[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 29 Orbital, Ocular, and Optic Nerve Tumors
Based on the incidence rates from the Surveillance, Epidemiology, and End Results Program of the National Cancer Institute, the American Cancer Society estimated that approximately 2390 new cases of all types of primary ocular and orbital malignant tumors were diagnosed in the United States in 2008.1 This demonstrates the rarity of ocular malignant tumors. Uveal melanoma is the single most common malignant tumor of the eye and orbit and makes up nearly 70% of all cancers of these tissues.1 Irradiation is used in the management of many ocular and orbital cancers, as well as in the management of several benign ocular processes. Understanding the pathophysiologic processes of ocular cancers and the results of radiation treatment can guide treatment recommendations.
Etiology and Epidemiology
Choroid Melanoma
Uveal melanoma is the most common primary malignant intraocular neoplasm.1 It is thought to arise from melanocytes of the uveal tract and in the United States occurs in approximately 1500 patients per year.1,2 Uveal melanomas are more common in lightly pigmented persons and are infrequent in nonwhite races. The average age at the time of diagnosis is 60 years in most large series,2–4 and the disease occurs with equal frequency in men and women. The tumors are rarely bilateral. Factors proposed as increasing the risk for the development of uveal melanoma are prolonged sustained sunlight exposure5,6 and lighter-colored irises.5,6 Certain diseases may predispose to melanoma, including xeroderma pigmentosum, which is an inherited disorder of DNA repair; oculodermal melanocytosis,7 in which the ocular surface and the uveal tract are hyperpigmented; and dysplastic nevus syndrome. There are some reports implying a possible rare genetic predisposition, although most melanomas occur as sporadic tumors.
Optic Nerve Tumors
Meningioma
Optic nerve sheath meningiomas arise from the meningothelial cap cells of arachnoid villi and can develop anywhere along the course of the optic nerve from the globe to the prechiasmal intracisternal optic nerve. They may be unilateral, bilateral, or multifocal. Patients with type 2 neurofibromatosis are predisposed to bilateral or multifocal lesions.8 Meningiomas from other locations can also extend to involve the optic nerve.
Optic nerve sheath meningiomas are more common in women. Dutton’s meta-analysis estimates a female preponderance of 61%.9 The typical age at presentation is 40 years. Bilateral cases appear to have an earlier age of onset.
Glioma
Optic nerve gliomas account for 1% to 5% of intracranial gliomas and 4% of orbital tumors. They occur most frequently in children, with 75% in the first decade and 90% in the first two decades of life.10 Approximately 25% of patients with optic gliomas also have type 1 neurofibromatosis. These tumors are increasingly being seen as markers of enhanced risk in patients and their families for the subsequent development of central nervous system tumors.11
Orbital Tumors
In a recent report of 1264 consecutive patients with orbital masses, 33% of patients had malignant lesions. Rhabdomyosarcoma was the most common malignant tumor in children, representing 3% of all orbital masses, and lymphoma was the most common malignant tumor in older patients, representing 10% of cases.12 In one large series, metastatic breast cancer comprised 4% of these lesions.
Lacrimal Gland Tumors
Lacrimal gland tumors are rare. The gender distributions are equal and patient age ranges from 10 to 73 years (mean of 46 years).13 Most of these lesions are malignant, with adenoid cystic carcinoma and mucoepidermoid cancer being the two most frequent ones. Benign pleomorphic adenomas also occur.
Lymphoma
Primary orbital lymphomas are rare and account for less than 1% of all lymphomas diagnosed.14 Lymphomas in the orbit can occur anteriorly in the conjunctival tissues or in the retrobulbar region. Patients with lymphoma at other sites can also present with eye involvement in the course of their disease.
Primary intraocular lymphoma may have malignant lymphoid cells involving the retina, vitreous, or optic nerve head, with or without concomitant central nervous system involvement. The incidence of primary intraocular lymphoma has been increasing over the past 15 years, in both immunocompetent and immunocompromised patients.15 The cause of this increased incidence is unknown. This is a disease of persons in their fifties and sixties, occurring more often in men than women.
Metastatic Tumors
The most common primary tumor to metastasize to the orbit is breast cancer.12 Other orbital metastases have been reported to occur from neuroblastoma, lung cancer, renal cell cancer, and prostate cancer.
Similarly, choroidal metastases are most frequently of breast cancer origin, followed by lung cancer. In one report of 420 patients, the uveal metastasis arose from a primary cancer of the breast in 196 patients (47%), the lung in 90 patients (21%), the gastrointestinal tract in 18 patients (4%), the kidney in 9 patients (2%), the skin in 9 patients (2%), the prostate in 9 patients (2%), and other cancers in 16 patients (4%).16
Benign Ocular Conditions
Pterygium
Most pterygia are located nasally and occur in patients aged 20 to 50 years.17 They are more commonly encountered in warm, dry climates or in patients who are chronically exposed to outdoor elements or smoky and dusty environments. Ultraviolet light exposure appears to be the most significant factor in the development of pterygia. Heredity may also be a factor.
Pterygia represent a degeneration of the conjunctival stroma with replacement by thickened, tortuous elastotic fibers.18 Activated fibroblasts in the leading edge of the pterygium invade and fragment Bowman’s layer. Pterygium development resembles actinic degeneration of the skin.
Pseudotumor
Orbital pseudotumor, or idiopathic orbital inflammatory syndrome, is a nonspecific inflammatory process of unknown etiology that may mimic orbital lymphoma.19 Because the disease mimics infectious or neoplastic processes, these must be ruled out. Autoimmune disease, endocrinopathies, and granulomatous processes must be excluded, as well as local infection with secondary involvement of the orbit, such as sinusitis and subperiosteal abscess. The diagnosis of pseudotumor is vague and remains one of exclusion.
Graves’ Ophthalmopathy
The pathogenesis of Graves’ ophthalmopathy is thought to be an autoimmune one in which activated T lymphocytes invade the orbit and stimulate glycosaminoglycan production in fibroblasts, resulting in tissue edema, lymphocyte infiltration, and marked enlargement of the extraocular muscles.20 Because lymphocytes and fibroblasts are sensitive to radiation, retrobulbar irradiation has been advocated as a treatment. The ophthalmopathy typically goes through three phases: progression, followed by stabilization, and perhaps some improvement. Residual signs of ophthalmopathy after the disease has reached a plateau are thought likely to be the result of fibrosis and other tissue changes rather than persistent inflammation.
Macular Degeneration
Age-related macular degeneration (AMD) is the leading cause of blindness in patients older than 65 years.21 The progressive loss of central vision is usually consequential to an atrophic process involving the retinal epithelium and choriocapillaries in the macula, which is classified as dry AMD. The wet form of AMD is an exudative process with subretinal neovascularization. Wet AMD generally progresses to destructive disciform scarring of the macula, with fibrovascular scars involving the choroid and sensory retina. Patients develop poor visual acuity.
Prevention and Early Detection
Because of the rarity of all ocular tumors, no prevention or early detection methods have been developed for most tumor types. However, in children born into a family with known retinoblastoma, early and careful ophthalmologic evaluation is warranted. Genetic counseling and evaluation of the siblings of patients with retinoblastoma is important, given the high hereditary likelihood.22 After the diagnosis of unilateral retinoblastoma, routine evaluation of the uninvolved eye is indicated because of the increased risk of contralateral disease (see Chapter 69).
Biologic Characteristics and Molecular Biology
Ocular Melanoma
Biologic characteristics that predict outcomes in patients with ocular melanoma include tumor cell type, tumor size, tumor location, and extension of the tumor outside the sclera.23 Cell type is discussed in the pathology section of this chapter, with patients with spindle cell tumors having a better survival rate than those with epithelioid cell tumors. Recent data suggest that specific chromosomal aberrations within the tumor may be associated with the length of survival. Many tumors are treated by means other than enucleation, and because biopsy is rarely performed, the cell type is often unknown.
Survival rates are adversely affected by increased size of the uveal melanoma. Although the earlier literature had various size classifications, the Collaborative Ocular Melanoma Study (COMS) has established a standard size classification.24–26 Small melanomas are 1 to 3 mm thick and less than 16 mm in largest dimension. Medium-sized melanomas are 3 to 8 mm thick and less than 16 mm in largest dimension, and large melanomas are more than 8 mm thick or more than 16 mm in largest dimension (Table 29-1).
TABLE 29-1 Various Other Definitions of Choroidal Melanoma Size
| Size | Measurement |
|---|---|
| Small | 1.5-2.4 mm height and 5-16 mm diameter |
| Medium | 2.5-10 mm height and <16 mm diameter |
| Large | >10 mm height and >16 mm diameter |
Prognosis is also affected by the location of the anterior tumor margin. Tumors located anterior to the equator of the eye in the anterior choroid or ciliary body have a worse prognosis than those located more posteriorly.23 Extraocular extension of a uveal melanoma has poor prognostic implications.24
Orbital Lymphoma
The biology of these tumors varies with the histologic classification within the Revised European-American Lymphoma system or the Working Formulation.27,28 Stage and histologic type play the most significant roles in outcome for orbital lesions.
Primary intraocular lymphoma is believed to be a subset of primary central nervous system lymphoma. It is usually a diffuse large B-cell non-Hodgkin’s lymphoma, and, less commonly, it may be T-cell lymphoma. Because pathologic diagnosis can be difficult, flow cytometry is often done to demonstrate monoclonal populations of B lymphocytes. Immunohistochemical assessments have also been used to demonstrate expression of BCL-6 and MUM1.29 These two markers have been revealed in systemic diffuse large B-cell lymphoma. Molecular analysis detecting immunoglobulin gene rearrangements and ocular cytokine levels showing elevated interleukin-10 (IL-10), with a ratio of IL-10 to IL-6 greater than 1, are helpful adjuncts for the diagnosis of intraocular lymphoma.14,30
Orbital Pseudotumor
The histopathologic type varies, and attempts to refine and subdivide patients have been advocated. The histopathologic spectrum of idiopathic orbital inflammation is typically nondiagnostic and diverse, ranging from the typical diffuse polymorphous infiltrate to the atypical granulomatous inflammation, tissue eosinophilia, and infiltrative sclerosis. One report suggested dividing this condition into three principal types: lymphoid, granulomatous, and sclerosing types.31 Several authors have endorsed the exclusion of reactive lymphoid hyperplasia and defined pseudotumor as a mixed inflammatory infiltrate with fibrosis of varying degrees.19
Pathology and Pathways of Spread
Skin Cancers
Basal cell cancers are seen in 90% of patients, followed by squamous cell cancers in 10%. For patients with squamous cell cancer, involved regional lymph nodes have been reported in as many as 24% of patients.32 Lymph node involvement is more common for larger tumors, recurrent tumors, and those with perineural invasion. Patients with recurrent lesions or perineural invasion may have tumor cells spread more peripherally than is clinically apparent. This may require wider radiation fields. Tumors located in the embryologic fusion planes of the face have been found to be more deeply infiltrating. This area around the medial canthus may affect the depth of treatment necessary for a successful outcome.
Ocular Melanoma
Uveal melanomas arise from melanocytes that are of neuroectodermal origin. Melanoma cells have a large nuclear to cytoplasmic ratio, prominent or multiple nucleoli, and frequent mitotic figures. Tumors are generally classified as composed of either spindle cells or epithelioid cells.33 Spindle cells have a fusiform shape and less pronounced atypia, whereas epithelioid cells are larger and more ovoid in shape and have more anaplastic characteristics. Uveal melanomas are classified as spindle cell melanomas if they are composed of only spindle cells, mixed cell melanomas if they contain both spindle cells and epithelioid cells, and epithelioid cell melanomas if they contain predominantly or exclusively epithelioid cells. Occasionally, the cells within the melanoma undergo necrosis, and these tumors are classified as necrotic melanomas.
The prognosis is more favorable in spindle cell melanoma, intermediate in mixed cell tumors, and poorest in epithelioid or necrotic melanoma. Other negative prognostic indicators include tumor dimension, thickness, and location of the anterior tumor margin.33 Melanomas tend to be slow-growing tumors, but the rate of growth can be quite variable. More rapidly growing tumors, which have a worse prognosis, tend to be anaplastic and composed of epithelioid cells.
At the time of diagnosis, most posterior uveal melanomas are confined inside the sclera of the eye. Extraocular extension of the melanoma can occur through emissary canals, through which normal ocular structures such as vessels and nerves enter and exit the sclera, and generally not by atrophy or thinning of the sclera. Extraocular extension is classified as microscopic or gross (visible on gross inspection), occurs in 8% to 14% of eyes harboring a melanoma, and is associated with a worse prognosis.34
Metastatic disease occurs in less than 3% of patients at the time of initial diagnosis. The most common sites of involvement include hematogenous spread to the liver and the lung. More unusual sites of spread include bone, skin and subcutaneous tissue, and brain and spinal cord tissue. Lymph node involvement was noted in 10% of patients dying from metastatic disease within the COMS.35
Choroidal Metastasis
The most common pathologic type is adenocarcinoma from the breast or non–small cell lung cancer. Most patients have other sites of metastatic disease. Disease extent can be localized to the choroid. The average number of lesions seen in the choroid in one large series was two.16 In this situation, involvement of the opposite choroid can occur in as many as 50% of patients. Orbital metastases can occur and are typically unilateral.
Orbital and Ocular Lymphoma
A wide range of non-Hodgkin’s lymphomas have been described involving the orbit and globe. In patients with orbital lymphomas, most of the lesions are of B-cell origin and classified as low or intermediate grade by the Working Formulation.36 The most common subtype of ocular adnexal lymphoma is a mucosa-associated lymphoid tissue (MALT) lymphoma. Intraocular lymphomas are usually of the aggressive, diffuse, large B-cell type.15
Orbital Meningioma
Meningiomas spread along paths of least resistance, remaining confined to the subarachnoid or intradural space of the intraorbital optic nerve. The tumor may invade the dura, adjacent orbital tissue, muscle, bone, and globe. These tumors may traverse the optic canal and affect the intracranial segment of the optic nerve or adjacent structures. Orbital meningiomas have histologic features similar to those of intracranial meningiomas. Most lesions demonstrate concentric formations of spindle cells or ovoid meningothelial cells with sheets of polygonal cells separated by vascular trabeculae.37
Lacrimal Gland Tumors
Pathologic findings are typically those of adenoid cystic cancer, followed by mucoepidermoid cancer and adenocarcinoma. Outcome seems to be related to the presence of necrosis, hemorrhage, perineural invasion, and high mitotic count.38
Optic Nerve Glioma
In the pediatric form of the disease, the cell of origin for optic nerve gliomas is unknown. Optic nerve gliomas are classified as grade I astrocytomas (pilocytic astrocytomas) in the World Health Organization (WHO) classification of gliomas, are slow growing, and do not tend to metastasize.39 Optic nerve gliomas develop in stages, from generalized hyperplasia of glial cells in the nerve to complete disorganization with loss of neural landmarks within the nerve and nerve sheath. A reactive meningeal hyperplasia may be incited, making it difficult to distinguish a glioma from a perioptic meningioma.
In pediatric patients with optic nerve glioma, 10% to 38% have type 1 neurofibromatosis, and 15% to 40% of children with neurofibromatosis 1 have an optic glioma.40 When bilateral, the lesions are pathognomonic for neurofibromatosis 1.
In the adult form of the disease, the optic nerve gliomas are more likely to be diffusely infiltrative, including astrocytoma (WHO grade II), anaplastic astrocytoma (WHO grade III), or glioblastoma multiforme (WHO grade IV).41 Nuclear atypia, mitoses, endothelial proliferation, and necrosis are potential histologic features.
Clinical Manifestations, Patient Evaluation, and Staging
The diagnostic algorithm for ocular, orbital, and lid tumors is given in Figure 29-1.
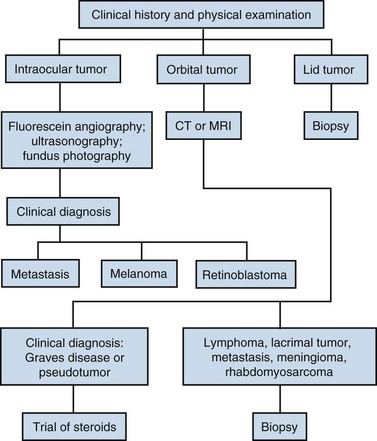
Ocular Melanoma
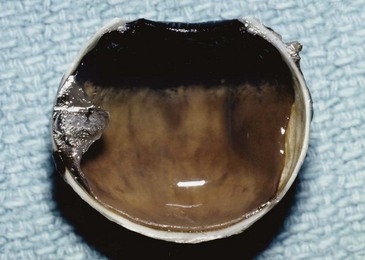
The diagnosis of melanoma is based primarily on ophthalmoscopic appearance and ancillary testing. The tumor typically appears as a unilateral, solitary, elevated, dark brown or gray, variably pigmented, dome-shaped mass (Fig. 29-2). Orange pigment (lipofuscin) is often found on the surface of the tumor and is thought to represent increased metabolic activity.42 Retinal detachment may be present and is more likely in larger tumors.
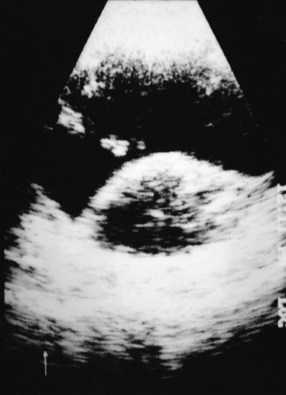
The initial ophthalmologic workup includes fundus photography, fluorescein angiography, and ultrasonography of the globe (Fig. 29-3). These tests facilitate making a correct diagnosis and are also useful in following the lesions for growth or regression after treatment. In a large prospective trial, 645 of 647 patients who underwent enucleation for melanoma were confirmed to be such at pathologic evaluation. Two of the patients had adenocarcinoma.24 In eyes with opaque media secondary to cataract or vitreous hemorrhage, ultrasonography of the globe and MRI of the orbits can allow an accurate diagnosis. Biopsy of a lesion suspicious for an ocular melanoma is used only in cases of diagnostic uncertainty. In patients requiring biopsy, histopathologic and immunohistochemical assessments can be useful in confirming the diagnosis.
To assess disease extent, obtain a general history and perform a physical examination with attention directed to skin lesions, weight loss, the lungs, and the liver. History of a prior cancer would indicate the possibility of a metastatic lesion in the eye rather than a primary melanoma. The most frequent site of metastasis is the liver, so the workup generally includes liver enzyme levels. Elevated levels will prompt evaluation with radiographic imaging. This approach was recently reported to have low sensitivity but high specificity and predictive value.43 Other sites of metastatic disease include the lung, and imaging with chest computed tomography (CT) scanning is most accurate for early detection of disease. The COMS report of sites of metastasis from melanoma demonstrated that sites included liver (91%), lung (28%), and bone (18%).43 Melanomas of the iris rarely metastasize. The brain is an infrequent site of metastasis; brain metastases were discovered in only 4% of patients at the time of death.44
Ocular Metastasis
Patients with uveal metastasis from breast cancer presented to ophthalmologists with visual symptoms in 93% of cases. Of 264 patients with uveal metastasis, 225 (85%) had choroidal metastasis, 8 (3%) had iris metastasis, 2 (<1%) had ciliary body metastasis, and 29 (11%) had metastasis in multiple uveal sites. Asymptomatic metastases were detected in 56% of patients in the opposite eye.45
Patients with orbital tumors more often present with diplopia, ocular motility disorder, and proptosis. Lesions are usually unilateral.46 Metastatic disease may be detected in a high proportion of the patients who present with this entity.
Orbital Lymphoma
Primary intraocular lymphoma is one of the most challenging intraocular tumors to diagnose. Cytologic examination of vitreal aspirates remains the gold standard for exclusion of neoplastic disease in patients with idiopathic uveitis.14 Patients with primary vitreous involvement should also have cerebrospinal fluid (CSF) examination and MRI of the brain and meninges. Eighty percent of patients with primary vitreous involvement have bilateral involvement.
Staging is done using the Ann Arbor staging system.47 A localized orbital lymphoma would be stage IeA.
Optic Nerve Meningioma
Patients typically present with visual loss, afferent papillary defect, color vision disturbance, visual field defect, proptosis, and optic disc edema. Ophthalmoscopic examination may reveal optic nerve head swelling, macular edema, nerve pallor, or choroidal folds. Optic disc swelling may also be observed. MRI is the procedure of choice for diagnosis of these lesions. Lesions appear slightly hypointense to brain and optic nerve tissue on T1-weighted images with fat suppression and gadolinium contrast. Thin CT scans may reveal regular or irregular thickening of the nerve sheath meninges. The nerve may appear normal in size or smaller as the result of circumferential compression or atrophy. Calcification has been described in 30% of patients and is associated with slower growth.48 Differential diagnosis includes sarcoid, demyelinating optic neuritis, orbital inflammatory disease, schwannoma, lymphoma, hemangiopericytoma, and optic nerve metastasis.
Usually these lesions are described as slow growing. Visual loss will generally occur in untreated eyes over a period of 5 to 10 years.49 Rapid growth has been observed during pregnancy, which may be mediated by hormone receptors.50 Other tumors have demonstrated rapid visual loss even in the absence of radiographic enlargement.
Optic Nerve Gliomas
Of optic pathway gliomas, 10% to 20% are confined to the orbit, with the remainder involving the intracranial compartment.10 MRI is the preferred method for evaluation of optic pathway glioma. Both the intraorbital lesion and its intracranial extent can be effectively characterized on MRI. When evaluating the orbit, gadolinium-enhanced T1-weighted images with fat saturation are effective at defining the extent of adult optic glioma. In the intracranial region, MRI provides superior evaluation of the optic nerve, chiasm, tracts, and geniculate body and of optic irradiation compared with CT scanning.
Orbital Pseudotumors
Patients present with proptosis, decreased ocular motility, soft tissue edema, and pain. Decreased visual acuity is not unusual. The condition may be unilateral or bilateral.51 CT scanning may reveal a unilateral focal or more diffuse mass. One review documented infiltration of the retrobulbar fat in 76% of patients, enlargement of the extraocular muscles in 57%, thickening of the optic nerve/sheath complex in 38%, contrast enhancement in 95%, and proptosis in 71%.52 Intracranial extension is unusual but has been documented in up to 8.8% of patients.53
Graves’ Ophthalmopathy
Signs and symptoms of Graves’ ophthalmopathy include bilateral exophthalmia, extraocular muscle dysfunction, diplopia, blurred vision, eyelid and periorbital edema, chemosis, lid lag and retraction, and compressive optic neuropathy. Imaging with CT scanning of the orbit will demonstrate changes consistent with enlargement of extraocular muscles, which are usually symmetric.54
Primary and Adjuvant Therapy
Eyelid Skin Cancers
In general, surgical excision is the preferred treatment. A large report of patients treated with Mohs’ surgery for basal cell cancer revealed a 0% recurrence rate following treatment for a newly diagnosed basal cell cancer, but a 7% recurrence rate for those treated for recurrent basal cell cancer.55 Another report of patients with squamous cell cancer demonstrated a recurrence rate of 3% at 5 years following Mohs’ surgery.56
If surgery is not feasible because of associated functional and cosmetic problems, treatment with radiation has been very successful for patients with either histologic type.57 In a series of more than 1000 patients with eyelid tumors treated with radiation, Fitzpatrick and colleagues58 noted a 5-year local control rate of 95% (basal cell carcinoma, 1009 of 1062 [95%]; squamous cell carcinoma, 97 of 104 [93%]).
Ocular Melanoma
The primary therapy of posterior uveal melanomas depends on many factors. Ocular factors involved in the selection of the treatment modality include tumor size, location, the presence or absence of extraocular extension, visual function in the involved eye, and visual function in the patient’s fellow eye. Systemic factors important in management decisions include patient age, presence or absence of metastatic disease, and general health of the patient. The treatment algorithm is shown in Figure 29-4.
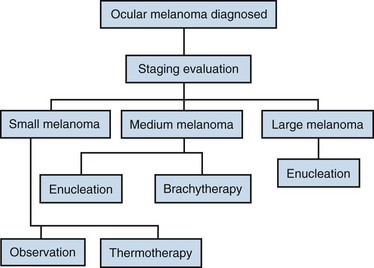
Observation without treatment is used for small melanomas with dormant characteristics. Patients managed by this strategy are often asymptomatic, and the lesion is discovered on routine ocular examination. Another subset of patients who may be managed by observation includes elderly patients with severe systemic health problems or with short life expectancy. In these situations, the treating physician and patient must weigh the risks of treatment (loss of vision and possible tumor dissemination) against the risks of withholding treatment with growth of the tumor and subsequent increased risk of metastatic disease. The COMS found that 21% of small melanomas managed initially by observation had grown by 2 years, with 31% having grown by 5 years.26
Enucleation has been the standard of care for the treatment of choroidal melanoma since the nineteenth century. Enucleation requires disinsertion of the extraocular muscles from the sclera, with removal of the entire globe with a long segment of optic nerve. It is the treatment of choice for large tumors, for eyes that are blind and/or painful, or where the tumor has invaded or is contiguous with the optic nerve. An increase in the death rate at 2 years following enucleation has been reported, putatively supporting the hypothesis that enucleation may induce dissemination of tumor cells (Zimmerman-McLean hypothesis), but this hypothesis remains controversial, with no strong evidence to support it.59 Although all eyes with uveal melanoma can be treated by enucleation, the obvious morbidity of immediate and permanent loss of the eye and of vision prompted the development of other techniques to treat these neoplasms.
Photocoagulation, transpupillary thermotherapy, and local resection have been used for treating selected patients.60 Transpupillary thermotherapy is probably the most common management for small melanomas and has produced excellent control in selected patients. Local resection is used rarely and is most appropriate for anterior tumors involving the ciliary body. Appropriate use of these techniques in the treatment of choroidal melanomas remains investigational.
Radiotherapy Options
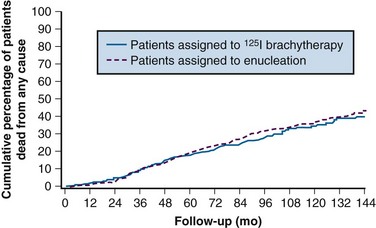
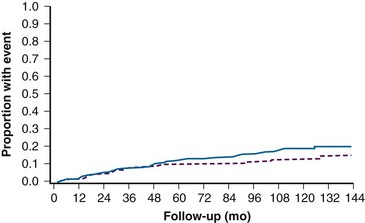
A randomized multicenter trial (COMS) enrolled 1317 patients with medium-size melanomas to treatment with enucleation or with brachytherapy with 125I plaques. This study demonstrated equivalent 5-year overall survival (OS) rates of 81% in both arms24 (Fig. 29-5). Visual acuity was found to decrease over time in a proportion of eyes treated with brachytherapy. Vision was 20/200 or worse in 43% of eyes. The risk of vision loss was associated with a history of diabetes, thick tumors, tumors close to or beneath the macula, tumors with secondary retinal detachments, and tumors that were not dome shaped. Five years after plaque therapy, enucleation was necessary in 10% of patients because of tumor growth and in 3% of patients secondary to complications such as radiation-induced ischemia.61 Complications leading to enucleation increased over time and consisted mainly of pain and/or loss of visual acuity62 (Fig. 29-6).
Charged particle radiotherapy is a less common technique for treating uveal melanoma. With this technique, tantalum rings are sutured to the external sclera at the margins of the tumor for localization, and charged particles (either protons or helium ions) are delivered in four or five equivalent fractions over a 7-day period. The standard target dose is 50 to 70 Gy. An advantage of charged particle therapy is the relatively uniform dose of radiation delivered to the entire tumor and the sharp reduction in dose outside the treated area.62
A systematic review published in 2009 found that as of 2007 there was one randomized control trial and 12 published case series using protons for eye melanoma.63 The randomized controlled trial was a dose-seeking trial to determine whether 50 Gy had fewer complications than 70 Gy, and it was a negative trial. The case series suggested favorable survival rates with significant complication rates. More trials with better design in the future will help to elucidate the role of protons for this disease.
Ocular Metastasis
Choroidal metastases are very sensitive to radiation, but overall survival rates are poor. In one study, external beam radiotherapy (EBRT) was used in 137 patients with uveal metastasis (52%), providing tumor control in 116 patients (85%) at a mean follow-up of 21 months. Using Kaplan-Meier estimates, the overall survival rate of all patients with uveal metastasis from breast cancer was 65% at 1-year, 34% at 3-year, and 24% at 5-year follow-up.45
In 1994, a prospective study of the German Cancer Society was initiated to examine the results of standardized EBRT for choroidal metastases with 40 Gy in 20 fractions.64 With a median follow-up of 5.8 months (range, 1 to 44 months), 41 of 50 patients were dead. The median survival time of all patients was 7 months, and for patients with breast cancer, 10 months. Of the 50 symptomatic eyes, visual acuity increased two or more lines in 36% (18 of 50 eyes), was stabilized in 50% (25 of 50 eyes), and decreased in 14% (7 of 50 eyes). No patient with asymptomatic metastasis (n = 15 eyes) developed ocular symptoms during follow-up. No patient with unilateral tumor and unilateral irradiation developed contralateral metastasis. Severe side effects, possibly related to tumor progression, occurred in three eyes (5%).
Ocular Lymphoma
With systemic disease, chemotherapy is the primary therapy of choice. Patients with solitary low-grade lymphoma can be successfully treated with local irradiation to the orbit. Patients who have intermediate-grade lymphoma may be treated with a combination of systemic chemotherapy and irradiation. Radiation therapy should be considered if a less than complete response in the orbit is observed following chemotherapy. Doses to the orbit or conjunctiva range from 20 to 36 Gy with 1.8 to 2 Gy per fraction in most series, with 89% to 100% local control rates. Follicular lymphomas are controlled with doses in the mid 20-Gy range; data suggest that MALT lymphoma should receive 30 to 36 Gy with 1.8 to 2 Gy/fraction.67 Complication rates are acceptably low with doses in the range of 20 to 30 Gy65,66,67,68; however, cataract formation is common without lens shielding.65 A recent publication suggested inferior results when only partial orbital irradiation was given compared with entire orbital irradiation with control rates of 66% and 100%, respectively.69
Primary Intraocular Lymphoma
Currently, most primary intraocular lymphomas are treated with systemic high-dose methotrexate-based chemotherapy.70 One report treated 10 patients with combined EBRT to the eye and chemotherapy. Complications of EBRT were high and included cataract in five patients (50%), dry eyes in four patients (40%), punctate keratopathy in two patients (20%), radiation retinopathy in two patients (20%), and optic atrophy in one patient (10%).71 Doses of EBRT in this report ranged from 20 to 60 Gy in 1.6- to 2-Gy fractions. Ocular recurrences are often treated with EBRT and increasingly with intraocular methotrexate.
Optic Nerve Meningioma
For most patients, fractionated EBRT is the technique most likely to achieve long-term preservation of visual function. Turbin and others50 studied 59 patients with visual loss at presentation. These patients were divided into four groups: observation, surgery, radiation therapy, or surgery and radiation therapy. Only the group treated with radiation alone did not have significant visual loss at the time of follow-up. Other series with shorter durations of follow-up have reported stable or improved vision in the majority of patients treated with more modern radiation therapy delivery techniques.72–75 More precise delivery of EBRT to smaller volumes will reduce the amount of normal tissue exposed to radiation. These techniques require better immobilization for reproducibility of treatment, CT treatment planning for better tumor localization, and conformal beam arrangements for optimizing treatment planning.
Treatment is usually initiated as soon as a decline in visual acuity or fields is documented. Tumor enlargement without loss of vision is often considered an indication for radiation therapy. It is advisable to treat these lesions with tight margins. Dose per fraction should be no more than 1.8 to 2 Gy, with total doses of 50 to 54 Gy.76 Complications can include radiation retinopathy, optic neuropathy, cataract formation, or dry eye, depending on the volume of normal tissue and dose gradients. Some authors have expressed concerns when treating a patient with diabetes because of potential vascular complications associated with irradiation.
Optic Nerve Glioma
The goals of treatment include survival improvement and stabilization of visual function. Children with isolated optic nerve tumors have a better prognosis than those whose lesions involve the chiasm or that extend along the visual pathway.77–79 Children with neurofibromatosis also have a better prognosis, especially when the tumor is found in asymptomatic patients at the time of screening.77 Observation is an option for patients with neurofibromatosis or slow-growing masses.77,78,80 Spontaneous regressions have been reported.81 For children with isolated optic nerve lesions and progressive symptoms, complete surgical resection or local radiation therapy may result in prolonged progression-free survival (PFS).76
One report of 24 patients detailed outcomes of patients treated with radiation therapy only if there was evidence of visual deterioration or other clinical or radiographic evidence of disease progression. Radiation doses ranged from 45 to 56.6 Gy (median, 54 Gy) in 1.8 Gy/fraction with up to a 17-year follow-up period (median, 6 years). The 6-year actuarial rates of freedom from disease progression and OS were 88% and 100%, respectively. Visual improvement or stabilization was seen in 21 patients (91%) after radiation therapy. A high incidence of endocrine abnormalities was reported, with 15 of the 18 patients evaluated after treatment showing growth hormone deficiency.82
Treatment with chemotherapy has been used in order to avoid surgery or irradiation. One study treated 50 patients with progressive gliomas with carboplatin on a phase II Pediatric Oncology Group (POG) study. Objective responses were found in 30% of patients.83
Orbital Pseudotumor
Patients with unilateral retrobulbar lesions are usually treated with a photon wedged-pair technique. Donaldson and colleagues84 suggested treatment with a central eye bar to shield the lens in each field. However, low-dose regions in the retrobulbar area do result from this technique.
Bilateral retrobulbar lesions are treated with opposed lateral fields with a half-beam block used to spare the anterior segment of the eye if it is uninvolved (Fig. 29-7).
Total EBRT doses are typically 20 Gy delivered in 2-Gy fractions. One report of nine patients treated with initial total doses of 3 Gy noted that five of these patients required further irradiation for durable control of orbital disease.85 Local control rates range from 74% to 100% for doses of 3.8 to 36 Gy.51,84–86
Lacrimal Gland Tumors
Primary therapy is surgical resection with adjuvant EBRT as needed for close surgical margins, perineural invasion, or adenoid cystic histology. Even with aggressive treatment, local recurrence rates were high following surgery and EBRT for adenoid cystic histologic types.87
Graves’ Disease
Peterson and others88 reported results in 311 patients treated with orbital irradiation, with 80% showing improvement of soft tissue symptoms, 75% showing improvement in corneal signs, 61% and 52% having improved extraocular dysfunction and proptosis, and 41% to 71% having improved visual acuity. After irradiation, 76% were able to discontinue corticosteroid therapy.
An older randomized controlled double-blinded trial treated 42 patients with EBRT to one eye and sham therapy to the other eye.89 Six months later, the therapies were reversed. There was no apparent benefit for the orbit that received initial irradiation. This study has been criticized because the patient characteristic included patients who had had onset of symptoms ranging from 0.2 to 16 years’ duration and may have included patients who had a more fibrotic reaction that would have been more stable compared with an inflammatory process.
A more recent controlled trial randomized 44 patients to orbital EBRT and 44 to sham irradiation. After 12 months, 52% of the irradiated patients had improvement compared with 27% of the sham-irradiated patients (p = .02).90
Long-term follow-up suggests that orbital irradiation is safe, with a similar incidence of cataract formation to the corticosteroid group but with a possibly increased risk of retinopathy for diabetic patients.91 Orbital irradiation is typically used in the setting of symptomatic Graves’ ophthalmopathy that is difficult to control with steroids, for patients with poor tolerance to steroids, or for patients for whom surgical orbital decompression has failed.
Pterygia
Treatment for pterygia involves surgical excision with removal of the fibrous tissue down to the level of Tenon’s capsule. Free conjunctival flaps may be grafted over the bare sclera. Recurrence rates have varied, with rates of 20% to 68% with surgery alone.92–94 Postoperative adjuvant therapy with irradiation, topical thiotepa, mitomycin, or other antimetabolic agents may diminish the chance of recurrence.
Multiple retrospective studies have been published that used irradiation with beta emitters started within 24 hours of surgery (Table 29-2). Doses range from 10 to 60 Gy given in one to six fractions.95,96,97–99 There does not appear to be a dose-response curve. Recurrence rates are more common for pterygia that are recurrent after initial surgery. Complication rates are low, but instances of scleral necrosis, cataract induction, corneal thinning, symblepharon, cataract, and corneal ulceration have been described. These are much more common in patients who have been reirradiated.99 Treatment of multiple fields with possible dose overlap may be responsible for some of these complications.99
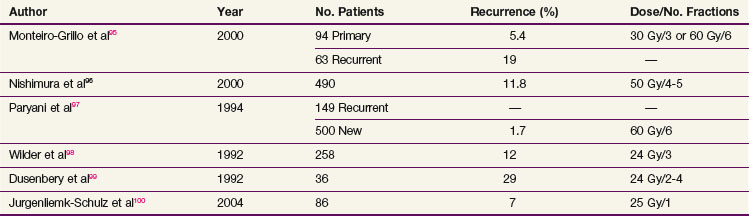
A randomized trial treated 96 patients with a single dose of 25 Gy using a strontium-90 (90Sr) ophthalmic applicator or sham irradiation within 24 hours of surgery. Crude control rates of 93% versus 33% were observed and no serious complications were observed (p = .004).100
Macular Degeneration
In the 1990s there was interest in investigating the effects of ionizing irradiation, which has the potential to destroy vascular tissue and prevent neovascularization. The rationale for using local irradiation for choroidal neovascular membrane is based on the radiosensitivity of proliferating endothelial cells, reduction in the inflammatory response, and possible occlusion of aberrant vessels. Various dose schedules have been given, ranging from 10 to 24 Gy, with 2- to 4-Gy fractions. Several small series suggested stabilization of vision and even improvement in vision.101–103 However, follow-up times were short. A randomized controlled trial from the Netherlands treated 34 patients with 24 Gy in four fractions compared with a control group. The time until vision decreased by at least three lines was prolonged.103
Other studies did not validate these results. A prospective trial on 95 eyes treated with 10 to 36 Gy did not reveal an effect of dose, and decrease in visual acuity was not prevented.104 Another group compared a historical group of patients treated with interferon with those who had been treated with radiation therapy. Irradiation did not prevent vision loss.105 Another report of 69 patients followed for at least 1 year who received 16 Gy in eight fractions failed to show any influence on vision or reading ability.106 A large prospective multicenter trial from Germany randomly assigned 205 patients to a dose of 16 Gy or sham irradiation. Radiation therapy did not prevent visual loss in this trial.107 Another randomized controlled trial of 203 patients from the United Kingdom demonstrated that an irradiation dose of 12 Gy versus no irradiation did not statistically improve visual outcomes at 12 or 24 months.108 Based on these data, there is little information at present that justifies radiation therapy for treatment of AMD outside of a clinical study.
Locally Advanced Disease and Palliation
Ocular Melanoma
Patients with large choroidal melanoma are usually treated with enucleation. The COMS enrolled 1003 patients to a study that randomized them to either enucleation or enucleation preceded by EBRT (20 Gy).25 Patients receiving preoperative radiation therapy had no significant difference in 5-year OS. The 5-year all-cause mortality rate for large melanomas was 40%. There was no difference in local control between the two groups.
Irradiation Techniques and Tolerance
Plaque Irradiation
Multiple different isotopes have been used for plaque therapy, including cobalt-60 (60Co), gold-198 (198Au), palladium-103 (103Pd), ruthenium-106 (106Ru), iodine-125 (125I), and iridium-192 (192Ir). The largest experience is with 125I. This isotope carries the advantage of low-energy gamma rays and therefore decreased shielding and dose to distant structures. Ruthenium-106 is a beta emitter that has few shielding concerns, but it has a penetration of only a short distance, which may limit its application.
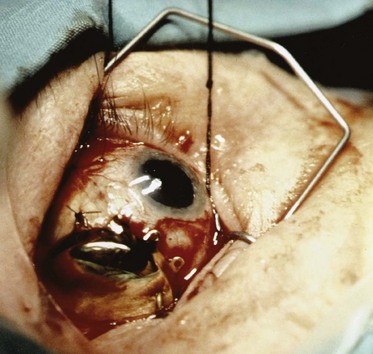
A variety of plaque applicators have been used for ocular melanomas, but the most commonly used ones are those developed for the COMS (Fig. 29-8). These plaques allow shielding of the posterior and lateral structures with a gold shield. A plaque that allows at least a 2-mm margin circumferentially around the tumor should be selected. Placement of the seeds within the applicator varies, depending on the height of the tumor. The prescription point should be the apex of the tumor or 5 mm, whichever is greater. A scleral thickness of 1 mm is added to the apical height. In the COMS, a total dose of 85 Gy was used and delivered to the apex of the tumor at a dose rate between 0.5 and 1.25 Gy/hour. The optimal dose of radiation therapy for treatment of choroidal melanoma is controversial. Equivalent local control, survival, and disease-free survival rates with less ocular morbidity and loss of vision have been reported with lower radiation therapy doses.109

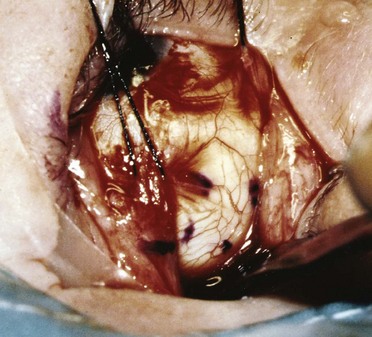
The plaque is placed by the ophthalmologist by marking the boundary of the tumor base with a surgical marking pencil or light cautery that can be seen when the tumor is transilluminated (Fig. 29-9). The plaque is then centered over the marks. Plaque placement may require detachment of the ocular rectus muscles. The plaque is then sutured to the sclera (Fig. 29-10). Good placement can be confirmed with ultrasonic evaluation of the eye. Occasionally, the melanoma is located adjacent to the optic nerve. The use of a notched plaque may allow better dosimetry and coverage of the tumor in this situation.
Skin Lesions
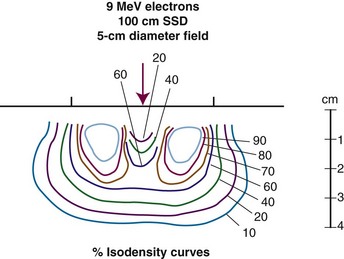
Treatment of skin lesions close to the eye can be difficult. The close proximity of normal structures must be balanced with the dosimetric constraints that are present with electrons. Most of the data supporting the use of radiation therapy came from series using kilovoltage x-ray beams. However, this type of x-ray equipment is no longer widely available. Electrons have a larger penumbra and area of beam constriction laterally, necessitating larger treatment fields than kilovoltage beams. One report demonstrated higher corneal doses with electrons compared with kilovoltage beams (doses were two to four times higher).110 Also, the 95% isodose area was 32% wider with kilovoltage beams than with 6-MeV electrons.110 Tungsten eye shields may provide better shielding of anterior eye structures than lead for 9-MeV electrons.111
Unilateral Eye Lesion
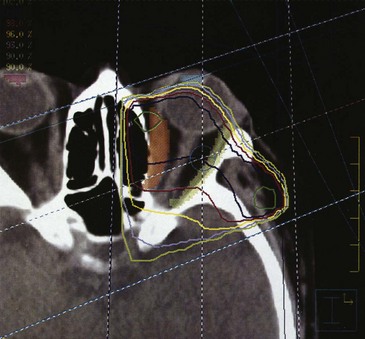
Treatment of one eye may be necessary for patients with choroidal or orbital metastasis or orbital lymphomas. This field arrangement is designed to limit the irradiation to the opposite eye. Typically, a wedged-pair technique is used if the entire orbit needs to be treated. CT planning can assist with location of the normal optic structures and the tumor volume that is to be included in the field (Fig. 29-11). Three-dimensional treatment planning may enable more treatment conformity around the structure to be treated and decrease normal tissue sequelae.
Bilateral Eye Lesions
Treatment of both eyes is usually done with opposed lateral fields. If it is possible to spare the lens, the central axis of the beam can be placed at the location of the lens (5 mm behind the anterior globe). A split-beam technique is used to block the anterior structures if possible. Graves’ disease, bilateral choroidal metastasis, and bilateral lymphomas are treated in this manner (see Fig. 29-7).
Anterior Lesions
Involvement of the conjunctiva may require treatment of the anterior structures alone. This can be accomplished with electron fields directed en face to the globe. Central lens shielding can be accomplished with a hanging block at which the patient is instructed to look (Fig. 29-12). En face electrons can be used with a lead contact lens if electrons of not more than 6 MeV are used111 (Fig. 29-13).

Intensity-Modulated Radiation Therapy
Techniques using three-dimensional treatment planning and intensity-modulated radiation therapy (IMRT) may be advantageous for treating optic nerve meningiomas or gliomas. All critical structures of the eye should be identified and appropriate dose constraints applied. Patient immobilization is crucial, along with CT simulation (Fig. 29-14).
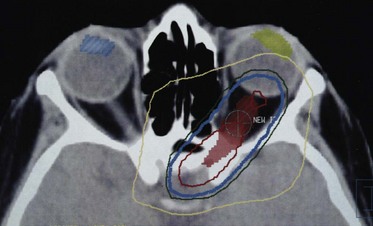
Tolerance Issues
In most human studies, total fractionated doses under 5 Gy have not produced visually significant lens opacities. Merriam and Focht112 reported cataract formation with EBRT fractions of 1.5 to 2 Gy, to a total dose of 12 Gy. When single-dose, total body irradiation was used, cataracts developed in 80% of survivors; the incidence decreased to 20% when fractionated EBRT was given to a total dose of 13 Gy.113 Reduction of dose to the lens may be possible with customized lens shields, pencil-beam lead blocks, or more complex treatment planning with IMRT. Lens extraction for cataract development can correct the vision in an otherwise functioning eye following irradiation.
Other anterior structures in the globe include the cornea and conjunctiva. The conjunctiva is a mucous membrane of nonkeratinized squamous epithelium with goblet cells overlying a thin substantia propria.114 It covers the inner surface of the eyelid and outer surface of the eye, extending to the peripheral cornea. The cornea is composed of nonkeratinized stratified squamous epithelium. Its outermost surface is composed of irregular microvilli made optically smooth by the precorneal tear film. The basal cells are attached to Bowman’s layer, which is made up of randomly dispersed collagen fibrils that may become opacified by scar tissue. The stroma, which makes up 90% of the total corneal thickness, is composed of fibroblasts and collagen lamellae.
Acute effects of fractionated radiotherapy include injection and erythema of the conjunctiva with irritation that is usually self-limited. Fractionated doses of radiation to a total dose above 40 to 50 Gy to the cornea can produce edema with punctate keratitis.115 Management consists of aggressive lubrication, patching, and antibiotic drops. Recurrent corneal erosions may progress to ulceration and infection leading, to either opacification or perforation. Ulcerations of the cornea have been reported with fractionated radiation therapy to doses above 40 Gy.65,116 Similarly, ulceration, telangiectasias, and keratinization can develop in the conjunctiva.
The eyelid skin is the thinnest in the body. The lids contain sebaceous, serous, and apocrine glands, which contribute to the tear film. There are puncta on the medial lid margins that form the opening to the nasolacrimal drainage system. Transient effects of eyelash loss and erythema occur at 30 to 40 Gy with conventional fractionation, whereas permanent lash loss occurs at doses above 50 Gy.117 Scarring and fibrosis can develop at doses above 50 Gy, which can result in ectropion or entropion of the lid as well as stenosis of the eyelid puncta.
There are several structures necessary for adequate tear film production, which include the lacrimal gland, goblet cells, meibomian glands, and accessory lacrimal glands. Dry eye syndrome can result from injury to any component of the tear film. Symptoms include burning, decreased vision, excessive tearing, and a foreign body sensation. Examination reveals corneal changes ranging from keratitis to corneal scarring and opacification. One report described 19% of patients who received doses less than or equal to 45 Gy in 25 fractions developing a dry eye syndrome compared with 100% for doses above or equal to 57 Gy in 30 fractions.118 Severe keratitis sicca can lead to corneal ulceration and perforation of the globe. Prevention of lacrimal damage requires consideration of the location of the lacrimal gland when designing fields for radiation therapy delivery.119
The sclera covers the posterior 80% of the globe. It is largely avascular and consists of collagen fibrils, fibroblast, and ground substance. It is very resistant to radiation and can withstand doses of over 150 Gy that are delivered with plaque radiotherapy used to treat choroidal melanomas. Scleral thinning and ulceration are unusual complications of high doses of radiation therapy.120
The retina consists of an extensive network of neural, glial, and vascular elements. The blood supply to the outer layers of the retina is via the choriocapillaries. The inner retinal layers are supplied by the branches of the central retinal artery, of which the largest branches are the temporal and nasal arcades. Pathology involving the region within the temporal arcades is more visually significant than that occurring elsewhere. Radiation retinopathy is caused by occlusive microangiopathy manifest by cotton wool spots, microaneurysms, telangiectasias, hemorrhages, macular edema, exudates, neovascularization, vitreous hemorrhage, and pigment changes. Visual symptoms secondary to radiation retinopathy depend on the area of the retina that is affected. A history of diabetes, collagen vascular disease, hypertension, and treatment with chemotherapy predisposes patients to radiation-induced injury.121 In one report, radiation retinopathy was not observed at doses below 45 Gy in 25 fractions but increased steadily above this dose.121 (Patients also seemed to be at increased risk if they were treated with doses per fraction greater than or equal to 1.9 Gy.121) The estimated tolerance for retinal damage is very steep, with a tolerance dose of 5% risk at 5 years (TD5/5) of 45 Gy and TD50/5 of 65 Gy.122
Optic nerve injury occurs secondary to ischemia. Damage begins with perivascular lymphocyte cuffing, loss of endothelial cells, and hyalinization, with fibrosis, thrombosis, and infarction of neural tissue. Optic nerve injury presents with painless monocular loss of vision. Fractionation is quite important in the development of optic nerve injury. Of patients who were treated to a total dose of 45 to 50 Gy and received a daily fraction size of more than 2.5 Gy, 18% developed visually significant changes, whereas those patients who received fractions below 2.5 Gy did not develop optic neuropathy.123 Stereotactic radiosurgery that delivers large fraction sizes can cause optic neuropathy with single doses of 8 Gy, although tiny volumes to high doses have been given without toxicity.124 With conventional fractionation, the TD5/5 of the optic nerve is considered to be 60 Gy.125
24 The Collaborative Ophthalmology Study Group. The Collaborative Ocular Melanoma Study randomized trial of iodine-125 brachytherapy for choroidal melanoma, III. Initial mortality findings. COMS report No. 18. Arch Ophthalmol. 2001;119:969.
25 Collaborative Ocular Melanoma Study Group. The Collaborative Ocular Melanoma Study randomized trial of pre-enucleation radiation of large choroidal melanoma, II. Initial mortality findings. COMS report No. 10. Am J Ophthalmol. 1998;125:779.
26 Collaborative Ocular Melanoma Study Group. Mortality in patients with small choroidal melanoma. COMS report No. 4. Arch Ophthalmol. 1997;115:886.
35 The Collaborative Ocular Melanoma Study Group. Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the Collaborative Ocular Melanoma Study (COMS). COMS report 15. Arch Ophthalmol. 2001;119:670.
39 Dutton JJ. Optic nerve gliomas and meningioma. Neurol Clin. 1991;9:163.
41 Dutton JJ. Gliomas of the anterior visual pathway. Surv Ophthalmol. 1994;38:427.
43 Diener-West M, Reynolds SM, Agugliaro DJ, et al. Screening for metastasis from choroidal melanoma. The Collaborative Ocular Melanoma Study Group Report 23. J Clin Oncol. 2004;22:2438.
44 The Collaborative Ocular Melanoma Study Group. Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the Collaborative Ocular Melanoma Study (COMS). COMS report 15. Arch Ophthalmol. 2001;119:670.
50 Turbin RE, Thompson CR, Kennerdell JS, et al. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophthalmology. 2002;109:890.
51 Mendenhall WM, Lessner AM. Orbital Pseudotumor. AJCO. Sept 4 2009. (epub ahead of print)
55 Malhotra R, Huilgol SC, Huynh NT, Selva D. The Australian Mohs database, part II. Periocular basal cell carcinoma outcome at 5-year follow-up. Ophthalmology. 2004;111:631.
56 Malhotra R, James CL, Selva D, et al. The Australian Mohs database. Periocular squamous carcinoma. Ophthalmology. 2004;111:617.
58 Fitzpatrick PJ, Thompson GA, Easterbrook WM, et al. Basal and squamous cell carcinoma of the eyelids and their treatment by radiotherapy. Int J Radiat Oncol Biol Phys. 1984;10:449.
61 Jampol LM, Moy CS, Murray TG, et al. The COMS Randomized Trial of Iodine 125 Brachytherapy for Choroidal Melanoma, IV. Local treatment failure and enucleation in the first 5 years after brachytherapy. COMS Report No. 19. Ophthalmology. 2002;109:2197-2206.
67 Fung C, Tarbell N, Lucarelli M, et al. Ocular Adnexal lymphoma: Clinical behaviour of distinct World Health Organization Classification subtypes. Int J Radiat Oncol Biol Phys. 2003;57:1382-1391.
69 Pfeffer MR, Rabin T, Tsvang L, et al. Orbital lymphoma. Is it necessary to treat the entire orbit? Int J Radiat Oncol Biol Phys. 2004;60(2):527-530.
87 Esmaeli B, Ahmadi MA, Youssef A, et al. Outcomes in patients with adenoid cystic carcinoma of the lacrimal gland. Ophthal Plast Reconstr Surg. 2004;20:22.
90 Prummel MF, Terwee CB, Gerding MN, et al. A randomized controlled trial of orbital radiotherapy versus sham irradiation in patients with mild Graves’ ophthalmopathy. J Clin Endocrinol Metab. 2004;89:15.
91 Wakelkamp IM, Tan H, Saeed P, et al. Orbital irradiation for Graves’ ophthalmopathy. Is it safe? A long-term follow-up study. Ophthalmology. 2004;111:1557.
96 Nishimura Y, Nakai A, Yoshimasu T, et al. Long-term results of fractionated strontium-90 radiation therapy for pterygia. Int J Radiat Oncol Biol Phys. 2000;46:137.
109 Saconn P, Gee C, McCoy T, et al. Alternative dose for choroidal melanoma treated with an iodine 125 radioactive plaque. A single institution retrospective study. Int J Radiat Oncol Biol Phys. 2010;78:844-848.
110 Amdur RJ, Kalbaugh KJ, Ewald LM, et al. Radiation therapy for skin cancer near the eye. Kilovoltage x-rays versus electrons. Int J Radiat Oncol Biol Phys. 1992;23:767.
111 Shiu AS, Tung SS, Gastorf RJ, et al. Dosimetric evaluation of lead and tungsten eye shields in electron beam treatment. Int J Radiat Oncol Biol Phys. 1996;35:599.
114 Gordon KB, Char DH, Sagerman RH. Late effects of radiation on the eye and ocular adnexa. Int J Radiat Oncol Biol Phys. 1995;31:1123-1139.
118 Parson JT, Bova FJ, Fitzgerald CR, et al. Severe dry-eye syndrome following external beam irradiation. Int J Radiat Oncol Biol Phys. 1994;30:775.
121 Parsons JT, Bova FJ, Fitzgeral CR, et al. Radiation retinopathy after external-beam irradiation. Analysis time-dose factors. Int J Radiat Oncol Biol Phys. 1994;30:765.
122 Emami B, Lyman J, Brown A, et al. Tolerance of normal tissue to therapeutic irradiation. Int J Radiat Oncol Biol Phys. 1991;21:109.
124 Stafford SL, Pollock BE, Leavitt JA, et al. A study on the radiation tolerance of the optic nerves and chiasm after stereotactic radiosurgery. Int J Radiat Oncol Biol Phys. 2003;55:1177.
125 Parsons JT, Bova FJ, Fitzgerald CR, et al. Radiation optic neuropathy after megavoltage external-beam irradiation. Analysis of time-dose factors. Int J Radiat Oncol Biol Phys. 1994;30:755.
1 American Cancer Society. Cancer Facts and Figures. Atlanta: American Cancer Society; 2008.
2 Singh AD, Topham A. Incidence of uveal melanoma in the United States: 1973. Ophthalmology. 2003;110:956.
3 Egan KM, Seddon JM, Glynn RJ, et al. Epidemiologic aspects of uveal melanoma. Surv Ophthalmol. 1988;32:239.
4 Iskovitch J, Ackerman C, Andrew H, et al. An epidemiologic study of posterior uveal melanomas in Israel, 1961. Int J Cancer. 1995;61:291.
5 Seddon JM, Gragoudas ES, Glynn RJ, et al. Hot factors, UV radiation, and risk of uveal melanoma. A case-controlled study. Arch Ophthalmol. 1990;108:1274-1280.
6 Tucker MA, Shields JS, Hartge P, et al. Sunlight exposure as a risk factor for intraocular melanoma. N Engl J Med. 1985;313:789.
7 Singh AD, De Potter P, Fijal BA, et al. Lifetime prevalence of uveal melanoma in white patients with oculo(dermal) melanocytosis. Ophthalmology. 1998;105:195.
8 Spencer WH. Primary neoplasms of the optic nerve and its sheaths. Clinical features and current concepts of pathogenic mechanisms. Trans Am Ophthalmol Soc. 1972;70:490.
9 Dutton JJ. Optic nerve gliomas and meningiomas. Neurol Clin. 1991;1:163.
10 Hollander MD, FitzPatrick M, O’Connor SG, et al. Optic gliomas. Radiol Clin North Am. 1999;37:59.
11 Walker D. Recent advances in optic nerve glioma with a focus on the young patient. Curr Opin Neurol. 2003;16:657.
12 Shields JA, Shields CL, Scartozzi R. Survey of 1264 patients with orbital tumors and simulating lesions. The 2002 Montgomery Lecture, part 1. Ophthalmology. 2004;111:997-1008.
13 Esmaeli B, Ahmadi MA, Youssef A, et al. Outcomes in patients with adenoid cystic carcinoma of the lacrimal gland. Ophthal Plast Reconstr Surg. 2004;20:22.
14 Chan CC, Buggage RR, Nussenblatt RB. Intraocular lymphoma. Curr Opin Ophthalmol. 2002;13:411.
15 Fain LJ, Chann CC. Primary intraocular lymphoma. Arch Pathol Lab Med. 2009;133:1228-1232.
16 Shields CL, Shields JA, Gross NE, et al. Survey of 520 eyes with uveal metastases. Ophthalmology. 1997;104:1265.
17 Coroneo MT, Di Girolamo N, Wakefield D. The pathogenesis of pterygia. Curr Opin Ophthalmol. 1999;10:282.
18 Dushku N, John MK, Schultz GS, Reid TW. Pterygia pathogenesis: corneal invasion by matrix metalloproteinase expressing altered limbal epithelial basal cells. Arch Ophthalmol. 2001;119:695.
19 Mombaerts I, Goldschmeding R, Schlingemann RO, Koornneef L. What is orbital pseudotumor? Surv Ophthalmol. 1996;41:66.
20 Zobian J, Maus M. Thyroid-related ophthalmology. Curr Opin Ophthalmol. 1998;9:105.
21 Bressler NM, Bressler SB, Fine SL. Age-related macular degeneration. Surv Ophthalmol. 1998;32:375.
22 Smith BJ, O’Brien JM. The genetics of retinoblastoma and current diagnostic testing. J Pediatr Ophthalmol Strabismus. 1996;33:120.
23 Diener-West M, Hawkins BS, Markowitz JA, Schachat AP. A review of mortality from choroidal melanoma. II. A meta-analysis of 5-year mortality rates following enucleation. 1966 through 1988. Arch Ophthalmol. 1992;110:245.
24 The Collaborative Ophthalmology Study Group. The Collaborative Ocular Melanoma Study randomized trial of iodine-125 brachytherapy for choroidal melanoma, III. Initial mortality findings. COMS report No. 18. Arch Ophthalmol. 2001;119:969.
25 Collaborative Ocular Melanoma Study Group. The Collaborative Ocular Melanoma Study randomized trial of pre-enucleation radiation of large choroidal melanoma, II. Initial mortality findings. COMS report No. 10. Am J Ophthalmol. 1998;125:779.
26 Collaborative Ocular Melanoma Study Group. Mortality in patients with small choroidal melanoma. COMS report No. 4. Arch Ophthalmol. 1997;115:886.
27 Simon R, Durrleman S, Hoppe RT, et al. The Non-Hodgkins Lymphoma Pathologic Classification Project. Long-term follow-up of 1153 patients with non-Hodgkins lymphomas. Ann Intern Med. 1988;109:939.
28 Harris N, Jaffe ES, Stein H, et al. A revised European-American classification of lymphoid neoplasms. A proposal from the International Lymphoma Study Group. Blood. 1994;84:1361.
29 Harris NL, Stein H, Coupland SE, et al. New approaches to lymphoma diagnosis. Hematology (Am Soc Hematol Educ Progr). 2001;94:194-200.
30 Wolf LA, Reed GF, Buggage RR, et al. Vitreous cytokine levels. Ophthalmology. 2003;110:426.
31 Fujii H, Fugisada H, Kondo T, et al. Orbital Pseudotumor: Histopathological Classification and Treatment. Basel, Switzerland: Ophthalmologica; 1985.
32 Faustina M, Diba R, Ahmadi MA, et al. Patterns of regional and distant metastasis in patients with eyelid and periocular squamous cell carcinoma. Ophthalmology. 2004;111:1930.
33 McLean IW, Saraiva VS, Burnier MNJr. Pathological and prognostic features of uveal melanomas. Can J Ophthalmol. 2004;39:343.
34 Singh AD, Shields CL, Shields JA. Prognostic factors in uveal melanoma. Melanoma Res. 2001;11:255.
35 The Collaborative Ocular Melanoma Study Group. Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the Collaborative Ocular Melanoma Study (COMS). COMS report 15. Arch Ophthalmol. 2001;119:670.
36 Bessell EM, Henk JM, Wright JE, et al. Orbital and conjunctival lymphoma. Treatment and prognosis. Radiother Oncol. 1988;13:237.
37 Saeed P, Rootman J, Nugent RA, et al. Optic nerve sheath meningiomas. Ophthalmology. 2003;110:2019.
38 Tellado MV, McLean IW, Specht CS, Varga J. Adenoid cystic carcinomas of the lacrimal gland in childhood and adolescence. Ophthalmology. 1997;104:1622.
39 Dutton JJ. Optic nerve gliomas and meningioma. Neurol Clin. 1991;9:163.
40 Czyzyk E, Jozwiak S, Roszkowski M, Schwartz RA. Optic pathway gliomas in children with and without neurofibromatosis. J Child Neurol. 2003;18:471.
41 Dutton JJ. Gliomas of the anterior visual pathway. Surv Ophthalmol. 1994;38:427.
42 Font RL, Simmerman LE, Armaly MF. The nature of orange pigment over a choroidal melanoma: histochemical and microscopic observations. Arch Ophthalmol. 1974;91:359.
43 Diener-West M, Reynolds SM, Agugliaro DJ, et al. Screening for metastasis from choroidal melanoma. The Collaborative Ocular Melanoma Study Group Report 23. J Clin Oncol. 2004;22:2438.
44 The Collaborative Ocular Melanoma Study Group. Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the Collaborative Ocular Melanoma Study (COMS). COMS report 15. Arch Ophthalmol. 2001;119:670.
45 Demirci H, Shields CL, Chao AN, Shields JA. Uveal metastasis from breast cancer in 264 patients. Am J Ophthalmol. 2003;136:264.
46 Demirci H, Shields CL, Shields JA, et al. Orbital tumors in the older adult population. J Clin Ophthalmol. 2002;109:243.
47 Carbone PP, Kaplan HS, Musshoff K. Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res. 1971;31:1860-1861.
48 Saeed P, Rootman J, Nugent RA, et al. Optic nerve sheath meningiomas. Ophthalmology. 2003;110:2019-2030.
49 Egan RA, Lessell S. A contribution to the natural history of optic nerve sheath meningiomas. Arch Ophthalmol. 2002;120:1505-1508.
50 Turbin RE, Thompson CR, Kennerdell JS, et al. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophthalmology. 2002;109:890.
51 Mendenhall WM, Lessner AM. Orbital Pseudotumor. AJCO. Sept 4 2009. (epub ahead of print)
52 Flanders AE, Mafee MF, Rao VM, et al. CT characteristics of orbital pseudotumors and other orbital inflammatory processes. J Comput Assist Tomogr. 1989;13:40.
53 Clifton AG, Borgstein RL, Moseley IF, et al. Intracranial extension of orbital pseudotumor. Clin Radiol. 1992;45:23.
54 Enzmann DR, Donaldson SS, Kriss JP. Appearance of Graves’ disease on orbital computed tomography. J Comput Assist Tomogr. 1979;3:815.
55 Malhotra R, Huilgol SC, Huynh NT, Selva D. The Australian Mohs database, part II. Periocular basal cell carcinoma outcome at 5-year follow-up. Ophthalmology. 2004;111:631.
56 Malhotra R, James CL, Selva D, et al. The Australian Mohs database. Periocular squamous carcinoma. Ophthalmology. 2004;111:617.
57 Leshin B, Yeatts P, Anscher M, et al. Management of periocular basal cell carcinoma. Mohs’ micrographic surgery versus radiotherapy. Surv Ophthalmol. 1993;38:193.
58 Fitzpatrick PJ, Thompson GA, Easterbrook WM, et al. Basal and squamous cell carcinoma of the eyelids and their treatment by radiotherapy. Int J Radiat Oncol Biol Phys. 1984;10:449.
59 Singh AD, Rennie IG, Kivela T, et al. The Zimmerman-McLean-Foster hypothesis. 25 years later. Br J Ophthalmol. 2004;88:962.
60 Augsberger JA, Damato BE, Bornfield N. Uveal melanoma. In: Yanoff M, Duker JS, editors. Ophthalmology. 2nd ed. St. Louis: Mosby; 2004:1053.
61 Jampol LM, Moy CS, Murray TG, et al. The COMS Randomized Trial of Iodine 125 Brachytherapy for Choroidal Melanoma, IV. Local treatment failure and enucleation in the first 5 years after brachytherapy. COMS Report No. 19. Ophthalmology. 2002;109:2197-2206.
62 Wilson MW, Hungerford JL. Comparison of episcleral plaque and proton beam radiation therapy for the treatment of choroidal melanoma. Ophthalmology. 1999;106:1579-1587.
63 Bekkering GE, Rutjes AW, Vlassov VV, et al. The effectiveness and safety of proton radiation therapy for indications of the eye. A systematic review. Strahlenther Onkol. 2009;185(4):211-221.
64 Wiegel T, Bottke D, Kreusel KM, et al. German Cancer Society. External beam radiotherapy of choroidal metastases-final results of a prospective study of the German Cancer Society (ARO 95-08). Radiother Oncol. 2002;64:13.
65 Bolek TW, Moyses HM, Marcus RB, et al. Radiotherapy in the management of orbital lymphoma. Int J Radiat Oncol Biol Phys. 1999;44:31.
66 Dunbar SF, Linggood RM, Doppke KP, et al. Conjunctival lymphoma. Results and treatment with a single anterior electron field. A lens sparing approach. Int J Radiat Oncol Biol Phys. 1990;19:249.
67 Fung C, Tarbell N, Lucarelli M, et al. Ocular Adnexal lymphoma: Clinical behaviour of distinct World Health Organization Classification subtypes. Int J Radiat Oncol Biol Phys. 2003;57:1382-1391.
68 Letschert JG, Gonzáles DG, Oskam J, et al. Results of radiotherapy in patients with stage I orbital non-Hodgkin’s lymphoma. Radiother Oncol. 1991;22:36.
69 Pfeffer MR, Rabin T, Tsvang L, et al. Orbital lymphoma. Is it necessary to treat the entire orbit? Int J Radiat Oncol Biol Phys. 2004;60(2):527-530.
70 Hormigo A, DeAngelis LM. Primary ocular lymphoma. Clinical features, diagnosis, and treatment. Clin Lymphoma. 2003;4:22.
71 Hoffman PM, McKelvie P, Hall AJ, et al. Intraocular lymphoma. A series of 14 patients with clinicopathological features and treatment outcomes. Eye. 2003;17:513.
72 Narayan S, Cornblath WT, Sander HM, et al. Preliminary visual outcomes after three-dimensional conformal radiation therapy for optic nerve sheath meningioma. Int J Radiat Oncol Biol Phys. 2003;56:537.
73 Andrews DW, Faroozan R, Yang BP, et al. Fractionated stereotactic radiotherapy for the treatment of optic nerve sheath meningiomas. Preliminary observations of 33 optic nerves in 30 patients with historical comparison to observation with or without prior surgery. Neurosurgery. 2002;51:890.
74 Narayan S, Cornblath WT, Sandler HM, et al. Preliminary visual outcomes after three-dimensional conformal radiation therapy for optic nerve sheath meningioma. Int J Radiat Oncol Biol Phys. 2003;5:537.
75 Moyer PD Golnik KC, Breneman J. Treatment of optic nerve sheath meningioma with three-dimensional conformal radiation. Am J Ophthalmol. 2000;129:694.
76 Turbin RE, Pokorny K. Diagnosis and treatment of orbital optic nerve sheath meningioma. Cancer Control. 2004;11:334.
77 Jenkin D, Angyalfi S, Becker L, et al. Optic glioma in children. Surveillance, resection, or irradiation? Int J Radiat Oncol Biol Phys. 1993;25:215.
78 Sovalic JJ, Grigsby PW, Shepard MJ, et al. Radiation therapy for gliomas of the optic nerve and chiasm. Int J Radiat Oncol Biol Phys. 1990;18:927.
79 Tao ML, Barnes PD, Billett AL, et al. Childhood optic chiasm gliomas. Radiographic response following radiotherapy and long-term clinical outcome. Int J Radiat Oncol Biol Phys. 1997;39:579.
80 Packer RJ, Ater J, Allen J, et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg. 1997;86:747.
81 Schmandt SM, Packer RJ, Vezina LG, et al. Spontaneous regression of low-grade astrocytomas in childhood. Pediatr Neurosurg. 2000;32:132.
82 Pierce SM, Barnes PD, Loeffler JS, et al. Definitive radiation therapy in the management of symptomatic patients with optic glioma. Survival and long-term effects. Cancer. 1990;65:45.
83 Mahoney DHJr, Cohen ME, Friedman HS, et al. Carboplatin is effective therapy for young children with progressive optic pathway tumors. A Pediatric Oncology Group phase II study. Neurooncology. 2000;2:213.
84 Donaldson SS, McDougall IR, Egbert PR, et al. Treatment of orbital pseudotumor by radiation therapy. Int J Radiat Oncol Biol Phys. 1980;6:79.
85 Notter M, Kern TH, Forrer A, et al. Radiotherapy of pseudotumor orbitae. Front Radiat Ther Oncol. 1997;30:180.
86 Austin Seymour MM, Donaldson SS, Egbert PR, et al. Radiotherapy of lymphoid disease of the orbit. Int J Radiat Oncol Biol Phys. 1985;11:371.
87 Esmaeli B, Ahmadi MA, Youssef A, et al. Outcomes in patients with adenoid cystic carcinoma of the lacrimal gland. Ophthal Plast Reconstr Surg. 2004;20:22.
88 Peterson IA, Kriss JP, McDougall IR, et al. Prognostic factors in the radiotherapy of Graves’ ophthalmopathy. Int J Radiat Oncol Biol Phys. 1990;19:259.
89 Gorman CA, Garrity JA, Fatourechi V, et al. A prospective randomized, double-blinded controlled study of orbital radiotherapy for Graves’ ophthalmopathy. Ophthalmology. 2001;108:1523-1534.
90 Prummel MF, Terwee CB, Gerding MN, et al. A randomized controlled trial of orbital radiotherapy versus sham irradiation in patients with mild Graves’ ophthalmopathy. J Clin Endocrinol Metab. 2004;89:15.
91 Wakelkamp IM, Tan H, Saeed P, et al. Orbital irradiation for Graves’ ophthalmopathy. Is it safe? A long-term follow-up study. Ophthalmology. 2004;111:1557.
92 Cameron ME. Pterygium Throughout the World. Springfield, Illinois: Charles C Thomas; 1965.
93 Bahrassa F, Datta R. Postoperative beta radiation treatment of pterygium. Int J Radiat Oncol Biol Phys. 1983;9:679.
94 Youngson RM. Recurrence of pterygium after excision. Br J Ophthalmol. 1972;56:120.
95 Monteiro-Grillo I, Gaspar L, Monteiro-Grillo M, et al. Postoperative irradiation of primary or recurrent pterygium: and sequelae. Int J Radiat Oncol Biol Phys. 2000;48:865.
96 Nishimura Y, Nakai A, Yoshimasu T, et al. Long-term results of fractionated strontium-90 radiation therapy for pterygia. Int J Radiat Oncol Biol Phys. 2000;46:137.
97 Paryani SB, Scott WP, Wells JW, et al. Management of pterygium with surgery and radiation therapy. North Florida Pterygium Study Group. Int J Radiat Oncol Biol Phys. 1994;28:101.
98 Wilder RB, Buatti JM, Kittelson JM, et al. Pterygium treated with excision and postoperative beta irradiation. Int J Radiat Oncol Biol Phys. 1992;23:533.
99 Dusenbery KE, Alul IH, Holland EJ, et al. Beta irradiation of recurrent pterygia. Results and complications. Int J Radiat Oncol Biol Phys. 1992;24:315.
100 Jurgenliemk-Schulz IM, Hartman LJ, Roesink JM, et al. Prevention of pterygium recurrence by postoperative single-dose beta-irradiation. A prospective randomized clinical double-blind trial. Int J Radiat Oncol Biol Phys. 2004;59:1138.
101 Chakravarthy U, Houston RF, Archer DB. Treatment of age-related subfoveal neovascular membranes by teletherapy. A pilot study. British Journal of Ophthalmology. 1993;77:265-273.
102 Sasai K, Murata R, Mandai M, et al. Radiation therapy for ocular choroidal neovascularization (phase I/II study). Preliminary report. Int J Radiat Oncol Biol Phys. 1997;39:173-178.
103 Bergink GJ, Hoyng CB, van-der-Maazen RW, et al. A randomized controlled clinical trial on the efficacy of radiation therapy in the control of subfoveal choroidal neovascularization in age-related macular degeneration: radiation versus observation. Graefes Arch Clin Exp Ophthalmol. 1998;236:321.
104 Tholen AM, Meister A, Bernasconi PP, Messmer EP. Radiotherapy for choroidal neovascularization (CNV) in age-related macular degeneration (AMD). Klin Monastbl Augenheilkd. 2000;216:112.
105 Spaide RF, Guyer DR, McCormick B, et al. External beam radiation therapy for choroidal neovascularization. Ophthalmology. 1998;105:24.
106 Gripp S, Stammen J, Petersen C, et al. Radiotherapy in age-related macular degeneration. Int J Radiat Oncol Biol Phys. 2002;52:489.
107 Holz FG, Engenhart-Cabillic R, Unnebrink K, et al. A prospective, randomized, double-masked trial on radiation therapy for neovascular age-related macular degeneration. Ophthalmology. 1999;106:2239-2247.
108 Hart PM, Chakravarthy U, Mackenzie G, et al. Visual outcomes in the subfoveal radiotherapy study. A randomized controlled trial of teletherapy for age-related macular degeneration. Arch Ophthalmol. 2002;120(8):1029-1038.
109 Saconn P, Gee C, McCoy T, et al. Alternative dose for choroidal melanoma treated with an iodine 125 radioactive plaque. A single institution retrospective study. Int J Radiat Oncol Biol Phys. 2010;78:844-848.
110 Amdur RJ, Kalbaugh KJ, Ewald LM, et al. Radiation therapy for skin cancer near the eye. Kilovoltage x-rays versus electrons. Int J Radiat Oncol Biol Phys. 1992;23:767.
111 Shiu AS, Tung SS, Gastorf RJ, et al. Dosimetric evaluation of lead and tungsten eye shields in electron beam treatment. Int J Radiat Oncol Biol Phys. 1996;35:599.
112 Merriam GR, Focht EF. A clinical study of radiation cataracts and the relationship to dose. AJR Am J Roentgenol. 1957;77:759.
113 Deeg HJ, Fluornoy N, Sullivan K, et al. Cataracts after TBI and marrow transplantation. A sparing effect of dose fractionation. Int J Radiat Oncol Biol Phys. 1984;10:957-964.
114 Gordon KB, Char DH, Sagerman RH. Late effects of radiation on the eye and ocular adnexa. Int J Radiat Oncol Biol Phys. 1995;31:1123-1139.
115 Merriam GRJr. The effects of B-irradiation on the eye. Radiology. 1956;66:240.
116 Kwok SK, Ho PC, Leung SF, et al. An analysis of the incidence and risk factors of developing severe keratopathy in eyes after megavoltage external beam radiation. Ophthalmology. 1998;105:2051.
117 Fitzpatrick PJ, Thompson GA, Easterbrook WM, et al. Basal and squamous cell carcinoma of the eyelids and their treatment by radiotherapy. Int J Radiat Oncol Biol Phys. 1984;10:449.
118 Parson JT, Bova FJ, Fitzgerald CR, et al. Severe dry-eye syndrome following external beam irradiation. Int J Radiat Oncol Biol Phys. 1994;30:775.
119 Metcalfe P, Chapman A, Arnold A, et al. Intensity-modulated radiation therapy. Not a dry eye in the house. Australas Radiol. 2004;48:35.
120 Brady LW, Shields J, Augusburger J, et al. Radiation tolerance of normal tissues. Front Radiat Ther Oncol. 1989;23:238.
121 Parsons JT, Bova FJ, Fitzgeral CR, et al. Radiation retinopathy after external-beam irradiation. Analysis time-dose factors. Int J Radiat Oncol Biol Phys. 1994;30:765.
122 Emami B, Lyman J, Brown A, et al. Tolerance of normal tissue to therapeutic irradiation. Int J Radiat Oncol Biol Phys. 1991;21:109.
123 Harris JR, Levene MB. Visual complications following irradiation for pituitary adenomas and craniopharyngiomas. Radiation. 1976;120:167-171.
124 Stafford SL, Pollock BE, Leavitt JA, et al. A study on the radiation tolerance of the optic nerves and chiasm after stereotactic radiosurgery. Int J Radiat Oncol Biol Phys. 2003;55:1177.
125 Parsons JT, Bova FJ, Fitzgerald CR, et al. Radiation optic neuropathy after megavoltage external-beam irradiation. Analysis of time-dose factors. Int J Radiat Oncol Biol Phys. 1994;30:755.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 29 Orbital, Ocular, and Optic Nerve Tumors
Based on the incidence rates from the Surveillance, Epidemiology, and End Results Program of the National Cancer Institute, the American Cancer Society estimated that approximately 2390 new cases of all types of primary ocular and orbital malignant tumors were diagnosed in the United States in 2008.1 This demonstrates the rarity of ocular malignant tumors. Uveal melanoma is the single most common malignant tumor of the eye and orbit and makes up nearly 70% of all cancers of these tissues.1 Irradiation is used in the management of many ocular and orbital cancers, as well as in the management of several benign ocular processes. Understanding the pathophysiologic processes of ocular cancers and the results of radiation treatment can guide treatment recommendations.
Etiology and Epidemiology
Choroid Melanoma
Uveal melanoma is the most common primary malignant intraocular neoplasm.1 It is thought to arise from melanocytes of the uveal tract and in the United States occurs in approximately 1500 patients per year.1,2 Uveal melanomas are more common in lightly pigmented persons and are infrequent in nonwhite races. The average age at the time of diagnosis is 60 years in most large series,2–4 and the disease occurs with equal frequency in men and women. The tumors are rarely bilateral. Factors proposed as increasing the risk for the development of uveal melanoma are prolonged sustained sunlight exposure5,6 and lighter-colored irises.5,6 Certain diseases may predispose to melanoma, including xeroderma pigmentosum, which is an inherited disorder of DNA repair; oculodermal melanocytosis,7 in which the ocular surface and the uveal tract are hyperpigmented; and dysplastic nevus syndrome. There are some reports implying a possible rare genetic predisposition, although most melanomas occur as sporadic tumors.
Optic Nerve Tumors
Meningioma
Optic nerve sheath meningiomas arise from the meningothelial cap cells of arachnoid villi and can develop anywhere along the course of the optic nerve from the globe to the prechiasmal intracisternal optic nerve. They may be unilateral, bilateral, or multifocal. Patients with type 2 neurofibromatosis are predisposed to bilateral or multifocal lesions.8 Meningiomas from other locations can also extend to involve the optic nerve.
Optic nerve sheath meningiomas are more common in women. Dutton’s meta-analysis estimates a female preponderance of 61%.9 The typical age at presentation is 40 years. Bilateral cases appear to have an earlier age of onset.
Glioma
Optic nerve gliomas account for 1% to 5% of intracranial gliomas and 4% of orbital tumors. They occur most frequently in children, with 75% in the first decade and 90% in the first two decades of life.10 Approximately 25% of patients with optic gliomas also have type 1 neurofibromatosis. These tumors are increasingly being seen as markers of enhanced risk in patients and their families for the subsequent development of central nervous system tumors.11
Orbital Tumors
In a recent report of 1264 consecutive patients with orbital masses, 33% of patients had malignant lesions. Rhabdomyosarcoma was the most common malignant tumor in children, representing 3% of all orbital masses, and lymphoma was the most common malignant tumor in older patients, representing 10% of cases.12 In one large series, metastatic breast cancer comprised 4% of these lesions.
Lacrimal Gland Tumors
Lacrimal gland tumors are rare. The gender distributions are equal and patient age ranges from 10 to 73 years (mean of 46 years).13 Most of these lesions are malignant, with adenoid cystic carcinoma and mucoepidermoid cancer being the two most frequent ones. Benign pleomorphic adenomas also occur.
Lymphoma
Primary orbital lymphomas are rare and account for less than 1% of all lymphomas diagnosed.14 Lymphomas in the orbit can occur anteriorly in the conjunctival tissues or in the retrobulbar region. Patients with lymphoma at other sites can also present with eye involvement in the course of their disease.
Primary intraocular lymphoma may have malignant lymphoid cells involving the retina, vitreous, or optic nerve head, with or without concomitant central nervous system involvement. The incidence of primary intraocular lymphoma has been increasing over the past 15 years, in both immunocompetent and immunocompromised patients.15 The cause of this increased incidence is unknown. This is a disease of persons in their fifties and sixties, occurring more often in men than women.
Metastatic Tumors
The most common primary tumor to metastasize to the orbit is breast cancer.12 Other orbital metastases have been reported to occur from neuroblastoma, lung cancer, renal cell cancer, and prostate cancer.
Similarly, choroidal metastases are most frequently of breast cancer origin, followed by lung cancer. In one report of 420 patients, the uveal metastasis arose from a primary cancer of the breast in 196 patients (47%), the lung in 90 patients (21%), the gastrointestinal tract in 18 patients (4%), the kidney in 9 patients (2%), the skin in 9 patients (2%), the prostate in 9 patients (2%), and other cancers in 16 patients (4%).16
Benign Ocular Conditions
Pterygium
Most pterygia are located nasally and occur in patients aged 20 to 50 years.17 They are more commonly encountered in warm, dry climates or in patients who are chronically exposed to outdoor elements or smoky and dusty environments. Ultraviolet light exposure appears to be the most significant factor in the development of pterygia. Heredity may also be a factor.
Pterygia represent a degeneration of the conjunctival stroma with replacement by thickened, tortuous elastotic fibers.18 Activated fibroblasts in the leading edge of the pterygium invade and fragment Bowman’s layer. Pterygium development resembles actinic degeneration of the skin.
Pseudotumor
Orbital pseudotumor, or idiopathic orbital inflammatory syndrome, is a nonspecific inflammatory process of unknown etiology that may mimic orbital lymphoma.19 Because the disease mimics infectious or neoplastic processes, these must be ruled out. Autoimmune disease, endocrinopathies, and granulomatous processes must be excluded, as well as local infection with secondary involvement of the orbit, such as sinusitis and subperiosteal abscess. The diagnosis of pseudotumor is vague and remains one of exclusion.
Graves’ Ophthalmopathy
The pathogenesis of Graves’ ophthalmopathy is thought to be an autoimmune one in which activated T lymphocytes invade the orbit and stimulate glycosaminoglycan production in fibroblasts, resulting in tissue edema, lymphocyte infiltration, and marked enlargement of the extraocular muscles.20 Because lymphocytes and fibroblasts are sensitive to radiation, retrobulbar irradiation has been advocated as a treatment. The ophthalmopathy typically goes through three phases: progression, followed by stabilization, and perhaps some improvement. Residual signs of ophthalmopathy after the disease has reached a plateau are thought likely to be the result of fibrosis and other tissue changes rather than persistent inflammation.
Macular Degeneration
Age-related macular degeneration (AMD) is the leading cause of blindness in patients older than 65 years.21 The progressive loss of central vision is usually consequential to an atrophic process involving the retinal epithelium and choriocapillaries in the macula, which is classified as dry AMD. The wet form of AMD is an exudative process with subretinal neovascularization. Wet AMD generally progresses to destructive disciform scarring of the macula, with fibrovascular scars involving the choroid and sensory retina. Patients develop poor visual acuity.
Prevention and Early Detection
Because of the rarity of all ocular tumors, no prevention or early detection methods have been developed for most tumor types. However, in children born into a family with known retinoblastoma, early and careful ophthalmologic evaluation is warranted. Genetic counseling and evaluation of the siblings of patients with retinoblastoma is important, given the high hereditary likelihood.22 After the diagnosis of unilateral retinoblastoma, routine evaluation of the uninvolved eye is indicated because of the increased risk of contralateral disease (see Chapter 69).
Biologic Characteristics and Molecular Biology
Ocular Melanoma
Biologic characteristics that predict outcomes in patients with ocular melanoma include tumor cell type, tumor size, tumor location, and extension of the tumor outside the sclera.23 Cell type is discussed in the pathology section of this chapter, with patients with spindle cell tumors having a better survival rate than those with epithelioid cell tumors. Recent data suggest that specific chromosomal aberrations within the tumor may be associated with the length of survival. Many tumors are treated by means other than enucleation, and because biopsy is rarely performed, the cell type is often unknown.
Survival rates are adversely affected by increased size of the uveal melanoma. Although the earlier literature had various size classifications, the Collaborative Ocular Melanoma Study (COMS) has established a standard size classification.24–26 Small melanomas are 1 to 3 mm thick and less than 16 mm in largest dimension. Medium-sized melanomas are 3 to 8 mm thick and less than 16 mm in largest dimension, and large melanomas are more than 8 mm thick or more than 16 mm in largest dimension (Table 29-1).
TABLE 29-1 Various Other Definitions of Choroidal Melanoma Size
| Size | Measurement |
|---|---|
| Small | 1.5-2.4 mm height and 5-16 mm diameter |
| Medium | 2.5-10 mm height and <16 mm diameter |
| Large | >10 mm height and >16 mm diameter |
Prognosis is also affected by the location of the anterior tumor margin. Tumors located anterior to the equator of the eye in the anterior choroid or ciliary body have a worse prognosis than those located more posteriorly.23 Extraocular extension of a uveal melanoma has poor prognostic implications.24
Orbital Lymphoma
The biology of these tumors varies with the histologic classification within the Revised European-American Lymphoma system or the Working Formulation.27,28 Stage and histologic type play the most significant roles in outcome for orbital lesions.
Primary intraocular lymphoma is believed to be a subset of primary central nervous system lymphoma. It is usually a diffuse large B-cell non-Hodgkin’s lymphoma, and, less commonly, it may be T-cell lymphoma. Because pathologic diagnosis can be difficult, flow cytometry is often done to demonstrate monoclonal populations of B lymphocytes. Immunohistochemical assessments have also been used to demonstrate expression of BCL-6 and MUM1.29 These two markers have been revealed in systemic diffuse large B-cell lymphoma. Molecular analysis detecting immunoglobulin gene rearrangements and ocular cytokine levels showing elevated interleukin-10 (IL-10), with a ratio of IL-10 to IL-6 greater than 1, are helpful adjuncts for the diagnosis of intraocular lymphoma.14,30
Orbital Pseudotumor
The histopathologic type varies, and attempts to refine and subdivide patients have been advocated. The histopathologic spectrum of idiopathic orbital inflammation is typically nondiagnostic and diverse, ranging from the typical diffuse polymorphous infiltrate to the atypical granulomatous inflammation, tissue eosinophilia, and infiltrative sclerosis. One report suggested dividing this condition into three principal types: lymphoid, granulomatous, and sclerosing types.31 Several authors have endorsed the exclusion of reactive lymphoid hyperplasia and defined pseudotumor as a mixed inflammatory infiltrate with fibrosis of varying degrees.19
Pathology and Pathways of Spread
Skin Cancers
Basal cell cancers are seen in 90% of patients, followed by squamous cell cancers in 10%. For patients with squamous cell cancer, involved regional lymph nodes have been reported in as many as 24% of patients.32 Lymph node involvement is more common for larger tumors, recurrent tumors, and those with perineural invasion. Patients with recurrent lesions or perineural invasion may have tumor cells spread more peripherally than is clinically apparent. This may require wider radiation fields. Tumors located in the embryologic fusion planes of the face have been found to be more deeply infiltrating. This area around the medial canthus may affect the depth of treatment necessary for a successful outcome.
Ocular Melanoma
Uveal melanomas arise from melanocytes that are of neuroectodermal origin. Melanoma cells have a large nuclear to cytoplasmic ratio, prominent or multiple nucleoli, and frequent mitotic figures. Tumors are generally classified as composed of either spindle cells or epithelioid cells.33 Spindle cells have a fusiform shape and less pronounced atypia, whereas epithelioid cells are larger and more ovoid in shape and have more anaplastic characteristics. Uveal melanomas are classified as spindle cell melanomas if they are composed of only spindle cells, mixed cell melanomas if they contain both spindle cells and epithelioid cells, and epithelioid cell melanomas if they contain predominantly or exclusively epithelioid cells. Occasionally, the cells within the melanoma undergo necrosis, and these tumors are classified as necrotic melanomas.
The prognosis is more favorable in spindle cell melanoma, intermediate in mixed cell tumors, and poorest in epithelioid or necrotic melanoma. Other negative prognostic indicators include tumor dimension, thickness, and location of the anterior tumor margin.33 Melanomas tend to be slow-growing tumors, but the rate of growth can be quite variable. More rapidly growing tumors, which have a worse prognosis, tend to be anaplastic and composed of epithelioid cells.
At the time of diagnosis, most posterior uveal melanomas are confined inside the sclera of the eye. Extraocular extension of the melanoma can occur through emissary canals, through which normal ocular structures such as vessels and nerves enter and exit the sclera, and generally not by atrophy or thinning of the sclera. Extraocular extension is classified as microscopic or gross (visible on gross inspection), occurs in 8% to 14% of eyes harboring a melanoma, and is associated with a worse prognosis.34
Metastatic disease occurs in less than 3% of patients at the time of initial diagnosis. The most common sites of involvement include hematogenous spread to the liver and the lung. More unusual sites of spread include bone, skin and subcutaneous tissue, and brain and spinal cord tissue. Lymph node involvement was noted in 10% of patients dying from metastatic disease within the COMS.35
Choroidal Metastasis
The most common pathologic type is adenocarcinoma from the breast or non–small cell lung cancer. Most patients have other sites of metastatic disease. Disease extent can be localized to the choroid. The average number of lesions seen in the choroid in one large series was two.16 In this situation, involvement of the opposite choroid can occur in as many as 50% of patients. Orbital metastases can occur and are typically unilateral.
Orbital and Ocular Lymphoma
A wide range of non-Hodgkin’s lymphomas have been described involving the orbit and globe. In patients with orbital lymphomas, most of the lesions are of B-cell origin and classified as low or intermediate grade by the Working Formulation.36 The most common subtype of ocular adnexal lymphoma is a mucosa-associated lymphoid tissue (MALT) lymphoma. Intraocular lymphomas are usually of the aggressive, diffuse, large B-cell type.15
Orbital Meningioma
Meningiomas spread along paths of least resistance, remaining confined to the subarachnoid or intradural space of the intraorbital optic nerve. The tumor may invade the dura, adjacent orbital tissue, muscle, bone, and globe. These tumors may traverse the optic canal and affect the intracranial segment of the optic nerve or adjacent structures. Orbital meningiomas have histologic features similar to those of intracranial meningiomas. Most lesions demonstrate concentric formations of spindle cells or ovoid meningothelial cells with sheets of polygonal cells separated by vascular trabeculae.37
Lacrimal Gland Tumors
Pathologic findings are typically those of adenoid cystic cancer, followed by mucoepidermoid cancer and adenocarcinoma. Outcome seems to be related to the presence of necrosis, hemorrhage, perineural invasion, and high mitotic count.38
Optic Nerve Glioma
In the pediatric form of the disease, the cell of origin for optic nerve gliomas is unknown. Optic nerve gliomas are classified as grade I astrocytomas (pilocytic astrocytomas) in the World Health Organization (WHO) classification of gliomas, are slow growing, and do not tend to metastasize.39 Optic nerve gliomas develop in stages, from generalized hyperplasia of glial cells in the nerve to complete disorganization with loss of neural landmarks within the nerve and nerve sheath. A reactive meningeal hyperplasia may be incited, making it difficult to distinguish a glioma from a perioptic meningioma.
In pediatric patients with optic nerve glioma, 10% to 38% have type 1 neurofibromatosis, and 15% to 40% of children with neurofibromatosis 1 have an optic glioma.40 When bilateral, the lesions are pathognomonic for neurofibromatosis 1.
In the adult form of the disease, the optic nerve gliomas are more likely to be diffusely infiltrative, including astrocytoma (WHO grade II), anaplastic astrocytoma (WHO grade III), or glioblastoma multiforme (WHO grade IV).41 Nuclear atypia, mitoses, endothelial proliferation, and necrosis are potential histologic features.
Clinical Manifestations, Patient Evaluation, and Staging
The diagnostic algorithm for ocular, orbital, and lid tumors is given in Figure 29-1.
Ocular Melanoma
The diagnosis of melanoma is based primarily on ophthalmoscopic appearance and ancillary testing. The tumor typically appears as a unilateral, solitary, elevated, dark brown or gray, variably pigmented, dome-shaped mass (Fig. 29-2). Orange pigment (lipofuscin) is often found on the surface of the tumor and is thought to represent increased metabolic activity.42 Retinal detachment may be present and is more likely in larger tumors.
The initial ophthalmologic workup includes fundus photography, fluorescein angiography, and ultrasonography of the globe (Fig. 29-3). These tests facilitate making a correct diagnosis and are also useful in following the lesions for growth or regression after treatment. In a large prospective trial, 645 of 647 patients who underwent enucleation for melanoma were confirmed to be such at pathologic evaluation. Two of the patients had adenocarcinoma.24 In eyes with opaque media secondary to cataract or vitreous hemorrhage, ultrasonography of the globe and MRI of the orbits can allow an accurate diagnosis. Biopsy of a lesion suspicious for an ocular melanoma is used only in cases of diagnostic uncertainty. In patients requiring biopsy, histopathologic and immunohistochemical assessments can be useful in confirming the diagnosis.
To assess disease extent, obtain a general history and perform a physical examination with attention directed to skin lesions, weight loss, the lungs, and the liver. History of a prior cancer would indicate the possibility of a metastatic lesion in the eye rather than a primary melanoma. The most frequent site of metastasis is the liver, so the workup generally includes liver enzyme levels. Elevated levels will prompt evaluation with radiographic imaging. This approach was recently reported to have low sensitivity but high specificity and predictive value.43 Other sites of metastatic disease include the lung, and imaging with chest computed tomography (CT) scanning is most accurate for early detection of disease. The COMS report of sites of metastasis from melanoma demonstrated that sites included liver (91%), lung (28%), and bone (18%).43 Melanomas of the iris rarely metastasize. The brain is an infrequent site of metastasis; brain metastases were discovered in only 4% of patients at the time of death.44
Ocular Metastasis
Patients with uveal metastasis from breast cancer presented to ophthalmologists with visual symptoms in 93% of cases. Of 264 patients with uveal metastasis, 225 (85%) had choroidal metastasis, 8 (3%) had iris metastasis, 2 (<1%) had ciliary body metastasis, and 29 (11%) had metastasis in multiple uveal sites. Asymptomatic metastases were detected in 56% of patients in the opposite eye.45
Patients with orbital tumors more often present with diplopia, ocular motility disorder, and proptosis. Lesions are usually unilateral.46 Metastatic disease may be detected in a high proportion of the patients who present with this entity.
Orbital Lymphoma
Primary intraocular lymphoma is one of the most challenging intraocular tumors to diagnose. Cytologic examination of vitreal aspirates remains the gold standard for exclusion of neoplastic disease in patients with idiopathic uveitis.14 Patients with primary vitreous involvement should also have cerebrospinal fluid (CSF) examination and MRI of the brain and meninges. Eighty percent of patients with primary vitreous involvement have bilateral involvement.
Staging is done using the Ann Arbor staging system.47 A localized orbital lymphoma would be stage IeA.
Optic Nerve Meningioma
Patients typically present with visual loss, afferent papillary defect, color vision disturbance, visual field defect, proptosis, and optic disc edema. Ophthalmoscopic examination may reveal optic nerve head swelling, macular edema, nerve pallor, or choroidal folds. Optic disc swelling may also be observed. MRI is the procedure of choice for diagnosis of these lesions. Lesions appear slightly hypointense to brain and optic nerve tissue on T1-weighted images with fat suppression and gadolinium contrast. Thin CT scans may reveal regular or irregular thickening of the nerve sheath meninges. The nerve may appear normal in size or smaller as the result of circumferential compression or atrophy. Calcification has been described in 30% of patients and is associated with slower growth.48 Differential diagnosis includes sarcoid, demyelinating optic neuritis, orbital inflammatory disease, schwannoma, lymphoma, hemangiopericytoma, and optic nerve metastasis.
Usually these lesions are described as slow growing. Visual loss will generally occur in untreated eyes over a period of 5 to 10 years.49 Rapid growth has been observed during pregnancy, which may be mediated by hormone receptors.50 Other tumors have demonstrated rapid visual loss even in the absence of radiographic enlargement.
Optic Nerve Gliomas
Of optic pathway gliomas, 10% to 20% are confined to the orbit, with the remainder involving the intracranial compartment.10 MRI is the preferred method for evaluation of optic pathway glioma. Both the intraorbital lesion and its intracranial extent can be effectively characterized on MRI. When evaluating the orbit, gadolinium-enhanced T1-weighted images with fat saturation are effective at defining the extent of adult optic glioma. In the intracranial region, MRI provides superior evaluation of the optic nerve, chiasm, tracts, and geniculate body and of optic irradiation compared with CT scanning.
Orbital Pseudotumors
Patients present with proptosis, decreased ocular motility, soft tissue edema, and pain. Decreased visual acuity is not unusual. The condition may be unilateral or bilateral.51 CT scanning may reveal a unilateral focal or more diffuse mass. One review documented infiltration of the retrobulbar fat in 76% of patients, enlargement of the extraocular muscles in 57%, thickening of the optic nerve/sheath complex in 38%, contrast enhancement in 95%, and proptosis in 71%.52 Intracranial extension is unusual but has been documented in up to 8.8% of patients.53
Graves’ Ophthalmopathy
Signs and symptoms of Graves’ ophthalmopathy include bilateral exophthalmia, extraocular muscle dysfunction, diplopia, blurred vision, eyelid and periorbital edema, chemosis, lid lag and retraction, and compressive optic neuropathy. Imaging with CT scanning of the orbit will demonstrate changes consistent with enlargement of extraocular muscles, which are usually symmetric.54
