the patient before the administration of anesthesia (Table 22-5). Somnolent patients with intracranial hypertension are at risk for aspiration pneumonitis and require a rapid-sequence induction of anesthesia.
In the presence of intracranial hypertension, the primary goal during induction is to minimize severe increases in ICP. The effects of anesthetic drugs on cerebral hemodynamics were discussed extensively earlier in this chapter and are summarized in Table 22-2. In general, most IV drugs decrease CBF, metabolism, and ICP. Thiopental (4 to 8 mg/kg) and propofol (2 to 5 mg/kg) have similar effects on cerebral hemodynamics and maintain tight coupling of CBF and cerebral metabolic rate. Patients at risk for aspiration pneumonitis (including certain patients with intracranial hypertension) should have rapid-sequence induction of anesthesia using cricoid pressure. An IV hypnotic drug, such as thiopental or propofol, is administered and followed immediately by a rapid-acting muscle relaxant. Rocuronium can be used when succinylcholine is contraindicated, such as for patients with spinal cord injuries or paretic extremities. In instances of preexisting neurologic injury (e.g., in a patient with a history of a stroke resulting in a weak extremity), succinylcholine can result in sudden, catastrophic hyperkalemia. Etomidate and ketamine are frequently used to induce anesthesia in hemodynamically compromised patients, because these drugs are less likely to cause hypotension than thiopental or propofol. However, CNS excitation and increased ICP have been associated with these drugs, respectively, and they may not be appropriate for many neurosurgical patients. Ketamine should be avoided because of its known ability to increase cerebral metabolism, CBF, and ICP.
Airway Management
Developmental changes in airway anatomy have a significant impact on management of the pediatric airway. Nasotracheal tubes may be best suited for situations when the patient will be prone, because orotracheal tubes can kink at the base of the tongue when the head is a flexed and result in airway obstruction. The timing of tracheal extubation may be challenging after neurosurgical procedures. Infants, particularly those with Chiari malformation (Cochrane et al., 1990) or older children after procedures in the posterior fossa (Cochrane et al., 1994), may exhibit intermittent apnea, vocal cord paralysis, or other irregularities before resuming a stable respiratory pattern. Significant airway edema and postoperative obstruction can complicate prone procedures or those involving significant blood losses and large volume replacement. Lingual or supraglottic swelling may require direct laryngoscopy to assess the airway. Head-up positioning and gentle forced diuresis usually improve airway edema within 24 hours.
Positioning
Patient positioning for surgery requires careful preoperative planning to allow adequate access to the patient for both the neurosurgeon and anesthesiologist. For the patient with altered physiologic states, various surgical positions can predispose to injury (Fig. 22-9 and Table 22-6). The prone position is commonly used for posterior fossa and spinal cord surgery. The sitting position may be more appropriate for obese patients, who may be difficult to ventilate in the prone position because of increased intrathoracic pressure. In addition to the physiologic sequelae of the sitting position, a spectrum of neurovascular compression and stretch injuries can occur. It is important to ensure free abdominal wall motion. Increased intraabdominal pressure can impair ventilation, cause venocaval compression, and increase epidural venous pressure and bleeding. Postoperative visual loss has been linked to surgery in the prone position. Therefore, the eyes must be free of any direct contact with the horseshoe or Mayfield head holder (Lee et al., 2006). Many neurosurgical procedures are performed with the head slightly elevated to facilitate venous and CSF drainage from the surgical site; however, superior sagittal sinus pressure decreases with increasing head elevation, and this increases the likelihood of VAE (Grady et al., 1986). Excessive rotation of the head can impede venous return via compression of the jugular veins and can lead to impaired cerebral perfusion, increased ICP, and venous bleeding. Flexion of the head can cause the tracheal tube to migrate into a main stem bronchus. Extreme head flexion can cause brainstem compression in patients with posterior fossa pathology, such as mass lesions or an Arnold-Chiari malformation and high cervical spinal cord ischemia. Likewise, extreme head extension may result in inadvertent tracheal extubation. Finally, all pressure points should be padded and peripheral pulses checked to prevent peripheral nerve and vascular compression and pressure injury.
| Position | Physiologic Effect |
| Head elevated | Enhanced cerebral venous drainage Decreased cerebral blood flow Increased venous pooling in lower extremities Postural hypotension |
| Head down | Increased cerebral venous and intracranial pressure Decreased functional residual capacity (lung function) Decreased lung compliance |
| Prone | Venous congestion of face, tongue, and neck Decreased lung compliance Increased abdominal pressure (can lead to venocaval compression) |
| Lateral decubitus | Decreased compliance of down-side lung |
Maintenance of Anesthesia
Specific drugs used for the maintenance of anesthesia have not been shown to affect the outcome of neurosurgical procedures when properly administered (Todd et al., 1993). The most frequently used technique during neurosurgery consists of an opioid (i.e., fentanyl, sufentanil, or remifentanil) along with inhaled nitrous oxide (70%) and low-dosage (0.2% to 0.5%) isoflurane. Routine administration of a preoperative benzodiazepine such as midazolam (0.5 mg/kg orally, or 0.1 mg/kg intravenously) should provide some degree of amnesia of perioperative events as well as minimize anxiety.
Chronic administration of anticonvulsant drugs, such as phenytoin and carbamazepine, induces rapid metabolism and clearance of neuromuscular blockers and opioids because of the enhanced activity of the hepatic P450 enzymes (Soriano and Martyn, 2004). Patients receiving chronic anticonvulsant therapy require larger dosages of muscle relaxants and narcotics because of the induced enzymatic metabolism of these agents (see Fig. 22-6) (Soriano et al., 2001). Muscle relaxants should be withheld or permitted to wear off when assessment of motor function during neurosurgery is planned.
Fluid and Electrolyte Management
Normal saline is commonly used as the maintenance fluid during neurosurgery because it is mildly hyperosmolar (308 mOsm/kg) and should minimize cerebral edema. Isotonic crystalloid solutions are commonly used during anesthesia and for cerebral resuscitation. However, rapid infusion of large quantities of normal saline (>60 mL/kg) can be associated with hyperchloremic acidosis (Scheingraber et al., 1999). The calculated maintenance rate of fluid administration depends on the weight of the patient (Holliday and Segar, 1957). These rates are based on normal physiologic conditions. Increases in insensible losses, blood loss, or other conditions such as diabetes insipidus or the syndrome of inappropriate antidiuretic hormone excretion, as noted later, should be considered when determining the proper amount of fluid administration. Depending on the extent and length of the surgical procedure and the exposure of vascular beds, additional fluid administration (3 to 10 mL/kg per hour) may be necessary. Unlike adults, children can become hypovolemic from scalp injuries and isolated traumatic brain injury. Hypotonic crystalloids should be avoided, and the role of colloids is controversial. In 2007, the SAFE study reported that patients with TBI who received fluid resuscitation with albumin had higher mortality rates than those who received fluids with crystalloids (Myburgh et al., 2007). The use of hydroxyethyl starch is discouraged because of its role in exacerbating coagulopathy. Hypertonic saline (0.1 to 1.0 mL/kg) may be used to increase CPP, but, in this setting, well-conducted studies show that there is no advantage to hypertonic saline compared with conventional prehospital fluid protocols (Cooper et al., 2004), and the very large, randomized SAFE trial found that there was no difference in 28-day survival between albumin and saline resuscitation for intensive care patients. However, in a subgroup of patients with TBI, fluid resuscitation with albumin was noted to have higher mortality rates than resuscitation with crystalloid (Myburgh et al., 2007).
Infants are at particular risk for perioperative hypoglycemia. Small premature neonates, with limited reserves of glycogen and limited gluconeogenesis, require continuous infusions of glucose at 5 to 6 mg/kg per minute to maintain serum levels. At the same time, the stress of critical illness and resulting insulin resistance can produce hyperglycemia that, in turn, is associated with neurologic injury (Jeremitsky et al., 2005; Van den Berghe et al., 2005) and poor outcomes in adults (Van den Berghe et al., 2001). However, it is unclear if tight glycemic control offers significant benefits to children (Branco and Tasker, 2007; Klein et al., 2007). Limited evidence now suggests that tight control may carry undue risk of hypoglycemia, and newer data are less supportive of very tight glycemic control (Van den Berghe et al., 2006). Retrospective studies from children suggest that both hyperglycemia (glucose level at 200 to 250 mg/100 mL) and hypoglycemia occur after TBI (Sharma et al., 2009), and that hyperglycemia is associated with poor outcome. Continuous intraoperative monitoring is not yet the standard of care. Hourly glucose checks are recommended.
Brain swelling can occur during neurosurgical procedures, with devastating consequences. Aggressive hyperventilation should be reserved for when herniation is impending and immediate life-saving maneuvers are required. However, a study in adults demonstrated that moderate hyperventilation may improve surgical conditions (Gelb et al., 2008). There is considerable evidence in patients with TBI that even mild hyperventilation leads to hypoperfusion (Coles et al., 2002). Although there are no data suggesting such a phenomenon in other neurosurgical patient populations, other methods of reducing intracranial pressure can be used. Elevation of the head above the heart and the use of hyperosmolar therapy (e.g., mannitol or hypertonic saline) are generally used initially.
Mannitol (0.25 to 0.5 g/kg) transiently alters cerebral hemodynamics and raises serum osmolality by 10 to 20 mOsm/kg (Soriano et al., 1996). However, repeated dosing can lead to extreme hyperosmolality, renal failure, and further brain edema. Hypertonic saline increases serum sodium, decreases ICP, and increases CPP titrated to a serum sodium rate change and brain edema (Khanna et al., 2000). Administration is typically 3 mL/kg of 3% (hypertonic) saline targeted to a serum sodium rate change of 0.05 mEq/hr and serum sodium levels of 155 to 160 mEq/L. However, the endpoints depend on the initial serum sodium and the degree of brain edema (typically 155 to 160 mEq/L). Three percent normal saline is generally administered centrally to avoid phlebitis and tissue necrosis, but hypertonic saline concentrations of 2% may be administrated peripherally. Theoretical risks include central pontine myelinolysis and renal failure. Furosemide is a useful adjunct to mannitol for decreasing acute cerebral edema and has been shown in vitro to prevent rebound swelling caused by mannitol (McManus and Soriano, 1998; Thenuwara et al., 2002).
Nonosmotic secretion of antidiuretic hormone (ADH) makes hyponatremia common after neurosurgery. Elevated ADH levels can result from a variety of stimuli, ranging from pain and nausea to fluid shifts and intravascular hypovolemia. Acute hyponatremia can provoke seizures and can be treated with hypertonic saline, fluid restriction, and diuretics (Porzio et al., 2000). The syndrome of cerebral salt wasting is also common in children and can be seen after head trauma and all manner of neurosurgical procedures. The syndrome has been diagnosed with increasing frequency and reported in association with meningitis (Celik et al., 2005), calvarial remodeling (Levine et al., 2001; Byeon and Yoo, 2005), tumor resection (Jimenez et al., 2006), and even hydrocephalus (Table 22-7). Although it is easily confused with other entities (Singh et al., 2002), a recent retrospective review put its incidence at 11.3 per 1000 procedures (Jimenez et al., 2006). In these patients, the mean duration of symptoms was 6 days, with a range of 1 to 5. Cerebral salt wasting, the result of excessively high atrial or brain natriuretic peptide levels (Berger et al., 2002), is marked by hyponatremia, hypovolemia, and excessive urinary excretion of sodium. Although the classic treatment involves saline administration, more rapid resolution has been achieved with fludrocortisone (Papadimitriou et al., 2007).
TABLE 22-7 Comparison of Syndrome of Inappropriate Antidiuretic Hormone (SIADH), Cerebral Salt Wasting (CSW), and Diabetes Insipidus
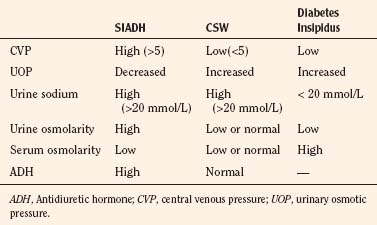
Diabetes insipidus is a well-known complication of surgical procedures involving or adjacent to the pituitary and hypothalamus. It is most frequently seen in association with craniopharyngioma, where it can be a manifesting symptom in 40% of cases (Ghirardello et al., 2006). Diabetes insipidus is recognized by a rising serum sodium (>145 mg/dL) accompanied by copious (>4 mL/kg per hour) output of dilute urine. Severe dehydration and hypovolemia may develop. One effective protocol employs maximal antidiuresis with IV vasopressin and strict limitation of IV fluids (Wise-Faberowski et al., 2004). This strategy avoids the pitfalls of titrating drug to urine output and recognizes that renal blood flow remains normal in the normovolemic, but maximally antidiuresed, child. Because urine output can be minimal (0.5 mL/kg per minute), other clinical markers of volume status must be used.
Monitoring
Patients undergoing major craniotomies are at risk of sudden hemodynamic instability resulting from hemorrhage, venous air emboli, herniation syndromes, or manipulation of cranial nerves. The potential massive blood loss warrants placement of an arterial cannula for continuous invasive blood pressure monitoring as well as for sampling serial blood gases, electrolytes, glucose levels, and hematocrit. The usefulness of central venous catheterization remains controversial. Cannulation of the jugular or subclavian veins with multiorificed catheters in adults is often preferred, particularly when VAE is anticipated. However, these catheters are too large for infants and most small children. In infants, even when VAE occurred, a central venous catheter was frequently not successful for aspirating air, presumably because of the high resistance of the small-gauge catheters used in these patients (Cucchiara and Bowers, 1982). Therefore, the risks of a central venous catheter may outweigh its benefits.
Venous Air Embolus
Venous air embolus can occur during craniotomies in infants and children, primarily because the head of a small child is large in relation to the rest of the body, and it rests above the heart in either the prone or the supine position. Venous sinuses have dural attachments that impede their ability to collapse. Other conduits for VAE include bone, bridging veins, and spinal epidural veins. When air enters the central circulation, right ventricular output is impeded, leading to pulmonary edema and bronchoconstriction and cor pulmonale, cardiovascular collapse, and death in the most severe cases. Air may gain access to the systemic circulation through right-to-left intracardiac shunts. Potential cardiac shunts exist in many healthy infants and children and may become clinically significant if pulmonary hypertension develops acutely after a large air embolism. Paradoxical air emboli may cause cerebral or coronary artery obstruction, with subsequent cerebral infarction or ventricular fibrillation. Although the incidence of VAE is greatest in the sitting position, it has been reported in the lateral, supine, and prone positions. The incidence of VAE in children undergoing suboccipital craniotomy in the sitting position is not significantly different from that in adults, but children appear to have a higher incidence of hypotension and a lower likelihood of successful aspiration of central (intravascular) air (Cucchiara and Bowers, 1982).
Venous air emboli pose a risk during craniotomies in infants and children, primarily because the head of a small child is large in relation to the rest of body and rests above the heart in both the prone and supine positions (Harris et al., 1987; Faberowski et al., 2000). Standard neurosurgical techniques may elevate the patient’s head even further by elevating the head of the table to improve cerebral venous drainage, which can increase the risk for air entrainment into the venous system through open venous channels in bone and sinuses (Grady et al., 1986). Patients with cardiac defects with potential for right-to-left shunting, such as patent foramen ovale or a patent ductus arteriosus, are at high risk for paradoxical air emboli leading to cerebral or myocardial infarction.
Prompt recognition of VAE is crucial to successful management (Fig. 22-10). Precordial Doppler ultrasonography has been demonstrated to be the earliest and most sensitive indicator of intracranial air. It enables diagnosis before the pathologic consequences occur. The Doppler probe is best positioned on the anterior chest, usually just over or to the right of the sternum at the fourth intercostal space (i.e., the nipple line). An alternative site between the scapulae on the posterior thorax can be used in infants in the prone position weighing approximately 6 kg or less (Soriano et al., 1994). Doppler positioning can be confirmed by listening for the characteristic change in sounds after rapid administration of a few milliliters of saline into a central or peripheral venous catheter. When VAE occurs, there is reflex pulmonary vasoconstriction and ventilation/perfusion mismatch caused by the air blocking passage of blood. This reflex results in increased dead-space ventilation and causes a sudden decrease in end-tidal CO2 concentration. Monitoring the end-tidal CO2 concentration is very useful in making the diagnosis and can be used to monitor the severity and duration of air emboli. An increase in end-tidal nitrogen concentration during continuous monitoring is a specific sign of air emboli, but it is usually of such small magnitude that it is difficult to detect with devices currently available in clinical practice. Increases in right atrial and pulmonary artery pressures correlate with the size of emboli, but these delayed findings alone should not be relied on for monitoring and diagnosis. Echocardiography (transthoracic or transesophageal) may be the most specific method for detecting small air emboli but is not easily used intraoperatively, especially in small infants and children. In addition to the characteristic changes in Doppler sounds, sudden decreases in end-tidal CO2, dysrhythmias, and ischemic changes in the electrocardiogram can occur with VAE.
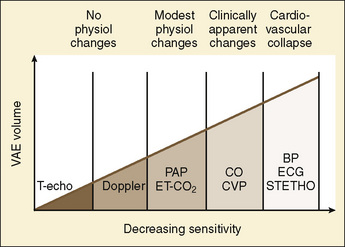
Once the patient has been stabilized, the procedure is resumed and a revised anesthesia plan is instituted. The decision to administer nitrous oxide after successful treatment of VAE is controversial. If reinstitution of nitrous oxide is associated with another decrease in end-tidal CO2 concentration or clinical deterioration, then residual air bubbles are probably present and nitrous oxide should be discontinued. If no changes occur, many clinicians reinstitute nitrous oxide unless repeated episodes of VAE occur. Hemodynamic support may be necessary to avoid hypotension, maintain adequate cerebral perfusion pressure, and minimize ischemic injury. Therefore, dopamine and epinephrine are effective in supporting systemic pressure and restoring cerebral blood flow, even in very low birth weight infants (Munro et al., 2004).
Neurophysiologic Monitoring
Recent advances in neurophysiologic monitoring have enhanced the ability to safely perform neurosurgical procedures in functional areas of the brain and spinal cord. However, the neurodepressant effects of many commonly used anesthetic agents limit the usefulness of these monitors. In general, electrocorticography (ECoG) and EEG can be used with low levels of volatile anesthetics. Somatosensory evoked potentials used during spinal and brainstem surgery can also be depressed by high levels of volatile agents and, to a lesser extent, nitrous oxide. An anesthetic technique using high-dosage opioids and minimal inhaled agents (<1 MAC) is the most appropriate type of monitoring. Alternatively, total IV anesthesia (TIVA) techniques can be used. Spinal cord and peripheral nerve surgery may require electromyography (EMG) and detection of muscle movement as an endpoint. Therefore, muscle relaxation should be avoided or discontinued after tracheal intubation to facilitate monitoring. Table 22-2 lists common anesthetics and their effects on various neurophysiologic monitors. Motor evoked potential (MEP) can be used to monitor the integrity of the motor tracts of the spinal cord by stimulating the motor cortex and detecting the action potentials in the corresponding muscle groups (Table 22-8). Volatile anesthetic agents, including nitrous oxide, have a dosage-dependent depressant effect on the MEP. IV anesthetics (propofol, opioids, ketamine, and dexmedetomidine) preserve the MEP.
| Brainstem Area | Signs | Changes in Monitor |
| CN V | Hypertension, bradycardia | Arterial pressure, ECG |
| CN VII | Facial muscle movement | EMG |
| CN X | Hypotension, bradycardia | Arterial pressure, ECG |
| Pons, medulla | Arrhythmias, hypotension or hypertension, tachycardia or bradycardia, irregular breathing pattern | ECG, arterial pressure, end-tidal CO2 monitor |
CN, Cranial nerve; ECG, electrocardiogram; EMG, electromyogram.
Monitors of Cerebral Oxygenation
Electroencephalography
The EEG is the most commonly used monitor for cerebral ischemia and has been intensively investigated during anesthesia. Electroencephalographic waveforms are the summation of excitatory and inhibitory postsynaptic potentials from the superficial layers of the cerebral cortex. Both regional and global ischemias result in profound depression of EEG activity, characterized by an attenuation of high-frequency activity and the appearance of slow waves in the corresponding area (Fig. 22-11). This feature makes the EEG the most reliable intraoperative monitor for focal cerebral ischemia. Direct analysis of the raw EEG by an electrophysiologist is the gold standard for monitoring cerebral ischemia. Conditions in which cerebral perfusion may be compromised can be monitored with scalp EEG electrodes or direct ECoG with subdural strip electrodes. Cerebrovascular disease is rare in infants and children, but two conditions in which EEG monitoring may be beneficial are temporary clipping of cerebral aneurysms and moyamoya disease. Cerebral aneurysm clipping can result in ischemia in the cerebral regions supplied by adjacent arteries. Direct ECoG in the region at risk can detect cerebral ischemic during test occlusion of the aneurysm and can serve as a guide for proper positioning of the aneurysm clip and institution of pharmacologic interventions to improve cerebral perfusion.
Jugular Bulb Catheters
Jugular bulb catheters may also be inserted retrograde into the internal jugular using angiocatheters for intermittent sampling, or fiberoptic catheters for continuous readings, and are a useful tool for determining cerebral oxygenation via jugular venous oxygen saturation (Sjvo2). Normal Sjvo2 ranges from 50% to 75%; values at either extreme reflect global ischemia or hyperemia. Sjvo2 monitoring can provide early diagnosis of global ischemia and is useful to guide decisions for optimizing hyperventilation therapy, perfusion pressure, fluid management, and oxygenation in head injury patients (Chan et al., 1992; Skippen et al., 1997). Matta and coworkers (1994) have demonstrated in adults that Sjvo2 monitoring detects critical intraoperative cerebral desaturation that would otherwise have been untreated, and Moss and coworkers (1995) have used Sjvo2 monitoring to determine the minimum blood pressure that should be maintained to avoid hypoperfusion during aneurysm surgery.
Cerebral Oximetry
Near-infrared spectroscopy (NIRS) provides a noninvasive assessment of cerebral intravascular oxyhemoglobin and deoxyhemoglobin (oxyhemoglobin and deoxyhemoglobin) and mitochondrial cytochrome oxygenation by measuring the abilities of these two chromophores to absorb near-infrared light. Quantitative measurement of changes in cerebral chromophore oxygen (O2) concentration is related to the overall optical attenuation of near-infrared light, and it has been used to assess O2 delivery and extraction, cerebral blood volume, CBF (by an indicator dye technique), and the redox state of the brain. NIRS correlated with mean arterial pressure and is effective in identifying premature infants with impaired cerebrovascular autoregulation (Tsuji et al., 2000). The equipment used in these studies is primarily a research prototype and is not commercially produced. Therefore, this technique has yet to find a niche in routine intraoperative monitoring. Regional cerebral oximetry (rSo2) (INVOS, Somanetics Corporation, Troy, MI) is a simpler NIRS modality that measures relative changes in oxygen extraction in the total blood volume of a small brain area. This technique uses two wavelengths to determine oxyhemoglobin and total cerebral hemoglobin concentrations. Because approximately 75% of the blood volume in the brain is venous, the cerebral oximeter measurement reflects venous oxyhemoglobin saturation and has been reported to correlate with jugular bulb saturation.
Nerve Root Monitoring
A specific modality for monitoring spinal cord reflexes is the Hoffman reflex (HR), which is a measure of motor neuron excitability and is significantly increased in children with spastic diplegia. This response can be evoked by directly stimulating the nerve root and recording the direct efferent muscle response and the HR (Fig. 22-12). The HR represents the reflex potential emanating from the motor neurons of the spinal cord (Logigian et al., 1994). An abnormal reflex would have an exaggerated HR amplitude. Inhalational agents, nitrous oxide, barbiturates, and benzodiazepines all depress the HR (Soriano et al., 1995). An opioid-based anesthetic with intermittent doses of ketamine or propofol does not appear to suppress the HR. Because the EMG is the observed endpoint of HR, muscle relaxation should be avoided.
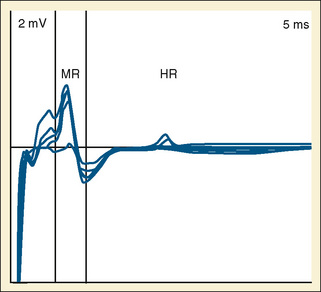
Specific pediatric neurosurgical conditions
Encephaloceles
Encephaloceles are neural tube defects that are protrusions of brain and CSF arising from the occiput to the frontal area (Fig. 22-13). They can even look like nasal polyps if they protrude through the cribriform plate. Large defects may present challenges to endotracheal intubation. Much blood loss can occur, especially if venous sinuses are entered. Adequate IV access should be ensured, and blood products should be available.
Myelodysplasia
Defects in the spine are known as spina bifida. If a bulge containing CSF without spinal tissue exists, it is called a meningocele. When neural tissue is also present in the lesion, the defect is called a meningomyelocele. Open neural tissue is known as rachischisis. Hydrocephalus is usually present when paralysis occurs below the lesion and is usually associated with an Arnold-Chiari malformation (see later) (Figs. 22-14 and 22-15).
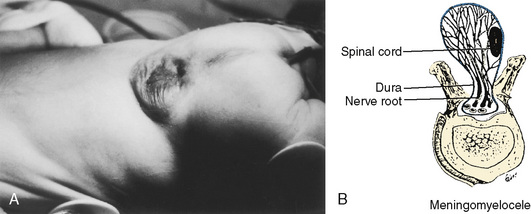
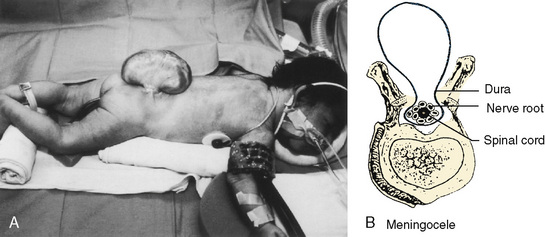
Most patients with a meningomyelocele present for primary closure within the first day or two of life to minimize the risk of infection. Many are now scheduled electively before birth for repair, because the defect is usually apparent on prenatal ultrasonography. Many neurosurgeons prefer to insert a ventriculoperitoneal shunt at the time of initial surgery. Alternatively, a shunt may be inserted a few days later or even occasionally deferred if there is no evidence of hydrocephalus at birth. A major anesthetic consideration is positioning the neonate for induction at surgery. In most cases, tracheal intubation can be performed with the infant in the supine position and the uninvolved portion of the patient’s back supported with towels (or a “donut” ring), so that there is no direct pressure on the meningomyelocele. For very large defects, it is occasionally necessary to place the infant in the lateral position for induction and intubation. Succinylcholine is rarely necessary in these situations, yet it does not appear to cause problems with hyperkalemia because the defect develops early in gestation (Dierdorf et al., 1986).
Patients with myelodysplasia are at high risk of developing allergic reactions to latex (Holzman, 1997). This is probably a result of repeated exposures to latex products encountered during surgery, including nonneurosurgical procedures required to correct coexisting orthopedic and urologic problems, or repeated bladder catheterizations. Patients who develop latex allergy appear to have an increased incidence of allergic reactions to other substances, including some antibiotics and foods, so anaphylaxis should always be anticipated in these situations. Postoperatively, respiratory status should be carefully assessed. Pulse oximetry is valuable during recovery from anesthesia because of difficulty with breathing after a tight closure, and because ventilatory responses to hypoxia are often diminished or absent in these patients when a Chiari malformation coexists (Waters et al., 1998).
Spinal dysraphism is the primary indication for laminectomies in pediatric patients. Patients with myelomeningocele suffer from multisystem diseases that result from a severe injury to the developing CNS early in gestation. The systems involved can include the musculoskeletal system, genitourinary system, and immune system in addition to the CNS. These patients suffer frequent exposures to latex products and are at high risk for developing a hypersensitivity to latex. All these patients should be treated with a latex alert, and a true anaphylaxis to latex must be considered when appropriate. Latex allergy can manifest itself by a severe anaphylactic reaction heralded by hypotension and wheezing with or without a rash and should be rapidly treated by removal of the source of latex and administration of IV fluid and vasopressors (Holzman, 1997). Insertion of an epidural catheter by the surgeon under direct vision can provide a conduit for the administration of local anesthetics and opioids for the management of postoperative pain.
Severe spasticity associated with cerebral palsy can be surgically alleviated by a selective dorsal rhizotomy, which reduces spasticity by surgically dividing dorsal rootlets to diminish the afferent input to motor neurons in the spinal cord, thus decreasing the hyperactive active reflexes associated with spastic diplegia. Pathologic rootlets are identified by direct stimulation and noting the corresponding muscle action potential with EMG. Exaggerated action potentials can be elicited in innervated as well as other distal muscle groups. These abnormal rootlets are partially sectioned to decrease afferent nerve conduction. However, these rootlets may contain sensory and proprioceptive fibers. Spinal cord reflexes can be quantified by measuring the HR (as noted earlier). The postoperative care of these patients is completed by severe somatic incisional pain, dysesthesia, and hyperesthesia of the affected limb and muscle spasms. A variety of postoperative pain management techniques have been advocated in these patients. These include IV morphine, midazolam/diazepam infusions, and epidural opioids (Geiduschek et al., 1997).
Chiari Malformations
Chiari malformations are generally anatomic abnormalities of the posterior fossa leading to cephalad displacement of the cerebellar vermis through the foramen magnum. There are four types of Chiari malformations (Fig. 22-16 and Box 22-1). Type I occurs in healthy children without myelodysplasia. These patients, most of them adolescents, generally have much milder symptoms, sometimes presenting with only headache or neck pain. The Arnold-Chiari malformation (type II) almost always coexists in children with myelodysplasia. This defect consists of a bony abnormality in the posterior fossa and upper cervical spine with caudal displacement of the cerebellar vermis and lower brainstem below the plane of the foramen magnum. Medullary cervical cord compression can occur. Vocal cord paralysis with stridor and respiratory distress, apnea, abnormal swallowing and pulmonary aspiration, opisthotonos, and cranial nerve deficits may be associated with the Arnold-Chiari malformation and usually appear during infancy. Patients of any age may have abnormal responses to hypoxia and hypercarbia because of cranial nerve and brainstem dysfunction (Ward et al., 1986). Extreme head flexion may cause brainstem compression in otherwise asymptomatic patients. Type III Chiari malformations are associated with encephaloceles and have the most severe symptoms and long-term disability. Type IV is associated with absent cerebellum, and large posterior fossa CSF spaces.
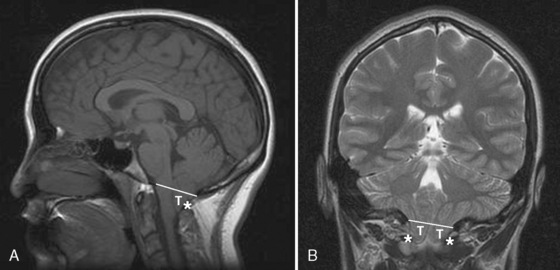
Tumors
Brain tumors are the most common solid tumors in children, which are exceeded only by the leukemias as the most common pediatric malignancy (Pollack, 1994). Supratentorial tumors account for about 25% to 40% of brain tumors in children. Supratentorial resection usually requires invasive monitoring and techniques to control elevated ICP (Fig. 22-17). The majority of brain tumors in children are infratentorial, in the posterior fossa. These include medulloblastomas, cerebellar astrocytomas, brainstem gliomas, and ependymomas of the fourth ventricle (Fig. 22-18). Because posterior fossa tumors usually obstruct CSF flow, increased ICP occurs early. Presenting signs and symptoms include early morning vomiting and irritability or lethargy. Cranial nerve palsies and ataxia are also common findings, with respiratory and cardiac irregularities usually occurring late. Sedation or general anesthesia may be required for radiologic evaluation or radiation therapy.
FIGURE 22-18 MR image of infratentorial tumors. A, Brainstem glioma. B, Ependymoma displacing the pons and medulla.
Arrhythmias and acute blood pressure changes may occur during surgical exploration, especially when the brainstem is manipulated, so careful observation of the electrocardiogram and arterial waveform tracing is important. Altered respiratory control is generally masked by muscle relaxants and mechanical ventilation. Intracranial compliance is presumed to decrease even when ICP is only marginally elevated, thus precautions to minimize increases in ICP should be taken. If ICP is markedly elevated or acutely worsens, a ventricular catheter may be inserted by the neurosurgeon prior to tumor resection to permit emergent drainage of CSF. Fixation pins used in small children can cause skull fractures, dural tears, and intracranial hematomas. Elevation of the bone flap can result in sinus tears, massive blood loss, or VAE. Surgical resection of tumors in the posterior fossa can also lead to brainstem and cranial nerve damage. Table 22-8 lists some of the signs of encroachment on these structures. VAE is a potentially serious complication that is not eliminated by the use of the prone or lateral position because head-up gradients of 10 to 20 degrees are frequently used to improve cerebral venous drainage. In infants and toddlers, large head size relative to body size accentuates this problem. Damage to the respiratory centers and cranial nerves can lead to apnea and airway obstruction after extubation of the patient’s trachea.
Tumors in the midbrain include craniopharyngiomas, optic gliomas, pituitary adenomas, and hypothalamic tumors and account for approximately 15% of intracranial tumors (Fig. 22-19). Hypothalamic tumors (hamartomas, gliomas, and teratomas) frequently appear with precocious puberty in children who are large for their chronological age. Craniopharyngiomas are the most common perisellar tumors in children and adolescents and may be associated with hypothalamic and pituitary dysfunction. Symptoms often include growth failure, visual impairment, and endocrine abnormalities. Signs and symptoms of hypothyroidism should be sought and thyroid function tests measured. Steroid replacement (dexamethasone or hydrocortisone) is generally administered because the integrity of the hypothalamic-pituitary-adrenal axis may be uncertain. In addition, diabetes insipidus occurs preoperatively in some patients and is a common postoperative problem. A careful history usually reveals this condition preoperatively, especially if attention is focused on nocturnal drinking and enuresis. Evaluation of serum electrolytes and osmolality, urine-specific gravity, and urine output is helpful, because hypernatremia and hyperosmolality, along with dilute urine, are typical findings. If diabetes insipidus does not exist preoperatively, it usually does not develop until the postoperative period. This occurs because the reserve of antidiuretic hormone in the posterior pituitary gland is enough to allow functioning for many hours even when the hypothalamic-pituitary stalk is damaged intraoperatively. Postoperative diabetes insipidus is marked by a sudden large increase in dilute urine output associated with a rising serum sodium concentration and osmolality. Treatment can initially be with dilute crystalloid solutions to replace the urine output, with careful attention to electrolyte measurements. However, urine output is usually so prodigious (up to 1 L/hr in an adult) that an infusion of aqueous vasopressin (1 to 10 mU/kg per hour) is best used, with fluid input then carefully restricted to match urine replacement and estimates of insensible losses. If diabetes insipidus persists, intranasal desmopressin can be used to replace IV pitressin, because desmopressin generally needs to be administered only twice daily. Return of antidiuretic hormone activity a few days postoperatively may cause a marked decrease in urinary output, water intoxication, seizures, and cerebral edema if desmopressin is not discontinued and fluid administration not adjusted appropriately.
Approximately 25% of intracranial tumors in children involve the cerebral hemispheres. These are primarily astrocytomas, oligodendrogliomas, ependymomas, and glioblastomas. Neurologic symptoms are more likely to include a seizure disorder or focal deficits. Succinylcholine should be avoided if motor weakness is present, as it can cause sudden massive hyperkalemia. Nondepolarizing muscle relaxants and narcotics may be metabolized more rapidly than usual in patients receiving chronic anticonvulsants. Choroid plexus papillomas are rare but occur most often in children younger than 3 years. They usually arise from the choroid plexus of the lateral ventricle and produce early hydrocephalus as a result of increased production of CSF and obstruction of CSF flow. Hydrocephalus usually resolves with surgical resection. When lesions lie near the motor or sensory strip, a special type of somatosensory evoked potential monitoring called phase reversal may also be used to delineate these important eloquent locations (Hatipoğlu et al., 2009). If cortical stimulation is planned to help identify motor areas, muscle relaxants must be permitted to wear off. Nitrous oxide and narcotics are usually sufficient to prevent patient movement during these periods.
Hydrocephalus
Hydrocephalus, the most common pediatric neurosurgical condition, involves a mismatch of CSF production and absorption, leading to increased intracranial CSF volume (Fig. 22-20). The majority of cases of hydrocephalus result from obstruction of CSF flow or inability to absorb CSF appropriately. Hemorrhage (neonatal intraventricular or subarachnoid), congenital problems (aqueductal stenosis), trauma, infection, or tumors (especially in the posterior fossa) can cause hydrocephalus. Hydrocephalus is classified as nonobstructive/communicating or obstructive/noncommunicating based on the ability of CSF to flow around the spinal cord in its usual manner. Unless the etiology of the hydrocephalus can be definitively treated, treatment entails surgical placement of a ventricular drain or ventriculoperitoneal shunt.
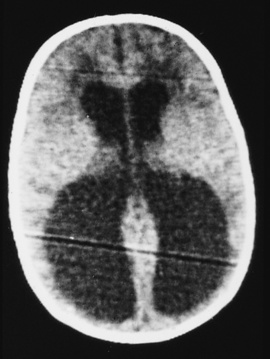
FIGURE 22-20 Infantile hydrocephalus. CT scan demonstrates a dilated ventricular system and thinning of the cortical mantle.
(Courtesy Division of Neuroradiology, University Health Center of Pittsburgh.)
Endoscopic third ventriculoscopy by way of a percutaneous flexible neuroendoscope is an alternative to extracranial shunt placement (Goumnerova and Frim, 1997). During these procedures, a ventriculostomy may be made to bypass an obstruction (such as aqueductal stenosis) by forming a communicating hole from one area of CSF flow to another using a blunt probe inserted through the neuroendoscope (Fig. 22-21). Common locations for a ventriculostomy are through the septum pellucidum (so the lateral ventricles can communicate) or through the floor of the third ventricle into the adjacent CSF cisterns. Complications such as damage to the basilar artery or its branches or neural injuries can be life threatening, and the anesthesiologist should be prepared for an emergency craniotomy during these procedures. Bradycardia and other arrhythmias have also been reported in conjunction with irrigation fluids and manipulation of the floor of the third ventricle (El-Dawlatly et al., 2000).
Slit ventricle syndrome develops in approximately 5% to 10% of patients with CSF shunts and is associated with overdrainage of CSF and small, slitlike lateral ventricular spaces. Patients with this condition do not have the usual amount of intracranial CSF to compensate for alterations in brain or intracranial blood volume. Administration of excess or hypotonic IV solutions should be avoided to minimize brain swelling, because postoperative cerebral herniation has been reported after uneventful surgical procedures in these children (Eldredge et al., 1997).
Craniosynostosis
Craniosynostosis is a congenital anomaly in which one or more cranial sutures close prematurely. It occurs in approximately one of every 2000 births, with males affected more frequently than females. It can involve one suture, or it may be very complex and be associated with a variety of syndromes. If left uncorrected, the deformed cranium can result in increased ICP and compression of the brain, with potential neurologic consequences (Inagaki et al., 2007). Surgical correction is usually performed within the first months of life to achieve the best cosmetic results, because brain growth is very rapid during infancy, “pushing” the skull into a normal shape. Repair of craniosynostosis may involve removal of one small strip of bone from the skull, or it may entail a complete reconstruction of the calvarium (see Chapter 25, Anesthesia for Plastic Surgery).
Although these operations are extradural procedures, significant blood loss from the scalp and cranium make them challenging. It is essential that adequate venous access for rapid blood administration be secured, especially if multiple sutures are involved. Antifibrinolytic drugs (tranexamic acid) may have some usefulness in these procedures. The incidence of VAE is also significant during these procedures (Faberowski et al., 2000). Precordial Doppler ultrasonography and end-tidal CO2 monitoring are useful for early detection of VAE. Because neuroendoscopic techniques are designed to minimize surgical incision, dissection, and blood loss, less aggressive fluid replacement and invasive hemodynamic monitoring is becoming the norm. The application of endoscopic techniques for craniosynostosis repair has resulted in significantly less morbidity. Endoscopic strip craniectomies are associated with decreased blood loss, decreased surgical time, and improved postoperative recovery time for neonates and infants (Jimenez and Barone, 1998; Jimenez et al., 2002). Tobias and colleagues (2001) reported a significant decrease in the incidence of VAE during this procedure.
Epilepsy
Epilepsy remains one of the most common neurologic disorders in children. Although new pharmacologic interventions have shown promise in the medical management of childhood epilepsy, a large number of children continue to have intractable seizures, and physicians resort to surgical interventions in such situations. Chronic administration of anticonvulsant drugs, such as phenytoin and carbamazepine, induces rapid metabolism and clearance of neuromuscular blockers and opioids by up-regulating hepatic P450 enzymes (Soriano and Martyn, 2004). General anesthetics can compromise the sensitivity of intraoperative neurophysiologic monitors that are used to guide the actual resection of the epileptogenic focus (Eldredge et al., 1995). If cortical stimulation is used to mimic the seizure pattern or identify areas on the motor strip, neuromuscular blockade should be antagonized.
Advances in neurophysiologic monitoring have enhanced the ability to safely perform more definitive neurosurgical resections in functional areas of the brain. A variety of techniques are used during the entire perioperative period to aid in localization of seizure foci. A major part of preoperative planning should include a thorough discussion of the modality and type of neurophysiologic monitoring to be used during the surgical procedure. In general, ECoG and an EEG can be used when the patient is on low levels of volatile anesthetics. Cortical stimulation of the motor cortex necessitates observation of motor movement of the specific area of the homunculus. Therefore, muscle relaxation should be avoided, or permitted to dissipate, during the monitoring period. ECoG is typically recorded continuously on a polygraph via a grid and strip electrodes placed on the surface of the brain after dural opening. Because some epileptogenic foci are in close proximity to cortical areas controlling speech, memory, and motor or sensory function, monitoring of patient and electrophysiologic responses is frequently used to minimize iatrogenic injury to these areas (Adelson et al., 1995; Ojemann et al., 2003). Cortical stimulation of the motor strip in a child under general anesthesia will require either EMG or direct visualization of muscle movement. Neuromuscular blockade should not be used in this situation. Cortical stimulation is possible using a dual channel stimulator. Epileptogenic activity may be evident by either clearly documented electrographic seizures or EEG spike activity, which consists of either interictal spikes of 50 to 80 msec or sharp waves of 80 to 200 msec. During anesthesia, the use of low concentrations of volatile anesthetics or nitrous oxide and opioids alone should avoid attenuating ECoG and EEG recordings.
In patients with generalized seizures, precise localization of the seizure focus is essential to minimize postoperative functional deficits. This involves serial craniotomies. The first is insertion of an intracranial grid and strip electrodes. EEG grids or strips are placed on the exposed cortical surface to accurately create a map that will localize the seizure focus. The patient is then observed in an electrophysiology unit to map the location of the seizure foci and provide a road map for the neurosurgeon for the resection. In some cases, the patient is monitored over several days. Once a seizure map is generated, the patient returns to the operating room for definitive resection. Depending on the location of seizure foci, age, and development of the patient, the resection is performed under general anesthesia or by an awake craniotomy technique. It is important to avoid administration of nitrous oxide until the dura is opened, because intracranial air can persist up to 3 weeks after a craniotomy, and nitrous oxide in these situations can cause rapid expansion of air cavities and result in tension pneumocephalus (Reasoner et al., 1994).
Neural function is best assessed in an awake and cooperative patient (Penfield, 1954). Positioning of the patient is critical for the success of this technique, as the patient must remain still for an extended period of time. The patient should be in a semilateral position to allow patient comfort as well as surgical and airway access to the patient. Motor and sensory cortices are localized by inducing motor movements or sensory changes with cortical stimulation. Language function is tested by eliciting speech arrest with cortical stimulation. Verbal memory is tested by stimulating the hippocampus or lateral temporal cortex. A variety of techniques have been advocated to facilitate intraoperative assessment of motor-sensory function and speech. These range from no sedation with local anesthesia, to asleep-awake-asleep techniques involving general anesthesia induced before and after functional testing (Sarang and Dinsmore, 2003; Brunson and Mayhew, 2005). The asleep-awake-asleep technique entails induction of general anesthesia. The airway is secured either by tracheal intubation or by placement of a laryngeal mask airway. General anesthesia is maintained during the craniotomy. The anesthetic is discontinued to permit functional neurophysiologic testing. Once the testing is completed, general anesthesia is reinstituted and the airway is secured. The advantage of this technique is that the patient is completely anesthetized during the most painful segments of the surgery. However, a major drawback is the depressant effects of residual anesthetics and the inability to predict the patient’s response to emerging from anesthesia under the surgical drapes. Therefore, monitored conscious sedation is the most frequently used technique in pediatric patients. Deep levels of sedation with a variety of IV anesthetic and sedative drugs (propofol and dexmedetomidine) combined with an opioid (fentanyl or remifentanil) have been shown to be efficient in several clinical reports (Ard et al., 2003; Keifer et al., 2005; Everett et al., 2006). An important factor in the choice of anesthetic drugs is its effect on the intraoperative ECoG. Therapeutic levels of propofol can potentially depress the ECoG. Propofol, when discontinued 20 minutes before monitoring, did not interfere with the ECoG, in awake cooperative children older than 10 years (Soriano et al., 2000a). However, it is imperative that candidates for an awake craniotomy be mature and psychologically prepared to participate in this procedure. Therefore, patients who are developmentally delayed or have a history of severe anxiety or psychiatric disorders should not be considered appropriate for an awake craniotomy.
Postoperative seizures are an uncommon but devastating complication. Prophylaxis in the perioperative period and aggressive treatment of new convulsions are well-recognized mainstays of care. Phenytoin is the agent used most commonly for prophylaxis, but maintaining therapeutic serum levels can be a challenge (Wolf et al., 2006). Levetiracetam (Keppra) is becoming increasingly common and in many instances supplants phenytoin as the choice for prophylaxis. Both drugs can be administered intravenously, but unlike phenytoin, administration of levetiracetam does not require following serum drug levels to monitor for toxicity. Alternative agents frequently used in pediatrics include phenobarbital, carbamazepine, and valproic acid. Status epilepticus can be treated effectively with lorezepam (0.1 mg/kg IV bolus over 2 minutes) or diazepam (0.5 mg/kg rectally). Lorezepam may be repeated after 10 minutes and accompanied by fosphenytoin (20 mg/kg IV or IM) if initial doses are ineffective. Phenobarbital (20 mg/kg) is also an effective first-line antiepileptic drug.
Vascular Malformations
Vascular anomalies are rare in infants and children. Most are congenital lesions that appear early in life. Large arteriovenous malformations (AVMs) in neonates may be associated with high-output congestive heart failure and require vasoactive support. Initial treatment of large AVMs often consists of several treatments involving intravascular embolization in the interventional radiology suite (Burrows and Robertson, 1998). Operative management is commonly associated with massive blood loss. Ligation of an AVM can lead to sudden hypertension with hyperemic cerebral edema (Morgan et al., 1999) and should be treated with vasodilators such as labetalol or nitroprusside.
Moyamoya syndrome is a rare chronic vasoocclusive disorder of the internal carotid arteries that appears as transient ischemic attacks or recurrent strokes (or both) in childhood. The etiology is unknown, but the syndrome can be associated with prior intracranial radiation, neurofibromatosis, Down syndrome, and a variety of hematologic disorders including sickle cell disease. Medical management consists of antiplatelet medications (e.g., aspirin or calcium channel blockers) and surgical management aims to improve blood flow to the ischemic area. The operation involves suturing a scalp artery onto the pial surface of the brain (pial synangiosis). The anesthetic management of these patients is directed at optimizing cerebral perfusion (Soriano et al., 1993); this includes ensuring generous preoperative hydration and maintaining the blood pressure close to the patient’s preoperative levels. Maintenance of normocapnia is essential as well, because both hypercapnia and hypocapnia can lead to steal from the ischemic region and further aggravate cerebral ischemia (Kuwabara et al., 1997). A nitrous oxide and narcotic-based anesthetic provides a stable level of anesthesia for these patients and is compatible with intraoperative EEG monitoring. Once the patient emerges from anesthesia, the same maneuvers that optimize cerebral perfusion should be extended into the postoperative period.
Neurotrauma
Neurotrauma primarily includes TBI and spinal cord injury (SCI). TBI is the leading cause of death and disability in children over 1 year of age. Approximately half of children with cervical spine injury have concomitant TBI, and the presence of TBI increases the risk of spine injury. After TBI, mortality is lower for children than for adults (10.4% versus 2.5%), but certain factors predict worse outcomes (Box 22-2). Because secondary injuries can progressively worsen outcome, basic life support algorithms, such as Pediatric Advanced Life Support (PALS) and Advanced Trauma Life Support (ATLS), and the principles of ATLS, including primary and secondary surveys, should be immediately applied. The spine cannot be “cleared” by radiographic examination alone; a child with normal cervical spine radiographs should be maintained with cervical spine immobilization until a thorough examination can be performed. In children, cervical spine fractures can occur without neurologic deficit, and neurologic deficit can occur without fracture. Neurologic deficit without fracture has been termed SCIWORA (SCI without radiologic abnormalities). Most children undergo an MRI evaluation. Blunt abdominal trauma and long-bone fractures may occur with TBI/SCI and can be major sources of blood loss. Craniotomies for the evacuation of either epidural or subdural hematomas are at high risk for massive blood loss and VAE. Infants with inflicted trauma often present with a myriad of chronic and acute subdural hematomas (Duhaime et al., 1998). Most inflicted injuries involving death involve TBI. Children with inflicted TBI (iTBI) commonly present with altered consciousness, coma, seizures, vomiting, or irritability, and either injuries out of proportion to history or developmental milestones or incomplete histories. Types of injuries include subdural hematoma, subarachnoid hemorrhage, skull fractures, and diffuse axonal injury with or without cerebral edema. Outcome is poor after iTBI.
Cerebral Hemodynamics after TBI
Cerebral Metabolic Rate, Blood Flow, Autoregulation, and CO2 Reactivity
During the first 6 to 12 hours after TBI, the brain may suffer poor perfusion and cerebral ischemia. This may be followed by hyperemia and ICP. Finally, vasospasm occurs in up to 19% of patients with TBI, and poor perfusion may occur (White et al., 2001). Compared with children without TBI, children with TBI have a lower VMCA, and cerebral hypoperfusion (CBF < 25 mL/100 g per minute) is associated with cerebral ischemia and poor outcome (Adelson et al., 1997; Skippen et al., 1997). However, after TBI, CBF and CMRo2 may not be matched, resulting in either cerebral ischemia or hyperemia. As noted before, there are age-related changes in VMCA, CBF, and CMRglu (see Fig. 22-3) (Kennedy and Sokoloff, 1957; Ogawa et al., 1987; Bode and Wais, 1988; Vink et al., 1988). One study of 30 children showed CO2 vasoreactivity changes of less than 2% to be associated with poor outcome (Adelson et al., 1997). Cerebral autoregulation is impaired more often after severe (42%) than after mild (17%) pediatric TBI (Stoyka and Schutz, 1975; Bouma et al., 1998; Vavilala et al., 2004, 2007), and autoregulation may be more impaired in children with iTBI (Vavilala et al., 2007). Hemispheric differences in cerebral autoregulation are common (40%) after focal TBI (Vavilala et al., 2008). If cerebral autoregulation is impaired, lower blood pressure may passively result in diminished CPP and CBF (Fig. 22-22). Autoregulation is not a static condition and may deteriorate in patients with initially intact autoregulatory capacity. Empirically increasing MAP to prevent cerebral ischemia in the presence of unilaterally impaired cerebral autoregulation in the presence of hyperemia could result in cerebral hemorrhage (Bruce et al., 1981; Aldrich et al., 1992; Mandera et al., 2002). Impaired cerebral autoregulation has been associated with poor outcome after pediatric TBI (Vavilala et al., 2006, 2007).
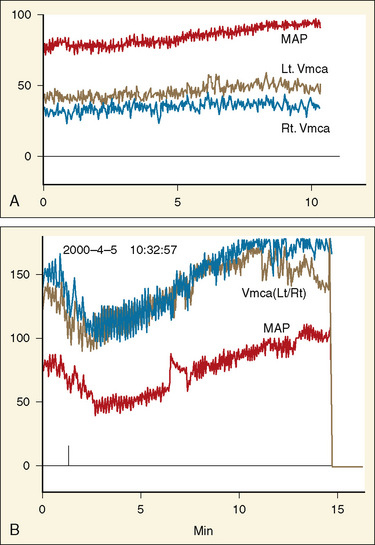
Cerebral Perfusion Pressure
In 2003, pediatric guidelines issued by a number of organizations recommended that a CPP of less than 40 mm Hg be avoided after severe TBI to prevent cerebral hypoperfusion leading to cerebral ischemia (Adelson et al., 2003). However, although it is not well understood, there is probably an age-dependent CPP threshold, with older children with TBI requiring higher CPP (Chambers et al., 2005, 2006). Furthermore, there may be variability (low and high) in CBFV, despite a CPP of greater than 40 mm Hg (Philip et al., 2009). These data suggest that empirical CPP management may not have predictable effects on CBF. However, today, either systolic blood pressure or CPP is used clinically to estimate cerebral perfusion. The presence of Cushing’s reflex and autonomic dysfunction might be the only indicators of increased ICP. Although hypotension is defined by a systolic blood pressure below the fifth percentile, in the absence of ICP monitoring and suspected increased ICP, supranormal systolic blood pressure may be needed to maintain CPP. At a minimum, MAP should not be allowed to decrease below values normal for age by using vasopressors. IV phenylephrine infusion is often used to maintain CPP above 50 mm Hg.
Intracranial Pressure and Intracranial Pressure Monitoring
The management of increased ICP in children is similar to that in adults (Fig. 22-23). The indications for ICP monitoring and the treatment threshold for increased ICP are given in the 2003 pediatric guidelines (Adelson et al., 2003). The use of ICP monitoring for infants and children with severe TBI and a Glasgow Coma Scale of 8 or less varies, but it is supported by several clinical studies (Adelson et al., 2003). Symptoms of increased ICP (>20 mm Hg) are nonspecific in children, and intermittent apnea may be its first sign in infancy (Sharples et al., 1995). ICP measurements from ventricular catheters and fiberoptic intraparenchymal transducers have a good correlation (Gambardella et al., 1993). Ventricular catheters also provide a conduit for withdrawing CSF.
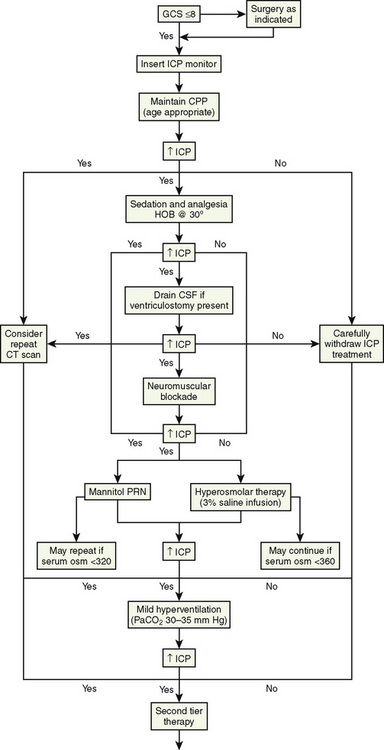
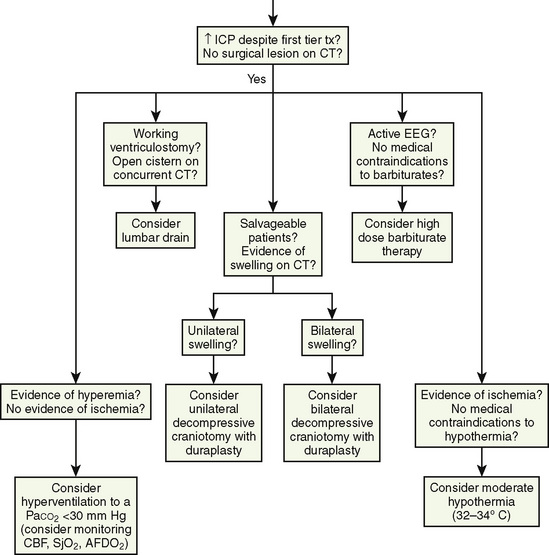
FIGURE 22-23 Algorithm for intracranial pressure management.
(Adapted from Adelson PD, Bratton SL, Carney NA, et al: Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents: critical pathway for the treatment of established intracranial hypertension in pediatric traumatic brain injury, Pediatr Crit Care Med 4[3 Suppl]:S65, 2003.)
Intracranial hypertension can be initially managed through elevation of the head, neuromuscular blockade, and hyperosmolar therapy. Mannitol can be given at a dosage of 0.25 to 1.0 g/kg intravenously. All diuretics interfere with the use of urine output as a guide to intravascular volume status. Hypertonic saline (3% NaCl) decreases ICP and increases CPP (Khanna et al., 2000). High-dosage barbiturate therapy can be titrated to produce a burst suppression pattern on the EEG, which results in a reduction in CMR. Refractory ICP can be treated with thiopental infusions, but volume loading and inotropic support may be needed to counter myocardial depression and hypotension (Adelson et al., 2003). If these maneuvers fail to control elevated ICP, decompressive craniectomy should be considered. Careful monitoring of blood gasses, minute ventilation, and end-tidal CO2 tensions are recommended. Current guidelines recommend maintaining normocapnia (Paco2, 35 to 40 mm Hg) except in the presence of impending herniation.
Anesthetic Management
The anesthetic approach to the traumatized child is based on the principles outlined in the 2003 pediatric guidelines for managing children with severe TBI (Table 22-9; see Fig. 22-23) and on ATLS guidelines and as previously described. Children with a Glasgow Coma Scale score of less than 9 require tracheal intubation for airway protection, and management of increased ICP. The most common approach to tracheal intubation is direct laryngoscopy and oral intubation with cricoid pressure after induction of anesthesia, ventilation with 100% oxygen, and neck in-line stabilization, without traction. Nasotracheal intubations are contraindicated in patients with basilar skull fractures. If increases in ICP occur despite hyperosmolar therapy during surgery, changing from less than 1 MAC volatile anesthesia to TIVA may be considered. Invasive arterial blood pressure monitoring should be used to guide blood pressure management and for hourly sampling of blood gases, glucose, and coagulation. Central access should be attempted by experienced personnel; intraosseous access is a second-line option if immediate vascular access is required (see Chapter 30, Anesthesia for the Pediatric Trauma Patient). Muscle relaxants should be used in unstable patients, and opioids should be used sparingly in patients with TBI if tracheal extubation is planned for the end of the case. For SCI, when neuromonitoring (including motor evoked potentials) is used, TIVA should be used to maintain anesthesia. Normothermia is currently maintained unless refractory ICP is present.
TABLE 22-9 Selected 2003 Pediatric Guidelines for the Management of Severe Brain Injury
| Physiologic Parameter | Recommendations |
| Blood glucose | Avoid dextrose-containing solutions Keep blood glucose level at < 200 to 250 mg/dL |
| Temperature | Avoid hyperthermia: cool patients to 36° to 37° C Hypothermia (32° to 34° C) may be considered for refractory ICP |
| Cerebral blood flow (CBF) and Paco2 | Avoid mild or prophylactic hyperventilation (Paco2 < 35 mm Hg) Mild hyperventilation if acute brainstem herniation exists Mild hyperventilation may be considered for refractory ICP |
| Systolic blood pressure (SBP) | Hypovolemia should be corrected ASAP SBP should be maintained at the 5th percentile, at least, for age May be beneficial to maintain SBP in normal range (≥50th percentile) |
| Cerebral perfusion pressure (CPP) | Keep CPP >40 mm Hg CPP 40 to 65 mm Hg may represent an age-related continuum for best treatment |
| Intracranial pressure (ICP) | Monitor if Glasgow Coma Scale score < 9 Treat ICP ≥20 mm Hg Ventriculostomy or intraparenchymal catheter |
| Hypertonic solutions | 3% saline, 0.1 to 1.0 mL/kg/hr Mannitol, 0.25 to 1.0 g/kg |
Adapted from Adelson PD, Bratton SL, Carney NA, et al; American Association for Surgery of Trauma; Child Neurology Society; International Society for Pediatric Neurosurgery; International Trauma Anesthesia and Critical Care Society; Society of Critical Care Medicine; World Federation of Pediatric Intensive and Critical Care Societies: Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents. Chapter 8: Cerebral perfusion pressure, Pediatr Crit Care Med 4(3 Suppl):S31, 2003.
Indications for Surgery
The major goal of surgery for patients with TBI is to optimize the recovery of viable brain. Most operations deal with the removal of mass lesions to prevent herniation, intracranial hypertension, or alterations in CBF. In general, unless small and deemed likely venous, epidural hematomas should be evacuated in comatose patients. Subdural hematomas that are associated with herniation thicker than 10 mm, or that produce a midline shift of greater than 5 mm, should be removed. Indications on intraparenchymal mass lesions include progressive neurologic deterioration referable to the lesion, signs of mass effect on CT, or refractory intracranial hypertension. Penetrating injury can often be managed with local débridement and watertight closure if not extensive, and if there is minimal intracranial mass effect (see Intracranial Pressure, earlier). Patients with severe brain swelling as manifest by cisternal compression or midline shift on CT, or intracranial hypertension by monitor, are potential candidates for decompressive craniectomy (Taylor et al., 2001). The relatively increased frequency of diffuse swelling in the pediatric population makes children more frequently candidates for such treatment. Generous decompressive craniectomy with duraplasty should be considered when intracranial hypertension reaches or approaches medical refractoriness in salvageable patients when the ICP elevation and its effects are believed to be the major threat to recovery. Unilateral craniectomy is appropriate for lateralized swelling; bifrontal decompression is selected for diffuse disease. In general, surgical removal of mass lesions in comatose patients should be performed as early as safely feasible. As this often involves incompletely resuscitated patients, close collaboration between surgery and anesthesia is critical. Bidirectional communication should be maintained regarding issues such as the stage of the procedure, anticipated and ongoing blood loss, systemic stability, and unanticipated events, so that the procedure can be altered or even terminated if necessary.
Induced Hypothermia
Head cooling and mild hypothermia have been demonstrated to be protective in asphyxiated neonates (Wyatt et al., 2007). However, induced hypothermia in adult with TBI has mixed results (Clifton et al., 2001). Recently, Adelson and colleagues (2005) demonstrated in a phase II trial that induced hypothermia can be a safe therapeutic option for children with TBI. A National Institutes of Health–sponsored phase III trial is underway. An international multicenter trial of induced hypothermia in pediatric patients reported that hypothermia did not improve the neurologic outcome and may increase mortality (Hutchison et al., 2008). Interestingly, the International Hypothermia in Aneurysm Surgery Trial failed to demonstrate any advantage of hypothermia over normothermia intraoperatively during surgical clipping of intracranial aneurysms (Todd et al., 2005).
Neuroendoscopy
Technological advances in minimally invasive endoscopic surgery have entered the neurosurgical arena. The anesthetic considerations for these evolving techniques are the same as those for other neurosurgical procedures discussed in this chapter. (See the discussion of neuroendoscopic techniques under Craniosynostosis, earlier.) Endoscopic third ventriculostomy is another approach for the treatment of obstructive hydrocephalus in infants and children (Rekate, 2004). A flexible fiberoptic scope inserted through a trocar provides a working channel that allows the neurosurgeon to insert a ventricular catheter or make fenestrations. Despite the relative safety of this procedure, bradycardia and other arrhythmias have been reported in conjunction with lack of egress of irrigation fluids and manipulation of the floor of the third ventricle (El-Dawlatly et al., 2000, 2001).
Summary
For questions and answers on topics in this chapter, go to “Chapter Questions” at www.expertconsult.com.
Aaslid R., Huber P., Nornes H. Evaluation of cerebrovascular spasm with transcranial Doppler ultrasound. J Neurosurg. 1984;60(1):37-41.
Adams R.W., Cucchiara R.F., Gronert G.A., et al. Isoflurane and cerebrospinal fluid pressure in neurosurgical patients. Anesthesiology. 1981;54(2):97-99.
Adelson P.D., Black P.M., Madsen J.R., et al. Use of subdural grids and strip electrodes to identify a seizure focus in children. Pediatr Neurosurg. 1995;22(4):174-180.
Adelson P.D., Bratton S.L., Carney N.A., et al. American Association for Surgery of Trauma; Child Neurology Society; International Society for Pediatric Neurosurgery; International Trauma Anesthesia and Critical Care Society; Society of Critical Care Medicine; World Federation of Pediatric Intensive and Critical Care Societies. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents. Chapter 8: Cerebral perfusion pressure. Pediatr Crit Care Med. 2003;4(Suppl 3):S31.
Adelson P.D., Clyde B., Kochanek P.M., et al. Cerebrovascular response in infants and young children following severe traumatic brain injury: a preliminary report. Pediatr Neurosurg. 1997;26(4):200-207.
Adelson P.D., Ragheb J., Kanev P., et al. Phase II clinical trial of moderate hypothermia after severe traumatic brain injury in children. Neurosurgery. 2005;56:740-754.
Albanese J., Durbec O., Viviand X., et al. Sufentanil increases intracranial pressure in patients with head trauma. Anesthesiology. 1993;79(3):493-497.
Albrecht R.F., Miletich D.J., Madala L.R. Normalization of cerebral blood flow during prolonged halothane anesthesia. Anesthesiology. 1983;58(1):26-31.
Albrecht R.F., Miletich D.J., Rosenberg R., et al. Cerebral blood flow and metabolic changes from induction to onset of anesthesia with halothane or pentobarbital. Anesthesiology. 1977;47(3):252-256.
Aldrich E.F., Eisenberg H.M., Saydjari C., et al. Diffuse brain swelling in severely head-injured children: a report from the NIH Traumatic Coma Data Bank. J Neurosurg. 1992;76(3):450-454.
Alexander S.C., Wollman H., Cohen P.J., et al. Cerebrovascular response to Paco2 during halothane anesthesia in man. J Appl Physiol. 1964;19:561-565.
Algotsson L., Messeter K., Nordström C.H., et al. Cerebral blood flow and oxygen consumption during isoflurane and halothane anesthesia in man. Acta Anaesthesiol Scand. 1988;32(1):15-20.
Alloul K., Whalley D.G., Shutway F., et al. Pharmacokinetic origin of carbamazepine-induced resistance to vecuronium neuromuscular blockade in anesthetized patients. Anesthesiology. 1996;84(2):330-339.
Anand K.J., Hickey P.R. Pain and its effects in the human neonate and fetus. N Engl J Med. 1987;317:1321-1329.
Anand K.J.S., Soriano S.G. Anesthetic agents and the immature brain: are these toxic or therapeutic agents? Anesthesiology. 2004;101:527-530.
Archer D.P., Labrecque P., Tyler J.L., et al. Cerebral blood volume is increased in dogs during administration of nitrous oxide or isoflurane. Anesthesiology. 1987;67(5):642-648.
Ard J., Doyle W., Bekker A. Awake craniotomy with dexmedetomidine in pediatric patients. J Neurosurg Anesthesiol. 2003;15(3):263-266.
Arieff A.I., Ayus J.C., Fraser C.L. Hyponatraemia and death or permanent brain damage in healthy children. BMJ. 1992;304:1218-1222.
Armstead W.M. Endothelin-1 contributes to normocapnic hyperoxic pial artery vasoconstriction. Brain Res. 1999;842(1):252-255.
Armstead W.M. Role of opioids in hypoxic pial artery dilation is stimulus duration dependent. Am J Physiol. 1998;275(3 Pt 2):H861-H867.
Artru A.A. Effects of halothane and fentanyl anesthesia on resistance to reabsorption of CSF. J Neurosurg. 1984;60(2):252-256.
Artru A.A. Effects of halothane and fentanyl on the rate of CSF production in dogs. Anesth Analg. 1983;62(6):581-585.
Artru A.A. Intracranial volume/pressure relationship during desflurane anesthesia in dogs: comparison with isoflurane and thiopental/halothane. Anesth Analg. 1994;79(4):751-760.
Artru A.A. Isoflurane does not increase the rate of CSF production in the dog. Anesthesiology. 1984;60(3):193-197.
Artru A.A. Nitrous oxide plays a direct role in the development of tension pneumocephalus intraoperatively. Anesthesiology. 1982;57(1):59-61.
Artru A.A. Relationship between cerebral blood volume and CSF pressure during anesthesia with halothane or enflurane in dogs. Anesthesiology. 1983;58(6):533-539.
Artru A.A. Relationship between cerebral blood volume and CSF pressure during anesthesia with isoflurane or fentanyl in dogs. Anesthesiology. 1984;60(6):575-579.
Baenziger O., Moenkhoff M., Morales C.G., et al. Impaired chemical coupling of cerebral blood flow is compatible with intact neurological outcome in neonates with perinatal risk factors. Biol Neonate. 1999;75(1):9-17.
Baker K.Z., Ostapkovich N., Sisti M.B., et al. Intact cerebral blood flow reactivity during remifentanil/nitrous oxide anesthesia. J Neurosurg Anesthesiol. 1997;9(2):134-140.
Bar-Joseph G., Guilburd Y., Tamir A., et al. Effectiveness of ketamine in decreasing intracranial pressure in children with intracranial hypertension. J Neurosurg Pediatr. 2009;4(1):40-46.
Bartels M., Althoff R.R., Boomsma D.I. Anesthesia and cognitive performance in children: no evidence for a causal relationship. Twin Res Hum Genet. 2009;12(3):246-253.
Bates D., Weinshilboum R.M., Campbell R.J., et al. The effect of lesions in the locus coeruleus on the physiological responses of the cerebral blood vessels in cats. Brain Res. 1977;136(3):431-443.
Berger T.M., Kistler W., Berendes E., et al. Hyponatremia in a pediatric stroke patient: syndrome of inappropriate antidiuretic hormone secretion or cerebral salt wasting? Crit Care Med. 2002;30(4):792-795.
Biagi L., Abbruzzese A., Bianchi M.C., et al. Age dependence of cerebral perfusion assessed by magnetic resonance continuous arterial spin labeling. J Magn Reson Imaging. 2007;25(4):696-702.
Bode H., Wais U. Age dependence of flow velocities in basal cerebral arteries. Arch Dis Child. 1988;63(6):606-611.
Bouma G.J., Muizelaar J.P., Fatouros P. Pathogenesis of traumatic brain swelling: role of cerebral blood volume. Acta Neurochir Suppl. 1998;71:272-275.
Boylan G.B., Young K., Panerai R.B., et al. Dynamic cerebral autoregulation in sick newborn infants. Pediatr Res. 2000;48:12-17.
Branco R.G., Tasker R.C. Glycemic level in mechanically ventilated children with bronchiolitis. Pediatr Crit Care Med. 2007;8:546-550.
Brian J.E.Jr, Faraci F.M., Heistad D.D. Recent insights into the regulation of cerebral circulation. Clin Exp Pharmacol Physiol. 1996;23(6–7):449-457.
Brown J.C., Charlton J.E., White D.J. A regional technique for the study of sensitivity to curare in human muscle. J Neurol Neurosurg Psychiatry. 1975;38(1):18-26.
Bruce D.A., Alavi A., Bilaniuk L., et al. Diffuse cerebral swelling following head injuries in children: the syndrome of “malignant brain edema,”. J Neurosurg. 1981;54(2):170-178.
Brunson C.D., Mayhew J.F. Laryngeal mask airway for awake craniotomy in pediatric patients. J Clin Anesth. 2005;17(2):149-150.
Brüssel T., Fitch W., Brodner G., et al. Effects of halothane in low concentrations on cerebral blood flow, cerebral metabolism, and cerebrovascular autoregulation in the baboon. Anesth Analg. 1991;73(6):758-764.
Burrows P.E., Robertson R.L. Neonatal central nervous system vascular disorders. Neurosurg Clin N Am. 1998;9:155-180.
Busija D.W., Orr J.A., Rankin J.H., et al. Cerebral blood flow during normocapnic hyperoxia in the unanesthetized pony. J Appl Physiol. 1980;48(1):10-15.
Byeon J.H., Yoo G. Cerebral salt wasting syndrome after calvarial remodeling in craniosynostosis. J Korean Med Sci. 2005;20:866-869.
Cafiero T., Di Minno R.M., Sivolella G., et al. Immediate postoperative pain management in patients undergoing major abdominal surgery after remifentanil-based anesthesia: sufentanil vs tramadol. Minerva Anestesiol. 2004;70(9):661-669.
Carlson A.P., Schermer C.R., Lu S.W. Retrospective evaluation of anemia and transfusion in traumatic brain injury. J Trauma. 2006;61:567-571.
Celik U.S., Alabaz D., Yildizdas D., et al. Cerebral salt wasting in tuberculous meningitis: treatment with fludrocortisone. Ann Trop Paediatr. 2005;25:297-302.
Chan K.H., Miller J.D., Dearden N.M., et al. The effect of changes in cerebral perfusion pressure upon middle cerebral artery blood flow velocity and jugular bulb venous oxygen saturation after severe brain injury. J Neurosurg. 1992;77:55-61.
Chambers I.R., Jones P.A., Lo T.Y. Critical thresholds of intracranial pressure and cerebral perfusion pressure related to age in paediatric head injury. J Neurol Neurosurg Psychiatry. 2006;77:234-240.
Chugani H.T., Phelps M.E., Mazziotta J.C. Positron emission tomography study of human brain functional development. Ann Neurol. 1987;22(4):487-497.
Clifton G.L., Miller E.R., Choi S.C., et al. Lack of effect of induction of hypothermia after acute brain injury. N Engl J Med. 2001;344:556-563.
Cochrane D.D., Adderley R., White C.P., et al. Apnea in patients with myelomeningocele. Pediatr Neurosurg. 1990;16:232-239.
Cochrane D.D., Gustavsson B., Poskitt K.P., et al. The surgical and natural morbidity of aggressive resection for posterior fossa tumors in childhood. Pediatr Neurosurg. 1994;20:19-29.
Coles J.P., Minhas P.S., Fryer T.D., et al. Effect of hyperventilation on cerebral blood flow in traumatic head injury: clinical relevance and monitoring correlates. Crit Care Med. 2002;30:1950-1959.
Cooper D.J., Myles P.S., McDermott F.T., et al. Prehospital hypertonic saline resuscitation of patients with hypotension and severe traumatic brain injury: a randomized controlled trial. JAMA. 2004;291(11):1350-1357.
Cooperman L.H. Succinylcholine-induced hyperkalemia in neuromuscular disease. JAMA. 1970;213(11):1867-1871.
Cooperman L.H., Strobel G.E.Jr, Kennell E.M. Massive hyperkalemia after administration of succinylcholine. Anesthesiology. 1970;32(2):161-164.
Cotev S., Shalit M.N. Effects on diazepam on cerebral blood flow and oxygen uptake after head injury. Anesthesiology. 1975;43(1):117-122.
Cottrell J.E., Gupta B., Rappaport H., et al. Intracranial pressure during nitroglycerin-induced hypotension. J Neurosurg. 1980;53(3):309-311.
Cottrell J.E., Hartung J., Giffin J.P., et al. Intracranial and hemodynamic changes after succinylcholine administration in cats. Anesth Analg. 1983;62(11):1006-1009.
Cowgill D.B., Mostello L.A., Shapiro H.M. Encephalitis and a hyperkalemic response to succinycholine. Anesthesiology. 1974;40(4):409-411.
Cucchiara R.F., Bowers B. Air embolism in children undergoing suboccipital craniotomy. Anesthesiology. 1982;57:338-339.
Cucchiara R.F., Theye R.A., Michenfelder J.D. The effects of isoflurane on canine cerebral metabolism and blood flow. Anesthesiology. 1974;40(6):571-574.
Dawson B., Michenfelder J.D., Theye R.A. Effects of ketamine on canine cerebral blood flow and metabolism: modification by prior administration of thiopental. Anesth Analg. 1971;50(3):443-447.
Dierdorf S.F., McNiece W.L., Rao C.C., et al. Failure of succinylcholine to alter plasma potassium in children with myelomeningocoele. Anesthesiology. 1986;64(2):272-273.
DiGeronimo R.J., Gegg C.A., Zuckerman S.L. Adenosine depletion alters postictal hypoxic cerebral vasodilation in the newborn pig. Am J Physiol. 1998;274(5 Pt 2):H1495-H1501.
DiGiovanni A.J., Goodrick J., Neigh J.L., et al. The effect of halothane anesthesia on intracranial pressure in the presence of intracranial hypertension. Anesth Analg. 1974;53(6):823-827.
Donegan M.F., Bedford R.F. Intravenously administered lidocaine prevents intracranial hypertension during endotracheal suctioning. Anesthesiology. 1980;52(6):516-518.
Dribben W.H., Creeley C.E., Wang H.H., et al. High dose magnesium sulfate exposure induces apoptotic cell death in the developing neonatal mouse brain. Neonatology. 2009;96(1):23-32.
Drummond J.C., Scheller M.S., Todd M.M. The effect of nitrous oxide on cortical cerebral blood flow during anesthesia with halothane and isoflurane, with and without morphine, in the rabbit. Anesth Analg. 1987;66(11):1083-1089.
Drummond J.C., Todd M.M. The response of the feline cerebral circulation to PaCO2 during anesthesia with isoflurane and halothane and during sedation with nitrous oxide. Anesthesiology. 1985;62(3):268-273.
Duhaime A.C., Christian C.W., Rorke L.B., et al. Nonaccidental head injury in infants: the “shaken-baby syndrome”. N Engl J Med. 1998;338(25):1822-1829.
El-Dawlatly A.A., Murshid W., Alshimy A., et al. Arrhythmias during neuroendoscopic procedures. J Neurosurg Anesthesiol. 2001;13:57-58.
El-Dawlatly A.A., Murshid W.R., Elshimy A., et al. The incidence of bradycardia during endoscopic third ventriculostomy. Anesth Analg. 2000;91(5):1142-1144.
Eldredge A., Soriano S.G., Rockoff M.A. Neuroanesthesia. Neurosurg Clin N Am. 1995;6(3):505-520.
Eldredge E.A., Rockoff M.A., Medlock M.D., et al. Postoperative cerebral edema occurring in children with slit ventricles. Pediatrics. 1997;99(4):625-630.
Ellingsen I., Hauge A., Nicolaysen G., et al. Changes in human cerebral blood flow due to step changes in Pao2 and Paco2. Acta Physiol Scand. 1987;129(2):157-163.
Endres M., Laufs U., Liao J.K., et al. Targeting eNOS for stroke protection. Trends Neurosci. 2004;27(5):283-289.
Engelhard K., Reeker W., Kochs E., et al. Effect of Remifentanil on intracranial pressure on cerebral blood flow velocity in head trauma. Acta Anesthesiol Scand. 2004;48:396-399.
Everett L.L., van Rooyen I.F., Warner M.H., et al. Use of dexmedetomidine in awake araniotomy in adolescents. Paediatr Anaesth. 2006;16(3):338-342.
Faberowski L.W., Black S., Mickle J.P. Incidence of venous air embolism during craniectomy for craniosynostosis repair. Anesthesiology. 2000;92(1):20-23.
Fairgrieve R., Rowney D.A., Karsli C., et al. The effect of sevoflurane on cerebral blood flow velocity in children. Acta Anaesthesiol Scand. 2003;47(10):1226-1230.
Faraci F.M., Heistad D.D. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78(1):53-97.
Ferrari L.R., editor. Anesthesia and pain management for the pediatrician. Baltimore: Johns Hopkins University Press, 1999.
Forster A., Juge O., Morel D. Effects of midazolam on cerebral blood flow in human volunteers. Anesthesiology. 1982;56(6):453-455.
Forster A., Van Horn K., Marshall L.F., et al. Anesthetic effects on blood-brain barrier function during acute arterial hypertension. Anesthesiology. 1978;49(1):26-30.
Fox P.T., Raichle M.E. Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory stimulation in human subjects. Proc Natl Acad Sci U S A. 1986;83(4):1140-1144.
Fraga M., Rama-Maceiras P., Rodiño S., et al. The effects of isoflurane and desflurane on intracranial pressure, cerebral perfusion pressure, and cerebral arteriovenous oxygen content difference in normocapnic patients with supratentorial brain tumors. Anesthesiology. 2003;98(5):1085-1090.
Gambardella G., Zaccone C., Cardia E., et al. Intracranial pressure monitoring in children: comparison of external ventricular device with the fiberoptic system. Childs Nerv Syst. 1993;9:470-473.
Gandhi I.C., Jindal M.N., Patel V.K. Mechaism of neuromuscular blockade with some antiepileptic drugs. Arzneimittelforschung. 1976;26(2):258-261.
Gardner A.E., Dannemiller F.J., Dean D. Intracranial cerebrospinal fluid pressure in man during ketamine anesthesia. Anesth Analg. 1972;51(5):741-745.
Gauthier A., Girard F., Boudreault D., et al. Sevoflurane provides faster recovery and postoperative neurological assessment than isoflurane in long-duration neurosurgical cases. Anesth Analg. 2002;95(5):1384-1388.
Geiduschek J.M., Haberkern C.M., McLaughlin J.F., et al. Pain management for children following selecive dorsal rhizotomy. Can J Anaesth. 1997;41(6):492-496.
Gelb A.W., Craen R.A., Rao G.S., et al. Does hyperventilation improve operating condition during supratentorial craniotomy? A multicenter randomized crossover trial. Anesth Analg. 2008;106:585-594.
Gerlach K., Uhlig T., Hppe M., et al. Postoperative analgesia after preincisional administration of remifentanil. Minerva Anestesiol. 2003;69(6):563-569. 569–573
Ghirardello S., Hopper N., Albanese A., et al. Diabetes insipidus in craniopharyngioma: postoperative management of water and electrolyte disorders. J Pediatr Endocrinol Metab. 2006;19:415-421.
Gotoh J., Kuang T.Y., Nakao Y., et al. Regional differences in mechanisms of cerebral circulatory response to neuronal activation. Am J Physiol Heart Circ Physiol. 2001;280(2):H821-H829.
Goumnerova L.C., Frim D.M. Treatment of hydrocephalus with third ventriculocisternostomy: outcome and CSF flow patterns. Pediatr Neurosurg. 1997;27(3):149-152.
Grady M.S., Bedford R.F., Park T.S. Changes in superior sagittal sinus pressure in children with head elevation, jugular venous compression, and PEEP. J Neurosurg. 1986;65(2):199-202.
Graham D.H. Monitoring neuromuscular block may be unreliable in patients with upper-motor-neuron lesions. Anesthesiology. 1980;52(1):74-75.
Griffin J.P., Cottrell J.E., Hartung J., et al. Intracranial pressure during nifedipine-induced hypotension. Anesth Analg. 1983;62(12):1078-1080.
Grosslight K., Foster R., Colohan A.R., et al. Isoflurane for neuroanesthesia: risk factors for increases in intracranial pressure. Anesthesiology. 1985;63(5):533-536.
Guy J., Hindman B.J., Baker K.Z., et al. Comparison of Remifentanil and fentanyl in patients undergoing craniotomy for supratentorial space-occupying lesions. Anesthesiology. 1997;86:514-524.
Hansen T.D., Warner D.S., Todd M.M., et al. The role of cerebral metabolism in determining the local cerebral blood flow effects of volatile anesthetics: evidence for persistent flow-metabolism coupling. J Cereb Blood Flow Metab. 1989;9(3):323-328.
Harper A.M., Bell R.A. The effect of metabolic acidosis and alkalosis on the blood flow through the cerebral cortex. J Neurol Neurosurg Psychiatry. 1963;26:341-344.
Harper A.M., Glass H.I. Effect of alterations in the arterial carbon dioxide tension on the blood flow through the cerebral cortex at normal and low arterial blood pressures. J Neurol Neurosurg Psychiatry. 1965;28(5):449-452.
Harris M.M., Yemen T.A., Davidson A., et al. Venous embolism during craniectomy in supine infants. Anesthesiology. 1987;67(5):816-819.
Hatipoğlu M.A., Prabhu S., Weinberg J., et al. Surgical treatment of supplementary motor area lesions. Turk Neurosurg. 2009;19(3):306-307.
Henriksen H.T., Jörgensen P.B. The effect of nitrous oxide on intracranial pressure in patients with intracranial disorders. Br J Anaesth. 1973;45(5):486-492.
Hernández-Palazón J., Martínez-Lage J.F., de la Rosa-Carrillo V.N., et al. Anesthetic technique and development of pneumocephalus after posterior fossa surgery in the sitting position. Neurocirugia (Astur). 2003;14(3):216-221.
Hernández-Palazón J., Tortosa J.A., Martínez-Lage J.F., et al. Rocuronium-induced neuromuscular blockade is affected by chronic phenytoin therapy. J Neurosurg Anesthesiol. 2001;13(2):79-82.
Herrick I.A., Gelb A.W., Manninen P.H., et al. Effects of fentanyl, sufentanil, and alfentanil on brain retractor pressure. Anesth Analg. 1991;72(3):359-363.
Hoffman W.E., Cunningham F., James M.K., et al. Effects of remifentanil, a new short-acting opioid, on cerebral blood flow, brain electrical activity, and intracranial pressure in dogs anesthetized with isoflurane and nitrous oxide. Anesthesiology. 1993;;79(1):107-113. discussion 26A
Holliday M.A., Segar W.E. The maintenance need for water in parenteral fluid therapy. Pediatrics. 1957;19:823-832.
Holzman R.S. Clinical management of latex-allergic children. Anesth Analg. 1997;85(3):529-533.
Hougaard K., Hansen A., Brodersen P. The effect of ketamine on regional cerebral blood flow in man. Anesthesiology. 1974;41(6):562-567.
Hudetz A.G., Shen H., Kampine J.P. Nitric oxide from neuronal NOS plays critical role in cerebral capillary flow response to hypoxia. Am J Physiol. 1998;274(3 Pt 2):H982-H989.
Hutchison J.S., Ward R.E., Lacroix J., et al. Hypothermia therapy after traumatic brain injury in children. N Engl J Med. 2008;358:2447-2456.
Iacopino D.G., Conti A., Battaglia C., et al. Transcranial Doppler ultrasound study of the effects of nitrous oxide on cerebral autoregulation during neurosurgical anesthesia: a randomized controlled trial. Neurosurgery. 2003;99(1):58-64.
Ikonomidou C., Bosch F., Miksa M., et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70-74.
Inagaki T., Kyutoku S., Seno T., et al. The intracranial pressure of the patients with mild form of craniosynostosis. Childs Nerv Syst. 2007;23(12):1455-1459.
Ishikawa T., Funatsu N., Okamoto K., et al. Blood-brain barrier function following drug-induced hypotension in the dog. Anesthesiology. 1983;59(6):526-531.
Ishikawa T., McDowall D.G. Electrical activity of the cerebral cortex during induced hypotension with sodium nitroprusside and trimethaphan in the cat. Br J Anaesth. 1981;53:605-611.
Iwatsuki N., Kuroda N., Amaha K., et al. Succinylcholine-induced hyperkalemia in patients with ruptured cerebral aneurysms. Anesthesiology. 1980;53(1):64-67.
Jennett W.B., Barker J., Fitch W., et al. Effect of anaesthesia on intracranial pressure in patients with space-occupying lesions. Lancet. 1969;1(7585):61-64.
Jeremitsky E., Omert L.A., Dunham C.M., et al. The impact of hyperglycemia on patients with severe brain injury. J Trauma. 2005;58:47-50.
Jevtovic-Todorovic V., Hartman R.E., Izumi Y., et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876-882.
Jimenez D.F., Barone C.M., Cartwright C.C., et al. Early management of craniosynostosis using endoscopic-assisted strip craniectomies and cranial orthotic molding therapy. Pediatrics. 2002;110(1 Pt 1):97-104.
Jimenez D.F., Barone C.M. Endoscopic craniectomy for early surgical correction of sagittal craniosynostosis. J Neurosurg. 1998;88(1):77-81.
Jimenez R., Casado-Flores J., Nieto M., et al. Cerebral salt wasting syndrome in children with acute central nervous system injury. Pediatr Neurol. 2006;35:261-263.
Jobes D.R., Kennell E.M., Bush G.L., et al. Cerebral blood flow and metabolism during morphine–nitrous oxide anesthesia in man. Anesthesiology. 1977;47(1):16-18.
Johnson J.O., Jimenez D.F., Barone C.M. Blood loss after endoscopic strip craniectomy for craniosynostosis. J Neurosurg Anesthesiol. 2000;12:60.
Jones S.C., Radinsky C.R., Furlan A.J., et al. Cortical NOS inhibition raises the lower limit of cerebral blood flow-arterial pressure autoregulation. Am J Physiol. 1999;276(4 Pt 2):H1253-H1262.
Kain Z.N., Caldwell-Andrews A.A., Krivutza D.M., et al. Trends in the practice of parental presence during induction of anesthesia and the use of preoperative sedative premedication in the United States, 1995–2002: results of a follow-up national survey. Anesth Analg. 2004;98(5):1252-1259.
Kalkman C.J., Peelen L., Moons K.G., et al. Behavior and development in children and age at the time of first anesthetic exposure. Anesthesiology. 2009;110(4):805-812.
Karsli C., Luginbuehl I., Bissonnette B. The cerebrovascular response to hypocapnia in children receiving propofol. Anesth Analg. 2004;99(4):1049-1052.
Karsli C., Luginbuehl I., Farrar M., et al. Cerebrovascular carbon dioxide reactivity in children anaesthetized with propofol. Paediatr Anaesth. 2003;13(1):26-31.
Karsli C., Luginbuehl I., Farrar M., et al. Propofol decreases cerebral blood flow velocity in anesthetized children. Can J Anaesth. 2002;49(8):830-834.
Keifer J.C., Dentchev D., Little K., et al. A retrospective analysis of a remifentanil/propofol general anesthetic for craniotomy before awake functional brain mapping. Anesth Analg. 2005;101(2):502-508.
Kennedy C., Sokoloff L. An adaptation of the nitrous oxide method to the study of the cerebral circulation in children; normal values for cerebral blood flow and cerebral metabolic rate in childhood. J Clin Invest. 1957;36(7):1130-1137.
Kennedy C., Grave G.D., Sokoloff L. Alterations of local cerebral blood flow due to exposure of newborn puppies to 80–90 per cent oxygen. Eur Neurol. 1971–1972;6(1):137-140.
Khanna S., Davis D., Peterson B., et al. Use of hypertonic saline in the treatment of severe refractory posttraumatic intracranial hypertension in pediatric traumatic brain injury. Crit Care Med. 2000;28:1144-1151.
Klein G.W., Hojsak J.M., Rapaport R. Hyperglycemia in the pediatric intensive care unit. Curr Opin Clin Nutr Metab Care. 2007;10:187-192.
Klimscha W., Ullrich R., Nasel C., et al. High-dose remifentanil does not impair cerebrovascular carbon dioxide reactivity in healthy male volunteers. Anesthesiology. 2003;99(4):834-840.
Koehler R.C., Traystman R.J. Bicarbonate ion modulation of cerebral blood flow during hypoxia and hypercapnia. Am J Physiol. 1982;243(1):H33-H40.
Kontos H.A., Raper A.J., Patterson J.L. Analysis of vasoactivity of local pH, PCO2 and bicarbonate on pial vessels. Stroke. 1977;8(3):358-360.
Kuwabara Y., Ichiya Y., Sasaki M., et al. Response to hypercapnia in moyamoya disease: cerebrovascular response to hypercapnia in pediatric and adult patients with moyamoya disease. Stroke. 1997;28:701-707.
Lacroix J., Hebert P.C., Hutchison J.S., et al. TRIPICU Investigators; Canadian Critical Care Trials Group; Pediatric Acute Lung Injury and Sepsis Investigators Network: Transfusion strategies for patients in pediatric intensive care units. N Engl J Med. 2007;356:1609-1619.
Lagace A., Karsli C., Luginbuehl I., et al. The effect of remifentanil on cerebral blood flow velocity in children anesthetized with propofol. Paediatr Anaesth. 2004;14(10):861-865.
Lam A.M., Matta B.F., Mayberg T.S., et al. Change in cerebral blood flow velocity with onset of EEG silence during inhalation anesthesia in humans: evidence of flow-metabolism coupling? J Cereb Blood Flow Metab. 1995;15(4):714-717.
Lanier W.L., Milde J.H., Michenfelder J.D. Cerebral stimulation following succinylcholine in dogs. Anesthesiology. 1986;64(5):551-559.
Lanier W.L., Milde J.H., Michenfelder J.D. The cerebral effects of pancuronium and atracurium in halothane-anesthetized dogs. Anesthesiology. 1985;63(6):589-597.
Lassen N.A. Cerebral blood flow and oxygen consumption in man. Physiol Rev. 1959;39:183-238.
Laughlin T.P., Newberg L.A. Prolonged myoclonus after etomidate anesthesia. Anesth Analg. 1985;64(1):80-82.
Lee L.A., Roth S., Posner K.L., et al. The American Society of Anesthesiologists Postoperative Visual Loss Registry: analysis of 93 spine surgery cases with postoperative visual loss. Anesthesiology. 2006;105(4):652-659.
Lenzi P., Zoccoli G., Walker A.M., et al. Cerebral circulation in REM sleep: is oxygen a main regulating factor? Sleep Res Online. 2000;3(2):77-85.
Leon J.E., Bissonnette B. Cerebrovascular responses to carbon dioxide in children anaesthetized with halothane and isoflurane. Can J Anaesth. 1991;38(7):817-825.
Levine J.P., Stelnicki E., Weiner H.L., et al. Hyponatremia in the postoperative craniofacial pediatric patient population: a connection to cerebral salt wasting syndrome and management of the disorder. Plast Reconstr Surg. 2001;108:1501-1508.
List W.F., Crumrine R.S., Cascorbi H.F., et al. Increased cerebrospinal fluid pressure after ketamine. Anesthesiology. 1972;36(1):98-99.
Lockhart C.H., Jenkins J.J. Ketamine-induced apnea in patients with increased intracranial pressure. Anesthesiology. 1972;37(1):92-93.
Loepke A.W., Istaphanous G.K., McAuliffe J.J.3rd, et al. The effects of neonatal isoflurane exposure in mice on brain cell viability, adult behavior, learning, and memory. Anesth Analg. 2009;108(1):90-104.
Loepke A.W., Soriano S.G. An assessment of the effects of general anesthetics on developing brain structure and neurocognitive function. Anesth Analg. 2008;106(6):1681-1707.
Logigian E.L., Wolinsky J.S., Soriano S.G., et al. H reflex studies in cerebral palsy patients undergoing partial dorsal rhizotomy. Muscle Nerve. 1994;17:539-549.
Losasso T.J., Muzzi D.A., Dietz N.M., et al. Fifty percent nitrous oxide does not increase the risk of venous air embolism in neurosurgical patients operated upon in the sitting position. Anesthesiology. 1992;77(1):21-30.
Lutz L.J., Milde J.H., Milde L.N. The response of the canine cerebral circulation to hyperventilation during anesthesia with desflurane. Anesthesiology. 1991;74(3):504-507.
Lutz L.J., Milde J.H. The cerebral functional, metabolic, and hemodynamic effects of desflurane in dogs. Anesthesiology. 1990;73(1):125-131.
Madsen J.B., Cold G.E., Hansen E.S., et al. The effect of isoflurane on cerebral blood flow and metabolism in humans during craniotomy for small supratentorial cerebral tumors. Anesthesiology. 1987;66(3):332-336.
Madsen P.L., Vorstrup S. Cerebral blood flow and metabolism during sleep. Cerebrovasc Brain Metab Rev. 1991;3(4):281-296.
Maekawa T., McDowall D.G., Okuda Y. Oxygen tension on the brain surface during hypotension induced by haemorrhage, trimetaphan or sodium nitroprusside. Acta Neurol Scand Suppl. 1977;64:504-505.
Magness A., Yashon D., Locke G., et al. Cerebral function during trimethaphan-induced hypotension. Neurology. 1973;23(5):506-509.
Mandera M., Larysz D., Wojtacha M. Changes in cerebral hemodynamics assessed by transcranial Doppler ultrasonography in children after head injury. Childs Nerv Syst. 2002;18(3–4):124-128. Epub 2002 Mar 20
Mann J.D., Butler A.B., Johnson R.N., et al. Clearance of macromolecular and particulate substances from the cerebrospinal fluid system of the rat. J Neurosurg. 1979;50(3):343-348.
Manohar M., Parks C. Porcine regional brain and myocardial blood flows during halothane-O2 and halothane-nitrous oxide anesthesia: comparisons with equipotent isoflurane anesthesia. Am J Vet Res. 1984;45(3):465-473.
Marsh M.L., Aidinis S.J., Naughton K.V., et al. The technique of nitroprusside administration modifies the intracranial pressure response. Anesthesiology. 1979;51(6):538-541.
Matta B.F., Lam A.M., Mayberg T.S., et al. A critique of the intraoperative use of jugular venous bulb catheters during neurosurgical procedures. Anesth Analg. 1994;79:745-750.
Mayberg T.S., Lam A.M., Eng C.C., et al. The effect of alfentanil on cerebral blood flow velocity and intracranial pressure during isoflurane-nitrous oxide anesthesia in humans. Anesthesiology. 1993;78(2):288-294.
Mazze R.I., Escue H.M., Houston J.B. Hyperkalemia and cardiovascular collapse following administration of succinylcholine to the traumatized patient. Anesthesiology. 1969;31(6):540-547.
Mazzoni P., Giffin J.P., Cottrell J.E., et al. Intracranial pressure during diltiazem-induced hypotension in anesthetized dogs. Anesth Analg. 1985;64(10):1001-1004.
McDowall D.G. Induced hypotension and brain ischaemia. Br J Anaesth. 1985;57(1):110-119.
McManus M.L., Soriano S.G. Rebound swelling of astroglial cells exposed to hypertonic mannitol. Anesthesiology. 1998;88:1586-1591.
McPherson R.W., Briar J.E., Traystman R.J. Cerebrovascular responsiveness to carbon dioxide in dogs with 1.4% and 2.8% isoflurane. Anesthesiology. 1989;70(5):843-850.
McPherson R.W., Kirsch J.R., Ghaly R.F., et al. Effect of nitric oxide synthase inhibition on the cerebral vascular response to hypercapnia in primates. Stroke. 1995;26(4):682-687.
McPherson R.W., Traystman R.J. Effects of isoflurane on cerebral autoregulation in dogs. Anesthesiology. 1988;69(4):493-499.
Mertens M.J., Engbers F.H., Burm A.G., et al. Predictive performance of computer-controlled infusion of remifentanil during propofol/remifentanil anaesthesia. Br J Anaesth. 2003;90(2):132-141.
Messer J., Haddad J., Bientz J., et al. Influence of anesthetics on cerebral blood flow velocity in infancy: effects of halothane versus thiopental-fentanyl. Dev Pharmacol Ther. 1989;13(2–4):145-149.
Meyer J.S., Teraura T., Sakamoto K., et al. Central neurogenic control of cerebral blood flow. Neurology. 1971;21(3):247-262.
Michenfelder J.D., Theye R.A. Canine systemic and cerebral effects of hypotension induced by hemorrhage, trimethaphan, halothane, or nitroprusside. Anesthesiology. 1977;46(3):188-195.
Michenfelder J.D. The interdependency of cerebral functional and metabolic effects following massive doses of thiopental in the dog. Anesthesiology. 1974;41(3):231-236.
Milde L.N., Milde J.H., Gallagher W.J. Effects of sufentanil on cerebral circulation and metabolism in dogs. Anesth Analg. 1990;70(2):138-146.
Miletich D.J., Ivankovich A.D., Albrecht R.F., et al. Absence of autoregulation of cerebral blood flow during halothane and enflurane anesthesia. Anesth Analg. 1976;55(1):100-109.
Miller R., Tausk H.C., Stark D.C. Effect of Innovar, fentanyl and droperidol on the cerebrospinal fluid pressure in neurosurgical patients. Can Anaesth Soc J. 1975;22(4):502-508.
Minto C.F., Schnider T.W., Shafer S.L. Pharmacokinetics and pharmacodynamics of remifentanil. II. Model application. Anesthesiology. 1997;86(1):24-33.
Minton M.D., Grosslight K., Stirt J.A., et al. Increases in intracranial pressure from succinylcholine: prevention by prior nondepolarizing blockade. Anesthesiology. 1986;65(2):165-169.
Minton M.D., Stirt J.A., Bedford R.F., et al. Intracranial pressure after atracurium in neurosurgical patients. Anesth Analg. 1985;64(11):1113-1116.
Misfeldt B.B., Jörgensen P.B., Spotoft H., et al. The effects of droperidol and fentanyl on intracranial pressure and cerebral perfusion pressure in neurosurgical patients. Br J Anaesth. 1976;48(10):963-968.
Mönkhoff M., Schwarz U., Gerber A., et al. The effects of sevoflurane and halothane anesthesia on cerebral blood flow velocity in children. Anesth Analg. 2001;92(4):891-896.
Moorthy S.S., Hilgenberg J.C. Resistance to non-depolarizing muscle relaxants in paretic upper extremities of patients with residual hemiplegia. Anesth Analg. 1980;59(8):624-627.
Morgan M.K., Sekhon L.H., Finfer S., et al. Delayed neurological deterioration following resection of arteriovenous malformations of the brain. J Neurosurg. 1999;90:695-701.
Morii S., Ngai A.C., Ko K.R., et al. Role of adenosine in regulation of cerebral blood flow: effects of theophylline during normoxia and hypoxia. Am J Physiol. 1987;253(1 Pt 2):H165-H175.
Moss E., Dearden N.M., Berridge J.C. Effects of changes in mean arterial pressure on SjO2 during cerebral aneurysm surgery. Br J Anaesth. 1995;75:527-530.
Moss E., McDowall D.G. I.c.p. increases with 50% nitrous oxide in oxygen in severe head injuries during controlled ventilation. Br J Anesth. 1979;51(8):757-761.
Moss E., Powell D., Gibson R.M., et al. Effect of etomidate on intracranial pressure and cerebral perfusion pressure. Br J Anaesth. 1979;51(4):347-352.
Moss E., Powell D., Gibson R.M., et al. Effects of fentanyl on intracranial pressure and cerebral perfusion pressure during hypocapnia. Br J Anaesth. 1978;50(8):779-784.
Muizelaar J.P., van der Poel H.G., Li Z.C., et al. Pial arteriolar vessel diameter and CO2 reactivity during prolonged hyperventilation in the rabbit. J Neurosurg. 1988;69(6):923-927.
Munro M.J., Walker A.M., Barfield C.P. Hypotensive extremely low birth weight infants have reduced cerebral blood flow. Pediatrics. 2004;114:1591-1596.
Myburgh J., Cooper D.J., Finfer S., et al. Saline or albumin for fluid resuscitation in patients with traumatic brain injury. SAFE Study Investigators; Australian and New Zealand Intensive Care Society Clinical Trials Group; Australian Red Cross Blood Service; George Institute for International Health. N Engl J Med. 2007;357(9):874-884.
Nakahashi K., Yomosa H., Matsuzawa N., et al. Effect on cerebral blood flow of midazolam during modified neurolept-anesthesia. Masui. 1991;40(12):1787-1792.
Nehls D.G., Todd M.M., Spetzler R.F., et al. A comparison of the cerebral protective effects of isoflurane and barbiturates during temporary focal ischemia in primates. Anesthesiology. 1987;66(4):453-464.
Newberg L.A., Michenfelder J.D. Cerebral protection by isoflurane during hypoxemia or ischemia. Anesthesiology. 1983;59(1):29-35.
Newberg L.A., Milde J.H., Michenfelder J.D. Systemic and cerebral effects of isoflurane-induced hypotension in dogs. Anesthesiology. 1984;60(6):541-546.
Ngai A.C., Meno J.R., Ko K.R., et al. Role of adenosine in cerebral vasodilator responses to sciatic nerve stimulation. J Cereb Blood Flow Metab. 1998;18(5):580-581.
Noguchi K.K., Walls K.C., Wozniak D.F., et al. Acute neonatal glucocorticoid exposure produces selective and rapid cerebellar neural progenitor cell apoptotic death. Cell Death Differ. 2008;15(10):1582-1592.
Nugent M., Artru A.A., Michenfelder J.D. Cerebral metabolic, vascular and protective effects of midazolam maleate: comparison to diazepam. Anesthesiology. 1982;56(3):172-176.
Ogawa A., Nakamura N., Sugita K., et al. Regional cerebral blood flow in children: normal value and regional distribution of cerebral blood flow in childhood. No To Shinkei. 1987;39(2):113-118.
Ojemann S.G., Berger M.S., Lettich E., et al. Localization of language function in children: results of electrical stimulation mapping. J Neurosurg. 2003;98(3):465-470.
Olney J.W., Young C., Wozniak D.F., et al. Anesthesia induced developmental neuroapoptosis: does it happen in humans? Anesthesiology. 2004;101:530-533.
Ornstein E., Matteo R.S., Schwartz A.E., et al. The effect of phenytoin on the magnitude and duration of neuromuscular block following atracurium or vecuronium. Anesthesiology. 1987;67(2):191-196.
Ornstein E., Matteo R.S., Young W.L., et al. Resistance to metocurine-induced neuromuscular blockade in patients receiving phenytoin. Anesthesiology. 1985;63(3):294-298.
Ornstein E., Young W.L., Fleischer L.H., et al. Desflurane and isoflurane have similar effects on cerebral blood flow in patients with intracranial mass lesions. Anesthesiology. 1993;79(3):498-502.
Ostapkovich N.D., Baker K.Z., Fogarty-Mack P., et al. Cerebral blood flow and CO2 reactivity is similar during remifentanil/N2O and fentanyl/N2O anesthesia. Anesthesiology. 1998;89(2):358-363.
Padayachee T.S., Kirkham F.J., Lewis R.R., et al. Transcranial measurement of blood velocities in the basal cerebral arteries using pulsed Doppler ultrasound: a method of assessing the circle of Willis. Ultrasound Med Biol. 1986;12(1):5-14.
Papadimitriou D.T., Spiteri A., Pagnier A., et al. Mineralocorticoid deficiency in post-operative cerebral salt wasting. J Pediatr Endocrinol Metab. 2007;20:1145-1150.
Paulson O.B., Strandgaard S., Edvinsson L. Cerebral autoregulation. Cerebrovasc Brain Metab Rev. 1990;2:161-191.
Pelligrino D.A., Miletich D.J., Hoffman W.E., et al. Nitrous oxide markedly increases cerebral cortical metabolic rate and blood flow in the goat. Anesthesiology. 1984;60(5):405-412.
Penfield W. Combined regional and general anesthesia for craniotomy and cortical exploration. Part I. Neurosurgical considerations. Anesth Analg. 1954;33:145-155.
Petersen K.D., Landsfeldt U., Cold G.E., et al. Intracranial pressure and cerebral hemodynamic in patients with cerebral tumors: a randomized prospective study of patients subjected to craniotomy in propofol-fentanyl, isoflurane-fentanyl, or sevoflurane-fentanyl anesthesia. Anesthesiology. 2003;98(2):329-336.
Philip S., Chaiwat O., Udomphorn Y., et al. Variation in cerebral blood flow velocity with cerebral perfusion pressure >40 mm Hg in 42 children with severe traumatic brain injury. Crit Care Med. 2009;37(11):2973-2978.
Phirman J.R., Shapiro H.M. Modification of nitrous oxide-induced intracranial hypertension by prior induction of anesthesia. Anesthesiology. 1977;46(2):150-151.
Pierce E.C.Jr, Lambertsen C.J., Deutsch S., et al. Cerebral circulation and metabolism during thiopental anesthesia and hyper-ventilation in man. J Clin Invest. 1962;41:1664-1671.
Pollack I.F. Brain tumors in children. N Engl J Med. 1994;331(22):1500-1507.
Porzio P., Halberthal M., Bohn D., et al. Treatment of acute hyponatremia: ensuring the excretion of a predictable amount of electrolyte-free water. Crit Care Med. 2000;28:1905-1910.
Prielipp R.C., Wall M.H., Tobin J.R., et al. Dexmedetomidine-induced sedation in volunteers decreases regional and global cerebral blood flow. Anesth Analg. 2002;95:1052-1059.
Pryds A., Tønnesen J., Pryds O., et al. Cerebral pressure autoregulation and vasoreactivity in the newborn rat. Pediatr Res. 2005;57(2):294-298.
Pryds O., Andersen G.E., Friis-Hansen B. Cerebral blood flow reactivity in spontaneously breathing, preterm infants shortly after birth. Acta Paediatr Scand. 1990;79(4):391-396.
Quint S.R., Scremin O.U., Sonnenschein R.R., et al. Enhancement of cerebrovascular effect of CO2 by hypoxia. Stroke. 1980;11(3):286-289.
Rampil I.J., Lockhart S.H., Zwass M.S., et al. Clinical characteristics of desflurane in surgical patients: minimum alveolar concentration. Anesthesiology. 1991;74(3):429-433.
Reasoner D.K., Todd M.M., Scamman F.L., et al. The incidence of pneumocephalus after supratentorial craniotomy: observations on the disappearance of intracranial air. Anesthesiology. 1994;80:1008-1012.
Régrigny O., Delagrange P., Scalbert E., et al. Effects of melatonin on rat pial arteriolar diameter in vivo. Br J Pharmacol. 1999;127(7):1666-1670.
Rekate H.L. Selecting patients for endoscopic third ventriculostomy. Neurosurg Clin N Am. 2004;15:39-49.
Renou A.M., Vernhiet J., Macrez P., et al. Cerebral blood flow and metabolism during etomidate anaesthesia in man. Br J Anaesth. 1978;50(10):1047-1051.
Rockoff M.A., Naughton K.V., Shapiro H.M., et al. Cerebral circulatory and metabolic responses to intravenously administered lorazepam. Anesthesiology. 1980;53(3):215-218.
Rosa G., Sanfilippo M., Orfei P., et al. The effects of pipecuronium bromide on intracranial pressure and cerebral perfusion pressure. J Neurosurg Anesthesiol. 1991;3(4):253-257.
Rossberg M.I., Armstead W.M. Role of cyclic nucleotides in vasopressin-induced piglet pial artery dilation and opioid release. Pediatr Res. 1997;41:498-504.
Roth S., Ebrahim Z.Y. Resistance to pancuronium in patients receiving carbamazepine. Anesthesiology. 1987;66(5):691-693.
Rovere A.A., Raynald A.C., Scremin O.U. On the mechanism of the amphetamine induced vasodilation at the rat’s cerebral cortex. Experientia. 1977;33(11):1461-1463.
Rowney D.A., Fairgrieve R., Bissonnette B. Cerebrovascular carbon dioxide reactivity in children anaesthetized with sevoflurane. Br J Anaesth. 2002;88:357.
Sakabe T., Kuramoto T., Inoue S., et al. Cerebral effects of nitrous oxide in the dog. Anesthesiology. 1978;48(3):195-200.
Sakabe T., Kuramoto T., Kumagae S., et al. Cerebral responses to the addition of nitrous oxide to halothane in man. Br J Anaesth. 1976;48(10):957-962.
Sakabe T., Maekawa T., Ishikawa T., et al. The effects of lidocaine on canine cerebral metabolism and circulation related to the electroencephalogram. Anesthesiology. 1974;40(5):433-441.
Sarang A., Dinsmore J. Anaesthesia for awake craniotomy: evolution of a technique that facilitates awake neurological testing. Br J Anaesth. 2003;90(2):161-165.
Scheingraber S., Rehm M., Sehmisch C., et al. Rapid saline infusion produces hyperchloremic acidosis in patients undergoing gynecologic surgery. Anesthesiology. 1999;90:1265.
Scheller M.S., Tateishi A., Drummond J.C., et al. The effects of sevoflurane on cerebral blood flow, cerebral metabolic rate for oxygen, intracranial pressure, and the electroencephalogram are similar to those of isoflurane in the rabbit. Anesthesiology. 1988;68(4):548-551.
Scheller M.S., Todd M.M., Drummond J.C., et al. The intracranial pressure effects of isoflurane and halothane administered following cryogenic brain injury in rabbits. Anesthesiology. 1987;67(4):507-512.
Schmetterer L., Findl O., Strenn K., et al. Role of NO in the O2 and CO2 responsiveness of cerebral and ocular circulation in humans. Am J Physiol. 1997;273:R2005-R2012.
Schwedler M., Miletich D.J., Albrecht R.F. Cerebral blood flow and metabolism following ketamine administration. Can Anaesth Soc J. 1982;29(3):222-226.
Settergren G., Lindblad B.S., Persson B. Cerebral blood flow and exchange of oxygen, glucose ketone bodies, lactate, pyruvate and amino acids in anesthetized children. Acta Paediatr Scand. 1980;69:457-465.
Shapiro K., Marmarou A., Shulman K. Characterization of clinical CSF dynamics and neural axis compliance using the pressure-volume index: I. The normal pressure-volume index. Ann Neurol. 1980;7:508.
Sharma D., Jelacic J., Chennuri R., et al. Incidence and risk factors for perioperative hyperglycemia in children with traumatic brain injury. Anesth Analg. 2009;108:81.
Sharples P.M., Stuart A.G., Matthews D.S., et al. Cerebral blood flow and metabolism in children with severe head injury. Part 1: Relation to age, Glasgow coma score, outcome, intracranial pressure, and time after injury. J Neurol, Neurosurg Psychiatry. 1995;58:145.
Shayevitz J.R., Matteo R.S. Decreased sensitivity to metocurine in patients with upper motoneuron disease. Anesth Anal. 1985;64(8):767-772.
Singh S., Bohn D., Carlotti A.P., et al. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med. 2002;30:2575.
Skahen S., Shapiro H.M., Drummond J.C., et al. Nitrous oxide withdrawal reduces intracranial pressure in the presence of pneumocephalus. Anesthesiology. 1986;65(2):192-195.
Skippen P., Seear M., Poskitt K., et al. Effect of hyperventilation on regional cerebral blood flow in head-injured children. Crit Care Med. 1997;25(8):1402-1409.
Slikker W.Jr, Zou X., Hotchkiss C.E., et al. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicological Sciences. 2007;98:145-158.
Smith A.L., Hoff J.T., Nielsen S.L., et al. Barbiturate protection in acute focal cerebral ischemia. Stroke. 1974;5(1):1-7.
Smith A.L., Marque J.J. Anesthetics and cerebral edema. Anesthesiology. 1976;45(1):64-72.
Smith R.B., Grenvik A. Cardiac arrest following succinylcholine in patients with central nervous system injuries. Anesthesiology. 1970;33(5):558-560.
Sollevi A., Ericson K., Eriksson L., et al. Effect of adenosine on human cerebral blood flow as determined by positron emission tomography. J Cereb Blood Flow Metab. 1987;7:673-678.
Soriano S.G., Eldredge E.A., Wang F.K., et al. The effect of propofol on intraoperative electrocorticography and cortical stimulation during awake craniotomies in children. Paediatr Anaesth. 2000;10(1):29-34.
Soriano S.G., Kaus S.J., Sullivan L.J., et al. Onset and duration of action of rocuronium in children receiving chronic anticonvulsant therapy. Paediatr Anaesth a. 2000;10:133-136.
Soriano S.G., Logigian E.L., Scott R.M., et al. Nitrous oxide depresses the H-reflex in children with cerebral palsy. Anesth Analg. 1995;80:239.
Soriano S.G., Martyn J.A. Antiepileptic-induced resistance to neuromuscular blockers: mechanisms and clinical significance. Clin Pharmacokinet. 2004;43:71.
Soriano S.G., McManus M.L., Sullivan L.J., et al. Cerebral blood flow velocity after mannitol infusion in children. Can J Anaesth. 1996;43:461.
Soriano S.G., McManus M.L., Sullivan L.J., et al. Doppler sensor placement during neurosurgical procedures for children in the prone position. J Neurosurg Anesthesiol. 1994;6:153.
Soriano S.G., Sethna N.F., Scott R.M. Anesthetic management of children with moyamoya syndrome. Anesth Analg. 1993;77:1066.
Soriano S.G., Sullivan L.J., Venkatakrishnan K., et al. Pharmacokinetics and pharmacodynamics of vecuronium in children receiving phenytoin or carbamazepine for chronic anticonvulsant therapy. Br J Anaesth. 2001;86:223.
Spacek A., Neiger F.X., Spiss C.K., et al. Chronic carbamazepine therapy does not influence mivacurium-induced neuromuscular block. Br J Anaesth. 1996;77(4):500-502.
Spacek A., Nickl S., Neiger F.X., et al. Augmentation of the rocuronium-induced neuromuscular block by the acutely administered phenytoin. Anesthesiology. 1999;90(6):1551-1555.
Sperry R.J., Bailey P.L., Reichman M.V., et al. Fentanyl and sufentanil increase intracranial pressure in head trauma patients. Anesthesiology. 1992;77(3):416-420.
Stange K., Greitz D., Ingvar M., et al. Global cerebral blood flow during infusion of adenosine in humans: assessment by magnetic resonance imaging and positron emission tomography. Acta Physiol Scand. 1997;160:117-122.
Stephan H., Gröger P., Weyland A., et al. The effect of sufentanil on cerebral blood flow, cerebral metabolism and the CO2 reactivity of the cerebral vessels in man. Anaesthesist. 1991;40(3):153-160.
Stirt J.A., Grosslight K.R., Bedford R.F., et al. “Defasciculation” with metocurine prevents succinylcholine-induced increases in intracranial pressure. Anesthesiology. 1987;67(1):50-53.
Stirt J.A., Maggio W., Haworth C., et al. Vecuronium: effect on intracranial pressure and hemodynamics in neurosurgical patients. Anesthesiology. 1987;67(4):570-573.
Stoyka W.W., Schutz H.H. The cerebral response to sodium nitroprusside and trimethaphan controlled hypotension. J Can Anesth Soc. 1975;22:275.
Takeshita H., Okuda Y., Sari A. The effects of ketamine on cerebral circulation and metabolism in man. Anesthesiology. 1972;36:69-75.
Talke P., Tong C., Lee H.W., et al. Effect of dexmedetomidine on lumbar cerebrospinal fluid pressure in humans. Anesth Analg. 1997;85:358-364.
Tarkkanen L., Laitinen L., Johansson G. Effects of d-tubocurarine on intracranial pressure and thalamic electrical impedance. Anesthesiology. 1974;40(3):247-251.
Tateishi A., Maekawa T., Takeshita H., et al. Clinical doses of midazolam and diazepam do not alter ICP diazepam and intracranial pressure. Anesthesiology. 1981;54(4):335-337.
Taylor A., Butt W., Rosenfeld J., et al. A randomized trial of very early decompressive craniectomy in children with traumatic brain injury and sustained intracranial hypertension. Childs Nerv Syst. 2001;17:154.
Thenuwara K., Todd M.M., Brian J.E.Jr. Effect of mannitol and furosemide on plasma osmolality and brain water. Anesthesiology. 2002;96:416.
Theye R.A., Michenfelder J.D. The effect of nitrous oxide on canine cerebral metabolism. Anesthesiology. 1968;29(6):1119-1124.
Thiagarajah S., Sophie S., Lear E., et al. Effect of suxamethonium on the ICP of cats with and without thiopentone pretreatment. Br J Anaesth. 1988;60(2):157-160.
Thomas E.T. Circulatory collapse following succinylcholine: report of a case. Anesth Analg. 1969;48(3):333-337.
Timmons S.D. The life-saving properties of blood: mitigating cerebral insult after traumatic brain injury. Neurocrit Care. 2006;5:1-3.
Tobey R.E. Paraplegia, succinylcholine and cardiac arrest. Anesthesiology. 1970;32(4):359-364.
Tobias J.D., Johnson J.O., Jimenez D.F., et al. Venous air embolism during endoscopic strip craniectomy for repair of craniosynostosis in infants. Anesthesiology. 2001;95:340.
Todd M.M., Drummond J.C. A comparison of the cerebrovascular and metabolic effects of halothane and isoflurane in the cat. Anesthesiology. 1984;60(4):276-282.
Todd M.M., Hindman B.J., Clarke W.R., et al. Mild intraoperative hypothermia during surgery for intracranial aneurysm. N Engl J Med. 2005;352:135.
Todd M.M., Warner D.S., Sokoll M.D., et al. A prospective, comparative trial of three anesthetics for elective supratentorial craniotomy. Anesthesiology. 1993;78:1005.
Tomiyama Y., Jansen K., Brian J.E.Jr, et al. Hemodilution, cerebral O2 delivery, and cerebral blood flow: a study using hyperbaric oxygenation. Am J Physiol. 1999;276:H1190-H1196.
Tong T.K. Succinylcholine-induced hyperkalemia in near-drowning. Anesthesiology. 1987;66(5):720.
Tontisirin N., Muangman S.L., Suz P., et al. Early childhood gender differences in anterior and posterior cerebral blood flow velocity and autoregulation. Pediatrics. 2007;119:610.
Tsuji M., Saul J.P., du Plessis A., et al. Cerebral intravascular oxygenation correlates with mean arterial pressure in critically ill premature infants. Pediatrics. 2000;106:625.
Turner J.M., Powell D., Gibson R.M., et al. Intracranial pressure changes in neurosurgical patients during hypotension induced with sodium nitroprusside or trimetaphan. Br J Anaesth. 1977;49(5):419-425.
Van den Berghe G., Schoonheydt K., Becx P., et al. Insulin therapy protects the central and peripheral nervous system of intensive care patients. Neurology. 2005;64:1348.
Van den Berghe G., Wilmer A., Milants I., et al. Intensive insulin therapy in mixed medical/surgical intensive care units: benefit versus harm. Diabetes. 2006;55:3151.
Van den Berghe G., Wouters P., Weekers F., et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359.
Van Hemelrijck J., Fitch W., Mattheussen M., et al. Effect of propofol on cerebral circulation and autoregulation in the baboon. Anesth Analg. 1990;71(1):49-54.
Vavilala M.S., Kincaid M.S., Muangman S.L., et al. Gender differences in cerebral blood flow velocity and autoregulation between the anterior and posterior circulations in healthy children. Peds Res. 2005;58:574.
Vavilala M.S., Lee L.A., Boddu K., et al. Cerebral autoregulation in pediatric traumatic brain injury. Pediatr Crit Care Med. 2004;5:257.
Vavilala M.S., Lee L.A., Lam A.M. Cerebral blood flow and vascular physiology. Anesthesiol Clin N Amer. 2002;20:247-264.
Vavilala M.S., Lee L.A., Lam A.M. The lower limit of cerebral autoregulation in children during sevoflurane anesthesia. J Neurosurg Anesthesiol. 2003;15:307.
Vavilala M.S., Muangman S., Tontisirin N., et al. Impaired cerebral autoregulation and 6-month outcome in children with severe traumatic brain injury: preliminary findings. Dev Neurosci. 2006;28:348.
Vavilala M.S., Muangman S., Waitayawinyu P., et al. Neurointensive care: impaired cerebral autoregulation in infants and young children early after inflicted traumatic brain injury: a preliminary report. J Neurotrauma. 2007;24(1):87-96.
Vavilala M.S., Newell D.W., Junger E., et al. Dynamic cerebral autoregulation in healthy adolescents. Acta Anaesthesiol Scand. 2002;46:393.
Vavilala M.S., Tontisirin N., Udomphorn Y., et al. Hemispheric differences in cerebral autoregulation in children with moderate and severe traumatic brain injury. Neurocrit Care. 2008;9(1):45-54.
Verhaegen M.J., Todd M.M., Warner D.S. A comparison of cerebral ischemic flow thresholds during halothane/N2O and isoflurane/N2O anesthesia in rats. Anesthesiology. 1992;76(5):743-754.
Vink R., Faden A.I., McIntosh T.K. Changes in cellular bioenergetic state following graded traumatic brain injury in rats: determination by phosphorus 31 magnetic resonance spectroscopy. J Neurotrauma. 1988;5:315.
Wagerle L.C., Degiulio P.A. Indomethacin-sensitive CO2 reactivity of cerebral arterioles is restored by vasodilator prostaglandin. Am J Physiol. 1994;266:H1332-H1338.
Ward S.L., Nickerson B.G., van der H.A., et al. Absent hypoxic and hypercapneic arousal responses in children with myelomeningocele and apnea. Pediatrics. 1986;78(1):44-50.
Warner D.S., Turner D.M., Kassell N.F. Time-dependent effects of prolonged hypercapnia on cerebrovascular parameters in dogs: acid-base chemistry. Stroke. 1987;18:142-149.
Waters K.A., Forbes P., Morielli A., et al. Sleep-disordered breathing in children with myelomeningocele. J Pediatr. 1998;132(4):672-681.
Weinstabl C., Mayer N., Richling B., et al. Effect of sufentanil on intracranial pressure in neurosurgical patients. Anaesthesia. 1991;46(10):837-840.
Weinstabl C., Mayer N., Spiss C.K. Sufentanil decreases cerebral blood flow velocity in patients with elevated intracranial pressure. Eur J Anaesthesiol. 1992;9(6):481-484.
Westerlind A., Larsson L.E., Häggendal J., et al. Effects of metabolic pH-alterations on cerebral blood flow and oxygen uptake following E. coli endotoxin in dogs. Acta Anaesthesiol Scand. 1994;38(2):130-135.
White J.R.M., Farukhi Z., Bull C., et al. Predictors of outcome in severely head injured children. Crit Care Med. 2001;29:534.
Wiegand C., Richards P. Measurement of intracranial pressure in children: a critical review of current methods. Dev Med Child Neurol. 2007;49:935.
Wilder R.T., Flick R.P., Sprung J., et al. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;110(4):796-804.
Winn H.R., Morii S., Berne R.M. The role of adenosine in autoregulation of cerebral blood flow. Ann Biomed Eng. 1985;13:321-328.
Winn H.R., Welsh J.E., Rubio R., et al. Brain adenosine production in rat during sustained alteration in systemic blood pressure. Am J Physiol. 1980;239:H636-H641.
Wintermark M., Lepori D., Cotting J., et al. Brain perfusion in children: evolution with age assessed by quantitative perfusion computed tomography. Pediatrics. 2004;113:1642.
Wise-Faberowski L., Soriano S.G., Ferrari L., et al. Lesson of the week: hyponatremic seizures and excessive intake of hypotonic fluids in young children. J Neurosurg Anesthesiol. 2004;16:14.
Wolf G.K., McClain C.D., Zurakowski D., et al. Total phenytoin concentrations do not accurately predict free phenytoin concentrations in critically ill children. Pediatr Crit Care Med. 2006;7:434.
Wollman H., Alexander S.C., Cohen P.J., et al. Cerebral circulation of man during halothane anesthesia: effects of hypocarbia and of d-tubocuranine. Anesthesiology. 1964;25:180-184.
Wong G.T., Luginbuehl I., Karsli C., et al. The effect of sevoflurane on cerebral autoregulation in young children as assessed by the transient hyperemic response. Anesth Analg. 2006;102:1051-1055.
Wyatt J.S., Gluckman P.D., Liu P.Y., et al. Determinants of outcomes after head cooling for neonatal encephalopathy. Pediatrics. 2007;119:912.
Wyte S.R., Shapiro H.M., Turner P., et al. Ketamine-induced intracranial hypertension. Anesthesiology. 1972;36(2):174-176.
Yaster M., Koehler R.C., Traystman R.J. Effects of fentanyl on peripheral and cerebral hemodynamics in neonatal lambs. Anesthesiology. 1987;66(4):524-530.
Yates H., Hamill M., Borel C.O., et al. Incidence and perioperative management of tension pneumocephalus following craniofacial resection. J Neurosurg Anesthesiol. 1994;6(1):15-20.
Yoon B.W., Morillo C.A., Cechetto D.F., et al. Cerebral hemispheric lateralization in cardiac autonomic control. Arch Neurol. 1997;54(6):741-744.
Young W.L. Effects of desflurane on the central nervous system. Anesth Analg. 1992;75(Suppl 4):S32-S37.
Zornow M.H., Fleisher J.E., Scheller M.S., et al. Dexmedetomidine, an alpha-2 adrenergic agonist, decreases cerebral blood flow in the isoflurane-anesthetized dog. Anesth Analg. 1990;70:624-630.
Zornow M.H., Maze M., Dyck J.B., et al. Dexmedetomidine decreases cerebral blood flow velocity in humans. J Cereb Blood Flow Metab. 1993;13:350-353.