68 Other Motor Neuron Diseases and Motor Neuropathies
Clinical Presentation
Spinal Muscular Atrophy Types I–IV
Of the multiple SMA phenotypes, the infantile and childhood forms are the most prevalent. SMA type I or Werdnig–Hoffman disease is the most severe form (Fig. 68-1). Clinical manifestations become evident within the first 6 months of life. In contrast to the latter three categories, afflicted children with SMA I never develop the capability of sitting independently. In some cases, recognition of reduced movement occurs in utero or within the first few days of life. Affected infants are hypotonic with a symmetric, generalized, or proximally predominant pattern of weakness. Like ALS, facial weakness is typically mild and extraocular muscles are spared. Fasciculations are seen in the tongue but rarely in limb muscles, presumably because of the ample subcutaneous tissue of neonates. Manual tremor, so characteristic of SMA types II and III, is rarely present. Deep tendon reflexes are typically absent. Abdominal breathing, a weak cry, and a poor suck are commonplace. Ventilation difficulties stem primarily from intercostal rather than diaphragmatic weakness. Pectus excavatum and a diminished anteroposterior diameter of the chest are seen. Mild contractures may occur but arthrogryposis is not part of the classic phenotype. Intellectual development is normal. Without mechanical ventilation, death is inevitable, almost always within a year or two. An earlier age of onset correlates with a shorter life expectancy.
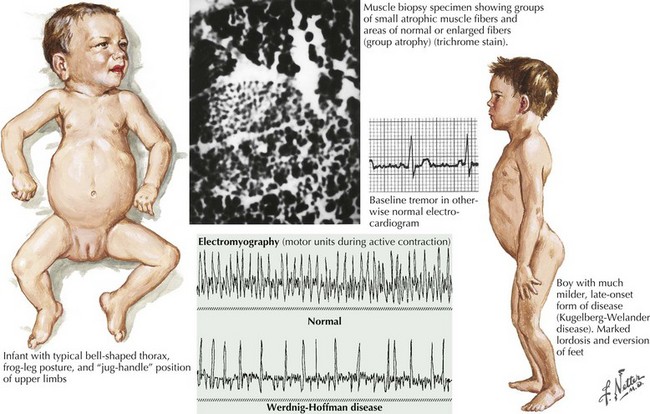
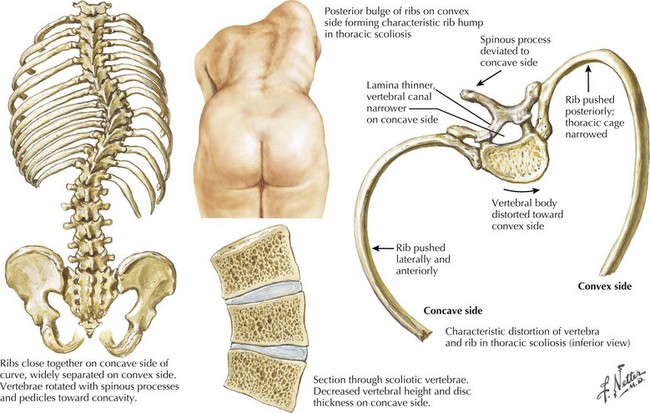
The intermediate form or SMA II typically begins between 6 and 18 months of age. The disorder is clinically defined by a child who sits independently but never walks. Postural hand tremor is the only significant phenotypic variance from Werdnig–Hoffman disease. Tongue fasciculations, areflexia, and a generalized to proximally predominant and symmetric pattern of weakness mimic the SMA I phenotype. Approximately 98% of these individuals survive to age 5 and two thirds to age 25. In view of the more protracted course and of wheelchair dependency SMA II and SMA III patients commonly acquire kyphoscoliosis and joint contractures (Fig. 68-2).
Juvenile Segmental Spinal Muscular Atrophy—Benign Focal Amyotrophy—Hirayama Disease
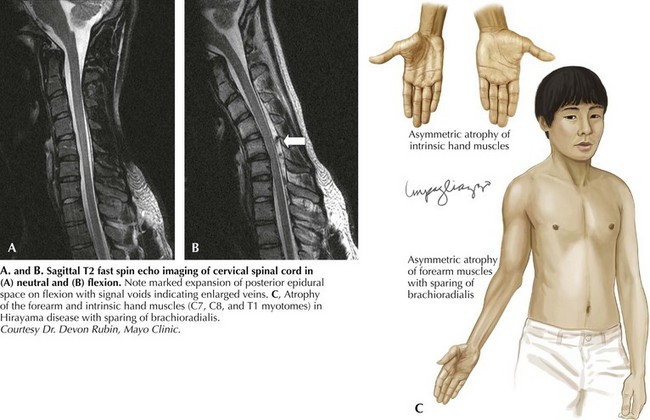
Hirayama disease is less frequently seen in Western populations. Ischemic changes in the cervical spinal cord of a single autopsied case of Hirayama disease led to the hypothesis of a compressive mechanism. In 2000, Hirayama reported the results of dynamic imaging in 73 patients and 20 controls. Ninety-four percent of patients had significant forward displacement and flattening of the posterior surface of the cervical cord during neck flexion (Fig. 68-3). The presumption is that the blood supply to the spinal cord is compromised, with the anterior horn representing the watershed and the most susceptible to ischemia. Other observations that supported this potential mechanism are the frequent asymmetric nature of spinal cord flattening in keeping with the asymmetric disease onset, and the lesser degree to which distortion occurred in older patients in whom progression had arrested. Nonetheless, this pathogenetic hypothesis is not universally accepted.
Distal SMA (Hereditary Motor Neuronopathy, Spinal Forms of Charcot–Marie–Tooth Disease)
Distal SMA (dSMA) is usually inherited in a dominant fashion in one third of cases but may have recessive or X-linked pattern as well. There are numerous genetic loci (Table 68-1). Like hereditary spastic paraparesis, distal SMA can be either “pure” or “complicated” based upon other neurologic system involvement. Complicated phenotypes may include diaphragmatic paralysis, vocal cord paralysis, and arthrogryposis.
| Classification | Chromosome | Gene |
|---|---|---|
| SMA I-IV | 5q12.2-q13.3 | SMN1 |
| SMARD I | 11q13.2-q13.4 | IGHMBP2 |
| SBMA (Kennedy) |
X | Androgen receptor gene |
| Juvenile segmental SMA (Hirayama) |
None identified | None identified |
| Scapuloperoneal (Davidenkow) |
17p11.2 | PMP 22 |
Harding and Thomas introduced the concept of dSMA in 1980. The dSMAs have been perceived as progressive, hereditary disorders producing distal symmetric weakness in the absence of either clinical or electrodiagnostic sensory loss. The dSMAs have also been referred to as hereditary motor neuropathies but are considered to be anterior horn cell disorders in view of their pure motor manifestations. Distal SMA strongly resembles Charcot–Marie–Tooth (CMT) disease without sensory involvement. In fact, at least three forms of dSMA are allelic to recessively inherited forms of CMT. Weakness in distal SMA typically predominates in ankle dorsiflexors and evertors and toe extensors. Foot deformities characteristic of CMT are also common. Hand muscles may eventually become involved. There are a number of recognized dSMA genotypes (see Table 68-1).
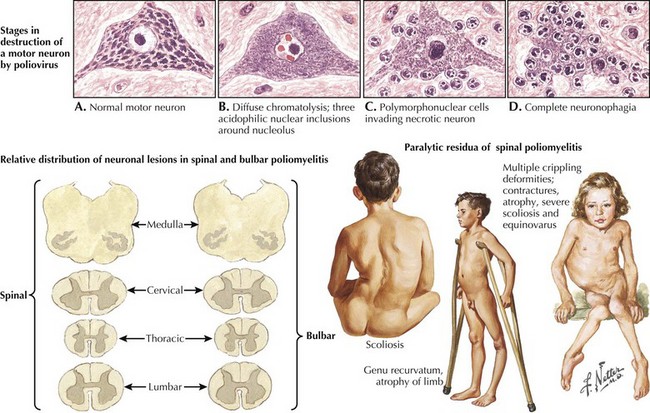
Poliomyelitis
Poliomyelitis is a viral infection with tropism for gray matter of the spinal cord and motor cranial nuclei. Poliomyelitis translates literally into inflammation of spinal cord gray matter. It is often used synonymously with paralytic polio caused by the polio virus. In this chapter, it will refer to any viral infection with a predilection for anterior horn cells or motor cranial nerve nuclei. Polio may be either a monophasic or biphasic disease. The initial symptoms are nonspecific, last 1–2 days, and are predominantly constitutional and/or gastrointestinal in nature. They consist of fever, malaise, pharyngitis, headache, nausea, vomiting, and abdominal cramping (Fig. 68-4). In the majority of infected individuals, the illness is self-limited and ends at this point. In individuals who fall victim to the “major” illness, symptoms of brain or spinal cord involvement develop 3 to 10 days subsequent to the initial symptoms. The major illness is defined by CNS involvement with meningoencephalitis, with or without an associated paralytic component. Stiff neck, back pain, and fever are prominent; encephalitis with altered mental status can also be seen.
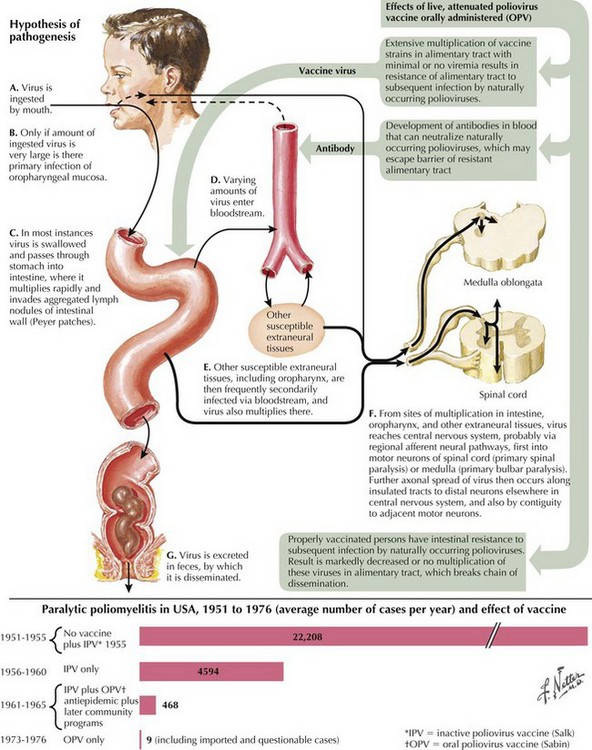
In individuals destined to develop paralytic disease, myalgias and cramping rapidly evolve into muscle weakness. The progression reaches its nadir within 48 hours of onset. The paralysis is typically asymmetric. It is confined to the limbs and trunk in half of the cases. There is a predilection for lumbosacral segments and proximal more than distal muscles (Fig. 68-5). Ten percent of cases have bulbar weakness only. Children are particularly susceptible to bulbar polio. Motor functions of the 7th, 9th and 10th cranial nerves are most likely to be affected. Ten percent of patients will manifest both spinal and bulbar weakness; ventilatory failure is more common in this group. Affected limbs are flaccid and areflexic. Like virtually all motor neuron disorders, the 3rd, 4th, and 6th cranial nerves are spared. Sensory signs and symptoms are atypical. In keeping with the known pathological involvement of the brainstem tegmentum and hypothalamus in cases with encephalitic components, clinical dysautonomia including fluctuating blood pressure, cardiac arrhythmia, and hyperhydrosis may occur.
Diagnostic Approach
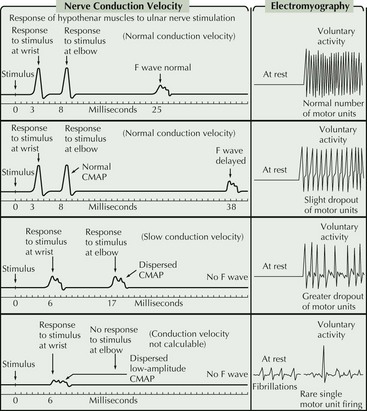
For the most part, the only EDX features that distinguish between the different motor neuron diseases are the distribution of abnormalities and the degree of active versus chronic denervation changes. More chronic disorders such as Kennedy disease or old polio are dominated by features of chronic denervation and reinnervation whereas ALS typically has prominent features of both active and chronic denervation. Kennedy disease is rather unique within the MND spectrum in that sensory nerve action potential amplitudes are often reduced or absent. A key diagnostic feature in MMN is the presence of demyelinating features on motor nerve conductions, particularly the presence of conduction block (Fig. 68-6). Unfortunately, there are a number of reasons why this feature is not always demonstrable.
Bertini E, Burghes A, Bushby K, et al. 134th ENMC International Workshop: Outcome measures and treatment of spinal muscular atrophy 11-13 February 2005. Naarden, The Netherlands. Neuromuscular Disorders. 2005;15:802-816.
Chahin N, Klein C, Mandrekar J, et al. Natural history of spinal-bulbar muscular atrophy. Neurology. 2008;70:1967-1971.
Harding AE. Inherited neuronal atrophy and degeneration predominantly of lower motor neurons. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF, editors. Peripheral Neuropathy. 3rd ed. Philadelphia: W. B. Saunders; 1993:1051-1064.
Irobi J, Dierick I, Jordanova A, et al. Unraveling the genetics of distal hereditary motor neuropathies. Neuromolec Med. 2006;8:131-146.






