Paget’s Disease of Bone
Incidence and Epidemiology
Studies of the incidence of Paget’s disease are inherently imprecise because many affected individuals are asymptomatic and have normal serum total alkaline phosphatase levels.1 On the basis of autopsy studies2,3 and review of radiographs, the prevalence of the disease is believed to be 3% or greater in individuals older than 40 years in countries where the disease is common.4
A striking feature of the epidemiology of Paget’s disease is the great variability in prevalence estimates in different regions of the world and even within a single country. A survey of hospital radiographs in patients older than 55 years in 31 British towns revealed a prevalence of Paget’s disease ranging from 2.3% in Aberdeen, Scotland, to 8.3% in Lancaster, England.5 A similar survey done throughout Europe found that only in France did the prevalence equal the lowest prevalence rates in Britain.6 Australia, New Zealand, and the United States7 have a relatively high prevalence, perhaps because of British migration. The disease appears to be rare in Asia. In Japan, the prevalence has been estimated to be 2.8 per million of the population.8 In most studies, the prevalence of Paget’s disease in men slightly exceeds that in women.2–7 Several studies suggest that the prevalence rate of Paget’s disease is decreasing in Britain,9 New Zealand,10 and the United States.11 Whether a decrease in prevalence has occurred or whether an ascertainment bias has been introduced by the use of automated serum chemistry panels about 30 years ago is unclear.
Much emphasis has been placed on the increase in prevalence of Paget’s disease with aging. It has been estimated that the prevalence is nearly 10% by the ninth decade.2–4 Conversely, the disease has rarely been reported in individuals younger than 20 years. Although Paget’s disease is most often recognized after age 50 years, it is probably misleading to conclude that the disease is rare in younger individuals. As is discussed later, the obvious manifestations, such as skeletal deformity, probably evolve over decades. Failure to diagnose the earlier phases of the disease is no doubt due to lack of symptoms and minimal use of the radiologic and biochemical tests that would lead to an early diagnosis in younger individuals.
Since 1883, it has been appreciated that Paget’s disease may affect more than one member of a family.12 In large studies, a family history of the disease has been obtained in 12.3% of 788 cases13 (United States), 13.8% of 407 cases14 (Great Britain), and 22.8% of 658 cases15 (Australia). In the former two studies, a 7- to 10-fold increase in Paget’s disease was noted in relatives of patients in comparison with control groups. In a small study in Spain in which relatives of patients were screened with bone scans, 40% of patients had at least one first-degree relative with Paget’s disease.16 Examination of the overall pattern of apparent transmission suggests an autosomal dominant mode of inheritance.12
The search for potential environmental factors in the pathogenesis of Paget’s disease has led to consideration of whether past ownership of dogs might be a risk factor,17 but this has not been confirmed by subsequent studies. Occupational exposure to lead has been proposed as a possible factor in Paget’s disease.18 In one study, levels of cortical bone lead were higher than those in control subjects, but trabecular bone lead was lower.19 The relevance of these findings is unknown.
Pathophysiology
Consideration of the radiologic and pathologic evolution of the lesions of Paget’s disease strongly suggests that the primary disturbance is localized acceleration of osteoclastic bone resorption. At the interface of normal bone and an advancing lesion, numerous osteoclasts are found in Howship’s lacunae in cortical or trabecular bone.2 Many of the osteoclasts are larger than normal and may have up to 100 nuclei in cross-section rather than the several found in normal osteoclasts.20 Examination of the ultrastructure of osteoclasts in specimens of Paget’s disease reveals a striking and characteristic feature: the presence of microfilaments in the nucleus and, less frequently, in the cytoplasm.21,22 These structures have not been observed in osteoclasts from normal subjects or in the bone of patients with primary or secondary hyperparathyroidism, osteoporosis, or osteomalacia. They also have not been observed in osteoblasts, osteocytes, or bone marrow cells in the lesions of Paget’s disease. The inclusions have been found in a small percentage of the multinucleated giant cells (osteoclasts) in giant cell tumors of bone and in the osteoclasts of some patients with osteopetrosis and pyknodysostosis.23 The structure of the microfilaments most closely resembles the nucleocapsids of viruses of the Paramyxoviridae family, a group of RNA viruses known to cause common childhood infections. Evidence of Paramyxoviridae nucleocapsid proteins24,25 and messenger RNA (mRNA)26,27 has been found in pagetic lesions. The full-length sequence of the measles virus nucleocapsid gene has been sequenced from a pagetic lesion of one patient.28 The relevance of these findings to the cause of Paget’s disease is discussed later.
As osteoclastic resorption progresses in the cortex, individual osteons widen and become confluent with adjacent osteons. The resorptive area thereby may extend to the endosteal and periosteal surfaces. In trabecular bone of the medullary cavities, the osteolytic process results in a marked reduction in bone volume. Associated with both early cortical and trabecular lesions is a remarkable proliferation of fibrous tissue that replaces normal fatty or hematopoietic bone marrow. The fibrous stroma is highly vascular, and although arteriovenous shunts were previously thought to be present, this feature has not been confirmed with radiolabeled albumin microspheres.29
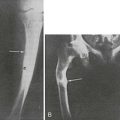
The earliest recognizable radiologic feature of Paget’s disease is a focal osteolytic lesion. The skull was first appreciated to be affected by circumscribed osteolytic lesions, and Schuller30 applied the term osteoporosis circumscripta to this finding. One or more osteolytic foci may be present, most often in the frontal and occipital regions, and may be observed to coalesce slowly over a period of years (Fig. 16-1). Pure osteolytic lesions also may be detected in other regions of the skeleton, but less frequently. They are seldom observed in the vertebral column or pelvis. In the long bones, osteolytic lesions usually develop at either end of the bone, less often in the diaphysis.31 Occasionally, osteolytic foci can be observed simultaneously at both ends of a bone. The junction of normal bone and the osteolytic lesion shows a characteristic appearance that is nearly diagnostic of Paget’s disease. The edge of the lesion usually assumes the shape of a flame or an inverted V (Fig. 16-2). Serial radiologic follow-up of untreated lesions has documented an average rate of extension of about 1 cm annually.31 Bone biopsies taken during this earliest phase of Paget’s disease not unexpectedly reveal a marked increase in osteoclastic activity and thinning of the trabeculae.32 Other striking features include a fibrovascular marrow, numerous osteoblasts lining the trabeculae, and prominent woven bone. Thus, a discrepancy is found in the results of radiologic and pathologic examination of an osteolytic lesion. Although focal density is decreased on radiographs, histology demonstrates very active bone formation, but not sufficient to overcome the remarkable degree of osteoclastic bone resorption.
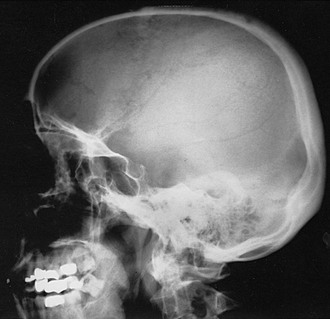
FIGURE 16-1 Radiograph demonstrating osteoporosis circumscripta of the skull affecting the frontal and temporal regions.
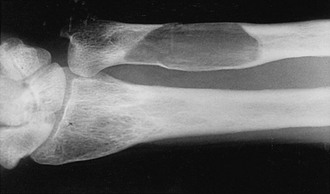
FIGURE 16-2 Radiograph of an osteolytic lesion of Paget’s disease that began in the diaphysis of the ulna and exhibits the characteristic flame-shaped or inverted-V extension toward the wrist. Note also the expansile nature of the lesion.
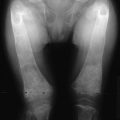
A more commonly observed stage of Paget’s disease is the mixed phase, in which osteoblastic (or osteosclerotic) features are intermixed with osteolytic features in an individual bone. This phase is best appreciated in long bones, in which one may observe the advancing osteolytic front adjacent to normal bone and, trailing this front, a heterogeneous region of osteosclerosis superimposed on the region that had previously been dominated by the osteolytic process (Fig. 16-3). Biopsies of the mixed phase reveal a characteristic abnormality of lamellar bone in both cortical and trabecular bone. The matrix is transformed into a bizarre “mosaic” pattern of irregularly juxtaposed pieces of lamellar bone separated by cement lines that have a scalloped outline. The irregularity probably reflects areas of previous osteoclastic resorption. The structure of the involved cortex is so disordered that complete osteons are rare, and the outer and inner circumferential lamellae and the interstitial lamella may be totally disrupted.
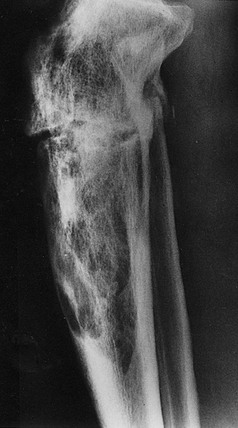
FIGURE 16-3 Radiograph of a tibia exhibiting a distal advancing osteolytic front with proximal sclerotic bone. A partially healed pathologic fracture is present proximally.
The same disordered matrix structure is seen in trabecular bone. Interspersed among the chaotic lamellae are patches of woven bone characterized by a random pattern of deposition of collagen fibers and a larger number of osteocytes per unit area of matrix. It has been suggested that the lacunae surrounding the osteocytes are larger than normal and that this finding represents osteocytic osteolysis.33 However, it is more likely that the increased size of the periosteocytic lacunae is simply a characteristic of woven bone and not a second type of bone resorption in Paget’s disease. At the surfaces of bone formation, plump osteoblasts are found in great number adjacent to abundant osteoid. This type of bone is seldom found in adults except when associated with rapid remodeling of bone, such as occurs after a fracture or in response to tumor invasion of bone. Studies using quantitative histomorphometry of bone documented the marked degree of cellular activity underlying the dramatic changes in bone structure in Paget’s disease.34 The total amount of osteoid and the percentage of the bone surface covered by osteoid may be increased fourfold to fivefold. The increase in osteoid is not associated with an increase in osteoid seam width because the rate of calcification also is increased, as established by double labeling of bone-forming surfaces with tetracycline. No dynamic means of defining the rate of bone resorption is available, but the extent of the total bone surface exhibiting evidence of bone resorption averages about sixfold that of normal individuals, and the number of osteoclasts may be increased as much as 10-fold. In the medullary cavities, the intense resorptive process may produce hemorrhagic cysts with encircling fibrous marrow containing macrophages filled with hemosiderin. These cysts are believed to result from rupture of multiple dilated vessels and ensuing microinfarctions.35 In the mixed phase of Paget’s disease, not only does patchy sclerosis of bone become apparent on radiography, but a bone also may be enlarged. If the osteolytic process extends to the subperiosteal layer, bone formation may be stimulated to such an extent that the thickness and circumference of the bone are increased as a result of periosteal new bone formation. When the skull is affected, this process can produce as much as a fourfold thickening of the calvarium, as was reported by Paget.36 A patchy form of sclerosis is often of a “cotton-wool” character (Fig. 16-4). The skull may be so severely affected that platybasia, or basilar impression, may be a complication. Long bones may be shortened, and typically lateral bowing of the femur or anterior bowing of the tibia or both may develop. Later in the course of the disease, the tibia may exhibit severe lateral bowing. The pathogenesis of the slowly progressive deformity is not known but must be related to the state of abnormal remodeling of the bone. Frequently, fissure fractures are associated with the bowing deformity. These fractures are linear transverse radiolucencies that usually are present in the cortex of the convex aspect of the deformed bone. They often are multiple and may remain stable in appearance for years. They can be found in either osteopenic or osteosclerotic cortices and may be present even in the absence of deformity. Histologic examination of these lesions suggests that they are incomplete fractures.2 Only a small percentage of these lesions progress to a complete transverse fracture, which has been seen more often in patients with a sclerotic cortex.
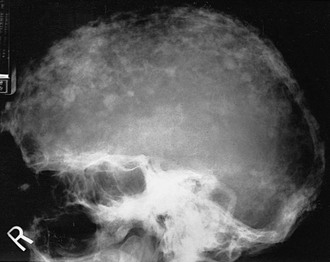
FIGURE 16-4 Radiograph of the sclerotic phase of Paget’s disease in the skull exhibiting a “cotton-wool” appearance.
The evolution of Paget’s disease can also be observed by administration of radioactive tracers and scanning of the entire skeleton or selected regions. Bone scans use technetium-labeled bisphosphonates, which after intravenous injection localize to skeletal sites in proportion to the relative blood flow and the rate of bone formation. The scans usually, but not always, demonstrate high uptake of radioactivity in the areas of the skeleton noted to be radiographically abnormal37 (Fig. 16-5). In a small proportion of patients, increased uptake may be seen when the radiograph is normal, thus illustrating the great sensitivity of bone scans. On the contrary, a small percentage of sclerotic lesions may not be picked up by a bone scan. These lesions appear to represent areas of inactive disease. Bone scans can be analyzed semiquantitatively or by computer and thus may be used to monitor response to treatment.
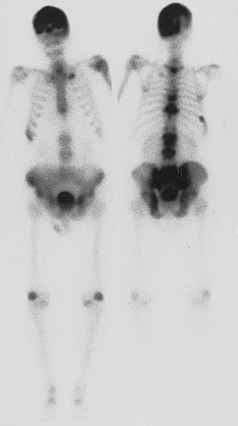
FIGURE 16-5 Anterior and posterior views of a bone scan in a patient with polyostotic Paget’s disease. Increased uptake of tracer can be noted in the skull, multiple vertebrae, and the pelvis, areas in which Paget’s disease was observed on radiographs. The other abnormal areas probably represent degenerative arthritis and a healed rib fracture.
Gallium scans, most often used to detect occult infection or tumors, also have been shown to delineate the lesions of Paget’s disease.38 Evidence indicates that tracer gallium is localized to the nuclei of osteoclasts.39 Therefore, the gallium scan may serve as a direct index of cellular activity in Paget’s disease.
Clinical Features
A considerable proportion of individuals with Paget’s disease have neither symptoms nor signs of the disease.1 The disease is accidentally discovered in these cases because of radiologic or biochemical abnormalities uncovered during investigation of another disorder.
Regional Manifestations
In the absence of an enlarged cranium, symptoms in the skull are uncommon. Even with an enlarged cranium, symptoms often are absent. Certainly, the most common symptom (30% to 50%) is hearing loss,40 which is slowly progressive in untreated patients. Vertigo or tinnitus or both are much less common. The main mechanism of hearing loss in Paget’s disease has been attributed to a reduction in bone mineral density of the cochlear capsule.41
Finally, severe skull disease may be associated with the vague findings of a withdrawn individual who is somnolent and weak. It has been suggested that this manifestation might be a consequence of shunting of blood from the brain vessels to the external carotid artery system, a possible pagetic steal syndrome.42 These symptoms also could represent a psychological response to disability, inasmuch as nearly 50% of patients in one study have been reported to have depression.43
The Jaws
Involvement of the mandible or maxilla may produce progressive root resorption leading to the loss of teeth.44 In the more advanced stages of Paget’s disease, excessive formation of the cementum is associated with absence of the lamina dura and periodontal membranes. Facial disfigurement may occur from enlargement of the maxilla or mandible or both and is associated with spreading of the teeth and malocclusion. Edentulous patients have difficulty acquiring properly fitting dentures. Oral surgery may be complicated by excessive intraoperative bleeding and postoperative osteomyelitis. Tooth extractions may prove difficult because of ankylosis resulting from hyperplasia of the cementum.
The Spine
Severe pain or impaired neurologic function or both may result from compression of the spinal cord or nerve roots.45 This complication can arise from enlargement of the vertebral bodies, pedicles, or laminae, as well as from compression fractures. It also has been suggested that shunting of blood may occur from the spinal arteries to the highly vascular bone.46 Neurologic syndromes are more likely to develop with thoracic involvement. Symptoms include back pain, difficult ambulation, numbness, paresthesias of the feet, and progressive paresis of the legs. Later problems can include impaired bladder and bowel function, as well as spastic paraparesis and loss of sensation. Computed tomography (CT) and magnetic resonance imaging (MRI) are particularly helpful in resolving the anatomic abnormalities producing the disturbed function.
A rare complication in the spine is the development of a discrete paraspinal mass consisting of a central marrow cavity surrounded by pagetic bone that extends from the vertebrae.47 It may appear that the lesion represents a neoplasm, but careful analysis of prior radiologic studies may reveal the chronic nature of the lesion and may make it unnecessary to perform a biopsy.
The other major causes of back pain in Paget’s disease are intervertebral disk disease and degenerative arthritis. No evidence indicates that disk degeneration is more common in patients with Paget’s disease, but it has been reported that the pagetic process can invade the disk and produce bony bridging across the disk space.48 Back pain in the lumbar region is frequently associated with degenerative arthritis,49 particularly when distortion of the facet joints is associated with Paget’s disease. Large osteophytes also may be found in association with enlarged vertebral bodies or where a compression fracture has occurred. A syndrome mimicking ankylosing spondylitis may occur in the presence of extensive osteophyte formation or with ossification of spinal ligaments,50 but the human leukocyte antigen (HLA)-B27 antigen is absent. However, classic ankylosing spondylitis has been found in association with Paget’s disease.49
The Pelvis and Extremities
The main symptoms associated with pelvic and lower extremity involvement are pain and impaired ambulation. Pain is seldom a significant symptom in the osteolytic phase of the disease. Hip pain is most common when both the acetabulum and the proximal end of the femur are affected by the sclerotic phase of the disease.50 Bowing of the femur and protrusio acetabuli are often associated with pain aggravated by weight bearing. Many patients are relatively comfortable when not weight bearing, unlike patients with bone pain, who usually have nocturnal discomfort.
Fractures of the lower extremity are more likely to affect the femur than the tibia. In the largest series of reported femoral fractures, the subtrochanteric region was the most common site of fracture (49 of 182), and the rate of nonunion was noted to be 40%, a figure considerably greater than was previously appreciated.51 Nonunion appears to be less common after tibial fractures.
Sarcoma, Giant Cell Tumors, and Nonskeletal Malignancies
The most feared complication in Paget’s disease is sarcoma. It has been estimated that 10% of patients with extensive disease may experience this problem,35 but if all affected individuals are considered, the incidence is probably less than 1%.52 Sarcomas have rarely been reported to develop in multiple members of families with Paget’s disease.
Patients in whom sarcomas develop usually have pain and swelling, always in an area previously affected with Paget’s disease. Occasionally, fracture at the tumor site may lead to discovery of the neoplasm. Tumors most often arise in the pelvis, femur, humerus, skull, and facial bones.53 Multifocal sarcomas are found only in patients with advanced and widespread polyostotic disease and are thought to represent tumors of independent origin rather than metastases.54
The histology of sarcomas is quite variable. Fibrosarcomas, chondrosarcomas, osteogenic sarcomas, and anaplastic sarcomas may be found.35 Variable numbers of multinucleated giant cells (probably osteoclasts) may be scattered throughout the tumor stroma and most likely are not neoplastic. The nuclear inclusions typical of Paget’s disease have been observed in giant cells but not in the tumor cells.55 It is not unusual for several histologic patterns to be present in a single tumor, which suggests that a common stem cell may give rise to a variety of better differentiated mesenchymal cells.
Lymphomas and multiple myelomas have been found in association with Paget’s disease52 but probably represent chance occurrence rather than a complication of the pathologic process.
The life expectancy for the average patient in whom a sarcoma develops is sadly brief, perhaps because of the difficulty associated with early detection. In one study, only 7.5% of patients survived 5 years, whereas in elderly patients free of underlying Paget’s disease, a 37% 5 year survival rate was noted.56
Giant cell tumors of bone, which usually follow a benign course and most often are found at the ends of long bones in otherwise normal individuals, may arise in the lesions of Paget’s disease.57 They appear to be much less common than sarcoma in Paget’s disease and have frequently been noted to originate in the skull and facial bones. Rarely, these tumors have been reported in multiple family members who have Paget’s disease.
These tumors are characterized by spindle-shaped cells with fusiform nuclei and clumped chromatin or nuclei and by scattered multinucleated giant cells. Mitoses are rarely found in either the mononuclear or giant cells. The giant cells contain the nuclear inclusions of Paget’s disease, but the stromal cells do not.57 The opinion has been expressed that many of the reported cases of giant cell tumor in Paget’s disease actually represent giant cell reparative granulomas, which are common lesions arising in the jaw.58
Surgery and radiation have been used to treat symptomatic giant cell tumors in Paget’s disease, and in one patient, high doses of dexamethasone were effective in shrinking an extraskeletal tumor.59
Biochemical Features
Indices of Bone Resorption
The increased bone resorption typical of active Paget’s disease might be expected to produce an increase in serum and urinary calcium levels, but in the absence of fractures or immobilization, hypercalcemia or hypercalciuria is not a prominent feature of Paget’s disease.60 It is generally believed that this finding is explained by a concomitant increase in bone formation that is demonstrable histologically and by kinetic analysis of plasma disappearance rates and skeletal uptake of radiocalcium60 or other skeletal tracers. A variety of bone collagen matrix breakdown products have been used as indices of bone resorption. These products include urinary hydroxyproline, total and free pyridinoline and deoxypyridinoline, type 1 collagen N-telopeptide, and type 1 collagen C-telopeptide. The telopeptide assays appear to be most specific for bone collagen resorption. Urinary N-telopeptide excretion is sensitive to therapeutic intervention.61 Measurement of non-isomerized fragments of collagen type 1 C-telopeptide may be the most sensitive means of evaluating bone resorption in patients with Paget’s disease.62
Indices of Bone Formation
Since 1929, it has been appreciated that serum total alkaline phosphatase activity may be increased in patients with Paget’s disease.63 The enzyme is localized at the plasma membrane in osteoblasts and may participate in the mineralization of bone matrix. In Paget’s disease, enzyme activity in the circulation correlates with the extent of disease on radiographic skeletal surveys,50 as well as with total urinary hydroxyproline.50 Effective treatment of Paget’s disease reduces total alkaline phosphatase activity by 50% or more.61
During long-term follow-up of untreated patients with Paget’s disease, alkaline phosphatase activity usually exhibits a gradual increase or no significant change.64 In some patients, major fluctuations may represent technical errors rather than changes in disease activity. In the presence of liver disease, hepatic alkaline phosphatase activity may interfere with an accurate assessment of Paget’s disease activity. In such patients, measurement of bone-specific alkaline phosphatase levels by immunoassay is preferable.65
Serum osteocalcin or bone Gla protein is a nearly specific product of osteoblasts that may be elevated in Paget’s disease, but not to the same degree as serum alkaline phosphatase activity.66 Paradoxically, treatment of patients with drugs that reduce turnover may produce a transient increase in osteocalcin levels.66 A potential explanation for these observations is that osteocalcin gene expression is reduced in active Paget’s disease.67
Serum levels of procollagen type 1 N-terminal peptide correlate significantly with quantitative bone scintigraphy and decline with treatment of Paget’s disease to a greater extent than bone-specific alkaline phosphatase.61
Calciotropic Hormones
Calcitonin secretion is normal in patients with Paget’s disease.68 Parathyroid hormone concentrations generally are within the normal range, but in subsets of patients, elevated concentrations have been found.69,70 This observation could be related to renal failure, vitamin D deficiency, or subtle hypocalcemia related to a higher rate of bone formation than bone resorption.
Patients with adequate vitamin D intake or sufficient ultraviolet light exposure or both have normal 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D levels. In two studies, serum 24,25-dihydroxyvitamin D was reported to be low and to correlate inversely with disease activity.71,72 The pathogenesis and clinical relevance of this finding are unknown.
Interleukin-6, an important local modulator of osteoclast function, has been reported to be elevated in the bone marrow of patients with Paget’s disease73 and in serum by some73–75 but not all investigators.76 Receptor activator of nuclear factor-κ beta ligand (RANK-L) and osteoprotegerin (OPG), a decoy receptor for RANK-L, are key modulators of osteoclast function. Serum measurements in patients with Paget’s disease have also been variable, but a recent study indicates that both RANK-L and OPG levels may be highly elevated, and that bisphosphonate therapy can increase OPG and to a lesser extent reduce RANK-L levels, thereby reducing the RANK-L/OPG ratio.77
Systemic Complications and Associated Diseases
Hypercalciuria, Hypercalcemia, and Primary Hyperparathyroidism
Urinary calcium excretion is usually normal in patients with Paget’s disease, and no compelling evidence has been presented that renal stone formation is increased over that in an age- and sex-matched control groups.60 However, hypercalciuria is readily provoked by immobilization after fracture78 or neurologic injury. In this setting, bone resorption increases while bone formation decreases.
Hypercalcemia is uncommon in patients with Paget’s disease but may occur as a consequence of immobilization,78 malignancy,79 or primary hyperparathyroidism.80 Despite some discussion that an increased incidence of primary hyperparathyroidism may occur in Paget’s disease, the presence of both diseases in one individual is very likely coincidental.
Hyperuricemia and Gout
Serum uric acid concentrations have been reported to be elevated primarily in men with relatively severe Paget’s disease.50 Nearly half of the hyperuricemic men had clinical episodes of gouty arthritis. Paget’s disease also was found to be present in 23% of a group of patients with gout.81 It is possible that a high turnover of nucleic acids in the lesions of Paget’s disease could increase the urate pool sufficiently to account for these observations.82
Cardiovascular Abnormalities
Increased cardiac output has been found to be present in patients who have at least 15% of their skeleton affected by Paget’s disease.83 This abnormality is often associated with left ventricular hypertrophy. The excessive vascularity of the soft tissue and adjacent pagetic bone must be factors contributing to these phenomena. In addition, a reduction in peripheral vascular resistance may lead to increased cardiac output.84 High-output cardiac failure may occur but seems to be unusual.
Calcific aortic stenosis appears to be four to six times more common in patients with Paget’s disease than in a control population.85,86 It is more likely to be found in patients with severe disease, which suggests that increased cardiac output producing turbulence across the valve may induce calcification. Intracardiac calcification also may occur in the interventricular septum and may produce a complete heart block.86,87
Drug Treatment
Indications for drug treatment of Paget’s disease are listed in Table 16-1. Perhaps the most common reason for treatment is bone pain. When Paget’s disease occurs adjacent to a joint, it may be difficult to distinguish bone pain from joint pain. In such cases, a therapeutic trial of drug therapy for 1 to 2 months may be particularly useful in clarifying the origin of the pain. Pretreatment of patients who require elective orthopedic surgery may prevent complications such as intraoperative or postoperative hemorrhage and immobilization hypercalcemia. Hypercalciuria and hypercalcemia can be reversed or prevented by drug treatment, but these indications are uncommon.
Table 16-1
Calcitonin
Salmon calcitonin was introduced into clinical use in the United States in 1975. This peptide hormone binds to calcitonin receptors on osteoclasts and rapidly inhibits bone resorption in vivo and in vitro. Salmon calcitonin, 50 to 100 U subcutaneously, 3 to 7 times per week, relieves bone pain in a high percentage of patients within 2 to 6 weeks, reduces cardiac output and vascularity of affected bones, reverses some neurologic deficits, and stabilizes hearing deficits.88 Patients treated preoperatively may have less hemorrhage from orthopedic procedures.89 An immediate decrease in bone resorption parameters is followed by a decrease in alkaline phosphatase activity in 1 month. Both types of parameters decrease by 50% in 3 to 6 months and return toward baseline months after treatment is stopped.
In patients with radiologically defined osteolytic lesions, restoration of a more normal bone structure occurs after long-term treatment.90 However, treatment must be continued indefinitely, or the osteolytic focus will recur. Bone scans91 and gallium scans38 show reduced activity of the pagetic lesions after long-term treatment. Reduced disease activity also is manifested in bone biopsies by a reduction in the number of bone cells, as well as by a decrease in the extent of woven bone and marrow fibrosis.92
As many as 26% of patients administered salmon calcitonin exhibit loss of biochemical responsiveness after an initial period of biochemical improvement.88 Nearly all these patients have high titers of antibodies specific to salmon calcitonin in the circulation. These patients can be treated successfully with any of the bisphosphonates.
Salmon calcitonin may cause a variety of side effects.88 The most common are nausea and facial flushing (10% to 20%). Less commonly, vomiting, abdominal pain, diarrhea, and polyuria may occur. Tetany and allergic reactions are very rare. Side effects are less common with a nasal spray mode of administration, but efficacy is reduced. Salmon calcitonin is now seldom used because of the availability of potent bisphosphonates.
Bisphosphonates
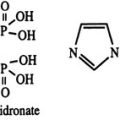
Bisphosphonates are analogues of inorganic pyrophosphate, a compound thought to participate in the mineralization of bone. By substituting a P-C-P bond for the naturally occurring P-O-P bond, a family of metabolically stable compounds has been produced that bind to hydroxyapatite and inhibit bone resorption and, secondarily, bone formation in experimental animals and humans. The earliest bisphosphonates, such as etidronate, induce osteoclast apoptosis by producing nonhydrolyzable analogues of adenosine triphosphate, whereas the more potent aminobisphosphonates inhibit farnesyl-diphosphate (FPP) synthase in the mevalonate pathway, which produces osteoclast apoptosis by inhibition of protein prenylation.93
Four oral bisphosphonates taken daily are etidronate94 (5 mg/kg body weight for 6 months), alendronate95 (40 mg for 6 months), tiludronate96 (400 mg for 3 months), and risedronate97 (30 mg for 2 months). Pamidronate is available in intravenous form and is commonly infused once over a period of several hours at a dose of 60 mg for patients with less than fivefold elevations of serum total alkaline phosphatase activity98 and at an intermittent dose of 60 or 90 mg on 2 or more days, depending on the level of alkaline phosphatase and the response to each infusion. Zoledronic acid, the most recently approved agent, is administered as a 5 mg dose intravenously over 15 minutes.99 Risedronate and the intravenous biphosphonates are probably the most common treatments used in the United States.
The bisphosphonates taken orally must be ingested with water only on an empty stomach because they are poorly absorbed. Generally, side effects are not a major problem and, when present, include abdominal distress, diarrhea, and a temporary increase in bone pain. Patients who receive pamidronate or zoledronic acid intravenously may experience fever and myalgias for about 24 hours. This side effect seldom occurs with subsequent infusions. Allergic reactions are rare and most often are inflammatory eye reactions associated with pamidronate use. Etidronate is the only bisphosphonate reported to produce significant osteomalacia, usually at a dose greater than 5 mg/kg body weight daily. Renal toxicity is not observed with intravenous zoledronic acid if the drug is infused as approved and the creatinine clearance is 35 cc/min or higher.100 Osteonecrosis of the jaw, an unusual event in cancer patients treated monthly with intravenous bisphosphonates, is extremely uncommon in patients with Paget’s disease or osteoporosis.101
The potent aminobisphosphonates can induce biochemical remissions in the great majority of patients with mild to moderate disease activity. Only zoledronic acid can consistently normalize biochemical parameters in the most severely affected patients,99 and the response can persist for at least 2 years.102 It remains to be seen whether long-term biochemical suppression, an achievable goal in most patients, can reduce the incidence of complications in patients who are most at risk. These individuals include those with skull, vertebral, pelvic, and lower extremity involvement.
Surgery
Certainly, the benefits of surgery for the appropriate indications in patients with Paget’s disease outweigh the potential complications of excessive hemorrhage and impaired healing. Probably the most common reason for orthopedic surgery is total hip replacement.103 The rate of success in relieving intractable hip pain and improving mobility is excellent. Heterotopic ossification may be somewhat more common postoperatively but is seldom a major problem. Total knee replacement also is now achieving good clinical results.104 Tibial and fibular osteotomies to correct varus deformity of the tibia are impressive in relieving knee and ankle pain associated with marked deformity.89 Because the rate of nonunion is relatively high in femoral fractures, open reduction and fixation of these fractures may prove necessary.
Much less commonly required are suboccipital craniectomy and upper cervical vertebral laminectomy in patients with symptomatic basilar impression. Equally uncommon is the need for ventricular shunting in patients with hydrocephalus. Attempted relief of hearing loss in patients with skull loss by stapes mobilization or stapedectomy has been of questionable benefit. Surgery to correct spinal stenosis or nerve root compression has generally been successful.4
Causes
In 2002, a mutation in the sequestosome 1 (SQSTM1) gene on chromosome 5 was reported in 11 of 24 French-Canadian families with Paget’s disease and in 18 of 112 patients with apparently sporadic disease (see Chapter 12).105 This was confirmed in different countries by the finding of at least 14 mutations of this gene.106 The mutations are mainly clustered around the ubiquitin-binding domain of the SQSTM1 protein. The SQSTM1 protein modulates activity of the nuclear factor kappa B (NF-κB) pathway, an important mediator of osteoclast function. It appears unlikely that mutations of this gene entirely account for a pagetic phenotype, because some of the family members of these pedigrees exhibit no evidence of Paget’s disease, yet harbor one of the mutations.106–108 It is unclear what role SQSTM1 mutations play in patients with sporadic disease, given the uncertainty of defining sporadic cases and the absence of these mutations as recently reported in 23 apparently sporadic cases.109 Because SQSTM1 mutations are found in only about 20% of Paget’s disease pedigrees, other genetic mutations have been sought. Linkage analyses suggest that chromosome 10p13 harbors a major locus in patients of British ancestry.110
A rare syndrome of Paget’s disease, inclusion body myositis and frontotemporal dementia, also has a genetic association. Mutations in valosine-containing protein are found in this syndrome.111 This protein also contains a ubiquitin-binding domain. Several rare heritable osteolytic disorders superficially resemble Paget’s disease.112 These are caused by mutations that affect RANK or OPG.
Slow Virus Infection
The presence of osteoclast nuclear and cytoplasmic microfilaments essentially identical in structure to nucleocapsids of the Paramyxoviridae virus family initially suggested the possibility that Paget’s disease was caused by a slow virus infection.21,22 Subsequent immunochemical studies24,25 and sequence analysis of nucleocapsid transcripts26–28 have added support to the hypothesis, although not all studies have been positive for a viral presence.113,114
The most compelling evidence to support a viral role in Paget’s disease comes from studies in transgenic mice in which bone histology typical of the disease was induced by inserting the measles virus nucleocapsid gene into precursor cells of the osteoclast lineage.115 In a separate study of transgenic mice, insertion of a mutated murine SQSTM1 gene into the endogenous mouse SQSTM1 gene increased the osteoclastogenic potential of the bone microenvironment but produced no histologic evidence of Paget’s disease.116
References
1. Eekhoff, EMW, van der Klift, M, Kroon, HM, et al. Paget’s disease of bone in the Netherlands: a population-based radiological and biochemical survey; the Rotterdam study. J Bone Miner Res. 2004;19:566–570.
2. Schmorl, G. Über Osteitis deformans Paget. Virchows Arch. 1932;283:694–751.
3. Collins, DH. Paget’s disease of bone: incidence and subclinical forms. Lancet. 1956;2:51–57.
4. Pygott, F. Paget’s disease of bone: The radiological incidence. Lancet. 1956;1:1170–1171.
5. Barker, DJP, Chamberlain, AT, Guyer, PB, et al. Paget’s disease of bone: the Lancashire focus. Br Med J. 1980;1:1105–1107.
6. Detheridge, FM, Guyer, PB, Barker, DJP. European distribution of Paget’s disease of bone. Br Med J. 1982;285:1005–1008.
7. Altman, RD, Bloch, DA, Hochberg, MC, et al. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res. 2000;15:461–465.
8. Hashimoto, J, Ohno, I, Nkatsuka, K, et al. Prevalence and clinical features of Paget’s disease of bone in Japan. J Bone Miner Metab. 2006;24:186–190.
9. Cooper, C, Harvey, NC, Dennison, E, et al. Update on the epidemiology of Paget’s disease of bone. J Bone Miner Res. 2006;21(Suppl 2):P3–8.
10. Cundy, T. Is the prevalence of Paget’s disease of bone decreasing? J Bone Miner Res. 2006;21(Suppl 2):P9–13.
11. Tiegs, RD, Lohse, CM, Wollan, PC, et al. Long-term trends in the incidence of Paget’s disease of bone. Bone. 2000;27:423–427.
12. McKusick, VA. Heritable Disorders of Connective Tissue. St. Louis: Mosby; 1972.
13. Siris, ES, Ottman, R, Flaster, E, et al. Familial aggregation of Paget’s disease of bone. J Bone Miner Res. 1991;6:495–500.
14. Sofoer, JA, Holloway, SM, Emery, AEH. A family study of Paget’s disease of bone. J Epidemiol Community Health. 1983;37:226–231.
15. Posen, S. Paget’s disease: current concepts. Aust N Z J Surg. 1992;62:17–23.
16. Morales-Piga, AA, Rey-Rey, JS, Corres-Gonzales, J, et al. Frequency and characteristics of familial aggregation of Paget’s disease of bone. J Bone Miner Res. 1995;10:663–670.
17. O’Driscoll, JB, Anderson, DC. Past pets and Paget’s disease. Lancet. 1985;2:919–921.
18. Spencer, H, O’Sullivan, V, Sontag, SJ. Does lead play a role in Paget’s disease of bone? A hypothesis. J Lab Clin Med. 1992;120:798–800.
19. Adachi, JD, Arlen, D, Webber, CE, et al. Is there any association between the presence of bone disease and cumulative exposure to lead? Calcif Tissue Int. 1998;63:429–432.
20. Rubinstein, MA, Smelin, A, Freedman, AL. Osteoblasts and osteoclasts in bone marrow aspiration. Arch Intern Med. 1953;92:684–696.
21. Rebel, A, Malkani, K, Basle, M. Anomalies nucleaires de la maladie osseuse de Paget. Nouv Presse Med. 1974;3:1299–1301.
22. Mills, BG, Singer, FR. Nuclear inclusions in Paget’s disease of bone. Science. 1976;194:201–202.
23. Singer, FR. Paget’s disease of bone: possible viral basis. Trends Endocrinol Metab. 1996;7:258–261.
24. Rebel, A, Basle, M, Pouplard, A, et al. Viral antigens in osteoclasts from Paget’s disease of bone. Lancet. 1980;2:344–346.
25. Mills, BG, Singer, FR, Weiner, LP, et al. Evidence for both respiratory syncytial virus and measles virus antigens in the osteoclasts of patients with Paget’s disease of bone. Clin Orthop. 1984;183:303–311.
26. Basle, MF, Fournier, JG, Rozenblatt, S, et al. Measles virus RNA detected in Paget’s disease bone tissue by in situ hybridization. J Gen Virol. 1986;67:907–913.
27. Gordon, MT, Sharpe, PT, Anderson, DC. Canine distemper virus localised in bone cells of patients with Paget’s disease. Bone. 1991;12:195–201.
28. Friedrichs, WE, Reddy, SV, Bruder, JM, et al. Sequence analysis of measles virus nucleocapsid transcripts in patients with Paget’s disease. J Bone Miner Res. 2002;17:145–151.
29. Rhodes, BA, Greyson, ND, Hamilton, CR, Jr., et al. Absence of anatomic arteriovenous shunts in Paget’s disease of bone. N Engl J Med. 1972;287:686–689.
30. Schuller, A. Ueber circumscripte Osteoporose des Schädels. Med Klin. 1929;25:631–632.
31. Maldague, B, Malghem, J. Dynamic radiologic patterns of Paget’s disease of bone. Clin Orthop. 1987;217:126–151.
32. Jacobs, P. Osteolytic Paget’s disease. Clin Radiol. 1974;25:137–144.
33. Belanger, LF, Jarry, L, Uhthoff, HK. Osteocytic osteolysis in Paget’s disease. Rev Can Biol. 1968;27:37–44.
34. Meunier, PJ, Coindre, JM, Edouard, CM, et al. Bone histomorphometry in Paget’s disease: quantitative and dynamic analysis of pagetic and non-pagetic bone tissue. Arthritis Rheum. 1980;23:1095–1103.
35. Jaffe, HL. Metabolic, Degenerative and Inflammatory Diseases of Bones and Joints. Philadelphia: Lea & Febiger; 1972.
36. Paget, J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans. 1877;60:37–64.
37. Vellenga, CJLR, Bijuoet, OLM, Pauwels, EKJ. Bone scintigraphy and radiology in Paget’s disease of bone: a review. Am J Physiol. 1988;3:154–168.
38. Waxman, AD, McKee, D, Siemsen, JK, et al. Gallium scanning in Paget’s disease of bone: effect of calcitonin. Am J Roentgenol. 1980;134:303–306.
39. Mills, BG, Masuoka, LS, Graham, CC, Jr., et al. Gallium-67 citrate localization in osteoclast nuclei of Paget’s disease of bone. J Nucl Med. 1988;29:1083–1087.
40. Nager, GT. Paget’s disease of the temporal bone. Ann Otol Rhinol Laryngol. 1975;84(Suppl 22):1–32.
41. Monsell, EM, Cody, DD, Bone, HG, et al. Hearing loss in Paget’s disease of bone: the relationship between pure tone thresholds and mineral density of the cochlear capsule. Hearing Res. 1995;83:114–120.
42. Blotman, F, Blard, J-M, Labauge, R, et al. Exploration ultrasonique de la circulation encephalique chez le Pagetique. Rev Rhum. 1975;42:647–651.
43. Gold, DT, Boisture, J, Shipp, KM, et al. Paget’s disease of bone and quality of life. J Bone Miner Res. 1996;11:1897–1904.
44. Smith, NHH. Monostotic Paget’s disease of the mandible presenting with progressive resorption of the teeth. Oral Surg Oral Med Oral Pathol. 1978;46:246–253.
45. Hadjipavlou, A, Lander, P. Paget’s disease of the spine. J Bone Joint Surg Am. 1991;73:1376–1381.
46. Douglas, DL, Duckworth, T, Kanis, JA, et al. Spinal cord dysfunction in Paget’s disease of bone. J Bone Joint Surg Br. 1981;63:495–503.
47. Samuels, MA, Schiller, AL. Case records of the Massachusetts General Hospital. N Engl J Med. 1981;304:1411–1421.
48. Lander, P, Hadjipavlou, A. Intradiscal invasion of Paget’s disease of the spine. Spine. 1991;16:46–51.
49. Altman, RD, Collins, B. Musculoskeletal manifestations of Paget’s disease of bone. Arthritis Rheum. 1980;23:1121–1127.
50. Franck, WA, Bress, NM, Singer, FR, et al. Rheumatic manifestations of Paget’s disease of bone. Am J Med. 1974;56:592–603.
51. Dove, J. Complete fractures of the femur in Paget’s disease of bone. J Bone Joint Surg Br. 1980;62:12–17.
52. Hadjipavlou, A, Lander, P, Srolovitz, H, et al. Malignant transformation in Paget disease of bone. Cancer. 1992;70:2802–2808.
53. Haibach, H, Farrell, C, Dittrich, BS. Neoplasms arising in Paget’s disease of bone: a study of 82 cases. Am J Clin Pathol. 1985;83:594–601.
54. Choquette, D, Haraoui, B, Altman, RD, et al. Simultaneous multifocal sarcomatous degeneration in Paget’s disease of bone. Clin Orthop. 1983;179:308–311.
55. Seret, P, Basle, MF, Rebel, A, et al. Sarcomatous degeneration in Paget’s bone disease. J Cancer Res Clin Oncol. 1987;113:392–399.
56. Huvos, AG. Osteogenic sarcoma of bones and soft tissues in older persons. Cancer. 1986;57:1442–1449.
57. Singer, FR, Mills, BG. Giant cell tumor in Paget’s disease of bone: recurrence after 36 years. Clin Orthop. 1993;293:293–301.
58. Upchurch, KS, Simon, LS, Schiller, AL, et al. Giant cell reparative granuloma of Paget’s disease of bone: a unique clinical entity. Ann Intern Med. 1983;98:35–40.
59. Ziambaras, K, Totty, WA, Teitelbaum, SL, et al. Extraskeletal osteoclastomas responsive to dexamethasone treatment in Paget bone disease. J Clin Endocrinol Metab. 1997;82:3826–3834.
60. Nagant de Deuxchaisnes, CN, Krane, SM. Paget’s disease of bone: clinical and metabolic observations. Medicine (Baltimore). 1964;43:233–266.
61. Alvarez, L, Guanabens, N, Peris, P, et al. Usefulness of biochemical markers of bone turnover in assessing response to the treatment of Paget’s disease. Bone. 2001;29:447–452.
62. Alexandersen, P, Peris, P, Guanabens, N, et al. Non-isomerized C-telopeptide fragments are highly sensitive markers for monitoring disease activity and treatment efficacy in Paget’s disease of bone. J Bone Miner Res. 2005;20:588–595.
63. Kay, HD. Plasma phosphatase in osteitis deformans and in other disease of bone. Br J Exp Pathol. 1929;10:253–256.
64. Woodard, HQ. Long term studies of the blood chemistry in Paget’s disease of bone. Cancer. 1959;12:1226–1237.
65. Panigrahi, K, Delmas, PD, Singer, F, et al. Characteristics of a two-site immunoradiometric assay for human skeletal alkaline phosphatase in serum. Clin Chem. 1994;40:822–828.
66. Papapoulos, SE, Frolich, M, Mudde, AH, et al. Serum osteocalcin in Paget’s disease of bone: basal concentrations and response to bisphosphonate treatment. J Clin Endocrinol Metab. 1987;65:189–194.
67. Naot, D, Bava, U, Matthews, B, et al. Differential gene expression in cultured osteoblasts and bone marrow stromal cells from patients with Paget’s disease of bone. J Bone Miner Res. 2007;22:298–309.
68. Kanis, JA, Heynen, G, Walton, RJ. Plasma calcitonin in Paget’s disease of bone. Clin Sci. 1977;52:329–332.
69. Chapuy, M-C, Zucchelli, P, Meunier, PJ. Parathyroid function in Paget’s disease of bone. Bone Miner Electrolyte Metab. 1981;6:112–118.
70. Siris, ES, Clemens, TP, McMahon, D, et al. Parathyroid function in Paget’s disease of bone. J Bone Miner Res. 1989;4:75–79.
71. Guillard-Cumming, DF, Beard, DJ, Douglas, DL, et al. Abnormal vitamin D metabolism in Paget’s disease of bone. Clin Endocrinol. 1985;22:559–566.
72. Castro-Errecaborde, N, de la Piedra, C, Rapado, A, et al. Correlation between serum osteocalcin and 24,25-dihydroxyvitamin D levels in Paget’s disease of bone. J Clin Endocrinol Metab. 1991;72:462–466.
73. Roodman, GD, Kurihara, N, Ohsaki, Y, et al. Interleukin 6: a potential autocrine/paracrine factor in Paget’s disease of bone. J Clin Invest. 1992;89:46–52.
74. Schweitzer, DH, Oostendorp-Van de Ruit, M, van der Plujim, G, et al. Interleukin-6 and the acute phase response during treatment of Paget’s disease with the nitrogen-containing bisphosphonate dimethylaminohydroxypropylidene bisphosphonate. J Bone Miner Res. 1995;10:956–962.
75. Rendina, D, Postiglione, L, Vuotto, P, et al. Clodronate treatment reduces serum levels of interleukin-6 soluble receptor in Paget’s disease of bone. Clin Exp Rheumatol. 2002;20:359–364.
76. Neale, SD, Schulze, E, Smith, R, et al. The influence of serum cytokines and growth factors on osteoclast formation in Paget’s disease. Q J Med. 2002;95:233–240.
77. Martini, G, Gennari, L, Merlotti, D, et al. Serum OPG and RANKL levels before and after intravenous bisphosphonate treatment in Paget’s disease of bone. Bone. 2007;40:457–463.
78. Reifenstein, EC, Jr., Albright, F. Paget’s disease: its pathologic physiology and the importance of this in the complications arising from fracture and immobilization. N Engl J Med. 1944;231:343–355.
79. Rosenkrantz, JA, Gluckman, EC. Coexistence of Paget’s disease of bone and multiple myeloma. Am J Roentgenol. 1957;78:30–38.
80. Gutteridge, DH, Gruber, HE, Kermode, DG, et al. Thirty cases of concurrent Paget’s disease and primary hyperparathyroidism: sex distribution, histomorphometry, and prediction of the skeletal response to parathyroidectomy. Calcif Tissue Int. 1999;65:427–435.
81. Lluberas-Acosta, G, Hansell, JR, Schumacher, HR, Jr. Paget’s disease of bone in patients with gout. Arch Intern Med. 1986;146:2389–2392.
82. Fennelly, JJ, Hogan, A. Pseudouridine excretion: a reflection of high RNA turnover in Paget’s disease. Ir J Med Sci. 1972;141:103–107.
83. Arnalich, F, Plaza, I, Sobrino, JA, et al. Cardiac size and function in Paget’s disease of bone. Int J Cardiol. 1984;5:491–505.
84. Morales-Piga, AA, Moya, JL, Bachiller, FJ, et al. Assessment of cardiac function by echocardiography in Paget’s disease of bone. Clin Exp Rheumatol. 2000;18:31–37.
85. Strickberger, SA, Schulman, SP, Hutchins, GM. Association of Paget’s disease of bone with calcific aortic valve disease. Am J Med. 1987;82:953–956.
86. Hultgren, HN. Osteitis deformans (Paget’s disease) and calcific disease of the heart valves. Am J Cardiol. 1998;81:1461–1464.
87. Harrison, CV, Lennox, B. Heart block in osteitis deformans. Br Heart J. 1948;10:167–176.
88. Waxman, AD, Ducker, S, McKee, D, et al. Evaluation of 99mTc diphosphonate kinetics and bone scans in patients with Paget’s disease before and after calcitonin treatment. Radiology. 1977;125:761–764.
89. Meyers, M, Singer, F. Osteotomy for tibia vara in Paget’s disease under cover of calcitonin. J Bone Joint Surg Am. 1978;60:810–814.
90. Nagant de Deuxchaisnes, C, Maldague, B, Malghem, J, et al. The action of the main therapeutic regimes on Paget’s disease of bone with a note on the effect of vitamin D deficiency. Arthritis Rheum. 1980;23:1215–1234.
91. Waxman, AD, Ducker, S, McKee, D. Paget’s disease of bone. In: Avioli LV, Krane SM, eds. Metabolic Bone Disease and Clinically Related Disorders. San Diego: Academic Press; 1998:545–605.
92. Fornasier, VL, Stapleton, K, Williams, CC. Histologic changes in Paget’s disease treated with calcitonin. Hum Pathol. 1978;9:455–461.
93. Rogers, MJ. New insights into the molecular mechanisms of action of bisphosphonates. Curr Pharm Des. 2003;9:2643–2658.
94. Khairi, MRA, Altman, RD, DeRosa, GP, et al. Sodium etidronate in the treatment of Paget’s disease of bone: a study of long-term results. Ann Intern Med. 1977;87:656–663.
95. Siris, E, Weinstein, RS, Altman, R, et al. Comparative study of alendronate versus etidronate for the treatment of Paget’s disease of bone. J Clin Endocrinol Metab. 1996;81:961–967.
96. Reginster, JY, Calson, F, Morlock, G, et al. Efficacy and safety of oral tiludronate in Paget’s disease of bone: a double-blind, multiple-dosage, placebo-controlled study. Arthritis Rheum. 1992;35:967–974.
97. Siris, ES, Chines, AA, Altman, RD, et al. Risedronate in the treatment of Paget’s disease of bone: an open label multicenter study. J Bone Miner Res. 1998;13:1032–1038.
98. Thiebaud, D, Jaeger, P, Gobelet, C, et al. A single infusion of the bisphosphonate AHPrBP (APD) as treatment of Paget’s disease of bone. Am J Med. 1988;85:207–212.
99. Reid, IR, Miller, P, Lyles, K, et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget’s disease. N Engl J Med. 2005;353:898–908.
100. Boonen, S, Sellmayer, DE, Lippuner, K, et al. Renal safety of annual zoledronic acid infusions in osteoporotic postmenopausal women. Kidney Int. 2008;74:641–648.
101. Rizzoli, R, Burlet, N, Cahall, D, et al. Osteonecrosis of the jaw and bisphophonate treatment for osteoporosis. Bone. 2008;42:841–847.
102. Hosking, D, Lyles, K, Brown, JP, et al. Long-term control of bone turnover in Paget’s disease with zoledronic acid and risedronate. J Bone Miner Res. 2007;22:142–148.
103. Sochart, DH, Porter, ML. Charnley low-friction arthroplasty for Paget’s disease of the hip. J Arthroplasty. 2000;15:210–219.
104. Lee, GC, Sanchez-Sotelo, J, Berry, DJ. Total knee arthroplasty in patients with Paget’s disease at the knee. J Arthroplasty. 2005;20:689–693.
105. Laurin, N, Brown, JP, Morissette, J, et al. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget’s disease of bone. Am J Hum Genet. 2002;70:1582–1588.
106. Morissette, J, Laurin, N, Brown, JP. Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget’s disease of bone. J Bone Miner Res. 2006;21(Suppl 2):P38–44.
107. Johnson-Pais, TL, Wisdom, JH, Weldon, KS, et al. Three novel mutations in SQSTM1 identified in familial Paget’s disease of bone. J Bone Miner Res. 2003;18:1748–1753.
108. Bolland, MJ, Tong, PC, Naot, D, et al. Delayed development of Paget’s disease in offspring inheriting SQSTM 1 mutations. J Bone Miner Res. 2007;22:411–415.
109. Matthews, BG, Naot, D, Bavra, U, et al. Absence of somatic SQTM1 mutations in Paget’s disease of bone. J Clin Endocrinol Metab. 2009;94:691–694.
110. Lucas, GJ, Riches, PL, Hocking, LJ, et al. Identification of a major locus for Paget’s disease on chromosome 10p13 in families of British descent. J Bone Miner Res. 2008;23:58–63.
111. Watts, GDJ, Wymer, J, Kovach, MJ, et al. Inclusion body myositis associated with Paget disease of bone and frontotemporal dementia is caused by a mutant valosin-containing protein. Nat Genet. 2004;36:377–381.
112. Ralston, SH. Juvenile Paget’s disease, familial expansile osteolysis and other genetic osteolytic disorders. Best Pract Res Clin Rheumatol. 2008;22:101–111.
113. Helfrich, MH, Hobson, RP, Grabowski, PS, et al. A negative search for a paramyxoviral etiology of Paget’s disease of bone: molecular, immunological, and ultrastructural studies in UK patients. J Bone Miner Res. 2000;15:2315–2329.
114. Matthews, BG, Afzal, MA, Minor, PD, et al. Failure to detect measles virus ribonucleic acid in bone cells from patients with Paget’s disease. J Clin Endocrinol Metab. 2008;93:1398–1401.
115. Kurihara, N, Zhou, H, Reddy, SV, et al. Expression of measles virus nucleocapsid protein in osteoclasts induces Paget’s disease–like bone lesions in mice. J Bone Miner Res. 2006;21:446–455.
116. Hiruma, Y, Kurihara, N, Subler, MA, et al. A SQSTM1/p62 mutation linked to Paget’s disease increases the osteoclastogenic potential of the bone microenvironment. Hum Mol Genet. 2008;17:3708–3719.