Chapter 3 Oogenesis
INTRODUCTION
Oogenesis is an area that has long been of interest in medicine, as well as biology, economics, sociology, and public policy. Almost four centuries ago, the English physician William Harvey (1578–1657) wrote ex ovo omnia—“all that is alive comes from the egg.” Oogenesis exemplifies many fundamental biologic processes, such as mitosis, meiosis, and intracellular and intercellular signaling. The process of oogenesis begins with migratory primordial germ cells (PGCs) and results in an ovulated egg containing genetic material, proteins, mRNA transcripts, and organelles that are essential to the early embryo.
The generation of healthy eggs with the correct genetic complement and the ability to develop into viable embryos requires that oocyte development and maturation be tightly regulated. In an effort to describe in detail our current understanding of the basic biologic mechanisms behind this process, this chapter discusses mammalian oogenesis as it is understood in the mouse model (Fig. 3-1) and highlights clinical conditions that are related to defects in oogenesis.
PRIMORDIAL GERM CELLS
Primordial germ cells are the founder cells of the gametes. In many species, including Drosophila, Danio rerio (zebrafish), and C. elegans, germ cells are derived from germ plasm, which is maternally derived cytoplasm that contains germ cell determinants. In contrast, mammalian PGC fate is thought to be induced by cell–cell interaction requiring the adhesion molecule E-cadherin and signaling from surrounding tissues.1,2
The migration of PGCs from the base of allantois to the genital ridges has been mapped out by alkaline phosphatase staining; more recently, their migration in vivo has been visualized by green fluorescent protein (GFP) expressed under the control of a truncated Oct-4 promoter (GFP:Oct-4) acting specifically in PGCs.3,4 Primordial germ cells are derived extragonadally from yolk sac endoderm and part of the allantois that arises from the posterior part of the primitive streak.
Primordial germ cells can be distinctly identified at 7.5 days post-coitum (dpc, a measure of embryonic age) during gastrulation by their high expression of alkaline phosphatase or GFP:Oct-4. At 7.5 dpc, PGCs are seen at the root of the allantois, from which they migrate along the endoderm via passive transfer mechanisms, to arrive at the epithelial layer of the hindgut at 8 dpc. Then, with the acquisition of motility via long pseudopodia, they traverse through the wall of the hindgut at 9 dpc.3
Specific Genes Expressed for Migration of Primordial Germ Cells
Several genes are known to be required for PGC cell fate determination, migration, and survival in the mouse embryo. Bone morphogenetic protein 4 (Bmp4) and Bmp8b are maximally expressed in the extraembryonic ectoderm adjacent to the proximal epiblast, where precursors of PGCs are localized at 6.5 dpc.5,6 These two proteins are thought to have essential roles in the differentiation of PGC precursors into PGCs because PGCs are absent in both Bmp4 and Bmp8b homozygous null mutant mouse models.6,7
Bone morphogenetic proteins are members of the transforming growth factor β (TGFβ) superfamily of growth factors, which also includes the TGFβ family and activins. These signaling molecules have diverse and often critical functions during embryonic development and in adult tissue homeostasis. They signal via formation of heterodimers or homodimers of the highly conserved carboxyl-terminal domain, which is characterized by six or seven cysteine residues that are critical to the structure, stability, and function of these proteins. TGFβ superfamily members signal by binding to serine/threonine kinase cell membrane receptors that leads to phosphorylation of proteins of the Smad family (named after the C. elegans gene sma and Drosophila gene Mad, reviewed in Shi and Massague8).
Smad complexes can bind DNA and mediate transcriptional activation or repression. Smad1 and Smad5 are known to act downstream of the Bmp signaling pathway and may have important functions in PGC development because loss of these proteins results in decreased size of the PGC population.9–11
Migration of PGCs through the hindgut and along the mesentery is mediated by the binding of c-Kit, a tyrosine kinase receptor expressed in PGCs, to its ligand, stem cell factor (Scf). Scf is expressed by somatic cells and its gradient along the migratory path is thought to direct the migration of PGCs. Mutations in the dominant white spotting (W) and Steel loci, which encode c-Kit and Scf, respectively, result in defects in gametogenesis. Although PGCs form, they do not proliferate, migrate to ectopic sites, and aggregate prematurely, so that the genital ridges are poorly populated by germ cells and the animals are sterile.12,13
Interestingly, these mutant mouse models also exhibit defects in melanogenesis and hematopoiesis because these genes are also required for the migration of melanocytes and mast cells.14 PGCs also express integrin subunits, which may potentially interact with laminin and fibronectin produced by cells in the genital ridges during the formation of the gonads. For example, integrin β1 is required for PGC colonization of the gonads, as the migration of PGCs is arrested at the gut endoderm in integrin β1 null mutants.15
OOGONIA AND THEIR MEIOTIC ENTRY TO BECOME OOCYTES
Colonization of the genital ridges by PGCs occurs concomitantly with the formation of the ovaries, at 12 to 13 dpc. Postmigratory PGCs, together with somatic epithelial cells that have derived from the coelomic epithelium and mesenchyme, form the ovarian cortical sex cords. Germ cell survival beyond 13 dpc requires the C2H2-type zinc-finger transcription factor Zpf148, which may activate tumor suppressor protein p53 in germ cells at this stage of development.16 Around the same time, PGCs differentiate into oogonia, which are sexually differentiated germ cells that would undergo the last rounds of mitotic divisions to establish the germ cell population at 11 to 13 dpc.17
Our understanding of mammalian oogonium function has been limited, perhaps due to their transient existence. Incomplete cytokinesis during synchronous, mitotic divisions of the oogonia results in the formation of clusters, or cysts, that are connected by intercellular bridges 0.5 to 1.0 μm in diameter.17 Oogonia become oocytes by entering meiosis I at 14 dpc; by 17 dpc, most oogonia have transitioned to oocytes. In the perinatal period, these germ cell cysts undergo breakdown, after which only a third of the oocytes survive and become enclosed in a single layer of granulosa cells to form primordial follicles.17 Thus, perinatal germ cell apoptosis appears to be a developmental process that is differentially regulated compared to the loss of oocytes during follicular atresia that occurs in the sexually mature animal. Pepling and Stradling suggested that cyst formation and subsequent breakdown may serve to ensure the incorporation of mitochondria from dying oocytes into those that survive.17
Oocyte Development
Prophase of Meiosis I
In zygotene, homologous chromosomes pair and begin to synapse. Their synapses are maintained by the synaptonemal complex, which is formed by many protein subunits, including synaptonemal complex protein 3 (Scp3) in the axial elements, Scp1 in the central element, and Scp2.18 Synapsis is completed in pachytene and continues to be maintained until diplotene, when homologous chromosomes are held together mainly at sites of chiasmata. The crossing-over and recombination of chromosomes occur over 4 days in the pachytene stage, prior to the formation of ovarian follicles.
MutL and MutS Families of Proteins
DNA mismatch recognition and repair by the MutL and MutS families of proteins are well conserved from yeast to humans.19 MutL and MutS heterodimeric protein complexes are thought to interact to activate DNA mismatch repair. Although the MutL and MutS vertebrate homologs (Mlh and Msh, respectively) are generally known for their roles in maintaining genomic stability against tumor formation, gene targeting in mice revealed that Mlh1, Mlh3, Msh4, and Msh5 have essential functions in meiosis in both males and females.20–24
Mlh1 and Mlh3 homozygous mutant females have similar infertility phenotypes, in which the newborn ovaries appear normal with a normal number of follicles at various stages of development. However, there is a significant decrease in the number of oocytes that can complete meiosis I, meiosis II, and undergo subsequent development to two-cell embryos.21,23 Both Mlh1 and Mlh3 colocalize to chromosomes in pachytene and are thought to have critical functions in meiotic recombination, as supported by the decreased number of chiasmata formed in the oocytes of these mutant animals.21,23,25
Although oocytes seem to enter and arrest at diplotene, chromosomes are unpaired and cannot stably attach to the bipolar spindle, which leads to abnormal meiotic I spindle formation, abnormal or incomplete meiosis I, and fertilization failure.25 Further, in both human and mouse pachytene oocytes, MLH1 and MLH3 serve as molecular markers of recombination nodules, which are necessary for the crossover structures that are present in the diplotene stage.26
Similarly, Msh4 and Msh5 have essential functions in pachytene of prophase I, although their phenotypes were more severe.20,22,24 Female null mutants of Msh4 and Msh5 have significant loss of oocytes by postnatal days 2 to 4, before meiotic arrest at the diplotene stage.20,22,24 Their oocytes enter leptotene to form synaptonemal complexes but fail to undergo complete pairing at zygotene and fail to enter diplotene. These oocytes undergo apoptosis, which results in atretic follicles, loss of follicular architecture, and premature ovarian failure. Both Msh4 and Msh5 localize to chromosomes, can form heterodimers in vitro, and are thought to function at the same time point during chromosomal synapsis.22 The relevance of these findings from the mouse model to human reproduction is further supported by the expression of MSH4 and MSH5 in the human testes and ovaries.27
We do not yet know the exact mechanisms by which these Mlh and Msh proteins act or how they bind to chromosomes. The oocyte apoptosis phenotype has been attributed to failure of chromosome pairing or formation of synapses. However, recent genetic analyses based on the Spo11/Msh or Spo11/Mlh double null mutants indicate that oocyte apoptosis is a consequence of failure to repair double-strand breaks.28 Spo11 initiates double-strand breaks, which are required for recombination. In the absence of Spo11, the phenotype of Mlh1 and Msh5 was less severe and resembled that of Spo11. Similarly, Dmc1, another protein involved in recombination, and ataxia-telengiectasia mutated (Atm), a DNA damage checkpoint protein, are both required for the repair of double-strand breaks; in their absence, persistent double-strand breaks lead to oocyte death via apoptosis.28
The functional mechanism of a protein is often elucidated by identifying proteins with which it associates and the nature of their interactions. The colocalization of Mlh1 proteins with Scp3 and cyclin-dependent kinase 2 (Cdk2) may suggest interaction or related functions, especially since Scp3 and Cdk2 have also been shown to be required in meiosis.29,30 Scp3 is a key protein subunit in the synaptonemal complex, which holds homologous chromosome pairs to facilitate synapsis. Female mice lacking Scp3 are subfertile, because many of their oocytes contain univalents (unpaired chromosomes), exhibit abnormal chromosomal segregation, and produce a decreased number of viable embryos. Interestingly, their fertility worsens with age, so that this model is thought to be potentially powerful in studying the age-related subfertility and aneuploidy that are major problems in human reproduction.30
Cdk Family of Proteins
Cyclin-dependent kinase (Cdk2) belongs to the family of Cdk proteins, which are considered master regulators of the cell cycle.31 However, Cdk2-null mutants are unexpectedly viable; thus, Cdk2 is not required for mitosis during development in vivo to be required for prophase I of meiosis in the oocytes instead. Oocyte loss and premature ovarian failure occur by the first few postnatal days. Although the precise molecular defects differ, a similar phenotype is seen in the null mutants of Dmc1, Msh4, Msh5, and Atm, presumably because these oocytes fail to enter or complete pachytene, and enter the common apoptotic pathway by default. In contrast, by postnatal day 5, all oocytes in wild type mice have progressed to the diplotene stage, where they remain arrested until peri-ovulation in the sexually mature adult, when they resume and complete meiosis.
OOCYTE DEVELOPMENT AND EARLY STAGES OF FOLLICULOGENESIS
The transition from pachytene to diplotene marks the beginning of folliculogenesis, which is closely associated with subsequent oocyte development. Whereas no pachytene oocytes are found within follicles, approximately 80% of diplotene oocytes are enclosed in primordial or more developmentally advanced follicles.32 Primordial follicles are formed by the encapsulation of a diplotene-arrested oocyte by a single flat layer of granulosa cells. Subsequently, the granulosa cells, still in a single layer, assume a cuboidal shape to form primary follicles.
Transcription Factors
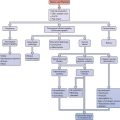
Our understanding of primordial and primary follicular development has recently been enlightened by the discovery of the requirement of two transcription factors at these stages of development (Fig. 3-2). Figα, a β-loop-helix-loop transcription factor, is required for oocyte survival into the primordial follicle stage.33 Based on its known role in the transcription of oocyte-specific genes zona pellucida 1 (zp1), zp2, and zp3, its role in primordial follicle formation is presumed to be the transcription of other oocyte-specific transcription factors.33
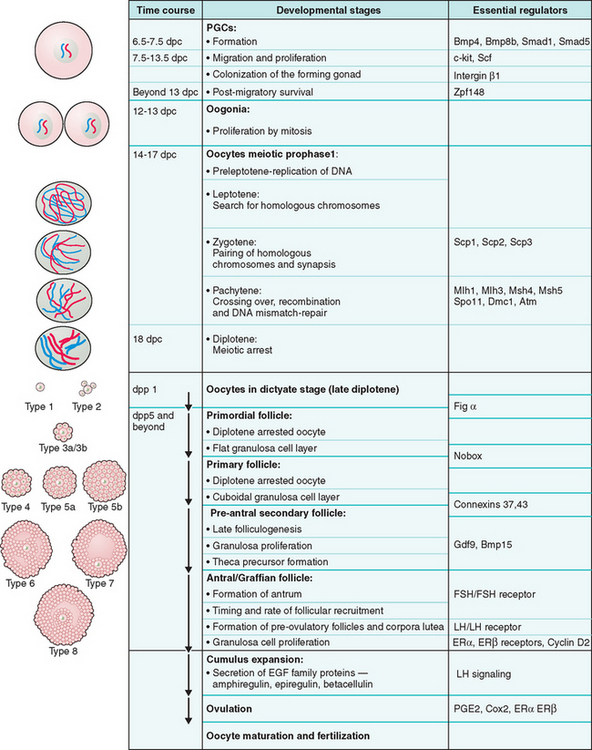
Figure 3-2 Schematic representation of oogenesis and folliculogenesis during development in the mouse model. This figure shows and correlates the developmental stages of the oocyte and follicle, and highlights examples of essential regulators at each stage. The “Types 1-8” nomenclature, as described by Pedersen and Peters191, is also shown to facilitate interpretation of the literature. PGCs, primordial germ cells; dpc, number of days postcoitum; dpp, number of days postpartum.
Another transcription factor, Nobox, which is an oocyte-specific homeobox protein, has a critical role in the transition of primordial to primary follicles.34 In the absence of Nobox, mouse ovarian follicles do not develop beyond the primordial stage, which results in massive oocyte loss in the early postnatal period.34 Identification of novel downstream target genes of Figα and Nobox in the future should provide insight into these early stages of folliculogenesis.
Zona Pellucida
The growing oocyte synthesizes zona pellucida glycoproteins, which comprise approximately 17% of the total cellular protein content. The zona pellucida proteins zp1, zp2, and zp3 are encoded by distinct genes and are coordinately expressed under the control of Figα during oocyte growth.33 These zona proteins form the zona pellucida or zona matrix that surrounds the growing oocyte, but each one has a critical role in proper oocyte and embryo development. zp1, the only zp protein that forms intermolecular disulfide bonds, contributes to 10% to 15% of the zona mass and is thought to provide structural integrity to the zona pellucida. zp3 serves as the receptor for sperm binding and the subsequent acrosome reaction; zp2 functions as a secondary receptor. Together, zp2 and zp3 mediate sperm binding and species specificity during fertilization.
These three glycoproteins are essential for fertilization and viability of growing oocytes and early embryos. Mice lacking zp3 cannot form the zona matrix and are sterile,35 and zp1 knockout mice have decreased fecundity and precocious hatching of early embryos from the structurally compromised zona matrix.36 Further, ectopic granulosa cells are seen between the oolemma and zona pellucida, and may increase the perivitelline space prior to ovulation.36 It is important to note that these may be four zona pellucida genes that are expressed in humans (ZP1, ZP2, ZP3, and zPB). Interestingly, abnormalities and lack of zona pellucida in human oocytes have been observed during IVF; however, the cause and implication of this defect in human reproduction is not well understood.
Regulation of Folliculogenesis by Oocyte Morphogens
Although bidirectional communication between the oocyte and follicular cells is required for the development of the meiotically competent oocyte, there is increasing support to view the oocyte as autonomous in determining its own fate by regulating follicular development via the secretion of various growth factors.37,38 The establishment of a morphogen gradient by the oocyte regulates follicular development via differential gene expression and function in granulosa cells. In the absence of these morphogens, follicle-stimulating hormone (FSH) induces all cumulus cells, which are directly adjacent to the oocyte and have distinct functions, to differentiate into membrane granulosa cells. Currently, several members of the TGFβ superfamily are the most well-understood candidate oocyte morphogens.
Transforming Growth Factor β
The mammalian oocyte expresses at least three TGFβ superfamily members: growth differentiation factor 9 (Gdf9), Bmp15, and Bmp6.38 Although Gdf9 is expressed throughout the reproductive tract and bone marrow,39 its oocyte expression is restricted to oocytes in follicles at the primary (type 3a), preantral, or more advanced stages of development.39,40 After fertilization, the level of Gdf9 transcripts decreases to low or undetectable in preimplantation embryos.41
Gdf9 is a critical regulator of follicular development. In Gdf9-deficient female mice, primordial and primary follicles appear normal, but development beyond type 3a follicles is blocked and the animals are sterile.42 Further, in the absence of Gdf9, theca cell precursors are absent around the follicles, and the expression of kit ligand and inhibin-α is upregulated in the granulosa cells of the primary follicles.43 The increased levels of kit ligand are thought to mediate excessive oocyte growth via interaction with the c-kit tyrosine kinase receptor on the oocyte, leading to abnormally large oocytes and their cell death.43 Interestingly, Gdf9 deficiency also causes failure of granulosa cells to proliferate or undergo cell death, which presumably leads to their abnormal differentiation into clusters of steroidogenic cells reminiscent of corpora lutea.43 Therefore, the oocyte can be viewed as the master regulator of early folliculogenesis via its secretion of Gdf9.
The oocyte expression pattern of Gdf9 is shared by its family member Bmp15, which is encoded by an X-linked gene.44,45 Bmp15 homozygous null mutant females are subfertile due to decreased rates of ovulation and early embryo survival. Further, an effect of gene dosage is observed in Gdf9+/− Bmp15−/− mice, which have a more severe phenotype than Bmp15−/− mice.46 In addition, cumulus–oocyte reconstitution and RNA interference (RNAi) experiments demonstrated that both proteins regulate cumulus expansion in vitro. Therefore, Gdf9 and Bmp15 are thought to have synergistic roles in the regulation of folliculogenesis.
Interestingly, naturally occurring mutations in GDF9 and BMP15 are common in certain strains of domestic sheep, perhaps as a result of breeding practices. Although sheep carrying mutations in both alleles of GDF9 or BMP15 are sterile, those carrying a single mutant allele have increased ovulation rates and are “super fertile.” Superovulation in the presence of heterozygosity of either of these genes is thought to be caused by decreased inhibin production by granulosa cells, which in turn leads to increased pituitary FSH secretion, development of more than one dominant follicle, and ultimately superovulation. Thus, the paradoxical increase in fertility when one allele is mutated exemplifies the impact of gene dosage on ovarian follicular function.45
RECRUITMENT OF OVARIAN FOLLICLES
Nongrowing mouse oocytes within primordial follicles have a diameter of approximately 12 μm and comprise the resting pool. Cohorts from the resting pool are continuously induced by ovarian paracrine signals to undergo coordinated oocyte and follicular growth during a process called initial recruitment (Fig. 3-347). The recruited, growing follicles are called primary follicles, but this growth phase is very protracted so that primordial and primary follicles may not be easily distinguishable. These growing follicles subsequently develop into secondary and antral follicles. (The timeline of different stages of follicular development in humans and rodents are shown in Fig. 3-4 and 3-5.47)

(From McGee EA, Hsueh AJW: Initial and cyclic recruitment of ovarian follicles. Endocrine Reviews 21:200–214, 2000; with permission. Copyright 2000, The Endocrine Society.47)

(From McGee EA, Hsueh AJW: Initial and cyclic recruitment of ovarian follicles. Endocrine Reviews 21:200–214, 2000; with permission. Copyright 2000, The Endocrine Society.47)

(From McGee EA, Hsueh AJW: Initial and cyclic recruitment of ovarian follicles. Endocrine Reviews 21:200–214, 2000; with permission. Copyright 2000, The Endocrine Society.47)
The oocyte grows from approximately 12 μm to approximately 80 μm in the fully grown state, which is achieved prior to formation of the antral follicle in sexually mature mice. In general, antral follicles tend to undergo follicular atresia, which is initiated by apoptosis in the granulosa cells, unless they are rescued by increased levels of FSH in the estrus or menstrual cycles in mice and humans, respectively. This cyclic recruitment of antral follicles results in the rescue and continued development of a few follicles, but usually only one dominant follicle prevails and develops to the periovulatory stage in the human menstrual cycle.47 Although cyclic recruitment is operative after puberty in humans, most antral follicles ultimately undergo atresia, which results in oocyte death.
Primordial follicles that have not been recruited are thought to remain dormant and be available for recruitment at a later time. Therefore, maintenance of the pool of nongrowing oocytes is necessary to sustain a normal reproductive life span. Müllerian inhibiting substance or antimüllerian hormone, a TGFβ superfamily member well-known for its regulatory role in the sexual differentiation of the reproductive tract during development, is expressed in granulosa cells of growing follicles starting in the perinatal period.48 During folliculogenesis, it inhibits the recruitment of primordial follicles to the pool of growing follicles by decreasing the responsiveness of granulosa cells to FSH.49 Antimüllerian hormone is recognized as an important clinical marker of the ovarian reserve of viable follicles.
Regulation of Folliculogenesis by Gonadotropins
Preantral follicle development, which involves oocyte growth with limited granulosa cell proliferation, is regulated predominantly by paracrine and autocrine signals. Although gonadotropins are not required for early follicle development in vivo, these follicles can develop in response to FSH in vitro.47 In contrast, gonadotropins are required for antrum formation and the rapid granulosa cell proliferation that results in the graafian or antral follicle.
FSHR comprises a large extracellular domain that binds the FSH hormone with high affinity, and a seven membrane-spanning domain that activates downstream cyclic adenylate cyclase signaling via the heterotrimeric G proteins.50 Mice deficient in the FSH β subunit, which normally forms heterodimers with the α subunits, are infertile due to arrested follicular development prior to formation of the antrum.51
Consistent with the critical role of FSH signaling via its receptor, follitropin receptor knockout (FORKO) mice are also infertile.52 At postnatal day 2, there are fewer nongrowing but more growing follicles, but by postnatal day 24, the numbers of resting and growing follicles are both decreased. Therefore, postnatal recruitment of resting follicles into the growing phase appears to be aberrantly accelerated, followed by arrest in further follicular recruitment later on.
Further, no antral follicles are observed. Although these mouse models confirm that FSH signaling is required for follicular development beyond the preantral stage, the phenotype of the FORKO mice also suggests that FSHR signaling is essential in the regulation of the timing and rate of follicular recruitment.52 In contrast, luteinizing hormone receptor knockout mice (LuRKO) females are infertile, presumably due to defects in the later stages of folliculogenesis because follicles up to the early antral stage are present, but preovulatory follicles and corpora lutea fail to form.53
FSH and FSHR-mediated downstream intracellular signaling results in gene and protein expression that exemplifies granulosa cell function. For example, antimüllerian hormone, which is implicated in primordial follicle recruitment, exhibits an aberrant expression pattern in FORKO mice.52 Although recruitment of theca cells and their expression of the LH receptor and P450 aromatase genes are independent of FSH signaling, expression of these genes in granulosa cells is dependent on FSH signaling.54 Similarly, the genes encoding for inhibin and activin subunits, whose homodimers and heterodimers are thought to be important paracrine and autocrine mediators of follicular growth, have decreased expression levels in granulosa cells in the FSHβ knockout model.54 Other genes that are dysregulated as a result of FSHβ deficiency include those encoding for androgen receptor (AR), estrogen receptor β (ERβ), and cyclin D2, all of which have been shown to be essential in the final stages of follicular development.54
Estrogen Receptors
Two estrogen receptors, ERα and ERβ, are expressed in granulosa cells in the antral follicle. Different knockout models have been generated for these two receptors based on different strategies of gene targeting, but they produced similar phenotypes.55 ERαKO females are infertile; ERβKO females are subfertile. In both ERαKO and ERβKO mice, folliculogenesis progresses normally until the large antral stage, when ERβ is critical for ER-mediated granulosa cell proliferation and subsequent ovulation.
In contrast, ERα is not critical for follicular growth but is required for ovulation. The ERαβKO females clarify the question of functional redundancy between the two ERs by demonstrating that in the absence of ERs, antral follicles only have single layers of granulosa and theca cells, multiple small pockets of antral fluid, and oocytes that are dissociated from cumulus cells.55
D-type Cyclin
Granulosa cell proliferation in late folliculogenesis also requires cyclin D2, which belongs to the D-type cyclin family of cell cycle regulators that promote the transition from G1 to S phases via binding to Cdk4 or Cdk6.56 Cyclin D2–deficient females have decreased FSH-mediated granulosa cell proliferation, which results in antral follicles that have significantly fewer layers of granulosa cells. This defect in granulosa cell proliferation also results in ovulation failure on stimulation by LH. However, the number of follicles is not decreased, luteinization of theca cells proceeds, and follicles differentiate to become corpora lutea.
We learn from the cyclin D2–deficient mutants that although these final stages of folliculogenesis and ovulation require cyclin D2 via FSHR-mediated activation, specifically of protein kinase A (PKA), oogenesis per se does not require these last rounds of granulosa cell proliferation. In fact, the oocyte not only appears normal, but it is also competent to undergo meiotic resumption and fertilization to produce viable embryos.56 Therefore, although oogenesis and folliculogenesis are closely intertwined, oocyte development is a cell-autonomous process to a certain extent, especially in the advanced stages and at periovulation.
THE ROLE OF CUMULUS CELLS IN OOGENESIS
Cumulus cells, also called corona radiata, are specialized granulosa cells that directly line the oocyte. In addition to their supportive role in cytoplasmic maturation of the oocyte, they have important functions in oocyte development, including the maintainenance of meiotic arrest and induction of ovulation. Ovulation is a broad term that encompasses the processes of follicular luteinization, follicle rupture, and meiotic resumption in the oocyte.
A critical step during LH-induced ovulation is the cumulus cell-mediated cumulus expansion. Mural granulosa cells express LH receptor, which allows them to respond to LH by secreting proteins of the epidermal growth factor (EGF) family—amphiregulin, epiregulin, and betacellulin—which are thought to act as paracrine signals that lead to cumulus expansion.57 During this process, cumulus cells disperse in the extracellular matrix, which comprises hyaluronic acid, tumor necrosis factor-stimulated gene 6 (TSG6), and serum-derived inter-α-inhibitor, all of which are essential for follicular rupture.58
The cumulus expression of Tsg6 and several other proteins implicated in cumulus expansion is regulated by prostaglandin E2 (PGE2) via its receptor EP2.58 Consistent with the critical role of PGE2 in cumulus expansion and ovulation, mice deficient in either EP2 or cycloxygenase-2 (Cox-2), which is the rate-limiting enzyme for PGE2, are infertile due to their inability to ovulate.58–60
Connexin and Gap Junctions
Cumulus cells mediate many of their important functions by communicating through gap junctions among themselves as well as with the oolemma. These gap junctions are intercellular channels formed by proteins of the connexin family for the diffusion of sugars, amino acids, lipid precursors, nucleotides, metabolites, and signaling molecules. Connexin family members share the same protein domains, including four membrane-spanning domains, two extracellular loops, a cytoplasmic loop, and cytoplasmic N- and C-terminals.61 In mice, there are at least 17 connexin proteins, whose distinct sequence or length in the cytoplasmic loops and C-terminal tails, as well as heterodimeric and homodimeric coupling, allow functional diversity.
In mice, connexin (Cx) 32, 37, 43, 45, and 57 are expressed in the cumulus–oocyte complex, where they are located between cumulus cells, on cumulus transzonal projections that anchor cumulus cells into the zona pellucida, or on the microvilli or plasma membrane of the oocyte.61
The critical role of gap junctions in oogenesis is exemplified by the sterility phenotype found in Cx37-deficient mice. Cx37 is made by both the oocyte and granulosa cells, but it may be the only connexin member in cumulus–oocyte gap junctions that is contributed by the oocyte.61 In the absence of Cx37, follicular development fails at the preantral-to-antral transition, so that most follicles are arrested at the primary follicle stage, and only a few small antral follicles are present. Further, ovulation does not occur despite the formation of numerous corpora lutea.62
Similarly, in vitro experiments show that Cx43, which is also expressed in granulosa cells, is required for folliculogenesis beyond the primary follicle stage.63 Most importantly, oocytes from these two mutant mouse models are meiotically incompetent, which may reflect the requirement of cumulus cell communication in the acquisition of meiotic competence.
These connexin-deficient experimental models demonstrate the critical role of gap junctions in folliculogenesis, but they do not explain the role of cumulus cells in the regulation of meiotic arrest and resumption in the oocyte. Numerous models and hypotheses attempt to explain how and to what extent cumulus cells mediate the high oocyte intracellular cAMP levels that are required to maintain meiotic arrest even after the oocyte has acquired meiotic competence.64 Although the complete story remains to be told, several key players have been identified.
MAINTENANCE OF MEIOTIC ARREST
GTP-binding Protein
The stimulatory heterotrimeric GTP-binding protein (Gs)-linked receptor 3 (GPR3) is expressed in the oocyte plasma membrane and is thought to mediate cumulus control of meiotic arrest. However, GPR3 is capable of activity independent of LH or cumulus cells also. The role of GPR3 is exemplified by ovarian sections from prepubertal GPR3−/− females in which meiotic stages beyond prophase I are seen in oocytes in intact early antral follicles in the absence of LH signaling or cumulus expansion.65
Similarly, inactivation of Gs by microinjection of an antibody specifically targeting Gs into the oocyte within an intact follicle allows the oocyte to resume meiosis even if cumulus cells remain adherent to the oocyte.65 GPR3 maintains activity of Gs in stimulating adenylyl cyclase to produce high cAMP levels, which in turn maintain PKA activity.65 Specifically, the isoform adenylyl cyclase 3 expressed by the oocyte is critical in generating sufficient cAMP.66 Acting downstream of the GPR3→Gs→cAMP→PKA signaling cascade, PKA-dependent phosphorylation of substrate proteins presumably inhibits meiotic resumption through as-yet unknown mechanisms that include suppression of M-phase promoting factor (MPF) activity; this represents an area of intense investigation.64
MEIOTIC RESUMPTION
Whereas LH triggers ovulation by the cumulus cell compartment and release of the oocyte from meiotic arrest, the resumption and completion of meiosis may be regulated intrinsically to a large extent by the fully grown oocyte. Resumption of meiosis requires both decreased levels of intracellular cAMP and activation of MPF.64 Decreased production and increased degradation of cAMP in the oocyte appear to contribute to meiotic resumption.
Decreased cAMP production is mediated by the inhibitory G protein, which in turn is activated by an oocyte-expressed G protein-coupled receptor called leucine-rich repeat-containing G protein-coupled receptor 8 (LGR8).67 Interestingly, the role of LH in meiotic resumption is demonstrated by this LGR8-mediated decrease in cAMP levels, because LGR8 is stimulated by insulin-like 3, which is expressed and secreted by ovarian theca cells in response to LH.67
Phosphodiesterase (PDE)
On the other hand, the degradation and inactivation of cAMP in the oocyte are mediated by the oocyte-expressed cyclic nucleotide phosphodiesterase family member, PDE3A. GV oocytes from PDE3A−/− mice fail to resume meiosis in vivo and remain in the GV stage even after ovulation. In addition, meiosis does not resume even when fully grown GV oocytes from these mice are isolated from cumulus cells and cultured in vitro.68 Thus, the GPR3 and PDE3A knockout mouse models demonstrate their critical and opposing roles in the regulation of meiotic arrest and resumption, respectively. More importantly, these models also prove that ovulation and meiotic resumption, though usually occurring synchronously, are independent processes that can be differentially regulated.
Meiotic Competence and Oocyte Maturation
Meiotic progression requires activation of MPF, which is composed of cyclin B and its protein kinase Cdk1 (also called p34cdc2).69 The requirement of MPF for the G→M transition in mitosis and meiosis is highly conserved across species. Activation and inactivation of many cell-cycle regulators are mediated via phosphorylation, a form of post-translational protein modification of specific residues. The dual phosphatase, Cdc25b, is required for the dephosphorylation of residues 14 and 15 of p34cdc2, which results in activating p34cdc2. Oocytes from Cdc25b−/− mice are unable to resume meiosis and remain arrested in prophase unless rescued by microinjection of Cdc25b mRNA.70 Active p34cdc2 accumulates rapidly through an autoamplification loop. Therefore, the rate-limiting step of MPF activation is thought to be the synthesis and accumulation of cyclin B. During oocyte growth, the level of active MPF increases and eventually reaches the threshold above which the oocyte becomes meiotically competent, which refers to its ability to undergo meiotic resumption.
In general, meiotic competence is related to oocyte size, presumably because cytoplasmic volume reflects the accumulation of synthesized proteins, including cyclin B, p34cdc2, and Cdc25b, which are essential for meiosis.71 Oocytes that are smaller than 70 to 80 μm have not reached their full growth potential and have low rates of meiotic competence, whereas those approximately 100 μm are considered fully grown and tend to be able to undergo meiosis. Another predictor of meiotic competence is the presence of a surround nucleolar (SN) chromosomal localization pattern that can be visualized in the live oocyte with culture media containing Hoescht stain.72,73
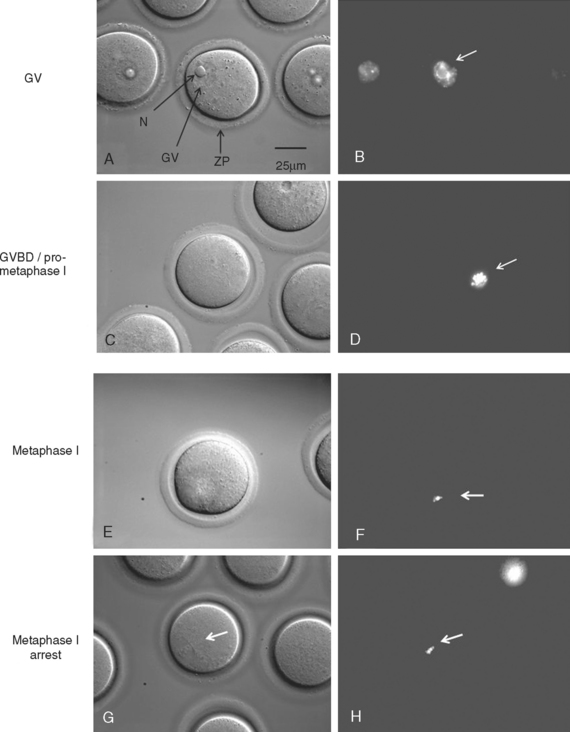
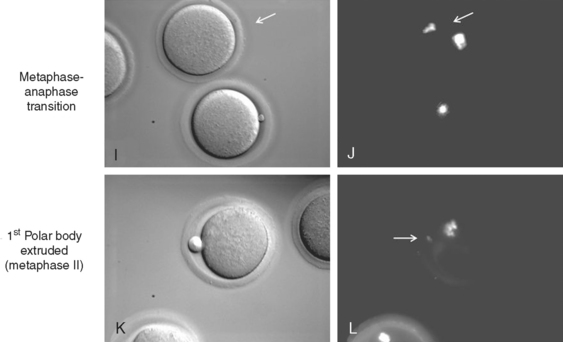
Germinal Vesicle Breakdown (GVBD)
At the anaphase–metaphase transition, chromosomes begin to segregate, and a cytoplasmic protrusion signifying late anaphase may be visible. MPF activity increases as the oocyte progresses from GV, GVBD, to metaphase I, but then declines transiently between metaphase I and metaphase II.74,75 However, progress beyond metaphase I may not occur in some oocytes if molecular prerequisites are not met.76
Metaphase–Anaphase Transition
The metaphase–anaphase transition is regulated by decreased MPF activity, suppression of protein kinase C (PKC) activity, and the activity of many other proteins.77–79 Decreased MPF activity is achieved by proteasome degradation of cyclin B, which is required for polar body extrusion (metaphase I–metaphase II) and at metaphase II exit on fertilization.80–83 Like many mitotic and meiotic cell cycle regulators, the proteolysis of cyclin B is mediated by ubiquitin and the anaphase-promoting complex (APC), which are well conserved and required for proteolysis of many proteins across species.
The anaphase-promoting complex is an E3 ubiquitin ligase that transfers ubiquitin “tags” from E2 ubiquitin conjugating enzymes to substrates, such as cyclin B, thereby “tagging” them for recognition and proteolysis by the 26S proteasome.82 The essential role of the proteasome in meiosis is further supported by the metaphase I arrest phenotype that is seen in mouse and rat oocytes cultured in media containing chemical inhibitors of the proteasome.84,85
Meiotic Spindle
Achievement of metaphase I depends on the formation of an intact meiotic spindle, which requires the cell cycle regulator, cell division cycle-6 homologue (Cdc6). Insight into this process is provided by oocytes in which Cdc6 expression is abrogated by the RNA interference technique.86 These oocytes undergo GVBD, but the meiotic spindle is not formed, and they fail to reach metaphase I. In the absence of the meiotic spindle, condensed chromosomes fail to form bivalents and do not align to form the metaphase plate. Instead, the chromosomes form a halolike structure and meiosis I is arrested with no cell division or extrusion of polar body.86
In mitosis, the centromeres of sister chromatids are held together by cohesin until the onset of anaphase, when cohesin cleavage by separase allows separation of sister chromatids.87–89 Inhibition of separase activity by securin prior to anaphase onset, and its subsequent activation at anaphase upon securin degradation via the APC, are critical in ensuring correct chromosomal segregation.90,91 The APC→securin→separase cascade is also required to ensure correct spindle function and homologous chromosome disjunction during the metaphase–anaphase transition in meiosis in the oocyte.92,93
Spindle Assembly Checkpoint (SAC)
Another mechanism that the oocyte has in common with mitotic cells to ensure the correct segregation of chromosomes is the spindle assembly checkpoint (SAC). SAC proteins detect whether chromosomes are properly aligned and whether kinetochores are attached to the spindle. SAC proteins control the onset of anaphase I through interactions with one another, the kinetochores of unaligned chromosomes, and Cdc20, which is a protein that mediates APC/C proteolysis. (Kinetochores are the sites of attachment of centromeres of the chromosomes to the microtubules of the spindle. Homologous chromosomes migrate to opposite poles through tension exerted by the spindle on the kinetochores.) If chromosome alignment is not complete, the SAC protein would inhibit APC/C to prevent precocious sister chromatid separation, which can cause aneuploidy in the resulting egg and embryo.
Mitotic SAC proteins that also have essential spindle checkpoint functions during maturation of the mouse oocyte include Mad2, BubR1, and Bub1. Interference of the function of these proteins results in accelerated completion of meiosis I, presumably because anaphase occurs prematurely.94 Further, the abrogation of Mad2 expression has specifically been shown to increase the rate of aneuploidy, whereas its overexpression inhibits disjunction of homologous chromosomes.95 Therefore, the expression and function of these cell-cycle regulators are candidates to be investigated for their potential involvement in age-related aneuploidy in human oocytes.
At the end of anaphase I, the first polar body is extruded, which is followed by reaccumulation of cyclin B levels. MPF reactivation is required for the synthesis of c-mos, which leads to activation of MAP kinase kinase and MAP kinase.96–100 This c-mos-mediated MAP kinase activity is critical for the maintenance of metaphase II arrest, as shown by the c-mos−/− mouse model. Unfertilized eggs from c-mos−/− mice fail to arrest at metaphase II, and undergo parthenogenetic activation, during which the second polar body is extruded in the absence of fertilization.101,102
NUCLEAR AND CYTOPLASMIC CHANGES DURING OOCYTE GROWTH
The growing oocyte undergoes nuclear and cytoplasmic changes that pertain to nuclear organization, epigenetic regulation, transcription, translation, ultrastructural morphology, and organelle function. These changes are sometimes referred to as nuclear or cytoplasmic maturation, broadly defined based on their association with oocyte maturation, subsequent fertilization, or embryo development. Many of the morphologic changes of the oocyte have been elegantly described at the cellular and ultrastructural levels.103 Further, expression, translational modification, and function of cell-cycle regulators, also considered indices of cytoplasmic maturation, are discussed in this chapter under “Meiotic Competence and Oocyte Maturation.” In this section, we highlight some biochemical features characteristic of the oocyte around the time of maturation.
Transcriptional Regulation
The growing oocyte is characterized by a high level of transcriptional activity that increases its total RNA content from approximately 0.2 ng in a small oocyte to approximately 0.6 ng in a fully grown oocyte.104 In growing oocytes, gonadotropin stimulation of follicles increases the proportion of transcriptionally inactive oocytes; thus granulosa cells are thought to have an important role in the regulation of transcriptional activity in the growing oocyte.105 The fully grown oocyte undergoes global transcriptional silencing, which is a prerequisite for meiotic resumption, oocyte maturation, and subsequent embryo development.106 Therefore, messenger RNAs (mRNAs) that are synthesized during oocyte growth are not only utilized for the translation of proteins during the growth phase, but are also stored for protein synthesis in the fully grown stage, during oocyte maturation, or later in the early embryo. Thus, precise control of translation is vital to the function and ultimate developmental potential of the oocyte and embryo.
Regulation of Translation
Translation requires modification of the mRNA transcript such as polyadenylation at the 3′ end (addition of a poly-A tail) and cap binding at the 5′ end. Such modifications are controlled by cis and trans elements. For example, the translation and accumulation of cyclin B1 is regulated via polyadenylation and cis-regulatory mechanisms in the 3′ untranslated region (3′UTR) of the cyclin B mRNA transcript.107
Cis elements in the 3′UTR consist of a highly conserved U-rich sequence (CPE) in conjunction with a downstream hexanucleotide cytoplasmic polydenylation element, AAUAAA. Cis elements mediate polyadenylation via binding to the phosphorylated form of the CPE binding protein (CPEB), which is a trans-acting factor specific to the oocyte. The critical function of CPEB in the oocyte was demonstrated in CPEB knockout mice.108 In the absence of CPEB oogenesis arrested at the pachytene stage on 16.5 dpc, presumably because translation of SCPs 1 and 3 did not occur, which led to death and resorption of the oocytes.108,109 However, the requirement of CPEB at this early stage also precluded the function of this protein during oocyte maturation to be ascertained.
Other regulatory mechanisms of translation involve inhibition of translation if the presence of certain proteins would interfere with oocyte maturation. For example, some mRNAs can be stored in the form of ribonuclear particles (RNPs), which do not support translation and prevent the misexpression of certain proteins.110
The importance of this mechanism is shown by the relative abundance of the Y-box protein family member, MSY2, which has an essential role in mediating translational silencing by RNPs and comprises 2% of the total protein content in the fully grown oocyte.111,112 In addition, poly-A-specific ribonuclease (PARN) mediates widespread deadenylation and subsequent degradation of mRNAs encoding for a variety of proteins, including zona pellucida proteins, actin, alpha tubulin, and Cx43, at the time of meiotic resumption.110 In contrast, the polyadenylation of certain cell-cycle proteins required during oocyte maturation (i.e., cyclin B1) is enhanced at meiotic resumption.113
Epigenetic Regulation
Chromosomes in the live mouse oocyte can be viewed using culture media containing Hoescht stain, a chemical that chelates with the minor groove of DNA and emits blue fluorescent light upon UV light absorption. Centromeres and pericentromeric heterochromatin are initially located at the periphery of the nucleus in oocytes in primordial and primary follicles.106 Then during oocyte growth, they are spread within the nucleus, followed by localization to the periphery of the nucleolus. This perinucleolar heterochromatin rim, or karyosphere, appears as a bright rim around the nucleolus and is also known as the surround nucleoli (SN) pattern.73,114
Although the SN pattern is associated with global transcriptional repression and high potential for meiotic competence and embryo development, we now know that chromatin remodeling and transcriptional repression are distinct processes controlled by different mechanisms.106 As in somatic cells, histone deacetylases were also found to be critical for the large-scale chromatin remodeling in the fully grown oocyte because their inhibition resulted in a breakdown of the SN architecture and aberrant configuration of the meiotic chromosomes and spindle.106
Another important epigenetic mechanism that has emerged is control of transcriptional activation and repression via demethylation and methylation, respectively. Whereas the genome of somatic cell precursors undergoes remethylation before gastrulation at 6.5 dpc, the genome of PGC precursors remains demethylated at 12.5 dpc. Then partial methylation occurs at 15.5 dpc and methylation is completed by 18.5 dpc.32 The embryonic genome is inactivated until the two-cell stage in mice (eight-cell stage in humans), when it becomes activated by global demethylation.
These global changes in methylation status are distinct from X-inactivation and genomic imprinting. Briefly, X-inactivation is a complex and random process in which one of the X chromosomes in XX mammalian females is inactivated to ensure an equivalent dosage of X-linked genes in XX females and XY males.115 Initiation of this process is controlled by a locus called X-inactivation center (Xic) at Xq13. This locus contains X-inactive specific transcript (Xist), which encodes a non-coding mRNA that coats the X-chromosome in cis to trigger inactivation115 (see Chapter 5).
Genomic Imprinting
Finally, a more gene-specific mechanism of epigenetic regulation is genomic imprinting, which mediates predesignated, differential silencing of the maternal and paternal alleles.116 During gametogenesis, certain genes on autosomes become hypermethylated, or silenced, based on their parental origin. Once established, these imprints are thought to be protected from genome-wide demethylation.
Imprinting possibly occurs at different time points in oogenesis, spermatogenesis, and embryogenesis for different genes. Congenital syndromes or cancer can result from a failure of the allele from the designated parental origin to be silenced or an effective null mutation based on a heterozygous mutation in the nonimprinted allele. These disease mechanisms and syndromes are further discussed in Chapter 5.
The mechanism of imprinting is an area of research that is particularly relevant to reproductive endocrinology and infertility treatment because of reports associating assisted reproductive technologies (ART) with possible increased risk of imprinting defects.117 In addition, it has been suggested that in the mouse model, oocyte maturation and embryo culture in vitro may interfere with imprinting.118,119 However, because defects in imprinting are rare in the general population and in children conceived through ART, it is difficult to determine the statistical significance in these studies. Consequently, whether there is a true association or a causal link remains an open question.
THE OOCYTE–EMBRYO TRANSITION
Maternal Effect Genes
It is not surprising, therefore, that several proteins encoded by maternal effect genes—oocyte-specific linker histone H1 (H1F00), nucleoplasmin 2 (Npm2), DNA methyltransferase1 oocyte isoform (Dnmt1o)—are involved in mechanisms of epigenetic gene regulation; these processes are initiated in the oocyte, but continue during early embryo development.120–124 All of these maternal effect genes, and potentially ones that have yet to be discovered, serve as critical links to demonstrate how the unfertilized oocyte can affect subsequent embryo development, besides contributing to the embryonic genome.
H1FOO is an oocyte-specific H1 linker histone that associates with chromatin during the GV and maturation stages; this association continues until the late two-cell to four-cell embryo stages when they are gradually replaced by somatic H1. Because somatic H1 linker histones are important in the regulation of chromatin function, the oocyte- and early embryo-specific expression of H1FOO suggests that it may be critical in chromatin remodeling during the oocyte–embryo transition.120,121
In contrast, Npm2, a protein that is present in the nuclei of oocytes and peri-implantation embryos, is proven to have an essential role in chromatin structure. Embryos of Npm2−/−mutant females lack Npm2, and die before implantation because Npm2 is required for the maintenance of normal nucleolar structures. Specifically, heterochromatin and deacetylated histone H3, which are usually localized to the rim of the nucleolus in oocytes and early embryos, are absent.122 Therefore, this mouse model also demonstrates the importance of chromatin regulation and nuclear architecture in early embryo development.
Another maternal effect gene that is important in epigenetic regulation is Dnmt1. The oocyte variant, Dnmt1o, is an isoform of the Dnmt1 protein that is expressed only in the growing oocyte and early embryo, under an oocyte-specific promoter. Embryos derived from Dnmt1o-deficient oocytes are also deficient in Dnmt1o and develop into fetuses that die during gestation.123 These embryos have defective imprinting, as shown by inappropriate methylation at certain gene loci and loss of allele-specific gene expression. The critical function of Dnmt1o is thought to occur at the eight-cell stage, when the protein transiently translocates from its usual location in the cytoplasm to the nucleus.123
Zar1-null mutant females appear to have normal oogenesis and fertilization appears to be initiated, but the two pronuclei remain separated and most embryos arrest at the one-cell stage.125 Further, of the small percentage of embryos lacking Zar1 that survive to the two-cell stage, there is decreased expression of proteins in the transcription-requiring complex, which indicates a defect in embryonic genome activation. Whereas Zar1 is critical for progression to the two-cell stage, Mater, another protein encoded by a maternal effect gene, is expressed in oocytes and preimplantation embryo and is critical for embryonic development beyond the two-cell stage.126 Investigation into the functions of these proteins will contribute to our understanding of mechanisms that control the merging of paternal and maternal genomes, embryonic genome activation, and early embryo development.
Polarity and Cytoplasmic Reorganization
Unlike other model organisms such as the fruit fly, the determination and establishment of cell polarity, or asymmetric distribution of cytoplasmic contents, in the mammalian oocyte and embryo is not well understood. Part of the difficulty is that although polarity in the form of an eccentric GV is observed in oocytes of some mammalian species, such as human, Rhesus monkey, bovine, and porcine, GV oocytes isolated from mouse and rat ovarian follicles do not demonstrate polarity, perhaps due to artificial effects of the in vitro culture system.127
The growing oocyte has one dominant microtubule organizing center (MTOC). The MTOC comprises centrosome, which organizes microtubules in an astral configuration. In mitotic cells, centrosomes are made up of a pair of centrioles, which are cylindrical structures containing nine triplets of microtubules that form the astral fibers required for cell division. However, centrosomes in the oocyte do not have centrioles and are thus called MTOCs. The fully grown, meiotic competent oocyte has multiple MTOCs that mediate migration of the GV from the center to an eccentric, cortical position during oocyte maturation. The eccentric position of the GV is thought to be related to or to determine the location of the meiotic spindle, which in turn determines the site of polar body extrusion.127
It is controversial whether the meiosis II cell division further determines the point of fusion between sperm and egg, the position of the first mitotic spindle, and even the spatial organization of the first mitotic division.128 The mos/MAP kinase pathway, which regulates the metaphase II arrest, is also required for transitioning the meiotic spindle from a cortical location back to the central location of a mitotic spindle.129 Therefore, determinants of oocyte polarity likely have as-yet unrecognized impact on the development and survival of the early embryo. Indeed, the cytoplasm undergoes drastic reorganization from the asymmetric oocyte and egg, to become an embryo that is capable of symmetric cell divisions.
APOPTOSIS: THE ULTIMATE CELL FATE FOR MOST OOCYTES
The first wave of apoptosis in the mouse model is thought to occur when germline cysts containing oogonia break down, and a similar process likely occurs in the human female fetus. The number of oocytes reaches a peak of approximately 7 million by midgestation in humans.32,130–133 Subsequently, oocytes that do not become encapsulated by granulosa cells undergo apoptosis. Further, growing follicles undergo atresia as they reach the antral stage, so that there are approximately 300,000 to 400,000 follicles at birth and about 200,000 follicles remaining by puberty.132,134,135
Environmental Factors
In addition to apoptosis that is part of “normal” development, environmental factors and genetic susceptibility can exacerbate depletion of the oocyte pool, which is often manifested clinically as premature ovarian failure or early menopause. Environmental factors that have demonstrated oocyte toxicity include organochlorine chemicals from pesticides and industrial processes,136 polycyclic aromatic hydrocarbons from tobacco smoke or fuel combustion,137,138 and chemotherapeutic agents (i.e., doxorubicin) that activate the apoptotic pathway via ceramide, a lipid that is generated by acid sphingomyelinase.139 The harmful effect of ceramide is further supported by the ability of its metabolite and antagonist, sphingosine-1-phosphate, to confer protection in preventing oocytes from undergoing apoptosis when exposed to chemotherapy.140,141
Genetic Factors
As in somatic cells, appropriate expression of pro-apoptotic proteins and those protecting against apoptosis is required to maintain the “normal” rate of attrition. Bcl-2 and Bcl-x proteins are expressed in the oocyte and granulosa cells and “protect” the oocyte from apoptosis. Although Bcl-2-deficient mice are fertile with normal litter size, there is a decrease in the number of primordial follicles as well as an increase in the number of abnormal primordial follicles that contain no oocyte or a dying oocyte.142 Similarly, Bcl-x hypomorphic mutant mice have significantly decreased numbers of primordial and primary follicles, but their phenotype is more severe and fertility is impaired.143 Therefore, these two genes are required for the maintenance of both the oocyte pool and follicular architecture. In contrast, targeted overexpression of Bcl-2 in granulosa cells results in enhanced folliculogenesis, larger litter size, and an increased incidence of germ cell tumors with age.144
Interestingly, female mice that are deficient in Bax are fertile and have significantly more primordial follicles than wild type mice even with advancing age. Further, they are able to superovulate in response to exogenous gonadotropins even at an advanced age, and these oocytes develop into viable embryos.145 Similar results were seen in the mouse model in which the pro-apoptotic gene caspase-2 is deleted. In addition, in the absence of caspase-2, oocytes do not undergo drug-induced apoptosis on exposure to the chemotherapeutic agent doxorubicin.146
CLINICAL RELEVANCE AND FUTURE DIRECTIONS
Many key challenges in clinical reproductive medicine today relate to the availability and function of human oocytes and the follicles that enclose them. However, we are very far from understanding what proportion of “unexplained infertility” is due to an oocyte-specific factor and even further from having any oocyte-specific treatment.147,148 Significant advances in our management of oocyte-related infertility will require a better understanding of the mechanisms through which maternal age, environmental effects, and patient-specific genetic susceptibility impact on oocyte function.
Premature Ovarian Failure
Of all oocyte- or ovarian follicle-related conditions, identification of human gene mutations has thus far largely focused on familial premature ovarian failure. Using cytogenetic and fluorescent in situ hybridization techniques, familial cases of premature ovarian failure associated with balanced translocation could be studied to identify candidate genes that are disrupted by the translocation breakpoint. For example, many autosomal dominant mutations in FOXL2, a transcription factor that is expressed only in granulosa and eyelid cells, have been identified as the cause of a syndrome of premature ovarian failure and eyelid malformation called blepharophimosis ptosis epicanthus inversus syndrome.149–153
Another genetic mutation that has been linked to familial hypergonadotropic gonadal failure in humans is localized to the X-linked gene BMP15.45,154 Further, the “critical region” on the long arm of the X chromosome (Xq13-Xq26) is proposed to contain several genes that are required for oogenesis or ovary development. A familial case of premature ovarian failure has been linked to a gene located in this critical region, DIA, which is the human homolog of the Drosophila diaphanous gene that is critical for oogenesis in Drosophila.155 Because mouse homologs of FOXL2 and BMP15, as well as the D. diaphanous gene, have critical functions in oogenesis or folliculogenesis, these examples underscore the value of connecting what we learn from animal models and scenarios encountered in the clinic.
Familial hypergonadotropic ovarian dysgenesis has also been identified in patients with autosomal recessive, inactivating point mutations in the FSHR gene,156–158 which presumably abrogated follicular response to FSH, resulting in atresia and premature ovarian failure. Conversely, there are also reports of various FSHR mutations that increase the sensitivity of the FSH receptor to FSH, human chorionic gonadotropin (hCG), and thyroid-stimulating hormone (TSH), which led to spontaneous ovarian hyperstimulation syndrome and spontaneous superovulation.159–165
These human gene mutations serve as spontaneous gene targeting models that demonstrate the role of these genes in human reproduction in vivo. Further, biochemical investigations of proteins encoded by these genes may provide insight into the pathogenesis of, and potential therapy for poor ovarian response to COH and the prevention of premature ovarian failure and ovarian hyperstimulation syndrome.
Aneuploidy
Abnormal chromosomal segregation in meiosis results in aneuploidy, which increases in incidence with advancing maternal age in humans. Aneuploidy, considered “the most important problem in human reproduction,” affects an estimated 25% to 50% of human eggs, accounts for at least 35% of spontaneous abortions and approximately 4% of stillbirths, and is the leading genetic cause of congenital mental retardation and developmental disabilities.166–172
The key risk factors for maternally derived aneuploidy in humans are maternal age and susceptible patterns of meiotic recombination, such as recombination sites that are close to the centromere or the telomere (reviewed in Lamb and colleagues173). Compared to the mouse model, there is an extreme variability in the location of recombination nodules in human oocytes,26 which may explain the high rates of aneuploidy in our eggs and embryos. However, although susceptible locations of recombination nodules are thought to be the main risk factor in young women, they do not explain the high aneuploidy rates seen with increasing maternal age.173 In fact, the association between these recombination patterns and aneuploidy decreases with increasing age. Therefore, as-yet undefined recombination-independent factors are proposed to be risk factors for older women.173
IN VITRO FERTILIZATION
Repeated IVF treatment failure in these patients would further identify them as the patient population that may benefit from having a molecular diagnosis and nonempirical treatment that specifically targets the molecular diagnosis. For example, recurrent or familial cases of zona pellucida abnormalities,174 maturation arrest,175–179 parthenogenetic activation,180 triploidy,181 abnormal fertilization,182 abnormal cell division in meiosis II,183 and empty follicle syndrome184,185 have been reported. Indeed, the development of molecular diagnosis and treatment for this subset of patients is a major challenge in reproductive medicine.
FUTURE DIRECTIONS
Outcome-based, systematic surveys of human oocyte and embryo phenotypes encountered in IVF treatment are critical in guiding clinicians and scientists to focus on conditions that are prevalent and relevant in patients seeking infertility treatment. Further, although mammalian oocyte biology research has traditionally been based on the candidate gene approach, complementary experimental strategies by more unbiased, forward genetic methods such as random chemical mutagenesis of the mouse genome followed by screening for infertility phenotype has begun to uncover novel genes that have critical functions in oogenesis.186 Newly identified genes with potentially critical functions in oogenesis can then be further studied by various gene targeting techniques.
Another forward genetic approach is chemical genetics in the form of high throughput small molecule (SM) library screening of processes such as oocyte maturation and follicular development. SM library screening followed by functional experiments have led to the identification of lead compounds in drug discovery and the identification of novel proteins in other areas of research.187–189 Although the technical challenges involved in the high throughput experimentation of the oocyte or follicle may seem quite overwhelming, they may not be truly insurmountable. We can look beyond medicine and biology, and engage bioengineers to bring microfluidics and other microsystems-based technologies to help us accelerate discovery in the field of oocyte biology and infertility.190 Such interdisciplinary effort will be instrumental in giving us the ability to treat or prevent oocyte-related reproductive problems. It will also facilitate the development of oocyte- or fertility-sparing pharmaceuticals and the identification of environmental aneugens to direct preventive strategies to preserve reproductive health.
New findings on functions and human mutations of ovarian genes can be searched online on several databases, including the Ovarian Kaleiodoscope (http://ovary.stanford.edu/) and Online Mendelian Inheritance in Man (OMIM) (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM),191 OMIM and Online Mendelian Inheritance in Man are trademarks of the Johns Hopkins University.
1 Tam PP, Zhou SX. The allocation of epiblast cells to ectodermal and germ-line lineages is influenced by the position of the cells in the gastrulating mouse embryo. Dev Biol. 1996;178:124-132.
2 Matsui Y, Okamura D. Mechanisms of germ-cell specification in mouse embryos. Bioessays. 2005;27:136-143.
3 Molyneaux K, Wylie C. Primordial germ cell migration. Int J Dev Biol. 2004;48:537-544.
4 Yoshimizu T, Sugiyama N, De Felice M, et al. Germline-specific expression of the Oct-4/green fluorescent protein (GFP) transgene in mice. Dev Growth Differ. 1999;41:675-684.
5 Lawson KA, Dunn NR, Bernard AJ, et al. Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 1999;13:424-436.
6 Ying Y, Liu X, Marble A, et al. Requirement of Bmp8b for the generation of primordial germ cells in the mouse. Mol Endocrinol. 2000;14:1053-1063.
7 Fujiwara T, Dunn NR, Hogan BL. Bone morphogenetic protein 4 in the extraembryonic mesoderm is required for allantois development and the localization and survival of primordial germ cells in the mouse. Proc Natl Acad Sci USA. 2001;98:13739-13744.
8 Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685-700.
9 Massague J, Chen YG. Controlling TGF-β signaling. Genes Dev. 2000;14:627-644.
10 Chang H, Matzuk MM. Smad5 is required for mouse primordial germ cell development. Mech Dev. 2001;104:61-67.
11 Tremblay KD, Dunn NR, Robertson EJ. Mouse embryos lacking Smad1 signals display defects in extra-embryonic tissues and germ cell formation. Development. 2001;128:3609-3621.
12 Cairns LA, Moroni E, Levantini E, et al. Kit regulatory elements required for expression in developing hematopoietic and germ cell lineages. Blood. 2003;102:3954-3962.
13 Godin I, Deed R, Cooke J, et al. Effects of the steel gene product on mouse primordial germ cells in culture. Nature. 1991;352:807-809.
14 Poole TW, Silvers WK. Capacity of adult steel (SI/SId) and dominant spotting (W/Wv) mouse skin to support melanogenesis. Dev Biol. 1979;72:398-400.
15 Anderson R, Fassler R, Georges-Labouesse E, et al. Mouse primordial germ cells lacking β1 integrins enter the germline but fail to migrate normally to the gonads. Development. 1999;126:1655-1664.
16 Takeuchi A, Mishina Y, Miyaishi O, et al. Heterozygosity with respect to Zfp148 causes complete loss of fetal germ cells during mouse embryogenesis. Nat Genet. 2003;33:172-176.
17 Pepling ME, Spradling AC. Mouse ovarian germ cell cysts undergo programmed breakdown to form primordial follicles. Dev Biol. 2001;234:339-351.
18 Page SL, Hawley RS. The genetics and molecular biology of the synaptonemal complex. Annu Rev Cell Dev Biol. 2004;20:525-558.
19 Wei K, Kucherlapati R, Edelmann W. Mouse models for human DNA mismatch-repair gene defects. Trends Mol Med. 2002;8:346-353.
20 de Vries SS, Baart E, Dekker M, et al. Mouse MutS-like protein Msh5 is required for proper chromosome synapsis in male and female meiosis. Genes Dev. 1999;13:523-531.
21 Edelmann W, Cohen P, Kane M, et al. Meiotic pachytene arrest in MLH1-deficient mice. Cell. 1996;85:1125-1134.
22 Kneitz B, Cohen P, Avdievichet E, et al. MutS homolog 4 localization to meiotic chromosomes is required for chromosome pairing during meiosis in male and female mice. Genes Dev. 2000;14:1085-1097.
23 Lipkin SM, Moens PB, Wang V, et al. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat Genet. 2002;31:385-390.
24 Edelmann W, Cohen PE, Kneitz B, et al. Mammalian MutS homologue 5 is required for chromosome pairing in meiosis. Nat Genet. 1999;21:123-127.
25 Woods LM, Hodges C, Baart E, et al. Chromosomal influence on meiotic spindle assembly: Abnormal meiosis I in female Mlh1 mutant mice. J Cell Biol. 1999;145:1395-1406.
26 Lenzi ML, Smith J, Snowden T, et al. Extreme heterogeneity in the molecular events leading to the establishment of chiasmata during meiosis i in human oocytes. Am J Hum Genet. 2005;76:112-127.
27 Paquis-Flucklinger V, Santucci-Darmanin S, Paul R, et al. Cloning and expression analysis of a meiosis-specific MutS homolog: The human MSH4 gene. Genomics. 1997;44:188-194.
28 Di Giacomo M, Barchi M, Baudatet F, et al. Distinct DNA-damage-dependent and -independent responses drive the loss of oocytes in recombination-defective mouse mutants. Proc Natl Acad Sci USA. 2005;102:737-742.
29 Ortega S, Prieto I, Odajima J, et al. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet. 2003;35:25-31.
30 Yuan L, Liu J, Hoja M, et al. Female germ cell aneuploidy and embryo death in mice lacking the meiosis-specific protein SCP3. Science. 2002;296:1115-1118.
31 Morgan DO. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261-291.
32 Matova N, Cooley L. Comparative aspects of animal oogenesis. Dev Biol. 2001;2:291-320.
33 Soyal SM, Amleh A, Dean J. FIGα, a germ cell-specific transcription factor required for ovarian follicle formation. Development. 2000;127:4645-4654.
34 Rajkovic A, Pangas S, Ballow D, et al. NOBOX deficiency disrupts early folliculogenesis and oocyte-specific gene expression. Science. 2004;305:1157-1159.
35 Rankin T, Soyal S, Dean J. The mouse zona pellucida: Folliculogenesis, fertility and pre-implantation development. Mol Cell Endocrinol. 2000;163:21-25.
36 Rankin T, Talbot P, Lee E, Dean J. Abnormal zonae pellucidae in mice lacking ZP1 result in early embryonic loss. Development. 1999;126:3847-3855.
37 Mazerbourg S, Hsueh AJ. Growth differentiation factor-9 signaling in the ovary. Mol Cell Endocrinol. 2003;202:31-36.
38 Lin SY, Morrison JR, Phillips DJ, den Kretser DM. Regulation of ovarian function by the TGF-β superfamily and follistatin. Reproduction. 2003;126:133-148.
39 Fitzpatrick SL, Sindoni DM, Shugyrue PJ, et al. Expression of growth differentiation factor-9 messenger ribonucleic acid in ovarian and nonovarian rodent and human tissues. Endocrinology. 1998;139:2571-2578.
40 Elvin JA, Yan C, Matzuk MM. Oocyte-expressed TGF-β superfamily members in female fertility. Mol Cell Endocrinol. 2000;159:1-5.
41 McGrath SA, Esquela AF, Lee SJ. Oocyte-specific expression of growth/differentiation factor-9. Mol Endocrinol. 1995;9:131-136.
42 Dong J, Albertini D, Nishimori K, et al. Growth differentiation factor-9 is required during early ovarian folliculogenesis. Nature. 1996;383:531-535.
43 Elvin JA, Yan C, Wang P, et al. Molecular characterization of the follicle defects in the growth differentiation factor 9-deficient ovary. Mol Endocrinol. 1999;13:1018-1034.
44 Yan C, Wang P, DeMayo J, et al. Synergistic roles of bone morphogenetic protein 15 and growth differentiation factor 9 in ovarian function. Mol Endocrinol. 2001;15:854-866.
45 Galloway SM, Gregan SM, Wilson T, et al. Bmp15 mutations and ovarian function. Mol Cell Endocrinol. 2002;191:15-18.
46 Su YQ, Wu X, O’Brien M, et al. Synergistic roles of BMP15 and GDF9 in the development and function of the oocyte–cumulus cell complex in mice: Genetic evidence for an oocyte–granulosa cell regulatory loop. Dev Biol. 2004;276:64-73.
47 McGee EA, Hsueh AJ. Initial and cyclic recruitment of ovarian follicles. Endocr Rev. 2000;21:200-214.
48 Rey R, Lukas-Croisier C, Lasala C, Bedecarrás P. AMH/MIS: What we know already about the gene, the protein and its regulation. Mol Cell Endocrinol. 2003;211:21-31.
49 Durlinger AL, Visser JA, Themmen AP. Regulation of ovarian function: The role of anti-Müllerian hormone. Reproduction. 2002;124:601-609.
50 Simoni M, Nieschlag E, Gromoll J. Isoforms and single nucleotide polymorphisms of the FSH receptor gene: Implications for human reproduction. Hum Reprod Update. 2002;8:413-421.
51 Kumar TR, Wang Y, Lu N, Matzuk M. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. 1997;15:201-204.
52 Balla A, Danilovich N, Yang Y, Sairam MR. Dynamics of ovarian development in the FORKO immature mouse: Structural and functional implications for ovarian reserve. Biol Reprod. 2003;69:1281-1293.
53 Lei ZM, Mishra S, Zou W, et al. Targeted disruption of luteinizing hormone/human chorionic gonadotropin receptor gene. Mol Endocrinol. 2001;15:184-200.
54 Burns KH, Yan C, Kumar TR, Matzuk M. Analysis of ovarian gene expression in follicle-stimulating hormone β knockout mice. Endocrinology. 2001;142:2742-2751.
55 Dupont S, Krust A, Gansmuller A, et al. Effect of single and compound knockouts of estrogen receptors α (ERα) and β (ERβ) on mouse reproductive phenotypes. Development. 2000;127:4277-4291.
56 Sicinski P, Donaher JL, Geng Y, et al. Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Nature. 1996;384:470-474.
57 Park JY, Su YQ, Ariga M, et al. EGF-like growth factors as mediators of LH action in the ovulatory follicle. Science. 2004;303:682-684.
58 Richards JS, Russell D, Ochsner S, Espey L. Ovulation: New dimensions and new regulators of the inflammatory-like response. Annu Rev Physiol. 2002;64:69-92.
59 Davis BJ, Lennard D, Lee C, et al. Anovulation in cyclooxygenase-2-deficient mice is restored by prostaglandin E2 and interleukin-1β. Endocrinology. 1999;140:2685-2695.
60 Hizaki H, Segi E, Sugimoto Y, et al. Abortive expansion of the cumulus and impaired fertility in mice lacking the prostaglandin E receptor subtype EP2. Proc Natl Acad Sci USA. 1999;96:10501-10506.
61 Kidder GM, Mhawi AA. Gap junctions and ovarian folliculogenesis. Reproduction. 2002;123:613-620.
62 Simon AM, Goodenough D, Li E, Paul D. Female infertility in mice lacking connexin 37. Nature. 1997;385:525-529.
63 Juneja SC, Barr K, Enders G, Kidder G. Defects in the germ line and gonads of mice lacking connexin43. Biol Reprod. 1999;60:1263-1270.
64 Conti M, Andersen C, Richard F, et al. Role of cyclic nucleotide signaling in oocyte maturation. Mol Cell Endocrinol. 2002;187:153-159.
65 Mehlmann LM, Saeki Y, Tanaka S, et al. The Gs-linked receptor GPR3 maintains meiotic arrest in mammalian oocytes. Science. 2004;306:1947-1950.
66 Horner K, Livera G, Hinckley M, et al. Rodent oocytes express an active adenylyl cyclase required for meiotic arrest. Dev Biol. 2003;258:385-396.
67 Kawamura K, Kumagai J, Sudo S, et al. Paracrine regulation of mammalian oocyte maturation and male germ cell survival. Proc Natl Acad Sci USA. 2004;101:7323-7328.
68 Masciarelli S, Horner K, Liu C, et al. Cyclic nucleotide phosphodiesterase 3A-deficient mice as a model of female infertility. J Clin Invest. 2004;114:196-205.
69 Gautier J, Minshull J, Lohka M, et al. Cyclin is a component of maturation-promoting factor from Xenopus.. Cell. 1990;60:487-494. Available at http://www.cell.com/content/article/abstract?uid=PII009286749090599A-aff4#aff4.
70 Lincoln AJ, Wickramasinghe D, Stein P, et al. Cdc25b phosphatase is required for resumption of meiosis during oocyte maturation. Nat Genet. 2002;30:446-449.
71 Mitra J, Schultz RM. Regulation of the acquisition of meiotic competence in the mouse: Changes in the subcellular localization of cdc2, cyclin B1, cdc25C and wee1, and in the concentration of these proteins and their transcripts. J Cell Sci. 1996;109:2407-2415.
72 Bouniol-Baly C, Hamraoui L, Guibert J, et al. Differential transcriptional activity associated with chromatin configuration in fully grown mouse germinal vesicle oocytes. Biol Reprod. 1999;60:580-587.
73 Mattson BA, Albertini DF. Oogenesis: Chromatin and microtubule dynamics during meiotic prophase. Mol Reprod Dev. 1990;25:374-383.
74 Verlhac MH, Kubiak JZ, Clarke HJ, Maro B. Microtubule and chromatin behavior follow MAP kinase activity but not MPF activity during meiosis in mouse oocytes. Development. 1994;120:1017-1025.
75 Choi T, Aoki F, Mori M, et al. Activation of p34cdc2 protein kinase activity in meiotic and mitotic cell cycles in mouse oocytes and embryos. Development. 1991;113:789-795.
76 Hampl A, Eppig JJ. Analysis of the mechanism(s) of metaphase I arrest in maturing mouse oocytes. Development. 1995;121:925-933.
77 Viveiros MM, Hirao Y, Eppig JJ. Evidence that protein kinase C (PKC) participates in the meiosis I to meiosis II transition in mouse oocytes. Dev Biol. 2001;235:330-342.
78 Viveiros MM, O’Brien M, Eppig JJ. Protein kinase C activity regulates the onset of anaphase I in mouse oocytes. Biol Reprod. 2004;71:1525-1532.
79 Spruck CH, de Miguel M, Smith A, et al. Requirement of Cks2 for the first metaphase/anaphase transition of mammalian meiosis. Science. 2003;300:647-650.
80 Holloway SL, Glotzer M, King RW, Murray AW. Anaphase is initiated by proteolysis rather than by the inactivation of maturation-promoting factor. Cell. 1993;73:1393-1402.
81 Ledan E, Polanski Z, Terret ME, Maro B. Meiotic maturation of the mouse oocyte requires an equilibrium between cyclin B synthesis and degradation. Dev Biol. 2001;232:400-413.
82 Peters JM. The anaphase-promoting complex: Proteolysis in mitosis and beyond. Mol Cell. 2002;9:931-943.
83 Winston NJ. Stability of cyclin B protein during meiotic maturation and the first mitotic cell division in mouse oocytes. Biol Cell. 1997;89:211-219.
84 Josefsberg LB-Y, Galiani D, Dantes A, et al. The proteasome is involved in the first metaphase-to-anaphase transition of meiosis in rat oocytes. Biol Reprod. 2000;62:1270-1277.
85 Mailhes JB, Hilliard C, Lowery M, London SN. MG-132, an inhibitor of proteasomes and calpains, induced inhibition of oocyte maturation and aneuploidy in mouse oocytes. Cell Chromosome. 2002;1:2.
86 Anger M, Stein P, Schultz RM. CDC6 requirement for spindle formation during maturation of mouse oocytes. Biol Reprod. 2005;72:188-194.
87 Michaelis C, Ciosk R, Nasmyth K. Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91:35-45.
88 Sumara I, Vorlaufer E, Gieffers C, et al. Characterization of vetebrate cohesion complexes and their regulation in prophase. J. Cell Biol. 2002;151:749-762.
89 Waizenegger IC, Giminez-Abian JF, Wemic D, Peters JM. Regulation of human separase by secruin binding and autocleavage. Curr Biol. 2000;12:1368-1378.
90 Ciosk R, Zachariae W, Michaelis C, et al. An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell. 1998;93:1067-1076.
91 Nasmyth K. Disseminating the genome: Joining, resolving, and separating sister chromatids during mitosis and meiosis. Annu Rev Genet. 2001;35:673-745.
92 Terret ME, Wassmann K, Waizenegger I, et al. The meiosis I-to-meiosis II transition in mouse oocytes requires separase activity. Curr Biol. 2003;13:1797-1802.
93 Herbert M, Levasseur M, Homer H, et al. Homologue disjunction in mouse oocytes requires proteolysis of securin and cyclin B1. Nat Cell Biol. 2003;5:1023-1025.
94 Tsurumi C, Hoffman S, Geley S, et al. The spindle assembly checkpoint is not essential for CSF arrest of mouse oocytes. J Cell Biol. 2004;167:1037-1050.
95 Homer HA, McDougall A, Levasseur M, et al. Mad2 prevents aneuploidy and premature proteolysis of cyclin B and securin during meiosis I in mouse oocytes. Genes Dev. 2005;19:202-207.
96 Ledan E, Polanski Z, Terret ME, Maro B. Meiotic maturation of the mouse oocyte requires an equilibrium between cyclin B synthesis and degradation. Dev Biol. 2001;232:400-413.
97 Hampl A, Eppig JJ. Analysis of the mechanisms of metaphase I arrest in maturing mouse oocytes. Development. 1995;121:925-933.
98 Winston N. Stability of cyclin B protein during meiotic maturation and the first meiotic cell cycle division in mouse oocyte. Biol Cell. 1997;89:211-219.
99 Gebauer F, Xu W, Cooper GM, Richter JD. Translational control by cytoplasmic polyadynenylation of c-mos mRNA is necessary for oocyte maturation in the mouse. Embo J. 1994;13:5712.
100 Verlhac M-H, Lefebvre C, Kubiak JZ, et al. Mos activates MAP kinase in mouse oocytes through two opposite pathways. Embo J. 2000;19:6065-6074.
101 Colledge WH, Carlton MBL, Udy GB, Evans MJ. Disruption of c-mos causes parthenogenetic development of unfertilized mouse eggs. Nature. 1994;370:65-67.
102 Hashimoto N, Watanabe M, Furuta Y, et al. Parthenogenetic activation of oocytes in c-mos-deficient mice. Nature. 1994;370:68-71.
103 Wassarman PM. Oogenesis. In: Adashi EY, Rosenwaks Z, editors. Reproductive Endocrinology, Surgery, and Technology. Philadelphia: Lippincott-Raven; 1996:342-357.
104 Wassarman PM, Letourneau GE. RNA synthesis in fully-grown mouse oocytes. Nature. 1976;261:73-74.
105 De La Fuente R, Eppig JJ. Transcriptional activity of the mouse oocyte genome: Companion granulosa cells modulate transcription and chromatin remodeling. Dev Biol. 2001;229:224-236.
106 De La Fuente R, Viveiros M, Burns K, et al. Major chromatin remodeling in the germinal vesicle (GV) of mammalian oocytes is dispensable for global transcriptional silencing but required for centromeric heterochromatin function. Dev Biol. 2004;275:447-458.
107 Tay J, Hodgman R, Richter JD. The control of cyclin B1 mRNA translation during mouse oocyte maturation. Dev Biol. 2000;221:1-9.
108 Tay J, Richter JD. Germ cell differentiation and synaptonemal complex formation are disrupted in CPEB knockout mice. Dev Cell. 2001;1:201-213.
109 Tay J, Hodgman R, Sarkissian M, Richter J. Regulated CPEB phosphorylation during meiotic progression suggests a mechanism for temporal control of maternal mRNA translation. Genes Dev. 2003;17:1457-1462.
110 Eichenlaub-Ritter U, Peschke M. Expression in in-vivo and in-vitro growing and maturing oocytes: Focus on regulation of expression at the translational level. Hum Reprod Update. 2002;8:21-41.
111 Yu J, Deng M, Medvedev S, et al. Transgenic RNAi-mediated reduction of MSY2 in mouse oocytes results in reduced fertility. Dev Biol. 2004;268:195-206.
112 Yu J, Hecht NB, Schultz RM. Requirement for RNA-binding activity of MSY2 for cytoplasmic localization and retention in mouse oocytes. Dev Biol. 2003;255:249-262.
113 Tay J, Hodgman R, Richter JD. The control of cyclin B1 mRNA translation during mouse oocyte maturation. Dev Biol. 2000;221:1-9.
114 Longo F, Garagna S, Merico V, et al. Nuclear localization of NORs and centromeres in mouse oocytes during folliculogenesis. Mol Reprod Dev. 2003;66:279-290.
115 Heard E. Recent advances in X-chromosome inactivation. Curr Opin Cell Biol. 2004;16:247-255.
116 Bestor TH. Cytosine methylation and the unequal developmental potentials of the oocyte and sperm genomes. Am J Hum Genet. 1998;62:1269-1273.
117 Niemitz EL, Feinberg AP. Epigenetics and assisted reproductive technology: A call for investigation. Am J Hum Genet. 2004;74:599-609.
118 Doherty AS, Mann M, Tremblay K, et al. Differential effects of culture on imprinted H19 expression in the preimplantation mouse embryo. Biol Reprod. 2000;62(6):1526-1535.
119 Fedoriw AM, Stein P, Svoboda P, et al. Transgenic RNAi reveals essential function for CTCF in H19 gene imprinting. Science. 2004;303:238-240.
120 Gao S, Young G, Parseghian M, et al. Rapid H1 linker histone transitions following fertilization or somatic cell nuclear transfer: Evidence for a uniform developmental program in mice. Dev Biol. 2004;266:62-75.
121 Tanaka M, Kihara M, Meczekalski B, et al. H100: A pre-embryonic H1 linker histone in search of a function. Mol Cell Endocrinol. 2003;202:5-9.
122 Burns KH, Viveiros M, Ren Y, et al. Roles of NPM2 in chromatin and nucleolar organization in oocytes and embryos. Science. 2003;300:633-636.
123 Howell CY, Bestor T, Feng D, et al. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell. 2001;104:829-838.
124 Mertineit C, Yoder J, Taketo T, et al. Sex-specific exons control DNA methyltransferase in mammalian germ cells. Development. 1998;125:889-897.
125 Wu X, Viveiros M, Eppid J, et al. Zygote arrest 1 (Zar1) is a novel maternal-effect gene critical for the oocyte-to-embryo transition. Nat Genet. 2003;33:187-191.
126 Tong ZB, Gold L, Pfeifer K, et al. Mater, a maternal effect gene required for early embryonic development in mice. Nat Genet. 2000;26:267-268.
127 Albertini DF, Barrett SL. The developmental origins of mammalian oocyte polarity. Semin Cell Dev Biol. 2004;15:599-606.
128 Vogel G. Embryologists polarized over early cell fate determination. Science. 2005;308:782-783.
129 Fan HY, Sun QY. Involvement of mitogen-activated protein kinase cascade during oocyte maturation and fertilization in mammals. Biol Reprod. 2004;70:535-547.
130 Tilly JL. Commuting the death sentence: How oocytes strive to survive. Nat Rev Mol Cell Biol. 2001;2:838-848.
131 Tilly JL, Kolesnick RN. Sphingolipids, apoptosis, cancer treatments and the ovary: Investigating a crime against female fertility. Biochim Biophys Acta. 2002;1585:135-138.
132 Forabosco A, Sforza C, De Pol A, et al. Morphometric study of the human neonatal ovary. Anat Rec. 1991;231:201-208.
133 Faddy MJ, Gosden RG, Gougeon A, et al. Accelerated disappearance of ovarian follicles in mid-life: Implications for forecasting menopause. Hum Reprod. 1992;7:1342-1346.
134 Block E. Quantitative morphological investigations of the follicular system in women; Variations at different ages. Acta Anat (Basel). 1952;14:108-123.
135 Block E. A quantitative morphological investigation of the follicular system in newborn female infants. Acta Anat (Basel). 1953;17:201-206.
136 Pocar P, Brevini T, Fischer B, Gandolfi F. The impact of endocrine disruptors on oocyte competence. Reproduction. 2003;125:313-325.
137 Matikainen T, Perez G, Jurisicova A, et al. Aromatic hydrocarbon receptor-driven Bax gene expression is required for premature ovarian failure caused by biohazardous environmental chemicals. Nat Genet. 2001;28:355-360.
138 Matikainen TM, Moriyama T, Morita Y, et al. Ligand activation of the aromatic hydrocarbon receptor transcription factor drives Bax-dependent apoptosis in developing fetal ovarian germ cells. Endocrinology. 2002;143:615-620.
139 Perez GI, Knudson M, Leykin L, et al. Apoptosis-associated signaling pathways are required for chemotherapy-mediated female germ cell destruction. Nat Med. 1997;3:1228-1232.
140 Morita Y, Perez G, Paris F, et al. Oocyte apoptosis is suppressed by disruption of the acid sphingomyelinase gene or by sphingosine-1-phosphate therapy. Nat Med. 2000;6:1109-1114.
141 Paris F, Perez G, Fuks Z, et al. Sphingosine 1-phosphate preserves fertility in irradiated female mice without propagating genomic damage in offspring. Nat Med. 2002;8:901-902.
142 Ratts VS, Flaws J, Kolp R, et al. Ablation of bcl-2 gene expression decreases the numbers of oocytes and primordial follicles established in the post-natal female mouse gonad. Endocrinology. 1995;136:3665-3668.
143 Rucker EB3rd, Dierisseau P, Wagner KU, et al. Bcl-x and Bax regulate mouse primordial germ cell survival and apoptosis during embryogenesis. Mol Endocrinol. 2000;14:1038-1052.
144 Hsu SY, Lai R, Finegold M, Hsueh A. Targeted overexpression of Bcl-2 in ovaries of transgenic mice leads to decreased follicle apoptosis, enhanced folliculogenesis, and increased germ cell tumorigenesis. Endocrinology. 1996;137:4837-4843.
145 Perez GI, Robles R, Knudson CM, et al. Prolongation of ovarian lifespan into advanced chronological age by Bax-deficiency. Nat Genet. 1999;21:200-203.
146 Morita Y, Maravei D, Bergeron L, et al. Caspase-2 deficiency prevents programmed germ cell death resulting from cytokine insufficiency but not meiotic defects caused by loss of ataxia telangiectasia-mutated (Atm) gene function. Cell Death Differ. 2001;8:614-620.
147 Krey LC, Grifo JA. Poor embryo quality: The answer lies (mostly) in the egg. Fertil Steril. 2001;75:466-468.
148 Ezra Y, Simon A, Laufer N. Defective oocytes: A new subgroup of unexplained infertility. Fertil Steril. 1992;58:24-27.
149 Beysen D, Vandesompele J, Messiaen L, et al. The human FOXL2 mutation database. Hum Mutat. 2004;24:189-193.
150 Crisponi L, Deiana A, Loi A, et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet. 2001;27:159-166.
151 De Baere E, Beysen E, Oley C, et al. FOXL2 and BPES: Mutational hotspots, phenotypic variability, and revision of the genotype–phenotype correlation. Am J Hum Genet. 2003;72:478-487.
152 Schmidt D, Ovitt C, Anlag K, et al. The murine winged-helix transcription factor Foxl2 is required for granulosa cell differentiation and ovary maintenance. Development. 2004;131:933-942.
153 Udar N, Yellore V, Chalukya M, et al. Comparative analysis of the FOXL2 gene and characterization of mutations in BPES patients. Hum Mutat. 2003;22:222-228.
154 Di Pasquale E, Beck-Peccoz P, Persani L. Hypergonadotropic ovarian failure associated with an inherited mutation of human bone morphogenetic protein-15 (BMP15) gene. Am J Hum Genet. 2004;75:106-111.
155 Bione S, Sala C, Manzini C, et al. A human homologue of the Drosophila melanogaster diaphanous gene is disrupted in a patient with premature ovarian failure: Evidence for conserved function in oogenesis and implications for human sterility. Am J Hum Genet. 1998;62:533-541.
156 Aittomaki K, Herva R, Stenman U, et al. Clinical features of primary ovarian failure caused by a point mutation in the follicle-stimulating hormone receptor gene. J Clin Endocrinol Metab. 1996;81:3722-3726.
157 Aittomaki K, Dieguez Lucena JL, Pakarinen P, et al. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell. 1995;82:959-968.
158 Doherty E, Pakarinen P, Tiitinen A, et al. A novel mutation in the FSH receptor inhibiting signal transduction and causing primary ovarian failure. J Clin Endocrinol Metab. 2002;87:1151-1155.
159 Delbaere A, Smits G, Olatunbosun O, et al. New insights into the pathophysiology of ovarian hyperstimulation syndrome. What makes the difference between spontaneous and iatrogenic syndrome? Hum Reprod. 2004;19:486-489.
160 Olatunbosun OA, Gilliland B, Brydon LA, Chizen DR. Spontaneous ovarian hyperstimulation syndrome in four consecutive pregnancies. Clin Exp Obstet Gynecol. 1996;23:127-132.
161 Smits G, Olatunbosun O, Delbaere A, et al. Ovarian hyperstimulation syndrome due to a mutation in the follicle-stimulating hormone receptor. NEJM. 2003;349:760-766.
162 Vasseur C, Rodien P, Beau I, et al. A chorionic gonadotropin-sensitive mutation in the follicle-stimulating hormone receptor as a cause of familial gestational spontaneous ovarian hyperstimulation syndrome. NEJM. 2003;349:753-759.
163 Al-Hendy A, Moshynska O, Saxena A, Feyles V. Association between mutations of the follicle-stimulating-hormone receptor and repeated twinning. Lancet. 2000;356:914.
164 Gromoll J, Simoni M. Follicle-stimulating-hormone receptor and twinning. Lancet. 2001;357:230-232.
165 Montgomery GW, Duffy D, Hall J, et al. Mutations in the follicle-stimulating hormone receptor and familial dizygotic twinning. Lancet. 2001;357:773-774.
166 Hassold TJ, Hunt PA. To err (meiotically) is human: The genesis of human aneuploidy. Nat Gen. 2001;2:280-291.
167 Hodges CA, LeMaire-Adkins R, Hunt PA. Coordinating the segregation of sister chromatids during the first meiotic division: Evidence for sexual dimorphism. J C Sci. 2001;114:2417-2426.
168 Hunt PA, Hassold TJ. Sex matters in meiosis. Science. 2002;296:2181-2183.
169 LeMaire-Adkins R, Radke K, Hunt PA. Lack of checkpoint control at the metaphase/anaphase transition: A mechanism of meiotic nondisjunction in mammalian females. J Cell Biol. 1997;139:1611-1619.
170 Hassold T, Chiu D. Maternal age-specific rates of numerical chromosome abnormalities with special reference to trisomy. Hum Genet. 1985;70:11-17.
171 Risch N, Stein Z, Kline J, Warburton D. The relationship between maternal age and chromosome size in autosomal trisomy. Am J Hum Genet. 1986;39:68-78.
172 Morton NE, Jacobs PA, Hassold T, Wu D. Maternal age in trisomy. Ann Hum Genet. 1988;52:227-235.
173 Lamb NE, Yu K, Shaffer J, et al. Association between maternal age and meiotic recombination for trisomy 21. Am J Hum Genet. 2005;76:91-99.
174 Alikani M, Noyes N, Cohen J, Rosenwaks Z. Monozygotic twinning in the human is associated with the zona pellucida architecture. Hum Reprod. 1994;9:1318-1321.
175 Bergere M, Lombroso R, Gombault M, et al. An idiopathic infertility with oocytes metaphase I maturation block: Case report. Hum Reprod. 2001;16:2136-2138.
176 Combelles CM, Albertini DF, Racowsky C. Distinct microtubule and chromatin characteristics of human oocytes after failed in-vivo and in-vitro meiotic maturation. Hum Reprod. 2003;18:2124-2130.
177 Levran D, Farhi J, Nahum H, et al. Maturation arrest of human oocytes as a cause of infertility: Case report. Hum Reprod. 2002;17:1604-1609.
178 Neal MS, Cowan L, Louis JP, et al. Cytogenetic evaluation of human oocytes that failed to complete meiotic maturation in vitro. Fertil Steril. 2002;77:844-845.
179 Schmiady H, Neitzel H. Arrest of human oocytes during meiosis I in two sisters of consanguineous parents: First evidence for an autosomal recessive trait in human infertility: Case report. Hum Reprod. 2002;17:2556-2559.
180 Oliveira FG, Dozortsev D, Diamond M, et al. Evidence of parthenogenetic origin of ovarian teratoma: Case report. Hum Reprod. 2004;19:1867-1870.
181 Pal L, Toth TL, Leykin L, Isaacson KB. High incidence of triploidy in in-vitro fertilized oocytes from a patient with a previous history of recurrent gestational trophoblastic disease. Hum Reprod. 1996;11:1529-1532.
182 van der Westerlaken L, Helmerhorst F, Verburg H, Naaktgeboren N. Successful intracytoplasmic sperm injection after failed in vitro fertilization due to multipronuclear oocytes. Fertil Steril. 2003;80:639-640.
183 Hardarson T, Lundin K, Hanson C. A human oocyte with two sets of MII/PB-structures. Hum Reprod. 2002;17:1892-1894.
184 La Sala GB, Ghirardini G, Cantarelli M, et al. Recurrent empty follicle syndrome. Hum Reprod. 1991;6:651-652.
185 Onalan G, Pabuçcu R, Önalan R, et al. Empty follicle syndrome in two sisters with three cycles: Case report. Hum Reprod. 2003;18:1864-1867.
186 Handel MA, Lessard C, Reinholdt L, et al. Mutagenesis as an unbiased approach to identify contraceptive targets. Mol Cell Endocrinol. 2006;250:201-205.
187 Yarrow JC, Feng Y, Perlman ZE, et al. Phenotypic screening of small molecule libraries by high throughput cell imaging. Comb Chem High Throughput Screen. 2003;6:279-286.
188 Ding S, Wu T, Brinker A, et al. Synthetic small molecules that control stem cell fate. Proc Natl Acad Sci USA. 2003;100:7632-7637.
189 Cheung A, Dantzig JA, Hollingworth S, et al. A small-molecule inhibitor of skeletal muscle myosin II. Nat Cell Biol. 2002;4:83-88.
190 Walker GM, Zeringue HC, Beebe DJ. Microenvironment design considerations for cellular scale studies. Lab Chip. 2004;4:91-97.
191 Pederson T, Peters H. Proposal for a classification of oocytes and follicles in the mouse ovary. J Reprod Fertil. 1968;17:555-557.