52
Other Genodermatoses
This chapter discusses genetic skin diseases that are not covered in other chapters, including conditions featuring extracutaneous tumorigenesis, enzyme deficiencies, premature aging, and ectodermal dysplasia.
Disorders Featuring Extracutaneous Tumorigenesis
Cowden Disease and Other Forms of PTEN Hamartoma Tumor Syndrome
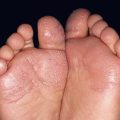
• Spectrum of autosomal dominant multisystem disorders that feature characteristic skin findings (Fig. 52.1), macrocephaly, hamartomatous overgrowth of a variety of tissues, and a predisposition to certain cancers (Table 52.1).
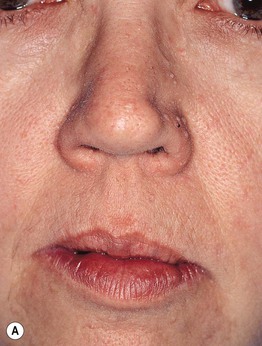
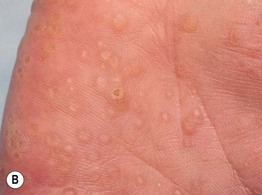
Fig. 52.1 Cowden disease. A Multiple skin-colored papules on the face, especially the nose, some of which are verrucous. B Multiple palmar keratoses, many with a glassy appearance or depression centrally. A, Courtesy, Kalman Watsky, MD; B, Courtesy, Joyce Rico, MD.
Table 52.1
Major clinical manifestations of PTEN hamartoma tumor syndrome.
This categorization reflects general tendencies, as the age of onset or recognition of these findings can vary. Criteria for PTEN gene testing and guidelines for surveillance in patients suspected to have Cowden disease are available at http://www.nccn.org/professionals/physician_gls/PDF/genetics_screening.pdf.
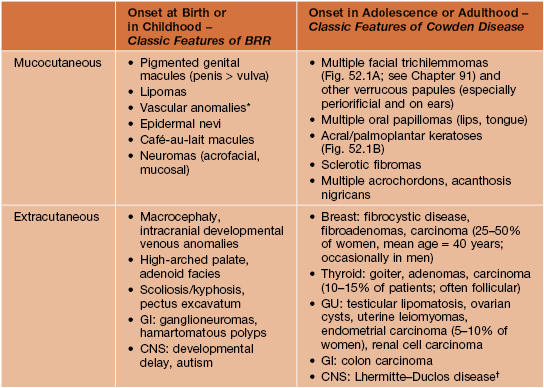
* Typically with fast-flow channels, intramuscular involvement, and ectopic fat; may have capillary, venous, and/or lymphatic components and associated soft tissue/bony overgrowth.
† Hamartomatous dysplastic gangliocytoma of the cerebellum.
BRR, Bannayan–Riley–Ruvalcaba syndrome.
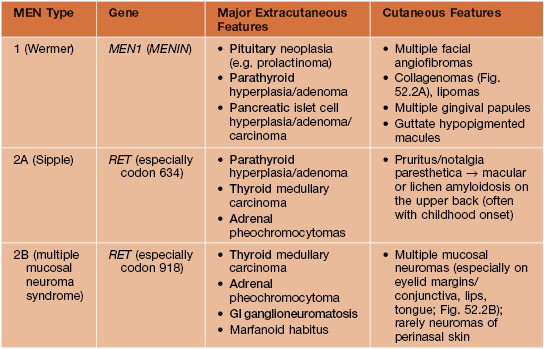
Multiple Endocrine Neoplasia Syndromes
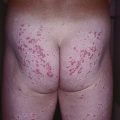
• Group of autosomal dominant disorders associated with neoplasia or hyperplasia in two or more endocrine organs, often presenting with mucocutaneous findings that serve as clues to the diagnosis (Table 52.2; Fig. 52.2).
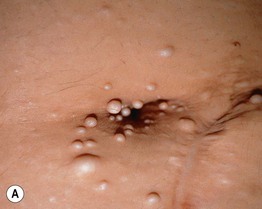
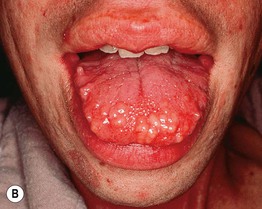
Muir–Torre Syndrome (MTS)
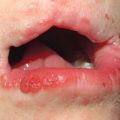
• Subtype of hereditary nonpolyposis colorectal cancer (Lynch) syndrome characterized by sebaceous neoplasms (e.g. sebaceous adenoma, sebaceoma, sebaceous carcinoma; Fig. 52.3, see Table 91.2) and keratoacanthomas (± sebaceous differentiation) as well as internal malignancies (Table 52.3).
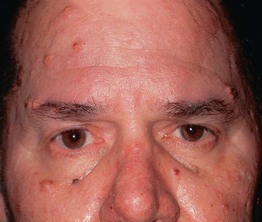
Table 52.3
Factors associated with a greater likelihood of Muir–Torre syndrome (MTS).

* Testing can be performed on paraffin-embedded tissue; diagnostic yield may be higher for colonic lesions.
** For example, other GI cancers (small bowel, gastric, biliary, pancreatic), genitourinary cancers (endometrial, bladder, ureteral, renal), and glioblastoma.
† Sensitivity and specificity have varied and the overall utility is debated.
IHC, immunohistochemical; HNPCC, hereditary nonpolyposis colorectal cancer.
Gardner Syndrome
Birt–Hogg–Dubé Syndrome
Enzyme Deficiency Disorders
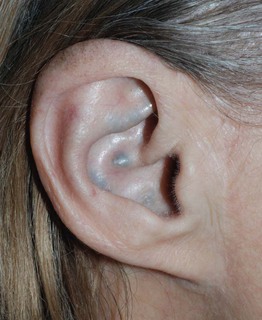
Alkaptonuria
• Rare autosomal recessive disorder due to homogentisic acid oxidase deficiency.
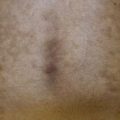
• Urine that darkens on standing and brown-black cerumen represent early findings.
• Blue-gray discoloration of the axillary skin, ear (Fig. 52.4), and sclera typically appears during early adulthood, when arthritis and valvular heart disease often develop.
Fabry Disease
• Angiokeratoma corporis diffusum presents as punctate dark red macules and papules clustered between the umbilicus and knees (‘bathing trunk’ distribution); serves as an early sign of Fabry disease, typically developing in childhood or adolescence along with paresthesias of the extremities, hypohidrosis, and whorled corneal opacities.
• Fig. 52.5 compares the clinical features of Fabry disease and fucosidosis, one of several other lysosomal storage disorders associated with angiokeratomas.
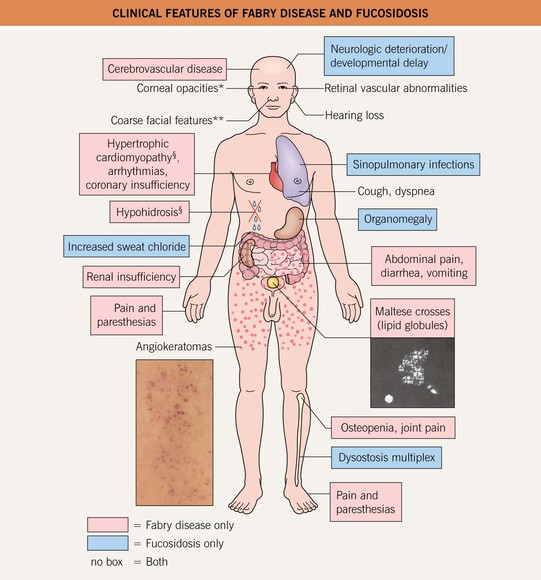
Fig. 52.5 Clinical features of Fabry disease and fucosidosis. *Also lenticular in Fabry disease. **Milder in Fabry disease. §Occasionally observed in fucosidosis. Angiokeratomas of Fabry disease, courtesy, Julie V. Schaffer, MD; photomicrograph of urinary sediment demonstrating Maltese crosses (via polarization), courtesy, Robert J. Desnick, MD, PhD.
Phenylketonuria
• Autosomal recessive disorder due to phenylalanine hydroxylase deficiency.
• Cutaneous findings can include diffuse pigmentary dilution (skin, hair, eyes), eczematous dermatitis, sclerodermatous skin changes favoring the proximal extremities (Fig. 52.6), and sweat with a musty odor; pigmentary banding of the hair can occur as a reflection of nonadherence to the diet.

Mitochondrial Disorders
• This heterogeneous group of conditions can present with a wide variety of cutaneous manifestations (Table 52.4) as well as neuromuscular and visceral dysfunction.

Premature Aging Disorders
Hutchinson–Gilford Progeria Syndrome
Werner Syndrome
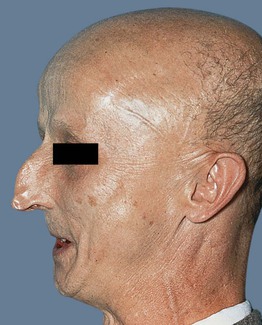
• Short stature, characteristic facies (e.g. thin with beaked nose; Fig. 52.7), atherosclerosis, osteoporosis, diabetes mellitus, and an increased risk of sarcomas are additional features; median survival ~50 years.
Ectodermal Dysplasias
• Major features of several classic forms of ectodermal dysplasia are presented in Table 52.5 and Figs. 52.8–52.11.
Table 52.5
Selected ectodermal dysplasia (ED) syndromes.
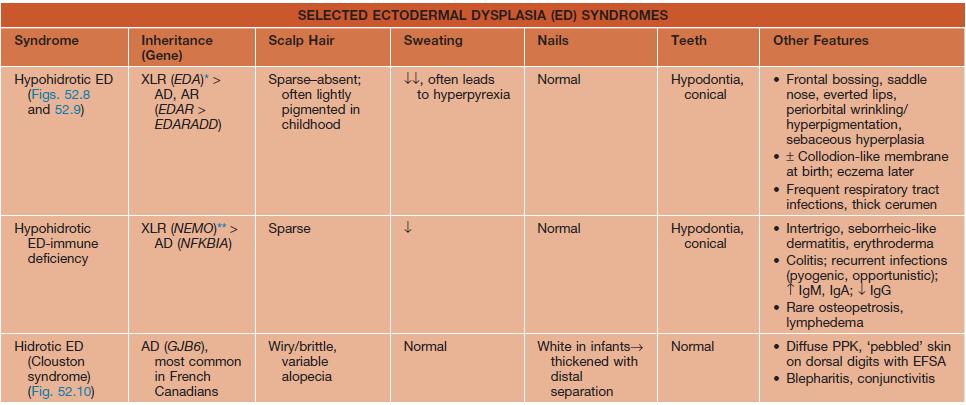
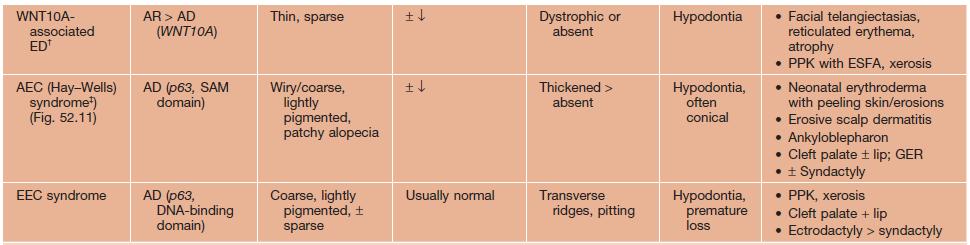
* Variable phenotype with mosaic pattern (e.g. hypohidrosis and hyperpigmentation along Blaschko’s lines) in female ‘carriers’ of XLR form.
** Female carriers may have mild features of incontinentia pigmenti (see Chapter 51).
† Includes odonto-onycho-dermal dysplasia and Schöpf–Schulz–Passarge syndrome, which also features eyelid hidrocystomas.
‡ Rapp–Hodgkin syndrome is now included in the AEC spectrum.
AD, autosomal dominant; AEC, ankyloblepharon-ED-clefting; AR, autosomal recessive; EDA, ectodysplasin A; EDAR, EDA receptor; EDARADD, EDAR-associated death domain; EEC, ED-ectrodactyly-clefting; ESFA, eccrine syringofibroadenomatosis; GER, gastroesophageal reflux; GJB6, gap junction β6 (encodes connexin 30); NEMO, NF-κB essential modulator; NFKBIA, NF-κB inhibitor-α; PPK, palmoplantar keratoderma; XLR, X-linked recessive.
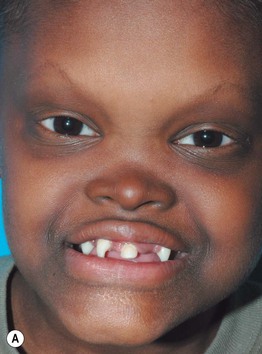
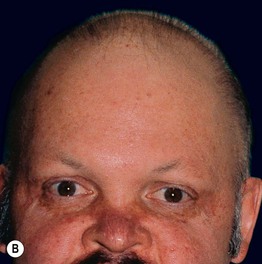
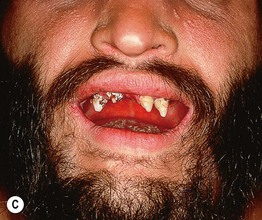
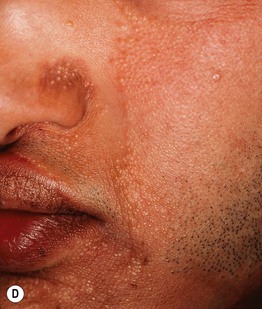
Fig. 52.8 Male patients with hypohidrotic ectodermal dysplasia. Note the flat nasal bridge, depressed nasal tip, sparse hair (scalp, eyebrows, eyelashes), peg-shaped teeth, full lips, and sebaceous hyperplasia. Also note the normal secondary hair in adults. A, Courtesy, Julie V. Schaffer, MD; B, D, Courtesy, Mary Williams, MD.
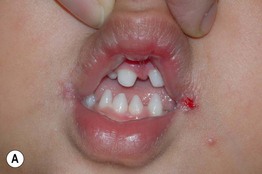

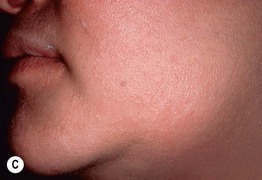
Fig. 52.9 Female patients with X-linked hypohidrotic ectodermal dysplasia. A Full lips, peg-shaped teeth, and hypodontia. B Periorbital hyperpigmentation. C Sebaceous hyperplasia. A, Courtesy, Julie V. Schaffer, MD.
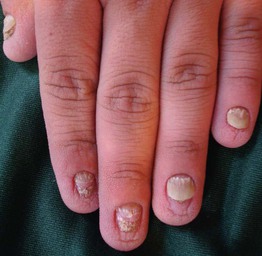
Fig. 52.10 Hidrotic ectodermal dysplasia (Clouston syndrome). Note the thickened, shortened nails with distal separation and the tiny papules in a regular distribution on the dorsal fingertips. Courtesy, Virginia Sybert, MD, and Alanna Bree, MD.
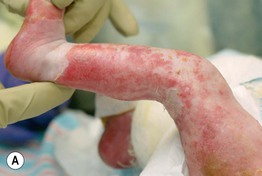
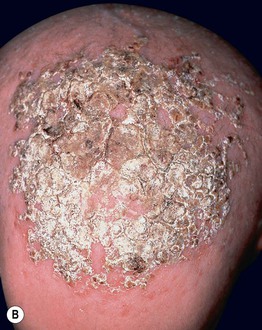
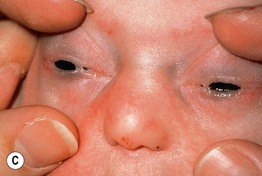
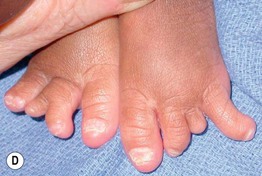
Fig. 52.11 Ankyloblepharon–ectodermal dysplasia–clefting syndrome. A Extensive erosions in an infant who succumbed to sepsis. B Chronic erosive scalp dermatitis with thick scale-crust and alopecia. C Strands of tissue between the eyelids (ankyloblepharon filiforme adnatum). D Digital abnormalities (including syndactyly) and nail dystrophy. A, C, D, Courtesy, Virginia Sybert, MD, and Alanna Bree, MD; B, Courtesy, Jean L. Bolognia, MD.
For further information see Ch. 63. From Dermatology, Third Edition.