Orthopaedic Pathology
section 1 Introduction
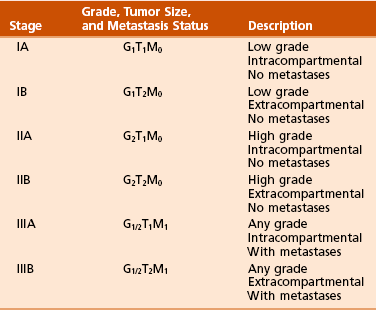
A Enneking system: This staging system can be synthesized into six distinct stages (Table 9-1).
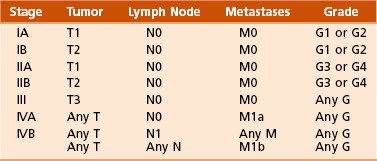
B Variables of AJCC system: The most recent edition of the AJCC system has become more popular than Enneking’s system among medical oncologists and many orthopaedic oncologists. A working knowledge of both systems is necessary for examinations. To use this system, the clinician must know the grade, the size, the presence or absence of discontinuous tumor (skip metastases), and the absence or presence of systemic metastases. The various stages are listed in Table 9-2. The order of importance for the variables of the AJCC staging system is as follows: stage (takes into account all factors), presence of metastases, discontinuous tumor, grade, and size.
Table 9-2
From American Joint Committee on Cancer: Bone. In Greene FL, et al, editor: AJCC cancer staging manual, New York, 2002, Springer-Verlag, pp 213-219.
A Most grading systems are based on three grades:
B The grade of the tumor is most strongly correlated with the potential for metastasis:
1. Grade I (low grade): less than 10% potential
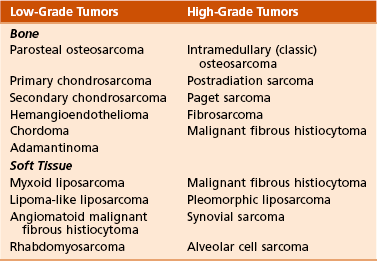
C Most malignant lesions are high grade (Enneking grade G2); low-grade malignant (Enneking grade G1) lesions are less common. Commonly graded lesions are shown in Table 9-3.
A Within the bone compartment (intracompartmental, or T1)
B Extending beyond the confines of the bone (extracompartmental, or T2)
A Chest radiograph and CT scan of the chest are obtained to search for pulmonary lesions.
B Technetium-labeled bone scan is obtained to exclude the presence of other bone lesions.
A Clinical presentation: Most patients with bone tumors present with musculoskeletal pain. However, the most common presentation of a benign bone tumor in childhood is as an incidental finding.
1. Pain is typically deep-seated and may resemble a that of a toothache.
2. Pain may initially be intermittent and related to activity, a work injury, or a sporting injury.
3. Pain usually progresses in intensity and becomes constant.
4. Patients experience pain at night.
5. Pain progresses, and it is not relieved by nonsteroidal anti-inflammatory drugs (NSAIDs) or weaker narcotics (such as acetaminophen [Tylenol] with codeine).
6. Patients with a high-grade sarcoma present with a 1- to 3-month history of pain.
7. With low-grade tumors, such as chondrosarcoma, adamantinoma, and chordoma, there may be a long history of mild to moderate pain (6 to 24 months).
B Physical examination: Patients with suspected bone tumors should be examined carefully.
1. Site is inspected for soft tissue masses, overlying skin changes, adenopathy, and general musculoskeletal condition.
2. When metastatic disease is suspected, the thyroid gland, abdomen, prostate, and breasts should be examined, as appropriate.
C Imaging studies: Radiographs in two planes are the first imaging studies to be performed. When the clinician suspects malignancy but the radiographs are normal, selected studies may follow.
1. Technetium-labeled bone scan is an excellent modality to search for occult bone involvement. In patients with myeloma for whom scan results may be negative, a skeletal survey is more sensitive.
2. MRI is an excellent modality for screening the spine for occult metastases, myeloma, or lymphoma.
3. A chest radiograph should be obtained for patients of any age when the clinician suspects a malignant lesion.
4. The radiographs must be carefully inspected to formulate a working diagnosis. The working diagnosis then guides the clinician during further evaluation and treatment. Formulation of the differential diagnosis is based on several clinical and radiographic parameters:
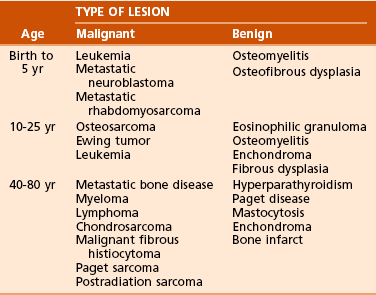
 Age of the patient: Knowledge of common diseases in defined age groups is the first step. Certain diseases are uncommon in particular age groups (Table 9-4).
Age of the patient: Knowledge of common diseases in defined age groups is the first step. Certain diseases are uncommon in particular age groups (Table 9-4).


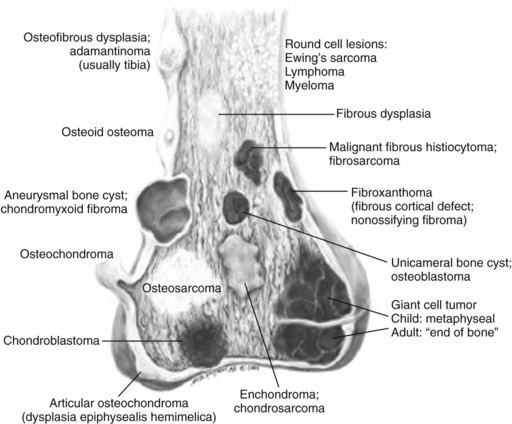
 Anatomic location within bone: Certain lesions have a predilection for occurring within a certain bone or a particular part of the bone (Figure 9-1; Table 9-5).
Anatomic location within bone: Certain lesions have a predilection for occurring within a certain bone or a particular part of the bone (Figure 9-1; Table 9-5).
Table 9-5
Most Common Musculoskeletal Tumors
| Tumor Type | Tumor Name |
| Soft tissue tumor (children) | Hemangioma |
| Soft tissue tumor (adults) | Lipoma |
| Malignant soft tissue tumor (children) | Rhabdomyosarcoma |
| Malignant soft tissue tumor (adults) | Malignant fibrous histiocytoma |
| Primary benign bone tumor | Osteochondroma |
| Primary malignant bone tumor | Osteosarcoma |
| Secondary benign lesion | Aneurysmal bone cyst |
| Secondary malignancies | Malignant fibrous histiocytoma Osteosarcoma Fibrosarcoma |
| Phalangeal tumor | Enchondroma |
| Sarcoma of the hand and wrist | Epithelioid sarcoma |
| Sarcoma of the foot and ankle | Synovial sarcoma |
Courtesy of Luke S. Choi, MD, Resident, Department of Orthopaedic Surgery, University of Virginia.

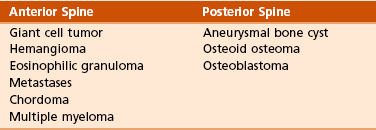
 Typical neoplasms of whole bone:
Typical neoplasms of whole bone:


 Typical neoplasms of part of bone (Table 9-6):
Typical neoplasms of part of bone (Table 9-6):
Table 9-6
Courtesy of Luke S. Choi, MD, Resident, Department of Orthopaedic Surgery, University of Virginia.




D Laboratory studies: Results of blood tests are often nonspecific. A set of routine studies should be obtained when the diagnosis is not obvious. Certain studies are appropriate for younger patients (up to age 40) and others for older patients (40 to 80 years; Box 9-1).
E Biopsy: Biopsy is generally performed after complete evaluation of the patient. It is of great benefit to both the pathologist and the surgeon to have a narrow working diagnosis because it allows accurate interpretation of the frozen-section analysis, and definitive treatment of some lesions can be based on the frozen section. Clinicians must follow several surgical principles:
1. The orientation and location of the biopsy tract are critical. If the lesion proves to be malignant, the entire biopsy tract must be removed with the underlying lesion. Transverse incisions should be avoided.
2. The surgeon must maintain meticulous hemostasis to prevent hematoma formation and subcutaneous hemorrhage. When possible, biopsy incisions are made through muscles so that the muscle layer can be closed tightly. Neurovascular structures should be avoided. Tourniquets are used to obtain tissue in a bloodless field and then are released so that bleeding points can be controlled. Avitene, Gelfoam, and Thrombostat sprays are used as necessary. If hemostasis cannot be achieved, a small drain should be placed at the corner of the wound to prevent hematoma formation. A compression dressing is routinely used on the extremities.
3. A frozen-section analysis is performed on all biopsy samples to ensure that adequate diagnostic tissue is obtained. Before biopsy, the surgeon should review the radiographs with the pathologist to plan the biopsy site. When possible, the soft tissue component rather than the bony component should be sampled.
4. All biopsy samples should be submitted for bacteriologic analysis if the frozen section does not reveal a neoplasm. Antibiotics should not be delivered until the cultures are obtained.
5. Needle biopsy is an excellent method for achieving a tissue diagnosis and providing minimum tissue disruption. Careful correlation of the small tissue sample with the radiographs often yields the correct diagnosis. When the nature of the lesion is obvious on the basis of the radiographic features and when adequate tissue can be obtained with a needle, the needle biopsy technique is safe to use. The pathologist must be experienced and comfortable with the small sample of tissue. When the diagnoses of needle biopsy and imaging studies are not concordant, an open biopsy should be performed to establish the diagnosis. Open biopsy is often necessary with low-grade tumors and when the needle biopsy does not provide a definitive diagnosis. Immunostains are helpful in diagnosis (Table 9-7).
Table 9-7
| Tumor | Immunostain |
| Langerhans cell histiocytosis | S100, +CD1A |
| Lymphoma | +CD20 |
| Ewing sarcoma | +CD99 |
| Chordoma | Keratin, S100 |
| Myeloma | +CD138 |
| Adamantinoma | Keratin |
Courtesy of Luke S. Choi, MD, Resident, Department of Orthopaedic Surgery, University of Virginia.
A Surgical procedures: The goal of the treatment of malignant bone tumors is to remove the lesion with minimal risk of local recurrence.
1. Limb salvage is performed when two essential criteria are met:


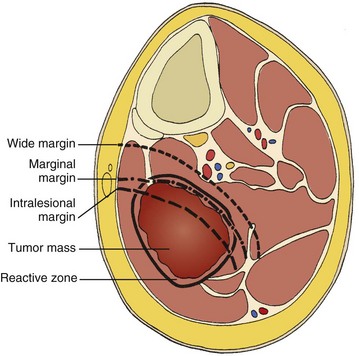
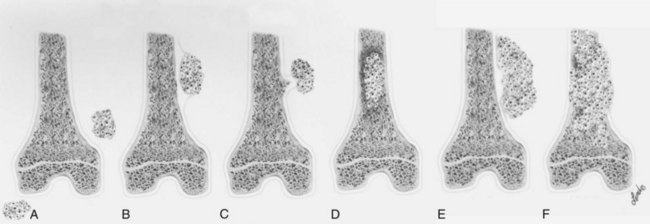
2. Surgical margins are graded according to the system of the Musculoskeletal Tumor Society (Figure 9-2).




 Multiagent chemotherapy has a significant effect on both the efficacy of limb salvage and disease-free survival for osteogenic sarcoma and Ewing tumor.
Multiagent chemotherapy has a significant effect on both the efficacy of limb salvage and disease-free survival for osteogenic sarcoma and Ewing tumor.
 The common mechanism of action of drugs is the induction of programmed cell death (apoptosis).
The common mechanism of action of drugs is the induction of programmed cell death (apoptosis).

 Patients undergo maintenance chemotherapy for 6 to 12 months.
Patients undergo maintenance chemotherapy for 6 to 12 months.

 The role of chemotherapy for soft tissue sarcoma remains more controversial.
The role of chemotherapy for soft tissue sarcoma remains more controversial.
 External beam irradiation is used in the following scenarios:
External beam irradiation is used in the following scenarios:
 For local control of Ewing tumor, lymphoma, myeloma, and metastatic bone disease
For local control of Ewing tumor, lymphoma, myeloma, and metastatic bone disease


 Postirradiation sarcoma: This is a devastating complication in which a spindle sarcoma occurs within the field of irradiation for a previous malignancy (e.g., Ewing tumor, breast cancer, Hodgkin disease). The histologic features are usually those of an osteosarcoma, a fibrosarcoma, or a malignant fibrous histiocytoma. Postirradiation sarcomas are probably more frequent in patients who undergo intensive chemotherapy (especially with alkylating agents) and irradiation.
Postirradiation sarcoma: This is a devastating complication in which a spindle sarcoma occurs within the field of irradiation for a previous malignancy (e.g., Ewing tumor, breast cancer, Hodgkin disease). The histologic features are usually those of an osteosarcoma, a fibrosarcoma, or a malignant fibrous histiocytoma. Postirradiation sarcomas are probably more frequent in patients who undergo intensive chemotherapy (especially with alkylating agents) and irradiation.

Several bone and soft tissue neoplasms have been associated with tumor suppressor genes or specific genetic defects. For osteosarcoma, the associations are the retinoblastoma tumor suppressor gene. For Ewing tumor, there is a balanced translocation of chromosomes 11 and 22. There is a gene fusion product from this balanced translocation: EWS-FLI1 (Table 9-8).
Table 9-8
Common Tumor-Associated Genetic Translocations
| Tumor Type | Genetic Translocation |
| Myxoid liposarcoma | t(12;16) |
| Ewing sarcoma | t(11;22) |
| Synovial sarcoma | t(X;18) |
| Myxoid chondrosarcoma | t(9;22) |
| Rhabdomyosarcoma | t(1;13) or t(2;13) |
Courtesy of Luke S. Choi, MD, Fellow, Orthopaedic Sports Medicine, Massachusetts General Hospital.
section 2 Soft Tissue Tumors
Soft tissue tumors are common. They may appear as small lumps or large masses.
A Classification: Soft tissue tumors can be broadly classified as benign or malignant (sarcoma) or characterized by reactive tumor-like conditions (Box 9-2). Lesions are classified according to the direction of differentiation of the lesion: the tumor tends to produce collagen (fibrous lesion), fat, or cartilage.
1. Benign soft tissue tumors: These tumors may occur in all age groups. The biologic behavior of these lesions varies from asymptomatic and self-limiting to growing and symptomatic. On occasion, benign lesions grow rapidly and invade adjacent tissues.
2. Malignant soft tissue tumors (sarcomas): Sarcomas are rare tumors of mesenchymal origin. In the United States, there are approximately 9000 new cases of soft tissue sarcoma each year.
 Diagnosis: Patients often experience an enlarging painless or painful soft tissue mass, which is the most common reason for seeking medical attention.
Diagnosis: Patients often experience an enlarging painless or painful soft tissue mass, which is the most common reason for seeking medical attention.
 Most sarcomas are large (>5 cm), deep, and firm.
Most sarcomas are large (>5 cm), deep, and firm.

 Initial radiographic evaluation begins with radiographs in two planes.
Initial radiographic evaluation begins with radiographs in two planes.





B Diagnosis: The evaluation of patients with soft tissue tumors must be systematic to avoid errors.
1. Unplanned removal of a soft tissue sarcoma is the most common error.
 Residual tumor may exist at the site of the operative wound, and repeat excision for all patients with an unplanned removal should be performed.
Residual tumor may exist at the site of the operative wound, and repeat excision for all patients with an unplanned removal should be performed.
2. Delay in diagnosis may also occur if the clinician does not recognize that the lesion is malignant.
3. Patients who have a new soft tissue mass or one that is growing or causing pain should undergo MRI.
4. The MRI scan should be carefully reviewed with a radiologist to characterize the nature of the mass. If it can be determined that the lesion is a benign process such as a lipoma, ganglionic cyst, or muscle tear, then it is classified as a determinate lesion, and treatment can be planned without a biopsy. In contrast, if the exact nature of a lesion cannot be determined, the lesion is classified as indeterminate, and either a needle or open biopsy is necessary to determine the exact diagnosis. Then treatment can be planned.
5. Excisional biopsy should not be performed when the clinician does not know the origin of a soft tissue tumor.
C Metastasis: Most soft tissue sarcomas metastasize to the lung.
A Calcifying aponeurotic fibroma
1. Manifests as a slow-growing, painless mass in the hands and feet in children and young adults 3 to 30 years of age.
2. Radiographs may reveal a faint mass with stippling.
3. Histologic examination reveals a fibrous tumor with centrally located areas of calcification and cartilage formation.
4. After local excision, the tumor often recurs (in up to 50% of cases); however, the condition appears to resolve with maturity.
1. Palmar (Dupuytren) and plantar (Ledderhose) fibromatosis: These disorders consist of firm nodules of fibroblasts and collagen that develop in the palmar and plantar fascia. The nodules and fascia become hypertrophic, producing contractures.
2. Extraabdominal desmoid tumor
 Most locally invasive of all benign soft tissue tumors.
Most locally invasive of all benign soft tissue tumors.
 Commonly occurs in adolescents and young adults.
Commonly occurs in adolescents and young adults.
 Patients with Gardner syndrome (familial adenomatous polyposis) have colonic polyps and a 10,000-fold increased risk of developing desmoid tumors.
Patients with Gardner syndrome (familial adenomatous polyposis) have colonic polyps and a 10,000-fold increased risk of developing desmoid tumors.
 On palpation, the tumor has a distinctive “rock-hard” character.
On palpation, the tumor has a distinctive “rock-hard” character.
 Multiple lesions may be present in the same extremity (10% to 25%).
Multiple lesions may be present in the same extremity (10% to 25%).

 Surgical treatment is aimed at resecting the tumor with a wide margin.
Surgical treatment is aimed at resecting the tumor with a wide margin.

 Radiotherapy has been used as an adjunctive treatment to prevent recurrence and progression.
Radiotherapy has been used as an adjunctive treatment to prevent recurrence and progression.

1. A common reactive lesion that manifests as a painful, rapidly enlarging mass in a young person (15 to 35 years of age).
2. Half of these lesions occur in the upper extremity.
3. Short, irregular bundles and fascicles; a dense reticulum network; and only small amounts of mature collagen characterize the lesion histologically. Mitotic figures are common, but atypical mitoses are not a feature.
4. Treatment consists of excision with a marginal line of resection.
D Malignant fibrous soft tissue tumors: Malignant fibrous histiocytoma and fibrosarcoma are the two malignant fibrous lesions.
 Similar clinical and radiographic manifestations; treatment methods are similar
Similar clinical and radiographic manifestations; treatment methods are similar
 Patients are generally between the ages of 30 and 80 years.
Patients are generally between the ages of 30 and 80 years.
 Most common manifestation is an enlarging, generally painless mass.
Most common manifestation is an enlarging, generally painless mass.






E Dermatofibrosarcoma protuberans
1. Rare, nodular, cutaneous tumor that occurs in early to middle adulthood
2. Low grade, with a tendency to recur locally, but it only rarely metastasizes (often after repeated local recurrence)
3. In 40% of the cases, it occurs on the upper or lower extremities. The tumor grows slowly but progressively.
4. The central portion of the nodules shows uniform fibroblasts arranged in a storiform pattern around an inconspicuous vasculature.
5. Wide-margin surgical resection is the best form of treatment.
A Lipomas: common benign tumors of mature fat
1. Occur in a subcutaneous, intramuscular, or intermuscular location
2. History of a mass is long, but sometimes the mass was only recently discovered.
4. Radiographs may show a radiolucent lesion in the soft tissues if the lipoma is deep within the muscle or between the muscle and bone.
5. CT scan or MRI shows a well-demarcated lesion with the same signal characteristics as those of mature fat on all sequences. On fat suppression sequences, the lipoma has a uniformly low signal. If the patient experiences no symptoms and the radiographic features are diagnostic of lipoma, no treatment is necessary.
6. If the mass is growing or causing symptoms, excision with a marginal line of resection or an intralesional margin is all that is necessary.
7. Local recurrence is uncommon.

 Commonly occurs in men (45 to 65 years of age)
Commonly occurs in men (45 to 65 years of age)














1. Type of sarcoma; direction of differentiation is toward fatty tissue
2. Heterogeneous group of tumors, having in common the presence of lipoblasts (signet ring–shaped cells) in the tissue
3. Liposarcomas virtually never occur in the subcutaneous tissues.
4. They are classified into the following types:
 Well-differentiated liposarcoma (low grade)
Well-differentiated liposarcoma (low grade)



 Myxoid liposarcoma (intermediate grade)
Myxoid liposarcoma (intermediate grade)



5. Liposarcomas metastasize according to the grade of the lesion:



A Neurilemoma (benign schwannoma)
2. Occurs in young to middle-aged adults (20 to 50 years of age)
3. Patients have no symptoms except for the presence of the mass.
4. Tumor grows slowly and may wax and wane in size (cystic changes).
5. MRI studies may demonstrate an eccentric mass arising from a peripheral nerve, or they may show only an indeterminate soft tissue mass (low signal on T1-weighted images and high signal on T2-weighted images).
6. Histologically, the lesion is composed of Antoni A and B areas.

 Compact spindle cells usually having twisted nuclei; indistinct cytoplasm; and occasionally clear, intranuclear vacuoles
Compact spindle cells usually having twisted nuclei; indistinct cytoplasm; and occasionally clear, intranuclear vacuoles
 There may be nuclear palisading, whorling of cells, and Verocay bodies.
There may be nuclear palisading, whorling of cells, and Verocay bodies.
 When the lesion is predominantly cellular (Antoni A), the tumor may be confused with a sarcoma.
When the lesion is predominantly cellular (Antoni A), the tumor may be confused with a sarcoma.
 Treatment: removing the eccentric mass while leaving the nerve intact
Treatment: removing the eccentric mass while leaving the nerve intact




1. Solitary or multiple (neurofibromatosis)
2. Superficial, slow-growing, and painless
3. When they involve a major nerve, they may expand it in a fusiform manner.
4. Histologic study shows interlacing bundles of elongated cells with wavy, dark-staining nuclei.
5. Cells are associated with wirelike strands of collagen.
6. Small to moderate amounts of mucoid material separate the cells and collagen
C Neurofibromatosis (von Recklinghausen disease)
1. Autosomal dominant trait (both peripheral and central forms)
2. Café au lait spots (smooth) and Lisch nodules (melanocytic hamartomas in the iris)
3. Variable skeletal abnormalities (metaphyseal fibrous defect [nonossifying fibroma], scoliosis, and long-bone bowing)
4. Malignant changes occur in 5% to 30% of affected patients.
5. Pain and an enlarging soft tissue mass may herald conversion to a sarcoma.
1. Manifests as a small nodule or a large extremity mass
1. The most common sarcoma in young patients; may grow rapidly
2. Composed of spindle cells in parallel bundles, multinucleated giant cells, and racquet-shaped cells
3. Cross-striations within the tumor cells (rhabdomyoblasts)
4. Rhabdomyosarcomas are sensitive to multiagent chemotherapy and wide-margin surgical resection after induction of chemotherapy. External beam irradiation plays a prominent role in treatment.
1. Commonly seen in children and adults
2. Cutaneous, subcutaneous, or intramuscular location
3. Large tumors have signs of vascular engorgement (aching, heaviness, swelling).
4. MRI scans demonstrate a heterogeneous lesion with numerous small blood vessels and fatty infiltration.
5. It is important to examine the patient in both the supine and standing positions. (The lower extremity often fills with blood after several minutes).
6. Radiographs may reveal small phleboliths.
7. Nonoperative treatment: NSAIDs, vascular stockings, and activity modification if local measures adequately control discomfort
8. Can be treated by application of a sclerosing agent such as alcohol
1. Out-pouching of the synovial lining of an adjacent joint
2. Common locations include the wrist, foot, and knee.
3. Filled with gelatinous, mucoid material
4. Paucicellular connective tissue without a true epithelial lining
5. MRI: homogeneously low signal on T1-weighted images and a very bright signal on T2-weighted images; contrast agent such as gadolinium is useful in differentiating a cyst from a solid neoplasm because cysts do not enhance (except for a small rim at the periphery) but active neoplasms usually do.
B Pigmented villonodular synovitis (PVNS)
1. Reactive condition (not a true neoplasm) characterized by an exuberant proliferation of synovial villi and nodules
2. May occur locally (within a joint) or diffusely
3. The knee is affected most often, followed in frequency by the hip and shoulder.
4. Manifests with pain and swelling in the affected joint
5. Recurrent, atraumatic hemarthrosis is the hallmark (arthrocentesis demonstrates a bloody effusion).
6. Cystic erosions may occur on both sides of the joint.
7. Highly vascular villi are lined with plump, hyperplastic synovial cells; hemosiderin-stained, multinucleated giant cells; and chronic inflammatory cells.
8. Treatment is aimed at complete synovectomy by arthroscopy for resection of all the intraarticular disease, followed by open posterior synovectomy to remove the posterior extraarticular extension.

9. Local recurrence is common (30% to 50% of cases) despite complete synovectomy.
10. External beam irradiation (3500 to 4000 cGy) can reduce the rate of local recurrence to 10% to 20%.
C Giant cell tumor of tendon sheath
1. Benign nodular tumor occurs along the tendon sheaths (hands/feet).
2. Moderately cellular (sheets of rounded or polygonal cells) zones; hypocellular, collagenized zones; multinucleated giant cells are common, as are xanthoma cells.
3. Treatment: resection with a marginal margin
4. Local recurrence is common (usually treated with repeat excision).
1. Synovial proliferative disorder that occurs within joints or bursae, ranging in appearance from metaplasia of the synovial tissue to firm nodules of cartilage
2. Typically affects young adults, who present with pain, stiffness, and swelling
3. The knee is the most common location.
4. Radiographs may demonstrate fine, stippled calcification.
1. Highly malignant, high-grade tumor that occurs near joints, most commonly around the knee
2. Although the name implies that it arises from synovial cells, it rarely arises from an intraarticular location.

3. May be present for years or may manifest as a rapidly enlarging mass
4. Lymph nodes may be involved.
5. Most common synovial sarcoma is in the foot.
6. Radiographs or CT scans may show mineralization within the lesion in up to 25% of cases (spotty mineralization may even resemble the peripheral mineralization seen in heterotopic ossification).

7. The tumor is often biphasic, with both epithelial and spindle cell components.
 The epithelial component may show epithelial cells that form glands or nests, or they may line cystlike spaces.
The epithelial component may show epithelial cells that form glands or nests, or they may line cystlike spaces.
9. The tumor may also be composed of a single type of cell (monophasic); the monophasic fibrous type is much more common than the monophasic epithelial type.
10. Translocation between chromosome 18 and the X chromosome—t(X;18)—is always present in tumor cells, and staining of the tumor cells yields positive results for keratin and epithelial membrane antigen.
11. The balanced translocation results in gene fusion products. The two most common are SYT-SSX1 and SYT-SSX2.
12. Wide-margin surgical resection with adjuvant radiotherapy is the most common method of treatment.
13. Metastases develop in 30% to 60% of cases.
14. Larger tumors (>5 to 10 cm) are more prone to distant spread.
1. Rare nodular tumor that commonly occurs in the upper extremities of young adults
2. May also occur about the buttock/thigh, knee, and foot
3. The most common sarcoma of the hand
4. May ulcerate and mimic a granuloma or rheumatoid nodule
5. Lymph node metastases are common.
6. Cells range in shape from ovoid to polygonal, with deeply eosinophilic cytoplasm (cellular pleomorphism is minimal).
7. Often misdiagnosed as benign processes.
8. Wide-margin surgical resection is necessary to prevent local recurrence.
1. Manifests as a slow-growing mass in association with tendons or aponeuroses
2. Usually occurs about the foot and ankle but may also involve the knee, thigh, and hand
3. Characterized by compact nests or fascicles of rounded or fusiform cells with clear cytoplasm; multinucleated giant cells are common
4. Wide-margin surgical resection with adjuvant irradiation is the treatment of choice.
1. Manifests as a slow-growing, painless mass in young adults (15 to 35 years of age)
2. Occurs in the anterior thigh
3. Dense, fibrous trabeculae dividing the tumor into an organoid or nestlike arrangement; cells are large and rounded and contain one or more vesicular nuclei with small nucleoli
4. Treatment: wide-margin surgical resection with adjuvant irradiation in selected cases
1. Hematoma may occur after trauma to the extremity.
2. Organizes and resolves with time
3. Sarcomas may spontaneously hemorrhage into the body of the tumor or after minor trauma and masquerade as a benign process. A lack of fascial plane tracking and subcutaneous ecchymosis suggests that the bleeding is contained by a pseudocapsule; this is an important physical examination finding.
4. Clinicians should monitor patients with hematomas at 6-week intervals until the mass resolves.
5. MRI scanning is often not able to distinguish a simple hematoma from a sarcoma with spontaneous hemorrhage.
B Myositis ossificans (heterotopic ossification)
1. Develops after single or repetitive episodes of trauma (occasionally, patients cannot recall the traumatic episode)
2. Most common locations are over the diaphyseal segment of long bones (in the middle aspect of the muscle bellies).
3. As maturation progresses, radiographs show peripheral mineralization with a central lucent area.
4. Lesion is not attached to the underlying bone, but in some cases, it may become fixed to the periosteal surface.
5. Zonal pattern, with mature, trabecular bone at the periphery and immature tissue in the center
6. Nonoperative treatment is all the management that is necessary.
section 3 Bone Tumors
A common classification system for bone tumors is shown in Table 9-9.
Table 9-9
Classification of Primary Tumors of Bone*
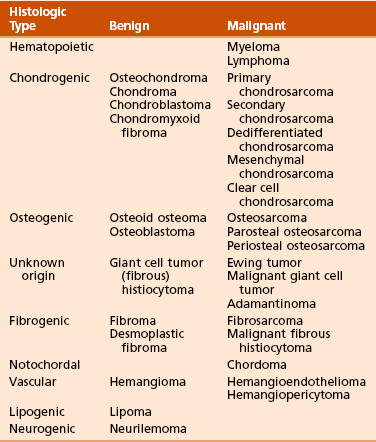
*Classification is based on that advocated by Lichtenstein L: Classification of primary tumors of bone, Cancer 4:335-341, 1951.
1. Malignant neoplasms of connective tissue (mesenchymal) origin
2. Exhibit rapid growth in a centripetal manner and invade adjacent normal tissues
3. Each year in the United States, about 2800 new bone sarcomas are diagnosed.
 Malignant bone tumors manifest most commonly with pain. This is in contrast to soft tissue tumors, which most commonly manifest as a painless mass.
Malignant bone tumors manifest most commonly with pain. This is in contrast to soft tissue tumors, which most commonly manifest as a painless mass.
4. High-grade, malignant bone tumors tend to destroy the overlying cortex and spread into the soft tissues.
5. Low-grade tumors are generally contained within the cortex or the surrounding periosteal rim.
6. Bone sarcomas metastasize primarily via the hematogenous route; the lungs are the most common site.
7. Osteosarcoma and Ewing sarcoma may also metastasize to other bone sites either at the initial manifestation or later in the disease.
Self-limiting benign bone lesion produces pain in young patients (5 to 30 years of age)
 Pain that increases with time, pain at night
Pain that increases with time, pain at night


 May produce painful nonstructural scoliosis, growth disturbances, and flexion contractures
May produce painful nonstructural scoliosis, growth disturbances, and flexion contractures
 Scoliosis caused by an osteoid osteoma results in a curve with the lesion on the concave side. This is thought to result fromj marked paravertebral muscle spasm.
Scoliosis caused by an osteoid osteoma results in a curve with the lesion on the concave side. This is thought to result fromj marked paravertebral muscle spasm.
 Common locations include the proximal femur, tibial diaphysis, and spine (Figure 9-3).
Common locations include the proximal femur, tibial diaphysis, and spine (Figure 9-3).
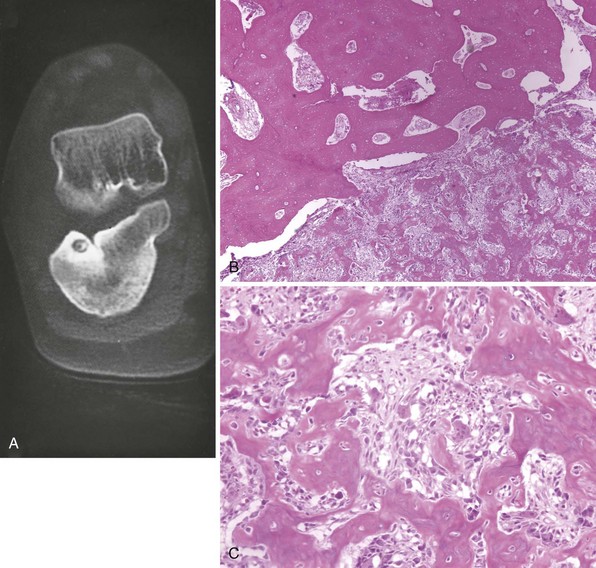
 Radiographs usually show intensely reactive bone and a radiolucent nidus (Figure 9-4). It may be possible to detect the lesion only with tomograms, CT scans, or MRI scans, because of the intense sclerosis.
Radiographs usually show intensely reactive bone and a radiolucent nidus (Figure 9-4). It may be possible to detect the lesion only with tomograms, CT scans, or MRI scans, because of the intense sclerosis.

 Technetium-labeled bone scans always yield positive findings and show intense focal uptake.
Technetium-labeled bone scans always yield positive findings and show intense focal uptake.


2. Patients can be treated with three different methods: NSAIDs, CT scan–guided radiofrequency ablation, and open surgical removal.




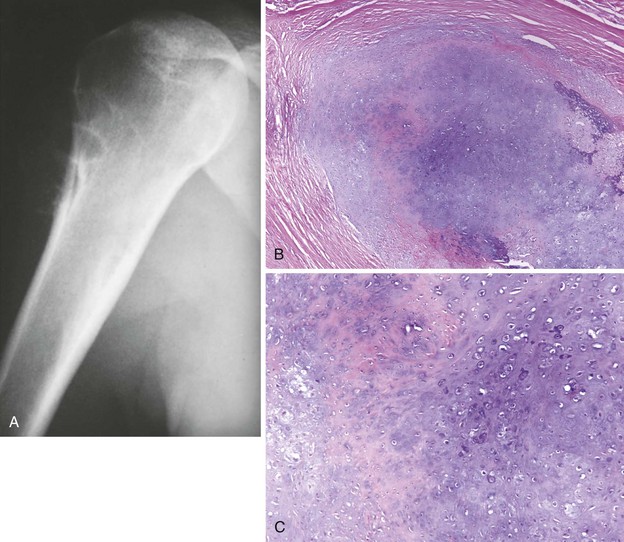
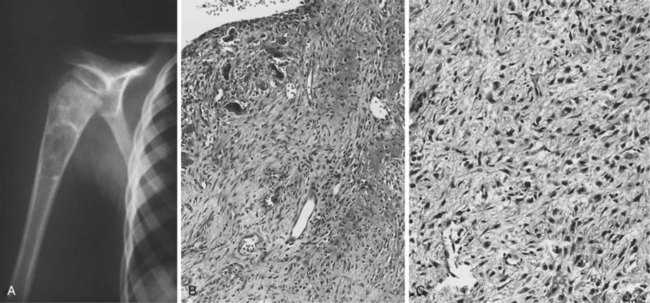
1. Rare bone-producing tumor that can attain a large size and is not self-limiting
2. Manifests with pain, and when the lesion involves the spine, neurologic symptoms may be present
3. Common locations include the spine, proximal humerus, and hip (Figure 9-5).
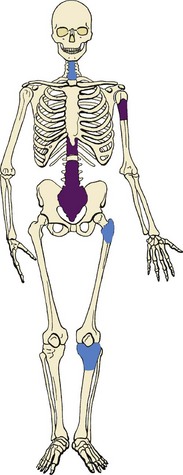
4. Causes bone destruction, with or without the characteristic reactive bone formation in osteoid osteoma
5. Area of bone destruction occasionally has a moth-eaten or permeative appearance simulating a malignancy.
6. Lesions show regularly shaped nuclei containing little chromatin but abundant cytoplasm (tissue is loosely arranged, with numerous blood vessels).
7. Tumor does not permeate the normal trabecular bone but instead merges with it.
8. Treatment: curettage or excision with a marginal line of resection
1. Spindle cell neoplasms that produce osteoid are arbitrarily classified as osteosarcoma.
2. There are many types of osteosarcoma (Box 9-3 and Figure 9-6).
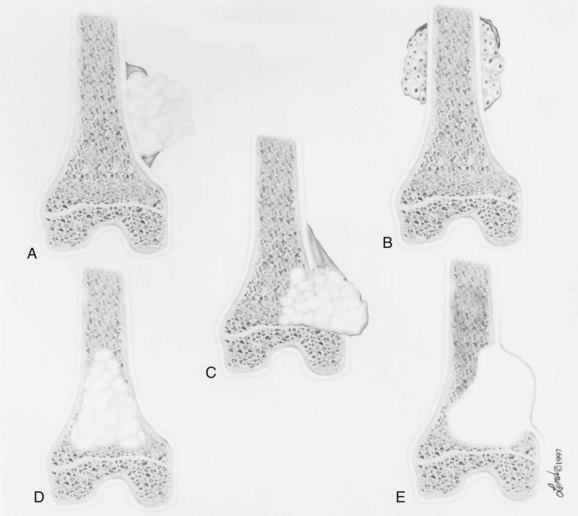
3. Lesions that must be recognized include high-grade intramedullary osteosarcoma (ordinary or classic osteosarcoma), parosteal osteosarcoma, periosteal osteosarcoma, telangiectatic osteosarcoma, osteosarcoma occurring with Paget disease, and osteosarcoma after irradiation.
4. Historically, osteosarcoma was treated by amputation; long-term studies demonstrated a survival rate of only 10% to 20%, the pulmonary system being the most common site of failure.
5. Multiagent chemotherapy has dramatically improved long-term survival and the potential for limb salvage.
 Doxorubicin (cardiac toxicity)
Doxorubicin (cardiac toxicity)
 Methotrexate (for cases of myelosuppression, also administer leucovorin)
Methotrexate (for cases of myelosuppression, also administer leucovorin)
6. Chemotherapy both kills the micrometastases that are present in 80% to 90% of the patients at presentation and sterilizes the reactive zone around the tumor.
7. Preoperative chemotherapy is delivered for 8 to 12 weeks, followed by resection of the tumor.

8. Rate of long-term survival is approximately 60% to 70%.
9. Prognostic factors that adversely affect survival include (1) expression of P-glycoprotein, high serum level of alkaline phosphatase, high lactic dehydrogenase level, vascular invasion, and no alteration of DNA ploidy after chemotherapy and (2) the absence of anti–shock protein-90 antibodies after chemotherapy.
10. Osteosarcoma is associated with an abnormality in the tumor suppressor genes Rb (retinoblastoma) and p53 (Li-Fraumeni syndrome).
 High-grade intramedullary osteosarcoma
High-grade intramedullary osteosarcoma
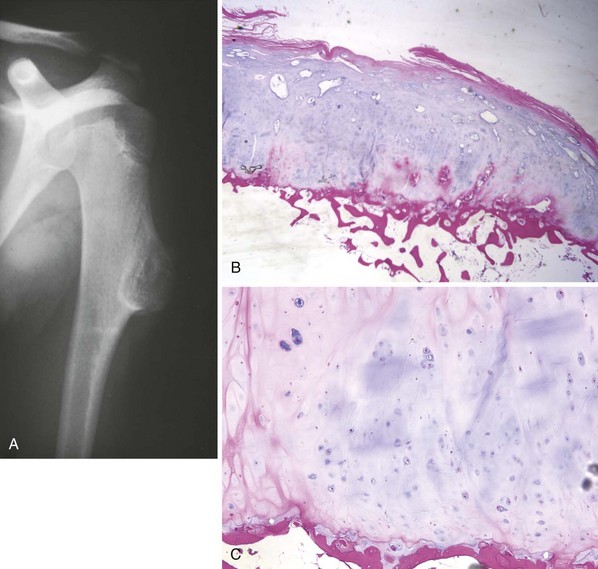
 Also called “ordinary” or “classic” osteosarcoma, this neoplasm is the most common type of osteosarcoma and usually occurs about the knee in children and young adults (Figure 9-7), but its incidence has a second peak in late adulthood.
Also called “ordinary” or “classic” osteosarcoma, this neoplasm is the most common type of osteosarcoma and usually occurs about the knee in children and young adults (Figure 9-7), but its incidence has a second peak in late adulthood.
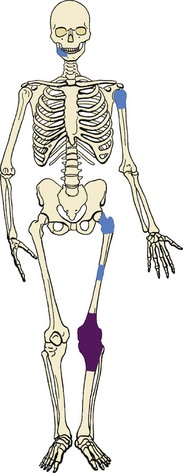
 Other common sites include the proximal humerus, proximal femur, and pelvis.
Other common sites include the proximal humerus, proximal femur, and pelvis.
 Patients present primarily with pain.
Patients present primarily with pain.

 About 10% to 20% of affected patients have pulmonary metastases at presentation.
About 10% to 20% of affected patients have pulmonary metastases at presentation.
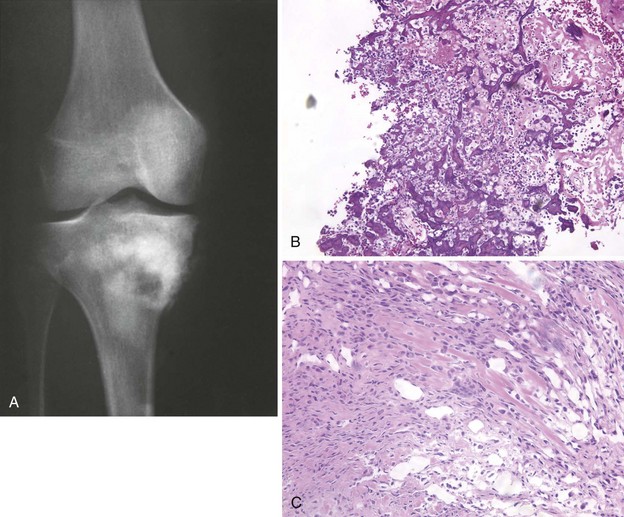
 Radiographs demonstrate a lesion in which there is bone destruction and bone formation (Figure 9-8). On occasion, the lesion is purely sclerotic or lytic. MRI and CT scans are useful for defining the anatomy of the lesion with regard to intramedullary extension, involvement of neurovascular structures, and muscle invasion.
Radiographs demonstrate a lesion in which there is bone destruction and bone formation (Figure 9-8). On occasion, the lesion is purely sclerotic or lytic. MRI and CT scans are useful for defining the anatomy of the lesion with regard to intramedullary extension, involvement of neurovascular structures, and muscle invasion.




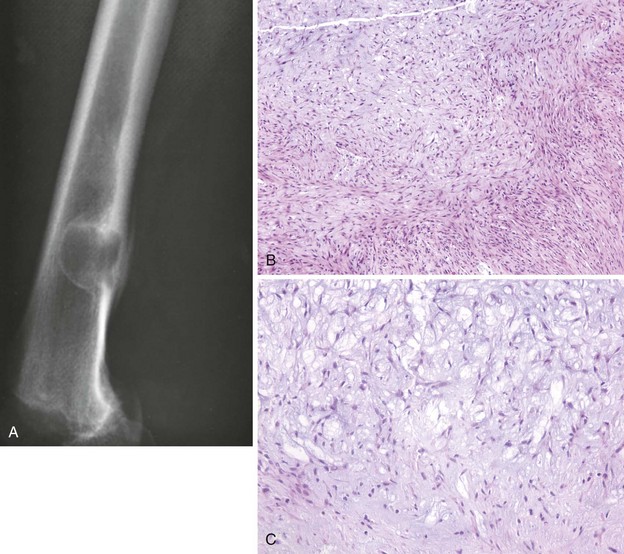
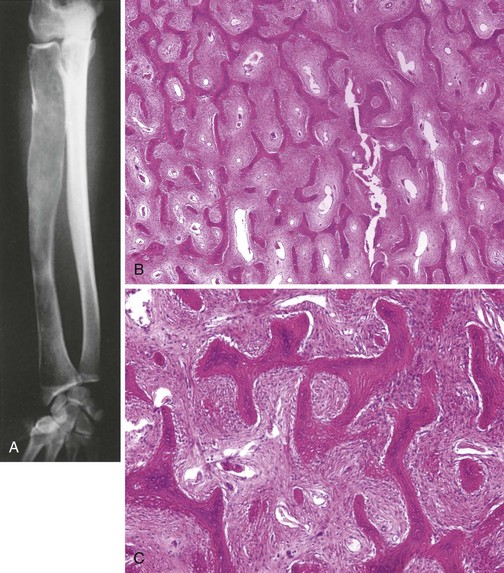
 Low-grade osteosarcoma that occurs on the surface of the metaphysis of long bones
Low-grade osteosarcoma that occurs on the surface of the metaphysis of long bones
 Affected patients often present with a painless mass.
Affected patients often present with a painless mass.
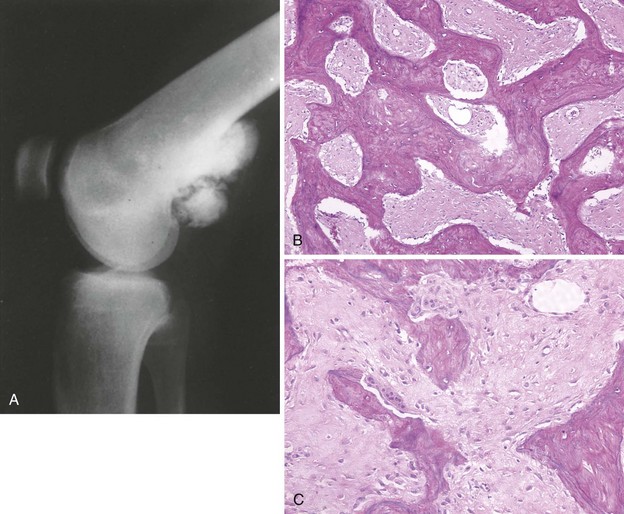
 Most common sites are the posterior aspect of the distal femur, proximal tibia, and proximal humerus (Figure 9-9).
Most common sites are the posterior aspect of the distal femur, proximal tibia, and proximal humerus (Figure 9-9).
 Characteristic radiographic appearance: a heavily ossified, often lobulated mass arising from the cortex (Figure 9-10)
Characteristic radiographic appearance: a heavily ossified, often lobulated mass arising from the cortex (Figure 9-10)

 Treatment: resection with a wide margin, which is usually curative
Treatment: resection with a wide margin, which is usually curative



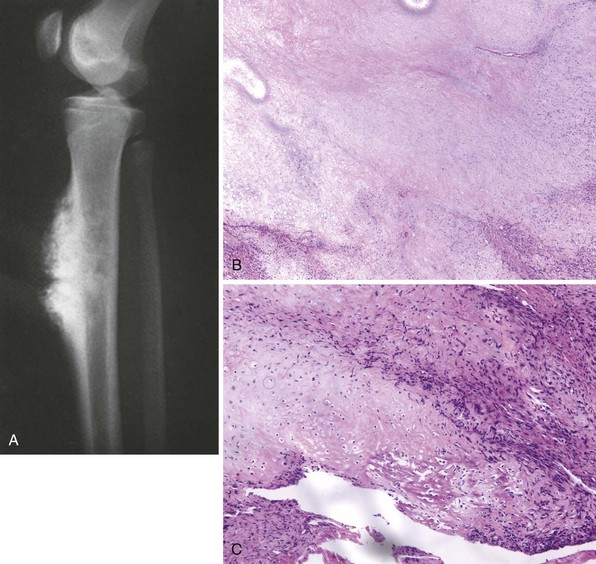
 Rare surface form of osteosarcoma occurs most often in the diaphysis of long bones (typically the femur or tibia; Figure 9-11).
Rare surface form of osteosarcoma occurs most often in the diaphysis of long bones (typically the femur or tibia; Figure 9-11).
 Radiographic appearance is fairly constant: a sunburst-type lesion rests on a saucerized cortical depression (Figure 9-12).
Radiographic appearance is fairly constant: a sunburst-type lesion rests on a saucerized cortical depression (Figure 9-12).



 The tissue of the lesion can be described as a bag of blood with few cellular elements.
The tissue of the lesion can be described as a bag of blood with few cellular elements.
 The radiographic features of telangiectatic osteosarcoma are those of a destructive, lytic, expansile lesion. Telangiectatic osteosarcomas occur in the same locations as aneurysmal bone cysts (Figure 9-13), and the radiographic appearances of both can be confused.
The radiographic features of telangiectatic osteosarcoma are those of a destructive, lytic, expansile lesion. Telangiectatic osteosarcomas occur in the same locations as aneurysmal bone cysts (Figure 9-13), and the radiographic appearances of both can be confused.
Appearances of these lesions are shown in Figure 9-14.
1. Histologic and radiographic features
 When benign cartilage tumors occur on the surface of the bone, they are called periosteal chondroma
When benign cartilage tumors occur on the surface of the bone, they are called periosteal chondroma
 They occur on the surfaces of the distal femur, proximal humerus, and proximal femur (Figure 9-15).
They occur on the surfaces of the distal femur, proximal humerus, and proximal femur (Figure 9-15).
 Appearance: usually a well-demarcated, shallow cortical defect and a slight periosteal chondroma
Appearance: usually a well-demarcated, shallow cortical defect and a slight periosteal chondroma
 Buttress of cortical bone at the edges of the lesion
Buttress of cortical bone at the edges of the lesion
 One third of the periosteal chondromas exhibit a mineralized cartilaginous matrix on the radiograph (Figure 9-16), whereas two thirds have no apparent radiographic mineralization.
One third of the periosteal chondromas exhibit a mineralized cartilaginous matrix on the radiograph (Figure 9-16), whereas two thirds have no apparent radiographic mineralization.
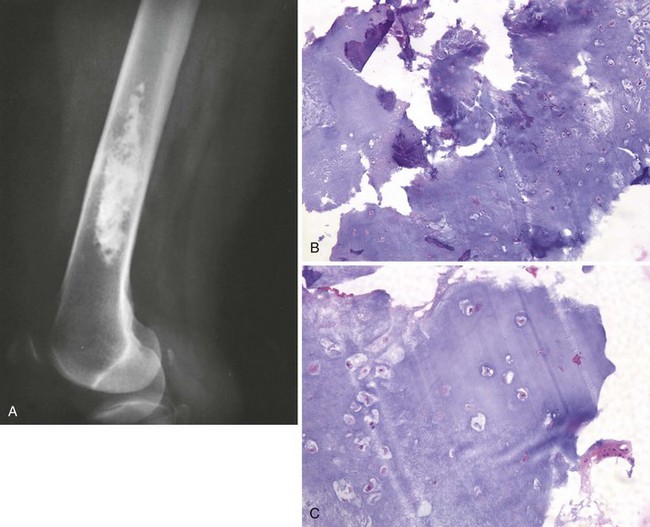
 In the medullary cavity in the metaphysis of long bones, especially the proximal femur and humerus and the distal femur, they are called enchondromas (Figure 9-17).
In the medullary cavity in the metaphysis of long bones, especially the proximal femur and humerus and the distal femur, they are called enchondromas (Figure 9-17).

 Most enchondromas in long bones are asymptomatic.
Most enchondromas in long bones are asymptomatic.
 Radiographically there may be a prominent stippled or mottled calcified appearance (Figure 9-18).
Radiographically there may be a prominent stippled or mottled calcified appearance (Figure 9-18).










 Benign surface lesions probably arise secondary to aberrant cartilage (from the perichondrial ring) on the surface of bone.
Benign surface lesions probably arise secondary to aberrant cartilage (from the perichondrial ring) on the surface of bone.
 They manifest with a painless mass after trauma, or the mass is discovered incidentally.
They manifest with a painless mass after trauma, or the mass is discovered incidentally.
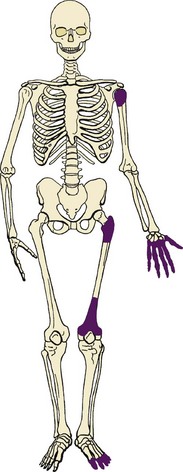
 Osteochondromas usually occur about the knee, proximal femur, and proximal humerus (Figure 9-19).
Osteochondromas usually occur about the knee, proximal femur, and proximal humerus (Figure 9-19).
 Characteristic appearance: a surface lesion in which the cortex of the lesion and the underlying cortex are continuous and the medullary cavity of the host bone also flows into (is continuous with) the osteochondroma (Figure 9-20).
Characteristic appearance: a surface lesion in which the cortex of the lesion and the underlying cortex are continuous and the medullary cavity of the host bone also flows into (is continuous with) the osteochondroma (Figure 9-20).
 Osteochondromas may have a narrow stalk (pedunculated) or a broad base (sessile).
Osteochondromas may have a narrow stalk (pedunculated) or a broad base (sessile).
 They typically occur at the site of tendon insertions, and the affected bone is abnormally wide.
They typically occur at the site of tendon insertions, and the affected bone is abnormally wide.


 When asymptomatic, these lesions are treated with observation only.
When asymptomatic, these lesions are treated with observation only.

 Pain in the absence of mechanical factors is a warning sign of malignant change.
Pain in the absence of mechanical factors is a warning sign of malignant change.
 The development of a sarcoma in an osteochondroma is rare, occurring in far fewer than 1% of cases.
The development of a sarcoma in an osteochondroma is rare, occurring in far fewer than 1% of cases.


 The lesion is termed a “secondary chondrosarcoma.”
The lesion is termed a “secondary chondrosarcoma.”
 The prognosis is usually excellent; these low-grade tumors seldom metastasize.
The prognosis is usually excellent; these low-grade tumors seldom metastasize.
3. Multiple hereditary exostoses
 The osteochondromas are often sessile and large. This is an autosomal dominant condition with mutations in the EXT1 and EXT2 gene loci. Approximately 10% of patients with multiple exostoses develop a secondary chondrosarcoma. The EXT1 mutation is associated with a greater burden of disease and higher risk of malignancy.
The osteochondromas are often sessile and large. This is an autosomal dominant condition with mutations in the EXT1 and EXT2 gene loci. Approximately 10% of patients with multiple exostoses develop a secondary chondrosarcoma. The EXT1 mutation is associated with a greater burden of disease and higher risk of malignancy.
1. Centered in the epiphysis in young patients, usually with open physes
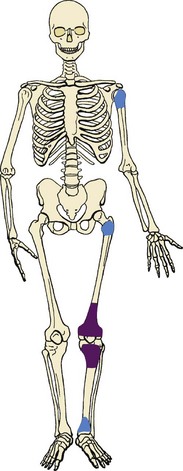
2. The most common locations are the distal femur, proximal tibia, and proximal humerus (Figure 9-21).
3. Lesion is usually in the epiphysis; it may also occur in an apophysis.
4. Manifests with pain referable to the involved joint.
5. It causes a central region of bone destruction that is usually sharply demarcated from the normal medullary cavity by a thin rim of sclerotic bone (Figure 9-22).
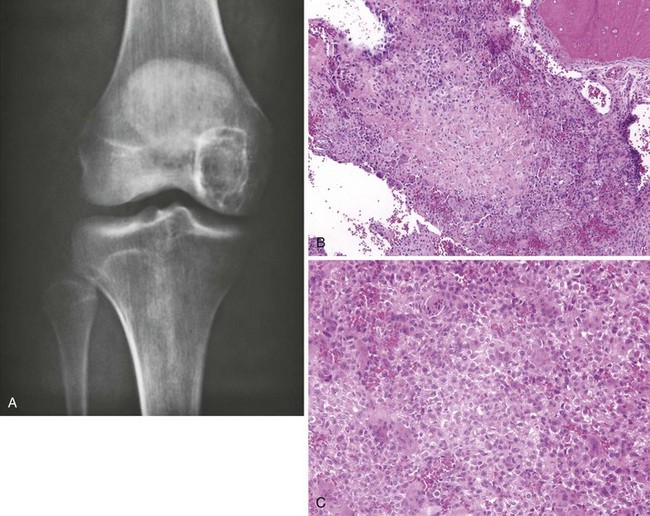
6. Mineralization may or may not occur within the lesion.
7. The basic proliferating cells are thought to be chondroblasts.
 Scattered multinucleated giant cells are found throughout the lesion.
Scattered multinucleated giant cells are found throughout the lesion.


8. Treatment: curettage (intralesional margin) and bone grafting
1. Rare, benign cartilage tumors that contain variable amounts of chondroid, fibromatoid, and myxoid elements
2. More common in boys and men
3. Tend to involve long bones (especially the tibia); the pelvis and distal femur are other common locations (Figure 9-23)
4. Manifest with pain of variable duration (months to years).
5. There is a lytic, destructive lesion that is eccentric and sharply demarcated from the adjacent normal bone (Figure 9-24).
6. It grows in lobules, and there is often a condensation of cells at the periphery of the lobules (concentration of chondroid element may vary from light to heavy).
1. Intramedullary chondrosarcoma
 This malignant neoplasm of cartilage occurs in older adults.
This malignant neoplasm of cartilage occurs in older adults.
 Most common locations include the shoulder and pelvic girdles, knee, and spine.
Most common locations include the shoulder and pelvic girdles, knee, and spine.
 Patients may have pain or a mass.
Patients may have pain or a mass.
 Radiographs usually show diagnostic findings, with bone destruction, thickening of the cortex, and mineralization consistent with cartilage within the lesion (Figure 9-25).
Radiographs usually show diagnostic findings, with bone destruction, thickening of the cortex, and mineralization consistent with cartilage within the lesion (Figure 9-25).

 Prominent cortical changes are present in 85% of affected patients.
Prominent cortical changes are present in 85% of affected patients.



 More than an occasional cell with two such nuclei
More than an occasional cell with two such nuclei
 Especially large cartilage cells with large single or multiple nuclei containing clumps of chromatin
Especially large cartilage cells with large single or multiple nuclei containing clumps of chromatin

 Chondromas of the hand (enchondromas)—the lesions in patients with Ollier disease and Maffucci syndrome—and periosteal chondromas may have atypical histopathologic features (Figure 9-26).
Chondromas of the hand (enchondromas)—the lesions in patients with Ollier disease and Maffucci syndrome—and periosteal chondromas may have atypical histopathologic features (Figure 9-26).


2. Dedifferentiated chondrosarcoma
 Most malignant cartilage tumor
Most malignant cartilage tumor
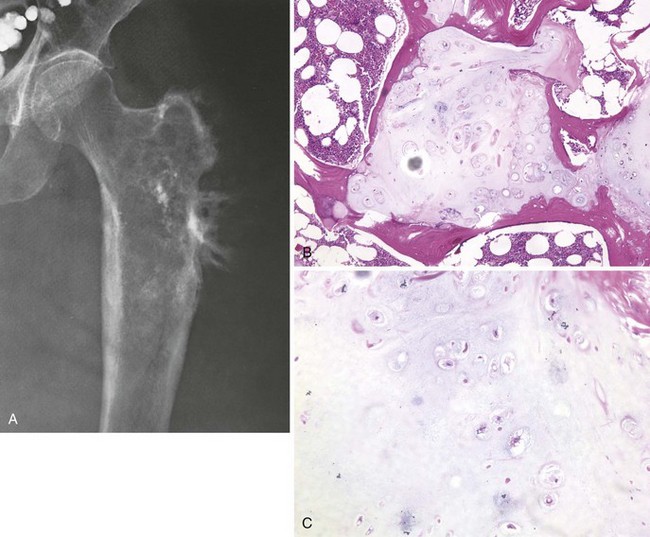
 Most common locations include the distal and proximal femur and the proximal humerus (Figure 9-27).
Most common locations include the distal and proximal femur and the proximal humerus (Figure 9-27).
 Bimorphic histologic and radiographic appearances
Bimorphic histologic and radiographic appearances

 More than 80% of the lesions are typical chondrosarcomas with a superimposed, highly destructive area (Figure 9-28).
More than 80% of the lesions are typical chondrosarcomas with a superimposed, highly destructive area (Figure 9-28).

 Prognosis is poor, and rate of long-term survival is less than 10%.
Prognosis is poor, and rate of long-term survival is less than 10%.
 Treatment: wide-margin surgical resection and multiagent chemotherapy
Treatment: wide-margin surgical resection and multiagent chemotherapy
A Metaphyseal fibrous defect (also known as nonossifying fibroma, nonosteogenic fibroma, and xanthoma)
2. Most such lesions resolve spontaneously and are probably not true neoplasms.
3. Most common locations are the distal femur, distal tibia, and proximal tibia.
4. These lesions are usually asymptomatic and discovered incidentally.
5. Characteristic radiographic appearance: a lucent lesion that is metaphyseal, eccentric, and surrounded by a sclerotic rim (Figure 9-29). The overlying cortex may be slightly expanded and thinned.
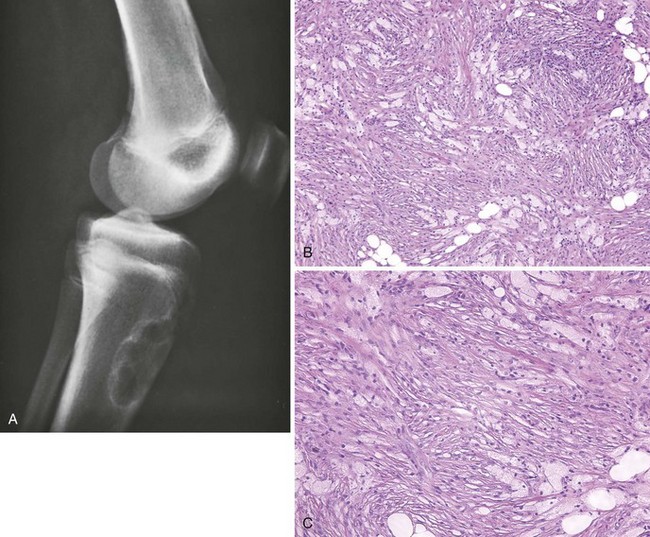

6. Cellular, fibroblastic connective tissue background, with the cells arranged in whorled bundles (numerous giant cells, lipophages, and various amounts of hemosiderin pigmentation).
7. Treatment: observation if the radiographic appearance is characteristic and the risk of pathologic fracture is not excessive.
8. If more than 50% to 75% of the cortex is involved and the patient has symptoms, curettage and bone grafting are performed.
1. Rare and low-grade but aggressive fibrous tumor of bone
3. When process is low grade, residual or reactive trabeculated (or corrugated) bone is often present.
4. Lesion is composed of abundant collagen and mature fibroblasts with no cellular atypia.
5. With wide-margin surgical resection, the risk of local recurrence is lowest, but the joint must be removed in young patients.
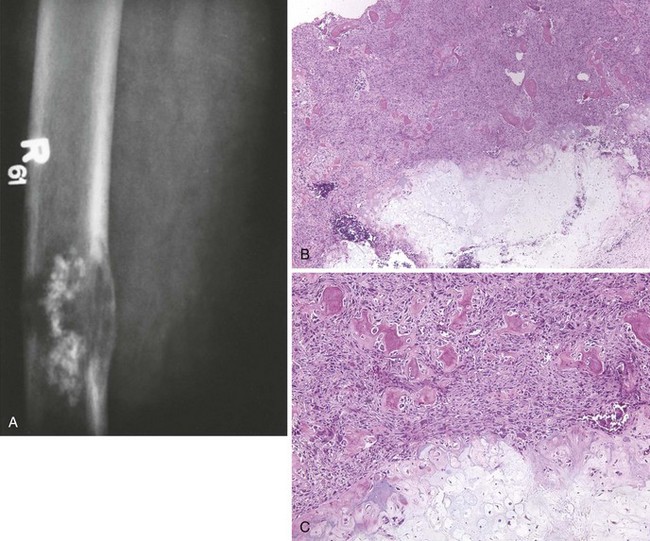
1. Presentation and localization are similar to those of osteosarcoma.
2. This tumor affects primarily older persons but does occur during all decades of life.
3. Lytic bone destruction is often in permeative pattern (Figure 9-30).
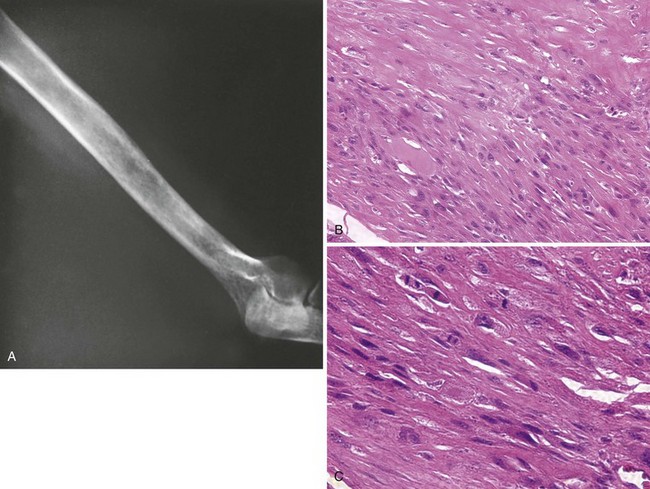
4. Spindle cells, variable collagen production, and a herringbone pattern
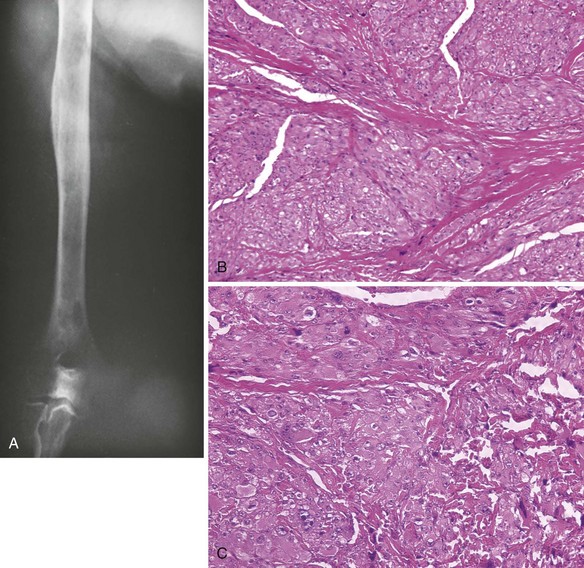
V MALIGNANT FIBROUS HISTIOCYTOMA
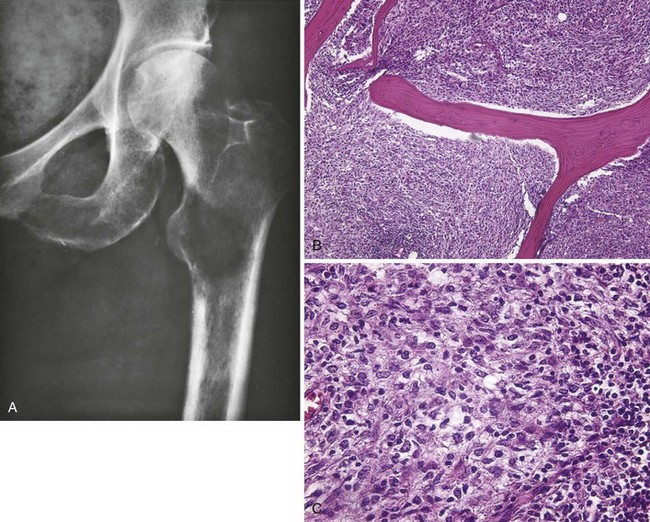
A Most common locations include the distal femur, proximal tibia, proximal femur, ilium, and proximal humerus (Figure 9-31).
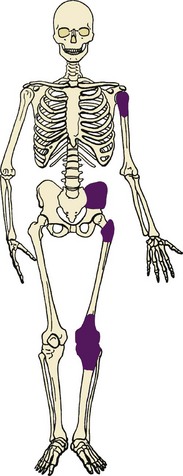
B Malignant bone tumors that have proliferating cells with a histiocytic quality
C Nuclei are often indented, the cytoplasm is usually abundant and may be slightly foamy, the nucleoli are often large, and multinucleated giant cells are usually a prominent feature.
D Variable amounts of fibrous tissue found within the lesion, and the fibrogenic areas have a storiform appearance
E Patients present with pain and swelling.
F This lesion is destructive, with either purely lytic bone destruction or a mixed pattern of bone destruction and formation (Figure 9-32).
A Chordoma is a malignant neoplasm in which the cell of origin is derived from primitive notochordal tissue.
B Occurs predominantly at the ends of the vertebral column (sacrococcygeal; Figure 9-33)
C About 10% of chordomas occur in the vertebral bodies (cervical, thoracic, and lumbar regions).
D Patients present with an insidious onset of pain. Lesions in the sacrum may manifest as pelvic pain, low-back pain, or hip pain or with primarily gastrointestinal symptoms (obstipation, constipation, loss of rectal tone). When vertebral bodies are involved, neurologic symptoms may vary widely because of nerve compression.
E Radiographs often do not reveal the true extent of sacrococcygeal chordomas. The sacrum is difficult to evaluate on plain radiographs because of overlying bowel gas and fecal material and the angulation of the sacrum away from the x-ray beam on the anteroposterior view. In addition, the anteroposterior pelvic view reveals bone destruction only at the sacral cortical margins and neural foramina; these areas are not typically involved early.
F CT scans show midline bone destruction and a soft tissue mass (Figure 9-34).
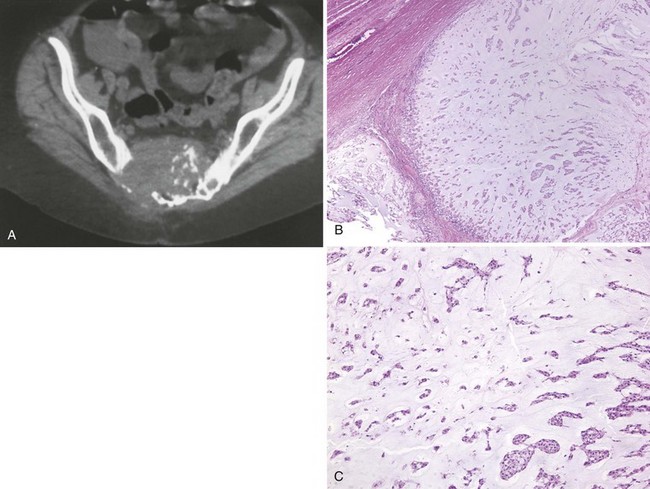

G MRI is an excellent modality for both detecting a chordoma and defining the anatomic features of the tumor.
1. Low signal on T1-weighted images
2. Very bright signal on T2-weighted images
3. Sacrum is often expanded, and the soft tissue mass may exhibit irregular mineralization.
4. In the vertebral bodies, areas of lytic bone destruction or a mixed pattern of both bone formation and bone destruction are often observed.
H The tumor grows in distinct lobules.
1. Chordoma cells sometimes have a vacuolated appearance and are called physaliferous cells.
2. Often arrayed in strands in a mass of mucus
3. Treatment: wide-margin surgical resection
4. Radiation therapy may be added if a wide margin is not achieved.
J Chordomas metastasize late in the course of the disease, and local extension can be fatal.
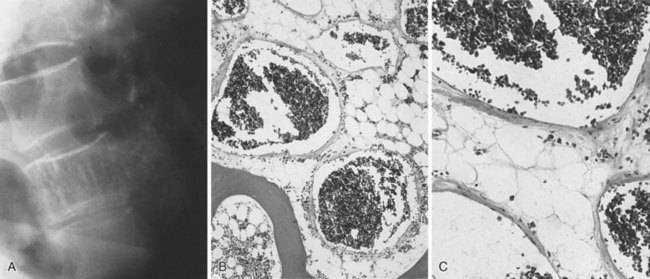
1. These tumors usually occur in vertebral bodies.
2. Patients may present with pain or pathologic fracture (often asymptomatic).
3. Vertebral hemangiomas have a characteristic appearance, with lytic destruction and vertical striations or a coarsened honeycomb appearance. On occasion, more than one bone is involved.
4. There are numerous blood channels. Most lesions are cavernous, although some may be a mixture of capillary and cavernous blood spaces.
1. May occur in any age group, and affected patients present with pain
2. Multifocal involvement of the bones of the same extremity is common
3. Predominantly oval lytic lesion with no reactive bone formation
4. The tumor cells form vascular spaces. The lesions range in structure from very well differentiated (easily recognizable vascular spaces) to very undifferentiated (difficult to recognize their vasoformative quality).
5. Low-grade multifocal lesions may be treated with radiation alone.
1. Lymphoma of bone is uncommon and occurs in three scenarios:
 As a solitary focus (primary lymphoma of bone)
As a solitary focus (primary lymphoma of bone)
 In association with other osseous sites and nonosseous sites (nodal disease and soft tissue masses)
In association with other osseous sites and nonosseous sites (nodal disease and soft tissue masses)

2. The most common locations include the distal femur, proximal tibia, pelvis, proximal femur, vertebra, and shoulder girdle (Figure 9-36).
4. Affected patients generally present with pain.
5. Images often show a lesion that involves a large portion of the bone (long lesion; Figure 9-37).


6. A mixed cellular infiltrate is usually present. Most lymphomas of bone are diffuse, large B-cell lymphomas.
7. Treatment generally combines multiagent chemotherapy and consolidative irradiation.
 Malignant plasma cell disorder that commonly occurs in patients between 50 and 80 years of age
Malignant plasma cell disorder that commonly occurs in patients between 50 and 80 years of age
 Manifests with bone pain, usually in the spine and ribs, or a pathologic fracture
Manifests with bone pain, usually in the spine and ribs, or a pathologic fracture
 Fatigue is a common complaint secondary to the associated anemia.
Fatigue is a common complaint secondary to the associated anemia.

 Serum creatinine levels are elevated in about 50% of affected patients.
Serum creatinine levels are elevated in about 50% of affected patients.
 Hypercalcemia is present in about 33% of affected patients.
Hypercalcemia is present in about 33% of affected patients.
 Radiographic appearance is of punched-out, lytic lesions (Figure 9-38), which may show expansion and a “ballooned” appearance. Osteopenia may be the only laboratory finding.
Radiographic appearance is of punched-out, lytic lesions (Figure 9-38), which may show expansion and a “ballooned” appearance. Osteopenia may be the only laboratory finding.

 Classic histologic appearance: sheets of plasma cells that appear monoclonal with immunostaining.
Classic histologic appearance: sheets of plasma cells that appear monoclonal with immunostaining.
 Well-differentiated plasma cells have an eccentric nucleus and a peripherally clumped, chromatic “clock face” (Figure 9-39).
Well-differentiated plasma cells have an eccentric nucleus and a peripherally clumped, chromatic “clock face” (Figure 9-39).
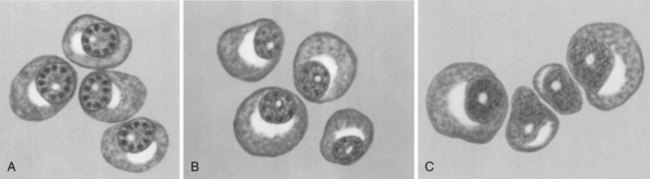
 There is a perinuclear clear zone (halo) that represents the Golgi apparatus.
There is a perinuclear clear zone (halo) that represents the Golgi apparatus.





2. Solitary plasmacytoma of bone
 A solitary lesion on skeletal survey
A solitary lesion on skeletal survey
 Histologic confirmation of plasmacytoma
Histologic confirmation of plasmacytoma
 Bone marrow plasmacyte count of 10% or less
Bone marrow plasmacyte count of 10% or less




 In approximately 50% to 75% of affected patients, solitary myeloma progresses to multiple myeloma.
In approximately 50% to 75% of affected patients, solitary myeloma progresses to multiple myeloma.
 Rare variant in which bone lesions are associated with a chronic inflammatory demyelinating polyneuropathy.
Rare variant in which bone lesions are associated with a chronic inflammatory demyelinating polyneuropathy.

 Sensory symptoms (tingling, pins and needles, coldness) are noted first, followed by motor weakness.
Sensory symptoms (tingling, pins and needles, coldness) are noted first, followed by motor weakness.




 Distinctive neoplasm that has poorly differentiated cells
Distinctive neoplasm that has poorly differentiated cells


 Most common in the epiphysis and metaphysis of long bones, and about 50% of lesions occur about the knee; the vertebra, sacrum, and distal radius are involved in about 10% of cases (Figure 9-40).
Most common in the epiphysis and metaphysis of long bones, and about 50% of lesions occur about the knee; the vertebra, sacrum, and distal radius are involved in about 10% of cases (Figure 9-40).
 The sacrum is the most common axial location of giant cell tumors of bone.
The sacrum is the most common axial location of giant cell tumors of bone.

 They are uncommon in children with open physes.
They are uncommon in children with open physes.
 Manifest with pain that is usually referable to the joint involved
Manifest with pain that is usually referable to the joint involved
 A purely lytic destructive lesion in the metaphysis that extends into the epiphysis and often borders the subchondral bone (Figure 9-41)
A purely lytic destructive lesion in the metaphysis that extends into the epiphysis and often borders the subchondral bone (Figure 9-41)
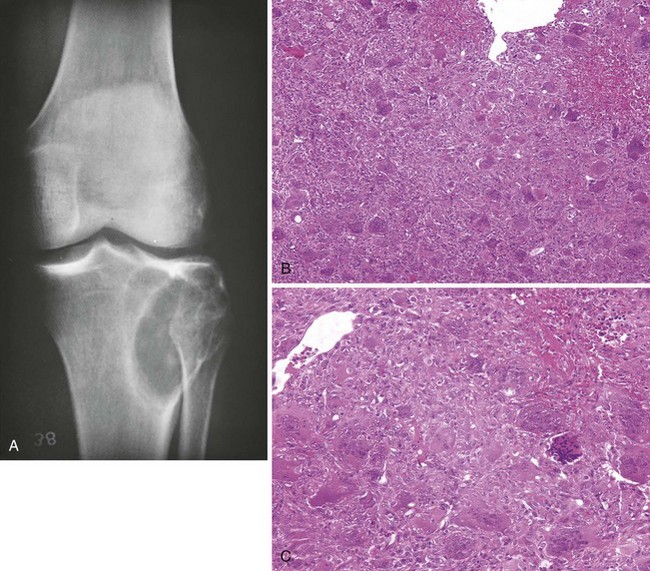



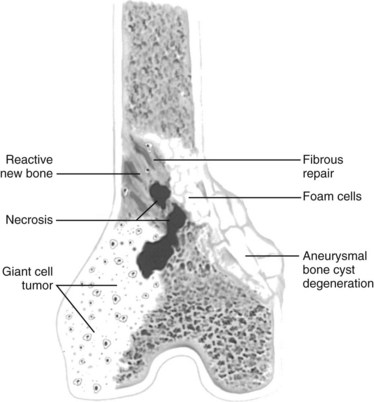
 Giant cell tumors may undergo a number of secondary degenerative changes, such as aneurysmal bone cyst formation, necrosis, fibrous repair, foam cell formation, and reactive new bone (Figure 9-42).
Giant cell tumors may undergo a number of secondary degenerative changes, such as aneurysmal bone cyst formation, necrosis, fibrous repair, foam cell formation, and reactive new bone (Figure 9-42).
 Treatment is aimed at removing the lesion, with preservation of the involved joint.
Treatment is aimed at removing the lesion, with preservation of the involved joint.
 Extensive exteriorization (removal of a large cortical window over the lesion)
Extensive exteriorization (removal of a large cortical window over the lesion)
 Curettage with manual and power instruments
Curettage with manual and power instruments
 Chemical cauterization with phenol
Chemical cauterization with phenol
 Area of defect is usually reconstructed with subchondral bone grafts, methylmethacrylate, or both.
Area of defect is usually reconstructed with subchondral bone grafts, methylmethacrylate, or both.
 Local control with this treatment regimen has a success rate of 85% to 90%.
Local control with this treatment regimen has a success rate of 85% to 90%.
2. Malignant forms: primary and secondary malignant giant cell tumors


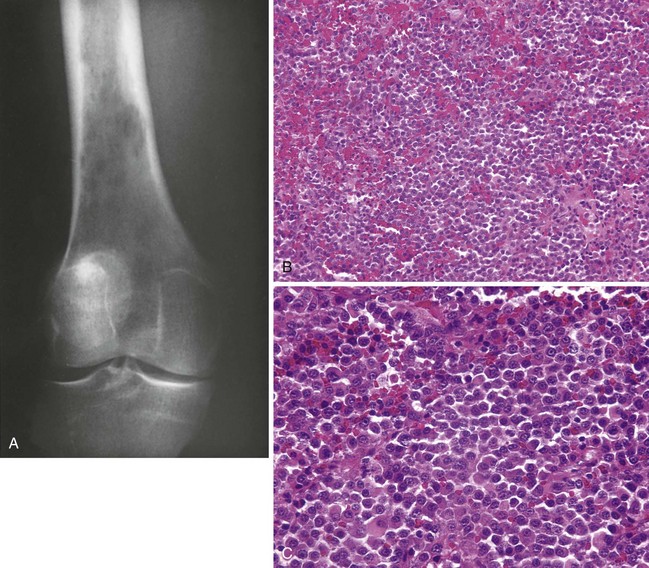
 Distinctive small, round cell sarcoma that occurs most often in children and young adults; most affected children are older than 5 years
Distinctive small, round cell sarcoma that occurs most often in children and young adults; most affected children are older than 5 years
 When a small blue cell tumor is found in a child younger than 5 years, metastatic neuroblastoma and leukemia should be confirmed or ruled out. In patients older than 30 years, metastatic carcinoma must be confirmed or ruled out (Table 9-10).
When a small blue cell tumor is found in a child younger than 5 years, metastatic neuroblastoma and leukemia should be confirmed or ruled out. In patients older than 30 years, metastatic carcinoma must be confirmed or ruled out (Table 9-10).
Table 9-10
Children
Adult
Courtesy of Luke S. Choi, MD, Resident, Department of Orthopaedic Surgery, University of Virginia.
 Most common locations include the pelvis, distal femur, proximal tibia, femoral diaphysis, and proximal humerus (Figure 9-43).
Most common locations include the pelvis, distal femur, proximal tibia, femoral diaphysis, and proximal humerus (Figure 9-43).
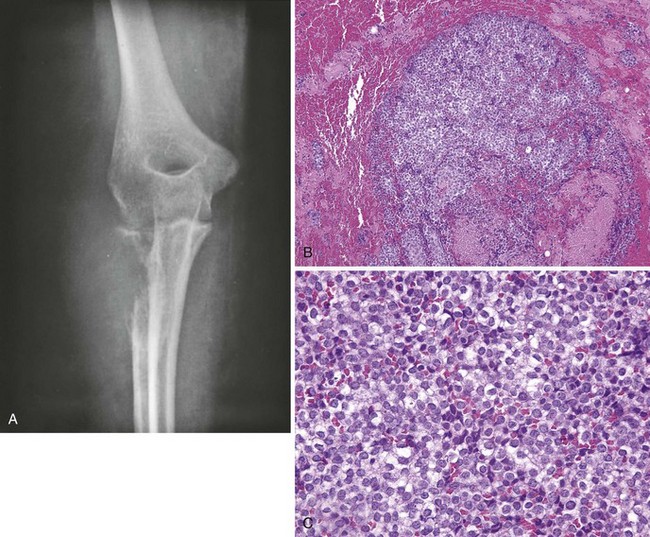
 Manifests with pain, and fever may be present.
Manifests with pain, and fever may be present.

 Radiographs often show a large, destructive lesion that involves the metaphysis and diaphysis.
Radiographs often show a large, destructive lesion that involves the metaphysis and diaphysis.
 The lesion may be purely lytic or may have variable amounts of reactive new bone formation (Figure 9-44).
The lesion may be purely lytic or may have variable amounts of reactive new bone formation (Figure 9-44).




2. Treatment: a multimodality approach with multiagent chemotherapy, irradiation, and surgical resection
 Most lesions have traditionally been treated with chemotherapy and irradiation, but the role of surgery is evolving.
Most lesions have traditionally been treated with chemotherapy and irradiation, but the role of surgery is evolving.











1. Rare low-grade, malignant tumor of long bones that contains epithelium-like islands of cells
2. The tibia is the most common site, although other long bones are infrequently involved (fibula, femur, ulna, radius; Figure 9-45).
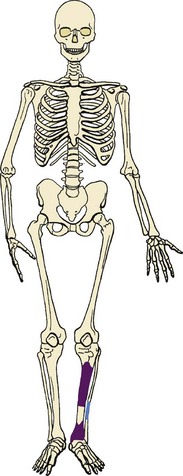
3. Affected patients are young adults and experience pain over months to years.
4. Radiographic appearance: multiple, sharply circumscribed, lucent defects of different sizes, with sclerotic bone interspersed between the zones and extending above and below the lucent zones (Figure 9-46; one of the lesions in the midshaft is the largest and is associated with cortical bone destruction).
5. The cells have an epithelial quality and are arranged in a palisading or glandular pattern; the epithelial cells occur in a fibrous stroma.
6. Treatment: wide-margin surgical resection
7. Tumor may metastasize either early or after multiple failed attempts at local control.
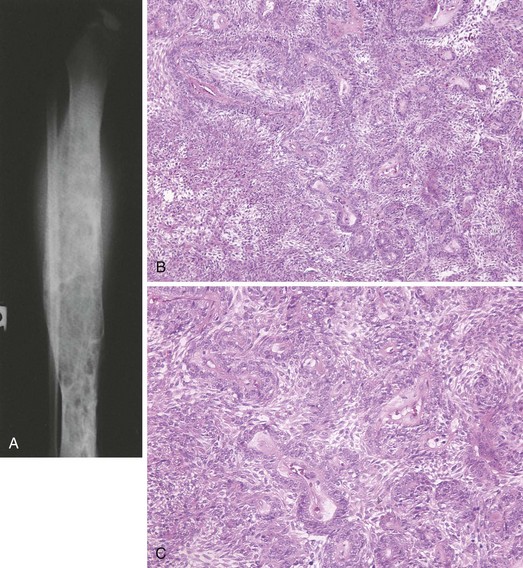
There are many lesions that simulate primary bone tumors and must be considered in the differential diagnosis (Box 9-4). These lesions range from metastases to reactive conditions.
1. Nonneoplastic reactive condition that may be aggressive in its ability to destroy normal bone and extend into the soft tissues
2. May arise primarily in bone or be found in association with other tumors, such as giant cell tumor, chondroblastoma, chondromyxoid fibroma, and fibrous dysplasia
3. Of the patients with an aneurysmal bone cyst, 75% are younger than 20 years.
4. Affected patients experience pain and swelling.
5. Characteristic radiographic finding: an eccentric, lytic, expansile area of bone destruction in the metaphysis
6. In classical cases, there is a thin rim of periosteal new bone surrounding the lesion (Figure 9-47).

7. Radiograph may demonstrate the periosteal bone if it is mineralized.
8. MRI scan usually shows the periosteal layer surrounding the lesion.
 Fluid-fluid levels visible on T2-weighted MRI scans are characteristic of aneurysmal bone cysts.
Fluid-fluid levels visible on T2-weighted MRI scans are characteristic of aneurysmal bone cysts.
 Essential histologic feature: cavernous blood-filled spaces without an endothelial lining.
Essential histologic feature: cavernous blood-filled spaces without an endothelial lining.
 There are thin strands of bone present in the fibrous tissue of the septa.
There are thin strands of bone present in the fibrous tissue of the septa.
 Benign giant cells may be numerous.
Benign giant cells may be numerous.


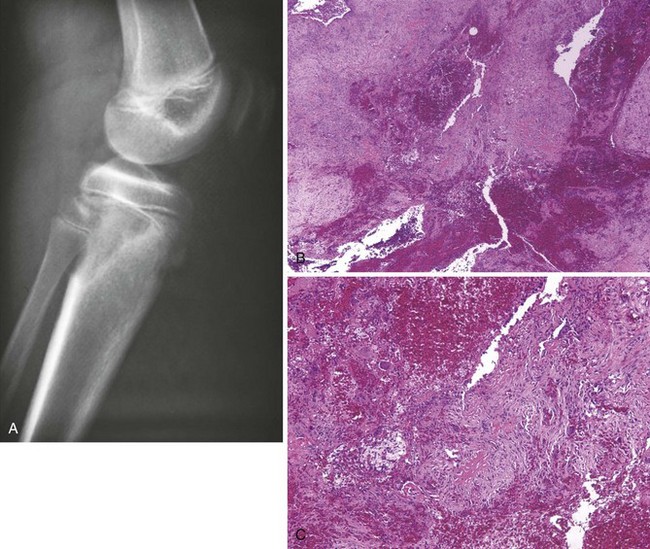
B Unicameral bone cyst (a.k.a., simple bone cyst)
1. Occurs most often in the proximal humerus; other sites are the proximal femur and distal tibia
2. Symmetric cystic expansion with thinning of the involved cortices
3. Manifests with pain, usually after a fracture caused by minor trauma (e.g., sporting event, throwing a baseball, wrestling)
4. Central lytic area and symmetric thinning of the cortices (Figure 9-48)







C Histiocytosis X (Langerhans cell histiocytosis)
1. Lichtenstein originally divided this disorder into three entities: eosinophilic granuloma (monostotic bone disease), Hand-Schüller-Christian disease (multiple bone lesions and visceral disease), and Letterer-Siwe disease (a fulminating condition in young children).
2. This disorder is now usually referred to as LCH.
3. The cellular abnormality is a proliferation of the Langerhans cells of the dendritic system.
4. Eosinophilic granuloma of bone is analogous to monostotic LCH, whereas Hand-Schüller-Christian disease could be called polyostotic LCH with visceral involvement.
 Eosinophilic granuloma of bone is the most common manifestation; only a single bone or, on occasion, multiple bones are involved.
Eosinophilic granuloma of bone is the most common manifestation; only a single bone or, on occasion, multiple bones are involved.
 Patients present with pain and swelling.
Patients present with pain and swelling.
 The lesion is highly destructive and has well-defined margins (Figure 9-49).
The lesion is highly destructive and has well-defined margins (Figure 9-49).
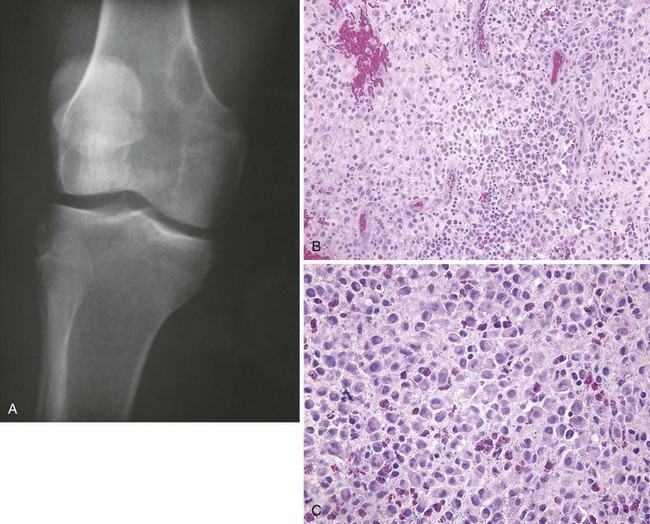


 There may be expansion of the involved bone.
There may be expansion of the involved bone.

 The proliferating Langerhans cell, with an indented or grooved nucleus, is the characteristic cell.
The proliferating Langerhans cell, with an indented or grooved nucleus, is the characteristic cell.

 Nuclear membrane has a crisp border.
Nuclear membrane has a crisp border.
 Mitotic figures may be common.
Mitotic figures may be common.
 Bilobed eosinophils with bright, granular, eosinophilic cytoplasm are present in large numbers.
Bilobed eosinophils with bright, granular, eosinophilic cytoplasm are present in large numbers.





5. Hand-Schüller-Christian disease:



6. Letterer-Siwe disease occurs in young children and is usually fatal.
1. Developmental abnormality of bone that is characterized by monostotic or polyostotic involvement
2. Yellow or brown patches of skin (café au lait spots with irregular borders) may accompany the bone lesions
3. Failure of the production of normal lamellar bone
4. Genetic mutation is an activating mutation of the GSα surface protein

5. When endocrine abnormalities (especially precocious puberty) accompany multiple bone lesions and skin abnormalities, the condition is called McCune-Albright syndrome.
6. Any bone may be involved; the proximal femur is the most commonly affected.
7. Variable appearance (looks highly lytic or like ground glass; Figure 9-50)

 Well-defined rim of sclerotic bone
Well-defined rim of sclerotic bone
 Proliferation of fibroblasts (produces a dense collagenous matrix)
Proliferation of fibroblasts (produces a dense collagenous matrix)
 Trabeculae of osteoid and bone within the fibrous stroma
Trabeculae of osteoid and bone within the fibrous stroma
 Cartilage may be present in variable amounts.
Cartilage may be present in variable amounts.

 Treatment: predicated on the presence of symptoms and the risk of fracture
Treatment: predicated on the presence of symptoms and the risk of fracture





E Osteofibrous dysplasia (also called ossifying fibroma or Campanacci lesion)
1. Primarily involves the tibia and is usually confined to the anterior tibial cortex.
2. Bowing is very common, and affected children may develop pathologic fractures.
3. Lesion typically manifests in children younger than 10 years.
5. Biopsy, when performed, reveals fibrous tissue stroma and a background of bone trabeculae with osteoblastic rimming.
6. Nonoperative treatment is preferred until the child reaches maturity.
7. These lesions usually regress and do not cause problems in adults.
1. Characterized by abnormal bone remodeling
2. Usually diagnosed during the fifth decade of life
4. Radiographs demonstrate coarsened trabeculae and remodeled cortices.
5. Coarsened trabeculae give the bone a blastic appearance.
6. Characteristic features: irregular, broad trabeculae; reversal or cement lines; osteoclastic activity; and fibrous vascular tissue between the trabeculae
8. Medical treatment: aimed at retarding the activity of the osteoclasts
9. Agents used include diphosphonates and calcitonin (pamidronate and zometa).
10. Affected patients may present with degenerative joint disease, fracture, or neurologic encroachment; joint degeneration is common in the hip and knee.
11. Patients undergoing arthroplasty should be treated with diphosphonates to decrease bleeding at the time of surgery.
12. Fewer than 1% of patients with Paget disease develop malignant degeneration with the formation of a sarcoma within a focus of a Paget lesion.



1. Bone infections that often simulate primary tumors
2. Occult infections may occur in all age groups.
3. Affected patients may present with fever, chills, bone pain, or a combination of these symptoms.
4. Affected patients usually present with bone pain but without systemic symptoms.
5. The radiograph findings may be nonspecific:
 Bone destruction and formation are the characteristic findings of chronic infections.
Bone destruction and formation are the characteristic findings of chronic infections.
 Acute infections often produce cortical bone destruction and periosteal elevation.
Acute infections often produce cortical bone destruction and periosteal elevation.





6. A chronic infection with long-standing wound drainage is occasionally complicated by a squamous cell carcinoma.
7. Material that has been sent for culture should be subjected to biopsy, and material that has been sent for biopsy should be subjected to culture.
8. Treatment: removal of all dead tissue and appropriate antibiotic therapy
A Most common entity that destroys the skeleton in older patients
B When a destructive bone lesion is found in a patient older than 40 years, metastases must be considered first.
C The five carcinomas that are most likely to metastasize to bone are those of the breast, lung, prostate, kidney, and thyroid. (Mnemonic: “BLT and a Kosher Pickle”)
D Most common locations of metastasis are the pelvis, vertebral bodies, ribs, and proximal limb girdles.
E The pathogenesis is probably related to Batson vertebral vein plexus.
1. Venous flow from the breast, lung, prostate, kidney, and thyroid drains into the vertebral vein plexus (Figure 9-51).
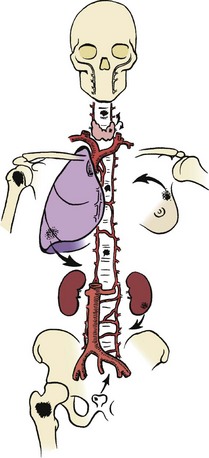
2. The plexus has intimate connections to the vertebral bodies, pelvis, skull, and proximal limb girdles.
F Radiographs demonstrate a destructive lesion that may be purely lytic, may have a mixed pattern of bone destruction and formation, or may be purely sclerotic (Figure 9-52).
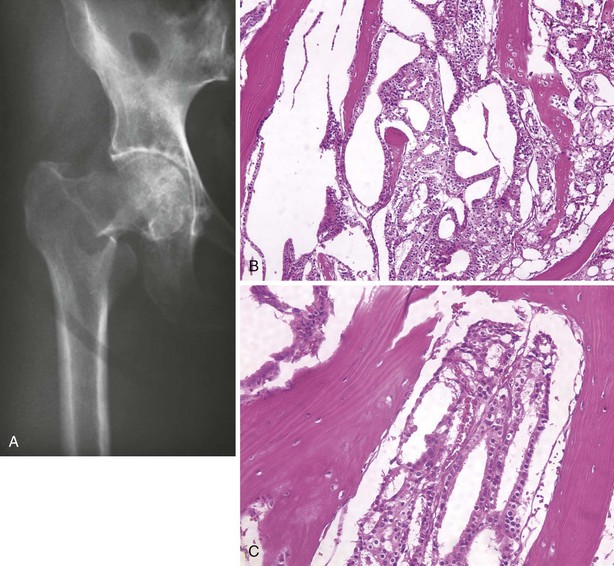
G Histologic hallmark: appearance of epithelial cells in a fibrous stroma; the epithelial cells are often arranged in a glandular pattern
H The bone destruction is caused not by the tumor cells themselves but by activation of osteoclasts (Figure 9-53).
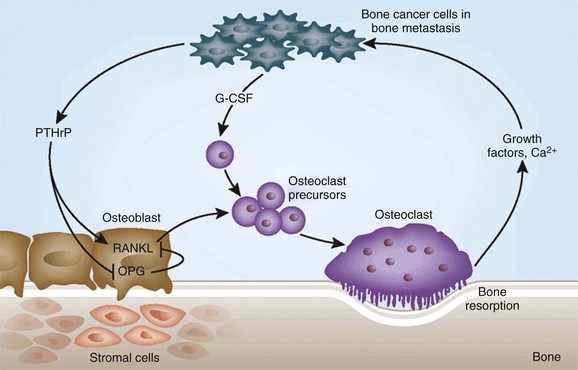
1. Tumor cells secrete parathyroid hormone–related peptide (PTHrP), which stimulates the release of the receptor activator for nuclear factor κB ligand (RANKL) from the osteoblasts and marrow stromal cells.
2. RANKL attaches to the receptor activator for nuclear factor κ (RANK) receptor on the osteoclast precursor cells.
3. In the presence of granulocyte colony–stimulating factor (G-CSF), the osteoclast precursor cells differentiate into active osteoclasts that resorb the trabecular and cortical bone.
4. With bone resorption, transforming growth factor-β, insulin-like growth factor-1, and calcium are released, and these factors stimulate the tumor cells to multiply and release more PTHrP.
5. This process has been termed as the “vicious cycle” of metastatic bone disease.
6. To combat the osteoclastic bone destruction, many patients are now treated with antiresorptive agents (diphosphonates) such as intravenous pamidronate and zoledronic acid.
7. Treatment: aimed at controlling pain and maintaining the independence of the patient
8. Prophylactic internal fixation is performed when impending fracture is deemed likely.
 In comparison with treatment of completed pathologic fractures, prophylactic fixation results in less blood loss, shorter hospital stays, greater likelihood of discharge to home, and greater likelihood of independent ambulation.
In comparison with treatment of completed pathologic fractures, prophylactic fixation results in less blood loss, shorter hospital stays, greater likelihood of discharge to home, and greater likelihood of independent ambulation.
9. There are many suggested criteria for fixation. The following conditions put the patient most at risk for fracture:





I Treatment of pathologic fractures is almost always surgical, inasmuch as these fractures rarely have the potential to heal.
J Surgical procedures should not rely on bony healing.
1. Most proximal femur fractures should be treated with cemented endoprosthesis. To protect the femoral shaft in patients with relatively long life expectancy, consideration should be given to using a long stem.
2. Risk factors for sudden death during insertion of a long-stem prosthesis: presence of breast cancer, hypovolemia, reduced pulmonary function
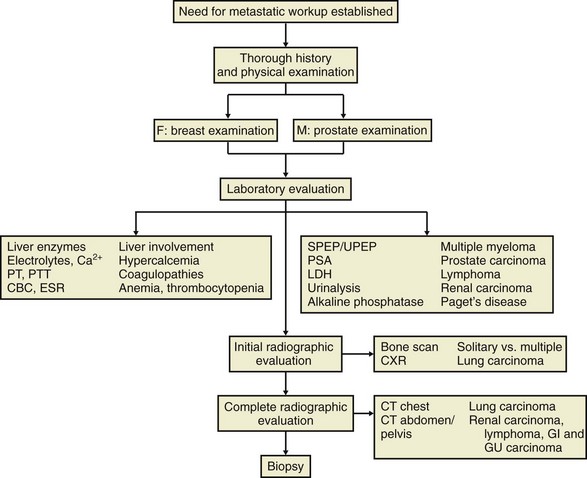
K In patients older than 40 years with a single destructive bone lesion but without a known primary tumor, metastatic disease must be considered present. Simon outlined a diagnostic strategy that identifies the primary lesion in up to 80% to 90% of patients (Figure 9-54).

L Histologically confirmed metastatic cancer for which a definitive primary site is not identified after a detailed medical examination is known as a carcinoma, unknown primary (CUP).