[level-membership-for-pulmolory-and-respiratory-category]9
Overview of Diffuse Parenchymal Lung Diseases
A large group of disorders affects the alveolar wall in a fashion that ultimately may lead to diffuse scarring or fibrosis. These disorders traditionally have been referred to as interstitial lung diseases, although the term is somewhat of a misnomer (see Chapter 8). The interstitium formally refers only to the region of the alveolar wall exclusive of and separating the alveolar epithelial and capillary endothelial cells. However, interstitial lung diseases affect all components of the alveolar wall: epithelial cells, endothelial cells, and cellular and noncellular components of the interstitium. In addition, the disease process often extends into the alveolar spaces and therefore is not limited to the alveolar wall. Many authors now prefer the expression diffuse parenchymal lung disease, which is the term generally used in this book. For practical purposes, however, the reader should recognize that the expressions diffuse parenchymal lung disease and interstitial lung disease typically refer to the same group of disorders causing inflammation and fibrosis of alveolar structures.
There are more than 150 diffuse parenchymal lung diseases. Table 9-1 lists the most common of these disorders, grouped by broad categories according to whether the underlying etiology of the disease is currently known or unknown. A third category of “mimicking disorders” is included in recognition of the fact that a number of additional well-defined clinical problems can produce diffuse parenchymal abnormalities on chest radiograph. Even though these mimicking disorders often are not included in traditional lists of diffuse parenchymal lung diseases, the clinician must remember to consider them in the appropriate clinical settings.
Table 9-1
CLASSIFICATION OF SELECTED DIFFUSE PARENCHYMAL LUNG DISEASES
KNOWN ETIOLOGY
Resulting from inhaled inorganic dusts (pneumoconiosis [e.g., asbestosis, silicosis])
UNKNOWN ETIOLOGY
MIMICKING DISORDERS
*Typically associated with focal or multifocal disease rather than diffuse disease.
Familiarity with all these diseases is difficult even for the pulmonary specialist, so the novice in pulmonary medicine cannot be expected to amass knowledge regarding each individual entity. Rather, the reader is urged to first develop an understanding of the pathologic, pathogenetic, pathophysiologic, and clinical features common to these disorders. This chapter is an overview of general aspects of diffuse parenchymal lung disease and refers to individual diseases only when necessary. Chapter 10 discusses disorders associated with an identifiable etiologic agent; approximately 35% of patients with diffuse parenchymal lung disease are in this category. Chapter 11 discusses diseases for which a specific etiologic agent has not been identified; the majority of patients with diffuse parenchymal lung disease belong in this second category. These chapters cover only a small number of the described types of diffuse parenchymal disease. The goal throughout is to consider those disorders the reader most likely will encounter. Included in the discussion of diseases of unknown etiology in Chapter 11 are several disorders that affect the lung parenchyma but do not characteristically have diffuse findings on chest radiograph. Examples of diseases in which the findings are more typically focal (or multifocal with more than one area of involvement) include Wegener granulomatosis and cryptogenic organizing pneumonia.
The diseases covered in these three chapters are primarily chronic (or sometimes subacute) diseases affecting the alveolar structures. Another group of diseases is associated with acute injury to various components of the alveolus. These latter disorders, which are of clinical importance as causes of acute respiratory failure, are discussed in Chapter 28.
Pathology
Typically, the diffuse parenchymal lung diseases, regardless of cause, have two major pathologic components: an inflammatory process in the alveolar wall and alveolar spaces (sometimes called an alveolitis) and a scarring or fibrotic process (Fig. 9-1). Both features frequently occur simultaneously, although the relative proportions of inflammation and fibrosis vary with the particular cause and duration of disease. The general presumption has been that active inflammation is the primary process and that fibrosis follows as a secondary feature. Idiopathic pulmonary fibrosis (IPF) is a major exception to this generalization. As discussed in Chapter 11, the primary process in IPF appears to be epithelial cell injury and fibrosis (representing an abnormal repair of injury) rather than alveolar inflammation (see Pathogenesis).
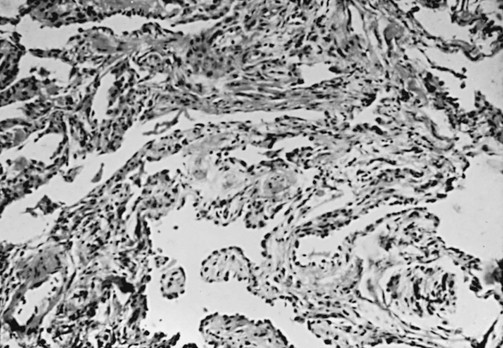
Figure 9-1 Photomicrograph of diffuse parenchymal lung disease shows markedly thickened alveolar walls. Cellular inflammatory process and fibrosis are present. Compare with appearance of normal alveolar walls in Figure 8-1.
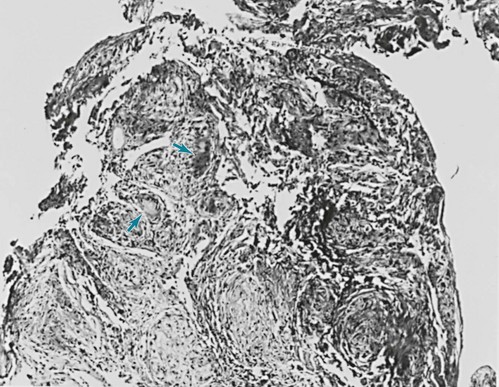
One of the most important pathologic features associated with several diffuse parenchymal lung diseases is the granuloma. A granuloma is a localized collection of cells called epithelioid histiocytes, which are tissue cells of the phagocytic or macrophage series (Fig. 9-2). These cells are generally accompanied by T lymphocytes within the granuloma, often forming a rim around it. When cellular necrosis is present in the center of a granuloma, the entity is termed a caseating granuloma, but diffuse parenchymal lung diseases are associated almost exclusively with noncaseating granulomas (i.e., granulomas in which the central area is not necrotic). In contrast, caseating granulomas are characteristically seen in infectious diseases, especially tuberculosis (see Chapter 24, Pathology). The granuloma typically also has multinucleated giant cells that result from fusion of several phagocytic cells into a single large cell with abundant cytoplasm and many nuclei (see Fig. 9-2). Examples of diffuse parenchymal lung disease in which granulomas are part of the pathologic process include sarcoidosis and hypersensitivity pneumonitis. Granulomas are often considered to reflect some underlying immune process, specifically an immune reaction to an exogenous agent. In the case of hypersensitivity pneumonitis, many agents have been identified. However, in the case of sarcoidosis, no specific exogenous agent has been identified. Granulomas in the lung have many other causes (e.g., tuberculosis, certain fungal infections, and foreign bodies), but they are not covered here because the granulomas are generally not associated with diffuse parenchymal lung disease.
Pathology of Idiopathic Interstitial Pneumonias
This chapter discusses seven pathologic entities subsumed under the broad term idiopathic interstitial pneumonias: (1) usual interstitial pneumonia, (2) desquamative interstitial pneumonia, (3) respiratory bronchiolitis interstitial lung disease, (4) nonspecific interstitial pneumonia, (5) acute interstitial pneumonia, (6) cryptogenic organizing pneumonia, and (7) lymphocytic interstitial pneumonia. This section briefly describes the pathologic characteristics defining these seven entities. Chapter 11 focuses on a broader consideration of the more important clinical counterparts of some of them. An important clinical branch point is distinguishing between UIP and the other conditions, because UIP typically carries a worse prognosis.
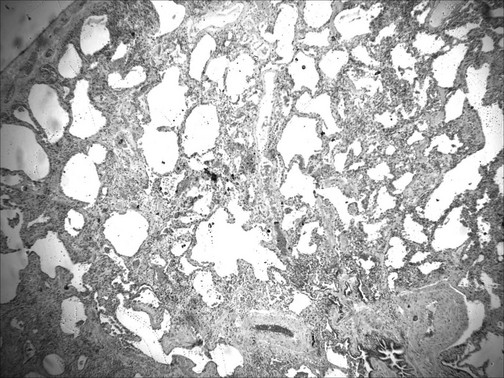
Usual interstitial pneumonia (UIP) is characterized by patchy areas of parenchymal fibrosis and interstitial inflammation interspersed between areas of relatively preserved lung tissue (Fig. 9-3). Fibrosis is the most prominent component of the pathology, with focal collections of proliferating fibroblasts called fibroblastic foci. The fibrosis often is associated with honeycombing, which represents cystic air spaces that result from retraction of surrounding fibrotic lung tissue. The inflammatory process in the alveolar walls is nonspecific and typically composed of a variety of cell types, including lymphocytes, macrophages, and plasma cells. Hyperplasia of type II pneumocytes (alveolar epithelial cells) presumably reflects an attempt to replenish damaged type I cells. Importantly, the pathology of UIP is characterized by the simultaneous presence of all stages of fibrosis: from early fibrosis with actively proliferating fibroblasts to end-stage acellular collagen scarring. It is thought the fibrotic process in UIP is ongoing and not related to a single event. By far the most important of the clinical disorders associated with the histopathologic pattern of UIP is idiopathic pulmonary fibrosis (IPF), and the terms are often used synonymously. However, the pathologic appearance of UIP can also result from exposure to certain inhaled dusts (especially asbestos), from a number of drug-induced lung diseases, and as a form of parenchymal lung disease associated with some systemic rheumatic (connective tissue) diseases. Remember that the term UIP refers to the histologic pattern seen under the microscope, whereas IPF refers to the clinical disease associated with idiopathic UIP.
Acute interstitial pneumonia (AIP) is believed to represent the organizing or fibrotic stage of diffuse alveolar damage, which is the histologic pattern seen in acute respiratory distress syndrome (ARDS) (see Chapter 28). In most cases of ARDS, an inciting cause is apparent, whereas in AIP, no initiating trigger for ARDS can be identified. The histology shows fibroblast proliferation and type II pneumocyte hyperplasia in the setting of what appears to be organizing diffuse alveolar damage.
End-Stage Diffuse Parenchymal Lung Disease
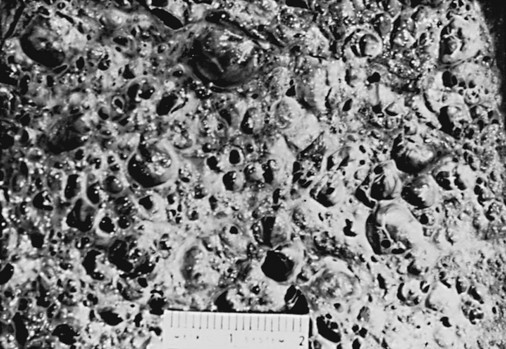
When diffuse parenchymal lung disease has been present for a fairly long time and is associated with significant fibrosis, any distinctive features of prior interstitial inflammation or alveolitis are often lost. For example, any of the granulomatous lung diseases may no longer demonstrate the characteristic granulomas after sufficient time has elapsed and a substantial degree of fibrosis has developed. Therefore, at a certain point all the diffuse parenchymal lung diseases, if sufficiently severe and chronic, follow a final common pathway toward end-stage diffuse parenchymal lung disease. Along with severe fibrosis, the lung at end stage exhibits a great deal of distortion that can be seen both grossly and microscopically, with areas of contraction and other areas showing formation of cystic spaces. In many cases, the result is “honeycomb lung,” in which the dense scarring and intervening cystic regions make areas of the lung resemble a honeycomb (Fig. 9-4).
Pathogenesis
A great deal of research during the last 2 decades attempted to clarify the pathogenetic sequence of events in various types of diffuse parenchymal lung disease. However, in most cases what initiates these diseases remains unknown, and our understanding of the cellular and biochemical events producing inflammation and fibrosis remains mostly at the descriptive level. This section outlines the general scheme of events thought to be operative in the production of parenchymal inflammation and fibrosis. Chapters 10 and 11 discuss specific diseases and provide additional information believed to be relevant to the pathogenesis of each disease. The general scheme outlined here has features similar to that of other forms of lung injury described elsewhere in this book (e.g., emphysema in Chapter 6 and ARDS in Chapter 28). A fundamental but unanswered question is what determines whether an injurious agent eventually leads to emphysema, acute lung injury (with acute respiratory distress syndrome), or chronic parenchymal inflammation and fibrosis.
Figure 9-5 summarizes the general sequence of events presumed to be common to many of the diffuse parenchymal lung diseases. The events can be divided into three stages: initiation, propagation, and final pathologic consequences. Each of these stages is considered in turn.
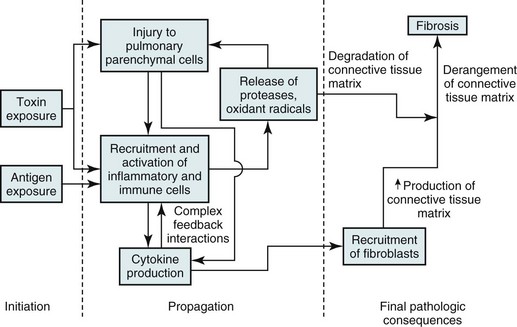
Pathophysiology
With minor exceptions and variations, the pathophysiologic features of the chronic diffuse parenchymal lung diseases are similar and therefore are discussed here as a single group. As a result of the inflammation and fibrosis affecting the alveolar walls, the following abnormalities are generally seen (Fig. 9-6): (1) decreased compliance (increased stiffness) of the lung, (2) generalized decrease in lung volumes, (3) loss of alveolar-capillary surface area resulting in impaired measured diffusion, (4) abnormalities in small airway function without generalized airflow obstruction, (5) disturbances in gas exchange, usually consisting of hypoxemia without CO2 retention, and (6) in some cases pulmonary hypertension. Each of these features is briefly considered in turn.
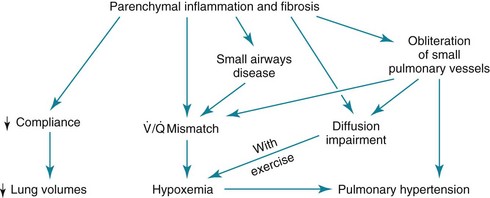
Decreased Compliance
Lung distensibility is significantly altered by processes involving inflammation and fibrosis of the alveolar walls. The lungs become much stiffer, have greatly increased elastic recoil, and therefore require greater distending (transpulmonary) pressures to achieve any given lung volume. The pressure-volume or compliance curve is shifted to the right (see Fig. 8-3), and at any given lung volume, a much higher elastic recoil pressure is found than in normal lungs. Because wider swings in transpulmonary pressure are required to achieve a normal tidal volume during inspiration, the patient’s work of breathing is increased. As a result, patients with diffuse parenchymal lung disease tend to breathe with smaller tidal volumes but increased respiratory frequency. This method allows the patient to expend less energy per breath but maintain adequate alveolar ventilation.
Decrease in Lung Volumes
Early in the course of diffuse parenchymal lung disease, lung volumes may be normal. However, in most cases some reduction in lung volumes is seen shortly thereafter, including a reduction in total lung capacity (TLC), vital capacity (VC), functional residual capacity (FRC), and to a lesser extent, residual volume (RV). The decreases in TLC, FRC, and RV are direct consequences of the change in lung compliance. At TLC, the force generated by the inspiratory muscles is balanced by the inward elastic recoil of the lung. Because the recoil pressure is increased, this balance is achieved at a lower lung volume or lower TLC. At FRC, the outward recoil of the chest wall is balanced by the inward elastic recoil of the lung. The balance is achieved at a lower lung volume or lower FRC because of the greater elastic recoil of the lung. As discussed in Chapter 1, RV is primarily determined by the strength of the expiratory muscles, but a small component is determined by the inward elastic recoil of the lungs. Because the elastic recoil is greater in diffuse parenchymal lung disease, the RV is slightly smaller. Generally, TLC is reduced more than RV, so it follows that VC (representing the difference between TLC and RV) is also decreased.
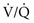 ) mismatching and hypoxemia are consequences. Evidence of more significant airflow obstruction may be seen in a few disorders causing diffuse parenchymal lung disease. This relatively infrequent problem sometimes results from severe fibrosis and airway distortion.
) mismatching and hypoxemia are consequences. Evidence of more significant airflow obstruction may be seen in a few disorders causing diffuse parenchymal lung disease. This relatively infrequent problem sometimes results from severe fibrosis and airway distortion.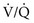 mismatch as the major contributor. The pathologic process in the alveolar walls is uneven, and normal matching of ventilation and perfusion is disrupted. In patients with small airways disease, dysfunction at this level probably also contributes to
mismatch as the major contributor. The pathologic process in the alveolar walls is uneven, and normal matching of ventilation and perfusion is disrupted. In patients with small airways disease, dysfunction at this level probably also contributes to 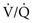 mismatch and hypoxemia. Characteristically, patients with diffuse parenchymal lung disease become even more hypoxemic with exercise. Again, the primary mechanism of oxygen desaturation associated with exertion is worsening
mismatch and hypoxemia. Characteristically, patients with diffuse parenchymal lung disease become even more hypoxemic with exercise. Again, the primary mechanism of oxygen desaturation associated with exertion is worsening 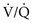 mismatch, but diffusion limitation also appears to be a contributing factor. The combination of impaired diffusion and decreased transit time of the red blood cell during exercise may prevent complete equilibration of P
mismatch, but diffusion limitation also appears to be a contributing factor. The combination of impaired diffusion and decreased transit time of the red blood cell during exercise may prevent complete equilibration of P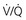 mismatch) and normal or decreased P
mismatch) and normal or decreased PDiagnostic Approach
The chest radiograph is the most important means for making the initial macroscopic assessment of diffuse parenchymal lung disease. The characteristic radiographic picture of diffuse parenchymal involvement that primarily involves alveolar walls is described as either reticular (increased linear markings) or reticulonodular (increased linear and small nodular markings; see Fig. 3-6). The pattern has also previously been called an “interstitial pattern” because it was believed to reflect a process limited to the alveolar walls. However, histopathology often indicates that some of these processes extend into alveolar spaces as well. Absence of radiographic abnormalities does not exclude the presence of diffuse parenchymal disease; entirely normal chest radiographic findings have been reported in up to 10% of patients. The pattern on chest radiograph is not particularly useful for gauging the relative amounts of inflammation versus fibrosis, each of which may result in a similar pattern. Reticular or reticulonodular changes are frequently diffuse throughout both lung fields, although individual causes of diffuse parenchymal lung disease may be more likely to result in either an upper or a lower lung field predominance of the abnormal markings. In addition to the reticular or reticulonodular pattern, certain diseases may reveal other associated findings on chest radiograph, such as hilar adenopathy or pleural disease. These additional features noted with some diseases are discussed in Chapters 10 and 11.
With long-standing and severe disease, the lungs may become grossly distorted. In addition, regions of cyst formation between scarred and retracted areas of lung may occur (see Fig. 9-4). A corresponding pattern of honeycombing on chest radiograph may be apparent. Cor pulmonale may be suspected on chest radiograph by the presence of right ventricular enlargement, best seen on the lateral view.
High-resolution computed tomography (HRCT) of the chest is an important step in the evaluation of diffuse parenchymal lung disease (see Fig. 3-9). Because of the quality of images of the pulmonary parenchyma, early changes that are not evident on routine chest radiography can often be seen by HRCT. In addition, the specific pattern of abnormality on an HRCT scan may be suggestive of a particular underlying diagnosis such as IPF and may help distinguish inflammation from fibrosis.
Treatment
Treatment considerations vary among the diseases. In general, patients with interstitial disease either do not respond well to any form of treatment or respond to immunosuppressive agents (e.g., corticosteroids, cyclophosphamide) to a variable extent. The rationale for immunosuppressive therapy is to reduce the alveolitis component of the disease; the fibrosis is generally considered irreversible. Other interesting therapeutic approaches involve targeting specific growth factors, cytokines, or oxidants involved in the inflammatory and fibrotic process within the lungs. However, these approaches are investigational at present. Specific aspects of treatment are covered in the discussion of individual diseases in Chapters 10 and 11.
American Thoracic Society and European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165:277–304.
Harari, S, Caminati, A. Update on diffuse parenchymal lung disease. Eur Respir Rev. 2010;19:97–108.
Lama, VN, Martinez, FJ. Resting and exercise physiology in interstitial lung diseases. Clin Chest Med. 2004;25:435–453.
Leslie, KO. Pathology of interstitial lung disease. Clin Chest Med. 2004;25:657–703.
Markart, P, Wygrecka, M, Guenther, A. Update in diffuse parenchymal lung disease. Am J Respir Crit Care Med. 2010;183:1316–1321.
Pipavath, S, Godwin, JD. Imaging of interstitial lung disease. Clin Chest Med. 2004;25:455–465.
Raghu, G, Nicholson, AG, Lynch, D. The classification, natural history and radiological/histological appearance of idiopathic pulmonary fibrosis and the other idiopathic interstitial pneumonias. Eur Respir Rev. 2008;17:108–115.
Rosenbloom, J, Castro, SV, Jimenez, SA. Narrative review: fibrotic diseases: cellular and molecular mechanisms and novel therapies. Ann Intern Med. 2010;152:159–166.
Silva, CI, Müller, NL. Idiopathic interstitial pneumonias. J Thorac Imaging. 2009;24:260–273.
Verschakelen, JA. The role of high-resolution computed tomography in the work-up of interstitial lung disease. Curr Opin Pulm Med. 2010;16:503–510.
Visscher, DW, Myers, JL. Histologic spectrum of idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3:322–329.
Wells, AU. The clinical utility of bronchoalveolar lavage in diffuse parenchymal lung disease. Eur Respir Rev. 2010;19:237–241.
Wittram, C. The idiopathic interstitial pneumonias. Curr Probl Diagn Radiol. 2004;33:189–199.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]9
Overview of Diffuse Parenchymal Lung Diseases
A large group of disorders affects the alveolar wall in a fashion that ultimately may lead to diffuse scarring or fibrosis. These disorders traditionally have been referred to as interstitial lung diseases, although the term is somewhat of a misnomer (see Chapter 8). The interstitium formally refers only to the region of the alveolar wall exclusive of and separating the alveolar epithelial and capillary endothelial cells. However, interstitial lung diseases affect all components of the alveolar wall: epithelial cells, endothelial cells, and cellular and noncellular components of the interstitium. In addition, the disease process often extends into the alveolar spaces and therefore is not limited to the alveolar wall. Many authors now prefer the expression diffuse parenchymal lung disease, which is the term generally used in this book. For practical purposes, however, the reader should recognize that the expressions diffuse parenchymal lung disease and interstitial lung disease typically refer to the same group of disorders causing inflammation and fibrosis of alveolar structures.
There are more than 150 diffuse parenchymal lung diseases. Table 9-1 lists the most common of these disorders, grouped by broad categories according to whether the underlying etiology of the disease is currently known or unknown. A third category of “mimicking disorders” is included in recognition of the fact that a number of additional well-defined clinical problems can produce diffuse parenchymal abnormalities on chest radiograph. Even though these mimicking disorders often are not included in traditional lists of diffuse parenchymal lung diseases, the clinician must remember to consider them in the appropriate clinical settings.
Table 9-1
CLASSIFICATION OF SELECTED DIFFUSE PARENCHYMAL LUNG DISEASES
KNOWN ETIOLOGY
Resulting from inhaled inorganic dusts (pneumoconiosis [e.g., asbestosis, silicosis])
UNKNOWN ETIOLOGY
MIMICKING DISORDERS
*Typically associated with focal or multifocal disease rather than diffuse disease.
Familiarity with all these diseases is difficult even for the pulmonary specialist, so the novice in pulmonary medicine cannot be expected to amass knowledge regarding each individual entity. Rather, the reader is urged to first develop an understanding of the pathologic, pathogenetic, pathophysiologic, and clinical features common to these disorders. This chapter is an overview of general aspects of diffuse parenchymal lung disease and refers to individual diseases only when necessary. Chapter 10 discusses disorders associated with an identifiable etiologic agent; approximately 35% of patients with diffuse parenchymal lung disease are in this category. Chapter 11 discusses diseases for which a specific etiologic agent has not been identified; the majority of patients with diffuse parenchymal lung disease belong in this second category. These chapters cover only a small number of the described types of diffuse parenchymal disease. The goal throughout is to consider those disorders the reader most likely will encounter. Included in the discussion of diseases of unknown etiology in Chapter 11 are several disorders that affect the lung parenchyma but do not characteristically have diffuse findings on chest radiograph. Examples of diseases in which the findings are more typically focal (or multifocal with more than one area of involvement) include Wegener granulomatosis and cryptogenic organizing pneumonia.
The diseases covered in these three chapters are primarily chronic (or sometimes subacute) diseases affecting the alveolar structures. Another group of diseases is associated with acute injury to various components of the alveolus. These latter disorders, which are of clinical importance as causes of acute respiratory failure, are discussed in Chapter 28.
Pathology
Typically, the diffuse parenchymal lung diseases, regardless of cause, have two major pathologic components: an inflammatory process in the alveolar wall and alveolar spaces (sometimes called an alveolitis) and a scarring or fibrotic process (Fig. 9-1). Both features frequently occur simultaneously, although the relative proportions of inflammation and fibrosis vary with the particular cause and duration of disease. The general presumption has been that active inflammation is the primary process and that fibrosis follows as a secondary feature. Idiopathic pulmonary fibrosis (IPF) is a major exception to this generalization. As discussed in Chapter 11, the primary process in IPF appears to be epithelial cell injury and fibrosis (representing an abnormal repair of injury) rather than alveolar inflammation (see Pathogenesis).
Figure 9-1 Photomicrograph of diffuse parenchymal lung disease shows markedly thickened alveolar walls. Cellular inflammatory process and fibrosis are present. Compare with appearance of normal alveolar walls in Figure 8-1.
One of the most important pathologic features associated with several diffuse parenchymal lung diseases is the granuloma. A granuloma is a localized collection of cells called epithelioid histiocytes, which are tissue cells of the phagocytic or macrophage series (Fig. 9-2). These cells are generally accompanied by T lymphocytes within the granuloma, often forming a rim around it. When cellular necrosis is present in the center of a granuloma, the entity is termed a caseating granuloma, but diffuse parenchymal lung diseases are associated almost exclusively with noncaseating granulomas (i.e., granulomas in which the central area is not necrotic). In contrast, caseating granulomas are characteristically seen in infectious diseases, especially tuberculosis (see Chapter 24, Pathology). The granuloma typically also has multinucleated giant cells that result from fusion of several phagocytic cells into a single large cell with abundant cytoplasm and many nuclei (see Fig. 9-2). Examples of diffuse parenchymal lung disease in which granulomas are part of the pathologic process include sarcoidosis and hypersensitivity pneumonitis. Granulomas are often considered to reflect some underlying immune process, specifically an immune reaction to an exogenous agent. In the case of hypersensitivity pneumonitis, many agents have been identified. However, in the case of sarcoidosis, no specific exogenous agent has been identified. Granulomas in the lung have many other causes (e.g., tuberculosis, certain fungal infections, and foreign bodies), but they are not covered here because the granulomas are generally not associated with diffuse parenchymal lung disease.
Pathology of Idiopathic Interstitial Pneumonias
This chapter discusses seven pathologic entities subsumed under the broad term idiopathic interstitial pneumonias: (1) usual interstitial pneumonia, (2) desquamative interstitial pneumonia, (3) respiratory bronchiolitis interstitial lung disease, (4) nonspecific interstitial pneumonia, (5) acute interstitial pneumonia, (6) cryptogenic organizing pneumonia, and (7) lymphocytic interstitial pneumonia. This section briefly describes the pathologic characteristics defining these seven entities. Chapter 11 focuses on a broader consideration of the more important clinical counterparts of some of them. An important clinical branch point is distinguishing between UIP and the other conditions, because UIP typically carries a worse prognosis.
Usual interstitial pneumonia (UIP) is characterized by patchy areas of parenchymal fibrosis and interstitial inflammation interspersed between areas of relatively preserved lung tissue (Fig. 9-3). Fibrosis is the most prominent component of the pathology, with focal collections of proliferating fibroblasts called fibroblastic foci. The fibrosis often is associated with honeycombing
[/not-level-membership-for-pulmolory-and-respiratory-category]
